 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thio-, seleno- and telluro-ether complexes of aluminium(III) halides: synthesis, structures and properties†
Kathryn
George
a,
Marek
Jura
b,
William
Levason
*a,
Mark E.
Light
a and
Gillian
Reid
a
aSchool of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: wxl@soton.ac.uk
bISIS, STFC, Harwell Innovation Campus, Didcot, Oxfordshire OX11 0QX, UK
First published on 7th January 2014
Abstract
The reaction of AlCl3 with Me2E (E = S, Se or Te) or nBu2E (E = Se or Te) in CH2Cl2 under rigorously anhydrous conditions gave the pseudo-tetrahedral complexes [AlCl3(R2E)]. The [AlX3(Me2E)] (X = Br or I, E = S; X = Br, E = Te) were made from toluene solution since attempted syntheses in CH2Cl2 resulted in substantial chloride incorporation. The synthesis of [(AlCl3)2{o-C6H4(CH2SEt)2}], in which the ligand bridges two tetrahedral aluminium centres, and of the six-coordinate trans-[AlX2{MeE(CH2)2EMe}2][AlX4] (X = Cl or Br, E = S, and X = Cl, E = Se) and cis-[AlI2{MeS(CH2)2SMe}2][AlI4] are reported. The tripodal thioether forms [AlCl3{MeC(CH2SMe)3}], which is a chain polymer with κ2-coordinated ligand and a tbp arrangement at Al(III). Chalcogenoether macrocycle complexes [AlCl3([9]aneS3)], [AlCl2([14]aneS4)][AlCl4] and [AlCl2([16]aneSe4)] [AlCl4] are also described. All complexes were characterised by microanalysis, IR and multinuclear NMR (1H, 27Al, 77Se or 125Te) spectroscopy as appropriate. In CH2Cl2 solution [AlCl3(Me2S)] with added Me2S forms [AlCl3(Me2S)2], and the [AlX2{MeS(CH2)2SMe}2][AlX4] exist as mixtures of cis and trans isomers which undergo rapid exchange at ambient temperatures. X-Ray crystal structures are reported for [AlCl3(Me2Se], [AlX3(Me2Te)] (X = Cl or Br), trans-[AlCl2{MeE(CH2)2EMe}2][AlCl4] (E = S or Se), cis-[AlI2{MeS(CH2)2SMe}2][AlI4], [AlCl3{MeC(CH2SMe)3}], and for the sulfonium salt [Me2SH][AlCl4]. The aluminium halide chalcogenoether chemistry is compared with the corresponding gallium and indium systems, and the relative Lewis acidities of the metals discussed. Attempts to use [AlCl3(nBu2E)] (E = Se or Te) as LPCVD reagents to form aluminium chalcogenide films were unsuccessful.
Introduction
Aluminium chloride is widely used both in the laboratory and industry as a Friedel–Crafts catalyst for alkylation and acylation reactions, and also catalyses condensation, isomerisation and polymerisation reactions.1 The reactions depend upon the strong Lewis acidity which produces incipient carbocations in combination with [AlCl4]− anions. Aluminium bromide and iodide promote similar chemistry and have some niche applications.1 The strong Lewis acidity of these three halides has resulted in the characterisation of very many adducts, mostly with N- or O-centres in solvents or donor ligands.2 In contrast, AlF3 is an inert polymer which forms few complexes.3 Surprisingly little is known about AlX3 adducts of chalcogenoethers, which are limited to early studies of adducts with Me2S or Et2S that reported phase diagrams, IR and 1H NMR spectra,4–6 and several studies of solution enthalpies.7 The only crystallographically characterised example of a neutral thioether coordinated to AlX3 is the recently reported8 [AlCl3(thianthrene)], although there are a few AlMe3 complexes with thiamacrocycles.9 There are no reports of studies with telluroethers, and the single complex of a selenium ligand is [AlCl3(selenoxan)], characterised only by microanalysis.10Recent studies of gallium(III) halides GaX3 (X = Cl, Br or I) with chalcogenoethers have established that most contain pseudo-tetrahedral gallium centres [GaX3L] (L = Me2S, Me2Se, Me2Te, etc.) or dinuclear [X3Ga(μ-L–L)GaX3] (L–L = RS(CH2)2SR, MeSe(CH2)2SeMe, MeTe(CH2)3TeMe etc.).11–14 Higher coordination numbers are rare in the gallium systems, but found with the thia-macrocycle [14]aneS4,‡ which binds exodentate via two sulfur centres, affording the chain polymer [GaCl3([14]aneS4)] with a trigonal bipyramidal geometry.15 The larger rings [16]aneS4 and [16]aneSe4 form octahedral cations trans-[GaCl2(macrocycle)][GaCl4].15 The larger indium centre forms 4-, 5- or 6-coordinate complexes of types [InX3(R2E)] (E = S, Se or Te), [InX3(R2S)2], [In2X6{RE(CH2)2ER2}2], trans-[InX2{RE(CH2)2ER2}2][InX4],11–16cis-[InCl2{[14]aneS4}][InCl4] and trans-[InCl2{[16]aneSe4}][InCl4].15,16
We have recently reported that [GaCl3(nBu2E)] (E = Se or Te) or [GaCl3{μ-nBuE(CH2)nEnBu)GaCl3] are effective single source precursors for low pressure chemical vapour deposition (LPCVD) of Ga2E3 thin films, and that preferential deposition occurs onto TiN in photolithographically patterned SiO2/TiN substrates.17 Here we report systematic studies of the reactions of AlX3 (X = Cl, Br or I) with a range of thio-, seleno- and telluro-ethers, detailed spectroscopic and structural data, and comparisons with their gallium and indium analogues. We also explored whether selected complexes would function as CVD reagents for deposition of aluminium chalcogenide films.
Results and discussion
Aluminium trihalides (AlX3 (X = Cl, Br or I), are strong hard Lewis acids with a very high affinity for water.2 Successful synthesis of their complexes with soft donor ligands such as thio-, seleno- or telluro-ethers requires anhydrous AlX3, rigorously anhydrous solvents and ligands and exclusion of water at all stages of the manipulations. Trace water displaces the neutral ligands and also generates [AlX4]− which are readily identified in the solids by their characteristic IR spectra ([AlCl4]−t2 = 498, [AlBr4]− 394, [AlI4]− 336 cm−1)18 and in solution by 27Al NMR spectroscopy (Table 1), where they have sharp characteristic resonances. The moisture sensitivity of the halides and the complexes is much greater than observed in the corresponding gallium(III) systems.12,13 The higher reactivity of the aluminium halides also affects the choice of solvent for the synthesis. Whilst complexes of AlCl3 are readily made in anhydrous CH2Cl2, use of this solvent for the AlBr3 or AlI3 reactions results in incorporation of substantial amounts of chloride into the products, and the heavier halides are best made from anhydrous toluene solution. Similar observations were made by Burford et al.19 in AlX3/R3PO/CH2Cl2 systems. However, the pre-isolated pure [AlX3L] (X = Br or I) react only slowly with CH2Cl2 (or CD2Cl2), which remains the NMR solvent of choice, as non-coordinating and useable down to 180 K. The reactivity of AlCl3 in CH2Cl2 has been ascribed to the formation of the intermediate carbenium ion [CH2Cl][AlCl4],8,20 and in previous studies of GaCl3 complexes of thioethers we observed the formation of [o-C6H4(SMeCH2Cl)2][GaCl4]2 when [(GaCl3)2{μ-o-C6H4(SMe)2}] is allowed to stand in CH2Cl2 solution for several days.13 Cleavage of C–Se or C–Te bonds is also observed, e.g. the formation of the selenonium cation in [o-C6H4(CH2)2SeMe][GaCl4] from o-C6H4(CH2SeMe)2.12 In the aluminium systems many of the complexes degrade on standing in solution at ambient temperatures (below), hence rapid isolation of the complexes from solution is advisable. Decomposition in solution is much slower at low temperatures, permitting growth of X-ray quality crystals overnight at −18 °C.| Compoundb | δ (27Al), 298 K | δ (27Al), 190 K | Comments |
|---|---|---|---|
| a Chemical shifts relative to [Al(H2O)6]3+ in H2O at pH = 1. b In CH2Cl2–CD2Cl2 solution at temperature specified. | |||
| [AlCl4]− | 103.5 | 103.6 | W 1/2 ∼ 5 Hz |
| [AlBr4]− | 80.6 | 81.4 | W 1/2 ∼ 15 Hz |
| [AlI4]− | −23.0 | W 1/2 ∼ 25 Hz | |
| [AlCl3(Me2S)] | 111.6 W1/2 ∼ 310 Hz | 111.2 | 73.8 with excess Me2S (298 K) |
| [AlBr3(Me2S)] | 81.2 W1/2 ∼ 250 Hz | 81.6 | No change with added Me2S |
| [AIl3(Me2S)] | 48.7 W1/2 ∼ 360 Hz | — | No change with added Me2S |
| [AlCl3(Me2Se)] | 110.5 W1/2 ∼ 300 Hz | 110.1 | No change with added Me2Se |
| [AlBr3(Me2Se)] | 99.7 W1/2 ∼ 170 Hz | — | |
| [AlCl3(Me2Te)] | 105.9 W1/2 ∼ 310 Hz | 103.4 | |
| [AlBr3(Me2Te)] | 92.6 W1/2 ∼ 320 Hz | — | Resonance lost on cooling |
| [AlCl3(nBu2Se)] | 109.9 W1/2 ∼ 490 Hz | — | |
| [AlCl3(nBu2Te)] | 92.6 W1/2 ∼ 555 Hz | — | |
| [AlCl2{MeS(CH2)2SMe}2][AlCl4] | 105.5, 37.4 | ||
| [AlBr2{MeS(CH2)2SMe}2][AlBr4] | 80.6 | 81.1 | Only anion resonance seen |
| [AlI2{MeS(CH2)2SMe}2][AlI4] | −23.0 | −23.0 | Only anion resonance seen |
| [AlCl2{MeSe(CH2)2SeMe}2][AlCl4] | 105.4 | — | |
| [(AlCl3)2{C6H4(CH2SEt)2}] | 111.3 W1/2 ∼ 900 Hz | — | |
[AlX3(R2E)] complexes
The reaction of AlCl3 in CH2Cl2 or AlX3 (X = Br or I) in toluene with Me2S affords [AlX3(Me2S)] in high yield. The chloro-complex is an oil at room temperature, the others crystalline solids. The complexes can also be made by condensing excess Me2S onto the appropriate AlX3 at −196 °C, allowing the mixture to thaw, when the halide dissolves to give a clear solution, and then removing excess Me2S in vacuo. The phase diagram of the AlCl3/Me2S system4 shows both [AlCl3(Me2S)] and [AlCl3(Me2S)2], but on removing the volatiles in vacuo from a mixture of AlCl3 and 3Me2S, only the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex was isolated.
:1 complex was isolated.
The IR spectrum of [AlCl3(Me2S)] shows features at 541 and 410 cm−1 assigned to the E and A1 modes of the pyramidal AlCl3 unit; both bands are quite broad and the E mode shows some evidence of further splitting. Similar observations were made in the spectra of many of the aluminium complexes in this work, and are probably due to solid state effects, such as lower site symmetry or cation–anion interactions. The 1H NMR spectrum of [AlCl3(Me2S)] in CD2Cl2 (295 K) shows a singlet at δ = 2.53 which does not change significantly on cooling the solution to 190 K. Addition of aliquots of Me2S to the solution produces progressive shifts in the single resonance to low frequency, and on cooling to 185 K, two broad resonances are resolved at δ = 2.24 and 2.20, suggesting ligand exchange is slowing, but that the low temperature limit has not been reached. The latter shift is similar to that of free Me2S (δ = 2.15), whilst the former is assigned to a new aluminium complex. The 27Al NMR spectra are more informative (Table 1). At ambient temperatures a broad singlet at δ = 111.6 is present in the spectrum of [AlCl3(Me2S)], which is little changed on cooling the solution to 190 K. However, addition of 1 mol. equivalent of Me2S to the solution generates a new resonance at δ = 73.5 (W1/2 = 650 Hz), and this resonance is unchanged upon addition of more Me2S and shows only a small low frequency drift on cooling the solution to 190 K. The new 27Al chemical shift is in the range expected for five-coordinate Al species.21 The combination of the 1H and 27Al NMR results show that in the presence of excess Me2S in CH2Cl2 solution the 2:1 complex [AlCl3(Me2S)2] forms; further addition of Me2S does not produce any evidence for a 3:1 complex. As noted above, work-up of the solution results in decomposition to reform [AlCl3(Me2S)]. Five-coordination is established in the solid state with aluminium–phosphine complexes, e.g. [AlI3(PEt3)2], which has a tbp geometry with axial phosphines.22
The complexes [AlX3(Me2S)] (X = Br or I) are generally similar to the chloride complex, and exhibit progressively lower frequency shifts in the 27Al NMR spectra as the halogen becomes heavier (Table 1). However, although the 1H NMR spectra show fast exchange with added Me2S in CH2Cl2 solution, no new resonances were evident in the 27Al NMR spectra in the presence of a large excess of Me2S, indicating that in these cases 2:1 complexes do not form.
The [AlX3(Me2E)] (X = Cl, Br, E = Se or Te) were obtained in high yields and their 1H NMR and IR spectroscopic properties are similar to those of the thioether analogues. The 27Al NMR spectra (Table 1) show only small low frequency shifts along the series E = S > Se > Te, and no new complexes are formed by adding excess Me2E to CH2Cl2 solutions of the appropriate [AlX3(Me2E)]. [AlCl3(Me2Se)] shows a 77Se NMR chemical shift of δ = −11.3, which corresponds to a small low frequency coordination shift (Δ = −11.3); this can be compared with small high frequency coordination shifts observed in [GaX3(Me2Se)].12 Although high frequency coordination shifts are seen in most transition metal selenoether (and telluroether) complexes, in p-block complexes both high and low frequency shifts are seen in different systems, and the causes are not understood.23 We were unable to observe a 77Se NMR resonance from [AlBr3(Me2Se)] or 125Te resonances from [AlX3(Me2Te)] over the temperature range 295–190 K, presumably due to fast exchange. The solutions of the selenoether and telluroether complexes develop new resonances on standing, some of which may be due to Me2E2, Me3E+ or Me2EX2 from their chemical shifts, but given the sensitivity of 77Se and 125Te chemical shifts to concentration, solvent etc.21,24 their identification was not pursued. They do, however, provide evidence of the fragility in solution of the AlX3 complexes with the heavier chalcogenoethers.
In view of the scarcity of aluminium complexes of the heavier chalcogenoethers, structures of three examples, [AlCl3(Me2Se)], [AlCl3(Me2Te)] and [AlBr3(Me2Te)] were determined. The structures (Fig. 1–3) are isomorphous (orthorhombic, Pbcm) and show the expected pseudo-tetrahedral geometry. The bond lengths are mostly unexceptional, although the Al–Te distances in [AlX3(Me2Te)] (X = Cl or Br) are the same within experimental error.
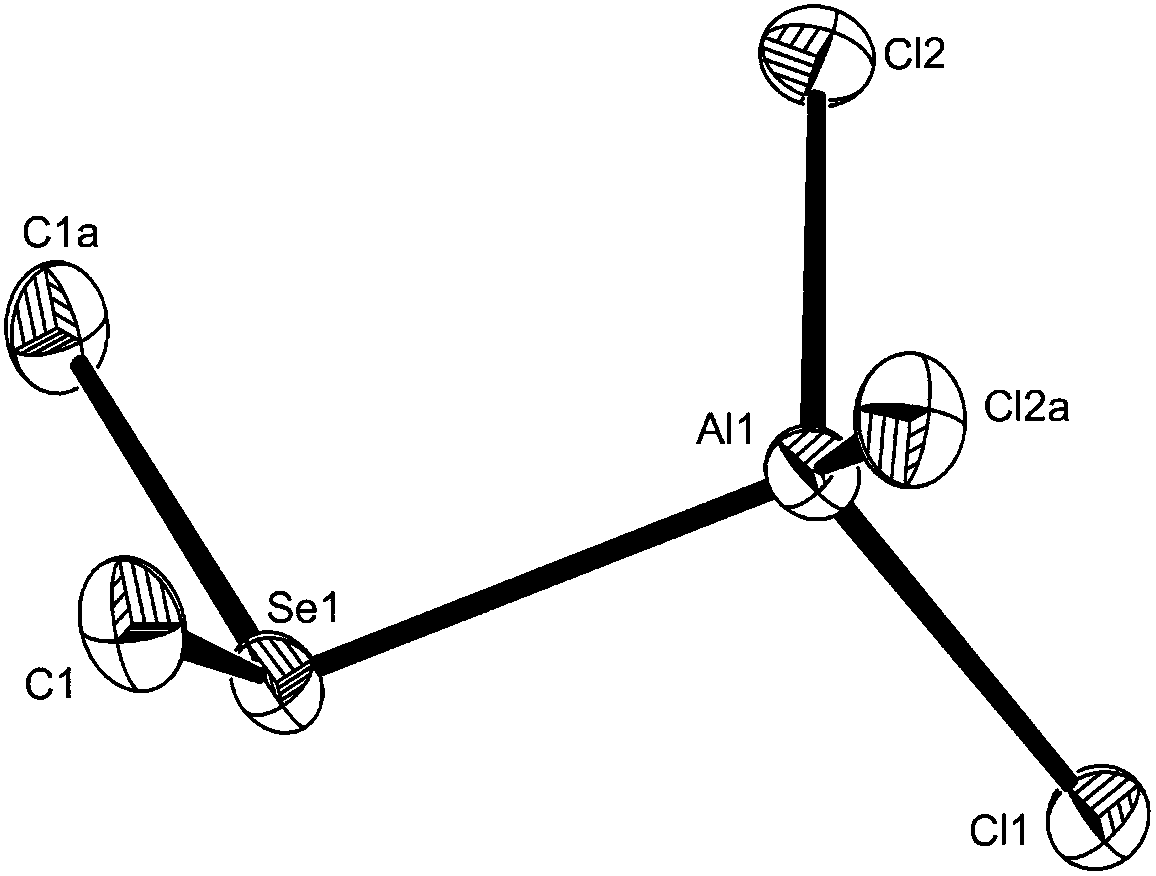 | ||
| Fig. 1 The structure of [AlCl3(Me2Se)] showing the numbering scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = x, y, 3/2 − z. Selected bond lengths (Å) and angles (°) Se1–Al1 = 2.486(2), Cl1–Al1 = 2.121(2), Cl2–Al1 = 2.1130(14), Cl2–Al1–Cl2a = 115.30(9), Cl2–Al1–Cl1 = 111.32(5), Cl2–Al1–Se1 = 106.47(5), Cl1–Al1–Se1 = 105.20(6). | ||
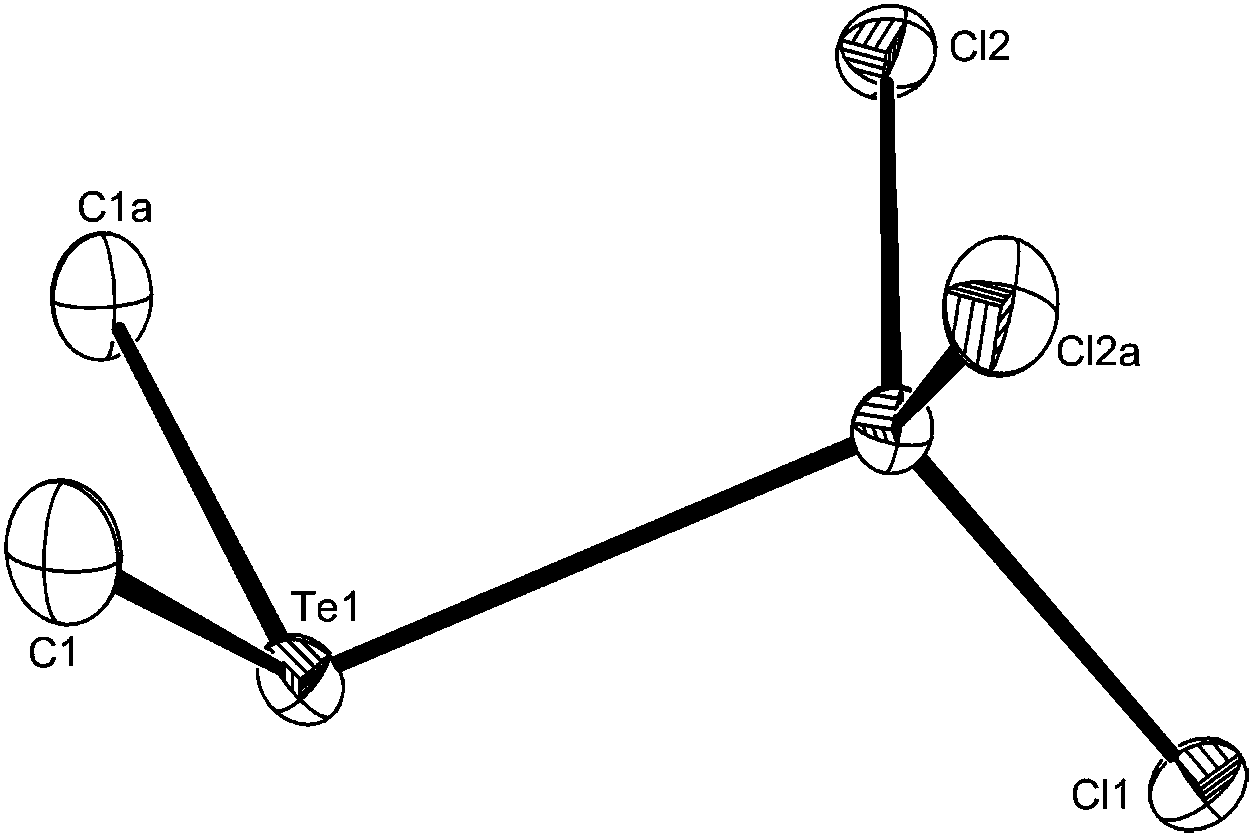 | ||
| Fig. 2 The structure of [AlCl3(Me2Te)] showing the atom numbering scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = x, y, 1/2 − z. Selected bond lengths (Å) and angles (°): Al1–Cl2 = 2.1207(6), Al1–Cl1 = 2.1295(10), Al1–Te1 = 2.6871(9), Cl2–Al1–Cl2a = 115.20(4), Cl2–Al1–Cl1 = 111.14(3), Cl2–Al1–Te1 = 106.25(2), Cl1–Al1–Te1 = 106.21(3). | ||
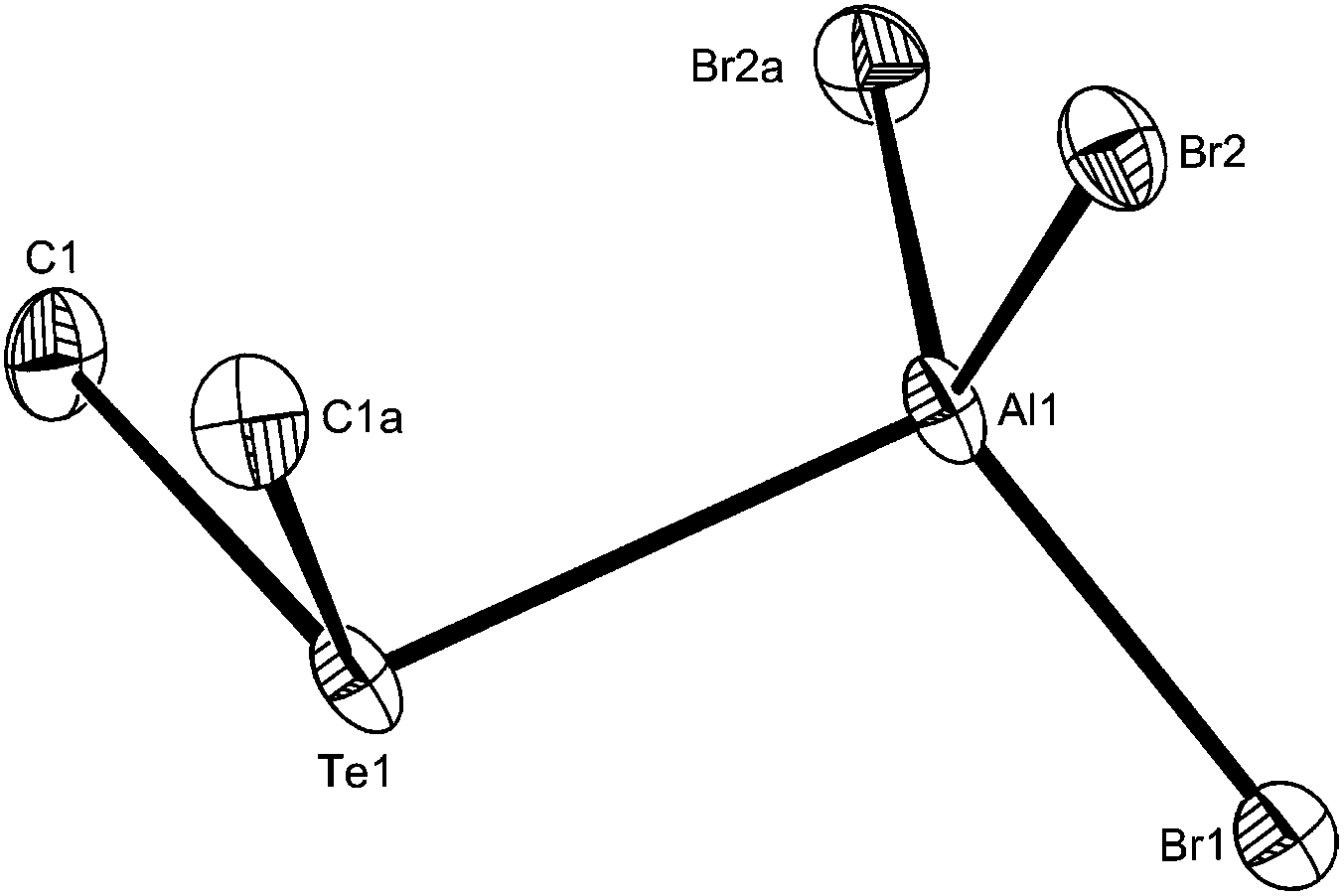 | ||
| Fig. 3 The structure of [AlBr3(Me2Te)] showing the atom numbering scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Symmetry operation: a = x, y, 3/2 − z. Selected bond lengths (Å) and angles (°): Al1–Br1 = 2.286(3), Al1–Br2 = 2.287(2), Al1–Te1 = 2.692(4), Br1–Al1–Br2 = 111.19(9), Br2–Al1–Br2a = 114.01(15), Br1–Al1–Te1 = 106.34(12), Br2–Al1–Te1 = 106.80(9). | ||
Storing a solution of [AlCl3(Me2S)] in CH2Cl2 in the refrigerator, produced a few small crystals which were identified as the sulfonium salt [Me2SH][AlCl4] (Fig. 4), by the X-ray structure solution, and probably formed by adventitious hydrolysis. Solid sulfonium salts are rare, but we have previously obtained examples from serendipitous hydrolysis of some niobium(V) fluoride–thioether complexes,25 the formation being promoted by “anhydrous” conditions and a large weakly coordinating anion. The S–H proton was identified in the difference map of residual electron density. A bulk sample of [Me2SH][AlCl4] was obtained by saturating a dry CH2Cl2 solution of [AlCl3(Me2S)] with HCl gas.
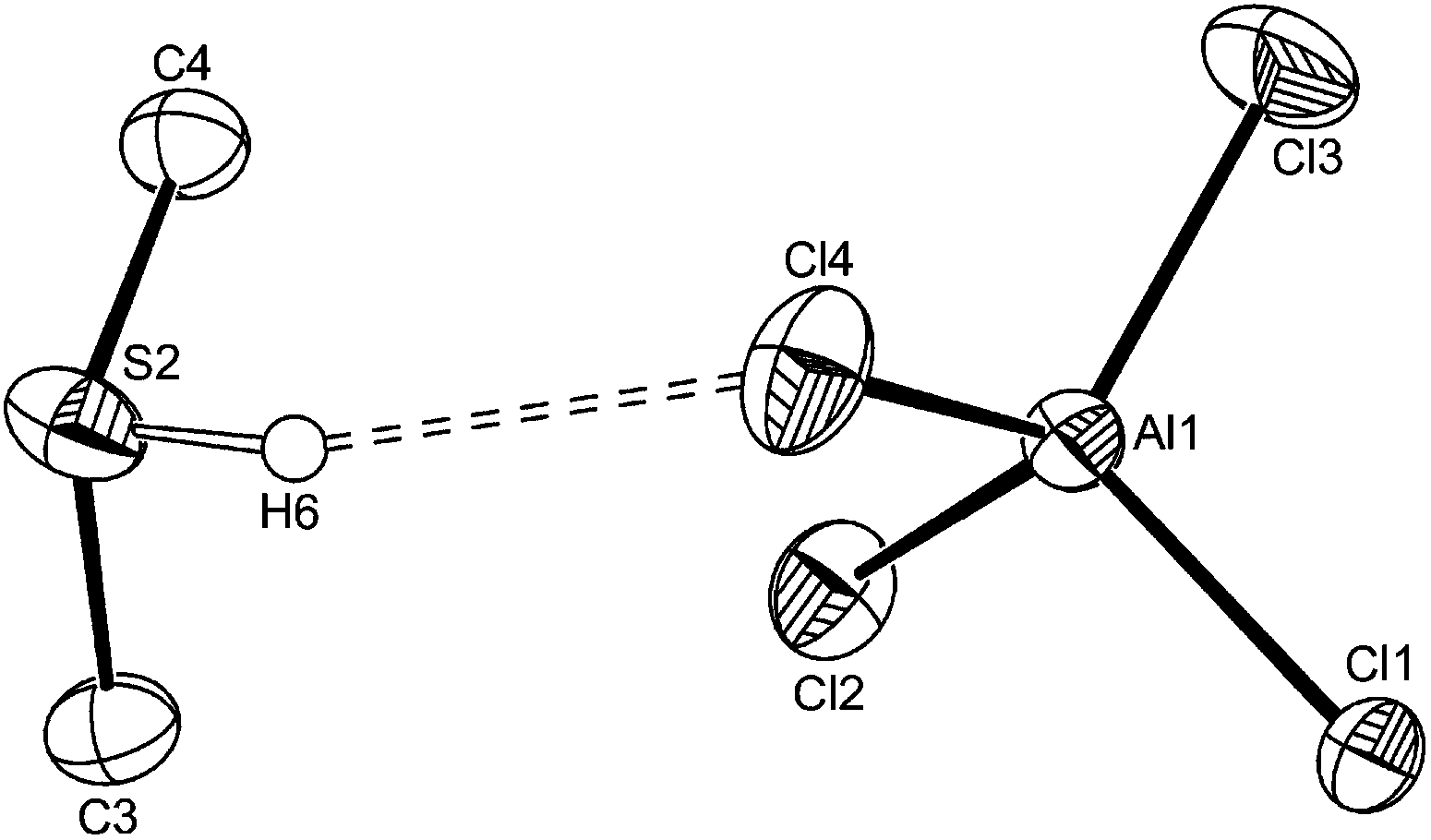 | ||
| Fig. 4 The structure of [Me2SH][AlCl4] showing the S2 centred cation and the H-bond (dashed bond) to an adjacent Cl atom. Ellipsoids are drawn at the 50% probability level and H atoms on C are not shown. The other crystallographically independent cation is similar. Selected bond lengths (Å): Al1–Cl3 = 2.1110(11), Al1–Cl4 = 2.1349(11), Al1–Cl2 = 2.1361(10), Al1–Cl1 = 2.1412(10), S2–H6 = 1.26(3), H6⋯Cl4 = 2.51(3). | ||
The n-butyl derivatives [AlCl3(nBu2E)] (E = Se, Te) were also synthesised as potential single source precursors for LPCVD as discussed below. Their characterisation data matched well with that of the equivalent methyl substituted complexes. The telluroether complexes are not stable when stored at room temperature over a period of days to weeks, even in an N2 filled glove box. They gradually become darker and a quantity of black solid (elemental Te) forms. Samples can be stored at −18 °C for a few weeks with minimal decomposition. A sample of [AlCl3(nBu2Te)] that had been stored in the glove box for several weeks and had visibly decomposed was analysed by ESI+ mass spectrometry, which showed the presence of [nBu3Te]+, and this ion was also present in the 125Te NMR spectrum (δ = 491).13
Complexes with bidentate ligands
By analogy with the corresponding GaX3 adducts,12 and considering the preference for four-coordination in [AlX3(R2E)] adducts described above, it was expected that flexible dithioethers or diselenoethers (L–L) would produce complexes of the type [X3Al(μ-L–L)AlX3]. In fact this was only true for the bulky o-xylyl-backboned dithioether, o-C6H4(CH2SEt)2, which gave yellow crystals of [(AlCl3)2{μ-o-C6H4(CH2SEt)2}]. The crystals were of modest quality and had the characteristics of a modulated structure (see Experimental), and thus comparison of the detailed bond lengths and angles is not warranted. However, they are isomorphous with the gallium(III) analogue,13 and serve as an example of this structure type (Fig. 5).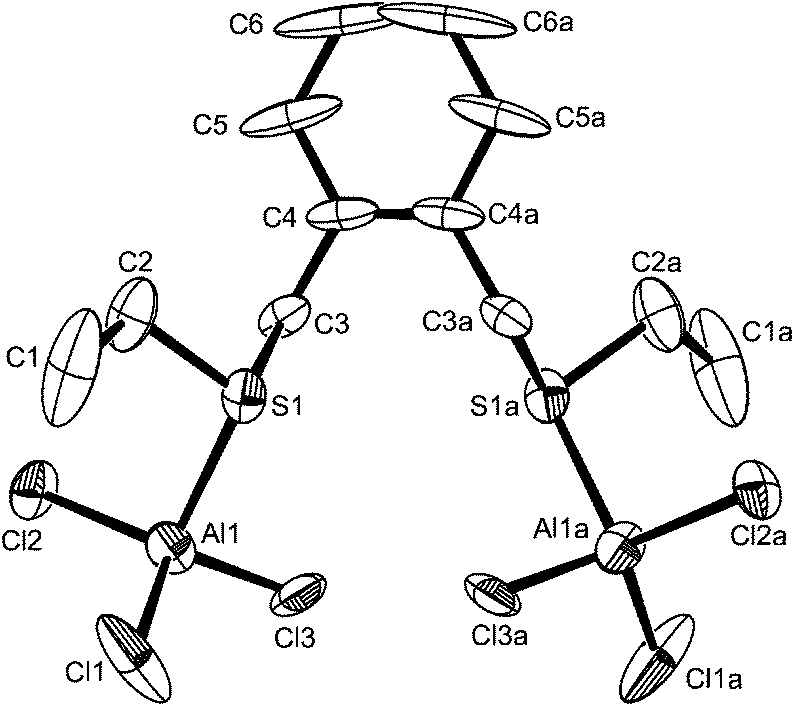 | ||
| Fig. 5 The structure of [(AlCl3)2{o-C6H4(CH2SEt)2}] showing the atom labelling scheme. The crystals had the properties of a modulated structure and the solution shown was derived from a smaller subcell. For this reason bond lengths and angles are less reliable than usual and not presented here. | ||
The spectroscopic data are consistent with four-coordinate aluminium in solution. The corresponding diselenoether o-C6H4(CH2SeMe)2 was completely converted to the selenonium cation [o-C6H4(CH2)2SeMe][AlCl4] upon reaction with AlCl3 in CH2Cl2 (see ESI†). The same selenonium cation is formed upon reaction of this ligand with GaCl3 or InCl3.12,15
Unexpectedly, reaction of AlX3 with MeE(CH2)2EMe (E = S or Se) failed to give [X3Al{μ-MeE(CH2)2EMe}AlX3], and the products had an AlX3:MeE(CH2)2EMe ratio of 1:1 irrespective of the ratio of reactants used. Crystals of three examples were grown and showed the presence of pseudo-octahedral cations and tetrahedral anions, [AlX2{MeE(CH2)2EMe}2][AlX4] (X = Cl, E = S or Se; X = I, E = S). The structures of [AlCl2{MeE(CH2)2EMe}2][AlCl4] (Fig. 6 and 7) reveal centrosymmetric cations (trans isomer) with identical d(Al–Cl), which are as expected longer than in the four-coordinate complexes. The d(Al–S) and d(Al–Se) differ by ∼0.14 Å, which approximates to the difference in covalent radii of the chalcogens.26 The bond angles around the aluminium show only small deviations from 90°.
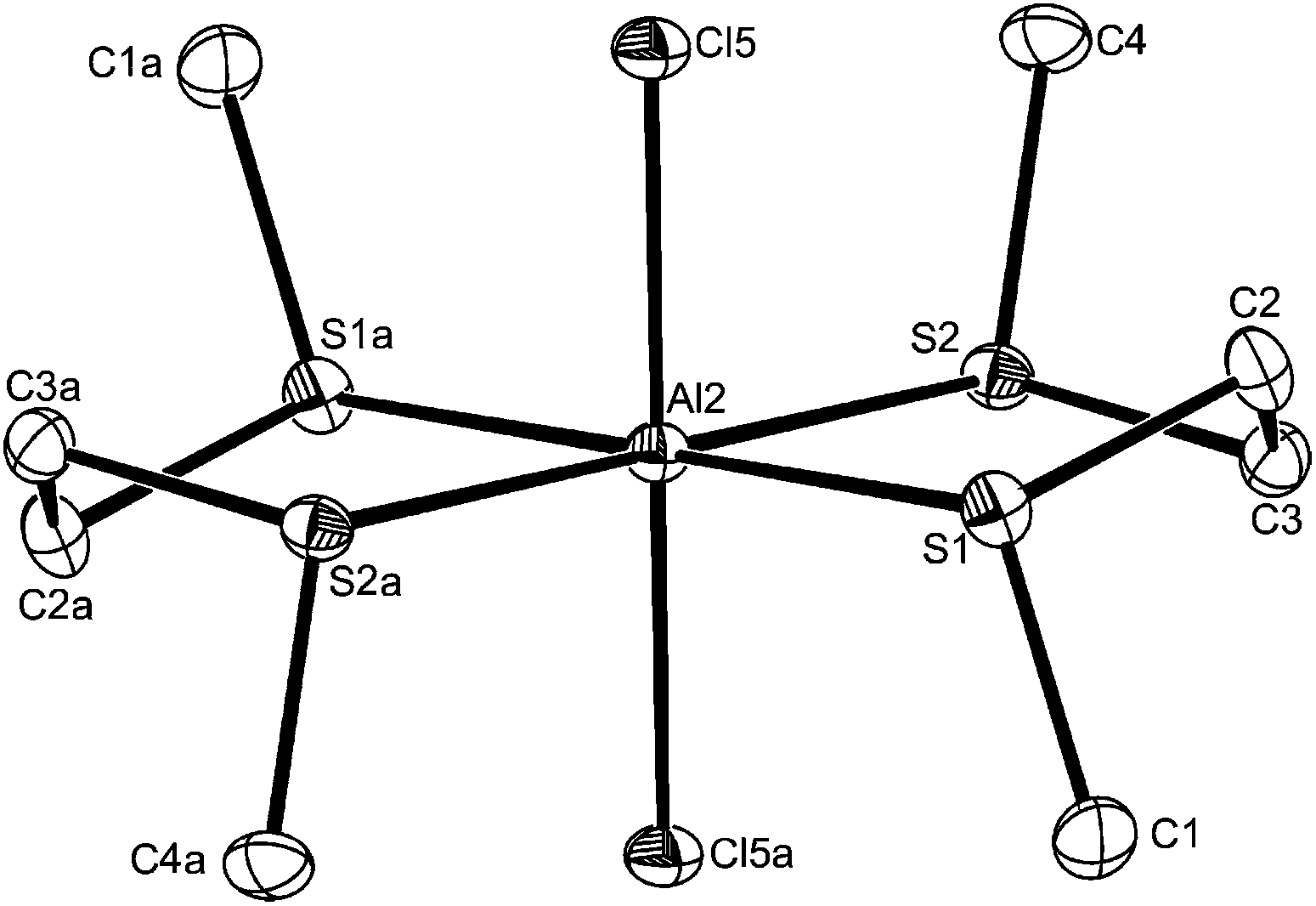 | ||
| Fig. 6 The structure of a cation in trans-[AlCl2{MeS(CH2)2SMe}2][AlCl4] showing the numbering scheme for the centrosymmetric Al2 centred cation. Ellipsoids are drawn at the 50% probability level and H atoms are not shown for clarity. The other cation is similar. Symmetry operation: a = 2 − x, −y, 1 − z. Selected bond lengths (Å) and angles (°): Al2–Cl5 = 2.2131(10), Al2–S1 = 2.4595(11), Al2–S2 = 2.4809(10), S1–Al2–S2 = 87.92(4), S1–Al2–S2a = 92.08(4), Cl5–Al–S1a = 94.49(2), Cl5–Al2–S1 = 85.51(2), Cl5–Al2–S2a = 86.50(3), Cl5–Al2–S2 = 93.50(3). | ||
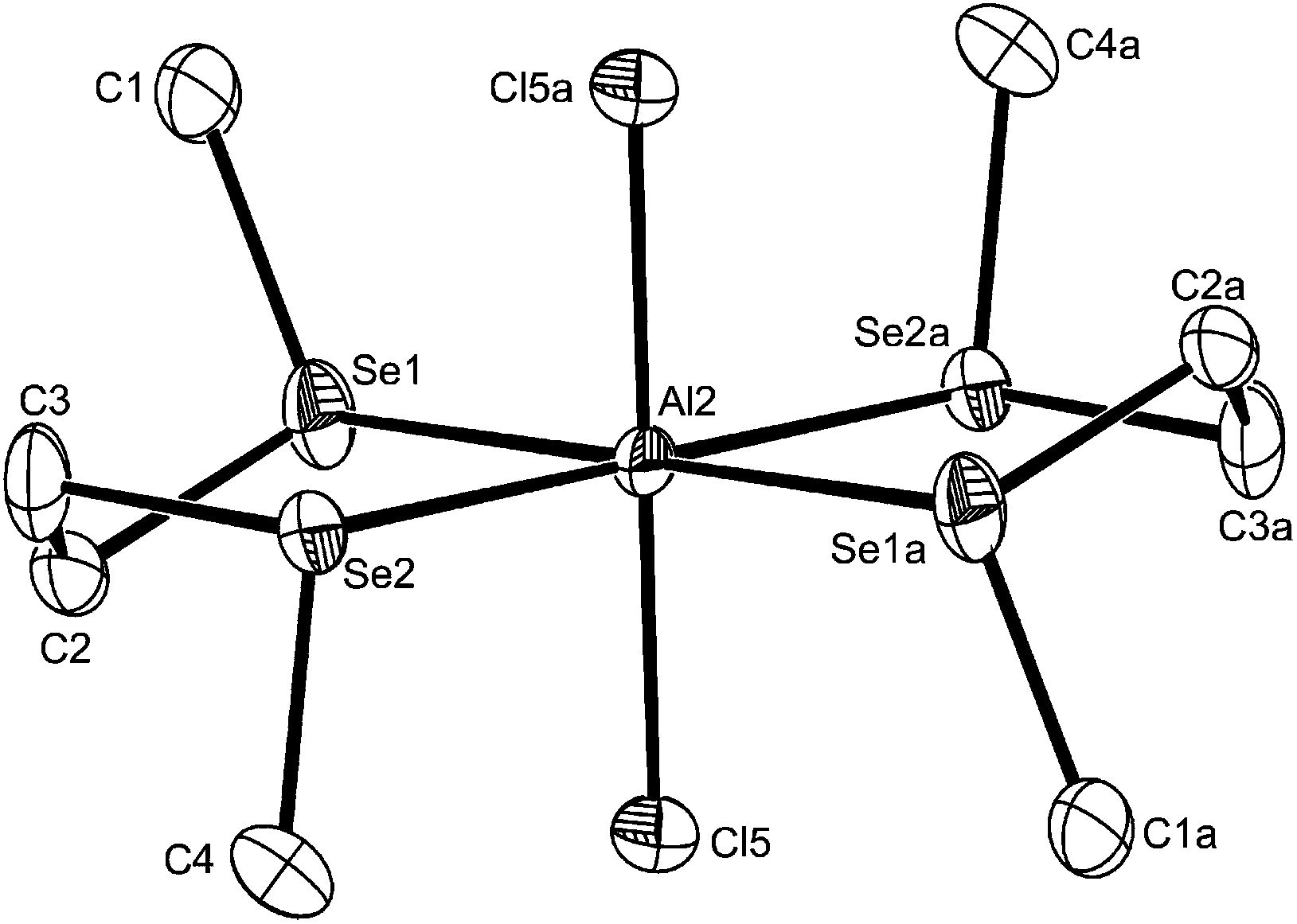 | ||
| Fig. 7 The structure of a cation in trans-[AlCl2{MeSe(CH2)2SeMe}2][AlCl4] showing the numbering scheme for the centrosymmetric Al2 centred cation. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. The other crystallographically independent cation is similar. Symmetry operation: a = 1 − x, 1 − y, 1 − z. Selected bond lengths (Å) and angles (°): Al2–Cl5 = 2.211(2), Al2–Se1 = 2.5950(13), Al2–Se2 = 2.6232(13), Se1–Al2–Se2 = 88.11(4), Se1–Al2–Se2a = 91.89(4), Cl5–Al2–Se1a = 94.50(6), Cl5–Al2–Se1 = 85.50(6), Cl5–Al2–Se2a = 86.51(6), Cl5–Al2–Se2 = 93.49(6). | ||
Although both of the structures contain the chalcogenoether in the DL conformation, the two crystals are not isomorphous and in the selenoether complex there are short contacts Se2⋯Se2′ (3.436 Å) and Se3⋯Se3′′ (3.542 Å). These link Al2-centred cations into chains through Se2 (along the a direction) and similarly, the Al3-centred cations are linked into chains through Se3 (again along the a direction) (Fig. 8).
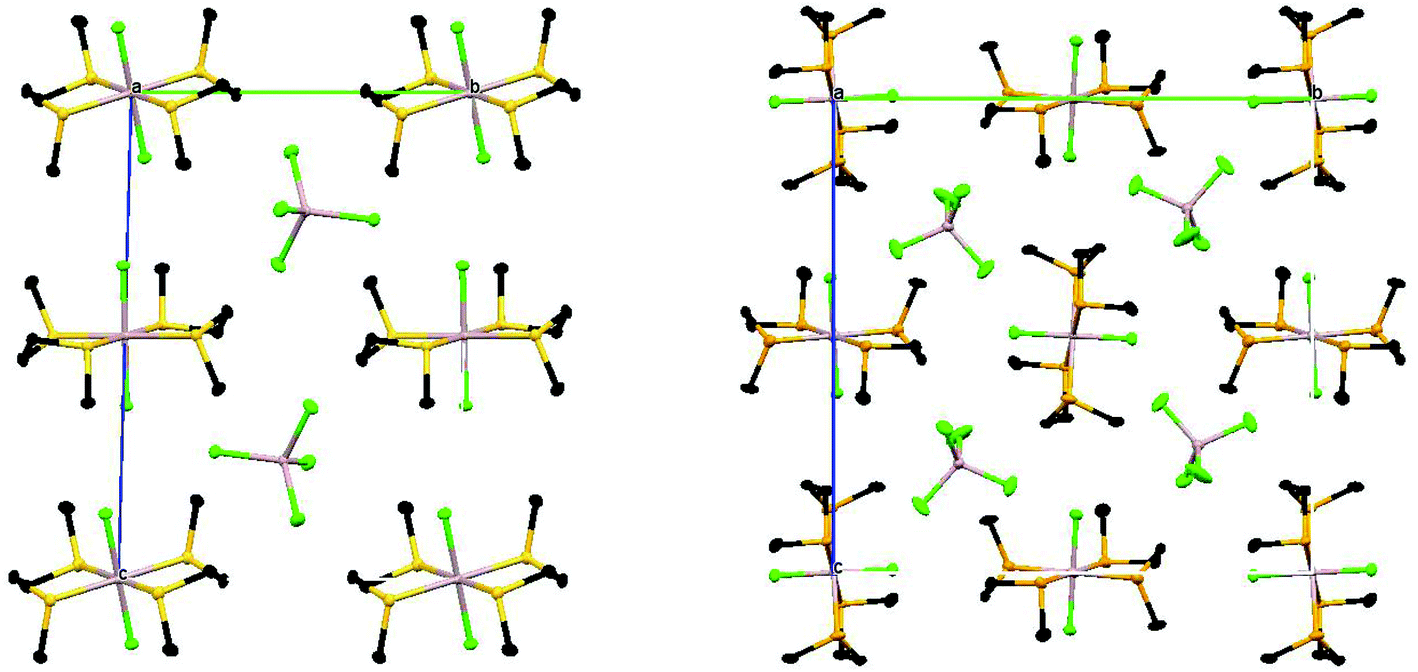 | ||
| Fig. 8 View along the a axis of the packing diagrams of [AlCl2{MeS(CH2)2SMe}2][AlCl4] (left) and [AlCl2{MeSe(CH2)2SeMe}2][AlCl4] (right) showing the different molecular packing. | ||
The structure of the [AlI2{MeS(CH2)2SMe}2][AlI4] (Fig. 9) also contains a pseudo-octahedral cation, but this has a cis-geometry and with the dithioethers in the meso form. In cis-[AlI2{MeS(CH2)2SMe}2]+ the Al–StransI are longer by ∼0.1 Å than Al–StransS, but the Al–StransS distances are not significantly different to those in trans-[AlCl2(MeSCH2CH2SMe)2][AlCl4].
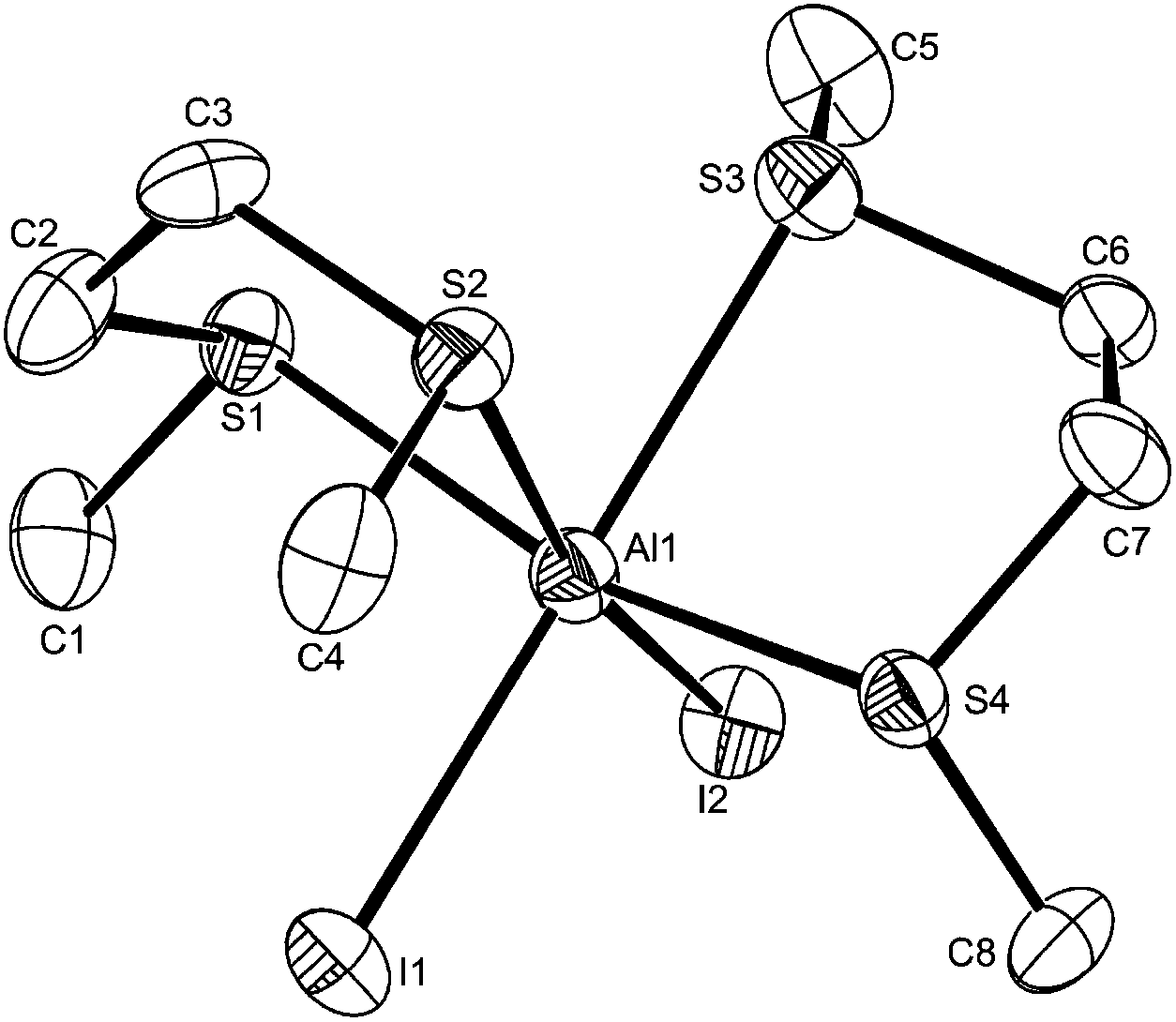 | ||
| Fig. 9 The structure of the cation in cis-[AlI2{MeS(CH2)2SMe}2][AlI4] showing the atom labelling scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–S1 = 2.431(4), Al1–S4 = 2.448(4), Al1–S3 = 2.533(5), Al1–S2 = 2.546(5), Al1–I1 = 2.612(4), Al1–I2 = 2.635(4), I1 Al1–I2 = 97.30(13), S1–Al1–S3 = 84.84(15), S4–Al1–S3 = 83.28(14), S1–Al1–S2 = 83.46(15), S4–Al1–S2 = 79.24(14), S3–Al1–S2 = 76.13(15), S1–Al1–I1 = 99.17(14), S4–Al1–I1 = 89.96(14), S2–Al1–I1 = 93.69(14), S1–Al1–I2 = 94.43(13), S4–Al1–I2 = 101.02(15), S3–Al1–I2 = 92.95(14). | ||
The Nujol mull IR spectra for these salts all confirm the presence of [AlX4]−, but the Al–X stretches of the cations could not be identified with certainty. The solution speciation is less clear, and all the complexes are extremely moisture sensitive in solution. All four complexes show 27Al resonances assignable to the [AlX4]− (Table 1), but only [AlCl2{MeS(CH2)2SMe}2][AlCl4] shows a second resonance (δ27Al = 37.4) which is assigned to the six-coordinate cation. For the other complexes it is likely that the increasing electric field gradients promote fast quadrupolar relaxation, resulting in the loss of the cation resonance.
At room temperature a CD2Cl2 solution of [AlCl2{MeS(CH2)2SMe}2][AlCl4] shows singlet CH3 (δ = 2.26) and CH2 (δ = 3.08) resonances. On cooling to 223 K the spectrum shows three CH3 resonances (δ = 2.26, 2.50, 2.66) and overlapping CH2 resonances (δ = 3.08–3.31), which we tentatively assign to a mixture of cis and trans isomers of [AlCl2{MeS(CH2)2SMe}2]+, the changes reversing on warming the solution. The bromo- and iodo-complexes behave similarly. Notably, none of the complexes show resonances due to free dithioether, which would seem to rule out significant amounts of [AlCl2{MeS(CH2)2SMe}]+, being present. At 185 K further splitting of the resonances is evident, which is probably due to slowing of the pyramidal inversion at S, leading to separate resonances for the individual invertomers. The solutions decompose slowly on standing.
The trans-[AlCl2{MeSe(CH2)2SeMe}2][AlCl4] exhibits a singlet 77Se NMR resonance at room temperature (δ = 95.5) which is a low frequency coordination shift (Δ = −25.5) and singlet CH3 (δ = 2.36) and CH2 (δ = 3.21) resonances in the 1H NMR spectrum. Cooling the solution to 190 K produces little change in the 1H NMR spectrum, although the 77Se resonance is lost below ∼240 K. In the selenoether complex only the trans isomer appears to be present in significant amounts. On standing, new resonances grow in due to decomposition. Attempts to record spectra in CD3CN solution resulted in displacement of the thio- or seleno-ethers by the nitrile.
Reaction of AlCl3 with the ditelluroether tBuTe(CH2)3TetBu produced a mixture of species resulting from ligand fragmentation. The 1H NMR spectrum of the product showed multiple resonances for the t-butyl groups and the CH2 units. Multiple signals were also observed in the 125Te NMR spectrum, whilst the 27Al NMR spectrum indicated the presence of [AlCl4]−. Very air sensitive, yellow crystals were isolated of one of the decomposition products, which proved to be [tBuTe(CH2)3Te(tBu)Te(CH2)3TetBu][AlCl4], derived from fragmentation of the ditelluroether (see ESI†).
Polydentates and macrocycles
The reaction of the tripodal trithioether MeC(CH2SMe)3 with AlCl3 in a 1:1 molar ratio in anhydrous CH2Cl2 gave colourless crystals whose structure (Fig. 10) showed a chain polymer with the ligand binding as a bridging bidentate with one uncoordinated –CH2SMe arm. The structure forms a chain in the a direction The geometry at aluminium is a distorted trigonal bipyramid with equatorial chlorines and there are two slightly different aluminium environments in the unit cell.
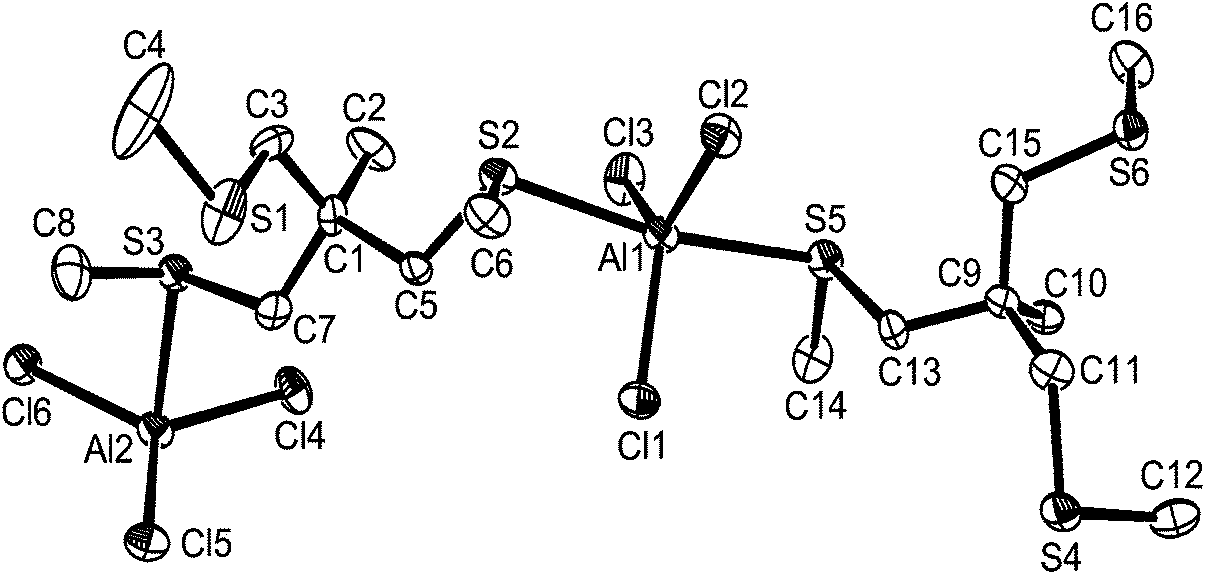 | ||
| Fig. 10 The structure of the asymmetric unit in [AlCl3{MeC(CH2SMe)3}] showing the atom labelling scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–Cl1 = 2.151(3), Al1–Cl2 = 2.152(3), Al1–Cl3 = 2.154(3), Al1–S5 = 2.473(3), Al1–S2 = 2.489(3), Al2–Cl5 = 2.158(3), Al2–Cl6 = 2.160(3), Al2–Cl4 = 2.169(3), Al2–S3 = 2.469(3), Al2–S6 = 2.488(3), Cl1–Al1–Cl2 = 117.05(12), Cl1–Al1–Cl3 = 118.80(13), Cl2–Al1–Cl3 = 124.15(13), Cl1–Al1–S5 = 94.98(11), Cl2–Al1–S5 = 88.33(11), Cl3–Al1–S5 = 86.31(10), Cl1–Al1–S2 = 95.32(11), Cl2–Al1–S2 = 89.27(11), Cl3–Al1–S2 = 86.49(10), S5–Al1–S2 = 169.38(11), Cl5–Al2–Cl6 = 117.44(13), Cl5–Al2–Cl4 = 119.86(12), Cl6–Al2–Cl4 = 122.70(12), Cl5–Al2–S3 = 94.48(10), Cl6–Al2–S3 = 89.38(11), Cl4–Al2–S3 = 86.84(11), Cl5–Al2–S6 = 90.27(10), Cl6–Al2–S6 = 92.63(10), Cl4–Al2–S6 = 86.71(11), S3–Al2–S6 = 173.30(12). | ||
In solution the 1H NMR spectrum shows single, sharp –CH2, –SMe and –CMe resonances consistent with fast exchange. Attempts to isolate complexes with higher AlCl3:tripod stoichiometries were unsuccessful. The [AlCl3{MeC(CH2SMe)3}] stoichiometry contrasts with the gallium complex which was [(GaCl3)3{MeC(CH2SMe)3}], assumed to contain tetrahedral gallium coordination.12
The reaction of the small ring thia-macrocycle [9]aneS3 with an equimolar amount of AlCl3 in toluene gave a white powder, [AlCl3([9]aneS3)]. The complex is insoluble in CH2Cl2 and the macrocycle is displaced by coordinating solvents like MeCN, precluding solution measurements. Both gallium and indium form similarly intractable 1:1 complexes with this ligand,11 and it seems likely that all three contain fac-octahedral structures. The larger macrocycles [14]aneS4 and [16]aneSe4 reacted with two molar equivalents of AlCl3 in anhydrous CH2Cl2 to form insoluble white complexes. The analytical data supported a formulation [AlCl2(macrocycle)][AlCl4] (macrocycle = [14]aneS4 and [16]aneSe4) and the far IR spectra confirmed the presence of [AlCl4]− anions. Poor quality crystals of [AlCl2([14]aneS4)][AlCl4] were obtained from the filtrate from the reaction solution and the structure shows the cation with endodentate macrocycle and a cis AlCl2 unit which serve to confirm the constitution (see ESI†). Although not isomorphous, the structure is similar to that of cis-[InCl2([14]aneS4}][InCl4].15 In contrast, gallium chloride forms an exodentate chain polymer [GaCl3([14]aneS4)] with trigonal bipyramidal coordination.13 The larger ring [16]aneSe4 forms trans-[MCl2([16]aneSe4)][MCl4] (M = Ga or In)13 and the aluminium complex probably has an analogous structure. The 16-membered ring macrocycle complexes were insoluble in CH2Cl2 and the macrocycle was displaced by stronger donor solvents.
LPCVD investigation
Following previous success in using neutral chalcogenoether adducts of GaCl3 to deposit thin films of crystalline Ga2Se3 and Ga2Te3,17 several coordination complexes of AlCl3 were synthesised as potential single source precursors to Al2E3 films. Ligands with nBu substituents were selected as these had previously been shown to be more effective than ligands with Me substituents, probably because they have the β-hydride elimination route available.17 [AlCl3(nBu2E)] (E = Se, Te) were synthesised as yellow oils as described above. LPCVD using both complexes was attempted at temperatures between 723 and 873 K, using the CVD equipment described previously.13 In all cases the precursor evaporated cleanly, having changed colour to dark brown during the evaporation. There was no deposition on the substrates and some elemental selenium or tellurium was deposited on the tube at the exit of the furnace. We conclude that these reagents are unsuitable for LPCVD under these conditions.Comparisons of Lewis acid behaviour of Al, Ga and In towards chalcogenoethers
There has been much recent interest in exploring Group 13 Lewis acid–Lewis base adducts both by modelling and experiment. The modelling work has mostly focussed on boron and aluminium complexes with O- or N-donor ligands, with trends sometimes extrapolated to gallium. The conclusions of these studies, although sometimes disputed in detail, were that Lewis acidity falls AlCl3 > AlBr3 > AlI3, and AlCl3 > AlBr3 > GaCl3 > GaBr3 (ref. 27–31 and references therein). One should note in passing that the order with halogen is reversed for boron. The modelling work is based upon gas phase molecules and does not take into account solid state effects (lattice energies, intermolecular interactions and packing effects), or the effects of lattice solvent, which may complicate the interpretation of experimental data, and in some cases lead to apparently anomalous results.30 The various contributions listed above, means that interpreting changes in metal–ligand bond lengths simply in terms of Lewis acidity must be done with care, and one might expect the occasional anomaly, but as a result of recent studies, there are sufficient data available to attempt some comparisons for Al/Ga/In–Group 16 donor complexes. Table 2 shows some illustrative data.| Compound | M–X/Å | M–E/Å | Coordination number of M | Reference |
|---|---|---|---|---|
| a Terminal M–X. | ||||
| [AlCl3(Me2Se)] | 2.121(2), 2.113(1) | 2.486(2) | 4 | This work |
| [AlCl3(Me2Te)] | 2.1207(6), 2.1295(10) | 2.6871(9) | 4 | This work |
| [AlBr3(Me2Te)] | 2.286(3), 2.287(2) | 2.692(4) | 4 | This work |
| trans-[AlCl2{MeS(CH2)2SMe}2]+ | 2.2131(10) | 2.4595(11), 2.4809(10) | 6 | This work |
| cis-[AlI2{MeS(CH2)2SMe}2]+ | 2.612(4), 2.635(4) | 2.431(4)–2.546(5) | 6 | This work |
| trans-[AlCl2{MeSe(CH2)2SeMe}2]+ | 2.211(2) | 2.5950(13), 2.6232(13) | 6 | This work |
| [(GaI3)2{MeS(CH2)2SMe}2] | 2.4997(7)–2.5316(7) | 2.4048(12) | 4 | 12 |
| [(GaCl3)2{o-C6H4(CH2SMe)2}] | 2.1478(14)–2.1650(15) | 2.3573(15) | 4 | 12 |
| [GaCl3(Me2Se)] | 2.1606(8), 2.1700(10) | 2.4637(7) | 4 | 12 |
| [GaI3(Me2Se)] | 2.5209(2)–2.5277(2) | 2.479(2) | 4 | 14 |
| [(GaCl3)2{nBuSe(CH2)2SenBu}] | 2.149(2)–2.166(2) | 2.468(1) | 4 | 12 |
| [(GaCl3)2{tBuTe(CH2)3TetBu}] | 2.158(3)–2.181(3) | 2.6378(14), 2.6356(13) | 4 | 17 |
| trans-[GaCl2([16]aneS4]+ | 2.276(3)–2.296(3) | 2.482(4)–2.518(4) | 6 | 13 |
| trans-[GaCl2([16]aneSe4]+ | 2.3058(11), 2.3220(12) | 2.5906(7)–2.6106(8) | 6 | 13 |
| [InBr3(Me2Se)] | 2.4901(12), 2.4981(7) | 2.6455(11) | 4 | 15 |
| [(InCl3)2(Me2Se)4] | 2.4243(1), 2.4457(1)a | 2.7722(6), 2.7802(6) | 6 | 14 |
| trans-[InCl2({iPrS(CH2)2SiPr}2]+ | 2.4237(18), 2.393(18) | 2.645(2)–2.696(2) | 6 | 15 |
| trans-[InBr2({iPrS(CH2)2SiPr}2]+ | 2.5775(5), 2.5918(5) | 2.6518(10)–2.6901(11) | 6 | 15 |
| trans-[InBr2{MeSe(CH2)2SeMe}2]+ | 2.6076(15), 2.6113(15) | 2.7370(16)–2.7762(15) | 6 | 15 |
| trans-[InCl2([16]aneSe4]+ | 2.475(2) | 2.7189(10), 2.7394(11) | 6 | 13 |
The data show firstly, that if one compares complexes of the same element with the same coordination number, the M–X distances seem unaffected by the specific chalcogen donor type present, which is consistent with the metal–halogen being the dominant interaction. A similar comparison of the M–E bond lengths shows that these increase (sometimes only marginally) with halide, Cl < Br < I, consistent with the trends deduced for lighter donor atoms. The M–X, and M–E bond lengths in comparable complexes of Al and Ga are also nearly identical, consistent with their almost identical covalent radii, resulting from the “3d block contraction”, i.e. the increased nuclear charge resulting from the 3d metals only partially screened by the d electron shell.26 As expected, In–X and In–E bonds are typically ∼0.2 Å longer. A very recent dft study31 suggested that whilst Ga and In halide complexes of Me2Se had a high degree of covalency in the M–Se bonds, those of aluminium had a markedly higher electrostatic component to the bonding. Our experimental data reported in the present paper, show no evidence for a significant change in the bonding type present along the series of group 16 donor complexes, the differences noted being due to the higher Lewis acidity of Al(III). We note that the dft calculations predict31 an Al–Se bond length (for the gas phase molecule) of 2.53 Å compared to the X-ray crystallographic result (for the solid) of 2.486(2) Å.
Our data also show the ready formation of six-coordinate cations with aluminium, [AlX2(L–L)2]+, contrasting with the reluctance of gallium to exceed four-coordination, except in macrocyclic compounds, cannot be due to steric effects, but must be a further consequence of the stronger Lewis acidity of aluminium. These differences must originate in the donor/acceptor orbital energy match (or mis-match) rather than in charge/radius effects. The larger indium centre has a less clear preference, easily accommodating four-, five- or six-coordination depending upon the ligand and reaction conditions.
Conclusions
Chalcogenoether complexes of aluminium(III) halides with four-, six- and (rarely) five-coordinate metal centres have been prepared, and their structures and properties compared with those of the heavier analogues GaX3 and InX3. The aluminium complexes are extremely moisture sensitive, and complexes with selenium or tellurium ligands are prone to slow E–C bond cleavage in solution. Nonetheless, the formation and structural characterisation of telluroether complexes of the hard AlX3 acceptors is notable. In contrast to the gallium complexes, it does not appear that the aluminium systems are suitable for LPCVD applications. The detailed study of Al(III) complexes with soft, modest donor chalcogenoethers has confirmed the trends in Lewis acidity observed with hard O or N donor ligands and are broadly in line with expectations based upon the dft calculations. The work further demonstrates that a significant range of chalcogenoether complexes with hard p-block Lewis acids are obtainable despite the hard/soft- acceptor/donor mismatch.Experimental
Infrared spectra were recorded as Nujol mulls between CsI plates using a Perkin-Elmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H NMR spectra were recorded using a Bruker AV300 spectrometer. 77Se{1H}, 125Te{1H} and 27Al NMR spectra were recorded using a Bruker DPX400 spectrometer and are referenced to external neat SeMe2, TeMe2 and aqueous [Al(H2O)6]3+ at pH = 1, respectively. Microanalyses were undertaken by Medac Ltd or London Metropolitan University. Solvents were dried prior to use: toluene by distillation from sodium benzophenone ketyl, CH2Cl2 from CaH2, and all preparations were undertaken using standard Schlenk techniques under a N2 atmosphere. Anhydrous aluminium(III) halides (Aldrich or Strem) were used as received. SMe2 and SeMe2 (Aldrich) were stored over molecular sieves. Other thio- seleno- and telluroethers were made by literature methods32–36 and stored under dinitrogen over molecular sieves.[AlCl3(Me2S)]: Me2S (0.09 g, 1.5 mmol) was added dropwise to a suspension of AlCl3 (0.20 g, 1.5 mmol) in anhydrous CH2Cl2 (15 mL) with stirring to give a colourless solution. After 30 minutes, all solvent was removed in vacuo to give a colourless oil. Yield 0.26 g, 89%. Anal. Calcd for C2H6AlCl3S: C, 12.3; H, 3.1. Found: C, 12.4; H, 3.1%. 1H NMR (CDCl3, 295 K): 2.53 (s). 27Al NMR (CD2Cl2, 295 K): 111.6. IR (cm−1): 541 (s), 410 (m). Raman (cm−1): 404 (m) Al–Cl.
[AlBr3(Me2S)]: AlBr3 (0.10 g, 0.37 mmol) was dissolved in toluene (10 mL) to give a pale yellow solution and Me2S (0.023 g, 0.37 mmol) was added dropwise when the solution became colourless. After stirring for 15 minutes, all solvent was removed in vacuo to give a white solid. Yield 0.10 g, 79%. Anal. Calcd for C2H6AlBr3S: C, 7.3; H, 1.8. Found: C, 6.4; H, 2.0%. 1H NMR (CD2Cl2, 295 K): 2.51 (s). 27Al NMR (CD2Cl2, 295 K): 81.2; (185 K): 81.6. IR (cm−1, Nujol): 392 (br) Al–Br.
[AlI3(Me2S)]: AlI3 (0.20 g, 0.5 mmol) was dissolved in toluene (10 mL) to give a pale yellow solution and Me2S (0.03 g, 0.5 mmol) was added dropwise. After stirring for 15 minutes, all solvent was removed in vacuo to give a pale yellow solid. Yield 0.17 g, 75%. Anal. Calcd for C2H6AlI3S: C, 5.1; H, 1.3. Found: C, 5.0; H, 1.3%. 1H NMR (CD2Cl2, 295 K): 2.44 (s). 27Al NMR (CD2Cl2, 295 K): 48.7. IR (cm−1, Nujol): 336 (br) Al–I.
[Me2SH][AlCl4]: Me2S (0.09 g, 1.5 mmol) was added dropwise to a suspension of AlCl3 (0.20 g, 1.5 mmol) in anhydrous CH2Cl2 (25 mL) with stirring to give a colourless solution. HCl was slowly bubbled through the solution for ∼30 seconds. After stirring for 30 minutes, some white precipitate formed. The white solid was collected by filtration and dried in vacuo. The volume of the pale yellow filtrate was reduced to ∼10 mL. Colourless crystals suitable for single crystal X-ray diffraction were obtained after storage of the filtrate at −18 °C for 24 hours. Yield 82%. Anal. Calcd for C2H7AlCl4S: C, 10.4; H, 3.0. Found: C, 10.5; H, 2.9%. IR (cm−1, Nujol): 2482 (br, w) SH, 488 (br), 467 (br), 395 (s) [AlCl4]−; the strong S–H⋯Cl–Al interactions causes extensive splitting of the t2 Al–Cl stretching mode in the anion.
[AlCl3(Me2Se)]: Me2Se (0.16 g, 1.5 mmol) was added dropwise to a suspension of AlCl3 (0.2 g, 1.5 mmol) in anhydrous CH2Cl2 (15 mL) with stirring to give a colourless solution. After 30 minutes, the volume of solvent was reduced in vacuo to ∼5 mL. Storage at −18 °C for 24 hours produced colourless crystals. Yield 0.30 g, 82%. Anal. Calcd for C2H6AlCl3Se: C, 9.9; H, 2.5. Found: C, 10.0; H, 2.6%. 1H NMR (CDCl3, 295 K): 2.92 (s); (283 K): 2.83 (s). 27Al NMR (CDCl3, 295 K): 110.5. 77Se NMR (CDCl3, 273 K): −11.3; (183 K): −14.0. IR (cm−1, Nujol): 531 (s), 399 (m). Raman (cm−1): 521 (w), 399 (s) Al–Cl.
[AlBr3(Me2Se)]: AlBr3 (0.10 g, 0.37 mmol) was dissolved in toluene (10 mL) to give a pale yellow solution and Me2Se (0.04 g, 0.37 mmol) was added dropwise when the solution became colourless. After stirring for 15 minutes, all solvent was removed in vacuo to give a white solid. Yield 0.11 g, 80%. Anal. Calcd for C2H6AlBr3Se: C, 6.4; H, 1.6. Found: C, 6.1; H, 1.9%. 1H NMR (CD2Cl2, 295 K): 2.47 (s). 27Al NMR (CD2Cl2, 295 K): 99.7. IR (cm−1, Nujol): 446 (s), 397 (s) Al–Br.
[AlCl3(nBu2Se)]: A solution of nBu2Se (0.29 g, 1.5 mmol) in anhydrous CH2Cl2 (7 mL) was added dropwise to a suspension of AlCl3 (0.20 g, 1.5 mmol) in anhydrous CH2Cl2 (8 mL) with stirring to give a colourless solution. After 30 minutes, all solvent was removed in vacuo to yield a pale yellow oil. Yield 0.36 g, 73%. Anal. Calcd for C8H18AlCl3Se: C, 29.4; H, 5.6. Found: C, 29.7; H, 5.8%. 1H NMR (CD2Cl2, 295 K): 3.03 (t, [4H]), 1.82 (m, [4H]), 1.46 (m, [4H]), 0.97 (t, [6H]). 27Al NMR (CD2Cl2, 295 K): 109.9. 77Se NMR (CDCl3, 295 K): 102.9. IR (cm−1): 539 (s), 399 (m) Al–Cl.
[AlCl3(Me2Te)]: Me2Te (0.12 g, 0.75 mmol) was added dropwise to a suspension of AlCl3 (0.10 g, 0.75 mmol) in anhydrous CH2Cl2 (10 mL) with stirring to give a colourless solution. After 30 minutes, the volume of solvent was reduced in vacuo to ∼5 mL. Storage at −18 °C for 24 hours produced yellow crystals. Yield: 0.135 g, 62%. Anal. Calcd for C2H6AlCl3Te: C, 8.3; H, 2.1. Found: C, 8.6; H, 2.1%. 1H NMR (CD2Cl2, 295 K): 2.19 (s). 27Al NMR (CD2Cl2, 295 K): 105.9. IR (cm−1, Nujol): 491 (s), 395 (m) Al–Cl.
[AlBr3(Me2Te)]: AlBr3 (0.10 g, 0.37 mmol) was dissolved in toluene (10 mL) to give a pale yellow solution. Me2Te (0.06 g, 0.37 mmol) was added dropwise to give a paler yellow solution. After stirring for 15 minutes, all solvent was removed in vacuo to give a yellow solid. Yield: 0.11 g, 68%. Anal. Calcd for C2H6AlBr3Te: C, 5.7; H, 1.4. Found: C, 5.7; H, 1.8%. 1H NMR (CD2Cl2, 295 K): 2.66 (s). 27Al NMR (CD2Cl2, 295 K): 92.6. IR (cm−1, Nujol): 404 (m), 391 (m) Al–Br.
[AlCl3(nBu2Te)]: A solution of nBu2Te (0.18 g, 0.75 mmol) in anhydrous CH2Cl2 (7 mL) was added dropwise to a suspension of AlCl3 (0.10 g, 0.75 mmol) in anhydrous CH2Cl2 (8 mL) with stirring to give a yellow solution. After 15 minutes, all solvent was removed in vacuo to yield a yellow oil. Yield 0.22 g, 78%. Anal. Calcd for C8H18AlCl3Te: C, 25.6; H, 4.8. Found: C, 26.1; H, 5.1%. 1H NMR (CD2Cl2, 295 K): 2.95 (t, [4H]), 1.83 (m, [4H]), 1.43 (m, [4H]), 0.96 (t, [6H]). 27Al NMR (CD2Cl2, 295 K): 106.8. IR (cm−1): 495 (s), 395 (m) Al–Cl.
[AlCl2{MeS(CH2)2SMe}2][AlCl4]: MeS(CH2)2SMe (0.18 g, 1.5 mmol) was added dropwise to a suspension of AlCl3 (0.20 g, 1.5 mmol) in anhydrous CH2Cl2 (15 mL) with stirring to give a colourless solution. After 30 minutes the volume of solvent was reduced in vacuo to ∼5 mL. A large quantity of white solid precipitated after storage at −18 °C for 24 hours. The solid was collected by filtration and dried in vacuo. Yield 0.26 g, 68%. Anal. Calcd for C8H20Al2Cl6S4: C, 18.8; H, 3.9. Found: C, 18.3; H, 3.8%. 1H NMR (CD2Cl2, 295 K): 3.07 (s, [4H]), 2.25 (s, [6H]). 27Al NMR (CDCl3, 295 K): 105.5, 37.4. IR (cm−1, Nujol): 489 (s), 463 (s), 346 (w). Raman (cm−1): 490 (w), 350 (s), 295 (s).
[AlBr2{MeS(CH2)2SMe}2][AlBr4]: MeS(CH2)2SMe (0.09 g, 0.75 mmol) was added dropwise to a pale yellow solution of AlBr3 (0.20 g, 0.75 mmol) in toluene (15 mL) with stirring to give a colourless solution and a white precipitate. After 30 minutes the reaction mixture was filtered and the white solid dried in vacuo. Yield 0.21 g, 72%. Anal. Calcd for C8H20Al2Br6S4: C, 12.3; H, 2.6. Found: C, 11.7; H, 2.9%. 1H NMR (CD2Cl2, 295 K): 3.10 (s, [4H]), 2.26 (s, [6H]). 27Al NMR (CD2Cl2, 295 K): 80.6. IR (cm−1, Nujol): 405 (s, br), 355 (s, br).
[AlI2{MeS(CH2)2SMe}2][AlI4]: MeS(CH2)2SMe (0.06 g, 0.5 mmol) was added dropwise to a pale yellow solution of AlI3 (0.20 g, 0.5 mmol) in toluene (15 mL) with stirring to give an orange solution and some pale yellow precipitate. After 30 minutes the reaction mixture was filtered and the pale yellow solid dried in vacuo. Crystals suitable for single crystal X-ray diffraction were obtained after storage of the filtrate at −18 °C for 48 hours. Yield 0.19 g, 74%. Anal. Calcd for C8H20Al2I6S4: C, 9.1; H, 1.9. Found: C, 8.9; H, 3.3%. 1H NMR (CD2Cl2, 295 K): 2.77 (s, [4H]), 2.17 (s, [6H]). 27Al NMR (CD2Cl2, 295 K): −23.0. IR (cm−1, Nujol): 338 (s), 304 (m).
[AlCl2{MeSe(CH2)2SeMe}2][AlCl4]: MeSe(CH2)2SeMe (0.16 g, 0.75 mmol) was added dropwise to a suspension of AlCl3 (0.10 g, 0.75 mmol) in anhydrous CH2Cl2 (10 mL) with stirring to give a yellow solution. After 30 minutes the volume of solvent was reduced in vacuo to ∼5 mL. Storage of the solution at −18 °C for 24 hours produced colourless crystals. Yield 0.18 g, 69%. Anal. Calcd for C8H20Al2Cl6Se4: C, 13.8; H, 2.9. Found: C, 12.8; H, 2.7%. 1H NMR (CDCl3, 295 K): 3.21 (s, [4H]), 2.36 (s, [6H]). 27Al NMR (CDCl3, 295 K): 105.4. 77Se NMR (CDCl3, 295 K): 95.4. IR (cm−1, Nujol): 488 (s), 442 (m).
[AlCl3{MeC(CH2SMe)3}]: MeC(CH2SMe)3 (0.16 g, 0.75 mmol) was added dropwise to a suspension of AlCl3 (0.10 g, 0.75 mmol) in anhydrous CH2Cl2 (10 mL) with stirring to give a colourless solution. After 30 minutes the volume of solvent was reduced in vacuo to ∼5 mL. Colourless crystals suitable for single crystal X-ray diffraction were obtained after storage at −18 °C for 48 hours. Yield 0.18 g, 70%. Anal. Calcd for C8H18AlCl3S3·1/2CH2Cl2: C, 26.4; H, 5.0. Found: C, 26.9; H, 4.9%. 1H NMR (CDCl3, 295 K): 2.76 (s, [6H]), 2.25 (s, [9H]), 1.16 (s, [3H]). IR (cm−1, Nujol): 534 (s), 491 (s).
[(AlCl3)2{C6H4(CH2SEt)2}]: o-C6H4(CH2SEt)2 (0.17 g, 0.75 mmol) was added dropwise to a suspension of AlCl3 (0.20 g, 1.5 mmol) in anhydrous CH2Cl2 (15 mL) with stirring to give a yellow solution. After 30 minutes the volume of solvent was reduced in vacuo to ∼5 mL. Storage of the solution at −18 °C for 24 hours produced yellow crystals. Yield 0.22 g, 60%. Anal. Calcd for C12H18Al2Cl6S2: C, 29.2; H, 3.7. Found: C, 29.3; H, 3.7%. 1H NMR (CDCl3, 295 K): 7.47 (m, [4H]), 4.37 (s, [4H]), 3.08 (q, [4H]), 1.49 (t, [6H]). 27Al NMR (CDCl3, 295 K): 111.3. IR (cm−1, Nujol): 565–500 (vbr, s), 394 (s). Raman (cm−1): 518 (m), 402 (m).
[AlCl3([9]aneS3)]: A solution of [9]aneS3 (0.14 g, 0.75 mmol) in anhydrous toluene (10 mL) was added to a solution of AlCl3 (0.10 g, 0.75 mmol) in anhydrous toluene (10 mL) with stirring to give a colourless solution and a large quantity of white precipitate. After 60 minutes, the precipitate was collected by filtration and dried in vacuo. Yield 0.16 g, 68%. Anal. Calcd for C6H12AlCl3S3: C, 23.0; H, 3.9. Found: C, 23.1; H, 3.9%. IR (cm−1, Nujol): 408 (s), 375 (m) Al–Cl.
[AlCl2([14]aneS4)][AlCl4]: A solution of [14]aneS4 (0.10 g, 0.37 mmol) in anhydrous CH2Cl2 (10 mL) was added to a suspension of AlCl3 (0.10 g, 0.75 mmol) in anhydrous CH2Cl2 (10 mL) with stirring to give a colourless solution. After approximately 5 minutes a large quantity of white precipitate formed. After 60 minutes, the precipitate was collected by filtration and dried in vacuo. Yield 0.14 g, 70%. Anal. Calcd for C10H20Al2Cl6S4: C, 22.4; H, 3.8. Found: C, 22.6; H, 3.7%. IR (cm−1, Nujol): 481 (br, s), 419 (m).
[AlCl2([16]aneSe4)][AlCl4]: was made similarly from a solution of [16]aneSe4 (0.18 g, 0.37 mmol) in anhydrous CH2Cl2 (10 mL) and AlCl3 (0.1 g, 0.75 mmol) in anhydrous CH2Cl2 (10 mL). The precipitate was collected by filtration and dried in vacuo. Yield 0.20 g, 70%. Anal. Calcd for C12H24Al2Cl6Se4: C, 19.2; H, 3.2. Found: C, 19.0; H, 3.1%. IR (cm−1): 483 (s), 434 (br).
X-Ray experimental
Details of the crystallographic data collection and refinement parameters are given in Table 3. Crystals suitable for single crystal X-ray analysis were obtained as described above. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (100 μm focus) with the crystal held at 100 K (N2 cryostream). Structure solution and refinement were straightforward,37,38 except as detailed below, with H atoms bonded to C being placed in calculated positions using the default C–H distance. For [AlCl3{MeC(CH2SMe)3}] the data were collected using the Rigaku automated routines which normally gives close to 100% of the data out to 2θ of 55°. For reasons that are not clear this did not happen in this case and it proved difficult to obtain more suitable crystals for a re-collection. Judged by the high Rint value, the data are of modest quality although the intensities seems satisfactory (80% exceed the Shelxl test, I > 2σ(I)). The structure that emerges from the analysis appears sound, with no unusual adp values or other causes for concern. For [(AlCl3)2{C6H4(CH2SEt)2}] the diffraction pattern exhibits many additional reflections in the 100 projection. These are likely due to a modulation. In fact, the Ga analogue13 exhibits the same behaviour and in that case it was possible to index the modulated cell. As with the Ga analogue, the data were indexed on a strong sub-cell making refinement of the basic structure possible; however, the ignored reflections result in unrealistic thermal parameters for many of the atoms. For both of these structures therefore, detailed comparisons of the geometric parameters require caution.| Compound | [AlCl3(Me2Se)] | [AlCl3(Me2Te)] | [AlBr3(Me2Te)] | [AlCl2{MeS(CH2)2SMe}2] [AlCl4] | [AlCl2{MeSe(CH2)2SeMe}2] [AlCl4] |
|---|---|---|---|---|---|
| Formula | C2H6AlCl3Se | C2H6AlCl3Te | C2H6AlBr3Te | C8H20Al2Cl6S4 | C8H20Al2Cl6Se4 |
| M | 242.36 | 291.00 | 424.38 | 511.14 | 698.74 |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic | Triclinic | Monoclinic |
| Space group | Pbcm (no. 57) | Pbcm (no. 57) | Pbcm (no. 57) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (no. 2) |
P21/c (no. 14) |
| a [Å] | 6.205(4) | 6.248(1) | 6.562(3) | 6.708(5) | 7.122(4) |
| b [Å] | 12.603(8) | 13.114(3) | 13.400(5) | 10.583(5) | 17.744(8) |
| c [Å] | 10.479(6) | 10.514(2) | 10.724(4) | 15.383(5) | 17.582(8) |
| α [°] | 90 | 90 | 90 | 90.070(5) | 90 |
| β [°] | 90 | 90 | 90 | 102.422(5) | 93.169(8) |
| γ [°] | 90 | 90 | 90 | 96.512(5) | 90 |
| U [Å3] | 819.6(8) | 861.4(3) | 943.0(6) | 1059.2(10) | 2218.5(18) |
| Z | 4 | 4 | 4 | 2 | 4 |
| μ(Mo Kα) [mm−1] | 5.564 | 4.388 | 15.870 | 1.276 | 7.405 |
| Total no. reflns | 4909 | 3839 | 8126 | 11612 |
10277 |
| Unique reflns | 1315 | 1312 | 1140 | 6072 | 5039 |
| R int | 0.103 | 0.018 | 0.297 | 0.026 | 0.114 |
| No. of params, restraints | 38, 0 | 38, 0 | 38, 0 | 188, 0 | 188, 0 |
| R 1 [Io > 2σ(Io)] | 0.055 | 0.018 | 0.065 | 0.046 | 0.077 |
| R 1 [all data] | 0.065 | 0.022 | 0.068 | 0.059 | 0.091 |
| wR2b [Io > 2σ(Io)] | 0.137 | 0.034 | 0.163 | 0.072 | 0.196 |
| wR2 [all data] | 0.148 | 0.035 | 0.168 | 0.077 | 0.207 |
| Compound | [AlI2{MeS(CH2)2SMe}2] [AlI4] | [{o-C6H4(CH2SEt)2}(AlCl3)2] | [AlCl3(MeC(CH2SMe)3)] | [Me2SH][AlCl4] |
|---|---|---|---|---|
| a Common items: temperature = 100 K; wavelength (Mo-Kα) = 0.71073 Å; θ(max) = 27.5°. b R 1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2/∑wFo4]1/2. | ||||
| Formula | C8H20Al2I6S4 | C12H18Al2Cl6S2 | C16H36Al2Cl6S6 | C2H7AlCl4S |
| M | 1059.84 | 493.04 | 687.47 | 231.92 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c (no. 14) | C2/c (no. 15) | P21/c (no. 14) | P21/c (no. 14) |
| a [Å] | 14.825(3) | 18.745(2) | 16.131(7) | 14.3783(7) |
| b [Å] | 12.181(2) | 10.810(10) | 8.310(3) | 10.9075(7) |
| c [Å] | 15.150(4) | 13.608(12) | 24.976(11) | 12.3742(5) |
| α [°] | 90 | 90 | 90 | 90 |
| β [°] | 108.449(8) | 125.779(1) | 107.156(8) | 90.260(3) |
| γ [°] | 90 | 90 | 90 | 90 |
| U [Å3] | 2595.3(10) | 2237(3) | 3199(2) | 1940.64(17) |
| Z | 4 | 4 | 4 | 8 |
| μ(Mo Kα) [mm−1] | 7.563 | 1.026 | 0.991 | 1.443 |
| Total no. reflns | 24085 |
8120 | 10176 |
11008 |
| Unique reflns | 5928 | 2199 | 4613 | 4432 |
| R int | 0.152 | 0.041 | 0.133 | 0.035 |
| No. of params, restraints | 185, 0 | 100, 3 | 279, 0 | 153, 0 |
| R 1 [Io > 2σ(Io)] | 0.057 | 0.108 | 0.085 | 0.045 |
| R 1 [all data] | 0.135 | 0.121 | 0.103 | 0.055 |
| wR2b [Io > 2σ(Io)] | 0.106 | 0.258 | 0.193 | 0.156 |
| wR2 [all data] | 0.115 | 0.277 | 0.207 | 0.189 |
Acknowledgements
We thank the CMSD at STFC and the EPSRC for support, and Dr M. Webster for assistance with the X-ray data analyses. We also thank Professor J. M. Dyke for helpful discussions.References
- Comprehensive organic synthesis, ed. B. M. Tost, Pergamon, Oxford, 1991, e.g. H. Heaney, vol. 2, ch. 3 and G. O. Olah, R. Krishnamurti and G. K. Surya, vol. 4, ch. 1.8 Search PubMed.
- (a) The chemistry of aluminium, gallium, indium and thallium, ed. A. J. Downs, Blackie, London, 1993 Search PubMed; (b) The group 13 metals aluminium, gallium, indium and thallium, ed. S. Aldridge and A. J. Downs, Wiley, NY, 2011 Search PubMed; (c) G. H. Robinson, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 3, p. 347 Search PubMed.
- S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460 RSC.
- R. L. Richards and A. Thompson, J. Chem. Soc. A, 1967, 1248 RSC.
- J. Lewis, J. R. Miller, R. L. Richards and A. Thompson, J. Chem. Soc., 1965, 5850 RSC.
- P. J. Ogren, L. Steenhoek, K. Greve and W. C. Hutton, J. Inorg. Nucl. Chem., 1975, 37, 293 CrossRef CAS.
- (a) L. A. Ganyushin, E. N. Guryanova, I. P. Romm and L. A. Lobanova, Zh. Obshch. Khim., 1979, 49, 2090 CAS; (b) L. A. Ganyushin, E. N. Guryanova and I. P. Romm, Zh. Obshch. Khim., 1978, 48, 2478 CAS; (c) I. P. Rom and E. N. Guryanova, Zh. Obshch. Khim., 1968, 38, 1927 Search PubMed.
- R. T. Tjahjanto, M. F. Peintinger, T. Bredow and J. Beck, Eur. J. Inorg. Chem., 2012, 3625 CrossRef CAS.
- (a) G. H. Robinson and S. A. Sangokara, J. Am. Chem. Soc., 1988, 110, 1494 CrossRef CAS; (b) G. H. Robinson and J. L. Atwood, Organometallics, 1987, 6, 887 CrossRef CAS.
- K. L. Baker and G. W. A. Fowles, J. Chem. Soc. A, 1968, 801 RSC.
- W. Levason, G. Reid and W. Zhang, Dalton Trans., 2011, 40, 8491 RSC.
- C. Gurnani, M. Jura, W. Levason, R. Ratnani, G. Reid and M. Webster, Dalton Trans., 2008, 6274 RSC.
- K. George, M. Jura, W. Levason, M. E. Light, L. P. Ollivere and G. Reid, Inorg. Chem., 2012, 51, 2231 CrossRef CAS PubMed.
- S. Mishra, E. Jeanneau and S. Daniele, Polyhedron, 2010, 29, 500 CrossRef CAS PubMed.
- C. Gurnani, M. Jura, W. Levason, R. Ratnani, G. Reid and M. Webster, Dalton Trans., 2009, 1611 RSC.
- (a) F. Fairbrother and N. Flitcroft, J. Less-Common Met., 1962, 4, 504 CrossRef CAS; (b) C. A. Evans and M. J. Taylor, J. Chem. Soc. A, 1969, 1343 RSC.
- K. George, C. H. de Groot, C. Gurnani, A. L. Hector, R. Huang, M. Jura, W. Levason and G. Reid, Chem. Mater., 2013, 25, 1829 CrossRef CAS.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, 4th edn, Wiley, NY, 1986 Search PubMed.
- N. Burford, B. W. Royan, R. E. v. H. Spence and T. S. Cameron, J. Chem. Soc., Dalton Trans., 1990, 1521 RSC.
- H. Bock and U. Lechner-Knoblauch, J. Organomet. Chem., 1985, 294, 295 CrossRef CAS.
- J. Mason, Multinuclear NMR, Plenum, NY, 1987 Search PubMed.
- A. Ecker and H. Schnoeckel, Z. Anorg. Allg. Chem., 1998, 624, 813 CrossRef CAS.
- W. Levason, S. D. Orchard and G. Reid, Coord. Chem. Rev., 2002, 225, 159 CrossRef CAS.
- N. P. Luthra and J. D. Odon, in The chemistry of organic selenium and tellurium compounds, ed. S. Patai and Z. Rappoport, Wiley, NY, 1986, ch. 6, vol. 1 Search PubMed.
- M. Jura, W. Levason, G. Reid and M. Webster, Dalton Trans., 2009, 7610 RSC.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reeves, J. Eschevaria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- A. Y. Timoshkin, A. V. Suvorov, H. F. Bettinger and H. F. Schaefer III, J. Am. Chem. Soc., 1999, 121, 5687 CrossRef CAS.
- A. Ogawa and H. Fujimoto, Inorg. Chem., 2002, 41, 4888 CrossRef CAS PubMed.
- F. Bessac and G. Frenking, Inorg. Chem., 2006, 45, 6956 CrossRef CAS PubMed.
- A. Y. Timoshkin, M. Bodensteiner, T. N. Sevastianova, A. S. Lisovenko, E. I. Davydova, M. Scheer, C. Grassl and A. V. Butlak, Inorg. Chem., 2012, 51, 11602 CrossRef CAS PubMed.
- T. I. Madzhikov and G. A. Chmutova, J. Phys. Chem. A, 2013, 117, 4011 CrossRef PubMed.
- R. J. Batchelor, F. W. B. Einstein, I. D. Gay, J.-H. Gu, B. D. Johnston and B. M. Pinto, J. Am. Chem. Soc., 1989, 111, 6582 CrossRef CAS.
- T. Kemmitt and W. Levason, Organometallics, 1989, 8, 1303 CrossRef CAS.
- W. Levason, M. Nirwan, R. Ratnani, G. Reid, N. Tsoureas and M. Webster, Dalton Trans., 2007, 439 RSC.
- D. J. Gulliver, E. G. Hope, W. Levason, S. G. Murray, D. M. Potter and G. L. Marshall, J. Chem. Soc., Perkin Trans. 2, 1984, 429 RSC.
- N. Kuhn, P. Faupel and E. Zauder, J. Organomet. Chem., 1986, 302, C4 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Structural data on [o-C6H4CH2Se(Me)CH2][AlCl4], [AlCl2([14]aneS4)][AlCl4] and [tBuTe(CH2)3Te(tBu)Te(CH2)3TetBu][AlCl4]. CCDC 966342–966353. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52991f |
| ‡ [14]aneS4 = 1,4,8,11-tetrathiacyclotetradecane, [16]aneSe4 = 1,5,9,13-tetraselanacyclohexadecane, [16]aneS4 = 1,5,9,13-tetrathiacyclohexadecane, [9]aneS3 = 1,4,7-trithiacyclononane. |
| This journal is © The Royal Society of Chemistry 2014 |