 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination chemistry of sterically encumbered pyrrolyl ligands to chromium(II): mono(pyrrolyl)chromium and diazachromocene formation†
Markus
Kreye
,
Constantin G.
Daniliuc
,
Matthias
Freytag
,
Peter G.
Jones
and
Marc D.
Walter
*
Institut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: mwalter@tu-bs.de; Fax: +49 531-391-5309
First published on 30th April 2014
Abstract
A series of diazachromocenes with sterically demanding pyrrolyl ligands, 2,5-(Me3C)2C4H2N (1), 2,5-(Me3C)2-3,4-Me2C4N (2) and 2,3,5-(Me3C)3C4HN (3), was prepared and investigated by various spectroscopic techniques, and in some cases by X-ray diffraction and magnetic susceptibility studies. The diazachromocenes exhibit an S = 1 ground state; no indication of a spin-equilibrium was obtained. With the same ligands mono(pyrrolyl)chromium(II) complexes are accessible, [{(κN-2,5-(Me3C)2C4H2N)Cr(thf)}2(μ-Cl)2] (1-CrCl(thf)), [(κN-2,5-(Me3C)2-3,4-Me2-C4H2N)Cr(Cl)(tmeda)] (2-CrCl(tmeda) and [(η5-2,3,5-(Me3C)3C4HN)Cr(μ-Cl)2] (3-CrCl), which show either η5- or η1-(κN) coordination depending on the substitution pattern. 1H NMR spectroscopy serves as a valuable tool to distinguish between these coordination modes. The Cr(II) atoms in the mono(pyrrolyl) complexes adopt a high spin configuration (S = 2) and in dimeric species antiferromagnetic coupling between the spin carriers was observed. However, none of these mono(pyrrolyl)chromium complexes is an effective or selective ethylene oligomerization catalyst on activation with MAO or AlMe3, supporting the importance of a Cr(I)/Cr(III)-based catalytic cycle.
Introduction
Transition metal complexes bearing heterocyclopentadienyl ligands have been of interest for some time.1–7 An interesting feature associated with these ligands is their ability to participate in η5 → η3 → η1 ring slippage processes. In homogeneous catalysis this isomerization might represent an advantageous feature. In the case of pyrrolyl, the N atom facilitates this switch,8 and pyrrolyl complexes with η5- and η1-(κN) coordination are well documented in the literature.6,7,9,10 However, in many cases the coordination mode is dictated by the Lewis-acidity of the metal atom.Surprisingly, while studies on pyrrolyl chromium(II) and chromium(III) complexes date back to 1966,11,12 structurally well-characterized species were first reported by Gambarotta in 1990 using pyrrolyl and 2,5-dimethylpyrrolyl as ligands, for which η1-(κN)-coordination was observed exclusively.13 Interestingly, chromium pyrrolide catalyst systems generated from Cr[O2CCH(Et)(Bu)]3, 2,5-dimethylpyrrole and AlEt2Cl/AlEt3 exhibit high activity and excellent selectivity in ethylene trimerization to 1-hexene.14,15 The origin of this selectivity, and also the question as to which intermediates are involved in this process, have been the focus of extensive studies.14,15 One important aspect was to establish which chromium species is responsible for the high selectivity of the trimerization, since under the employed reaction conditions catalytic cycles involving Cr(I)/Cr(III) or Cr(II)/Cr(IV) intermediates are feasible. DFT calculations have been undertaken, but could support either possibility.16 However, more recent in situ EPR monitoring of the trimerization process supports the formation of Cr(I) and Cr(II) species, whereby only Cr(I) is catalytically relevant.17 Furthermore, Gambarotta and Duchateau used the more sterically encumbered 2,5-di-tert-butylpyrrolyl for this reaction and prepared the Cr(III) complex [(η5-2,5-(Me3C)2C4H2N)CrCl2(thf)]. On addition of AlEt3 at low temperature the dinuclear Cr(III) species [{(η5-2,5-(Me3C)2C4H2N)CrEt}2(μ-Cl)2] was formed, which acts as a single-component, self-activating precatalyst for ethylene trimerization. The authors also isolated the catalytically inactive dinuclear Cr(II) complex [{(κN-2,5-(Me3C)2C4H2N)Cr(thf)}2(μ-Cl)2] from the reaction of [{(η5-2,5-(Me3C)2C4H2N)CrEt}2(μ-Cl)2] with AlEt3.18 Both observations further strengthen the hypothesis of a Cr(I)/Cr(III) catalytic cycle.
Nevertheless, from a structural point of view the κN-coordination for the latter compound is surprising, since tert-butyl (tBu) groups in 2,5-positions of the pyrrolyl framework generally sterically disfavour η1-coordination but instead η5-coordination is observed.6,7,19,20 Recently, we have initiated a research program on sterically encumbered pyrrolyl systems with an emphasis on comparing them with their cyclopentadienyl analogues, with respect to their ability to stabilize half-sandwich complexes.21–23 Herein, we report our observations on the coordination chemistry of bulky pyrrolyls (Chart 1) with Cr(II) and briefly discuss the reactivity of pyrrolyl chromium(II) half-sandwich complexes in the polymerization/oligomerization of ethylene.
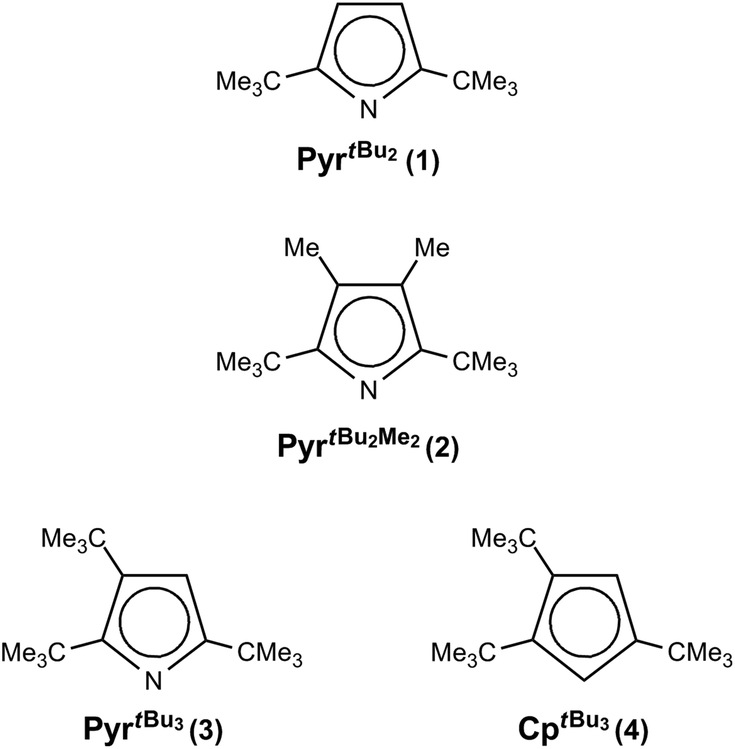 | ||
| Chart 1 | ||
Results and discussion
Synthesis and characterization of diazachromocenes
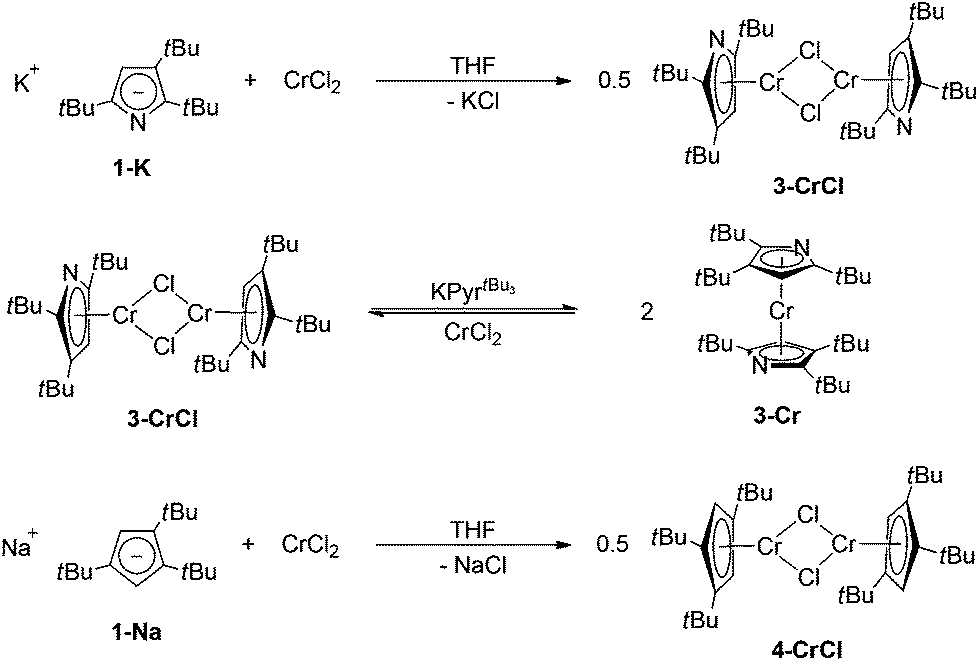
For Cr(II), η5-coordination of pyrrolyl ligands is rarely observed and requires the functionalization of the N-lone pair, e.g., by methylation24 or coordination by Lewis-acids such as AlMe3.25 However, based on our previous investigations on azatrozircenes22 and diazaferrocenes23 we anticipated that the sterically encumbered pyrrolyl ligands 1–3 would behave similarly to their cyclopentadienyl analogues. Indeed, the reaction of pyrrolides 1-Na, 2-K and 3-K with CrCl2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in THF yields the corresponding red bis(pyrrolyl)chromium(II) complexes 1-Cr, 2-Cr and 3-Cr, respectively (Scheme 1).
:1 ratio in THF yields the corresponding red bis(pyrrolyl)chromium(II) complexes 1-Cr, 2-Cr and 3-Cr, respectively (Scheme 1).
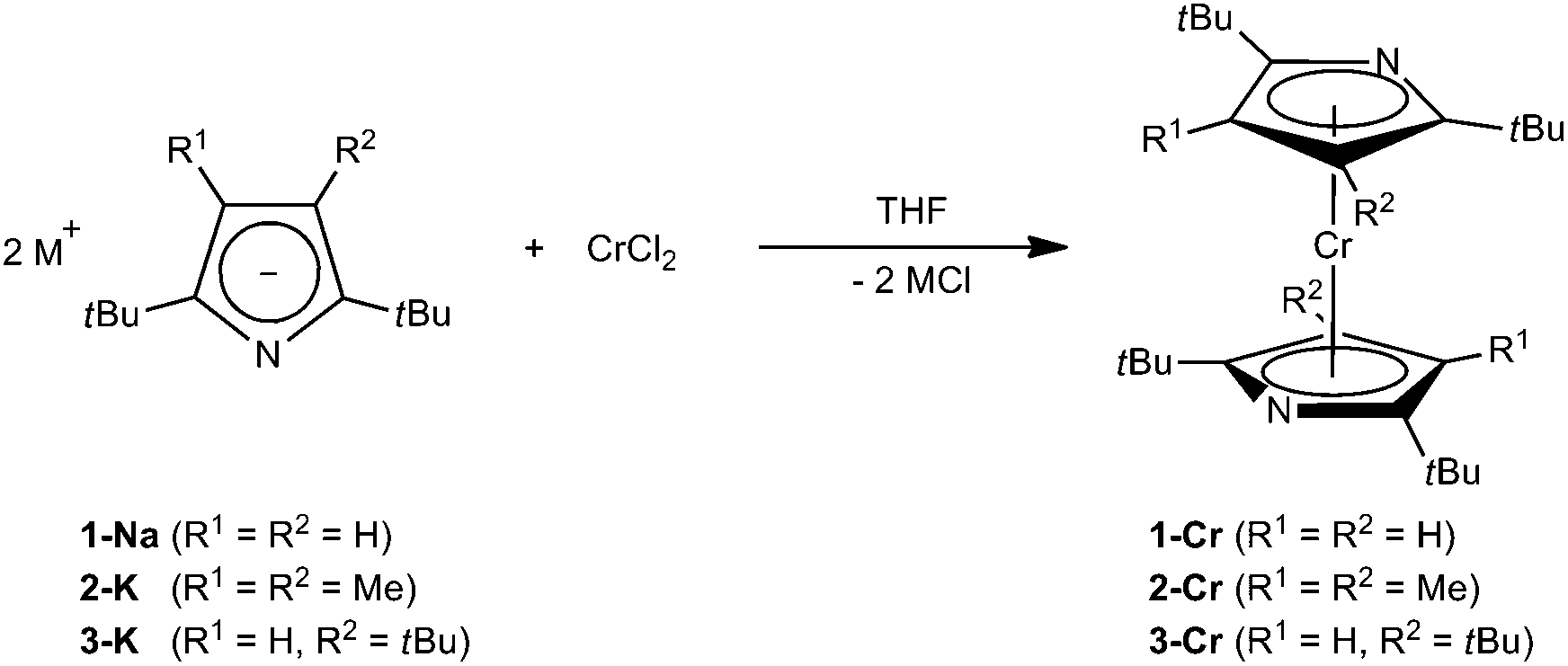 | ||
| Scheme 1 | ||
These complexes are very soluble in aliphatic hydrocarbons, in which they form red solutions, and crystals of 1-Cr and 2-Cr suitable for X-ray diffraction were grown from concentrated pentane or hexane solutions at −24 °C (Table 1).
| Compound reference | 1-Cr | 2-Cr | 1-CrCl(thf) | 2-CrCl(tmeda) | 4-CrCl |
| Chemical formula | C24H40CrN2 | C28H48CrN2 | C32H56Cl2Cr2N2O2 | C20H40ClCrN3 | C34H58Cl2Cr2 |
| Formula mass | 408.58 | 464.68 | 675.69 | 410.00 | 641.70 |
| Crystal system | Tetragonal | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| a/Å | 8.6668(2) | 9.0267(5) | 8.0202(2) | 9.6784(4) | 10.2638(4) |
| b/Å | 8.6668(2) | 32.0929(16) | 15.7066(4) | 17.7933(7) | 12.3091(6) |
| c/Å | 30.8408(12) | 10.4615(6) | 14.2953(4) | 12.8412(5) | 13.8743(6) |
| α/° | 90.00 | 90.00 | 90.00 | 90.00 | 89.431(4) |
| β/° | 90.00 | 114.962(7) | 103.847(4) | 93.221(4) | 88.919(4) |
| γ/° | 90.00 | 90.00 | 90.00 | 90.00 | 89.248(4) |
| Unit cell volume/Å3 | 2316.56(12) | 2747.5(3) | 1748.45(8) | 2207.81(15) | 1752.31(13) |
| Temperature/K | 100(2) | 130(2) | 100(2) | 100(2) | 100(2) |
| Space group | P(−4)21c | P21/c | P21/n | P21/n | P(−1) |
| No. of formula units per unit cell, Z | 4 | 4 | 2 | 4 | 2 |
| Radiation type | Mo Kα | Cu Kα | Mo Kα | Cu Kα | Mo Kα |
| Absorption coefficient, μ/mm−1 | 0.504 | 3.522 | 0.803 | 5.414 | 0.792 |
| No. of reflections measured | 87304 |
59471 |
68193 |
46931 |
49059 |
| No. of independent reflections | 2764 | 5706 | 4169 | 4578 | 7693 |
| R int | 0.0438 | 0.0990 | 0.0477 | 0.0618 | 0.0554 |
| Final R1 values (I > 2σ(I)) | 0.0265 | 0.0587 | 0.0287 | 0.0745 | 0.0302 |
| Final wR(F2) values (I > 2σ(I)) | 0.0658 | 0.1482 | 0.0617 | 0.0732 | 0.0601 |
| Final R1 values (all data) | 0.0279 | 0.0651 | 0.0358 | 0.0355 | 0.0569 |
| Final wR(F2) values (all data) | 0.0663 | 0.1538 | 0.0644 | 0.0770 | 0.0625 |
| Goodness of fit on F2 | 1.131 | 1.044 | 1.046 | 1.042 | 0.838 |
| Δρ/e Å−3 | 0.266/−0.337 | 1.210/−0.707 | 0.340/−0.277 | 0.346/−0.313 | 0.334/−0.296 |
However, because of its high solubility, crystals of 3-Cr had to be grown from a saturated hexamethyldisiloxane solution at −24 °C. The 1H NMR spectra of these complexes feature broad resonances between δ 5 and −1 ppm at ambient temperature. Nevertheless, for 1-Cr and 3-Cr only the tBu resonances were observed, while the methine CH protons are too broad to be detected (see the Experimental section for details). For comparison, in the 1H NMR spectrum of [(η5-1,3-(Me3C)2C5H3)2Cr] the tBu-groups resonate at δ 0.2 (ν1/2 = 250 Hz) and no methine resonances were found.26 Furthermore, the UV/vis spectra of 1-Cr, 2-Cr and 3-Cr resemble each other closely, consistent with the notion that the pyrrolyl ligands 1–3 exhibit similar ligand field strengths (see ESI† for details).
The molecular structures of 1-Cr (which displays crystallographic twofold symmetry) and 2-Cr (which displays approximately twofold symmetry, r.m.s.d. 0.20 Å) are shown in Fig. 1 and 3, and selected bond distances and angles are listed in the figure caption. The electron delocalization within a pyrrolyl system can be readily deduced from the individual CC bond distances, which are intermediate between the distances of isolated single (d0(C–C) = 1.54 Å)27 and double (d0(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) = 1.34 Å)27 bonds. To quantify the extent of delocalization within the heterocyclic ring, the parameter τ28–30 was defined as the normalized quotient of the single and double bond lengths (eqn (1)).
C) = 1.34 Å)27 bonds. To quantify the extent of delocalization within the heterocyclic ring, the parameter τ28–30 was defined as the normalized quotient of the single and double bond lengths (eqn (1)).
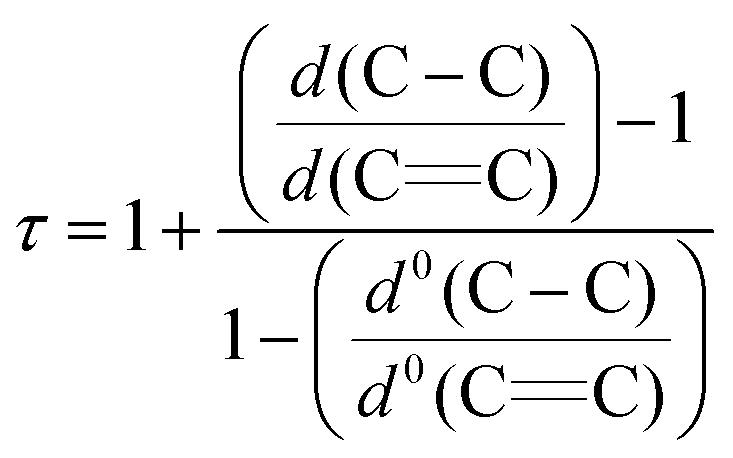 | (1) |
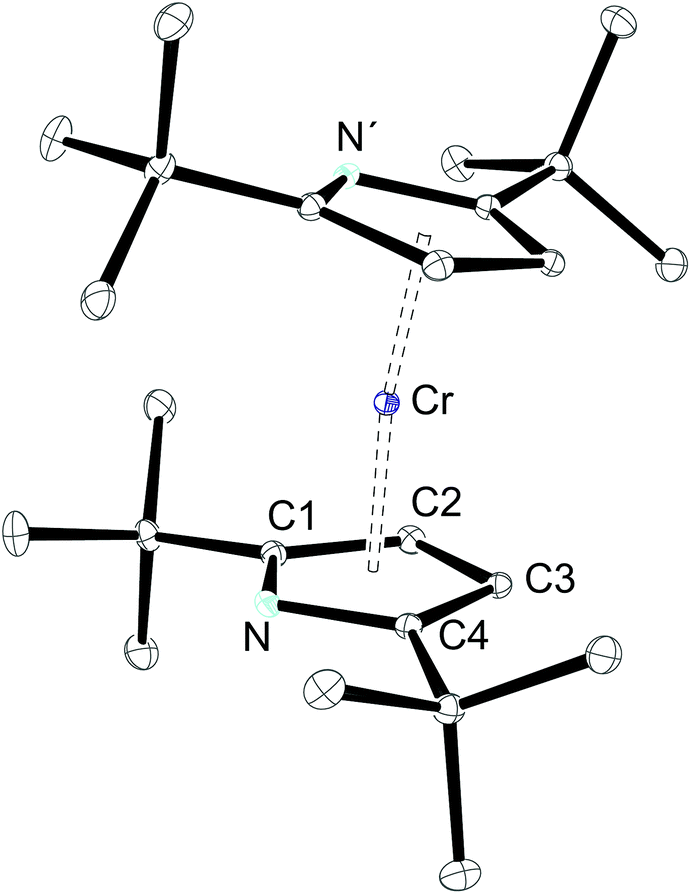 | ||
| Fig. 1 ORTEP diagram of 1-Cr with thermal displacement parameters drawn at 50% probability. The hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): N–C1 1.3769(18), C1–C2 1.4254(19), C2–C3 1.4288(19), C3–C4 1.4208(19), C4–N 1.3874(17), Cr–N 2.1902(12), Cr–C1 2.1945(13), Cr–C2 2.1552(15), Cr–C3 2.1240(14), Cr–C4 2.1411(14), Pyr(cent)–Cr 1.80, Pyr(cent)–Cr–Pyr(cent) 172.3; τ 0.97; N–Pyr(cent)⋯Pyr′(cent)–N′ −85.3°. | ||
A completely delocalized system such as benzene adopts a τ value of 1, while τ approaches 0 for a localized system.28–30 For 1-Cr and 2-Crτ values are 0.97 and 1.00, respectively, suggesting complete electron delocalization. Furthermore, as expected from the NMR spectroscopy, the molecular structure of 1-Cr is closely related to that of the cyclopentadienyl derivative [(η5-1,3-(Me3C)2C5H3)2Cr],26 which shows a Cp(cent)–Cr distance of 1.81 Å. This bond distance is also similar to that (1.80 Å) of [(η5-1,2,4-(Me2CH)3C5H2)2Cr]. Structurally it is also related to the hexaphosphachromocene, [(η5-2,3-(Me3C)2C2P3)2Cr].31 All these system complexes have an S = 1 ground state. For comparison, Cr(II) high spin (S = 2) complexes have significantly longer Cp(cent)–Cr distances, as shown for the mixed-sandwich complex [(η5-C5Me5)Cr(Tp)] (Tp = hydrotris(pyrazolyl)borate) (2.01 Å).32,33
However, because of intramolecular ligand repulsion, bulky Cp ligands can also favour longer metal–carbon bonds associated with the high spin state, e.g., [(η5-1,2,3,4-(Me2CH)4C5H)2Cr] is a low spin (S = 1) molecule in solution at ambient temperature,34 but in the solid state a gradual spin-crossover from the S = 1 to the S = 2 state is observed.35 To exclude this possibility for 1-Cr, the solid state magnetic susceptibility was recorded between 2 and 360 K and the χTvs. T plot is shown in Fig. 2. The magnetic moment of μeff = 3.0μB (χT ≈ 1.12 emu K) is temperature independent and consistent with an S = 1 ground state, and no evidence of spin-crossover behaviour can be gathered.
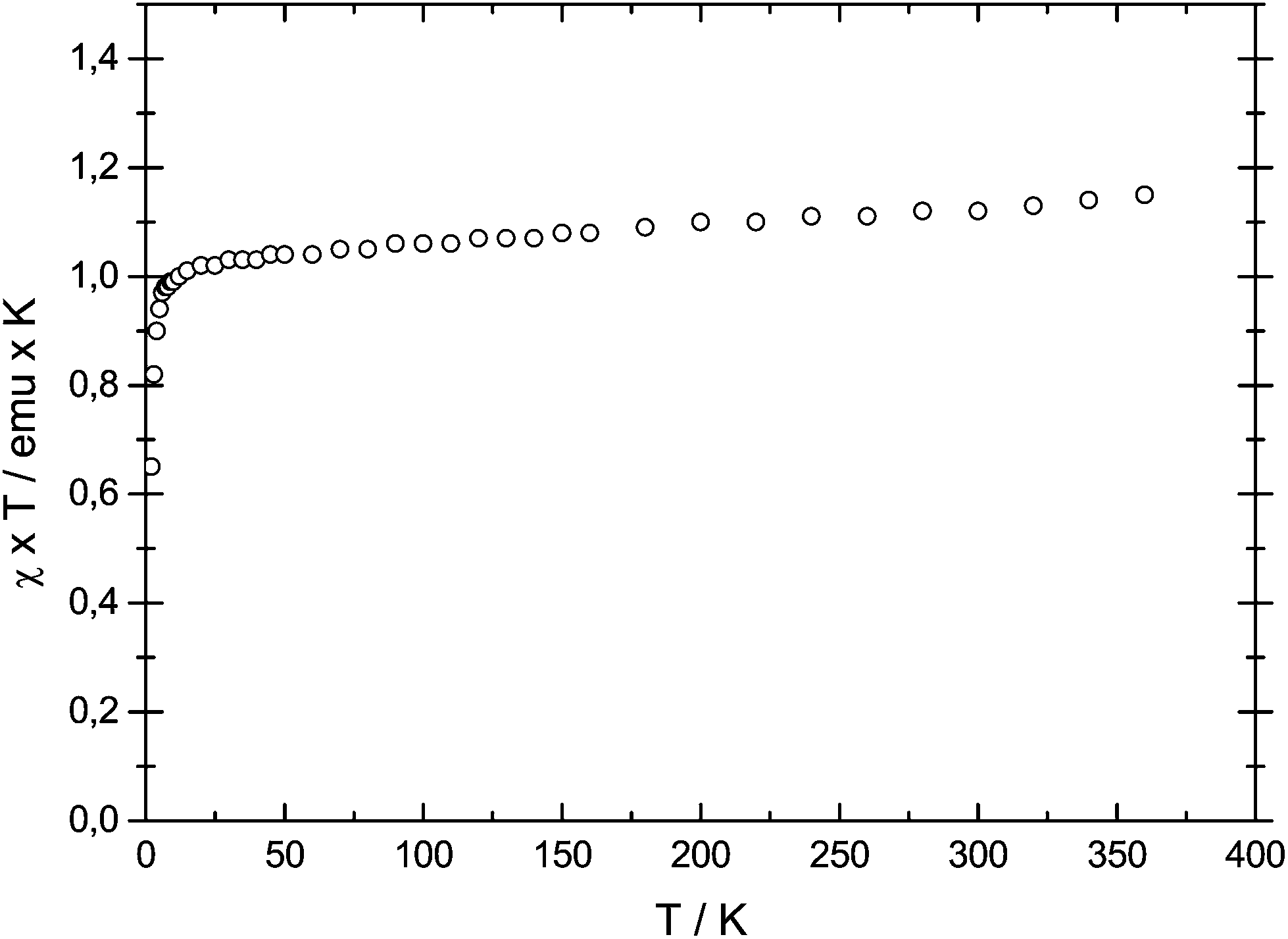 | ||
| Fig. 2 χ T vs. T plot for 1-Cr. | ||
The introduction of the more sterically encumbered pyrrolyl 2 has only minor effects on the overall molecular structure and the Pyr(cent)–Cr distance (1.82 Å) is only slightly elongated (Fig. 3). Nevertheless, a closer inspection also reveals that the N-atom is better shielded in 2-Cr than in 1-Cr, since the additional methyl substituents in the 3,4-positions push the adjacent tert-butyl groups towards the nitrogen atoms, as shown by the average C(tBu)–C–N angles of 119.3° and 120.6° for 2-Cr and 1-Cr, respectively.
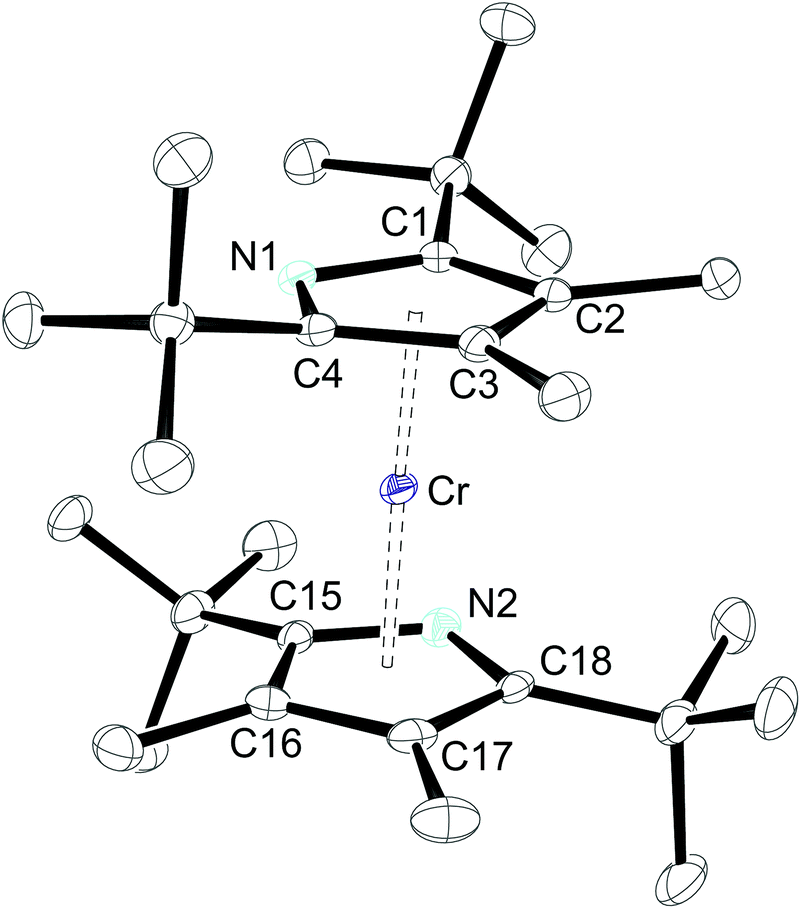 | ||
| Fig. 3 ORTEP diagram of 2-Cr with thermal displacement parameters drawn at 50% probability. The hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): N–C1 1.395(3), C1–C2 1.421(3), C2–C3 1.422(4), C3–C4 1.426(3), C4–N 1.397(3), N2–C15 1.385(3), C15–C16 1.431(4), C16–C17 1.426(4), C17–C18 1.417(4), C18–N2 1.396(3), Cr–N1 2.095(2), Cr–C1 2.165(2), Cr–C2 2.259(2), Cr–C3 2.232(2), Cr–C4 2.132(2), Cr–N2 2.092(2), Cr–C15 2.148(2), Cr–C16 2.258(2), Cr–C17 2.259(2), Cr–C18 2.149(2), Pyr(cent)–Cr 1.82, Pyr(cent)–Cr–Pyr(cent) 174.3; τ 1.00; N1–Pyr1(cent)⋯Pyr2(cent)–N2 99.9°. | ||
X-ray investigations also established the qualitative molecular structure for 3-Cr, but this could not be refined satisfactorily because no unambiguous differentiation between the N- and CH-positions within the pyrrolyl ring was possible.
Synthesis and characterization of mono(pyrrolyl)chromium complexes
The next question to be addressed is the influence of the pyrrolyl substitution pattern on the coordination modes in mono(pyrrolyl)chromium(II) complexes. The reaction of 1-Na with 1 equiv. of CrCl2 in THF results in a blue solution, from which the previously reported complex [{(κN-2,5-(Me3C)2C4H2N)Cr(thf)}2(μ-Cl)2] (1-CrCl(thf))18 can be isolated in good yield. The complex 1-CrCl(thf) is only sparingly soluble in aliphatic and aromatic hydrocarbons, but has a good solubility in THF. Alternatively, 1-CrCl(thf) can be prepared from the reaction of 1-Cr with 1 equiv. of CrCl2 (Scheme 2). | ||
| Scheme 2 | ||
The different coordination modes in 1-CrCl(thf) and 1-Cr are immediately obvious when the 1H NMR spectra of both compounds are compared. Whereas the tBu-groups in 1-Cr are observed at δ 1.99 (ν1/2 = 121 Hz), they experience a dramatic downfield shift to δ 55.4 (ν1/2 = 2580 Hz). This behaviour can be readily understood when one assumes that the paramagnetic chemical shift is dominated by the pseudo-contact term; that is, resonances closest to the paramagnetic centre display the greatest paramagnetic shifts and are also broader than those further away from the paramagnetic centre.36 This also rationalizes the fact that the methine resonances are now observable at δ −28.4 (ν1/2 = 399 Hz). Overall, 1H NMR spectroscopy offers a powerful tool for distinguishing κN- and η5-coordination in these molecules. When 1-CrCl(thf) is exposed to a dynamic vacuum for a prolonged period of time, the colour changes from blue to green and a poorly soluble material was obtained, precluding NMR spectroscopic analysis; but on addition of THF the blue colour is re-established. Unfortunately, at present we can only speculate on the identity of the green material, but the colour change might be related to a switch in the pyrrolyl coordination mode (from κN- to η5-coordination).
Crystals of 1-CrCl(thf) suitable for X-ray diffraction were grown by vapour diffusion of pentane into a concentrated THF solution at ambient temperature. The complex 1-CrCl(thf) crystallized in the monoclinic space group P21/n (Table 1), which is different from the previously reported rhombohedric space group R-3.18 The conformation and orientation of the thf ligands are different in both polymorphs, whereas the bond distance and angles vary only slightly (see below).
An ORTEP diagram is shown in Fig. 4 and selected bond distances and angles are listed in the figure caption. The environment around the Cr2+ atom is square planar (sum of the angles: 360.7(1)°) and the two halves of the dimer are related by an inversion centre located in the middle of the Cr2Cl2-core. For steric reasons the pyrrolyl ligands are twisted out of the Cr2Cl2 plane by 87.7°. The Cr atom lies 0.8 Å outside the plane defined by the pyrrolyl moiety; in the rhombohedric polymorph this displacement is only 0.39 Å.18 Two relatively short non-bonding Cr⋯C distances (2.91 and 2.97 Å for Cr–C6 and Cr–C10, respectively) between one methyl group of the pyrrolyl tBu-groups are observed, providing additional steric protection of the fifth and the sixth coordination site of the Cr atom. The Cr–N distance of 2.0176(11) Å compares well with those of other reported Cr(II) amides.13,37,38 A slightly shorter Cr–N distance (2.0121(19) Å) was found for the other polymorph.18 In contrast to 1-Cr and 2-Cr the τ value of 0.87 for 1-CrCl(thf) suggests a more localized electronic structure, as one would expect from κN-coordination, and it can be compared to that of the unsubstituted pyrrole (τ = 0.830).39 The long Cr⋯Cri distance of 3.49 Å precludes any Cr–Cr bond, and therefore electron exchange between the Cr(II) centres has to occur via the bridging Cl-atoms. Solid state magnetic susceptibility studies revealed that the Cr(II) atoms with a d4 electron configuration adopt a high spin (S = 2) configuration and only a weak antiferromagnetic coupling is observed at low temperature. Fig. 5 shows the experimental data and the fit to the effective spin Hamilitonian Ĥ = −2J(Ŝ1·Ŝ2) (Fig. 5).
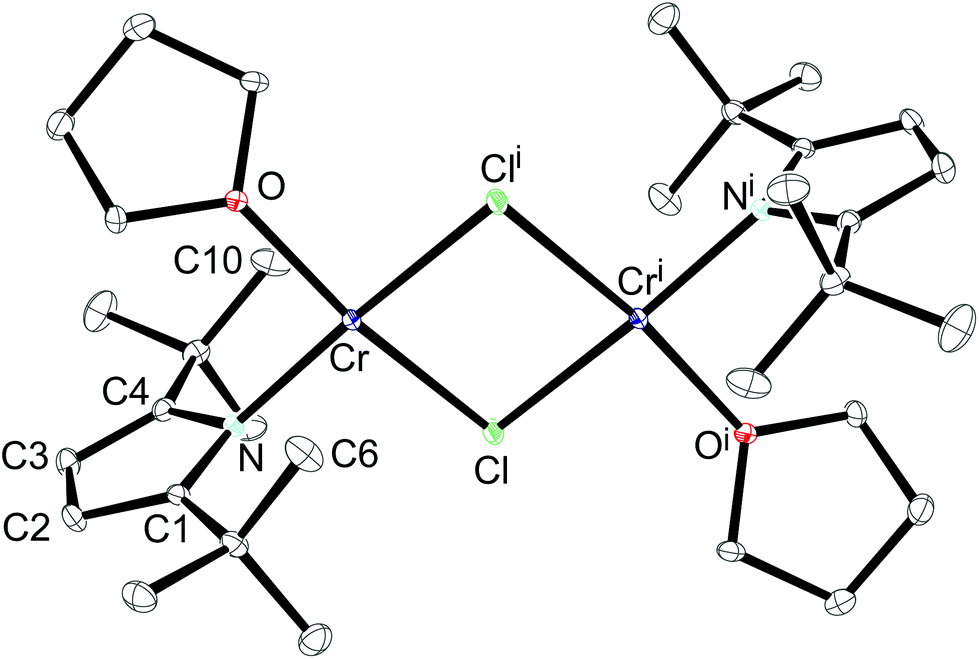 | ||
| Fig. 4 ORTEP diagram of 1-CrCl(thf) with thermal displacement parameters drawn at 50% probability. The hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Cr–N 2.0176(11), Cr–O 2.0733(10), Cr–Cl 2.3902(4), Cr–Cli 2.3961(4), N–C1 1.3909(17), C1–C2 1.3813(19), C2–C3 1.411(2), C3–C4 1.381(2), C4–N 1.3864(17), Cr⋯Cri 3.49, Cr⋯C6 2.91, Cr⋯C10 2.97, O–Cr–Cl 171.93(3), N–Cr–Cli 172.76(4), N–Cr–Cl 93.76(3), Cl–Cr–Cli 86.257(14), O–Cr–Cli 88.55(3), N–Cr–O 92.17(4); τ 0.87. | ||
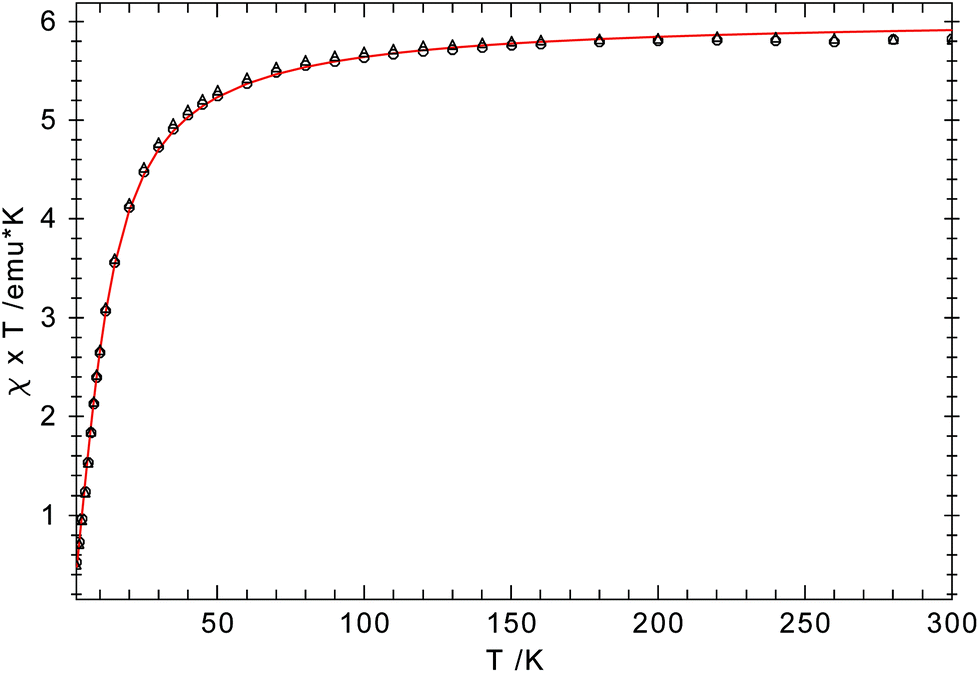 | ||
| Fig. 5 χ T vs. T plot for 1-CrCl(thf). Simulation: S1 = S2 = 2; g1 = g2 = 2.01; J = 1.17 cm−1. | ||
As pointed out above, the addition of two methyl groups at the 3,4-positions of the pyrrolyl systems increases the steric demand around the N-atom and therefore reduces its ability to coordinate in a κN-fashion. When 2-K is treated with 1 equiv. of CrCl2 in THF a steel-blue solution is formed, suggesting the formation of a mono(pyrrolyl) species analogous to 1-CrCl(thf). However, when the solvent is removed, an additional colour change to red-brown is observed and only the diazachromocene 2-Cr can be isolated on pentane extraction. However, on tmeda addition to the reaction mixture the monomeric Cr(II) complex 2-CrCl(tmeda) can be crystallized from a concentrated THF solution at −24 °C (Scheme 3).
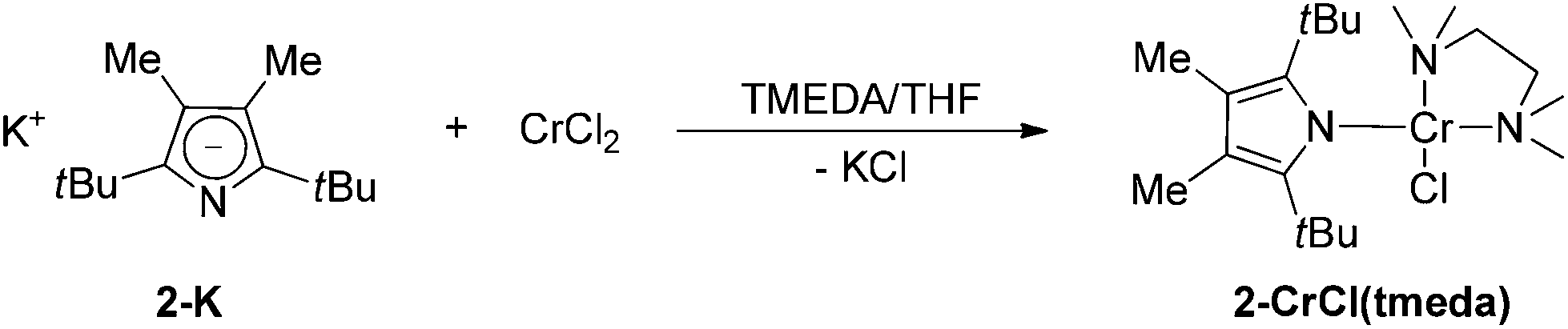 | ||
| Scheme 3 | ||
The 1H NMR spectrum of 2-CrCl(tmeda) exhibits similar features to that of 1-CrCl(thf), confirming the κN-coordination. This assumption was further substantiated by X-ray diffraction. The molecular structure of 2-CrCl(tmeda) is depicted in Fig. 6 and selected bond distances and angles are listed in the figure caption. The Cr-atom is coordinated by one Cl-atom, the bidentate tmeda ligand and pyrrolyl 2 in a square planar geometry. Two short non-bonding Cr⋯C distances (2.87 and 2.81 Å for Cr–C8 and Cr–C14, respectively) provide additional steric protection of the Cr atom, and the Cr–N(pyrrolyl) distance (2.0557(13) Å) is slightly longer than in 1-CrCl(thf).
 | ||
| Fig. 6 ORTEP diagram of 2-CrCl(tmeda) with thermal displacement parameters drawn at 50% probability. The hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Cr–N1 2.0557(13), Cr–N2 2.2142(13), Cr–N3 2.1892(13), Cr–Cl 2.3646(4), N1–C1 1.3880(19), C1–C2 1.395(2), C2–C3 1.422(2), C3–C4 1.391(2), C4–N1 1.3921(19), Cr⋯C8 2.87, Cr⋯C6 2.81, N1–Cr–N2 95.86(5), N3–Cr–Cl 89.68(4), N1–Cr–N3 174.54(5), N2–Cr–N3 81.20(5), N1–Cr–Cl 93.31(4), N2–Cr–Cl 170.83(4), τ 0.86. | ||
Nevertheless, the most sterically encumbered pyrrolyl 3 in this series behaves differently. Deep-blue crystals were isolated from a 1:1 mixture of CrCl2 and KPyrtBu3 in THF (Scheme 4), but the 1H NMR spectrum is distinctly different from those of 1-CrCl(thf) and 2-CrCl(tmeda). Three resonances attributable to the three inequivalent tBu-groups are observed at δ 2.69, 6.68 and 12.17 ppm with similar line-width at half-height (ν1/2) of 573, 533 and 566 Hz, respectively. This suggests that in 3-CrCl the pyrrolyl ligand is too sterically encumbered to allow κN-coordination; instead it shows η5-coordination, as in the diazachromocene 3-Cr. In addition, elemental analysis and EI mass spectrometry confirm the solvent-free, dimeric structure. Complex 3-CrCl can also be prepared from 3-Cr and CrCl2.
 | ||
| Scheme 4 | ||
Whereas X-ray investigations on 3-CrCl confirmed the η5-bonding mode, the N- and CH-positions within the pyrrolyl ring could not be unambiguously assigned during the refinement. To overcome this crystallographic difficulty, we decided to prepare the analogue half-sandwich complex with the 1,2,4-(Me3C)3C5H2 (4) ligand, which we expected to be isostructural to 3-CrCl. The reaction between CrCl2 and NaCptBu3 forms a green THF solution, and after work-up deep blue crystals are isolated from very concentrated pentane solutions at −24 °C (Scheme 4). Elemental analysis and EI MS data agree with the proposed structure for 4-CrCl, but one particular observation for this complex is a solvent dependence of its solution colour and its 1H NMR spectrum (see the Experimental section for details). In pentane and aromatic solvents green solutions are obtained, whereas it forms a blue solution in THF and CH2Cl2. Fig. 7 shows the molecular structure of 4-CrCl, and the figure caption lists some selected bond distances and angles. Key features of the molecular structure include the long Cp(cent)–Cr distances of 1.95 Å and short Cr1⋯Cr2 distance of 3.181 Å, which is in the range of Cr–Cr bond distances (3.185–3.471 Å).40 In principle, the Cr2Cl2-core can adopt two different structures, either a planar arrangement (A) or a butterfly geometry (B) (Chart 2). In the latter, the displacement of the two Cr atoms towards each other suggests a (weak) bonding interaction between the two Cr atoms.
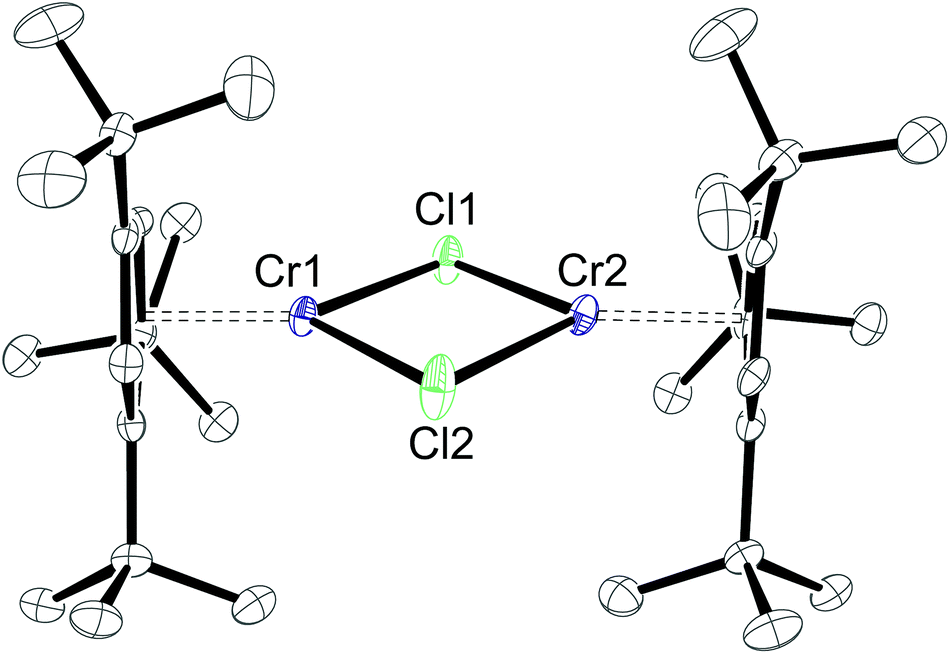 | ||
| Fig. 7 ORTEP diagram of 4-CrCl with thermal displacement parameters drawn at 50% probability. The hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Cr1–Cl1 2.3542(6), Cr1–Cl2 2.3128(5), Cr2–Cl1 2.3093(5), Cr2–Cl2 2.3704(6), Cp(cent)–Cr 1.95, Cr1⋯Cr2 3.181; Cr1–Cl1–Cr2 86.019(18), Cl1–Cr2–Cl2 93.94(2), Cr1–Cl2–Cr2 85.567(19), Cl1–Cr1–Cl2 94.27(2). | ||
 | ||
| Chart 2 | ||
Both arrangements have literature precedents. The butterfly structure is adopted in [(η5-C5Me5)Cr(μ-X)]2 (X = Cl, Me),41 [(η5-C5Me5)Cr(μ-Et)(μ-Ph)]2 (X = Cl, Me),41 [(η5-C5H5)Cr(μ-I)]2,42 and [(η5-C5H5)Cr(μ-OtBu)]243 and has direct consequences for the electronic structure of these dimers, which show strong antiferromagnetic coupling between the two Cr-atoms (S = 1).41 In a more recent report, Layfield and Scheer describe a planar arrangement for [(η5-C5H5)Cr(μ-X)]2 (X = P(SiMe3)2, As(SiMe3)2), in which two high spin (S = 2) Cr(II) centres couple antiferromagnetically.40 To investigate the electronic structure in 4-CrCl, magnetic susceptibility studies were undertaken between 5 and 400 K. The χTvs. T plot is shown in Fig. 8; and χT is close to zero at 5 K and then slowly increases to 2.6 emu K at 400 K. While this value is significantly less than the spin-only value of 6.0 emu K expected for two non-interacting high spin Cr(II) atoms (assuming g = 2), it is clearly higher than the expected value for two non-interacting S = 1 Cr(II) centers (2.0 emu K), suggesting antiferromagnetic coupling. The magnetic data can also be simulated by the spin-Hamiltonian Ĥ = −2J(Ŝ1·Ŝ2) with S1 = S2 = 2; g1 = g2 = 2.41; J = −65.8 cm−1; TIP = 2.189 × 10−4 emu K and ρ = 0.004 (S = 2 impurity). These values are similar to those found for [(η5-C5H5)Cr(μ-As(SiMe3)2)]2.40
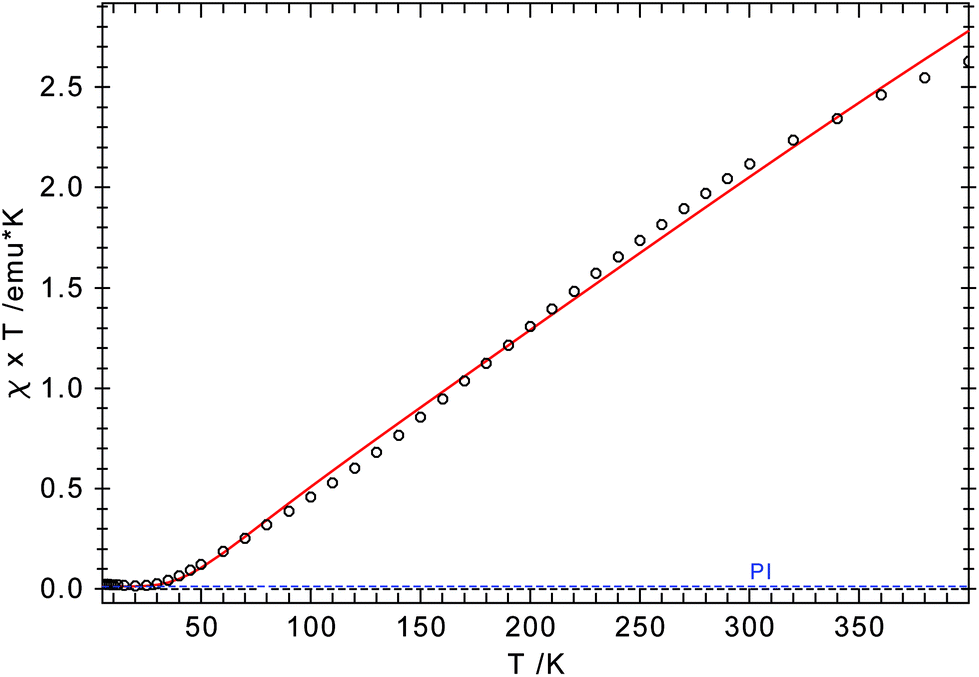 | ||
| Fig. 8 χ T vs. T plot for 4-CrCl. The red line represents a theoretical fit of the experimental data (see text for parameters). | ||
Catalytic screening in ethylene oligo-/polymerisation
With these Cr(II) complexes in hand, the activity of 2-CrCl(tmeda), 3-CrCl and 4-CrCl in ethylene oligomerisation and polymerisation was investigated. This study should also provide insights into the influence of the pyrrolyl coordination mode on the catalytic activity. The complexes were activated by addition of MAO or AlMe3 in a 1:100 and 1:30 ratio, respectively. Furthermore 2 equiv. of dodecyltrimethylammonium chloride was added in the AlMe3 reactions, since an external chloride source has some advantageous effects on the catalytic performance.44 These mixtures were then heated to 50 °C under ethylene (30 bar). As shown in the previous study on 1-CrCl(thf),18 these Cr(II) complexes are only poor catalysts in the selective ethylene polymerization and only trace amounts of oligomers are formed. However, there is a marked difference between the η5-coordinate complexes 3-CrCl and 4-CrCl and the κN-bound complex 2-CrCl(tmeda). The former two complexes exhibit identical catalytic performance in olefin oligomerization (no selectivity and low activity of ca. 2 g h−1) and no polyethylene formation regardless of the activator. In contrast 2-CrCl(tmeda) forms polyethylene (ca. 12 g h−1) on activation with MAO, but on activation with AlMe3 no reaction occurs at 50 °C. However, when the temperature is raised to 75 °C a very slow reaction is initiated with very low ethylene consumption (<2 g h−1) and most of the ethylene is converted to polyethylene. In conclusion, Cr(II) complexes with sterically encumbered pyrrolyl ligands are not suitable for the selective ethylene oligomerization, supporting the previous in situ EPR study, which established that only Cr(I) is relevant for the ethylene trimerization.17
Conclusions
Diazachromocenes with an S = 1 ground state are readily accessible from CrCl2 and sterically encumbered pyrrolyl ligands. No indication of spin equilibrium was found based on solid state magnetic susceptibility studies for 1-Cr. Reaction of the diazachromocenes with an additional equivalent of CrCl2 forms mono(pyrrolyl) complexes, in which the Cr(II) atoms adopt a high spin (S = 2) ground state. In addition while κN-coordination is observed for 1-CrCl(thf) and 2-CrCl(tmeda), this coordination mode is strongly disfavoured for the more sterically demanding PyrtBu3 ligand and therefore η5-coordination is observed in 3-CrCl. To distinguish these coordination modes 1H NMR spectroscopy proved to be a very valuable tool. None of the mono(pyrrolyl)chromium complexes is a very active or selective ethylene oligomerization catalyst after activation with MAO or AlMe3, strongly indicating that Cr(I)/Cr(III) are the important oxidation states in catalysis.Further studies on the coordination chemistry of these pyrrolyl ligands are ongoing and will be reported in due course.
Experimental section
General
All synthetic and spectroscopic manipulations were carried out under an atmosphere of purified nitrogen, either in a Schlenk apparatus or in a glovebox. Solvents were dried and deoxygenated either by distillation under a nitrogen atmosphere from sodium benzophenone ketyl (THF) or by an MBraun GmbH solvent purification system (all other solvents). NMR data were recorded on a Bruker DPX 200, a Bruker DRX 400, a Bruker Avance III 400 or a Bruker Avance II 300 spectrometer at ambient temperature unless stated otherwise. The residual solvent signal was used as a chemical shift reference (δH = 7.16 for benzene, 7.26 for chloroform, 3.58 for α-H of THF) for the 1H spectra and the solvent signal (δC = 128.06 ppm for benzene, 77.17 for chloroform, 67.21 for α-C of THF) for the 13C spectra. Elemental analyses were performed by combustion and gas chromatographic analysis with an Elementar varioMICRO instrument. Magnetic measurements were conducted in a 7 T Quantum Design MPMS magnetometer utilizing a superconducting quantum interference device (SQUID). Between 10 and 25 mg of the sample were sealed in evacuated quartz tubes held in place with ∼5 mg of quartz wool. This method provided a very small and reliable container correction, typically of about −2 × 10−5 emu mol−1. The data were also corrected for the overall diamagnetism of the molecule using Pascal constants.45 For a more detailed description see ref. 46. The program package JulX was used for spin-Hamiltonian simulations and fitting of the data by a full-matrix diagonalization approach.47 NaPyrtBu2 (1-Na),48 KPyrtBu2Me2 (2-K),22 KPyrtBu3 (3-K)22 and NaCptBu3 (4-Na)49 were prepared according to the literature procedures.X-ray diffraction studies
Data were recorded at 100 K on Oxford Diffraction diffractometers using monochromated Mo Kα or mirror-focussed Cu Kα radiation (Table 1). Absorption corrections were performed on the basis of multi-scans. The structures were refined anisotropically using the SHELXL-97 program.50 Hydrogen atoms were included using rigid idealised methyl groups or a riding model. Special features and exceptions: 1-Cr was refined as a racemic twin (Flack parameter 0.17(2)). 2-Cr was measured at 130 K because the crystals shatter at 100 K.Acknowledgements
MDW acknowledges the financial support by the Deutsche Forschungsgemeinschaft (DFG) through the Emmy Noether program (WA 2513/2). We thank Prof. Richard A. Andersen for the access to SQUID and Prof. Uwe Rosenthal and his co-workers at LIKAT, Rostock, for the catalytic screening.Notes and references
- K. H. Pannell, B. L. Kalsotra and C. Párkányi, J. Heterocycl. Chem., 1978, 15, 1057–1081 CAS.
- L. Kollar and G. Keglevich, Chem. Rev., 2010, 110, 4257–4302 CAS.
- A. J. Ashe and S. A. Al-Ahmad, Adv. Organomet. Chem., 1996, 39, 325–353 CAS.
- (a) F. Mathey, Nouv. J. Chim., 1987, 11, 285 Search PubMed; (b) D. Carmichael and F. Mathey, Top. Curr. Chem., 2002, 220, 27–51 CAS, and references cited therein; (c) P. Le Floch, Coord. Chem. Rev., 2006, 250, 627–681 CAS.
- K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus. The carbon copy; from organophosphorus to phospha-organic chemistry, John Wiley, Chichester, 1998 Search PubMed.
- N. Kuhn, Bull. Soc. Chim. Belg., 1990, 99, 707–715 CAS.
- C. Janiak and N. Kuhn, Adv. Nitrogen Heterocycl., 1996, 2, 179–210 CAS.
- D. L. Kershner and F. Basolo, J. Am. Chem. Soc., 1987, 109, 7396–7402 CrossRef CAS.
- R. V. Bynum, H. M. Zhang, W. E. Hunter and J. L. Atwood, Can. J. Chem., 1986, 64, 1254–1257 CrossRef.
- F. Nief, Eur. J. Inorg. Chem., 2001, 891–904 CrossRef CAS.
- D. Tille, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1966, 21, 1239 CAS.
- D. Tille, Z. Anorg. Allg. Chem., 1971, 384, 136–146 CrossRef CAS.
- J. J. H. Edema, S. Gambarotta, A. Meetsma, B. F. Van, A. L. Spek and W. J. J. Smeets, Inorg. Chem., 1990, 29, 2147–2153 CrossRef CAS.
- W. M. Woodard, W. M. Ewert, H. D. Hensley, M. E. Lashier, B. E. Kreischer, G. D. Cowan, J. W. Freeman, R. V. Franklin, R. D. Knudsen, R. L. Anderson and L. R. Kallenbach, Process and catalyst system for the trimerization of olefins, WO9919280A1, 1999 Search PubMed.
- J. T. Dixon, M. J. Green, F. M. Hess and D. H. Morgan, J. Organomet. Chem., 2004, 689, 3641–3668 CrossRef CAS PubMed.
- R. W. J. Van, C. Grove, J. P. Steynberg, K. B. Stark, J. J. Huyser and P. J. Steynberg, Organometallics, 2004, 23, 1207–1222 CrossRef.
- I. Y. Skobelev, V. N. Panchenko, O. Y. Lyakin, K. P. Bryliakov, V. A. Zakharov and E. P. Talsi, Organometallics, 2010, 29, 2943–2950 CrossRef CAS.
- S. Licciulli, K. Albahily, V. Fomitcheva, I. Korobkov, S. Gambarotta and R. Duchateau, Angew. Chem., Int. Ed., 2011, 50, 2346–2349 CrossRef CAS PubMed.
- N. Kuhn, G. Henkel, J. Kreutzberg, S. Stubenrauch and C. Janiak, J. Organomet. Chem., 1993, 456, 97–106 CrossRef CAS.
- C. Janiak, N. Kuhn and R. Gleiter, J. Organomet. Chem., 1994, 475, 223–227 CrossRef CAS.
- M. Kreye, J. W. Runyon, M. Freytag, P. G. Jones and M. D. Walter, Dalton Trans., 2013, 42, 16846–16856 RSC.
- M. Kreye, A. Glöckner, C. G. Daniliuc, M. Freytag, P. G. Jones, M. Tamm and M. D. Walter, Dalton Trans., 2013, 42, 2192–2200 RSC.
- M. Kreye, D. Baabe, P. Schweyen, M. Freytag, C. G. Daniliuc, P. G. Jones and M. D. Walter, Organometallics, 2013, 32, 5887–5898 CrossRef CAS.
- P. Crewdson, S. Gambarotta, M.-C. Djoman, I. Korobkov and R. Duchateau, Organometallics, 2005, 24, 5214–5216 CrossRef CAS.
- I. Korobkov and S. Gambarotta, Organometallics, 2009, 28, 5560–5567 CrossRef CAS.
- M. Schultz, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m3085 CAS.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaka, New York, 1960 Search PubMed.
- A. Hildebrandt, D. Schaarschmidt, R. Claus and H. Lang, Inorg. Chem., 2011, 31, 10623–10632 CrossRef PubMed.
- A. Hildebrandt and H. Lang, Dalton Trans., 2011, 40, 11831–11837 RSC.
- D. Miesel, A. Hildebrandt, M. Korb, P. J. Low and H. Lang, Organometallics, 2013, 32, 2993–3002 CrossRef CAS.
- R. Bartsch, P. B. Hitchcock and J. F. Nixon, J. Organomet. Chem., 1988, 356, C1–C4 CrossRef CAS.
- T. J. Brunker, J. C. Green and D. O'Hare, Inorg. Chem., 2002, 41, 1701–1703 CrossRef CAS PubMed.
- T. J. Brunker, J. C. Green and D. O'Hare, Inorg. Chem., 2003, 42, 4366–4381 CrossRef CAS PubMed.
- J. S. Overby, N. J. Schoell and T. P. Hanusa, J. Organomet. Chem., 1998, 560, 15–19 CrossRef CAS.
- (a) H. Sitzmann, M. Schär, E. Dormann and M. Kelemen, Z. Anorg. Allg. Chem., 1997, 623, 1850–1852 CrossRef CAS; (b) H. Sitzmann, Coord. Chem. Rev., 2001, 214, 287–327 CrossRef CAS.
- R. D. Fischer, in NMR of Paramagnetic Molecules, ed. G. N. La Mar, W. D. Horrocks and R. H. Holm, Academic Press, New York, 1973, pp. 521–553 Search PubMed.
- D. C. Bradley, M. B. Hursthouse, C. W. Newing and A. J. Welch, J. Chem. Soc., Chem. Commun., 1972, 567–568 RSC.
- J. N. Boynton, W. A. Merrill, W. M. Reiff, J. C. Fettinger and P. P. Power, Inorg. Chem., 2012, 51, 3212–3219 CrossRef CAS PubMed.
- C. W. Bird and G. W. H. Cheeseman, in Comprehensive Heterocycles, ed. A. R. Katritzky and C. W. Rees, Pergamon Press, Oxford, 1984, pp. 1–10 Search PubMed.
- S. Scheuermayer, F. Tuna, E. M. Pineda, M. Bodensteiner, M. Scheer and R. A. Layfield, Inorg. Chem., 2013, 52, 3878–3883 CrossRef CAS PubMed.
- R. A. Heintz, R. L. Ostrander, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 1994, 116, 11387–11396 CrossRef CAS.
- M. E. Burin, M. V. Smirnova, G. K. Fukin, E. V. Baranov and M. N. Bochkarev, Eur. J. Inorg. Chem., 2006, 2006, 351–356 CrossRef.
- M. H. Chisholm, F. A. Cotton, M. W. Extine and D. C. Rideout, Inorg. Chem., 1979, 18, 120–125 CrossRef CAS.
- B. H. Müller, N. Peulecke, S. Peitz, B. R. Aluri, U. Rosenthal, M. H. Al-Hazmi, F. M. Mosa, A. Wöhl and W. Müller, Chem. – Eur. J., 2011, 17, 6935–6938 CrossRef PubMed.
- C. J. O'Connor, in Progress in Inorganic Chemistry, ed. S. J. Lippard, J. Wiley & Sons, New York, 1982, vol. 29, pp. 203–285 Search PubMed.
- M. D. Walter, M. Schultz and R. A. Andersen, New J. Chem., 2006, 30, 238–246 RSC.
- E. Bill, http://www.cec.mpg.de/forschung/molekulare-theorie-und-spektroskopie-molecular-theory-and-spectroscopy/moessbauer-mcd.html.
- H. Schumann, J. Winterfeld, H. Hemling and N. Kuhn, Chem. Ber., 1993, 126, 2657–2659 CrossRef CAS.
- M. D. Walter, C. D. Sofield, C. H. Booth and R. A. Andersen, Organometallics, 2009, 28, 2005–2019 CrossRef CAS.
- (a) G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structure from Diffraction Data, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis spectra of 1-Cr, 2-Cr, 3-Cr, 1-Cr(thf), 2-Cr(tmeda), 3-CrCl and 4-CrCl, experimental details for catalytic testing. CCDC 987421–987425. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00533c |
| This journal is © The Royal Society of Chemistry 2014 |