DOI:
10.1039/C3DT53477D
(Paper)
Dalton Trans., 2014,
43, 4653-4667
Red emitting [Ir(C^N)2(N^N)]+ complexes employing bidentate 2,2′:6′,2′′-terpyridine ligands for light-emitting electrochemical cells†
Received
11th December 2013
, Accepted 19th January 2014
First published on 30th January 2014
Abstract
2,2′:6′,2′′-Terpyridine (tpy), 4′-(4-HOC6H4)-2,2′:6′,2′′-terpyridine (1), 4′-(4-MeOC6H4)-2,2′:6′,2′′-terpyridine (2), 4′-(4-MeSC6H4)-2,2′:6′,2′′-terpyridine (3), 4′-(4-H2NC6H4)-2,2′:6′,2′′-terpyridine (4) and 4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine (4) act as N^N chelates in complexes of the type [Ir(C^N)2(N^N)][PF6] in which the cyclometallating ligand, C^N, is derived from 2-phenylpyridine (Hppy) or 3,5-dimethyl-1-phenyl-1H-pyrazole (Hdmppz). The single crystal structures of eight complexes have been determined, and in each iridium(III) complex cation, the non-coordinated pyridine ring of the tpy unit is involved in a face-to-face π-stacking interaction with the cyclometallated ring of an adjacent ligand. Solution NMR spectra of the [Ir(ppy)2(N^N)]+ complexes are consistent with the presence of a non-classical hydrogen bond between the non-coordinated N-donor of the tpy domain and a CH unit of one pyridine ring of an adjacent ppy− ligand; the presence of the N⋯HC interaction was confirmed in one of the solid-state structures. The pendant pyridine ring of the coordinated tpy undergoes hindered rotation on the NMR timescale at 295 K. In CH2Cl2, the complexes are orange or red emitters, with λemmax in the range 580 to 642 nm; photoluminescence quantum yields (PLQY) are <10%, and lifetimes range from 54 to 136 ns. N-Methylation of the pendant 4′-(4-pyridyl) group in [Ir(dmppz)2(pytpy)][PF6] essentially quenches the emission. Light-emitting electrochemical cells (LECs) have been fabricated in a thin film configuration; the emission spectra of the LECs are red-shifted with respect to the PL spectra of the corresponding complex in thin film configuration. For the device incorporating [Ir(ppy)2(pytpy)][PF6], the PL to EL red-shift is extremely large and this is indicative of a different emitting state being involved. The most efficient devices used [Ir(ppy)2(1)][PF6], [Ir(ppy)2(2)][PF6] or [Ir(ppy)2(3)][PF6] in the emissive layer; the devices exhibited rapid turn-on times, but showed relatively low efficiencies in accordance with the solid state photoluminescence quantum yields.
Introduction
The luminescent properties of cationic iridium(III) complexes of the type [Ir(C^N)2(N^N)]+ in which C^N is a cyclometallating ligand (e.g. the conjugate base of 2-phenylpyridine, Hppy) and N^N is 2,2′-bipyridine (bpy) or a derivative thereof have inspired their use as the ionic-transition metal complex (iTMC) component in light-emitting electrochemical cells (LECs).1,2 In [Ir(C^N)2(N^N)]+ cations, the character of the highest occupied molecular orbital (HOMO) is a combination of iridium dπ and C^N ligand orbitals, while the lowest unoccupied molecular orbital (LUMO) is localized on the N^N ligand. This localization of orbital character allows the energies of the HOMO and LUMO (and hence the HOMO–LUMO gap), and thus the emission colour, to be tuned by judicious choice of substituents on the C^N and/or N^N ligands.2 While an ability to colour-tune the emission of the iTMC is essential to its application in a LEC, achieving a long-lived device is also crucial. We have approached the latter by designing [Ir(C^N)2(N^N)]+ complexes in which there are inter-ligand π-stacking interactions.3–6 The introduction of an aryl substituent into the 6-position of the bpy ligand results in the evolution of an intramolecular face-to-face π-interaction between the aryl ring and the phenyl ring of one of the C^N ligands.3,7–10 A LEC incorporating the iTMC [Ir(ppy)2(6-Phbpy)]+ (Fig. 1, 6-Phbp = 6-phenyl-2,2′-bipyridine) exhibits a device lifetime of more than 3000 hours at an average luminance of 200 cd m−2 (driving voltage of 3 V),3 and this is attributed to the formation of a cage-like arrangement of ligands that prevents expansion of the coordination sphere in the excited state, thereby inhibiting attack at the iridium centre by water with subsequent quenching. Since our initial report of this effect,3 N^N ligands with pendant 6-substituted aryl groups have become a standard design feature of our iTMCs.11–15 Interestingly, modifying the substitution pattern of the bpy ligand to permit two π-stacking interactions within the coordination sphere does not further enhance the device lifetime.16
 |
| | Fig. 1 Schematic representation of [Ir(ppy)2(6-Phbpy)]+ and structural figure showing the face-to-face stacking of the phenyl substitution and phenyl ring of one [ppy]− ligand. | |
In contrast to the wide range of bpy- or substituted bpy-containing [Ir(C^N)2(N^N)]+ complexes reported,2 only two well-characterized examples of [Ir(C^N)2(N^N)]+ cations in which N^N is a derivative of 2,2′:6′,2′′-terpyridine (tpy) have been described.7,17 Room temperature, degassed MeCN solutions of [Ir(ppy)2(4′-tolyltpy)][PF6] (4′-tolyltpy = 4′-(4-tolyl)-2,2′:6′,2′′-terpyridine) show an emission maximum in the red-orange region (λemmax = 625 nm) which shifts to 560 nm at 77 K in MeOH–EtOH. The related complex [Ir(ppy)2(4′-HO2CC6H4tpy)][PF6] emits at 632 nm in MeCN at room temperature, and at 615 nm in a solid film.17 No structural data are available for these compounds, and a search of the Cambridge Structural Database18 (v. 5.34 with November 2012 updates, using Conquest v. 1.1519) reveals no structures of complexes containing an {Ir(ppy)2(tpy)}-unit. The preferred octahedral geometry of iridium(III) should dictate a bidentate mode for the tpy ligand in [Ir(C^N)2(tpy)]+ cations, leaving a pendant pyridine ring that can engage in a face-to-face π-interaction analogous to that shown for the phenyl rings in Fig. 1.
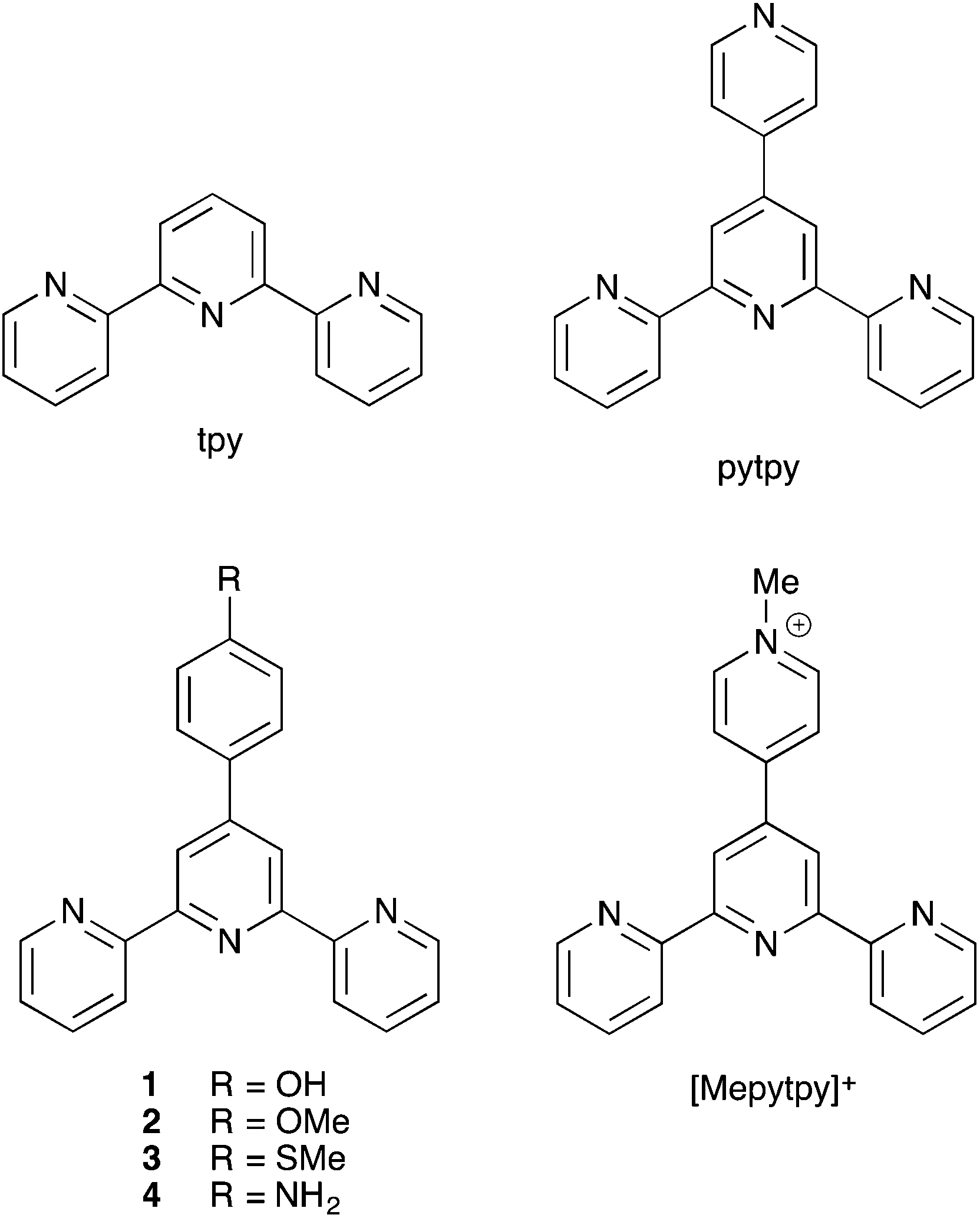
In this paper, we describe a series of [Ir(ppy)2(N^N)]+ complexes in which N^N is a tpy ligand. With the known orange-red emission7 of [Ir(ppy)2(4′-tolyltpy)]+ as a starting point, this family of complexes appeared suited for achieving deep red emitters. To date, red-emitting [Ir(C^N)2(N^N)]+ complexes which have been reported include those with photoluminescence emission maxima (in solution) of ≈650,20 65621 and 687 nm,22 and more recently, two red-emitting [Ir(C^N)2(N^N)]+ complexes containing 1,3,4-oxadiazole ligands and giving high efficiency LECs.23 To achieve a smaller HOMO–LUMO gap and a longer wavelength emission, the HOMO should be destabilized and/or the LUMO stabilized.2 In the first series of complexes discussed here, the C^N ligand is maintained as [ppy]−, leading to an effectively constant HOMO level. The energy of the LUMO may be lowered by introducing electron-withdrawing substituents into the N^N ligand or extending the π-conjugation in the latter. The N^N ligands (Scheme 1) selected for this study were the parent tpy, ligands 1–4 which contain an extended π-system and a remote electron-withdrawing group, and 4′-pyridyl-2,2′:6′,2′′-terpyridine (pytpy) in which the π-system extends to the additional pyridine ring. For [Ir(C^N)2(tpy)]+ and [Ir(C^N)2(pytpy)]+, we also investigate the effects of changing the C^N ligand from [ppy]− to [dmppz]− (Hdmppz = 3,5-dimethyl-1-phenyl-1H-pyrazole).
 |
| | Scheme 1 Structures of the N^N ligands. | |
Experimental
General
1H and 13C NMR spectra were recorded using a Bruker Avance III-500 NMR spectrometer at 295 K unless otherwise stated; chemical shifts were referenced to residual solvent peaks with respect to δ(TMS) = 0 ppm. Electrospray ionization (ESI) mass spectra were recorded on a Bruker esquire 3000plus instrument. Solution absorption and emission spectra were recorded on an Agilent 8453 spectrophotometer and a Shimadzu RF-5301 PC spectrofluorometer, respectively. A Shimadzu 8400S instrument with Golden Gate accessory (solid samples) was used to record FT-IR spectra. Quantum yields were measured using a Hamamatsu absolute PL quantum yield spectrometer C11347 Quantaurus-QY. Lifetimes were measured using a Hamamatsu Compact Fluorescence lifetime spectrometer C11367 Quantaurus-Tau; an LED light source with an excitation wavelength of 280 nm was used. Microwave reactions were carried out in a Biotage Initiator 8 reactor.
Electrochemical measurements were recorded using a CH Instruments 900B potentiostat with glassy carbon, a platinum wire and a silver wire as the working, counter, and reference electrodes, respectively. Samples were dissolved in HPLC grade MeCN (≈ 10−4 mol dm−3) containing 0.1 mol dm−3 [nBu4N][PF6] as the supporting electrolyte; all solutions were degassed with argon. Cp2Fe was used as an internal reference.
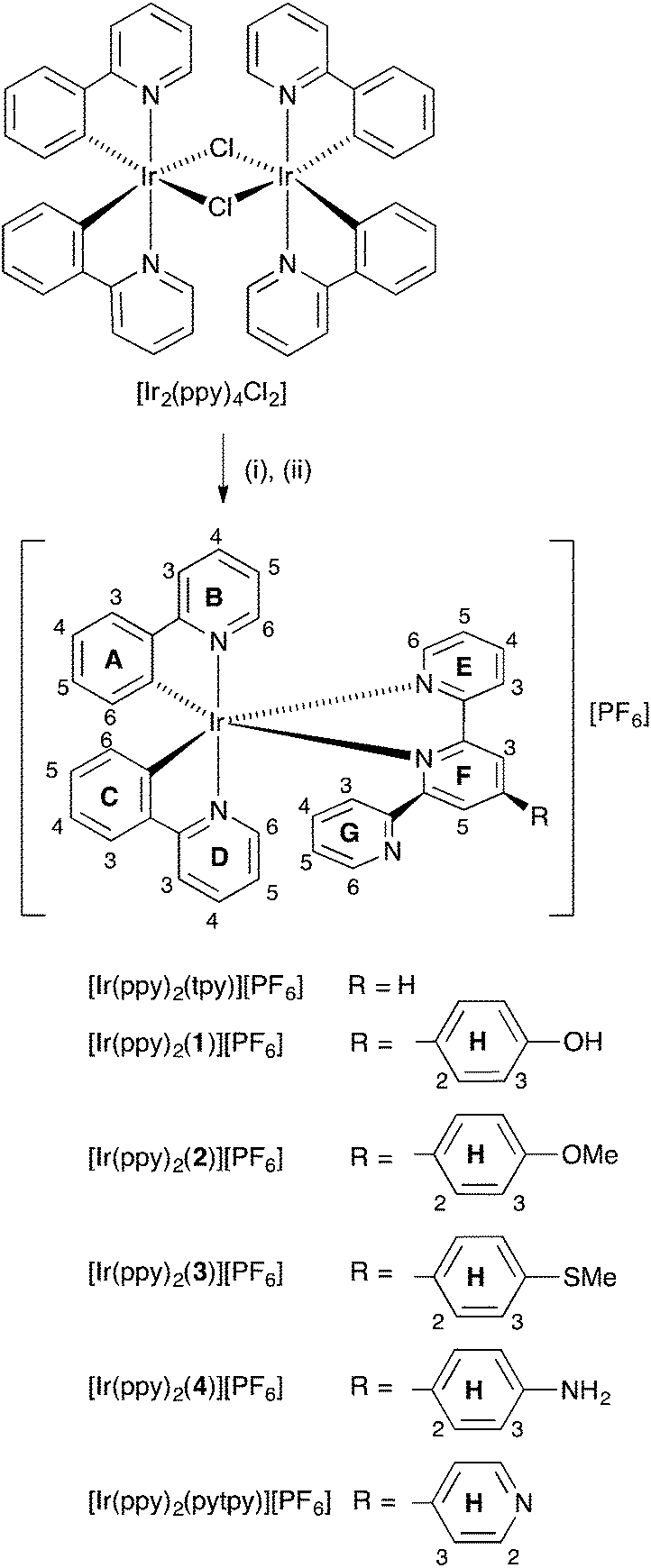
Literature procedures were followed for the preparation of tpy,24 pytpy,25 [Mepytpy][PF6],261,252,253,27 and 4,28 and the dimers [Ir2(ppy)4Cl2] and [Ir2(dmppz)4Cl2] were prepared by a standard method.29
[Ir(ppy)2(tpy)][PF6].
A yellow suspension of [Ir2(ppy)4Cl2] (100.0 mg, 0.0933 mmol) and tpy (43.8 mg, 0.188 mmol) in MeOH (15 mL) was heated in a microwave reactor for 30 min at 120 °C (P = 13 bar). The orange solution was then cooled to room temperature, and an excess of solid NH4PF6 was added. The mixture was stirred for 30 min at room temperature and then evaporated to dryness. The product was purified using column chromatography (Merck alumina 90; CH2Cl2 changing to CH2Cl2–MeOH = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), and was then redissolved in CH2Cl2–MeOH and over-layered with Et2O. Pure product precipitated over a few days and [Ir(ppy)2(tpy)][PF6] was isolated as an orange solid (78.4 mg, 0.0892 mmol, 47.6%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.87 (br, 1H, HB6), 8.54 (overlapping d, 2H, HF3+E3), 8.21 (m, 2H, HG6+E4/F4), 8.16 (td, J = 8.1, 1.6 Hz, 1H, HF4/E4), 7.89 (m, 1H, HB3), 7.84 (d, J = 7.9 Hz, 1H, HD3), 7.81–7.77 (m, 3H, HB4+D4+E6), 7.62 (dd, J = 7.8, 1.2 Hz, 1H, HA3), 7.45 (dd, J = 7.7, 1.2 Hz, 1H, HF5), 7.39 (m, 2H, HD6+E5), 7.34 (dd, J = 7.8, 1.2 Hz, 1H, HC3), 7.15 (m, 2H, HB5+G4), 6.97 (m, 3H, HA4+D5+G5), 6.76 (td, J = 7.5, 1.4 Hz, 1H, HA5), 6.64 (m, 1H, HC4), 6.53 (d, J = 7.7 Hz, 1H, HG3), 6.33 (td, J = 7.5, 1.3 Hz, 1H, HC5), 5.89 (dd, J = 7.7, 0.9 Hz, 1H, HA6), 5.45 (m, 1H, HC6). 13C NMR (126 MHz, CD2Cl2) δ/ppm 168.7 (CD2), 167.0 (CB2), 163.4 (CF6), 157.5 (CE2/F2), 157.4 (CE2/F2), 156.4 (CG2), 152.9 (CB6), 150.9 (CE6), 150.4 (CC1), 148.7 (CG6), 148.4 (CD6), 146.7 (CA1), 143.6 (CA2), 142.9 (CC2), 140.4 (CF4), 139.9 (CE4), 138.7 (CB4/D4), 138.6 (CB4/D4), 136.9 (CG4), 132.9 (CC6), 131.1 (CA6), 131.0 (CA5), 130.5 (CC5), 129.5 (CF5), 128.2 (CE5), 125.8 (CE3), 125.2 (CA3), 124.7 (CG5), 124.6 (CF3), 124.4 (CC3), 123.3 (CB5), 123.2 (2C, CA4+D5), 123.1 (CG3), 121.3 (CC4), 119.9 (CB3/D3), 119.8 (CB3/D3). IR (ν/cm−1) 3090 w, 2921 w, 2852 w, 2350 w, 1778 w, 1608 m, 1583 m, 1478 m, 1452 m, 1420 m, 1317 w, 1270 w, 1155 s, 1137 s, 1065 w, 1030 w, 991 w, 955 w, 886 w, 836 s, 825 s, 775 m, 757 s, 739 m, 729 m, 722 m, 697 m, 630 m, 602 s. ESI MS m/z 734.2 [M − PF6]+ (calc. 734.2). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 2.50 × 10−5 mol dm−3): 261 (48700), 375 (6100). Emission (CH2Cl2, 2.50 × 10−5 mol dm−3, λexc = 260 nm) λemmax = 590 nm. Found C 50.31, H 3.16, N 7.97; C37H27F6IrN5P requires C 50.57, H 3.10, N, 7.97%.
:5), and was then redissolved in CH2Cl2–MeOH and over-layered with Et2O. Pure product precipitated over a few days and [Ir(ppy)2(tpy)][PF6] was isolated as an orange solid (78.4 mg, 0.0892 mmol, 47.6%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.87 (br, 1H, HB6), 8.54 (overlapping d, 2H, HF3+E3), 8.21 (m, 2H, HG6+E4/F4), 8.16 (td, J = 8.1, 1.6 Hz, 1H, HF4/E4), 7.89 (m, 1H, HB3), 7.84 (d, J = 7.9 Hz, 1H, HD3), 7.81–7.77 (m, 3H, HB4+D4+E6), 7.62 (dd, J = 7.8, 1.2 Hz, 1H, HA3), 7.45 (dd, J = 7.7, 1.2 Hz, 1H, HF5), 7.39 (m, 2H, HD6+E5), 7.34 (dd, J = 7.8, 1.2 Hz, 1H, HC3), 7.15 (m, 2H, HB5+G4), 6.97 (m, 3H, HA4+D5+G5), 6.76 (td, J = 7.5, 1.4 Hz, 1H, HA5), 6.64 (m, 1H, HC4), 6.53 (d, J = 7.7 Hz, 1H, HG3), 6.33 (td, J = 7.5, 1.3 Hz, 1H, HC5), 5.89 (dd, J = 7.7, 0.9 Hz, 1H, HA6), 5.45 (m, 1H, HC6). 13C NMR (126 MHz, CD2Cl2) δ/ppm 168.7 (CD2), 167.0 (CB2), 163.4 (CF6), 157.5 (CE2/F2), 157.4 (CE2/F2), 156.4 (CG2), 152.9 (CB6), 150.9 (CE6), 150.4 (CC1), 148.7 (CG6), 148.4 (CD6), 146.7 (CA1), 143.6 (CA2), 142.9 (CC2), 140.4 (CF4), 139.9 (CE4), 138.7 (CB4/D4), 138.6 (CB4/D4), 136.9 (CG4), 132.9 (CC6), 131.1 (CA6), 131.0 (CA5), 130.5 (CC5), 129.5 (CF5), 128.2 (CE5), 125.8 (CE3), 125.2 (CA3), 124.7 (CG5), 124.6 (CF3), 124.4 (CC3), 123.3 (CB5), 123.2 (2C, CA4+D5), 123.1 (CG3), 121.3 (CC4), 119.9 (CB3/D3), 119.8 (CB3/D3). IR (ν/cm−1) 3090 w, 2921 w, 2852 w, 2350 w, 1778 w, 1608 m, 1583 m, 1478 m, 1452 m, 1420 m, 1317 w, 1270 w, 1155 s, 1137 s, 1065 w, 1030 w, 991 w, 955 w, 886 w, 836 s, 825 s, 775 m, 757 s, 739 m, 729 m, 722 m, 697 m, 630 m, 602 s. ESI MS m/z 734.2 [M − PF6]+ (calc. 734.2). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 2.50 × 10−5 mol dm−3): 261 (48700), 375 (6100). Emission (CH2Cl2, 2.50 × 10−5 mol dm−3, λexc = 260 nm) λemmax = 590 nm. Found C 50.31, H 3.16, N 7.97; C37H27F6IrN5P requires C 50.57, H 3.10, N, 7.97%.
[Ir(ppy)2(1)][PF6].
A yellow suspension of [Ir2(ppy)4Cl2] (107.2 mg, 0.100 mmol) and 1 (64.7 mg, 0.199 mmol) in MeOH (15 mL) was heated in a microwave reactor for 2 h at 120 °C (P = 13 bar). The orange solution was cooled to room temperature, and excess solid NH4PF6 was added. The mixture was stirred for 30 minutes at room temperature, but no precipitate formed. An excess of AgPF6 was added, the mixture was stirred for 1 h and then evaporated to dryness. The crude material was purified twice by column chromatography (Merck alumina 90, CH2Cl2 changing to CH2Cl2–MeOH–MeCN = 100:5:5, followed by Fluka silica 60; CH2Cl2 changing to CH2Cl2–MeOH = 100:3). [Ir(ppy)2(1)][PF6] was isolated as an orange solid (154.0 mg, 0.158 mmol, 79.5%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.98 (br, 1H, HB6), 8.68 (d, J = 8.3 Hz, 1H, HE3), 8.58 (d, J = 2.0 Hz, 1H, HF3), 8.48 (br, 1H, HOH), 8.21 (br, 1H, HG6), 8.16 (ddd, J = 8.2, 7.7, 1.6 Hz, 1H, HE4), 7.89 (m, 1H, HB3/D3), 7.86 (m, 1H, HB3/D3), 7.84–7.78 (m, 3H, HB4+D4+E6), 7.59 (dd, J = 7.8, 1.2 Hz, 1H, HA3), 7.56 (m, 2H, HH2), 7.54 (d, J = 6.0 Hz, 1H, HD6), 7.43 (d, J = 1.9 Hz, 1H, HF5), 7.38 (m, 2H, HE5+C3), 7.17 (m, 2H, HB5+G4), 7.02–6.95 (m, 3H, HD5+G5+A4), 6.80 (td, J = 7.5, 1.4 Hz, 1H, HA5), 6.71 (d, J = 8.6 Hz, 2H, HH3), 6.66 (ddd, J = 7.8, 7.3, 1.2 Hz, 1H, HC4), 6.56 (br, 1H, HG3), 6.38 (td, J = 7.5, 1.2 Hz, 1H, HC5), 5.91 (m, 1H, HA6), 5.53 (d, J = 7.5 Hz, 1H, HC6). 13C NMR (126 MHz, CD2Cl2) δ/ppm 168.7 (CD2), 167.2 (CB2), 160.4 (CF6), 160.0 (CH4), 157.5 (CF2), 157.3 (CE2), 155.5 (CG2), 151.9 (CB6), 151.1 (CF4), 150.9 (CC1), 150.8 (CE6), 149.1 (CD6), 148.2 (CG6), 147.4 (CA1), 143.7 (CA2), 143.2 (CC2), 139.8 (CE4), 138.7 (CB4/D4), 138.6 (CB4/D4), 137.2 (CG4), 132.5 (CC6), 131.0 (CA5), 130.9 (CA6), 130.5 (CC5), 129.4 (CH2), 128.2 (CE5), 126.0 (CH1), 125.9 (CF5), 125.8 (CE3), 125.1 (2C, CA3+G5), 124.6 (CC3), 123.9 (CG3), 123.8 (CB5), 123.3 (CA4), 123.2 (CD5), 121.3 (CC4), 120.9 (CF3), 120.0 (2C, CB3+D3), 117.5 (CH3). IR (ν/cm−1) 3043 (w, νOH), 2601 w, 2162 w, 1663 w, 1607 s, 1583 s, 1520 m, 1477 s, 1438 m, 1419 m, 1404 m, 1367 w, 1319 w, 1285 m, 1270 m, 1233 m, 1163 m, 1042 m, 1031 m, 1025 m, 1009 m, 830 s, 828 s, 805 s, 790 s, 755 s, 739 s, 730 s, 627 s, 618 s. ESI MS m/z 826.3 [M − PF6]+ (calc. 826.2). UV-Vis (CH2Cl2, 1.00 × 10−5 mol dm−3): λ/nm (ε/dm3 mol−1 cm−1) 256 (52700), 364 (26600). Emission (CH2Cl2, 1.00 × 10−5 mol dm−3, λexc = 360 nm): λemmax = 590 nm. Found C 48.92, H 3.37, N 6.61; C43H31F6IrN5OP·4H2O requires C 49.52, H 3.77, N 6.71%.
[Ir(ppy)2(2)][PF6].
The method was the same as that for [Ir(ppy)2(1)][PF6] starting with [Ir2(ppy)4Cl2] (107.2 mg, 0.100 mmol) and 2 (67.5 mg, 0.199 mmol). The product was purified twice by column chromatography (Merck alumina 90 and then Fluka silica 60; CH2Cl2 changing to CH2Cl2–MeOH = 100:2 for both columns). [Ir(ppy)2(2)][PF6] was isolated as an orange solid (159 mg, 0.162 mmol, 81.2%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.89 (br, 1H, HB6), 8.67 (d, J = 2.0 Hz, 1H, HF3), 8.65 (d, J = 8.3 Hz, 1H, HE3), 8.22 (broadened d, 1H, HG6), 8.17 (ddd, J = 8.2, 7.7, 1.6 Hz, 1H, HE4), 7.88–7.83 (overlapping m, 3H, HB3+D3+H2), 7.78 (m, 3H, HB4+D4+E6), 7.62 (m, 2H, HA3+F5), 7.45 (d, J = 5.8 Hz, 1H, HD6), 7.39 (ddd, J = 7.6, 5.5, 1.2 Hz, 1H, HE5), 7.36 (dd, J = 7.8, 1.1 Hz, 1H, HC3), 7.14 (m, 2H, HB5+G4), 7.09 (m, 2H, HH3), 6.97 (m, 2H, HA4+D5), 6.93 (ddd, J = 7.6, 4.9, 0.9 Hz, 1H, HG5), 6.77 (td, J = 7.5, 1.3 Hz, 1H, HA5), 6.64 (m, 1H, HC4), 6.55 (broadened d, 1H, HG3), 6.34 (td, J = 7.4, 1.3 Hz, 1H, HC5), 5.90 (dd, J = 7.8, 0.8 Hz, 1H, HA6), 5.47 (dd, J = 7.6, 0.6 Hz, 1H, HC6), 3.88 (s, 3H, HMe). 13C NMR (126 MHz, CD2Cl2) δ/ppm 168.8 (CD2), 167.1 (CB2), 163.6 (CF6), 162.9 (CH4), 157.8 (CF2), 157.5 (CE2), 156.6 (CG2), 152.8 (CB6), 151.5 (CF4), 150.9 (CE6), 150.6 (CC1), 148.5 (CD6), 148.3 (CG6), 147.3 (CA1), 143.7 (CA2), 142.8 (CC2), 139.8 (CE4), 138.6 (CB4/D4), 138.5 (CB4/D4), 137.1 (CG4), 132.8 (CC6), 131.1 (CA6), 131.0 (CA5), 130.5 (CC5), 129.4 (CH2), 128.2 (CE5), 127.5 (CH1), 125.9 (CF5), 125.7 (CE3), 125.1 (CA3), 124.6 (CG5), 124.3 (CC3), 123.3 (3C, CA4+B5+G3), 123.2 (CD5), 121.3 (CC4), 121.1 (CF3), 119.9 (CB3/D3), 119.8 (CB3/D3), 115.6 (CH3), 56.2 (CMe). IR (ν/cm−1) 3042 w, 2344 w, 1700 m, 1603 s, 1583 s, 1517 m, 1476 s, 1459 m, 1437 m, 1424 m, 1420 m, 1410 m, 1399 m, 1366 w, 1303 m, 1268 m, 1247 s, 1231 m, 1181 m, 1164 m, 1122 w, 1095 w, 1065 w, 1030 m, 1003 m, 994 w, 879 w, 828 s, 828 s, 804 s, 788 s, 753 s, 727 s. ESI MS m/z 840.3 [M − PF6]+ (calc. 840.2). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 1.00 × 10−5 mol dm−3) 258 (42200), 274 (42700), 365 (15300), 386 (10500). Emission (CH2Cl2, 1.00 × 10−5 mol dm−3, λexc = 320 nm) λemmax = 589 nm. Found C 52.55, H 3.40, N 6.92; C44H33F6IrN5OP·H2O requires C 52.69, H 3.52, N, 6.98%.
[Ir(ppy)2(3)][PF6].
The method was the same as that for [Ir(ppy)2(1)][PF6] starting with [Ir2(ppy)4Cl2] (107.2 mg, 0.100 mmol) and 3 (70.7 mg, 0.199 mmol). Chromatographic purification was the same as that for [Ir(ppy)2(2)][PF6]. [Ir(ppy)2(3)][PF6] was isolated as a pale orange solid (180.0 mg, 0.179 mmol, 90.2%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.86 (br, 1H, HB6), 8.70 (d, J = 1.9 Hz, 1H, HF3), 8.66 (d, J = 8.2 Hz, 1H, HE3), 8.23 (d, J = 4.7 Hz, 1H, HG6), 8.17 (td, J = 8.0, 1.6 Hz, 1H, HE4), 7.88 (m, 2H, HB3+D3), 7.80 (m, 5H, HB4+D4+E6+H2), 7.65 (s, 1H, HF5), 7.62 (dd, J = 7.9, 1.1 Hz, 1H, HA3), 7.46 (d, J = 5.5 Hz, 1H, HD6), 7.41 (m, 3H, HC3+H3), 7.37 (dd, J = 7.8, 1.0 Hz, 1H, HE5), 7.16 (m, 2H, HB5+G4), 6.98 (m, 3H, HA4+D5+G5), 6.78 (td, J = 7.5, 1.3 Hz, 1H, HA5), 6.65 (td, J = 7.8, 1.2 Hz, 1H, HC4), 6.57 (broadened d, 1H, HG3), 6.35 (td, J = 7.5, 1.2 Hz, 1H, HC5), 5.90 (dd, J = 7.7, 0.9 Hz, 1H, HA6), 5.49 (d, J = 7.8 Hz, 1H, HC6), 2.55 (s, 3H, HMe). 13C NMR (126 MHz, CD2Cl2, 298 K) δ/ppm 168.7 (CD2), 167.1 (CB2), 157.9 (CF2), 157.4 (CE2), 152.8 (CG2), 152.6 (CB6), 151.3 (CF4), 150.9 (CE6), 150.5 (CC1), 148.6 (CD6), 148.1 (CG6), 147.2 (CA1), 144.3 (CH4), 143.7 (CA2), 142.9 (CC2), 139.8 (CE4), 138.7 (CB4/D4), 138.6 (CB4/D4), 136.9 (CG4), 132.9 (CC6), 131.3 (CH1), 131.1 (CA6), 131.0 (CA5), 130.5 (CC5), 128.4 (CE5), 128.1 (CH2), 126.8 (CH3), 126.3 (CF5), 125.8 (CE3), 125.1 (CA3), 124.8 (CG5), 124.4 (CC3), 123.4 (2C, CB5+G3), 123.3 (CA4), 123.2 (CD5), 121.3 (2C, CC4+F3), 119.9 (CB3/D3), 119.8 (CB3/D3), 15.3 (CMe); signal for CF6 not resolved. IR (ν/cm−1) 3038 w, 1700 w, 1607 s, 1583 s, 1565 m, 1476 s, 1420 m, 1393 m, 1364 w, 1317 w, 1307 w, 1269 w, 1255 w, 1228 w, 1164 m, 1099 m, 1064 w, 1030 m, 993 w, 960 w, 879 w, 831 s, 831 s, 785 m, 753 s, 727 s, 710 m, 666 w, 627 s, 610 s, 603 m. ESI MS m/z 856.3 [M − PF6]+ (calc. 856.2). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 1.00 × 10−5 mol dm−3) 256 (52700), 288 (41200), 343 (26100), 364 (26600), 372 (25700), 394 (18000). Emission (CH2Cl2, 1.00 × 10−5 mol dm−3, λexc = 325 nm) λemmax = 595 nm. Found C 51.41, H 3.27, N 6.81; C44H33F6IrN5PS·H2O requires C 51.86, H 3.46, N, 6.87%.
[Ir(ppy)2(4)][PF6].
A yellow suspension of [Ir2(ppy)4Cl2] (107.2 mg, 0.100 mmol) and 4 (65.7 mg, 0.201 mmol) in MeOH (15 mL) was heated in a microwave reactor for 2 h at 120 °C (P = 15 bar). The orange solution was then cooled to room temperature, and an excess of solid NH4PF6 was added. The mixture was stirred for 1 h at room temperature and then the solvent was evaporated to dryness. The crude orange product was dissolved in CH2Cl2 and the solution layered with Et2O. After 2 days, the precipitate that had formed was separated by decanting the solvent, and [Ir(ppy)2(4)][PF6] was isolated as an orange-red solid (190 mg, 0.196 mmol, 97.2%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.90 (br, 1H, HB6), 8.62 (m, 2H, HE3+F3), 8.22 (broadened d, 1H, HG6), 8.14 (m, 1H, HE4), 7.87 (m, 2H, HB3+D3), 7.79 (m, 3H, HB4+D4+E6), 7.70 (m, 2H, HH2), 7.62 (dd, J = 7.9, 1.2 Hz, 1H, HA3), 7.56 (d, J = 2.0 Hz, 1H, HF5), 7.45 (d, J = 5.7 Hz, 1H, HD6), 7.36 (m, 2H, HE5+C3), 7.14 (m, 1H, HB5), 7.12 (td, J = 7.8, 1.8 Hz, 1H, HG4), 6.96 (m, 2H, HA4+D5), 6.92 (ddd, J = 7.6, 4.9, 1.1 Hz, 1H, HG5), 6.80 (m, 2H, HH3), 6.77 (m, 1H, HA5), 6.64 (td, J = 7.7, 1.2 Hz, 1H, HC4), 6.54 (broadened d, 1H, HG3), 6.33 (m, 1H, HC5), 5.91 (dd, J = 7.7, 0.9 Hz, 1H, HA6), 5.47 (dd, J = 7.6, 0.7 Hz, 1H, HC6), 3.65 (br, HNH2). 13C NMR (126 MHz, CD2Cl2) δ/ppm 168.8 (CD2), 167.1 (CB2), 163.3 (CF6), 157.7 (CE2), 157.5 (CF2), 156.7 (CG2), 152.7 (CB6), 151.6 (CF4), 150.8 (2C, CE6+H4), 150.7 (CC1), 148.6 (CG6), 148.4 (CD6), 147.5 (CA1), 143.7 (CA2), 142.8 (CC2), 139.7 (CE4), 138.6 (CB4/D4), 138.5 (CB4/D4), 136.6 (CG4), 132.8 (CC6), 131.1 (CA6), 130.9 (CA5), 130.4 (CC5), 129.2 (CH2), 128.0 (CE5), 125.5 (CE3), 125.1 (CA3), 124.9 (CF5), 124.5 (CG5), 124.3 (CC3), 123.9 (CH1), 123.3 (CB5), 123.2 (CD5), 123.1 (2C, CA4+G3), 121.2 (CC4), 120.1 (CF3), 119.9 (CB3/D3), 119.8 (CB3/D3), 115.7 (CH3). IR (ν/cm−1) 3393 w, 3048 w, 2356 w, 1700 m, 1597 m, 1583 m, 1521 w, 1474 m, 1412 w, 1364 w, 1305 w, 1254 w, 1228 w, 1189 w, 1164 w, 1064 w, 1030 w, 1003 w, 942 w, 879 w, 828 s, 803 m, 788 m, 753 m, 728 m, 668 m, 602 s. ESI MS m/z 825.4 [M − PF6]+ (calc. 825.2). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 1.00 × 10−5 mol dm−3) 259 (52100), 339 (24800), 382 (25500), 475 (2100). Emission (CH2Cl2, 1.00 × 10−5 mol dm−3, λexc = 255 nm) λemmax = 580 nm. Found C 51.87, H 3.48, N 8.38; C43H32F6IrN6P·H2O requires C 52.28, H 3.47, N, 8.51%.
[Ir(ppy)2(pytpy)][PF6].
A yellow suspension of [Ir2(ppy)4Cl2] (107.6 mg, 0.100 mmol) and pytpy (62.4 mg, 0.201 mmol) in MeOH (15 mL) was heated in a microwave reactor for 30 min at 120 °C (P = 14 bar). The red solution was then cooled to room temperature, and an excess of solid NH4PF6 was added. The mixture was stirred for 1 h at room temperature, and the precipitate that formed was separated by filtration and washed with water (3 × 25 mL) and Et2O (3 × 25 mL). The product was dissolved in CH2Cl2–MeOH and overlayered with n-hexane to give a red solid. This process was repeated, and the solid was then washed with Et2O. [Ir(ppy)2(pytpy)][PF6] was isolated as a red solid (145 mg, 0.152 mmol, 76.1%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.94 (br, 1H, HB6), 8.80 (d, J = 2.0 Hz, 1H, HF3), 8.79 (m, 2H, HH2), 8.75 (d, J = 8.3 Hz, 1H, HE3), 8.25 (broadened d, 1H, HG6), 8.19 (ddd, J = 8.1, 7.7, 1.6 Hz, 1H, HE4), 7.91 (m, 1H, HB3), 7.86 (m, 1H, HD3), 7.84–7.76 (m, 5H, HB4+D4+E6+H3), 7.67 (d, J = 1.9 Hz, 1H, HF5), 7.64 (dd, J = 7.9, 1.2 Hz, 1H, HA3), 7.44 (ddd, J = 5.9, 1.2, 0.5 Hz, 1H, HD6), 7.42 (ddd, J = 7.6, 5.5, 1.2 Hz, 1H, HE5), 7.37 (dd, J = 7.8, 1.1 Hz, 1H, HC3), 7.15 (m, 2H, HB5+G4), 6.97 (m, 3H, HA4+D5+G5), 6.77 (m, 1H, HA5), 6.65 (ddd, J = 7.8, 7.3, 1.2 Hz, 1H, HC4), 6.60 (broadened d, 1H, HG3), 6.34 (m, 1H, HC5), 5.90 (dd, J = 7.7, 0.8 Hz, 1H, HA6), 5.47 (dd, J = 7.6, 0.8 Hz, 1H, HC6). 13C NMR (126 MHz, CD2Cl2) δ/ppm 168.6 (CD2), 167.0 (CB2), 164.2 (CF6), 158.6 (CF2), 156.9 (CE2), 156.2 (CG2), 152.9 (CB6), 151.1 (CH2), 150.9 (CE6), 150.3 (CC1), 149.3 (CH4), 148.9 (CG6), 148.5 (CD6), 146.9 (CA1), 143.6 (2C, CA2+F4), 142.9 (CC2), 140.0 (CE4), 138.7 (CB4/D4), 138.6 (CB4/D4), 136.7 (CG4), 132.9 (CC6), 131.1 (CA6), 131.0 (CA5), 130.5 (CC5), 128.5 (CE5), 126.9 (CF5), 126.3 (CE3), 125.2 (CA3), 124.8 (CG5), 124.3 (CC3), 123.4 (2C, CA4+B5), 123.3 (CD5), 123.2 (CG3), 122.2 (CH3), 122.1 (CF3), 121.4 (CC4), 120.0 (CB3/D3), 119.9 (CB3/D3). IR (ν/cm−1) 3043 w, 2344 w, 1607 m, 1583 m, 1535 w, 1477 m, 1439 w, 1419 m, 1398 m, 1316 w, 1270 w, 1228 w, 1164 w, 1065 w, 1031 w, 993 w, 879 w, 828 s, 788 m, 754 m, 728 m, 668 m, 624 m, 602 m. ESI MS m/z 811.3 [M − PF6]+ (calc. 810.9). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 2.50 × 10−5 mol dm−3): 258 (68100), 350 (11400). Emission (CH2Cl2, 2.50 × 10−5 mol dm−3, λexc = 250 nm) λemmax = 640 nm. Found C 51.85, H 3.23, N 8.64; C42H32F6IrN6P·H2O requires C 51.80, H 3.31, N, 8.63%.
[Ir(dmppz)2(tpy)][PF6].
The method and purification were the same as that for [Ir(ppy)2(tpy)][PF6], starting with [Ir2(dmppz)4Cl2] (114.0 mg, 0.100 mmol) and tpy (46.9 mg, 0.201 mmol). [Ir(dmppz)2(tpy)][PF6] was isolated as an orange solid (179.2 mg, 196.2 mmol, 98.1%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.55 (dd, J = 8.2, 1.3 Hz, 1H, HF3), 8.51 (d, J = 8.2 Hz, 1H, HE3), 8.24 (t, J = 7.9 Hz, 1H, HF4), 8.09 (m, 2H, HE4+G6), 7.76 (ddd, J = 5.5, 1.6, 0.7 Hz, 1H, HE6), 7.48 (dd, J = 7.7, 1.3 Hz, 1H, HF5), 7.35 (ddd, J = 7.6, 5.5, 1.2 Hz, 1H, HE5), 7.29 (dd, J = 8.2, 1.0 Hz, 1H, HA3), 7.14 (td, J = 7.7, 1.7 Hz, 1H, HG4), 6.97 (ddd, J = 8.1, 7.4, 1.4 Hz, 1H, HA4), 6.92 (dd, J = 8.2, 0.8 Hz, 1H, HC3), 6.85 (m, 2H, HG5), 6.73 (m, 1H, HA5), 6.57 (ddd, J = 8.2, 7.4, 1.4 Hz, 1H, HC4), 6.42 (td, J = 7.4, 1.1 Hz overlapping br signal, 2H, HC5+G3), 6.15 (s, 1H, HD4), 6.05 (s, 1H, HB4), 5.99 (dd, J = 7.6, 1.4 Hz, 1H, HA6), 5.74 (dd, J = 7.5, 1.3 Hz, 1H, HC6), 2.79 (s, 3H, HMe-D3), 2.69 (s, 3H, HMe-B3), 1.81 (s, 3H, HMe-B5), 1.52 (s, 3H, HMe-D5). 13C NMR (126 MHz, CD2Cl2) δ/ppm 164.4 (CF6), 157.8 (CE2/F2), 157.6 (CE2/F2), 154.9 (CG2), 150.8 (CB5), 150.5 (CD5), 150.4 (CE6), 148.9 (CG6), 144.6 (CA2/C2), 144.4 (CA2/C2), 142.0 (CB3), 141.5 (CD3), 140.4 (CF4), 139.7 (CE4), 135.7 (CG4), 134.4 (CC6), 133.2 (CC1), 133.0 (CA6), 130.8 (CA1), 130.3 (CF5), 128.0 (CE5), 126.2 (CA5), 125.3 (CE3), 124.9 (CC5), 124.6 (CG5), 123.9 (2C, CA4+F3), 123.1 (CG3), 121.9 (CC4), 113.2 (CA3), 112.6 (CC3), 110.8 (CB4/D4), 110.5 (CB4/D4), 15.1 (CMe-D3), 14.8 (CMe-B3), 12.9 (CMe-D5), 12.5 (CMe-B5). IR (ν/cm−1) 2920 w, 2366 w, 1988 w, 1696 w, 1604 w, 1568 w, 1553 m, 1471 m, 1445 w, 1428 w, 1397 w, 1372 w, 1312 w, 1296 w, 1274 w, 1234 w, 1192 w, 1167 w, 1150 w, 1122 w, 1075 w, 1036 w, 992 w, 925 w, 906 w, 884 w, 835 s, 823 m, 771 w, 744 m, 732 m, 717 m, 691 w, 636 m. ESI m/z 768.2 [M − PF6]+ (calc. 768.2). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 2.50 × 10−5 mol dm−3) 250 (53000), 290 (33300), 350 (7200). Emission (CH2Cl2, 2.50 × 10−5 mol dm−3, λexc = 250 nm) λemmax = 599 nm. Found C 48.15, H 3.60, N 10.55; C37H33F6IrN7P·0.5H2O requires C 48.21, H 3.72, N, 10.64%.
[Ir(dmppz)2(pytpy)][PF6].
The method was the same as that for [Ir(ppy)2(1)][PF6] starting with [Ir2(dmppz)4Cl2] (114.0 mg, 0.100 mmol) and pytpy (62.4 mg, 0.201 mmol). The product was purified twice by column chromatography (Merck alumina 90 and then Fluka silica 60; CH2Cl2 changing to CH2Cl2–MeOH 100:5). [Ir(dmppz)2(pytpy)][PF6] was isolated as a red solid (135.0 mg, 0.136 mmol, 68.0%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 8.83 (m, 2H, HH2), 8.79 (d, J = 2.0 Hz, 1H, HF3), 8.72 (d, J = 8.2 Hz, 1H, HE3), 8.13 (m, 2H, HE4+G6), 7.82 (m, 2H, HH3), 7.79 (ddd, J = 5.5, 1.6, 0.7 Hz, 1H, HE6), 7.72 (d, J = 1.9 Hz, 1H, HF5), 7.39 (m, 1H, HE5), 7.31 (dd, J = 8.2, 1.0 Hz, 1H, HA3), 7.16 (td, J = 7.7, 1.7 Hz, 1H, HG4), 6.98 (ddd, J = 8.2, 7.4, 1.4 Hz, 1H, HA4), 6.93 (dd, J = 8.2, 0.8 Hz, 1H, HC3), 6.88 (m, 1H, HG5), 6.74 (m, 1H, HA5), 6.58 (ddd, J = 8.2, 7.4, 1.4 Hz, 1H, HC4), 6.50 (br, 1H, HG3), 6.44 (td, J = 7.4, 1.1 Hz, 1H, HC5), 6.15 (s, 1H, HD4), 6.06 (s, 1H, HB4), 6.00 (dd, J = 7.6, 1.4 Hz, 1H, HA6), 5.76 (dd, J = 7.5, 1.3 Hz, 1H, HC6), 2.79 (s, 3H, HMe-D3), 2.70 (s, 3H, HMe-B3), 1.90 (s, 3H, HMe-B5), 1.54 (s, 3H, HMe-D5). 13C NMR (126 MHz, CD2Cl2) δ/ppm 165.1 (CF6), 159.0 (CF2), 157.3 (CE2), 154.7 (CG2), 151.4 (CH2), 150.8 (CB5), 150.6 (CE6), 150.5 (CD5), 149.4 (CH4), 149.1 (CG6), 144.6 (CA2/C2), 144.4 (CA2/C2), 143.5 (CF4), 142.1 (CB3), 141.5 (CD3), 139.9 (CE4), 135.7 (CG4), 134.5 (CC6), 133.2 (CC1), 133.0 (CA6), 130.8 (CA1), 128.3 (CE5), 127.7 (CF5), 126.3 (CA5), 125.6 (CE3), 125.0 (CC5), 124.8 (CG5), 124.0 (CA4), 123.2 (CG3), 122.1 (CH3), 122.0 (CC4), 121.4 (CF3), 113.3 (CA3), 112.6 (CC3), 110.9 (CB4/D4), 110.5 (CB4/D4), 15.1 (CMe-D3), 14.8 (CMe-B3), 13.0 (CMe-D5), 12.8 (CMe-B5). IR (ν/cm−1) 3131 w, 3054 w, 2162 w, 1700 m, 1585 m, 1549 m, 1541 m, 1471 s, 1442 m, 1420 m, 1398 s, 1373 m, 1276 m, 1263 m, 1239 m, 1168 w, 1143 m, 1068 w, 1037 m, 995 m, 903 m, 860 m, 830 s, 820 s, 792 s, 786 s, 753 s, 742 s, 730 s, 717 s, 703 s, 694 s, 659 m, 622 s, 612 s, 608 s. ESI MS m/z 845.3 [M − PF6]+ (calc. 845.3). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 5.00 × 10−5 mol dm−3) 253 (64600), 320 (20900), 350 (8400). Emission (CH2Cl2, 2.50 × 10−5 mol dm−3, λexc = 256 nm) λemmax = 642 nm. Found C 48.64, H 3.53, N 10.53; C42H36F6IrN8P·2H2O requires C 49.17, H 3.93, N, 10.92%.
[Ir(dmppz)2(Mepytpy)][PF6]2.
The method was the same as that for [Ir(ppy)2(1)][PF6] starting with [Ir2(dmppz)4Cl2] (114.0 mg, 0.100 mmol) and [Mepytpy][PF6] (98.8 mg, 0.21 mmol) with a reaction time of 90 min. The crude material was purified twice by column chromatography (Merck alumina 90 followed by Fluka silica 60; CH2Cl2 changing to CH2Cl2–MeOH = 100:4). [Ir(dmppz)2(Mepytpy)][PF6]2 was isolated as a red solid (124.0 mg, 0.127 mmol, 63.6%). 1H NMR (500 MHz, CD2Cl2) δ/ppm 9.03 (d, J = 2.0 Hz, 1H, HF3), 8.88 (d, J = 8.3 Hz, 1H, HE3), 8.80 (d, J = 6.9 Hz, 2H, HH2), 8.60 (d, J = 7.0 Hz, 2H, HH3), 8.14 (m, 2H, HE4+G6), 7.85 (d, J = 1.9 Hz, 1H, HF5), 7.79 (m, 1H, HE6), 7.39 (ddd, J = 7.6, 5.5, 1.1 Hz, 1H, HE5), 7.32 (dd, J = 8.2, 1.0 Hz, 1H, HA3), 7.22 (m, 1H, HG4), 6.98 (m, 1H, HA4), 6.92 (m, 2H, HC3+G5), 6.73 (m, 1H, HA5), 6.63 (s, 1H, HG3), 6.59 (m, 1H, HC4), 6.45 (m, 1H, HC5), 6.14 (s, 1H, HD4), 6.07 (s, 1H, HB4), 5.99 (dd, J = 7.6, 1.3 Hz, 1H, HA6), 5.76 (dd, J = 7.5, 1.3 Hz, 1H, HC6), 4.47 (s, 3H, HMe-H1), 2.78 (s, 3H, HMe-D3), 2.71 (s, 3H, HMe-B3), 1.93 (s, 3H, HMe-B5), 1.53 (s, 3H, HMe-D5). 13C NMR (126 MHz, CD2Cl2, 298 K) δ/ppm 165.5 (CF6), 160.0 (CF2), 156.9 (CE2), 154.0 (CG2), 152.1 (CH4), 151.1 (CB5), 150.6 (CD5), 150.4 (CE6), 149.1 (CG6), 146.3 (CH2), 144.7 (CF4), 144.5 (CA2/C2), 144.3 (CA2/C2), 142.1 (CB3), 141.6 (CD3), 140.1 (CE4), 136.0 (CG4), 134.6 (CC6), 133.0 (2C, CA6+C1), 130.4 (CA1), 128.6 (CE5), 128.1 (CF5), 127.6 (CH3), 126.5 (CE3), 126.3 (CA5), 125.1 (2C, CC5+G5), 124.1 (CA4), 123.4 (CG3), 122.2 (CC4), 122.1 (CF3), 113.3 (CA3), 112.6 (CC3), 110.9 (CB4), 110.6 (CD4), 49.4 (CMe-H1), 15.2 (CMe-D3), 14.8 (CMe-B3), 13.1 (CMe-B5), 13.0 (CMe-D5). IR (ν/cm−1) 3657 w, 3131 w, 3054 w, 1646 w, 1617 w, 1570 w, 1553 w, 1528 w, 1471 s, 1444 w, 1414 w, 1398 w, 1375 w, 1337 w, 1276 w, 1270 w, 1225 w, 1195 w, 1146 w, 1122 w, 1093 w, 1059 w, 1035 w, 993 w, 822 s, 816 s, 789 s, 773 s, 743 s, 731 w, 719 w, 692 w, 657 w, 601 s. ESI MS m/z 1005.3 [M − PF6]+ (calc. 1005.3), 535.2 [Ir(dmppz)2]+ (calc. 535.2), 430.2 [M − 2PF6]2+ (calc. 430.2). UV-Vis λ/nm (ε/dm3 mol−1 cm−1) (CH2Cl2, 2.50 × 10−5 mol dm−3) 256 (64000), 315 (19000), 400 (6000). Emission (CH2Cl2, 2.50 × 10−5 mol dm−3, λexc = 245 nm) λemmax = 359 nm. Satisfactory elemental analysis was not obtained.
Crystallography
Data were collected on either a Bruker-Nonius KappaAPEX or a Stoe IPDS diffractometer with data reduction, solution and refinement using the program APEX230 or Stoe IPDS31 software and SHELXL97.32 Structures were analysed using Mercury v. 3.0.133,34 and the program was also used to draw the ORTEP-type figures. Crystallographic data are listed in Table 1.
Table 1 Crystallographic data for the iridium(III) complexes
| Compound |
4{[Ir(ppy)2(tpy)][PF6]}·2.5Et2O·1.5MeCN·3H2O |
4{[Ir(ppy)2(2)][PF6]}·Et2O·2MeCN |
2{[Ir(ppy)2(3)][PF6]}·MeCN·H2O |
[Ir(ppy)2(4)][PF6]·2CH2Cl2 |
| Formula |
C161H143.5F24Ir4N21.5O5.5P4 |
C184H148F24Ir4N22O5P4 |
C90H73F12Ir2N11O2P2S2 |
C45H36Cl4F6IrN6P |
| Formula weight |
3816.16 |
4096.00 |
2079.12 |
1139.79 |
| Crystal colour and habit |
Yellow block |
Yellow block |
Orange block |
Orange plate |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c |
P21/n |
P21/n |
|
a, b, c/Å |
13.770(2) |
55.632(4) |
14.5443(10) |
10.2803(4) |
| 17.615(3) |
10.5213(11) |
14.7612(10) |
12.4470(4) |
| 18.315(2) |
34.013(2) |
19.8943(14) |
33.7717(12) |
|
α, β, γ/° |
85.763(12) |
90 |
90 |
90 |
| 85.821(11) |
124.848(4) |
104.707(4) |
93.369(2) |
| 67.441(11) |
90 |
90 |
90 |
|
U/Å3 |
4086.4(10) |
16338(2) |
4131.2(5) |
4313.9(3) |
|
Dc /Mg m−3 |
1.548 |
1.665 |
1.668 |
1.755 |
|
Z
|
1 |
4 |
2 |
4 |
|
μ(Mo-Kα)/mm−1 |
3.372 |
3.381 |
3.392 |
3.448 |
|
T/K |
173 |
173 |
123 |
173 |
| Refln. collected (Rint) |
93421 (0.1519) |
163250 (0.0969) |
134040 (0.0425) |
31698 (0.0542) |
| Unique refln. |
16046 |
20080 |
12026 |
8021 |
| Refln. for refinement |
13628 |
18711 |
10753 |
6667 |
| Parameters |
1048 |
1220 |
570 |
622 |
| Threshold |
I > 2.0σ |
I > 2.0σ |
I > 2.0σ |
I > 2.0σ |
|
R
1 (R1 all data) |
0.0523 (0.0605) |
0.0286 (0.0320) |
0.0194 (0.0247) |
0.0476 (0.0637) |
| wR2 (wR2 all data) |
0.1393 (0.1464) |
0.0605 (0.0617) |
0.0444 (0.0471) |
0.0993 (0.1155) |
| Goodness of fit |
1.080 |
1.195 |
1.047 |
1.151 |
| CCDC deposition‡ |
974016
|
974020
|
974021
|
974019
|
| Compound |
2{[Ir(ppy)2(pytpy)][PF6]}·Et2O·CH2Cl2 |
[Ir(dmppz)2(tpy)][PF6] |
[Ir(dmppz)2(pytpy)][PF6]·CH2Cl2 |
[Ir(dmppz)2(Mepytpy)][PF6]2·2CH2Cl2 |
| Formula |
C89H72Cl2F12Ir2N12OP2 |
C37H33F6IrN7P |
C43H38Cl2F6IrN8P |
C45H43Cl4F12IrN8P2 |
| Formula weight |
2070.87 |
912.89 |
1074.90 |
1319.83 |
| Crystal colour and habit |
Orange needle |
Yellow block |
Orange block |
Red block |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
P |
P21/n |
P |
P |
|
a, b, c/Å |
10.641(2) |
12.7713(7) |
10.8170(7) |
9.0099(16) |
| 18.025(4) |
21.4055(16) |
13.5418(8) |
14.026(3) |
| 21.629(4) |
12.8079(8) |
14.6844(9) |
20.222(5) |
|
α, β, γ/° |
88.86(3) |
90 |
94.081(2) |
101.420(16) |
| 82.15(3) |
94.874(5) |
100.297(2) |
96.000(16) |
| 81.97(3) |
90 |
103.316(2) |
99.536(14) |
|
U/Å3 |
4069.3(14) |
3488.7(4) |
2045.2(2) |
2445.4(8) |
|
D
c
/Mg m−3 |
1.690 |
1.738 |
1.745 |
1.792 |
|
Z
|
2 |
4 |
2 |
2 |
|
μ(Mo-Kα)/mm−1 |
3.457 |
3.944 |
8.510 |
3.104 |
|
T/K |
173 |
173 |
123 |
173 |
| Refln. collected (Rint) |
50153 (0.2647) |
93669 (0.0726) |
46149 (0.0286) |
26234 (0.0704) |
| Unique refln. |
14413 |
10141 |
7238 |
10145 |
| Refln. for refinement |
9184 |
9710 |
7171 |
8551 |
| Parameters |
1084 |
473 |
554 |
682 |
| Threshold |
I > 2.0σ |
I > 2.0σ |
I > 2.0σ |
I > 2.0σ |
|
R
1 (R1 all data) |
0.0995 (0.1535) |
0.0234 (0.0251) |
0.0227 (0.0229) |
0.0442 (0.0600) |
| wR2 (wR2 all data) |
0.1963 (0.2328) |
0.0575 (0.0582) |
0.0596 (0.0598) |
0.0879 (0.0926) |
| Goodness of fit |
1.043 |
1.167 |
1.100 |
1.042 |
| CCDC deposition‡ |
974018
|
974017
|
974022
|
974023
|
Device preparation
The solvents were supplied by Aldrich. The thickness of films was determined using an Ambios XP-1 profilometer. ITO-coated glass plates (15 Ω □−1) were patterned by conventional photolithography (http://www.naranjosubstrates.com) and substrates were cleaned by sonication in water-soap, water, and 2-propanol baths, in that order. After drying, the substrates were placed in a UV-ozone cleaner (Jelight 42-220) for 20 min.
To prepare the electroluminescence devices, an 80 nm layer of PEDOT:PSS (CLEVIOS™ P VP AI 4083, aqueous dispersion, 1.3–1.7% solid content, Heraeus) was first spin-coated onto the ITO glass substrate to improve device reproducibility and to prevent the formation of pinholes. Transparent films (90 nm thick) of the Ir complexes and the ionic liquid 1-butyl-3-methylimidazolium hexafluoridophosphate ([BMIM][PF6], >98.5%, Sigma-Aldrich) in a 4 to 1 molar ratio were spin-coated from 20 mg cm−3 MeCN solution at 1000 rpm for 20 s. The devices were transferred in an inert atmosphere glovebox (<0.1 ppm O2 and H2O, MBraun) and were annealed at 100 °C for 1 h. The Al electrode (70 nm) was thermally evaporated using a shadow mask under a vacuum (<1 × 10−6 mbar) with an Edwards Auto500 evaporator integrated into the glovebox. The area of the device was 6.5 mm2. The devices were not encapsulated and were characterized inside the glovebox at room temperature.
LEC characterization
Electroluminescence spectra were recorded using an Avantes fibre-optics photo-spectrometer Avaspec-2048. Device lifetimes were measured by applying pulsed currents and monitoring the voltage or current and luminance by a True Colour Sensor MAZeT (MTCSiCT Sensor) with a Botest OLT OLED Lifetime-Test System (for AC measurements) and a Keithley 2400 source meter and a photodiode coupled to a Keithley 6485 picometer using a Minolta LS100 to calibrate the photocurrent (for DC measurements).
Results and discussion
Syntheses of complexes and solution mass spectrometric and NMR spectroscopic characterizations
The complexes [Ir(ppy)2(N^N)][PF6] (N^N = tpy, pytpy, 1–4) and [Ir(dmppz)2(N^N)][PF6] (N^N = tpy, pytpy) were prepared35 by treating [Ir2(ppy)4Cl2] or [Ir2(dmppz)4Cl2] with two equivalents of the appropriate N^N ligand (Schemes 2 and 3). Yields were variable, ranging from 47.6% for [Ir(ppy)2(tpy)][PF6] to 98.1% for [Ir(dmppz)2(tpy)][PF6]. In the ESI mass spectrum of each complex, the dominant peak envelope arose from the [M − PF6]+ ion and the observed isotope pattern matched that calculated.
 |
| | Scheme 2 Syntheses of [Ir(ppy)2(N^N)][PF6] complexes. Conditions: (i) N^N ligand, MeOH, microwave reactor 120 °C, time (see the Experimental section); (ii) NH4PF6. Atom and ring labels used for NMR spectroscopic assignments are shown. | |
 |
| | Scheme 3 Syntheses of [Ir(dmppz)2(N^N)][PF6]n complexes. Conditions: (i) N^N ligand, MeOH, microwave reactor 120 °C; (ii) NH4PF6. Atom and ring labels used for NMR spectroscopic assignments are shown. | |
The 1H and 13C NMR spectra of CD2Cl2 solutions of the complexes were recorded at room temperature, and were assigned using COSY, HMQC, HMBC and NOESY techniques. The aromatic regions of the 1H NMR spectra of [Ir(ppy)2(N^N)][PF6] with N^N = tpy, pytpy and ligands 1–4 appeared to be similar to one another and a representative example is shown in Fig. 2. An additional broad signal at δ 8.48 ppm in the aromatic region of the 1H NMR spectrum of [Ir(ppy)2(1)][PF6] was assigned to the OH proton of ligand 1. The presence of the pendant pyridine ring (ring G) in each complex leads to non-equivalence of the two [ppy]− ligands. In Fig. 2, the two lowest frequency doublets arise from HA6 and HC6 of the cyclometallated rings, and the observed chemical shifts of these signals are typical of those observed in related complexes.13 We have previously described the solution dynamic behaviour of complexes of type [Ir(ppy)2(PhN^N)]+,13 [Ir(dfppz)2(PhN^N)]+ (Hdfppz = 1-(2,4-difluorophenyl)-1H-pyrazole),14 and [Ir(msppz)2(PhN^N)]+ (Hmsppz = 1-(4-(methylsulfonyl)phenyl)-1H-pyrazole)15 in which PhN^N is a 6-phenyl-2,2′-bipyridine ligand. In these complexes, the 6-phenyl substituent undergoes hindered rotation on the NMR timescale and the corresponding signals are typically broad or very broad at 298 K. In the solution 1H NMR spectra of [Ir(ppy)2(tpy)][PF6], [Ir(ppy)2(pytpy)][PF6] and [Ir(ppy)2(4)][PF6], the signals for HG3 and HG6 are slightly broadened; signals for HG4 and HG5 overlap with other resonances. More noticeable line broadening is observed in the spectra of [Ir(ppy)2(1)][PF6], [Ir(ppy)2(2)][PF6] and [Ir(ppy)2(3)][PF6] (Fig. 2). The second significant difference between the 1H NMR spectra of the [Ir(ppy)2(PhN^N)]+ (ref. 13–15) and [Ir(ppy)2(tpy)]+ families is that the signal for HB6 is broad in the latter (Fig. 2) and is shifted to higher frequency with respect to the corresponding signal in [Ir(ppy)2(PhN^N)]+. This is rationalized by the formation of an N⋯HC hydrogen bond between the N atom of ring G and HB6. Since ring G undergoes hindered rotation on the NMR timescale at room temperature, proton HB6 experiences more than one environment as the hydrogen bond is sequentially formed and broken. Note that no other signal for ring B protons is broadened. The presence of the N⋯HC interaction is confirmed in the solid-state structures of four of the complexes described below.
 |
| | Fig. 2 500 MHz 1H NMR spectrum of [Ir(ppy)2(3)][PF6] in CD2Cl2 (295 K); the signal for the Me group is omitted. See Scheme 2 for atom labelling. | |
The essential differences between the aromatic regions of the 1H NMR spectra of [Ir(dmppz)2(N^N)][PF6] (N^N = tpy or pytpy) and [Ir(ppy)2(N^N)][PF6] (N^N = tpy, pytpy, 1–4) are in signals associated with rings B and D. Singlets at δ 6.15 and 6.05 ppm or at δ 6.15 and 6.06 ppm are assigned to HD4 and HB4 in [Ir(dmppz)2(tpy)][PF6] and [Ir(dmppz)2(pytpy)][PF6], respectively. No hydrogen-bonding interaction analogous to that seen in [Ir(ppy)2(N^N)]+ is possible in [Ir(dmppz)2(N^N)]+ because of the presence of the methyl substituents in ring B (but see the structural description below).
Crystal structure determinations
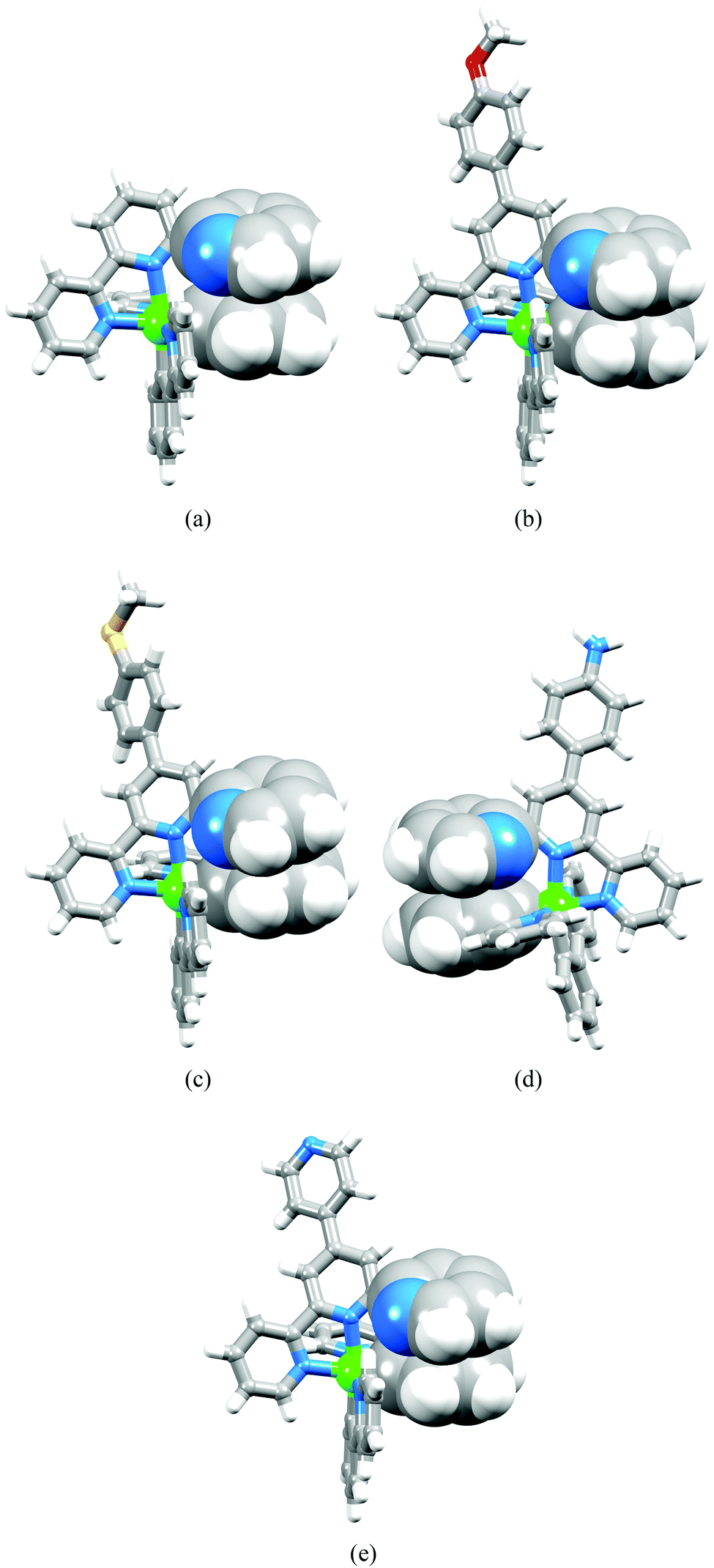
The single crystal structures of 4{[Ir(ppy)2(tpy)][PF6]}·2.5Et2O·1.5MeCN·3H2O, 4{[Ir(ppy)2(2)][PF6]}·Et2O·2MeCN, 2{[Ir(ppy)2(3)][PF6]}·MeCN·H2O, [Ir(ppy)2(4)][PF6]·2CH2Cl2 and 2{[Ir(ppy)2(pytpy)][PF6]}·Et2O·CH2Cl2 were determined. Each complex crystallizes in a centrosymmetric space group with both enantiomers of the octahedral [Ir(ppy)2(N^N)]+ cation in the unit cell. Since the structures of the cations are similar, they are discussed together; ORTEP diagrams are shown in Fig. S1–S5.† In each [Ir(ppy)2(N^N)]+ cation, the N-donors of the cyclometallated ligands are mutually trans, and the bond distances within the iridium(III) coordination sphere (see captions to Fig. S1–S5†) are unexceptional. Each tpy ligand binds to the iridium(III) ion in a chelating manner and the third pyridine ring is involved in a face-to-face stacking interaction with the phenyl ring of one of the [ppy]− domains (Fig. 3). The parameters that define the π-interactions are given in Table 2 and confirm efficient contacts.36 With the exception of [Ir(ppy)2(4)]+, the tpy domain in the cation adopts a cis,cis-conformation. The cis-arrangement of one pair of pyridine rings is necessitated by the chelating mode of the ligand. The cis-arrangement of the middle and pendant pyridine rings is a consequence of an intramolecular CH⋯N hydrogen bond. Fig. 4a shows the interaction in [Ir(ppy)2(tpy)]+, and hydrogen bond parameters for analogous contacts in the four complexes are listed in Table 3. Fig. 4b illustrates that in [Ir(ppy)2(4)][PF6]·2CH2Cl2, centrosymmetric pairs of cations associate through a combination of NHamine⋯Npyridine hydrogen bonds (N⋯H = 2.68 Å) and face-to-face interactions between the aminophenylpyridine units of adjacent ligands 4 (interplane separation = 3.37 Å). Note that the NMR spectroscopic data discussed above are consistent with intramolecular hydrogen bonding in all five complexes.
 |
| | Fig. 3 Intra-cation π-interactions in (a) [Ir(ppy)2(tpy)]+ (one of two independent cations), (b) [Ir(ppy)2(2)]+ (one of two independent cations), (c) [Ir(ppy)2(3)]+, (d) [Ir(ppy)2(4)]+ and (e) [Ir(ppy)2(pytpy)]+ (one of two independent cations). Nitrogen atoms are shown in blue. | |
 |
| | Fig. 4 (a) Intramolecular CH⋯N hydrogen bond in one of the independent [Ir(ppy)2(tpy)]+ cations in 4{[Ir(ppy)2(tpy)][PF6]}·2.5Et2O·1.5MeCN·3H2O. (b) Association of centrosymmetric pairs of [Ir(ppy)2(4)]+ cations in [Ir(ppy)2(4)][PF6]·2CH2Cl2 through π-stacking and NH⋯N hydrogen bonds. | |
Table 2 Comparison of the twist angle of the pendant pyridine ring and the structural parameters (in last two columns of the table) that define the face-to-face π-interactions in the [Ir(ppy)2(N^N)]+ cations (N^N = tpy, 2, 3, 4 and pytpy)
| Cation |
Twist angle between the middle and pendant py rings of the tpy unit/° |
Angle between least squares planes of rings/° |
Distance between ring centroids/Å |
| [Ir(ppy)2(tpy)]+ |
64.7 |
11.0 |
3.50 |
| (2 independent cations) |
62.0 |
7.2 |
3.45 |
| [Ir(ppy)2(2)]+ |
64.4 |
3.4 |
3.49 |
| (2 independent cations) |
60.5 |
8.0 |
3.42 |
| [Ir(ppy)2(3)]+ |
41.9 |
12.1 |
3.60 |
| [Ir(ppy)2(4)]+ |
64.2 |
9.8 |
3.52 |
| [Ir(ppy)2(pytpy)]+ |
50.7 |
8.3 |
3.54 |
| (2 independent cations) |
47.8 |
14.5 |
3.56 |
Table 3 Comparison of intramolecular hydrogen bond parameters (see Fig. 4) in the [Ir(ppy)2(N^N)]+ cations (N^N = tpy, 2, 3, 4 and pytpy)
| Cation |
CH⋯N/Å |
C⋯N/Å |
C–H⋯N/° |
| [Ir(ppy)2(tpy)]+ |
2.57 |
3.18(1) |
122 |
| (2 independent cations) |
2.63 |
3.249(8) |
123 |
| [Ir(ppy)2(2)]+ |
2.55 |
3.116(5) |
119 |
| (2 independent cations) |
2.55 |
3.185(4) |
125 |
| [Ir(ppy)2(3)]+ |
2.40 |
3.040(2) |
124 |
| [Ir(ppy)2(pytpy)]+ |
2.44 |
3.12(2) |
128 |
| (2 independent cations) |
2.58 |
3.09(3) |
114 |
Single crystals of [Ir(dmppz)2(tpy)][PF6] and [Ir(dmppz)2(pytpy)][PF6]·CH2Cl2 were grown from dichloromethane solutions of the compounds. The two complexes crystallize in the P space group with both enantiomers of the octahedral cation in the unit cell. ORTEP representations of the [Ir(dmppz)2(tpy)]+ and [Ir(dmppz)2(pytpy)]+ cations are given in Fig. S6 and S7;† Ir–C and Ir–N bond distances are listed in the figure captions.† Consistent with expectations, the N-atoms of the cyclometallated [dmppz]− ligands are trans to one another, and the non-coordinated pyridine ring of the tpy domain is twisted with respect to the two coordinated rings to facilitate an intra-cation π-interaction (Fig. 5). The twist angles are 69.6° in [Ir(dmppz)2(tpy)]+ and 62.7° in [Ir(dmppz)2(pytpy)]+. For the face-to-face contacts between pyridine and cyclometallated rings, the angles between the ring planes and the distances between the ring centroids are 10.1° and 3.41 Å in [Ir(dmppz)2(tpy)]+, and 8.0° and 3.55 Å in [Ir(dmppz)2(pytpy)]+. These values compare favourably with those in Table 2. Fig. 5a also highlights a short CHMe⋯N contact (2.70 Å) between the non-coordinated pyridine ring and a methyl substituent of one [dmppz]− ligand. The corresponding separation in [Ir(dmppz)2(pytpy)]+ is significantly longer (3.46 Å). The [Ir(dmppz)2(pytpy)]+ cations in [Ir(dmppz)2(pytpy)][PF6]·CH2Cl2 assemble into chains running along the c-axis supported by CHdmppz⋯Npy contacts (Npy = 4′-pyridyl group) at a separation of 2.70 Å.
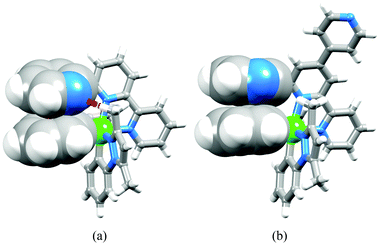 |
| | Fig. 5 Structures of (a) [Ir(dmppz)2(tpy)]+ showing π-stacking and CHMe⋯N (in red) interactions, and (b) [Ir(dmppz)2(pytpy)]+ showing intramolecular π-stacking. | |
Synthesis and characterization of [Ir(dmppz)2(Mepytpy)][PF6]2
The absorption and emission spectra of iron(II), ruthenium(II), osmium(II) and iridium(III) complexes containing pytpy ligands can be dramatically affected by protonation or methylation of the pendant pyridyl substituents.37,38 We therefore prepared the N-methylated complex [Ir(dmppz)2(Mepytpy)]2+ for comparison with its non-methylated analogue. [Ir(dmppz)2(Mepytpy)][PF6]2 was synthesized (Scheme 3) by a reaction of [Ir2(dmppz)4Cl2] with [Mepytpy][PF6].26 After work-up, the product was isolated in 63.6% yield. The electrospray mass spectrum exhibited peak envelopes at m/z 1005.3 and 430.2 corresponding to [M − PF6]+ and [M − 2PF6]2+. Additionally, a peak at m/z 535.2 arising from [Ir(dmppz)2]+ was observed. The 1H and 13C NMR spectra (assigned by 2D methods) were in accord with the structure shown in Scheme 3. The solution 1H NMR spectrum of [Ir(dmppz)2(Mepytpy)][PF6]2 (Fig. S8†) exhibits a singlet at δ 4.47 ppm assigned to the N-Me group. Otherwise, the most significant changes on going from the spectrum of [Ir(dmppz)2(pytpy)][PF6] to [Ir(dmppz)2(Mepytpy)][PF6]2 are shifts to higher frequency for the signals arising from protons HH3 (δ 7.82 to 8.60 ppm), HF3 (δ 8.79 to 9.03 ppm), and HF5 (δ 7.72 to 7.85 ppm).
Single crystals of [Ir(dmppz)2(Mepytpy)][PF6]2·2CH2Cl2 were grown from a dichloromethane solution of the complex. Crystallization in the P space group leads to both enantiomers of the octahedral cation being present in the unit cell. Fig. S9† shows an ORTEP representation of the [Ir(dmppz)2(Mepytpy)]2+ cation, and gives Ir–C and Ir–N bond distances.† The structural determination confirms that the N-atoms of the cyclometallated ligands are mutually trans, and that the non-coordinated pyridine ring of the tpy unit twists through 78.2°, enabling it to engage in a π-stacking interaction with one of the cyclometallated phenyl rings (Fig. 6a); the angle between the ring planes is 8.4°, and the inter-centroid separation is 3.44 Å (both values are similar to those in [Ir(dmppz)2(pytpy)]+, see above). A short CHMe⋯N contact (2.69 Å) mimics that in [Ir(dmppz)2(tpy)]+ (compare Fig. 5 and 6a). The N-methylpyridinio unit is directed into the V-shaped cavity formed by two of the cyclometallating ligands of an adjacent cation, and is involved in close CHMe⋯π contacts (the shortest CH⋯centroid distances are 2.68 and 2.84 Å). This results in the assembly of chains (Fig. 6b) that run obliquely through the unit cell.
 |
| | Fig. 6 (a) Structure of the [Ir(dmppz)2(Mepytpy)]2+ cation showing face-to-face π-stacking and CHMe⋯N short contact (in red). (b) Cations assemble into chains through π⋯HMe contacts. | |
Electrochemistry
The iridium(III) complexes are all electrochemically active and Table 4 summarizes their redox behaviour. Each complex undergoes a quasi-reversible (Fig. S10†) or irreversible oxidation, assigned to an iridium-based process. The oxidation of [Ir(ppy)2(tpy)]+ occurs at slightly higher potential than that of [Ir(ppy)2(bpy)]+ (0.84 V).39 Introduction of the 4′-aryl substituent on going from tpy to ligands 1–4 lowers the oxidation potential, consistent with the electron-releasing properties of the aromatic substituent. Oxidation of [Ir(ppy)2(pytpy)]+ occurs at a similar potential to that of [Ir(ppy)2(tpy)]+. Introduction of the electron-rich [dmppz]− ligands in place of [ppy]− lowers Eox1/2 by 0.08 V for [Ir(C^N)2(tpy)]+ and by 0.09 V for [Ir(C^N)2(pytpy)]+. The increase in Eox1/2 on going from [Ir(dmppz)2(pytpy)]+ to [Ir(dmppz)2(Mepytpy)]2+ is consistent with the increase in overall positive charge on the complex. At least one ligand-based reduction is observed for each complex. These processes are expected to be based on the tpy domain since the LUMO of the complex is localized on the N^N ligand.
Table 4 Cyclic voltammetric data with respect to Fc/Fc+; CH2Cl2 solutions with [nBu4N][PF6] supporting electrolyte, and scan rate of 0.1 V s−1 (ir = irreversible; qr = quasi-reversible)
| Compound |
E
ox1/2/V |
E
red1/2/V |
ΔE1/2/V |
| [Ir(ppy)2(tpy)][PF6] |
+0.90ir |
−1.82qr |
2.72 |
| [Ir(ppy)2(1)][PF6] |
+0.75ir |
−1.86qr, −2.15ir |
2.61 |
| [Ir(ppy)2(2)][PF6] |
+0.77qr |
−1.85qr |
2.62 |
| [Ir(ppy)2(3)][PF6] |
+0.84ir |
−1.82qr |
2.66 |
| [Ir(ppy)2(4)][PF6] |
+0.74ir |
−1.74ir, −2.05qr |
2.48 |
| [Ir(ppy)2(pytpy)][PF6] |
+0.88ir |
−1.71qr |
2.59 |
| [Ir(dmppz)2(tpy)][PF6] |
+0.82ir |
−1.87qr |
2.69 |
| [Ir(dmppz)2(pytpy)][PF6] |
+0.79qr |
−1.71qr |
2.50 |
| [Ir(dmppz)2(Mepytpy)][PF6]2 |
+0.88qr |
−1.30qr, −2.07ir |
2.18 |
Solution photophysical properties
The electronic absorption spectra of the [Ir(ppy)2(N^N)]+, [Ir(dmppz)2(N^N)]+ and [Ir(dmppz)2(Mepytpy)]2+ complexes are shown in Fig. 7. The spectra are dominated by intense bands in the UV region assigned to C^N and N^N ligand-based π*←π and π*←n transitions. The presence of the 4′-aryl or 4′-pyridyl unit extends the π-conjugation, leading to enhanced intensities of absorptions in [Ir(ppy)2(N^N)]+ (N^N = 1–4, pytpy) and [Ir(dmppz)2(N^N)]+ (N^N = pytpy, [Mepytpy]+) compared to those in [Ir(ppy)2(tpy)]+ and [Ir(dmppz)2(tpy)]+.
 |
| | Fig. 7 Absorption spectra of CH2Cl2 solutions of (a) [Ir(ppy)2(N^N)][PF6] and (b) [Ir(dmppz)2(N^N)][PF6]n (n = 1 for N^N = tpy and pytpy; n = 2 for N^N = [Mepytpy]+). | |
The photoluminescence (PL) spectra of CH2Cl2 solutions of [Ir(ppy)2(N^N)][PF6] and [Ir(dmppz)2(N^N)][PF6] are shown in Fig. 8, and PL data are given in Table 5. The iridium(III) complexes containing neutral, tpy-based ligands are orange or red emitters, and the emission bands are broad and unstructured. The parent complex [Ir(ppy)2(tpy)]+ emits at 590 nm, but exhibits a very low quantum yield and a lifetime of 68 ns (Table 5). The emission maximum is little shifted on introducing 4′-C6H4X (X = OH, OMe, SMe, NH2) substituents into the tpy domain or by replacing [ppy]− by [dmppz]−. However, the quantum yields and lifetimes are enhanced (Table 5), the two best being 9.9% and 125 ns for [Ir(ppy)2(4)][PF6], and 7.5% and 136 ns for [Ir(dmppz)2(tpy)][PF6]. A significant red shift in the emission is observed on going from [Ir(ppy)2(tpy)][PF6] to [Ir(ppy)2(pytpy)][PF6] (Δλ = 50 nm), or from [Ir(dmppz)2(tpy)][PF6] to [Ir(dmppz)2(pytpy)][PF6] (Δλ = 43 nm). This is consistent with the observed decrease in the electrochemical bandgaps for these pairs of compounds (Table 4). The effect of N-methylation on going from [Ir(dmppz)2(pytpy)][PF6] to [Ir(dmppz)2(Mepytpy)][PF6]2 is to virtually quench the emission.
 |
| | Fig. 8 Solution photoluminescence spectra of CH2Cl2 solutions of (a) [Ir(ppy)2(N^N)][PF6] and (b) [Ir(dmppz)2(N^N)][PF6]. | |
Table 5 Solution (degassed CH2Cl2) photoluminescence data for the iridium(III) complexes
| Compound |
λ
exc/nm |
λ
emmax/nm |
τ
1/2
/ns |
QY/% |
|
Solutions were degassed with argon for 15 min; lifetime measurements were made under argon.
|
| [Ir(ppy)2(tpy)][PF6] |
260 |
590 |
68 |
1.7 |
| [Ir(ppy)2(1)][PF6] |
360 |
590 |
93 |
5.8 |
| [Ir(ppy)2(2)][PF6] |
320 |
589 |
98 |
6.1 |
| [Ir(ppy)2(3)][PF6] |
325 |
595 |
87 |
6.8 |
| [Ir(ppy)2(4)][PF6] |
255 |
580 |
125 |
9.9 |
| [Ir(ppy)2(pytpy)][PF6] |
250 |
640 |
54 |
2.9 |
| [Ir(dmppz)2(tpy)][PF6] |
250 |
599 |
136 |
7.5 |
| [Ir(dmppz)2(pytpy)][PF6] |
256 |
642 |
119 |
6.2 |
Electroluminescence and device performances
LECs were prepared of all complexes in the thin film configuration described in the Experimental section. However, for [Ir(dmppz)2(pytpy)][PF6], film formation was not sufficiently good to permit a detailed investigation. Even though the performance of some of the devices was poor, especially when compared with that of the archetypal iTMC [Ir(ppy)2(6-Phbpy)]+ (see Fig. S11†), we were able to obtain the electroluminescent spectra for all the complexes and these are shown in Fig. 9. Electroluminescence (EL) maxima are compared with PL data in Table 6. The emission spectra of all LECs are red-shifted with respect to the PL spectra of the corresponding Ir-iTMC in thin film configuration. It is noteworthy that the EL maximum is red-shifted with respect to the archetypal iTMC [Ir(ppy)2(6-Phbpy)][PF6]3 leading to interesting red-emitting devices. Such a red shift is frequently observed in LECs and may be related to the location of the emission zone in the devices. However, the shift observed in the device based on [Ir(ppy)2(pytpy)][PF6] is exceptionally large which indicates a different emitting state involved.40 Note that that particular device showed weak electroluminescence even at higher applied current densities. Due to this, no further investigation as to the origin of the large shift in the emission spectrum was carried out.
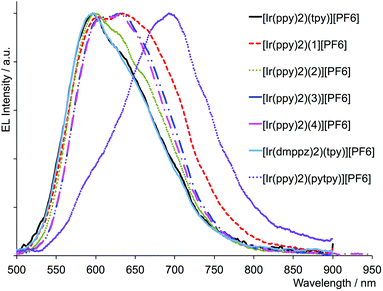 |
| | Fig. 9 Electroluminescence spectra for ITO/PEDOT:PSS/[Ir-iTMC]/IL(4:1)/Al LECs biased by a pulsed current of 100 A m−2 (1000 Hz, 50% duty cycle, block wave), except for devices employing complexes [Ir(ppy)2(tpy)][PF6], [Ir(ppy)2(4)][PF6], [Ir(dmppz)2(tpy)][PF6] (measured at 150 A m−2) and [Ir(ppy)2(pytpy)][PF6] (measured at 300 A m−2). | |
Table 6 Photoluminescence in thin film (iTMC:IL 4:1) and electroluminiscence data for LECs: ITO/PEDOT:PSS/iTMC:IL 4:1/Al biased by a pulsed current of 100 A m−2 (1000 Hz, 50% duty cycle, block wave). PL spectra are shown in Fig. S12
| Compound |
PL λmax/nm |
PLQY/% |
Turn-on time (ton)/min |
Luminancemax/cd m−2 |
EL λmax/nm |
EQE/% |
|
Measured under an average current density of 300 A m−2.
Measured under an average current of 150 A m−2.
|
| [Ir(ppy)2(tpy)][PF6] |
590 |
11.8 |
<1 |
11 |
598b |
<0.1 |
| [Ir(ppy)2(1)][PF6] |
593 |
11.3 |
<1 |
60 |
625 |
0.4 |
| [Ir(ppy)2(2)][PF6] |
591 |
23.5 |
3.6 |
54 |
602 |
0.3 |
| [Ir(ppy)2(3)][PF6] |
604 |
15.8 |
6 |
29 |
620 |
0.2 |
| [Ir(ppy)2(4)][PF6] |
600 |
4.6 |
<1 |
1 |
632b |
<0.1 |
| [Ir(ppy)2(pytpy)][PF6] |
641 |
4.8 |
— |
— |
695a |
— |
| [Ir(dmppz)2(tpy)][PF6] |
597 |
8.7 |
30 |
11 |
600b |
<0.1 |
| [Ir(dmppz)2(pytpy)][PF6] |
637 |
6.6 |
— |
— |
— |
— |
The most efficient devices were those employing the complexes [Ir(ppy)2(1)][PF6], [Ir(ppy)2(2)][PF6] and [Ir(ppy)2(3)][PF6]. The transient luminance and driving voltage of these devices are depicted in Fig. 10. For all three devices the turn-on is very rapid, in part due to the pulsed current driving applied and in part due to the presence of the ionic liquids in the emitting layer.41,42 The voltage required to sustain the applied current density of 100 A m−2 decreases rapidly over the first few minutes. This demonstrates the reduction of the injection barrier for electrons and/or holes as the ions accumulate at the device interface. With increasing time, the voltage continues to decrease but at a slower speed. This coincides with a gradual decrease in the luminance with a similar slope as that observed for the voltage decrease. Hence, this is a signature of progression of the doped regions towards the interior of the emissive film.43 This broadening of the doped regions leads to decreasing intrinsic film thickness. Hence, it requires a lower voltage to maintain the applied current density but also more excitons are quenched by the increased number of doping sites. The efficiency of the LECs is not very high, in spite of the reasonable PLQY observed for some of the complexes, measured in device configuration of iTMC:IL 4:1 molar ratio. This low efficiency indicates that the device performance is not optimal, and this is probably related to an imbalance in charge carriers.
 |
| | Fig. 10 (a) Luminance and (b) average voltage versus time for ITO/PEDOT:PSS/[Ir-iTMC]/IL(4:1)/Al LECs biased by a pulsed current of 100 A m−2 (1000 Hz, 50% duty cycle, block wave). | |
Conclusions
We have prepared and characterized a series of [Ir(C^N)2(N^N)][PF6] complexes in which the cyclometallating ligand, C^N, is either ppy− or dmppz−, and the N^N chelate is a tpy or a derivative thereof. Solution NMR spectroscopic data reveal that (i) the pendant pyridine ring of the coordinated tpy undergoes hindered rotation and (ii) in the [Ir(ppy)2(N^N)]+ complexes, the non-coordinated N-donor of the pendant pyridine ring forms a non-classical hydrogen bond with a CH unit of one pyridine ring of an adjacent ppy− ligand. The single crystal structures of eight complexes have been elucidated, and confirm that a general feature is intra-cation face-to-face π-stacking of the non-coordinated pyridine ring of the tpy domain with the cyclometallated ring. In CH2Cl2, the complexes are orange or red emitters, with λemmax in the range 580 to 642 nm. Quantum yields are <10%, and the emission lifetimes range from 54 to 136 ns. LECs have been fabricated using the new Ir-iTMCs, and the emission spectra of the LECs are red-shifted with respect to the photoluminescence spectra of the corresponding complex in thin film configuration. For the device incorporating [Ir(ppy)2(pytpy)][PF6], the PL to EL red-shift is especially large, indicating that a different emitting state is involved. The most efficient devices investigated were [Ir(ppy)2(1)][PF6], [Ir(ppy)2(2)][PF6] and [Ir(ppy)2(3)][PF6]; the devices exhibited rapid turn-on times, but displayed relatively low efficiencies.
Acknowledgements
We thank the European Research Council (Advanced Grant 267816 LiLo), the Swiss National Science Foundation, and the University of Basel for financial support, the Spanish Ministry of Economy and Competitiveness (MINECO) (MAT2011-24594, CTQ2009-08790 and Consolider-Ingenio CSD2007-00010), the Generalitat Valenciana (PROMETEO/2012/053) and the European Union (CELLO, STRP 248043; https://www.cello-project.eu/) for financial support. C.R. acknowledges the Spanish Ministry of Education, Culture, and Sport for an FPU grant. A.P. also acknowledges MINECO for an FPI grant. Nik Hostettler and Jonas Schönle are thanked for recording 500 MHz NMR spectra.
Notes and references
- E. Baranoff, J.-H. Yum, M. Graetzel and Md. K. Nazeeruddin, J. Organomet. Chem., 2009, 694, 2661 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- H. J. Bolink, E. Coronado, R. D. Costa, E. Ortí, M. Sessolo, S. Graber, K. Doyle, M. Neuburger, C. E. Housecroft and E. C. Constable, Adv. Mater., 2008, 20, 3910 CrossRef CAS.
- S. Graber, K. Doyle, M. Neuburger, C. E. Housecroft, E. C. Constable, R. D. Costa, E. Ortí, D. Repetto and H. J. Bolink, J. Am. Chem. Soc., 2008, 130, 14944 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft, M. Neuburger, S. Schaffner and E. C. Constable, Chem. Commun., 2009, 2029 RSC.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft and E. C. Constable, Adv. Funct. Mater., 2010, 20, 1511 CrossRef CAS.
- F. Neve, A. Crispini, S. Campagna and S. Serroni, Inorg. Chem., 1999, 38, 2250 CrossRef CAS.
- F. Neve and A. Crispini, Eur. J. Inorg. Chem., 2000, 1039 CrossRef CAS.
- M. Lepeltier, T. K.-M. Lee, K. K.-W. Lo, L. Toupet, H. Le Bozec and V. Guerchais, Eur. J. Inorg. Chem., 2005, 110 CrossRef CAS.
- J. O. Huh, M. H. Lee, H. Jang, K. Y. Hwang, J. S. Lee, S. H. Kim and Y. Do, Inorg. Chem., 2008, 47, 6566 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft and E. C. Constable, J. Am. Chem. Soc., 2010, 132, 5978 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft and E. C. Constable, Chem. Commun., 2011, 47, 3207 RSC.
- A. M. Bünzli, H. J. Bolink, E. C. Constable, C. E. Housecroft, M. Neuburger, A. Pertegás and J. A. Zampese, Eur. J. Inorg. Chem., 2012, 3780 CrossRef.
- E. Baranoff, H. J. Bolink, E. C. Constable, M. Delgado, D. Häussinger, C. E. Housecroft, M. K. Nazeeruddin, M. Neuburger, E. Ortí, G. E. Schneider, D. Tordera, R. M. Walliser and J. A. Zampese, Dalton Trans., 2013, 42, 1073 RSC.
- D. Tordera, A. M. Bünzli, A. Pertegás, J. M. Junquera-Hernández, E. C. Constable, J. A. Zampese, C. E. Housecroft, E. Ortí and H. J. Bolink, Chem.–Eur. J., 2013, 19, 8597 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, S. Schaffner, M. Neuburger, C. E. Housecroft and E. C. Constable, Adv. Funct. Mater., 2009, 19, 3456 CrossRef CAS.
- F. Neve, M. La Deda, F. Puntoriero and S. Campagna, Inorg. Chim. Acta, 2006, 359, 1666 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef PubMed.
- L. He, J. Qiao, L. Duan, G. Dong, D. Zhang, L. Wang and Y. Qiu, Adv. Funct. Mater., 2009, 19, 2950 CrossRef CAS.
-
H.-C. Su, H.-F. Chen, F.-C. Fang, C.-C. Liu, C.-C. Wu, K.-T. Wong, Y.-H. Liu and S.-M. Peng, J. Am. Chem. Soc, 2008, 130, 3413 Search PubMed.
- J. L. Rodríguez-Redondo, R. D. Costa, E. Ortí, A. Sastre-Santos, H. J. Bolink and F. Fernández-Lázaro, Dalton Trans., 2009, 9787 RSC.
- J. Zhang, L. Zhou, H. A. Al-Attar, K. Shao, L. Wang, D. Zhu, Z. Su, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2013, 23, 4667 CAS.
- K. T. Potts, P. Ralli, G. Theodoridis and P. Winslow, Org. Synth., 1986, 64, 189 CAS.
- J. Wang and G. S. Hanan, Synlett, 2005, 1251 CAS.
- E. C. Constable, C. E. Housecroft, M. Neuburger, D. Phillips, P. R. Raithby, E. Schofield, E. Sparr, D. A. Tocher, M. Zehnder and Y. Zimmermann, J. Chem. Soc., Dalton Trans., 2000, 2219 RSC.
- E. C. Constable, C. E. Housecroft, E. Medleycott, M. Neuburger, F. Reinders, S. Reymann and S. Schaffner, Inorg. Chem. Commun., 2008, 11, 518 CrossRef CAS PubMed.
- G. D. Storrier, S. B. Colbran and D. C. Craig, J. Chem. Soc., Dalton Trans., 1997, 3011 RSC.
-
S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1984, 106, 6647 Search PubMed.
- APEX2, version 2 User Manual, M86-E01078, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2006.
-
Stoe & Cie, IPDS software v 1.26, Stoe & Cie, Darmstadt, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- See for example ref. 7 and references cited therein.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- See for example: E. C. Constable and A. W. M. Cargill Thompson, J. Chem. Soc., Dalton Trans., 1994, 1409 RSC; S. Silvi, E. C. Constable, C. E. Housecroft, J. E. Beves, E. L. Dunphy, M. Tomasulo, F. M. Raymo and A. Credi, Chem.–Eur. J., 2009, 15, 178 CrossRef CAS PubMed; M. G. Lobello, S. Fantacci, A. Credi and F. De Angelis, Eur. J. Inorg. Chem., 2011, 1605 CrossRef.
- K. J. Arm, W. Leslie and J. A. G. Williams, Inorg. Chim. Acta, 2006, 359, 1222 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, D. Tordera, A. Pertegás, H. J. Bolink, S. Graber, C. E. Housecroft, L. Sachno, M. Neuburger and E. C. Constable, Adv. Energy Mater., 2011, 1, 282 CrossRef CAS.
- H. J. Bolink, L. Cappelli, S. Cheylan, E. Coronado, R. D. Costa, N. Lardies, M. K. Nazeeruddin and E. Orti, J. Mater. Chem., 2007, 17, 5032 RSC.
- S. T. Parker, J. D. Slinker, M. S. Lowry, M. P. Cox, S. Bernhard and G. G. Malliaras, Chem. Mater., 2005, 17, 3187 CrossRef CAS.
- D. Tordera, S. Meier, M. Lenes, R. D. Costa, E. Orti, W. Sarfert and H. J. Bolink, Adv. Mater., 2012, 24, 897 CrossRef CAS PubMed.
- M. Lenes, G. García-Belmonte, D. Tordera, A. Pertegás, J. Bisquert and H. J. Bolink, Adv. Funct. Mater., 2011, 21, 1581 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7, Fig. S9: ORTEP representations of complexes; Fig. S8. 1H NMR spectrum of [Ir(ppy)2(Mepytpy)][PF6]2; Fig. S10. Representative cyclic voltammograms; Fig. S11. Device performance of [Ir(ppy)2(6-Phbpy)][PF6] for comparison; Fig. S12. Thin film PL spectra. CCDC 974016–974023. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt53477d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a