 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, properties and structures of NbOF3 complexes and comparisons with NbOCl3 analogues†
William
Levason
*,
Gillian
Reid
,
Jonathan
Trayer
and
Wenjian
Zhang
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: wxl@soton.ac.uk; Tel: +44 (0)23 80593792
First published on 3rd January 2014
Abstract
The first series of complexes of niobium(V) oxide trifluoride, [NbOF3(OPR3)2] (R = Me or Ph), [NbOF3(dppmO2)] (dppmO2 = Ph2P(O)CH2P(O)Ph2), [NbOF3(dmso)2], [NbOF3(tmeda)] (tmeda = Me2N(CH2)2NMe2) and [NbOF3(diimine)] (diimine = 2,2′-bipy, 1,10-phen) have been prepared, either by reaction of the corresponding complexes of NbF5 and hexamethyldisiloxane (HMDSO) in CH2Cl2–MeCN solution, or directly from NbF5, ligand and HMDSO. They were characterised by IR, 1H, 31P{1H} and 19F{1H} NMR spectroscopy, and X-ray crystal structures are reported for [NbOF3(OPR3)2] (R = Me or Ph) and [NbOF3(dppmO2)]. Complexes of NbOCl3, [NbOCl3(OPPh3)2], [NbOCl3(dppmO2)], [NbOCl3(dppeO2)] (dppeO2 = Ph2P(O)(CH2)2P(O)Ph2), [NbOCl3(tmeda)] and [NbOCl3(diimine)] were made from NbCl5 and HMDSO in MeCN (which forms [NbOCl3(MeCN)2] in situ), followed by addition of the neutral ligand. Their properties are compared with the oxide fluoride analogues. X-ray structures are reported for [NbOCl3(dppmO2)], [NbOCl3(dppeO2)], [NbOCl3(tmeda)] and [NbOCl3(2,2′-bipy)]. The synthesis and spectroscopic characterisation of [MF5L] (M = Nb or Ta; L = OPR3, OAsPh3) and [MF4(diimine)2][MF6] are also described, and the key properties of the four series of complexes compared.
Introduction
The fluorides and oxide fluorides of early transition metals in high oxidation states are strong Lewis acids and form a substantial range of complexes with F− and with hard N- or O-donor ligands, whilst their more limited chemistry with soft donor ligands (P, S etc.) sometimes includes redox chemistry at the metal centre and oxidation/fluorination of the heteroatom donor, in addition to adduct formation.1 The properties of the metal centre are significantly altered by the small very electronegative fluoride ligands, and the chemistry of these fluorides/oxide fluorides is often very different to that of the chloride analogues.1Within Group V, the coordination chemistry of the oxide fluorides VOF3,2 and VO2F2,3 has been studied in some detail recently, whilst that of VF5 is completely unexplored. In contrast, an extensive series of complexes of MF5 (M = Nb or Ta) with both hard N- and O-donor1,4 and soft S-donor5 ligands are known, but the oxide-fluorides, MOF3, are intractable and very little studied.6,7 Here we report the synthesis, spectroscopic and structural characterisation of a series of adducts of NbOF3. Complexes of NbOCl3 have long been known, originally obtained by adventitious hydrolysis, or O-abstraction from the solvent or ligand in reactions of NbCl5.8 More systematic syntheses used the reaction of NbCl5 with siloxanes or occasionally direct reaction with the polymeric NbOCl3,9 and selected examples have been re-examined in the present work to provide comparisons with the NbOF3 complexes. NbOF3 is obtained by heating NbF5 with NbO2F in argon, and has a structure based upon six-coordinate niobium (SnF4 type), but the O/F disorder is only partially understood.6 It decomposes on heating above 180 °C, hydrolyses in air in a few hours, and is insoluble in organic solvents, making it completely unsuitable as a synthon to explore the coordination chemistry. TaOF3, which is formed similarly from TaO2F and TaF5, is also disordered and unreactive.6
We describe here a convenient alternative route to NbOF3 complexes involving F/O exchange from the corresponding NbF5 adducts, using hexamethyldisiloxane (HMDSO). Similar halogen/oxygen exchange has proved to be a useful route for the preparation of complexes of polymeric oxide halides, including, for example, MO2X2 (M = Mo or W; X = Cl or Br),10 although it has rarely been used for oxide fluoride complexes.1
Results and discussion
MF5 complexes
The reaction of NbF5 with OPR3 (R = Me or Ph) in rigorously anhydrous CH2Cl2 solution gave [NbF5(OPR3)] as white powders, easily soluble in halocarbon solvents. The complexes have been mentioned before, but with limited characterisation data.4d,11 The 19F{1H} NMR spectra show two singlets with relative intensities 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 and the 31P{1H} NMR spectra are singlets with large, high frequency coordination shifts (Table 1), consistent with their formulation as octahedral monomers. They also show broad singlet 93Nb NMR resonances‡δ ∼ −1530. The IR spectra show strong terminal Nb–F vibrations in the range 630–570 cm−1, and ν(PO) are markedly lower than the “free” ligand values (Table 1). The [NbF5(OAsPh3)] was made similarly from cold (0 °C) CH2Cl2 solution, but must be isolated rapidly, otherwise significant decomposition occurs, forming Ph3AsF2 (δ(19F) = −89.4),3a [NbF6]− (identified by in situ19F NMR spectroscopy)5a and other unidentified products. The tantalum complexes [TaF5(OPR3)] and [TaF5(OAsPh3)] were prepared similarly, and show corresponding trends in their spectroscopic properties (Table 1). However, the large quadrupole moment of 181Ta (I = 7/2, Q = 3 × 10−28 m2) results in fast quadrupolar relaxation and hence 181Ta NMR resonances are not observable.
:4 and the 31P{1H} NMR spectra are singlets with large, high frequency coordination shifts (Table 1), consistent with their formulation as octahedral monomers. They also show broad singlet 93Nb NMR resonances‡δ ∼ −1530. The IR spectra show strong terminal Nb–F vibrations in the range 630–570 cm−1, and ν(PO) are markedly lower than the “free” ligand values (Table 1). The [NbF5(OAsPh3)] was made similarly from cold (0 °C) CH2Cl2 solution, but must be isolated rapidly, otherwise significant decomposition occurs, forming Ph3AsF2 (δ(19F) = −89.4),3a [NbF6]− (identified by in situ19F NMR spectroscopy)5a and other unidentified products. The tantalum complexes [TaF5(OPR3)] and [TaF5(OAsPh3)] were prepared similarly, and show corresponding trends in their spectroscopic properties (Table 1). However, the large quadrupole moment of 181Ta (I = 7/2, Q = 3 × 10−28 m2) results in fast quadrupolar relaxation and hence 181Ta NMR resonances are not observable.
| Complex | 19F{1H}a | 31P{1H}a | ν(P/AsO)b | ν(Nb/TaX)b/cm−1 | ν(NbO)b/cm−1 |
|---|---|---|---|---|---|
| a CH2Cl2–CD2Cl2 solution 298 K. b Nujol mull. c Ligand δ(P) = +28.0, ν(PO) = 1195. d Ligand δ(P) = +35.0, ν(PO) = 1160. e Ligand δ(P) = +25.0, ν(PO) = 1187. f Ligand δ(P) = +35.0, ν(PO) = 1174 cm−1 data from ref. 24. | |||||
| [NbF5(OPPh3)] | 161.8(s, [F]), 128.6(s, [4F]) | 53.9c | 1061(vs)c | 624(sh), 608(vs, br) | — |
| [NbF5(OPMe3)] | 157.6(s, [F]), 134.5(s, [4F]) | 75.6d | 1092(vs)d | 615(vs, br), 582(m) | — |
| [NbF5(OAsPh3)] | 145.0(s, [F]), 110.5(s, [4F)] | — | 845(s) | 620(sh), 600(vs, br) | — |
| [NbF4(2,2′-bipy)2][NbF6] | 139.7(s, [4F]), 103.2 (10 lines, J = 335 Hz) | — | — | 615(vs), 603(s), 585(vs) | — |
| [NbF4(1,10-phen)2][NbF6] | 138.0(s, [4F]), 103.4 (10 lines, J = 335 Hz) | — | — | 608(vs), 586(vs), 565(sh) | — |
| [NbOF3(OPPh3)2] | 49.5(s, [F]), 37.8(s, [2F]) | 45.0(s, [P]), 36.0(s, [P]) | 1155(m), 1067(s) | 602(m), 579(s) | 941(s) |
| [NbOF3(OPMe3)2] | 41.5(s, [F]), 30.6(s, [2F]) | 67.1(s, [P]), 53.3(s, [P]) | 1140(m), 1087(s) | 614(s), 582(m), 555(s) | 958(s) |
| [NbOF3(dppmO2)] | 55.7(s, [F]), 36.4(s, [2F]) | 46.6(d, [P])e, 36.8(d, [P])f | 1156(s), 1088(s)f | 608(vs), 582(s) | 944(s) |
| [NbOF3(dmso)2] | 50.4(s, [F]), 38.0(s, [2F]) | — | — | 590(s), 564(s) | 920(s) |
| [NbOF3(2,2′-bipy)] | 49.0(s, [F]), 42.8(s, [2F]) | — | — | 612(vs), 579(s) | 959(s) |
| [NbOF3(1,10-phen)] | Insol | — | — | 610(sh), 594(s), 583(s) | 970(s) |
| [NbOF3(tmeda)] | Insol | — | — | 587(s), 557(s) | 920(s) |
| [TaF5(OPPh3)] | 84.2(s, [F]), 54.7(s, [4F]) | 53.2(s) | 1078(s) | 617(sh), 592(vs, br) | — |
| [TaF5(OPMe3)] | 82.5(s, [F]), 55.9(s, [4F]) | 76.9(s) | 1092(vs) | 601(sh), 583(vs, br) | — |
| [TaF5(OAsPh3)] | 62.5(s, [F]), 48.6(s, [4F]) | — | 845(s) | 617(sh), 592(vs, br) | — |
| [TaF4(2,2′-bipy)2][TaF6] | 68.1(s, [4F]), 38.0(s, [6F]) | — | 605(sh), 581(vs) | — | |
| [TaF4(1,10-phen)2][TaF6] | 66.1(s, [4F]), 37.9(s, [6F]) | — | — | 605(sh), 576(s) | — |
| [NbOCl3(OPPh3)2] | — | 50.0(s, [P]), 38.8(s, [P]) | 1159(s), 1074(s) | 325(s), 294(m) | 936(s) |
| [NbOCl3(dppmO2)] | — | 48.5(d, [P]), 36.8(d, [P]) | 1157(s), 1095(s) | 327(s), 296(m) | 928(s) |
| [NbOCl3(dppeO2)] | — | 56.7(s, [P]), 44.9(s, [P]) | 1172(s), 1066(s) | 320(s), 293(w) | 943(s) |
| [NbOCl3(2,2′-bipy)] | — | — | — | 349(s), 338(s) | 943(s) |
| [NbOCl3(1,10-phen)] | — | — | — | 338(br) | 944(s) |
| [NbOCl3(tmeda)] | — | — | — | 341(s), 320(sh) | 945(s) |
The reaction of NbF5 with 2,2′-bipyridyl or 1,10-phenanthroline in CH2Cl2 solution gave very poorly soluble complexes with a 1:1 NbF5:diimine composition, originally assumed12 to be seven-coordinate monomers. We found them to be sufficiently soluble in CD2Cl2 solution to obtain 1H and 19F{1H} NMR spectra after long accumulations, which show equivalent pyridyl rings and two 19F resonances with intensity ratio of 2:3. The more intense resonance is the characteristic 10 line multiplet of [NbF6]−,5a leading to the revised formulation, [NbF4(diimine)2][NbF6], with an eight-coordinate cation, as found in other adducts with chelating bidentate ligands.1 The [TaF4(diimine)2][TaF6] were made similarly and were even less soluble. Eight-coordination is also found in the diimine complexes of Zr and Hf (M′), [M′F4(diimine)2].13 The very poor solubility of the isolated [MF4(diimine)2][MF6] complexes made them unsuitable as synthons for the O/F exchange reactions, and hence studies were switched to using in situ syntheses, although the data on the isolated MF5 adducts are useful for comparison purposes (Table 1).
NbOF3 complexes
Treatment of an anhydrous CH2Cl2 solution of [NbF5(OPPh3)] with one mol. equivalent of OPPh3, followed by one mol. equivalent of HMDSO, resulted in slow formation of a white precipitate, identified as [NbOF3(OPPh3)2]. We subsequently found that “one-pot” syntheses were possible and more convenient, although the sequence of addition of the reactants and the time-scales are key to obtaining pure complexes (Scheme 1). The addition of NbF5 and two mol. equivalents of OPPh3 to anhydrous CH2Cl2 yields a colourless solution, which was stirred for 20 min. and then one mol. equivalent of HMDSO and a small amount of MeCN were added. After 24 h the mixture, now containing much white precipitate, was concentrated in vacuo, and the [NbOF3(OPPh3)2] isolated. If the HMDSO was added before, or simultaneously with, the OPPh3, very impure products resulted, and several hours seem necessary to complete the O/F exchange. The role of the MeCN is not entirely clear, but its presence seems necessary to obtain pure samples from the in situ preparations. In the syntheses of [WO2Cl2{RS(CH2)2SR}] from WCl6 or WOCl4, RS(CH2)2SR and HMDSO, use of MeCN–CH2Cl2 as solvent prevents the precipitation of polymeric WO2Cl2, by forming the nitrile adduct in situ.10c While a similar role may be present in the niobium systems, we note that attempts to isolate nitrile complexes failed (see below). The complexes [NbOF3(OPMe3)2] and [NbOF3(dppmO2)] were prepared similarly to [NbOF3(OPPh3)2], but all attempts to obtain [NbOF3(OAsPh3)2] gave mixtures containing [NbF5(OAsPh3)], [NbF6]− and Ph3AsF2 (identified based upon in situ19F and 93Nb NMR spectra). [NbOF3(OAsPh3)] was originally reported to be formed from adding OAsPh3 to a solution of Nb2O5 in conc. aqueous HF, although identified only by an IR spectrum.7b Using a 4:2:1 molar ratio of OPPh3:HMDSO:NbF5 in CH2Cl2–MeCN resulted only in isolation of [NbOF3(OPPh3)2], further O/F exchange did not occur. The complex [NbOF3(dmso)2] was also isolated in high yield from reaction of NbF5, dmso and HMDSO. As noted above, the very poor solubility of [NbF4(diimine)2][NbF6] made it impossible to redissolve them in CH2Cl2 for conversion to oxide-fluoride complexes. However, combination of the diimine and NbF5 in a large volume of CH2Cl2 (which gave a opalescent solution), followed by addition of HMDSO, did give [NbOF3(diimine)]. The [NbOF3(tmeda)] was made in high yield as an air-stable white powder by sequential reaction of NbF5, tmeda and HMDSO.
 | ||
| Scheme 1 Synthesis of NbOF3 adducts. | ||
In contrast, reaction of NbF5 with ethers, including thf and MeO(CH2)2OMe or with MeCN in CH2Cl2 followed by addition of HMDSO, gave white insoluble powders, which showed only traces of organic ligand in the IR spectra, and had very broad, ill-defined bands in the IR spectra, similar to those reported for NbOF3.6,7a The attempted reaction of NbF5, MeS(CH2)2SMe and HMDSO also failed. Ether, nitrile and thioether adducts of NbF5 are well characterised,1,4,5 but it seems that these ligands are too weakly bound to the “NbOF3” to prevent polymerisation and precipitation of ligand-free NbOF3. Similar behaviour was observed with VO2F,3 and the niobium system seems to be a further example of the metal centre preferring to form oxide/fluoride bridges rather than coordinate to weak, neutral donor groups.1 Thus far, attempts to isolate TaOF3 complexes from TaF5, ligand (ligand = OPR3, dmso or 2,2′-bipy) and HMDSO under similar reaction conditions, have been unsuccessful.
The solid [NbOF3(OPR3)2], [NbOF3(dmso)2] and [NbOF3(dppmO2)] complexes are white powders, relatively air-stable in the solid state (some appear hygroscopic on prolonged exposure), although hydrolysed by wet solvents. They are easily soluble in CH2Cl2, whereas the [NbOF3(diimine)] are very poorly soluble, and [NbOF3(tmeda)] is insoluble. The 1H and 31P{1H} NMR spectra (Table 1) of [NbOF3(OPMe3)2] show two phosphine oxide environments, and the 19F{1H} NMR spectrum contains two singlets with integrals in the ratio 1:2, which is consistent with mer-fluorines and one OPMe3trans to O and one trans to F. Attempts to record a 93Nb NMR spectrum were unsuccessful (an effect observed for all the NbOF3 adducts), contrasting with the ready observation of resonances from the NbF5 adducts described above. The low symmetry at the niobium centre will result in a large electric field gradient, and unobservably broad lines due to fast quadrupolar relaxation. The different trans-influences of Nb–F and Nb![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in these complexes are also shown by the difference in 31P chemical shifts for the trans disposed OPMe3 ligands (∼14 ppm), and similar differences are seen in the ν(PO) frequencies in the IR spectra which differ by >50 cm−1. A strong band in the range 970–920 cm−1 is assignable to the terminal NbO vibrations.
O groups in these complexes are also shown by the difference in 31P chemical shifts for the trans disposed OPMe3 ligands (∼14 ppm), and similar differences are seen in the ν(PO) frequencies in the IR spectra which differ by >50 cm−1. A strong band in the range 970–920 cm−1 is assignable to the terminal NbO vibrations.
Confirmation of the geometry of [NbOF3(OPMe3)2] comes from the X-ray crystal structure (Fig. 1).
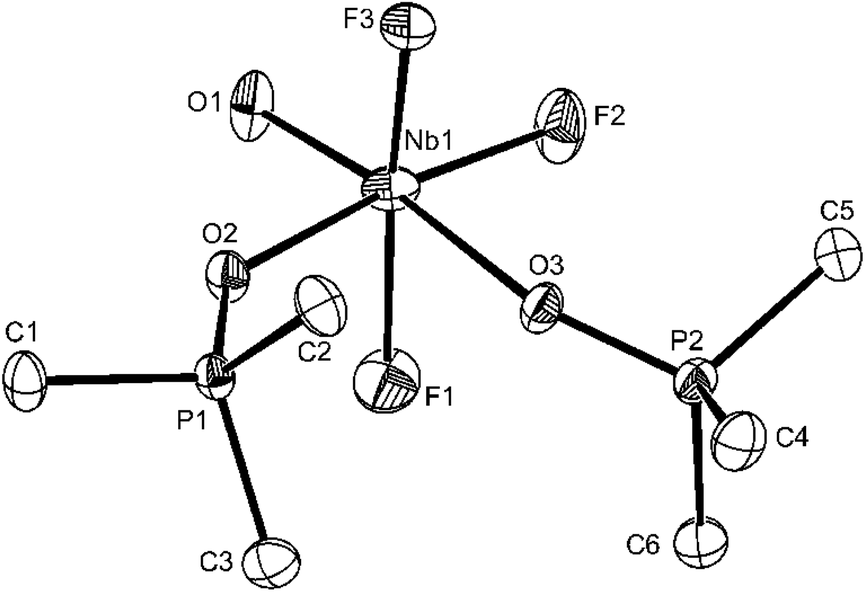 | ||
| Fig. 1 The structure of the Nb1 centred molecule in [NbOF3(OPMe3)2]·1/3CH2Cl2 showing the atom labelling scheme. Displacement ellipsoids are drawn at the 50% probability level and H-atoms and the solvate molecule are omitted for clarity. The second Nb2 centred molecule is similar with the third (Nb3) being disordered. Selected bond lengths (Å) and angles (°): Nb1–O1 = 1.773(2), Nb1–F2 = 1.868(2), Nb1–F3 = 1.9184(19), Nb1–F1 = 1.935(2), Nb1–O2 = 2.104(2), Nb1–O3 = 2.205(2), P1–O2 = 1.526(2), P2–O3 = 1.521(2), O1–Nb1–F2 = 98.58(10), O1–Nb1–F3 = 97.00(11), F2–Nb1–F3 = 92.20(10), O1–Nb1–F1 = 95.09(11), F2–Nb1–F1 = 92.72(10), F3–Nb1–F1 = 166.12(9), O1–Nb1–O2 = 91.75(9), F3–Nb1–O2 = 86.21(9), F1–Nb1–O2 = 86.63(9), F2–Nb1–O3 = 87.90(8), F3–Nb1–O3 = 84.76(9), F1–Nb1–O3 = 82.46(9), O2–Nb1–O3 = 81.80(8). | ||
There is no evidence in this molecule for O/F disorder in plane, which is a common problem in this area of chemistry (cf. [VOF3(OPPh3)2]2b). The niobium is in a distorted octahedral environment with the axial F–Nb–F unit bent away from the oxido-ligand. The Nb–Ftrans F are longer than Nb–Ftrans O by ∼0.06 Å and the NbO of 1.773(2) Å is consistent with the expected multiple bond character. The Nb–O(P)trans F distances of 2.104(2) Å and Nb–O(P)trans O = 2.205(2) Å show the disparate effects of the trans donor and parallel the spectroscopic evidence. Curiously, d(P–O) in the two phosphine oxide ligands are only slightly different. The spectroscopic data on [NbOF3(OPPh3)2] (Table 1) are very similar to those of the OPMe3 complex discussed, but in this case the X-ray structure (Fig. 2) shows F/O disorder trans to OPPh3, and the bond length and angle data are correspondingly unreliable, although the identity of the complex is confirmed.
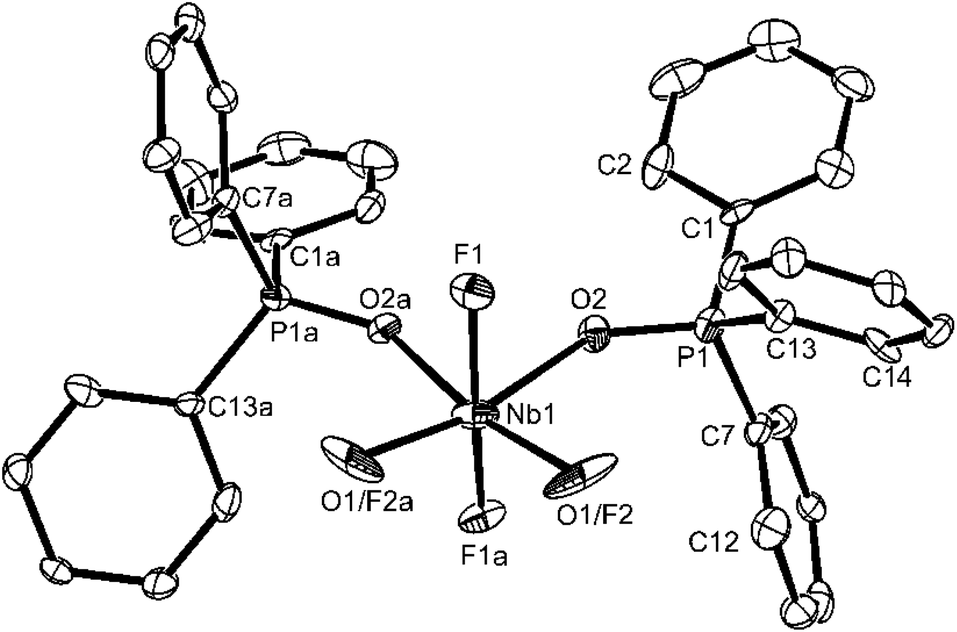 | ||
| Fig. 2 The structure of [NbOF3(OPPh3)2] showing the atom numbering scheme. The phenyl rings are numbered cyclically starting at the ipso C atom. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. The molecule has two-fold symmetry. Notice the disorder at the atom site O1/F2. Symmetry operation: a = 1 − x, −y, z. Selected bond lengths (Å) and angles (°): Nb1–O1 = 1.850(4), Nb1–F2 = 1.850(4), Nb1–F1 = 1.932(4), Nb1–O2 = 2.189(4), P1–O2 = 1.532(4), O1–Nb1–F2 = 108.2(4), O1–Nb1–F1 = 92.06(19), F2–Nb1–F1 = 95.36(18), F1–Nb1–F1a = 167.3(2), O1–Nb1–O2 = 86.8(2), F1–Nb1–O2 = 84.23(16), O2–Nb1–O2 = 78.3(2). | ||
The structural parameters of [NbOF3(dppmO2)] are generally similar to those already discussed above, and this complex seems free of O/F disorder (Fig. 3).
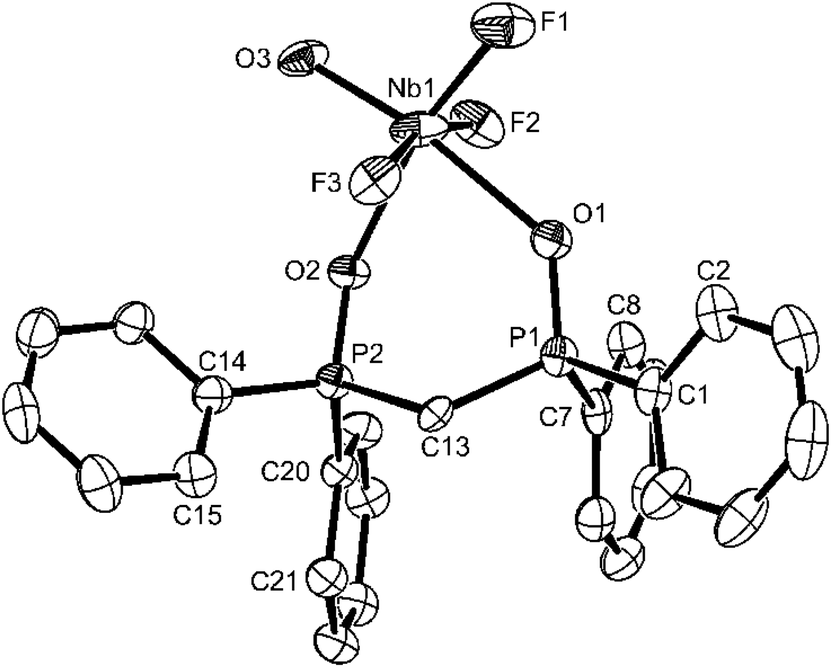 | ||
| Fig. 3 The structure of [NbOF3(dppmO2)] showing the atom labelling scheme. Displacement ellipsoids are drawn at the 50% probability level and H-atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Nb1–O3 = 1.782(4), Nb1–F1 = 1.850(3), Nb1–F2 = 1.912(3), Nb1–F3 = 1.970(3), Nb1–O2 = 2.171(3), Nb1–O1 = 2.257(4), P1–O1 = 1.508(4), P2–O2 = 1.509(3), O3–Nb1–F1 = 100.96(17), O3–Nb1–F2 = 98.04(16), F1–Nb1–F2 = 95.28(15), O3–Nb1–F3 = 95.34(16), F1–Nb1–F3 = 94.41(15), O3–Nb1–O2 = 91.31(15), F2–Nb1–O2 = 83.99(13), F3–Nb1–O2 = 83.24(13), F1–Nb1–O1 = 87.03(15), F2–Nb1–O1 = 84.16(14), F3–Nb1–O1 = 80.84(13), O2–Nb1–O1 = 80.66(13), F2–Nb1–F3 = 161.67(13). | ||
The [NbOF3(tmeda)] is insoluble in non-coordinating solvents and MeCN, and is partially decomposed by dmf or dmso which prevented solution measurements. However, the [NbOF3(diimine)], although very poorly soluble in chlorocarbons or MeCN (a property shared with the NbF5 analogues above, and also the ZrF4, HfF4, VOF3 and VO2F diimine complexes),2,3,13 gave 1H NMR spectra showing inequivalent pyridyl rings, and hence that the diimine was trans to O/F. The 19F{1H} NMR spectrum of [NbOF3(2,2′-bipy)] (Table 1) shows two resonances in the ratio 1:2 consistent with a mer arrangement of the fluorines, and the chemical shifts are ∼100 ppm to low frequency of those observed for the [NbF4(diimine)2]+. The [NbOF3(1,10-phen)] was very poorly soluble in weakly coordinating solvents and a convincing 19F{1H} NMR spectrum was not obtained. The diimine complexes are readily hydrolysed in solution in CH2Cl2 or MeCN forming [NbF6]− ions, based upon 19F NMR evidence and also shown by attempts to obtain crystals of [NbOF3(2,2′-bipy)] for an X-ray study which produced a few poor quality crystals of [2,2′-bipyH][NbF6]. The solids also hydrolyse slowly on exposure to the atmosphere.
NbOCl3 complexes
Solid NbOCl3 contains dimeric [Cl2Nb(O)(μ-Cl)2Nb(O)Cl2] units linked into chains via unsymmetrical oxide bridges, giving six-coordinate niobium.14 The syntheses of the [NbOCl3(L–L)] (L–L = 2,2′-bipy, 1,10-phen, dppmO2, dppeO2, tmeda and 2 × OPPh3) were carried out in anhydrous MeCN solution, with the reversed order of reagent addition to that used for the oxide-fluoride syntheses, i.e. reacting NbCl5 with HMDSO to form ‘NbOCl3’ in situ, followed by addition of the neutral ligand (Scheme 2). The initially yellow solution of NbCl5 in MeCN rapidly pales on addition of HMDSO, indicating formation of [NbOCl3(MeCN)2] in situ,9b,15 which was converted into near colourless [NbOCl3(L–L)] upon addition of the neutral ligand. Once isolated, the [NbOCl3(tmeda)] is essentially insoluble in non-coordinating solvents, although crystals were grown adventitiously from the reaction filtrate. The other complexes are soluble in CH2Cl2 or MeCN. The IR spectra of the complexes (Table 1) show strong ν(NbO) in the region 920–950 cm−1 and ν(NbCl) 290–350 cm−1 with disparate ν(PO) vibrations for the phosphine oxide groups trans to Cl and trans to O. In solution, the 1H and 31P{1H} NMR spectra of [NbOCl3(L–L)] (L–L = dppmO2, dppeO2, 2 × OPPh3) show the expected inequivalence of the neutral donor groups, but attempts to record 93Nb spectra were unsuccessful; as with the oxide-fluorides this is attributed to fast quadrupolar relaxation in the low symmetry electric fields.
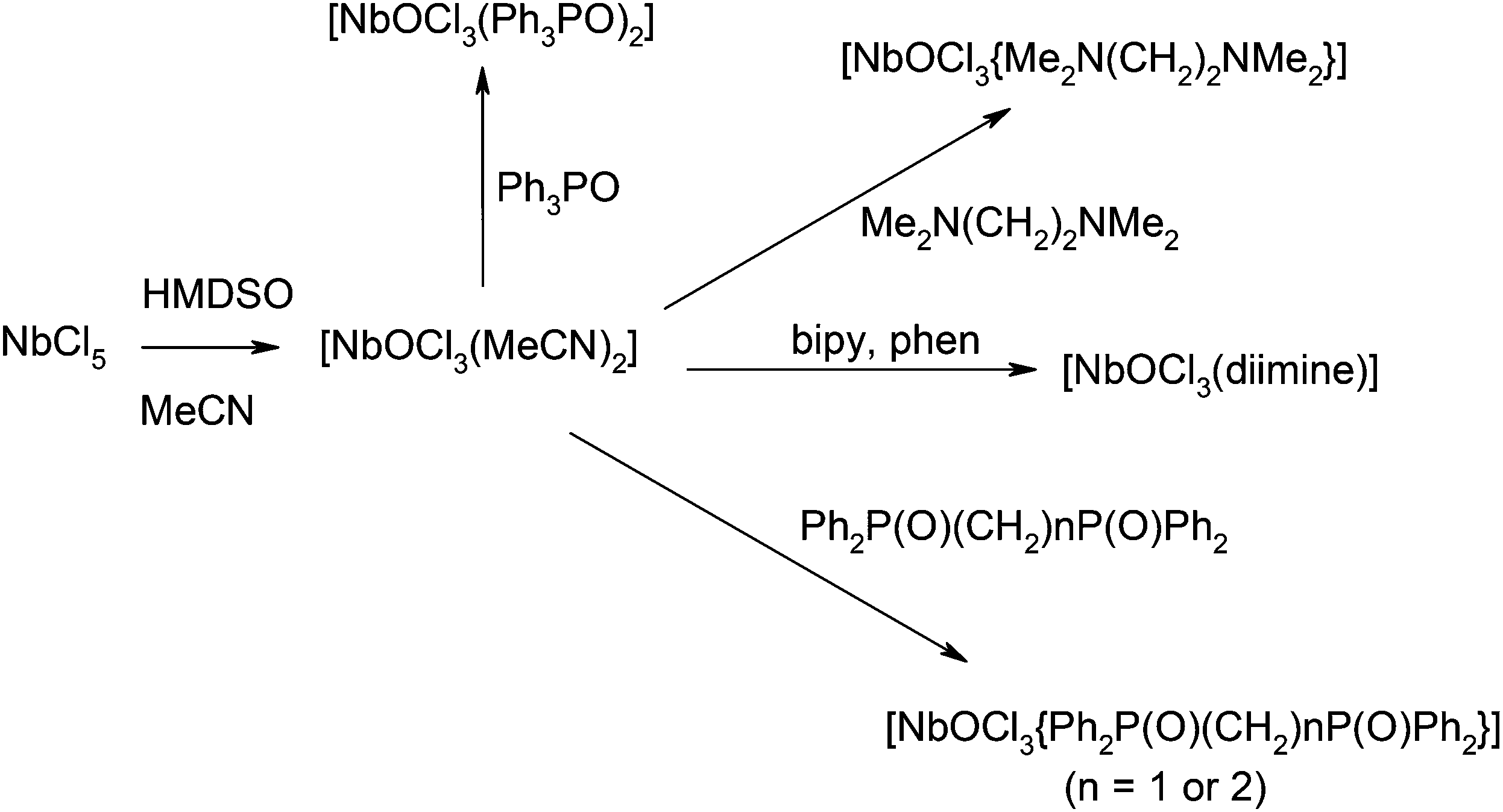 | ||
| Scheme 2 Synthesis of NbOCl3 adducts. | ||
X-Ray crystal structures were obtained for five of the complexes. The structure of [NbOCl3(OPPh3)2] has been reported previously and shows16mer-chlorines, and cis OPPh3 groups, with O/Cl disorder trans to OPPh3. The crystal structures of the two diphosphine dioxide complexes (Fig. 4 and 5) show d(NbO) slightly shorter by ∼0.1 Å compared to the oxide fluoride complexes, but with similarly disparate d(Nb–O(P)) suggesting the trans influence of F and Cl are similar in these complexes. The d(NbO) and d(Nb–Cl) distances in a range of NbOCl3 adducts cover quite a narrow range,8,9,15,16 suggesting that these are the dominant bonding interactions, with the neutral ligands completing the coordination sphere, but having little influence on the NbO and Nb–Cl bonds.
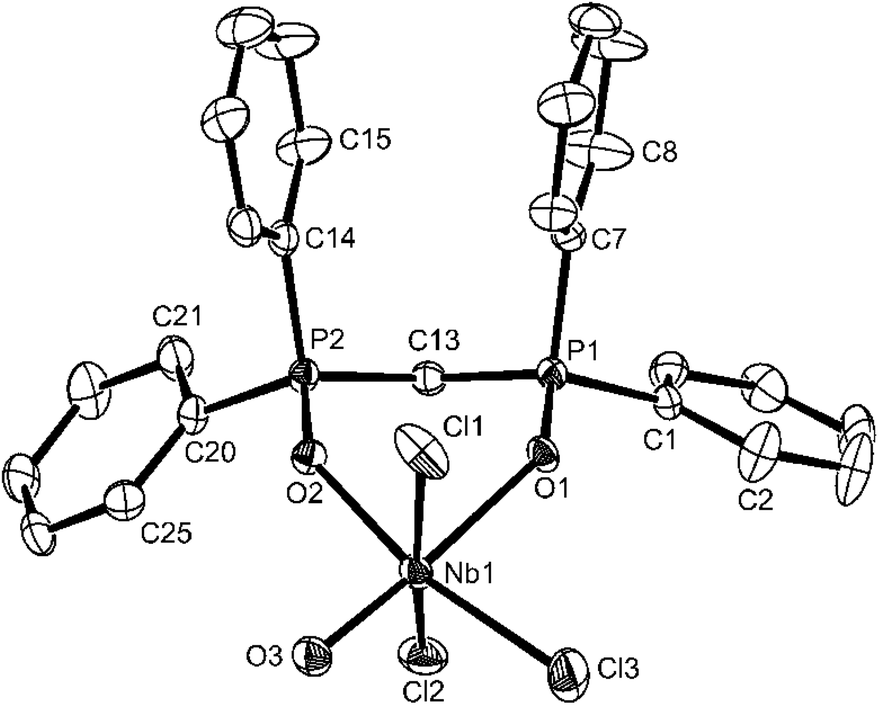 | ||
| Fig. 4 The structure of [NbOCl3(dppmO2)]·nMeCN showing the atom labelling scheme. Displacement ellipsoids are drawn at the 50% probability level and H-atoms are omitted for clarity. The solvate acetonitrile is also omitted. The phenyl rings are numbered cyclically starting at the ipso-C atom. Selected bond lengths (Å) and angles (°): Nb1–O3 = 1.706(3), Nb1–O2 = 2.095(3), Nb1–O1 = 2.266(3), Nb1–Cl3 = 2.3463(13), Nb1–Cl1 = 2.3815(13), Nb1–Cl2 = 2.4203(12), O3–Nb1–O2 = 94.47(12), O2–Nb1–O1 = 80.48(10), O3–Nb1–Cl3 = 98.84(10), O1–Nb1–Cl3 = 86.17(7), O3–Nb1–Cl1 = 97.53(10), O2–Nb1–Cl = 86.85(8), O1–Nb1–Cl1 = 84.56(7), Cl3–Nb1–Cl1 = 92.45(6), O3–Nb1–Cl2 = 93.91(10), O2–Nb1–Cl2 = 85.00(8), O1–Nb1–Cl2 = 83.41(7), Cl3–Nb1–Cl2 92.98(5), Cl1–Nb1–Cl2 = 166.43(4). | ||
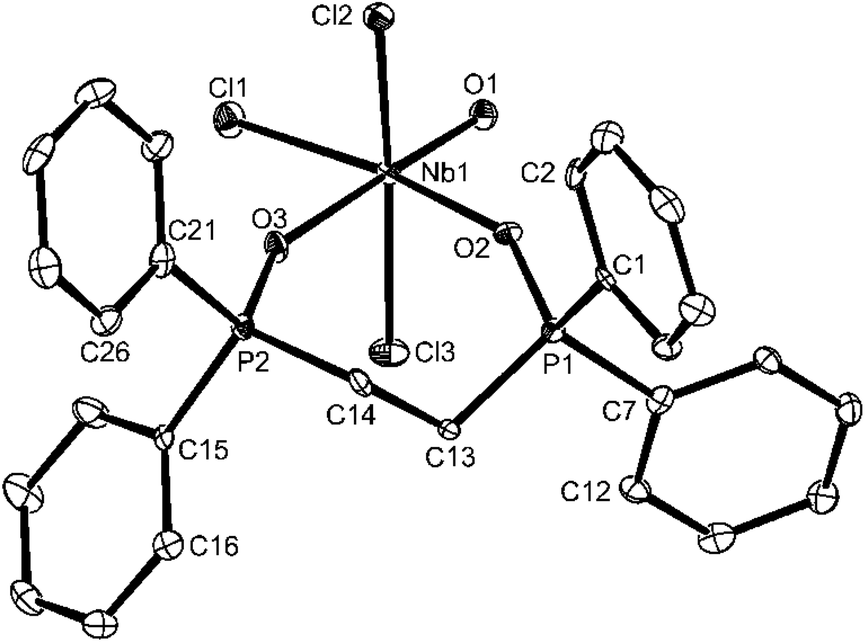 | ||
| Fig. 5 The structure of [NbOCl3(dppeO2)]·nMeCN showing the atom labelling scheme. Displacement ellipsoids are drawn at the 50% probability level and H-atoms are omitted for clarity. The phenyl rings are numbered cyclically starting at the ipso-C atom. The solvate acetonitrile is also omitted. Selected bond lengths (Å) and angles (°): Nb1–O1 = 1.702(3), Nb1–O2 = 2.077(3), Nb1–O3 = 2.219(3), Nb1–Cl1 = 2.3602(12), Nb1–Cl3 = 2.4136(12), Nb1–Cl2 = 2.4210(13), O1–Nb1–O2 = 93.09(12), O2–Nb1–O3 = 82.31(10), O1–Nb1–Cl1 = 97.74(10), O3–Nb1–Cl1 = 86.86(8), O1–Nb1–Cl3 = 97.02(10), O2–Nb1–Cl3 = 86.17(8), O3–Nb1–Cl3 = 84.91(8), Cl1–Nb1–Cl3 = 92.43(4), O1–Nb1–Cl2 = 94.82(10), O2–Nb1–Cl2 = 86.11(8), O3–Nb1–Cl2 = 82.73(8), Cl1–Nb1–Cl2 93.00(4), Cl3–Nb1–Cl2 = 166.19(4). | ||
The structure of [NbOCl3(tmeda)] (Fig. 6) shows the same features as those of the oxygen donor complexes, although the carbon atoms about N2 show some disorder; there is no evidence for O/Cl disorder. The dimensions in the structure of [NbOCl3(2,2′-bipy)] (Fig. 7) are also unexceptional, although the octahedron about the niobium is more distorted due to the small chelate bite of the 2,2′-bipyridyl (<N1–Nb1–N2 = 69.52(21)°).
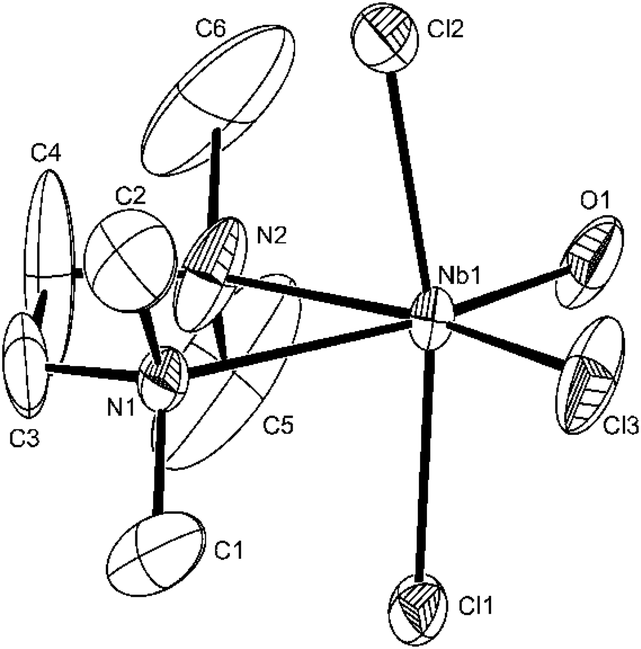 | ||
| Fig. 6 The structure of [NbOCl3(tmeda)] showing the atom labelling scheme. The carbon atoms associated with N2 show some disorder. Displacement ellipsoids are drawn at the 50% probability level and H-atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Nb1–O1 = 1.798(5), Nb1–N2 = 2.331(8), Nb1–N1 = 2.518(5), Nb1–Cl3 = 2.328(3), Nb1–Cl2 = 2.348(3), Nb1–Cl1 = 2.358(2), O1–Nb1–N2 = 92.5(3), O1–Nb1–Cl3 = 103.5(2), O1–Nb1–Cl2 = 94.3(3), N2–Nb1–Cl2 = 86.1(3), Cl3–Nb1–Cl2 = 91.21(16), O1–Nb1–Cl1 = 96.5(3), N2–Nb1–Cl1 = 87.6(3), Cl3–Nb1–Cl1 = 91.87(10), N2–Nb1–N1 = 74.1(2), Cl3–Nb1–N1 = 89.86(16), Cl2–Nb1–N1 = 84.00(15), Cl1–Nb1–N1 = 84.09(15), Cl2–Nb1–Cl1 = 167.70(10). | ||
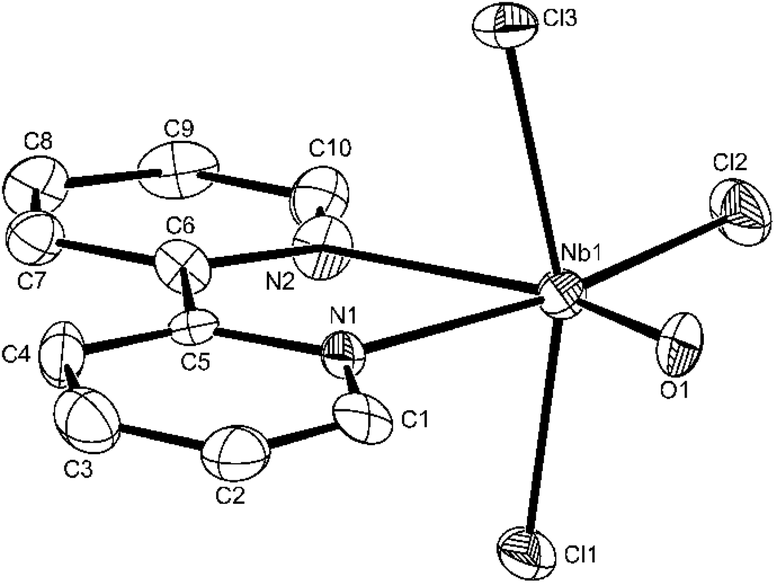 | ||
| Fig. 7 The structure of the Nb1 centred molecule in [NbOCl3(2,2′-bipy)] showing the atom labelling scheme. This molecule has no crystallographic symmetry whereas the Nb2 centred molecule has 2-fold symmetry. Displacement ellipsoids are drawn at the 50% probability level and H-atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Nb1–O1 = 1.694(6), Nb1–Cl1 = 2.363(2), Nb1–Cl2 = 2.356(2), Nb1–Cl3 = 2.372(2), Nb–N1 = 2.262(6), Nb1–N2 = 2.385(6), O1–Nb1–N1 = 89.3(3), O1–Nb1–Cl2 = 104.8(2), O1–Nb1–Cl1 = 98.0(2), O1–Nb1–Cl3 = 97.7(2), Cl2–Nb1–N2 = 96.41(15), N1–Nb1–N2 = 69.5(2), Cl1–Nb1–N1 = 84.50(16), Cl2–Nb1–Cl1 = 94.18(7), Cl3–Nb1–Cl2 = 92.76(7), Cl3–Nb1–N1 = 84.37(16), N2–Nb1–Cl1 = 81.08(15), Cl3–Nb1–N2 = 80.13(15). | ||
Attempts to obtain a complex of NbOCl3 with 1,4,7-trimethyl-1,4,7-triazacyclononane (Me3-tacn) gave a mixture of products. Recrystallisation of the mixture from MeCN gave a few crystals identified as [(Me3-tacn)H]2[NbOCl5]. The anion has been structurally characterised with a variety of cations, but often the niobium is on a high symmetry site which results in O/Cl disorder.17 In the present case the structure appears to free of such disorder and the data are presented as ESI.†
Comparisons of NbF5, NbOF3 and NbOCl3 complexes
Comparison of the spectroscopic data in Table 1 for [NbF5(OPR3)] and [NbOF3(OPR3)2] shows very significant differences due to replacement of two fluoride ligands by the oxo-group. The 19F and 31P chemical shifts are very different, with those of [NbF5(OPR3)] at much higher frequency for each nucleus. Similar differences are apparent in the 19F chemical shifts for the two series of N-donor complexes. The data demonstrate that the presence of the strong π-donating oxo-group significantly changes the electron density at the Nb(V) centre, making it much less electron poor, and hence a weaker Lewis acid. Within the NbOF3 complexes there is also a large trans influence of the oxo-group which results in significantly longer bonds to the trans ligand than for those groups trans to fluorine. The d(PO) show very small differences, although the relative trans influence is clear in the ν(PO) vibrations in the IR spectra. Comparing the structural and spectroscopic data on corresponding NbOF3 and NbOCl3 complexes reveals rather small differences. The d(NbO) in these and in literature examples of the oxide chloride complexes8,9,15,16 show they occur in a narrow range, ∼1.7–1.8 Å, irrespective of the halide or neutral co-ligands present. Similarly, d(Nb–Cl)trans L are relatively insensitive to the nature of L (the ligand types are too restricted to make a similar comparison for the fluoride).
The bond angles about the niobium centres also show significant deviations from those expected for a regular octahedral geometry. The factors determining the geometry adopted by ML6 complexes of transition metals as a function of ligand types (σ-donor only, or σ and π donor), dn count and ligand architecture have been discussed in several articles,18 and the fact that [MF6]n− species are Oh and [M(CH3)6]n− (n = 0, 1, 2 etc.) are trigonal prisms has been rationalised in terms of electronic factors by MO calculations.19 The niobium oxide halide structures discussed in the present work (12e, d0 complexes) are based upon distorted octahedral geometries, as would be expected, given the presence of dominant σ and π donor ligands. As observed in many early transition metal complexes containing MO bonds, the angles involving the latter, OM–L and OM–X are larger than X–M–X, X–M–L, or L–M–L, in effect the electron rich multiply bonded MO unit occupies more space about the metal centre. Superimposed upon this are smaller effects arising from the steric demands of the X and L groups and constraints of neutral ligand geometries, such as chelate bites in the bidentates. In the cis-MOX3L2 unit the axial X–M–X group bends away from the MO and towards the neutral co-ligands.2,3,5,8–10
Comparing the IR data within the two series of NbOX3 complexes shows ν(NbO) lying in a range ∼920–970 cm−1, and the ν(PO) in corresponding phosphine oxide adducts also show little difference. Hence we conclude that the NbOX3 core has the dominant structural and spectroscopic effects in these complexes.
The differences between NbOF3 and NbOCl3 as acceptors towards weaker donor ligands such as ethers or nitriles, where the latter forms complexes with thf, MeO(CH2)2OMe, MeCN, etc.,4,9,15 but attempts to isolate analogues with NbOF3 result in intractable, ligand-free products (NbOF3 polymer). This can be ascribed to the preference of the niobium centre to form fluoride bridges over weak Nb–L bonds, and is seen in other fluoride and oxide fluoride systems.1
Finally, these niobium complexes can be compared with those of the 3d analogue, vanadium. VOF3 forms similar phosphine oxide, diimine and diamine complexes to NbOF3, but also complexes with ethers, thioethers and nitriles.2 The differences are again readily rationalised by the niobium's preference for fluorine bridges; NbOF3 is an inert, very strongly bridged polymer (above), whereas VOF3 although (weakly) F-bridged in the solid,20 easily vapourises as a monomer on heating and dissolves in most organic solvents. The complexes of VOCl3 with neutral ligands are thermally and often photochemically unstable, and extremely readily hydrolysed and reduced (often spontaneously) to V(IV) or V(III) compounds,21 whereas the NbOCl3 adducts remain pentavalent, unless specifically treated with reducing agents.
Conclusions
The O/F exchange reaction between complexes of the binary fluoride NbF5 and a siloxane have been shown to produce complexes of the otherwise intractable oxide-fluoride, NbOF3, in good yield. However, further O/F exchange to form derivatives of NbO2F did not occur under similar conditions. Comparison of the spectroscopic properties of the NbF5 and NbOF3 complexes demonstrates the substantial effect on the metal centre of replacing two fluoride by the stronger π-donor oxido-group.The HMDSO/MFn route may well offer a synthetic pathway to oxide fluoride complexes of other high valent early metal complexes, e.g. those of Mo, W, Ti or Zr. TaOF3 complexes are not formed under analogous reaction conditions; further studies are required to develop a suitable route to these.
Experimental
Infrared spectra were recorded as Nujol mulls between CsI plates using a Perkin-Elmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H, 19F{1H}, 31P{1H} and 93Nb NMR spectra were recorded using a Bruker DPX400 spectrometer and are referenced to the protio resonance of the solvent, external CFCl3, 85% H3PO4, and [NEt4][NbCl6] in CD3CN, respectively. Microanalyses were undertaken by Medac Ltd or London Metropolitan University. Solvents were dried prior to use: THF, Et2O and MeOCH2CH2OMe by distillation from sodium benzophenone ketyl, MeCN and CH2Cl2 from CaH2. OPMe3 was sublimed in vacuo, OPPh3, OAsPh3, 2,2′-bipy, 1,10-phen were heated in vacuo, and tmeda distilled from BaO. All preparations were undertaken using standard Schlenk techniques under a N2 atmosphere.[NbF5(OPPh3)]: A solution of OPPh3 (0.262 g, 1.0 mmol) in CH2Cl2 (20 mL) was added to finely powdered NbF5 (0.188 g, 1.0 mmol), and vigorously stirred to give a clear solution. This was filtered to remove any residual solid and concentrated in vacuo to ∼5 mL. On standing a white powdered separated, which was filtered off and dried in vacuo. Yield 0.40 g, 85%. Anal: required for C18H15F5NbOP (466.2): C, 46.4; H, 3.2. Found: C, 46.9; H, 3.6%. 1H NMR (CD2Cl2, 293 K): 7.1–7.6 (m). 19F{1H} NMR (CD2Cl2, 293 K): +161.8 (s, [F]), +128.6 (s, [4F]); (210 K): +157.0 (s, [F]), +125.7 (s, [4F]). 31P{1H} NMR (CD2Cl2, 293 K): 53.9 (s). 93Nb NMR (CD2Cl2, 293 K): −1530 (br s). IR (Nujol/cm−1): 1061 (vs) PO, 624 (sh), 608 (vs, br) NbF.
[NbF5(OPMe3)]: Made similarly to the OPPh3 adduct. Yield 75%. Anal: required for C3H9F5NbOP (280.0): C, 12.9; H, 3.2. Found: C, 13.2; H, 3.5%. 1H NMR (CD2Cl2, 293 K): 1.9 (d, 2JPH = 15 Hz). 19F{1H} NMR (CD2Cl2, 293 K): 157.6 (s, [F]), 134.5 (s, [4F]). 31P{1H} NMR (CD2Cl2, 293 K): +75.6 93Nb NMR (CD2Cl2, 293 K): −1530 (br, s). IR (Nujol/cm−1): 1092 (vs) PO, 615 (vs, br), 582 (m) NbF.
[NbF5(OAsPh3)]: Prepared as for the OPPh3 analogue except that the complex was prepared in ice-bath and solution stirred for 5 min. It was then concentrated in vacuo and the precipitated solid isolated immediately. If the solid is left in solution a yellow and then brown colour develops and in situ NMR data shows formation of Ph3AsF2, [NbF6]− and other unidentified impurities. The pure solid seems stable for some weeks in a freezer. Yield 55%. Anal: required for C18H15AsF5NbO (510.1): C, 42.4; H, 3.0. Found: C, 42.4; H, 3.0%. 1H NMR (CD2Cl2, 293 K): 7.2–7.6 (m). 19F{1H} NMR (CD2Cl2, 293 K): +145.0 (s, [F]), +110.5 (s, [4F)]. 93Nb NMR (CD2Cl2, 293 K): −1511 (br, s). IR (Nujol/cm−1): 845 (m) AsO, 620 (sh), 600 (vs, br) NbF.
[NbF4(2,2′-bipy)2][NbF6]: NbF5 (0.188 g, 1.0 mmol) was added to CH2Cl2 (20 mL) and vigorously stirred, whilst a solution of 2,2′-bipy (0.16 g, 1.0 mmol) in CH2Cl2 (10 mL) was added, resulting in rapid precipitation of a fine white powder. After 2 h the solid was isolated by filtration, rinsed with diethyl ether (5 mL) and dried in vacuo. Yield 0.30 g, 86%. Anal: required for C20H16F10N4Nb2 (688.2): C, 34.9; H, 2.3; N, 8.1. Found: C, 34.7; H, 2.2, N, 8.1%. 1H NMR (CD2Cl2, 293 K): 9.34 (d, [2H], J = 9 Hz), 8.63 (d, [2H], J = 9 Hz), 8.40 (m, [2H]), 7.78 (m, [2H]). 19F{1H} NMR (CD2Cl2, 293 K): +139.7 (s, [4F]), +103.2 (10 lines, J = 335 Hz). IR (Nujol/cm−1): 615 (vs), 603 (s), 585 (vs) NbF.
[NbF4(1,10-phen)2][NbF6]: was made similarly in 89% yield. Anal: required for C24H16F10N4Nb2 (736.2): C, 39.2; H, 2.2; N, 7.6. Found: C, 39.2; H, 2.3, N, 7.4%. 1H NMR (CD3CN, 293 K): 9.20 (d, [2H], J = 9 Hz), 8.96 (d, [2H], J = 9 Hz), 8.27 (m, [2H]), 8.17 (m, [2H]). 19F{1H} NMR (CD3CN, 293 K): +138.0 (s, [4F]), +103.4 (10 lines, J = 335 Hz). IR (Nujol/cm−1): 608 (vs), 586 (vs), 565 (sh) NbF.
[NbOF3(OPPh3)2]: NbF5 (0.19 g, 1 mmol) and OPPh3 (0.56 g, 2 mmol) were added to dry CH2Cl2 (25 mL) and the mixture stirred for 20 min. Hexamethyldisiloxane (0.16 g, 1 mmol) and MeCN (0.5 mL) were added and the mixture stirred overnight at room temperature. The solvents were removed in vacuo leaving a slightly sticky white powder which was stirred with dry diethyl ether (40 mL) when it became a fine white powder. This was filtered off, rinsed with diethyl ether (10 mL) and dried in vacuo. Yield 0.41 g, 57%. Refrigeration of the filtrate gave small crystals used for the X-ray data collection. Anal: required for C36H30F3NbO3P2 (722.5): C, 59.9; H, 4.2. Found: C, 59.6; H, 4.3%. 1H NMR (CD2Cl2, 293 K): 7.1–7.7 (m). 19F{1H} NMR (CD2Cl2, 293 K): 49.5 (s, [F]), 37.8 (s, [2F]). 31P{1H} NMR (CD2Cl2, 293 K): 45.0 (s, [P]), 36.0 (s, [P]). 93Nb NMR (CD2Cl2, 293 K): not observed. IR (Nujol/cm−1): 1155 (m), 1067 (s) PO, 941 (s) NbO, 602 (m), 579 (s) NbF.
[NbOF3(OPMe3)2]: was made similarly Yield 50.5%. Crystals were obtained by refrigeration overnight of the filtrate from the synthesis solution. Anal: required for C6H18F3NbO3P2·CH2Cl2 (435.0): C, 19.3; H, 4.6. Found: C, 18.7; H, 4.3%. 1H NMR (CD2Cl2, 293 K): 1.60 (d, [H] 2JPH = 13 Hz), 1.86 (d, [H] 2JPH = 13 Hz). 19F{1H} NMR (CD2Cl2, 293 K): 41.5 (s, [F]), 30.6 (s, [2F]). 31P{1H} NMR (CD2Cl2, 293 K): 67.1 (s, [P]), 53.3 (s, [P]). 93Nb NMR (CD2Cl2, 293 K): not observed. IR (Nujol/cm−1): 1140 (m), 1087 (s) (PO), 958 (s), NbO, 614 (s), 555 (s) NbF.
[NbOF3(2,2′bipy)]: NbF5 (0.19 g, 1 mmol) was dissolved in CH2Cl2 (200 mL) and dry 2,2′-bipy (0.16 g, 1 mmol) in CH2Cl2 (10 mL) was added with stirring. After 15 min. hexamethyldisiloxane (0.16 g, 1 mmol) and MeCN (0.5 mL) were added and the mixture stirred overnight at room temperature, producing a white precipitate. The mixture was concentrated to ∼5 mL in vacuo, the white solid filtered off, rinsed with diethyl ether and dried in vacuo. Yield 0.27 g, 83%. Anal: required for C10H8F3N2NbO (322.1): C, 37.3; H, 2.5; N, 8.7. Found: C, 37.5; H, 2.4; N, 8.6%. 1H NMR (CD2Cl2, 293 K): 9.28 (s, [H]), 9.17 (s, [H]), 8.54 (m, [H]), 8.36 (m, [H]), 8.32 (s, [2H]), 7.85 (s, [H]), 7.72 (s, [H]). 19F{1H} NMR (CD2Cl2, 293 K): 49.0 (s, [F]), 42.8 (s, [2F]). IR (Nujol/cm−1): 959 (s) NbO, 612 (vs), 579 (s) NbF.
[NbOF3(1,10-phen)]: NbF5 (0.19 g, 1 mmol) was dissolved in CH2Cl2 (200 mL) and dry 1,10-phen (0.18 g, 1 mmol) in CH2Cl2 (10 mL) added with stirring, producing some fine white precipitate. After 5 min hexamethyldisiloxane (0.16 g, 1 mmol) and MeCN (0.5 mL) were added and the mixture stirred for 48 h. at room temperature, producing a dense white precipitate. The precipitate was filtered off, rinsed with diethyl ether (10 mL) and dried in vacuo. Yield 0.30 g, 86%. Anal: required for C12H8F3N2NbO (346.1): C, 41.6; H, 2.3; N, 8.1. Found: C, 41.4; H, 2.3; N, 7.9%. 1H NMR (CD2Cl2, 293 K): 9.36 (s, [H]), 9.28 (s, [H]), 8.77 (m, [H]), 8.56 (m, [H]), 8.19 (s, [H]), 8.13 (s, [H]), 8.02 (s, [H]), 7.90 (s, [H]). 19F{1H} NMR (CD2Cl2, 293 K): insufficiently soluble. IR (Nujol/cm−1): 970 (s) NbO, 610 (sh), 594 (s), 583 (s) NbF.
[NbOF3(dppmO2)]: NbF5 (0.19 g, 1 mmol) and dppmO2 (0.41 g, 1 mmol) were added to dry CH2Cl2 (25 mL) and the mixture stirred for 20 min. Hexamethyldisiloxane (0.16 g, 1 mmol) and MeCN (0.5 mL) were added and the mixture stirred overnight at room temperature. The solvents were removed in vacuo leaving a slightly sticky cream powder which was extracted with CH2Cl2 (20 mL), filtered to remove some undissolved solid, and concentrated to ∼5 mL. Dry diethyl ether (20 mL) was added slowly and the cream precipitate filtered off and dried in vacuo. Yield 0.34 g, 45%. Refrigeration of the filtrate for 5 d. gave crystals suitable for the X-ray data collection. Anal: required for C25H22F3NbO3P2 (582.3): C, 51.6; H, 3.8. Found: C, 51.5; H, 3.9%. 1H NMR (CD2Cl2, 293 K): 7.82–7.15 (m, [10H]), 3.70 (m, [H], J = 13 Hz). 19F{1H} NMR (CD2Cl2, 293 K): 55.7 (s, [F]), 36.4 (s, [2F]). 31P{1H} NMR (CD2Cl2, 293 K): 46.4(d, [P], 2Jpp = 17 Hz), 36.8 (s, [P], 2Jpp = 17 Hz). IR (Nujol/cm−1): 1156 (s), 1088 (s) PO, 944 (s) NbO, 608 (vs), 582 (s) NbF.
[NbOF3(dmso)2]: NbF5 (0.19 g, 1 mmol) was added to dry CH2Cl2 (25 mL), followed by dry dmso (0.5 mL) and the mixture stirred for 20 min. producing a clear colourless solution. Hexamethyldisiloxane (0.16 g, 1 mmol) and MeCN (0.5 mL) were added and the mixture stirred for 6 h at room temperature, during which a fine microcrystalline solid was deposited. The solid was filtered off, rinsed by diethyl ether (5 mL) and dried in vacuo. Yield 0.25 g, 78%. Anal: required for C4H12F3NbO3S2 (322.2): C, 14.9; H, 3.8. Found: C, 15.1; H, 3.9%. 1H NMR (CD2Cl2, 293 K): 2.65 (br); (253 K): 2.59 ([6H]), 2.55 ([6H]). 19F{1H} NMR (CD2Cl2, 293 K): 50.4 (s, [F]), 38.0 (s, [2F]). IR (Nujol/cm−1): 1039 (s), 1005 (s) Me2SO, 920 (s) NbO, 590 (s), 564 (s) NbF.
[NbOF3(tmeda)]: NbF5 (0.19 g, 1 mmol) was added to dry CH2Cl2 (200 mL), followed by dry tmeda (0.12 g, 1 mmol) and the mixture stirred for 20 min. producing a cloudy suspension. Hexamethyldisiloxane (0.16 g, 1 mmol) and MeCN (0.5 mL) were added and the mixture stirred overnight at room temperature, during which a fine white powder was deposited. The solid was filtered off, rinsed by diethyl ether (5 mL) and dried in vacuo. Yield 0.24 g, 85%. Anal: required for C6H16F3N2NbO·CH2Cl2 (367.0): C, 21.9; H, 4.9; N, 7.6. Found: C, 21.4; H, 5.5; N, 7.9%. Insoluble in non-donor solvents. IR (Nujol/cm−1): 920 (s) NbO, 587 (s) 557 (s) NbF.
[NbOCl3(2,2′-bipy)]: NbCl5 (0.067 g, 0.25 mmol) was dissolved into acetonitrile (4 mL) to give a bright yellow-green solution. Hexamethyldisiloxane (0.040 g, 0.25 mmol) was added and the mixture was stirred for 10 min. during which time the solution turned very pale. 2,2′-Bipy (0.039 g, 0.25 mmol) in acetonitrile (4 mL) was added slowly with stirring. After 30 min. the solution was concentrated in vacuo and the white precipitate filtered off, and dried in vacuo. Yield 0.048 g, 52%. Crystals of [NbOCl3(2,2′-bipy)] were grown from acetonitrile solution in the freezer. Anal: required for C10H8Cl3N2NbO (371.4): C, 32.3; H, 2.2; N, 7.5. Found: C, 32.3; H, 2.1; N, 7.7%. 1H NMR (CD2Cl2, 295 K): 8.98 (s, [H]), 8.91 (s, [H]), 8.31 (br m, [4H]), 7.79 (s, [H]), 7.73 (s, [H]). IR (Nujol/cm−1): 943 (s) NbO, 349 (s), 338 (s) NbCl.
[NbOCl3(1,10-phen)]: The white compound was made in an analogous way to [NbOCl3(2,2′-bipy)]. Yield 61%. Anal: required for C12H8Cl3N2NbO (395.4): C, 36.4; H, 2.0; N, 7.1. Found: C, 36.3; H, 2.0; N, 7.1%. 1H NMR (CD2Cl2, 295 K): 9.86 (s, [H]), 9.75 (s, [H]), 8.88 (m, [H]), 8.74 (m, [H]), 8.17 (s, [2H]), 8.08 (s, [2H]). IR (Nujol/cm−1): 944 (s) NbO, 338 (vbr, s) NbCl.
[NbOCl3(tmeda)]: NbCl5 (0.270 g, 1.0 mmol) was dissolved into acetonitrile (10 mL) and hexamethyldisiloxane (0.244 g, 1.5 mmol) was added. After 10 min. tmeda (0.14 g, 1.2 mmol) in dichloromethane (4 mL) was added slowly to the reaction mixture with stirring. After 2 h the mixture was concentrated in vacuo and the resulting precipitate was filtered off and dried in vacuo. Yield 0.055 g, 17%. Single crystals of [NbOCl3(tmeda)] were grown from the filtrate in the freezer. Anal: required for C6H16Cl3N2NbO (331.4): C, 21.7; H, 4.9; N, 8.5. Found: C, 21.6; H, 4.8; N, 8.4%. 1H NMR (CD2Cl2, 295 K): insoluble. IR (Nujol/cm−1): 945 (s) NbO, 341 (s) 320 (sh) NbCl.
[NbOCl3(OPPh3)2]: NbCl5 (0.270 g, 1.0 mmol) was dissolved in acetonitrile (5 mL) whilst stirring and hexamethyldisiloxane (0.162 g, 1.0 mmol) was added. After 10 min. OPPh3 (0.556 g, 2 mmol) was added producing a milky white mixture. The reaction was left to stir for 2 h and the white solid filtered off and dried in vacuo. Yield: 0.450 g, 58%. Anal: required for C36H30O3Cl3NbP2 (771.8): C, 56.0; H, 3.9. Found: C, 55.7; H, 3.6%. 1H NMR (CD2Cl2, 295 K): 7.7–7.2 (m). 31P{1H} NMR (CDCl3, 298 K): 50.0 (s, [P]), 38.8 (s, [P]). IR (Nujol/cm−1): 1159 (s), 1074(s) PO, 936 (s) NbO, 325(s), 294(m) NbCl.
[NbOCl3(dppeO2)]: NbCl5 (0.068 g, 0.25 mmol) was dissolved in acetonitrile (5 mL) and hexamethyldisiloxane (0.062 g, 0.38 mmol) was added. The mixture was left to stir for 15 min. and then dppeO2 (0.108 g, 0.25 mmol) was added and the reaction was left to stir overnight. The precipitate was filtered off, rinsed with small amount of CH2Cl2 and dried in vacuo. Yield 0.102 g, 63%. Crystals of [NbOCl3(dppeO2)] were grown from CH2Cl2 solution in the freezer. Anal: required for C26H24Cl3NbO3P2 (645.6): C, 48.4; H, 3.8. Found: C, 48.6; H, 4.0%. 1H NMR (CD2Cl2, 295 K): 7.89–7.48 (m [10H]), 2.84 (m, [H]), 2.62 (m, [H]). 31P{1H} NMR (CDCl3, 298 K): 56.7 (s), 44.9 (s). IR (Nujol/cm−1): 1172 (s), 1066 (s) PO, 943 (s) NbO, 320 (s), 293 (w) NbCl.
[NbOCl3(dppmO2)]: was made similarly to [NbOCl3(dppeO2)]. Yield 67%. Crystals of [NbOCl3(dppmO2)] were grown from a saturated dichloromethane solution in the freezer. Anal: required for C25H22Cl3NbO3P2 (631.6): C, 46.7; H, 3.5. Found: C, 46.8; H, 3.9%. 1H NMR (CD2Cl2, 295 K): 7.75–7.35 (m, [10H]), 3.80 (t, [H], 2JPH = 15 Hz). 31P{1H} NMR (CH2Cl2–CDCl3, 298 K): 48.5 (d, 2JPP = 19 Hz) 36.8 (d, 2JPP = 19 Hz). IR (Nujol/cm−1): 1157 (s), 1095 (s) PO, 928 (s) NbO, 327 (s), 294 (m) NbCl.
[TaF5(OPPh3)]: A solution of OPPh3 (0.26 g, 1.0 mmol) in CH2Cl2 (20 mL) was added to finely powdered TaF5 (0.28 g, 1.0 mmol), and vigorously stirred to give a clear solution. This was filtered to remove any residual solid and concentrated in vacuo to ∼2 mL. On standing a white powder separated, which was filtered off and dried in vacuo. Yield 0.45 g, 81%. Anal: required for C18H15F5OPTa (554.2): C, 39.0; H, 2.7. Found: C, 38.5; H, 2.9%. 1H NMR (CD2Cl2, 293 K): 7.2–7.6 (m). 19F{1H} NMR (CD2Cl2, 293 K): 84.2 (s, [F)], 54.7 (s, [4F]; (210 K): 81.8 (s, [F]), 56.3 (s, [4F]). 31P{1H} NMR (CD2Cl2, 293 K): 53.2(s). IR (Nujol/cm−1): 1078 (vs) PO, 617 (sh), 592 (vs, br) TaF.
[TaF5(OAsPh3)]: was made similarly, from OAsPh3 (0.32 g, 1.0 mmol) and TaF5 (0.28 g, 1.0 mmol), except that the reaction was worked up and the solid isolated after 20 min. Yield 0.50 g, 85%. Anal: required for C18H15AsF5OTa (598.2): C, 36.2; H, 2.5. Found: C, 37.3; H 2.6%. 1H NMR (CD2Cl2, 293 K): 7.2–7.6 (m). 19F{1H} NMR (CD2Cl2, 293 K): 62.5 (s, [F]), 48.6 (s, [4F]), weak resonances at 38.6 ([TaF6]−) and −89.4 (Ph3AsF2). IR (Nujol/cm−1): 845 (s) AsO, 620 (sh), 581 (vs, br) TaF.
[TaF5(OPMe3)]: A solution of OPMe3 (0.092 g, 1.0 mmol) in CH2Cl2 (10 mL) was added to finely powdered TaF5 (0.276 g, 1.0 mmol), and vigorously stirred to give a clear solution. This was filtered to remove any residual solid and concentrated in vacuo to ∼5 mL. A white powder separated, which was filtered off and dried in vacuo. Yield 0.25 g, 65%. Anal: required for C3H9F5OPTa (368.0): C, 9.8; H, 2.5. Found: C, 10.2; H, 2.3%. 1H NMR (CD2Cl2, 293 K): 1.9 (d, 2JPH = 15 Hz). 19F{1H} NMR (CD2Cl2, 293 K): 82.5 (s, [F]), 55.9 (s, [4F]). 31P{1H} NMR (CD2Cl2, 293 K): 76.9 (s). IR (Nujol/cm−1): 1092 (vs) PO, 601 (sh), 583 (vs, br) TaF.
[TaF4(2,2′-bipy)2][TaF6]: TaF5 (0.28 g, 1.0 mmol) was added to CH2Cl2 (50 mL) and vigorously stirred, whilst a solution of 2,2′-bipy (0.16 g, 1.0 mmol) in CH2Cl2 (10 mL) was added, resulting in rapid precipitation of a fine white powder. After 24 h the solid was isolated by filtration, rinsed with diethyl ether (5 mL) and dried in vacuo. Yield 0.35 g, 86%. Anal: required for C20H16F10N4Ta2 (864.2): C, 27.8; H, 1.9; N, 6.5. Found: C, 27.9; H,1.9; N, 6.4%. 1H NMR (CD2Cl2, 293 K): 9.34 (d, [2H], J = 9 Hz), 8.50 (d, [2H], J = 8 Hz), 8.37 (m, [2H]), 7.81 (m, [2H]). 19F{1H} NMR (CD2Cl2, 293 K): 68.1 (s, [4F]), 38.0 (s, [6F]). IR (Nujol/cm−1): 605 (sh), 581 (vs) TaF.
[TaF4(1,10-phen)2][TaF6]: was made similarly. Yield 83%. Anal: required for C24H16F10N4Ta2 (912.3): C, 31.6; H, 1.8; N, 6.1. Found: C, 31.5; H, 1.8; N, 6.0%. 1H NMR (CD2Cl2, 293 K): 9.15 (s, [2H]), 8.63 (d, [2H], J = 10 Hz), 8.09 (s, [2H]), 7.92 (m, [2H]). 19F{1H} NMR (CD2Cl2, 293 K): 66.1 (s, [4F]), 37.9 (s, [6F]). IR (Nujol/cm−1): 605 (sh), 576 (vs) TaF.
X-Ray experimental
Details of the crystallographic data collection and refinement parameters are given in Table 2. Crystals suitable for single crystal X-ray analysis were obtained as described above. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (100 μm focus) with the crystal held at 100 K (N2 cryostream). Structure solution and refinement were straightforward,22,23 except as detailed below, with H atoms bonded to C being placed in calculated positions using the default C–H distance. Several cases of O/X disorder have been discussed in the text. Three of the carbon atoms in [NbOCl3(tmeda)], C4, C5 and C6 were elongated, suggesting some disorder, but attempts to split these over two positions were unsuccessful. For [NbOCl3(2,2′-bipy)] Nb2 was initially placed on the two-fold axis but showed a very elongated ellipsoid with two large Q peaks close to Nb2. A subsequent model displaced Nb2 by a few tenths of an Å from the axis and this gave a better fit to the data, R1 reduced from ∼0.08 to ∼0.05.| Compound | [NbOF3(OPPh3)2] | [NbOF3(OPMe3)2]·0.33CH2Cl2 | [NbOF3(dppmO2)] | [NbOCl3(dppmO2)·0.3MeCN |
|---|---|---|---|---|
| Formula | C36H30F3NbO3P2 | C6H18F3NbO3P2·0.33CH2Cl2 | C25H22F3NbO3P2 | C25H22Cl3NbO3P2·0.3CH3CN |
| M | 722.45 | 378.36 | 582.28 | 643.94 |
| Crystal system | Orthorhombic | Triclinic | Monoclinic | Monoclinic |
| Space group (no.) | Fdd2 (no. 43) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no 2) (no 2) |
P21/n (no. 14) | P21/n (no. 14) |
| a/Å | 18.762(9) | 7.8890(15) | 13.154(8) | 10.694(2) |
| b/Å | 33.289(14) | 14.584(3) | 10.967(6) | 15.640(4) |
| c/Å | 10.152(5) | 20.046(4) | 17.248(10) | 17.181(4) |
| α/° | 90 | 102.497(4) | 90 | 90 |
| β/° | 90 | 99.803(4) | 97.173(19) | 105.721(6) |
| γ/° | 90 | 97.324(2) | 90 | 90 |
| U/Å3 | 6340(5) | 2186.3(7) | 2469(3) | 2766.0(13) |
| Z | 8 | 6 | 4 | 4 |
| μ(Mo-Kα)/mm−1 | 0.534 | 1.191 | 0.665 | 0.867 |
| F(000) | 2944 | 1140 | 1176 | 1298 |
| Total number reflns | 7136 | 22109 |
15969 |
11311 |
| R int | 0.0849 | 0.0250 | 0.0915 | 0.0386 |
| Unique reflns | 3000 | 9978 | 4794 | 5288 |
| No. of params, restraints | 204, 20 | 556, 15 | 307, 0 | 320, 2 |
| R 1, wR2 [I > 2σ(I)]b | 0.0719, 0.1062 | 0.0359, 0.0847 | 0.0770, 0.1725 | 0.0514, 0.0946 |
| R 1, wR2 (all data) | 0.0888, 0.1148 | 0.0430, 0.0880 | 0.0982, 0.1854 | 0.0742, 0.1045 |
| Compound | [NbOCl3(dppeO2)]·0.5MeCN | [NbOCl3(2,2′-bipy)] | [NbOCl3(tmeda)] |
|---|---|---|---|
| a Common items: T = 100 K; wavelength (Mo-Kα) = 0.71073 Å; θ(max) = 27.5°. b R 1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑wFo4]1/2. | |||
| Formula | C26H24Cl3NbO3P2·0.5CH3CN | C10H8Cl3N2NbO | C6H16Cl3N2NbO |
| M | 666.18 | 371.44 | 331.47 |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic |
| Space group (no.) | P21/n (no. 14) | Fdd2 (no. 43) | Pna21 (no. 33) |
| a/Å | 10.752(2) | 12.4975(4) | 14.352(7) |
| b/Å | 14.367(3) | 21.6322(8) | 7.368(4) |
| c/Å | 18.048(4) | 29.090(2) | 11.781(6) |
| α/° | 90 | 90 | 90 |
| β/° | 92.519(10) | 90 | 90 |
| γ/° | 90 | 90 | 90 |
| U/Å3 | 2785.1(10) | 7864.3(6) | 1245.8(11) |
| Z | 4 | 24 | 4 |
| μ(Mo-Kα)/mm−1 | 0.864 | 1.512 | 1.578 |
| F(000) | 1348 | 4368 | 664 |
| Total number reflns | 14141 |
5418 | 3500 |
| R int | 0.0555 | 0.0243 | 0.0196 |
| Unique reflns | 5451 | 2991 | 2135 |
| No. of params, restraints | 338, 2 | 240, 16 | 119, 2 |
| R 1, wR2 [I > 2σ(I)]b | 0.0591, 0.0921 | 0.0469, 0.1169 | 0.0449, 0.1102 |
| R 1, wR2 (all data) | 0.0721, 0.0968 | 0.0506, 0.1204 | 0.0521, 0.1162 |
Acknowledgements
We thank EPSRC for support (EP/1033394/1) and Drs M. Webster and M. E. Light for assistance with the X-ray data analyses.References
- S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460 RSC.
- (a) J. Sala-Pala and J. E. Guerchais, J. Mol. Struct., 1974, 20, 169 CrossRef CAS; (b) M. F. Davis, W. Levason, J. Paterson, G. Reid and M. Webster, Eur. J. Inorg. Chem., 2008, 802 CrossRef CAS; (c) M. D. Hoops and B. S. Ault, J. Mol. Struct., 2002, 616, 91 CrossRef CAS.
- (a) A. J. Edwards, D. R. Slim, J. Sala-Pala and J. E. Guerchais, J. Chem. Soc., Dalton Trans., 1977, 984 RSC; (b) M. F. Davis, M. Jura, A. Leung, W. Levason, B. Littlefield, G. Reid and M. Webster, Dalton Trans., 2008, 6265 RSC; (c) P. DeBurgomaster and J. Zubieta, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m1303 CAS.
- (a) F. Marchetti and G. Pampaloni, Chem. Commun., 2012, 48, 635 RSC; (b) R. Bini, C. Chiappe, F. Marchetti, G. Pampaloni and S. Zacchini, Inorg. Chem., 2010, 49, 339 CrossRef CAS PubMed; (c) F. Marchetti, G. Pampaloni and S. Zacchini, Inorg. Chem., 2008, 47, 365 CrossRef CAS PubMed; (d) F. Marchetti, G. Pampaloni and S. Zacchini, J. Fluorine Chem., 2010, 131, 21 CrossRef CAS PubMed; (e) F. Marchetti, G. Pampaloni and S. Zacchini, Dalton Trans., 2009, 8096 RSC; (f) F. Marchetti, G. Pampaloni and S. Zacchini, Dalton Trans., 2009, 6759 RSC; (g) M. Bortoluzzi, F. Marchetti, G. Pampaloni, M. Puchino and S. Zacchini, Dalton Trans., 2013, 42, 13054 RSC.
- (a) M. Jura, W. Levason, R. Ratnani, G. Reid and M. Webster, Dalton Trans., 2010, 39, 883 RSC; (b) S. L. Benjamin, A. Hyslop, W. Levason and G. Reid, J. Fluorine Chem., 2012, 137, 77 CrossRef CAS PubMed.
- J. Köhler, A. Simon, L. van Wüllen, S. Cordier, T. Roisnel, M. Poulain and M. Somer, Z. Anorg. Allg. Chem., 2002, 628, 2683 CrossRef.
- (a) T. Funaioli, F. Marchetti, G. Pampoloni and S. Zacchini, Dalton Trans., 2013, 42, 14168 RSC; (b) J. Sala-Pala, J. Y. Calves and J. Guerchais, J. Inorg. Nucl. Chem., 1975, 37, 1296 CrossRef.
- L. G. Hubert-Pfalzgraf, M. Postel and J. G. Reiss, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, vol. 3, p. 585 Search PubMed.
- (a) W. A. Herrmann, W. R. Thiel and E. Herdtweck, Chem. Ber., 1990, 123, 271 CrossRef CAS; (b) V. C. Gibson and T. P. Kee, J. Chem. Soc., Dalton Trans., 1993, 1657 RSC; (c) V. C. Gibson, T. P. Kee and A. Shaw, Polyhedron, 1988, 7, 2217 CrossRef CAS; (d) V. C. Gibson, T. P. McKee, R. M. Sorrell, A. P. Bashall and M. McPartlin, Polyhedron, 1988, 7, 2221 CrossRef CAS; (e) F. Marchetti, G. Pampaloni and S. Zacchini, Chem. Commun., 2008, 3651 RSC.
- (a) K. Dreisch, C. Andersson and C. Stalhandske, Polyhedron, 1991, 10, 2417 CrossRef CAS; (b) M. F. Davis, W. Levason, R. Ratnani, G. Reid, T. Rose and M. Webster, Eur. J. Inorg. Chem., 2007, 306 CrossRef CAS; (c) M. F. Davis, W. Levason, M. E. Light, R. Ratnani, G. Reid, K. Saraswat and M. Webster, Eur. J. Inorg. Chem., 2007, 1903 CrossRef CAS.
- Yu. A. Buslaev, E. G. Ilyin, M. E. Ignatov, I. S. Butorina and T. A. Mastryukova, J. Fluorine. Chem., 1978, 12, 381 CAS.
- (a) C. Djordjevic and V. Katovic, J. Chem. Soc. A, 1970, 3382 RSC; (b) M. E. Ignatov, D. B. Grebshekov and E. G. Il'lin, Russ. J. Inorg. Chem., 1983, 28, 617 CAS.
- S. L. Benjamin, W. Levason, D. Pugh, G. Reid and W. Zhang, Dalton Trans., 2012, 41, 12548 RSC.
- M. Stroebele and H.-J. Meyer, Z. Anorg. Allg. Chem., 2002, 628, 488 CrossRef CAS.
- C. Chavant, J. C. Daran, Y. Jeannin, G. Constant and R. Morancho, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 1828 CrossRef.
- V. S. Sergienko, M. A. Porai-Koshits, A. A. Konovalova and V. V. Kovalev, Koord. Khim., 1984, 10, 1116 CAS.
- (a) M. Weishaupt and J. Straehle, Z. Anorg. Allg. Chem., 1977, 429, 261 CrossRef CAS; (b) J. Poitras and A. L. Beauchamp, Can. J. Chem., 1994, 72, 1675 CrossRef CAS; (c) U. Mueller and I. Lorenz, Z. Anorg. Allg. Chem., 1980, 463, 110 CrossRef CAS.
- (a) D. L. Kepert, Prog. Inorg. Chem., 1977, 23, 1 CrossRef CAS; (b) R. Hoffmann, J. M. Howell and A. R. Rossi, J. Am. Chem. Soc., 1976, 98, 2484 CrossRef CAS.
- (a) S. El-Kurdi and K. Seppelt, Chem.–Eur. J., 2011, 17, 3956 CrossRef CAS PubMed; (b) K. Seppelt, Acc. Chem. Res., 2003, 36, 147 CrossRef CAS PubMed.
- J. Supel, U. Abram, A. Hagenbach and K. Seppelt, Inorg. Chem., 2007, 46, 5591 CrossRef CAS PubMed.
- C. D. Beard, R. J. Barrie, J. Evans, W. Levason, G. Reid and M. D. Spicer, Eur. J. Inorg. Chem., 2006, 4391 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- S. M. Corcoran, W. Levason, R. Patel and G. Reid, Inorg. Chim. Acta, 2005, 358, 1263 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The crystallographic data and selected bond lengths and angles for [Me3TACNH]2[NbOCl5] are also available in the ESI. CCDC 973570–973577. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C3DT53322K |
| ‡ 93Nb 100% abundance, I = 9/2, Ξ = 24.44 MHz, Q = −0.2 × 10−28 m2, Dc = 2740 is one of the more sensitive nuclei and, despite the medium size quadrupole moment, is readily observed in many systems. The zero reference is [NbCl6]− in MeCN. |
| This journal is © The Royal Society of Chemistry 2014 |