 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characterization and biological evaluation of a clioquinol–ruthenium complex with copper-independent antileukaemic activity†
Martina
Gobec‡
a,
Jakob
Kljun‡
bc,
Izidor
Sosič‡
a,
Irena
Mlinarič-Raščan
a,
Matija
Uršič
c,
Stanislav
Gobec
*a and
Iztok
Turel
*bc
aFaculty of Pharmacy, University of Ljubljana, Aškerčeva 7, SI-1000 Ljubljana, Slovenia. E-mail: stanislav.gobec@ffa.uni-lj.si
bEN-FIST Centre of Excellence, Dunajska 156, SI-1000 Ljubljana, Slovenia. E-mail: iztok.turel@fkkt.uni-lj.si
cFaculty of Chemistry and Chemical Technology, University of Ljubljana, Aškerčeva 5, SI-1000 Ljubljana, Slovenia
First published on 4th April 2014
Abstract
In this study, we present the synthesis, biological characterization, and first crystal structure of an organometallic–clioquinol complex. Combining ruthenium with the established apoptotic agent and 8-hydroxyquinoline derivative, clioquinol, resulted in a complex that induces caspase-dependent cell death in leukaemia cells. This activity is copper independent and is improved compared to the parent compound, clioquinol. The study of the mode of action reveals that this clioquinol–ruthenium complex does not intercalate between DNA base pairs. Additionally, this clioquinol–ruthenium complex shows proteasome-independent inhibition of the NFκB signalling pathway, with no effects on cell-cycle distribution. These data suggest a mechanism of action that involves a target profile that is different from that for clioquinol alone.
1. Introduction
After the clinical success of cisplatin, the development of novel transition-metal-based compounds has received great attention.1 This field of inorganic medicinal chemistry will certainly continue to develop in the future, as there are several metal complexes currently in clinical trials. These include the new generation of antitumor platinum complexes,2,3 the antimalarial ferrocene–quinoline conjugates,4 and the ruthenium complexes NAMI-A and KP-1019 that show potent antineoplastic actions.5Ruthenium-based compounds have also attracted increasing interest as potential antitumor agents. The ruthenium ion can be either reactive, when it bears halide ligands that can dissociate in aqueous solution to confer multifunctional potential to the target complexes, or act solely as a building block without being involved in direct interactions with biological target(s), thus acting as a scaffold to organize various well-established bioactive organic compounds around its metal center.6 Indeed, these compounds show promise for significant advances due to their high delivery to cancer cells, as well as the low occurrence of side effects and the favourable toxicological profile.5 It is known that some ruthenium compounds bind to DNA more strongly and are less affected by cell repair mechanisms compared to cisplatin.7 However, as recently described, some ruthenium-based compounds can induce cell death through other mechanisms.8
Clioquinol (CqH) shows a wide range of biological activities. It has been used as an antimicrobial agent for many years, and recently, very encouraging data have been reported for clioquinol use in the treatment of Alzheimer's9 and Parkinson's diseases.10 Clioquinol was studied up to a pilot phase 2 study, but due to difficulties in preventing diiodo 8-hydroxyquinoline contamination upon large scale chemical synthesis no further phase II/III studies have been carried out.9,11 Although the mechanism of action has not been fully elucidated, the biological activity of clioquinol has been ascribed to its ability to cross the blood–brain barrier and to its chelation of metal ions, such as Cu(II) and Zn(II), which are associated with protein aggregation and degeneration processes in the brain.12 Moreover, it has been shown that in the presence of copper(II) ions, clioquinol inhibits the proteasomal activity and proliferation in cultured human cancer cells.13–15 In a similar study, clioquinol was fully characterized as an inducer of cell death in leukaemia and myeloma cell lines, where its actions are copper dependent and are also due to copper-dependent inhibition of the proteasome.16 With transition metal ions, such as zinc(II) and copper(II), clioquinol forms stable [M(Cq)2] complexes, however the active copper(II)–clioquinol species and its mechanism of action have not yet been determined.12,17,18 Nowadays, topical formulations of clioquinol are still available for the treatment of topical fungal and parasitic infections.16
A way to extend or improve the biological activity of known bioactive compounds is to bind them to a metal centre, which can result in synergistic/improved activity.19 The most advanced example of this concept relates to ferroquine, a ferrocenyl analogue of the antimalarial drug chloroquine.20 Ferroquine is an antimalarial candidate that completed a phase IIb clinical trial in 2011, and it is active against parasites that have acquired resistance to chloroquine and other antimalarial agents.21 This concept has been extended to ruthenium–arene derivatives of chloroquine, which inhibit the growth of colon cancer cells.22
We recently reported on our studies of ruthenium complexes with the quinolone antibacterial agents23–26 that show increased toxicities against selected cancer cell lines and enhanced inhibition of the cathepsins, the enzymes of the cysteine protease family that are involved in the development and progression of many diseases, including cancer and arthritis.27 Herein, we present the synthesis and biological evaluation of a clioquinol–ruthenium complex (1) (Fig. 1). In our efforts to ‘teach an old dog new tricks’, we combined the potential of an established apoptotic agent, clioquinol, with an organoruthenium moiety. The main goal was to obtain an agent with improved anticancer properties in comparison with the parent compound, clioquinol. Even though the synthesis of this clioquinol–ruthenium complex was reported recently, we have considerably improved the synthetic procedure in terms of the yield and the synthesis time, and we have also proceeded with its full physicochemical characterization in the solid state and in solution.28 The stability of the clioquinol–ruthenium complex in dimethylsulfoxide (DMSO) and in aqueous solution was studied, and its crystal structure was also determined.
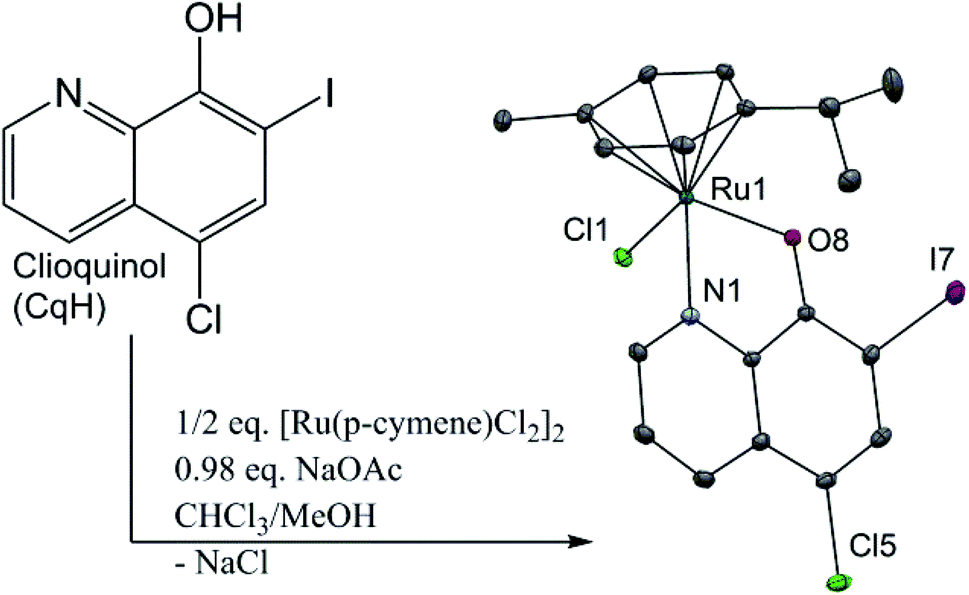 | ||
| Fig. 1 Synthetic route and crystal structure of the [Ru(η6-p-cymene)Cl(Cq)] complex (1) with heteroatom labelling. The hydrogen atoms are omitted for clarity. The thermal ellipsoids are shown at the 30% probability level. CqH, clioquinol. Selected bond lengths and angles: Ru–Cl1, 2.427(1) Å; Ru–N1, 2.106(3) Å; Ru–O8, 2.076(3) Å; Ru–(cymene centroid), 1.671(2) Å; N1–Ru1–O8, 78.27(12)°. | ||
2. Results
2.1. Synthesis and characterization of [Ru(η6-p-cymene)Cl(Cq)] (1)
The clioquinol–ruthenium complex (1) was prepared by reacting the ligand clioquinol with the dimeric ruthenium precursor [Ru(η6-p-cymene)(μ-Cl)Cl]2 in the presence of a weak base (NaOAc) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CHCl3 and MeOH. After filtration of the by-product NaCl, the product was precipitated from a CH2Cl2 solution by addition of cold hexane. Crystals of 1 suitable for X-ray analysis were obtained by slow evaporation of the 1:1 CH2Cl2/n-hexane solution. The clioquinol–ruthenium complex (1) adopts the pseudo-octahedral geometry typical of organoruthenium species, in which the p-cymene ring is π-bonded to the ruthenium ion, while the remaining three coordination sites are occupied by the chlorido ligand and the N1 and O8 atoms from the clioquinolato (Cq) ligand in its deprotonated form (Fig. 1 and ESI†).
:1 mixture of CHCl3 and MeOH. After filtration of the by-product NaCl, the product was precipitated from a CH2Cl2 solution by addition of cold hexane. Crystals of 1 suitable for X-ray analysis were obtained by slow evaporation of the 1:1 CH2Cl2/n-hexane solution. The clioquinol–ruthenium complex (1) adopts the pseudo-octahedral geometry typical of organoruthenium species, in which the p-cymene ring is π-bonded to the ruthenium ion, while the remaining three coordination sites are occupied by the chlorido ligand and the N1 and O8 atoms from the clioquinolato (Cq) ligand in its deprotonated form (Fig. 1 and ESI†).
Interestingly, the Cambridge structural database currently reports crystal structures of only nine clioquinol metal complexes: [ReOCl2(Cq)(PPh3)],29trans-[Zn(Cq)2(OH2)], trans-[Cu(Cq)2],17trans-[Ni(Cq)2], (Me2NH2)[Ni(Cq)3],30 (CqH2)[PdCl2(Cq)],31trans-[Pd(Cq)2],32 [PtCl(Cq)(dmso)]33 and [Ce(Cq)4].34 In all cases, clioquinol acts as a bidentate ligand in its deprotonated form, as is usual for simple 8-hydroxyquinoline ligands. It is a very versatile ligand for the construction of novel molecules, as among the above listed compounds we can find square-planar, pyramidal, octahedral and dodecahedral complexes. Here, we report the first crystal structure of an organometallic clioquinol complex. Moreover, more than 60 crystal structures of ruthenium coordination compounds with 8-hydroxyquinoline ligands are reported, most of which contain the parent ligand 8-hydroxyquinoline;35–38 however, the crystal structure of the 2-methyl-5,7-dichloro-8-hydroxyquinoline and 5,7-dimethyl-8-hydroxyquinoline complexes with an analogous chemical structure to the clioquinol–ruthenium complex (1) is also reported.39
Complex 1 links into supramolecular dimers about a centre of inversion (Fig. 2). The inter-ruthenium distance Ru1–Ru1′ in the dimers is 5.409(4) Å, which is in line with the observations from our previous study on organoruthenium complexes with β-diketonate ligands.40
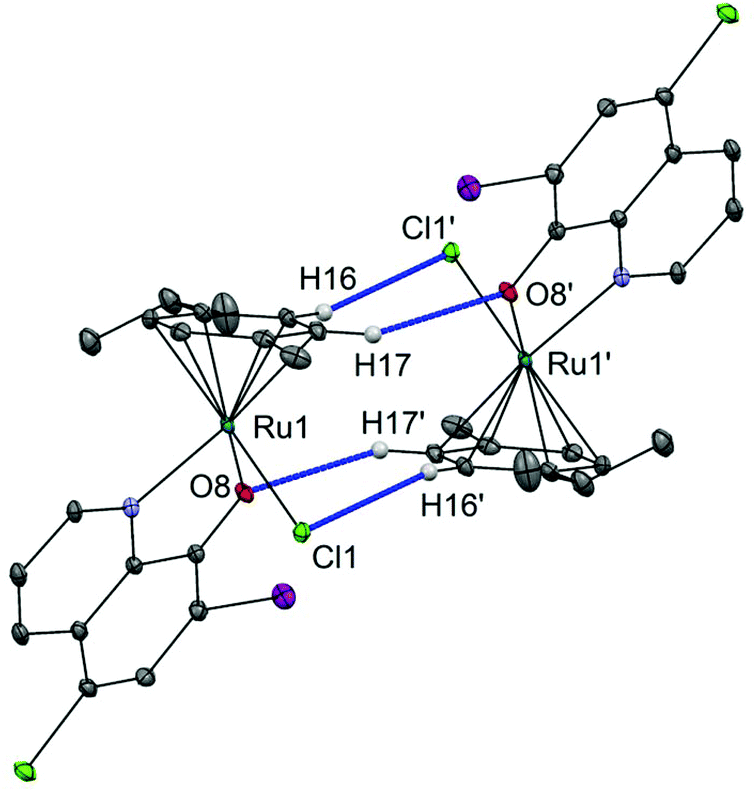 | ||
| Fig. 2 Supramolecular interactions in the crystal structure of 1. Dimers of the clioquinol–ruthenium complex (1) linking through CCym–H⋯O and CCym–H⋯Cl bonds. Hydrogen atoms that are not involved in H-bonding are omitted for clarity. Ellipsoids are shown at the 30% probability level. Selected distances: Cl1–H16′ = 2.78 Å, O8–H17′ = 2.49 Å. | ||
Intrigued by the findings of Gasser et al.,41 who reported deleterious effects of the commonly used bioassay co-solvent DMSO on the stability of organoruthenium compounds, we explored the stability of the clioquinol–ruthenium complex (1) in DMSO-d6 and in 10% DMSO-d6/90% H2O solution using 1H NMR (see ESI†). Monitoring these solutions along the time, no changes were observed in the NMR signals, and the integrity of the complex in DMSO solution as well as the aqua species in 10% DMSO aqueous solution was intact within the time-frame of the biological experiments. The stability of the complex in the presence of phosphate buffers in solutions with pH values 8.0, 8.5, and 9.0 was monitored by UV-Vis spectroscopy (see ESI†). The spectra remain unchanged after 24 hours. Both experiments show a remarkable stability of complex 1, as no release of ligand Cq was observed.
2.2. Selective cytotoxicity of the clioquinol–ruthenium complex (1) towards leukaemia cell lines
To evaluate the antiproliferative activity of 1, we performed metabolic activity assays with six human cancer cell lines of different tissues of origin. Breast cancer (MCF-7), prostate cancer (PC-3), osteosarcoma (HOS), Burkitt's lymphoma (Raji), acute T-cell leukaemia (Jurkat) and acute monocytic leukaemia (Thp-1) cells were treated for 24 h with different concentrations of clioquinol and 1. The IC50 values were determined (see ESI†) and are summarized in Table 1. Clioquinol and 1 showed comparable effects on MCF-7, PC-3 and HOS cells. On the other hand, for the leukaemic cells, in comparison with clioquinol, significantly lower concentrations of 1 were needed to reduce cell viability by 50%. As already established in the Introduction, clioquinol requires copper(II) ions to show its apoptotic and anticancer activities. Therefore, in the next step, we investigated whether this is also the case for the clioquinol–ruthenium complex (1); the data in Fig. 3 demonstrate that this cytotoxicity of 1 is copper independent.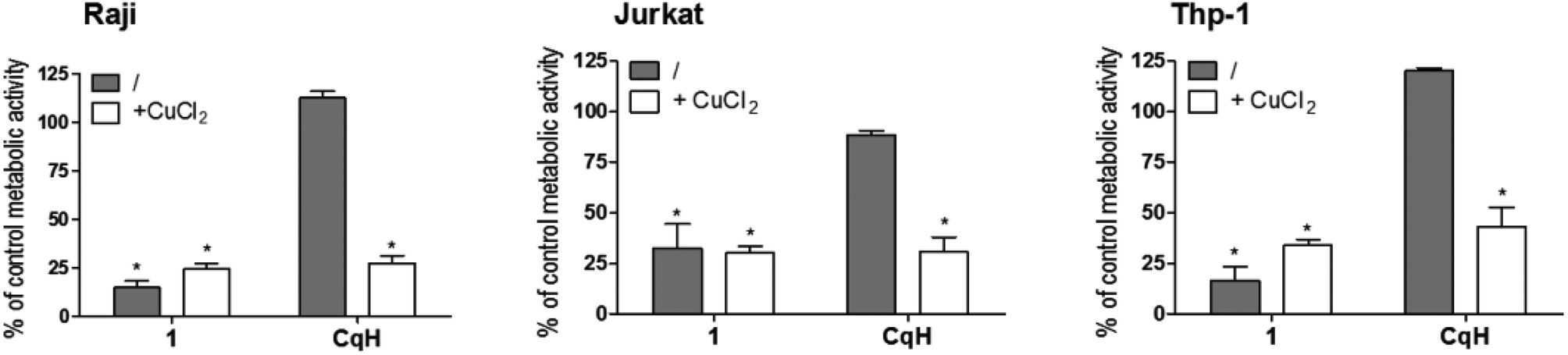 | ||
| Fig. 3 Copper-independent effects of the clioquinol–ruthenium complex (1). Raji, Jurkat and Thp-1 cells were treated for 24 h with clioquinol (10 μM) or 1 (10 μM) in the absence and presence of CuCl2 (10 μM). CqH, clioquinol. Data are means ± SD (n = 3), expressed as percentages of untreated control cells. *p < 0.05, versus control cells. | ||
| Agent | IC50 against selected cancer cell lines (μM) | |||||
|---|---|---|---|---|---|---|
| MCF-7 | PC-3 | HOS | Raji | Thp-1 | Jurkat | |
| a Cells were treated with different concentrations of clioquinol or 1 for 24 h. Cell viability was determined using the MTS assay. IC50 values are presented as the means ± SD from three independent experiments. | ||||||
| Clioquinol | >50 | 45 ± 7 | 28 ± 4 | 25 ± 5 | 24 ± 6 | 22 ± 5 |
| 1 | >50 | 42 ± 10 | 27 ± 2 | 6 ± 3 | 6 ± 5 | 5 ± 2 |
2.3. The clioquinol–ruthenium complex (1) induces caspase-dependent apoptosis
To explore the mode of cell death induced by 1, we incubated Raji cells with clioquinol (25 μM) or 1 (5 μM, 10 μM, 15 μM) for 24 h. Apoptosis was determined by detection of the externalized phospholipid phosphatidylserine using the annexin V/propidium iodide assay and by detection of caspase 3/7 activity. As shown in Fig. 4A, a concentration-dependent increase in apoptotic cells was seen for 1. A 5-fold higher concentration of clioquinol was needed to achieve the same effect as for the clioquinol–ruthenium complex (1). The apoptotic process was further shown through increased caspase 3/7 activity (Fig. 4B). To confirm that this process is caspase-dependent, the Raji cells were pre-treated with the pan-caspase inhibitor zVAD.fmk (50 μM). The presence of zVAD.fmk completely blocked the effects of 1 (see ESI†). Taken together, these data demonstrate that the clioquinol–ruthenium complex (1) induces apoptosis in Raji cells in a caspase-dependent manner.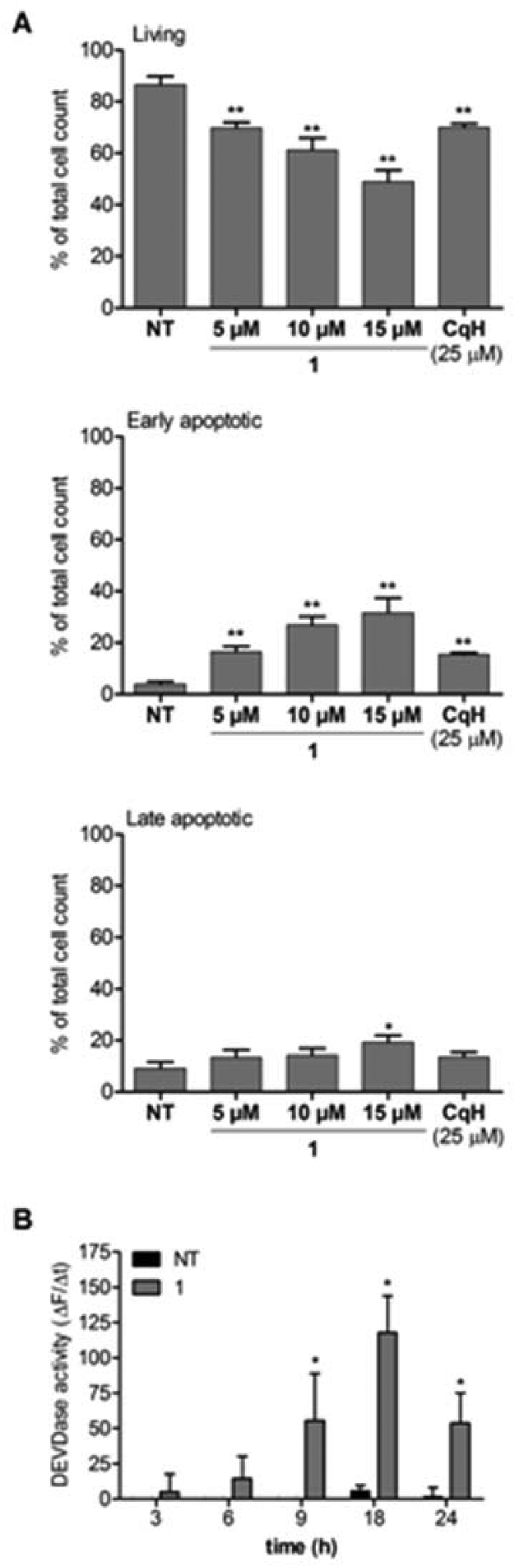 | ||
| Fig. 4 The complex 1 induces apoptosis in a concentration-dependent manner. (A) Raji cells were treated with clioquinol (25 μM) or 1 (5 μM, 10 μM, 15 μM) for 24 h. The cells were then stained with annexin V/propidium iodide. (B) Caspase 3/7 activity was determined in cell lysates of Raji cells treated for 3, 6, 9, 18, and 24 h with 1 (10 μM). Cleavage of Ac-DEVD-AFC in whole-cell lysates was determined spectrofluorometrically. The data are presented as changes in the fluorescence as a function of time. CqH, clioquinol. Data are means ± SD (n = 3), expressed as percentages of total cells. *p < 0.05; **p < 0.01, versus nontreated cells. | ||
Clioquinol is known to intercalate between DNA base pairs30 therefore we investigated whether 1 has the same properties. A ctDNA intercalation assay was performed with a fixed concentration of 1 (15 μM) to which five varying concentrations of the ctDNA were added until the concentration ratio between ctDNA and 1 reached 10. The UV–visible spectra were recorded from 230 nm to 400 nm. For the absorption maxima, determined at 285 nm, no decreased intensity or shift to higher wavelengths could be observed (see ESI†). This suggests that the clioquinol–ruthenium complex (1) most probably does not act as a DNA intercalator.
Very recently, it was shown that clioquinol inhibits histone deacetylase activity, which leads to the arrest of leukaemic cells in the G0/G1 phase of the cell cycle.42 The analysis of the cell cycle of Raji cells treated for 24 h with either clioquinol or 1 showed that unlike clioquinol, the clioquinol–ruthenium complex (1) had no effects on the cell-cycle distribution (Fig. 5).
 | ||
| Fig. 5 Cell-cycle analysis. Raji cells were incubated with clioquinol (25 μM) or 1 (5 μM, 10 μM, 15 μM) for 24 h. Propidium iodide staining was performed. CqH, clioquinol. Data are means ± SD (n = 3), expressed as percentages of total viable cells. *p < 0.05; NT, non-treated cells. | ||
2.4. Nonproteasomal NFκB inhibition
As leukaemic cells are known to constitutively express the important cell survival signal NFκB, we investigated the involvement of NFκB signalling modulation in the induction of cell death by the complex 1.43 This was investigated in the Ramos Blue™ leukaemic cell line, which stably expresses an NFκB/AP-1-inducible secreted embryonic alkaline phosphate (SEAP) reporter construct that allows the detection of modulation of the NFκB signalling pathway. The Ramos Blue™ cells were pre-treated with a nontoxic concentration of clioquinol (10 μM) or with 1 (2.5 μM) for 1 h, and then stimulated with a known NFκB activating stimulus: recombinant tumour necrosis factor (TNF)-α (50 ng mL−1). Determination of the SEAP activity in the supernatant after 16 h demonstrated that clioquinol and 1 reduced the TNF-α-induced NFκB activity by >50% (Fig. 6). Additionally, inhibition of NFκB activity by 1 was not mediated by inhibition of the proteasome (as has been established for clioquinol). This was demonstrated in a cell-free proteasome activity assay, where no inhibition of the proteasome by 1 was observed (see ESI†). These data thus show that in these leukaemia cells, the increased cell death induced by the clioquinol–ruthenium complex (1) appears to be due to nonproteasomal inhibition of NFκB activity, followed by the disruption of pro-survival pathways.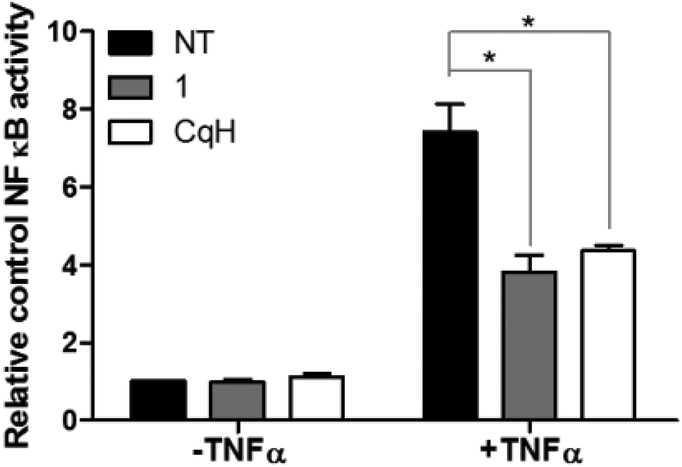 | ||
| Fig. 6 Inhibition of NFκB activity. Determination of the NFκB transcriptional activity. Secreted embryonic alkaline phosphate (SEAP) activity was measured in Ramos-Blue™ cells after their pre-treatment with clioquinol (10 μM) or 1 (2.5 μM) for 1 h, and the subsequent addition of TNF-α (50 ng mL−1). CqH, clioquinol. Data are means ± SD (n = 3), expressed relative to the untreated control cells. *p < 0.05, versus TNF-α-treated cells. | ||
3. Discussion
Complexing clioquinol with an organoruthenium moiety enhanced the induction of apoptosis of the resulting clioquinol–ruthenium complex (1). The data also show that clioquinol and 1 have different antitumor activities and selectivities. Upon coordination to ruthenium, the cytotoxicity of 1 against leukaemia cell lines is considerably improved, while the effects on solid tumour cell lines (MCF-7, PC-3, HOS cells) are seen at the same concentrations as for clioquinol.Moreover, the complex 1 shows copper-independent reduction of cell viability. It has previously been shown that clioquinol-methoxy derivatives that cannot chelate copper have no anticancer activity.15 It has also been established that copper ions are a prerequisite for the proteasome inhibition of clioquinol and the consequent induction of apoptosis. The clioquinol–ruthenium complex presented here shows these cytotoxic effects in the absence of copper, with concentration-dependent cell death by apoptosis that is mediated through caspase activation. Additionally, the concentration needed to initiate apoptosis is significantly lower for 1 (5 μM) than for clioquinol (25 μM) (Fig. 4). The mechanism of cell death induced by 1 does not appear to require reactive oxygen species, as these did not show increased production (see ESI†).
When we addressed the mode of action of the clioquinol–ruthenium complex, we hypothesized that it maintains similar targets to the parent clioquinol. First, possible intercalation-based interactions of 1 with ctDNA were investigated, as it was recently shown that clioquinol can intercalate between DNA base pairs.22 The absorption pattern of the 15 μM clioquinol–ruthenium complex was followed upon addition of increasing amounts of ctDNA (0–150 μM). The absorption maximum at 285 nm was not shifted to higher wavelengths, and the intensity was not decreased (see ESI†), which suggests that the clioquinol–ruthenium complex (1) does not act as a DNA intercalator. The results of this preliminary experiment, however, do not absolutely exclude the possibilities of other types of interactions of 1 with DNA (especially the possibility of covalent interactions).
It has recently been shown that clioquinol inhibits histone deacetylase activity, which leads to the arrest of leukaemic cells in the G0/G1 phase of the cell cycle.42 Surprisingly, while clioquinol resulted in severe arrest of Raji cells in the G0/G1 phase of the cell cycle, complex 1 had no effect on the cell-cycle distribution (Fig. 5), which indicates that it has a different target profile. This was further demonstrated in a cell-free proteasome assay. Here, clioquinol is known to have an antiproliferative action that is mediated through copper-dependent inhibition of the proteasome.13–15 The clioquinol–ruthenium complex (1) does not inhibit the chymotrypsin-like activity of the proteasome; however, as we showed in the NFκB transcriptional activity assay, complex 1 appears to modulate the NFκB pathway, as it can reduce TNF-α-induced activation of NFκB by >50% (Fig. 6). Moreover, lower concentrations of the clioquinol–ruthenium complex are needed to observe this effect, compared to clioquinol (2.5 μM vs. 10 μM, respectively).
Taken together, these data indicate that the clioquinol–ruthenium complex (1) has different targets in comparison with clioquinol; it does, however, inhibit the NFκB pathway, although not via proteasome inhibition. Constitutive NFκB activation is a common feature of many haematological malignancies, and is believed to be crucial for the survival of these malignant leukaemia cells.43 Therefore, targeting any cascade in the NFκB signalling pathway is a valid anticancer strategy.
4. Conclusion
We have synthesized and fully characterized the clioquinol–ruthenium complex (1) and determined its crystal structure. The cytotoxicity studies show that 1 has antiproliferative activity in leukaemia cell lines, which is mediated through caspase activation. Moreover, we have demonstrated that the cytotoxic activity in these leukaemia cell lines is improved, compared with clioquinol, and is copper independent. The mechanism-of-action studies show that 1 does not intercalate between DNA base pairs. The clioquinol–ruthenium complex (1) shows proteasome-independent inhibition of the NFκB signalling pathway, and has no effects on the cell-cycle distribution, which suggests a mechanism of action that is different from that of the 8-hydroxyquinoline derivative, clioquinol. These findings provide an important step towards the elucidation of the precise mode of action of 1, the knowledge of which will provide a solid basis for future structure-based optimization efforts.5. Experimental section
5.1. Synthesis of [Ru(η6-p-cymene)Cl(Cq)] (1)
30.0 mg of ruthenium precursor (P1, 0.049 mmol), 29.9 mg of clioquinol (1.00 eq., 0.098 mmol), and 13.1 mg of NaOAc·3H2O (1.00 eq., 0.098 mmol) were dissolved in 20 mL of 1:1 MeOH/CHCl3 mixture, and the solution was refluxed for 3 h. The solvent was rotary-evaporated, replaced by CH2Cl2, and NaCl was removed by filtration. The solution was concentrated and precipitated by the addition of n-hexane. The orange precipitate was collected by filtration and washed with n-hexane (η = 85%). Crystals of 1 suitable for X-ray analysis were obtained by slow evaporation of a 1:1 CHCl3/n-hexane solution.
NMR: 1H NMR (500 MHz, CDCl3): δ 8.91 (d, J = 4.1 Hz, 1H, C2H), 8.36 (dd, J = 8.5, 0.9 Hz, 1H, C4H), 7.80 (s, 1H, C6H), 7.46 (dd, J = 8.6, 4.9 Hz, 1H, C3H), 5.66 (d, J = 6.0 Hz, 1H, Ar-H cym), 5.44 (d, J = 6.0 Hz, 2H, Ar-H cym), 5.38 (d, J = 5.9 Hz, 1H, Ar-H cym), 2.88 (hept, J = 6.8 Hz, 1H, Ar-CH(CH3)2 cym), 2.34 (s, 3H, Ar-CH3 cym), 1.30 (d, J = 6.9 Hz, 3H, Ar-CH(CH3)2 cym), 1.18 (d, J = 6.9 Hz, 3H, Ar-CH(CH3)2 cym).
1H NMR (500 MHz, DMSO-d6): δ 9.30 (dd, J = 4.9, 1.0 Hz, 1H, C2H), 8.36 (dd, J = 8.6, 1.0 Hz, 1H, C4H), 7.79 (s, 1H, C6H), 5.93 (d, J = 6.0 Hz, 1H, Ar-H cym), 5.78 (d, J = 5.9 Hz, 1H, Ar-H cym), 5.71 (d, J = 5.9 Hz, 1H, Ar-H cym), 5.64 (d, J = 6.0 Hz, 1H, Ar-H cym), 2.71 (hept, J = 6.9 Hz, 1H, Ar-CH(CH3)2 cym), 2.19 (s, 3H, Ar-CH3 cym), 1.17 (d, J = 6.9 Hz, 3H, Ar-CH(CH3)2 cym), 1.09 (d, J = 6.9 Hz, 3H, Ar-CH(CH3)2 cym).
13C NMR (126 MHz, CDCl3): δ 167.08 (C8), 149.24 (C2), 141.82 (C9), 137.15 (C6), 135.09 (C4), 127.42 (C10), 122.64 (C3), 112.57 (C5), 102.16 (Ar-C cym), 97.63 (Ar-C cym), 82.73 (Ar-C cym), 81.98 (Ar-C cym), 81.63 (Ar-C cym), 80.97 (C7), 80.69 (Ar-C cym), 77.26 (CDCl3), 77.01 (CDCl3), 76.76 (CDCl3), 31.00 (Ar-C cym), 22.34 (Ar-C cym), 22.18 (Ar-C cym), 18.52 (Ar-C cym).
15N (through 1H–15N HMBC, CDCl3): δ 238.17 (N1).
Selected IR resonances (cm−1, ATR): 1572, 1542, 1481, 1441, 1369, 1360, 1250, 1109, 877, 751.
CHN: Calc. for C19H18Cl2INORu: C, 39.67; H, 3.15; N, 2.44. Found C, 39.67; H, 2.90; N, 2.25.
UV/Vis (λ) [nm] (ε) [L mol−1 cm−1], c = 1 × 10−4 mol L−1, MeOH: 285 (22500), 341 (3200), 355 (3300), 418 (2600), 467 (2200).
ESI-HRMS (CH3CN) m/z (found for [M − Cl]+ (calcd)): 539.9155 (539.9165).
Acknowledgements
This study was supported by the Slovenian Research Agency (Research Programmes P1-0208 and P1-0175, Research Project J1-4131). We thank Sara Seršen for help with NMR data.References
- Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH Verlag & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- E. Gallerani, J. Bauer, D. Hess, S. Boehm, C. Droege, S. Jeckelmann, M. Miani, R. Herrmann, S. Marsoni, S. Sperka and C. Sessa, Acta Oncol., 2011, 50, 1105–1110 CrossRef CAS PubMed.
- F. Dubar, J. Khalife, J. Brocard, D. Dive and C. Biot, Molecules, 2008, 13, 2900–2907 CrossRef CAS PubMed.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- T. Gianferrara, I. Bratsos and E. Alessio, Dalton Trans., 2009, 7588–7598 RSC.
- V. Brabec and O. Novakova, Drug Resist. Updat., 2006, 9, 111–122 CrossRef CAS PubMed.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- P. A. Adlard, R. A. Cherny, D. I. Finkelstein, E. Gautier, E. Robb, M. Cortes, I. Volitakis, X. Liu, J. P. Smith, K. Perez, K. Laughton, Q. X. Li, S. A. Charman, J. A. Nicolazzo, S. Wilkins, K. Deleva, T. Lynch, G. Kok, C. W. Ritchie, R. E. Tanzi, R. Cappai, C. L. Masters, K. J. Barnham and A. I. Bush, Neuron, 2008, 59, 43–55 CrossRef CAS PubMed.
- D. Kaur, F. Yantiri, S. Rajagopalan, J. Kumar, J. O. Mo, R. Boonplueang, V. Viswanath, R. Jacobs, L. Yang, M. F. Beal, D. DiMonte, I. Volitaskis, L. Ellerby, R. A. Cherny, A. I. Bush and J. K. Andersen, Neuron, 2003, 37, 899–909 CrossRef CAS.
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- A. I. Mot, A. G. Wedd, L. Sinclair, D. R. Brown, S. J. Collins and M. W. Brazier, Expert Rev. Neurother., 2011, 11, 1717–1745 CrossRef PubMed.
- K. G. Daniel, D. Chen, S. Orlu, Q. C. Cui, F. R. Miller and Q. P. Dou, Breast Cancer Res. Treat., 2005, 7, R897–R908 CrossRef CAS PubMed.
- D. Chen, Q. C. Cui, H. J. Yang, R. A. Barrea, F. H. Sarkar, S. J. Sheng, B. Yan, G. P. V. Reddy and Q. P. Dou, Cancer Res., 2007, 67, 1636–1644 CrossRef CAS PubMed.
- S. M. Zhai, L. Yang, Q. C. Cui, Y. Sun, Q. P. Dou and B. Yan, J. Biol. Inorg. Chem., 2010, 15, 259–269 CrossRef CAS PubMed.
- X. Mao, X. Li, R. Sprangers, X. Wang, A. Venugopal, T. Wood, Y. Zhang, D. A. Kuntz, E. Coe, S. Trudel, D. Rose, R. A. Batey, L. E. Kay and A. D. Schimmer, Leukemia, 2009, 23, 585–590 CrossRef CAS PubMed.
- M. Di Vaira, C. Bazzicalupi, P. Orioli, L. Messori, B. Bruni and P. Zatta, Inorg. Chem., 2004, 43, 3795–3797 CrossRef CAS PubMed.
- M. J. Pushie, K. H. Nienaber, K. L. Summers, J. J. H. Cotelesage, O. Ponomarenko, H. K. Nichol, I. J. Pickering and G. N. George, J. Inorg. Biochem., 2014, 133, 50–56 CrossRef CAS PubMed.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- N. J. Farrer and P. J. Sadler, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH Verlag & Co. KGaA, Weinheim, Germany, 2011, pp. 1–48 Search PubMed.
- C. Supan, G. Mombo-Ngoma, M. P. Dal-Bianco, C. L. O. Salazar, S. Issifou, F. Mazuir, A. Filali-Ansary, C. Biot, D. Ter-Minassian, M. Ramharter, P. G. Kremsner and B. Lell, Antimicrob. Agents Chemother., 2012, 56, 3165–3173 CrossRef CAS PubMed.
- C. S. K. Rajapakse, A. Martinez, B. Naoulou, A. A. Jarzecki, L. Suarez, C. Deregnaucourt, V. Sinou, J. Schrevel, E. Musi, G. Ambrosini, G. K. Schwartz and R. A. Sanchez-Delgado, Inorg. Chem., 2009, 48, 1122–1131 CrossRef CAS PubMed.
- I. Turel, J. Kljun, F. Perdih, E. Morozova, V. Bakulev, N. Kasyanenko, J. A. W. Byl and N. Osheroff, Inorg. Chem., 2010, 49, 10750–10752 CrossRef CAS PubMed.
- J. Kljun, A. K. Bytzek, W. Kandioller, C. Bartel, M. A. Jakupec, C. G. Hartinger, B. K. Keppler and I. Turel, Organometallics, 2011, 30, 2506–2512 CrossRef CAS PubMed.
- R. Hudej, J. Kljun, W. Kandioller, U. Repnik, B. Turk, C. G. Hartinger, B. K. Keppler, D. Miklavcic and I. Turel, Organometallics, 2012, 31, 5867–5874 CrossRef CAS.
- J. Kljun, I. Bratsos, E. Alessio, G. Psomas, U. Repnik, M. Butinar, B. Turk and I. Turel, Inorg. Chem., 2013, 52, 9039–9052 CrossRef CAS PubMed.
- M. M. Mohamed and B. F. Sloane, Nat. Rev. Cancer, 2006, 6, 764–775 CrossRef CAS PubMed.
- W. Fuyi, L. Qun, L. Shuang, W. Kui, H. Wenbing, J. Liyun and H. Yumiao, Eur. Pat. Off. CN200918461420090522, 2010 Search PubMed.
- Y. Miyashita, T. Ohashi, A. Imai, N. Amir, K. Fujisawa and K. Okamoto, Sci. Technol. Adv. Mater., 2005, 6, 660–666 CrossRef CAS PubMed.
- I. Potocnak and P. Vranec, Monatsh. Chem., 2012, 143, 217–226 CrossRef CAS.
- P. Vranec and I. Potocnak, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, M1508–M1509 CAS.
- P. Vranec, I. Potocnak and V. Kavecansky, Polyhedron, 2012, 39, 118–123 CrossRef CAS PubMed.
- C. Martin Santos, S. Cabrere, C. Rios-Luci, J. M. Padron, I. Lopez Solera, A. G. Quiroga, M. A. Medrano, C. Navarro-Ranninger and J. Aleman, Dalton Trans., 2013, 42, 13343–13348 RSC.
- Z. F. Chen, J. H. Wei, Y. C. Liu, M. Liu, Y. Q. Gu, K. B. Huang, M. Wang and H. Liang, Eur. J. Med. Chem., 2013, 68, 454–462 CrossRef CAS PubMed.
- C. Gemel, R. John, C. Slugove, K. Mereiter, R. Schmid and K. Kirchner, J. Chem. Soc., Dalton Trans., 2000, 2607–2612 RSC.
- R. Schuecker, R. O. John, M. A. Jakupec, V. B. Arion and B. K. Keppler, Organometallics, 2008, 27, 6587–6595 CrossRef CAS.
- J. G. Malecki, M. Jaworska, R. Kruszynski and J. Klak, Polyhedron, 2005, 24, 3012–3021 CrossRef CAS PubMed.
- S. L. Nongbri, B. Therrien and K. M. Rao, Inorg. Chim. Acta, 2011, 376, 428–436 CrossRef CAS PubMed.
- T. T. Thai, B. Therrien and G. Suss-Fink, J. Organomet. Chem., 2009, 694, 3973–3981 CrossRef CAS PubMed.
- S. Seršen, J. Kljun, F. Požgan, B. Štefane and I. Turel, Organometallics, 2013 Search PubMed.
- M. Patra, T. Joshi, V. Pierroz, K. Ingram, M. Kaiser, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, Chem. – Eur. J., 2013, 19, 14768–14772 CrossRef CAS PubMed.
- B. Cao, J. Li, J. Zhu, M. Shen, K. Han, Z. Zhang, Y. Yu, Y. Wang, D. Wu, S. Chen, A. Sun, X. Tang, Y. Zhao, C. Qiao, T. Hou and X. Mao, J. Biol. Chem., 2013, 288, 34181–34189 CrossRef CAS PubMed.
- A. Keutgens, I. Robert, P. Viatour and A. Chariot, Biochem. Pharmacol., 2006, 72, 1069–1080 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, crystallographic and spectroscopic data (Table S1 and Fig. S1–S9), bioassays (Fig. S10–S13). CCDC 977981. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00463a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |