 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of mononuclear early transition-metal complexes of κ3cyclo-tetrametaphosphate ([P4O12]4−) and cyclo-trimetaphosphate ([P3O9]3−)†
Cesar M.
Manna
a,
Mostafa Y.
Nassar
ab,
Daniel
Tofan
a,
Khetpakorn
Chakarawet
a and
Christopher C.
Cummins
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: ccummins@mit.edu
bDepartment of Chemistry, Faculty of Science, Benha University, Benha 13518, Egypt
First published on 7th November 2013
Abstract
We herein report the preparation of several mononuclear-metaphosphate complexes using simple techniques and mild conditions with yields ranging from 56% to 78%. Treatment of cyclo-tetrametaphosphate ([TBA]4[P4O12]·5H2O, TBA = tetra-n-butylammonium) with various metal sources including (CH3CN)3Mo(CO)3, (CH3CN)2Mo(CO)2(η3-C3H5)Cl, MoO2Cl2(OSMe2)2, and VOF3, leads to the clean and rapid formation of [TBA]4[(P4O12)Mo(CO)3]·2H2O, [TBA]3[(P4O12)Mo(CO)2(η3-C3H5)], [TBA]3[(P4O12)MoO2Cl] and [TBA]3[(P4O12)VOF2]·Et2O salts in isolated yields of 69, 56, 68, and 56% respectively. NMR spectroscopy, NMR simulations and single crystal X-ray studies reveal that the [P4O12]4− anion behaves as a tridentate ligand wherein one of the metaphosphate groups is not directly bound to the metal. cyclo-Trimetaphosphate-metal complexes were prepared using a similar procedure i.e., treatment of [PPN]3[P3O9]·H2O (PPN = bis(triphenylphosphine)iminium) with the metal sources (CH3CN)2Mo(CO)2(η3-C3H5)Cl, MoO2Cl2(OSMe2)2, MoOCl3, VOF3, WOCl4, and WO2Cl2(CH3CN)2 to produce the corresponding salts, [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)], [PPN]2[(P3O9)MoO2Cl], [PPN]2[(P3O9)MoOCl2], [PPN]2[(P3O9)VOF2]·2CH2Cl2, and [PPN]2[(P3O9)WO2Cl] in isolated yields of 78, 56, 75, 59, and 77% respectively. NMR spectroscopy, NMR simulations and single-crystal X-ray studies indicate that the trianionic ligand [P3O9]3− in these complexes also has κ3 connectivity.
1. Introduction
Bimetallated cyclo-tetrametaphosphate ([P4O12]4−) and monometallated cyclo-trimetaphosphate ([P3O9]3−) moieties have a wide range of applications including use as pigments, catalysts, food additives, and fluorescent materials.1–6 The cyclo-tetraphosphate anion is of special interest due to its flexible eight-membered ring structure that can produce complexes with different conformations.1,7–9 Currently, two different strategies are employed for the preparation of bimetallic complexes of cyclo-tetrametaphosphate. One strategy is based on acidification of MCl2 (M = Co, Fe, Mn, Ni, Cu) by [H2PO4]−, followed by thermal treatment at high temperatures (≥600 °C), leading to binary oligomeric systems of the general formula M2P4O12.2–4,10–19A second strategy developed by Kamimura et al. comprises stirring a solution containing a salt of the cyclo-tetrametaphosphate ligand with the desired noble transition metal (such as rhodium, iridium, ruthenium, and palladium) at room temperature overnight. The desired product can then be obtained by further extraction and crystallization from dichloromethane and diethyl ether.7 They have also described the synthesis of di- and trinuclear μ-oxo titanium(IV) cyclo-tetraphosphate complexes.1 Even though the preparation of noble and non-noble metal complexes using this ligand has been reported, a strategy for the straightforward preparation of mononuclear complexes of cyclo-tetrametaphosphate using mild conditions and non-noble metals represents a vacancy in the literature. In addition to filling a vacancy, we recognize that metals such as molybdenum and vanadium are useful in oxidation catalysis, such that cyclophosphate complexes of these metals may turn out to be valuable as precatalysts.
The chemistry of complexes incorporating the trianionic cyclo-trimetaphosphate ligand [P3O9]3− has also been investigated, specifically in studies of catalytic applications such as vinylidene rearrangement of general internal alkynes via the 1,2-migration of alkyl, aryl, and acyl groups.5,6 The methodology used for the synthesis of metal-cyclo-trimetaphosphate compounds was first reported by Klemperer and co-workers in 1981, and since then only a handful of publications reporting the preparation of mononuclear cyclo-trimetaphosphate-transition-metal complexes has been published.5,6,20–29 Additionally, the coordination chemistry of early transition-metal-[P3O9] complexes has not been explored extensively. Our group has recently reported the preparation of the first tridentate-cyclo-tetrametaphosphate complex.8 Montag et al. reported the preparation of [Na][(P4O12)Co(TACN)] (TACN = 1,4,7-triazacyclononane) in aqueous media. The structure determination of this salt revealed that the ligand is coordinated to the metal in a κ3 fashion in which one of the phosphate groups is not directly connected to the metal center.
Herein we report the coordination chemistry of mononuclear complexes of cyclo-tetrametaphosphate and cyclo-trimetaphosphate ligands that incorporate non-noble metals (Fig. 1) along with the development of a simple synthetic procedure, using readily accessible precursors and common glove-box techniques.
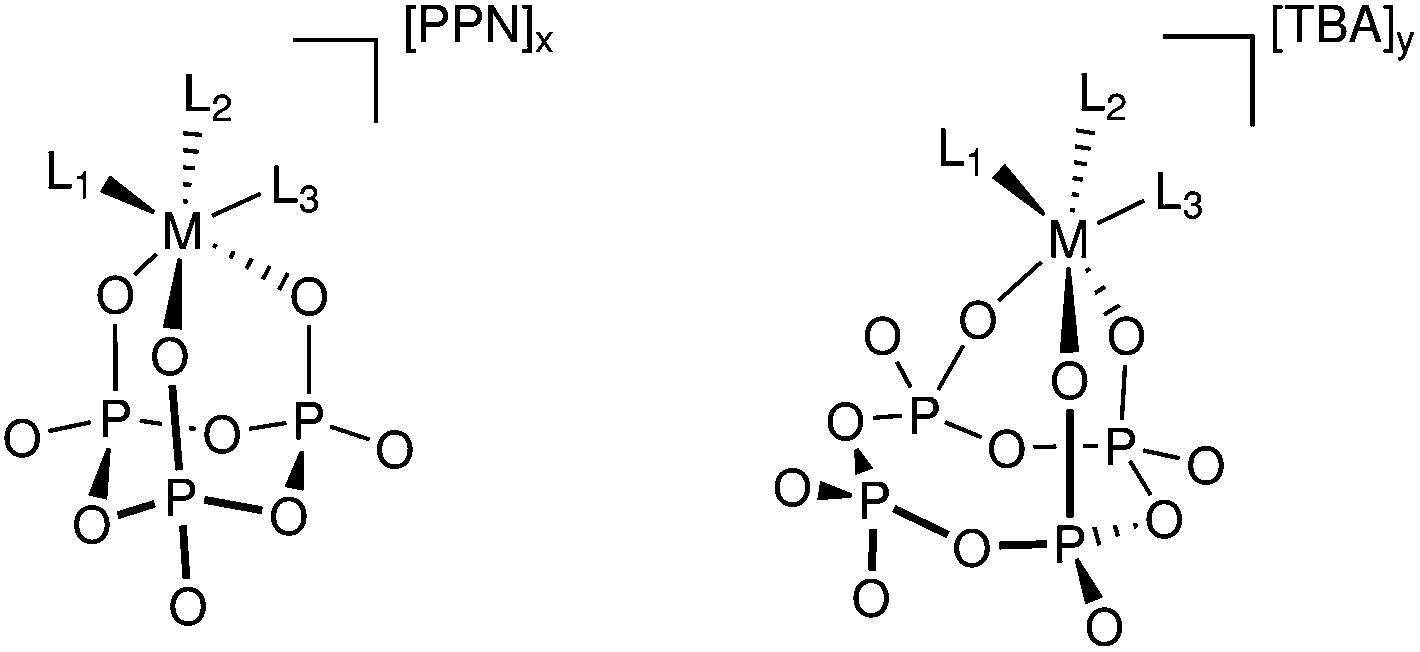 | ||
| Fig. 1 General structure of cyclo-tetrametaphosphate (right) and cyclo-trimetaphosphate (left) salts. The letter M represents one of the transition metals used in this work (M = V, W or Mo) and Ln represents the remainder of the metal's coordination sphere. | ||
2. Results and discussion
2.1. Synthesis and characterization
[TBA]4[P4O12]·5H2O (1) was prepared from the known salt [Na]4[P4O12]·5H2O30 by dissolving the latter in deionized water and passing the solution through a DOWEX® column which was previously conditioned with [TBA][OH]. The eluent was evaporated to afford a white solid which was dissolved in acetonitrile and then precipitated with diethyl ether to yield the desired compound. In general, salts 2, 3, 4 and 5 were prepared by dissolving [TBA]4[P4O12]·5H2O in dry acetonitrile and adding an appropriate transition-metal reagent (Fig. 2). Typically, the reaction mixture was stirred for one hour, leading to complete and clean conversion into the desired complex with isolated yields ranging from 56 to 69%. The observed less-than-quantitative yields are attributed mainly to losses incurred in the purification process (crystallization).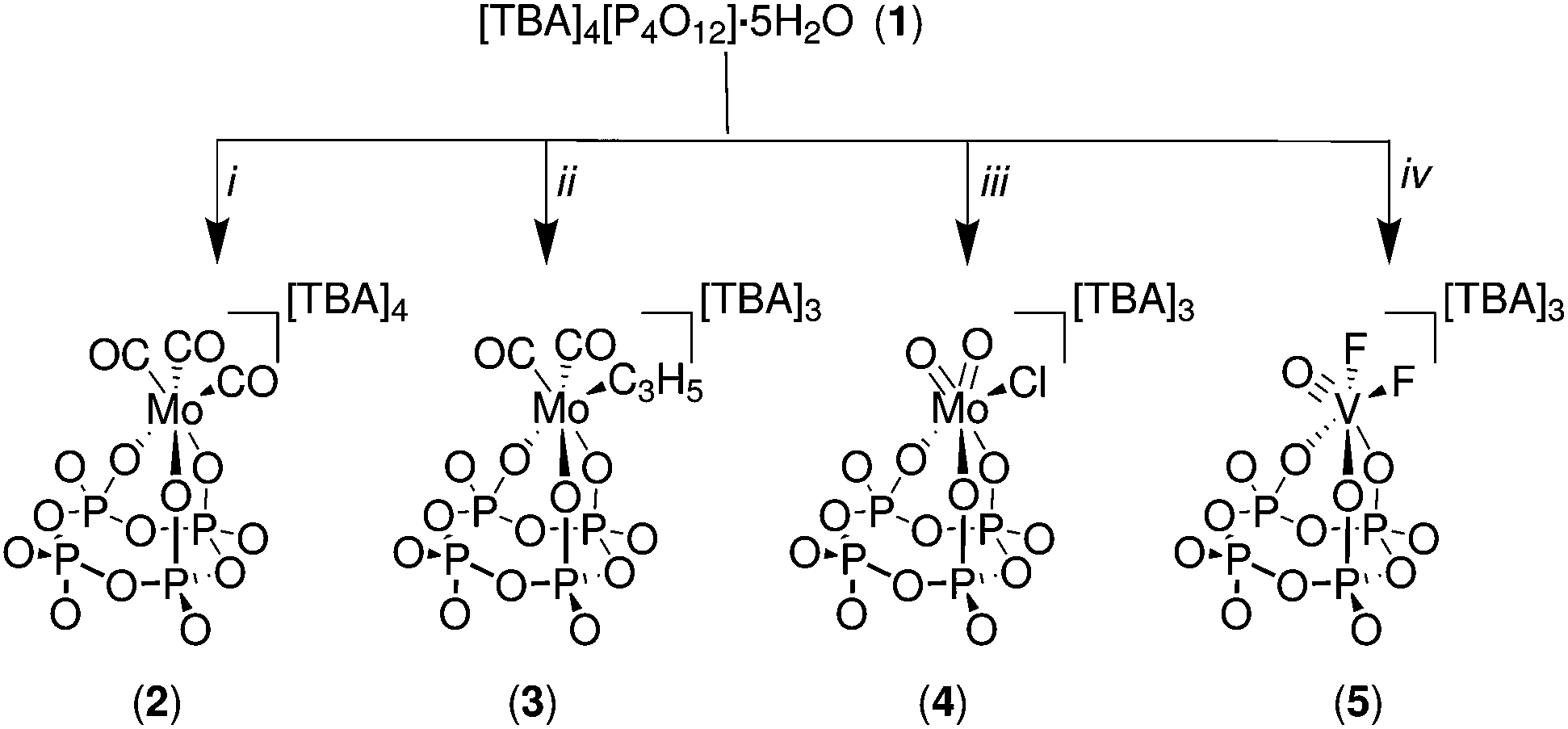 | ||
| Fig. 2 Synthesis of molybdenum and vanadium cyclo-trimetaphosphate salts. All reactions were carried out in a glovebox using dry solvents. i = (CH3CN)3Mo(CO)3, ii = (CH3CN)2Mo(CO)2(η3-C3H5)Cl, iii = MoO2Cl2(OSMe2)2, iv = VOF3. | ||
The salts 7, 8, 9, 10 and 11 were obtained by adding the corresponding metal precursors to a stirring solution of [PPN]3[P3O9]·H2O in dry acetonitrile, affording complete conversion to the final products (Fig. 3). The compounds [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)] (7) and [PPN]2[(P3O9)MoO2Cl] (8) spontaneously precipitate ca. two minutes after the addition of the corresponding precursor to the acetonitrile solution of the metaphosphate ligand. The isolated yields of these complexes are very similar to those obtained in the case of the cyclo-tetrametaphosphate complexes, varying from 56 to 78%.
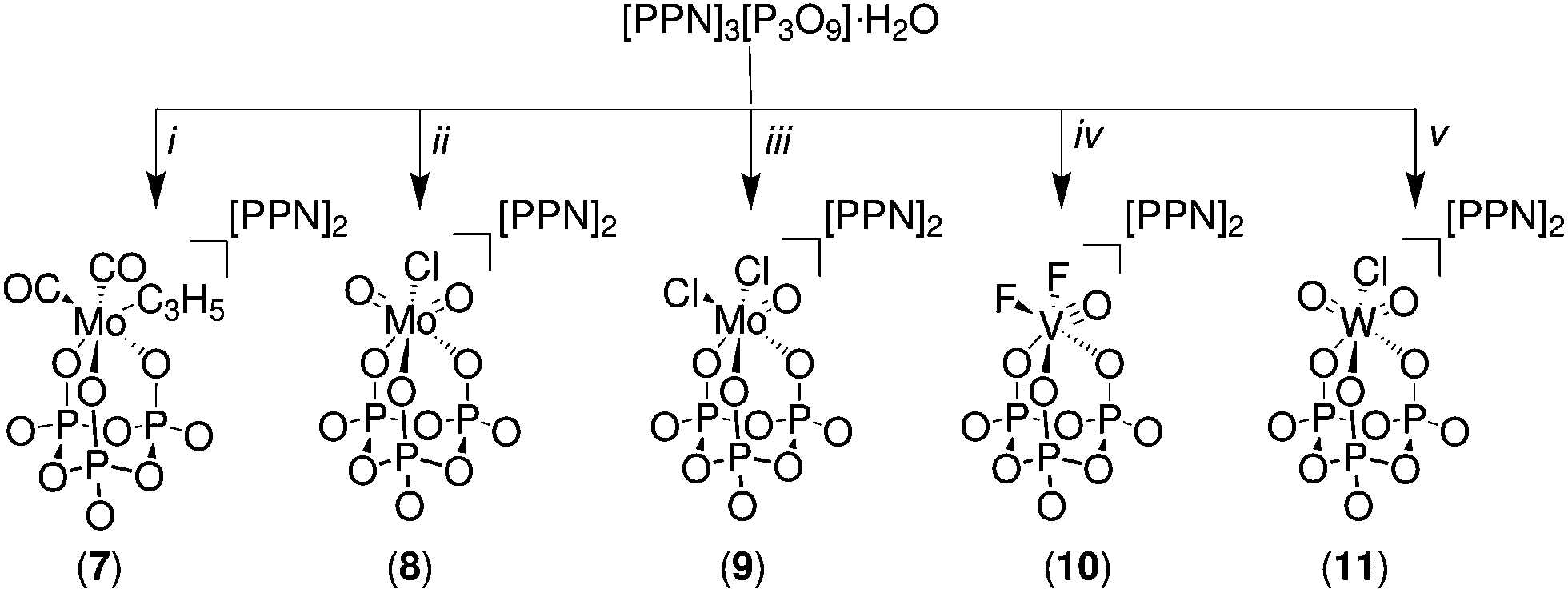 | ||
| Fig. 3 Synthesis of molybdenum, tungsten, and vanadium cyclo-trimetaphosphate salts. All reactions were carried out in a glovebox using dry solvents. i = (CH3CN)2Mo(CO)2(η3-C3H5)Cl, ii = MoO2Cl2(OSMe2)2, iii = MoOCl3, iv = VOF3, v = WOCl4. | ||
The products were characterized by solid-state FT-IR spectroscopy; stretching frequencies for signature oscillators of cyclo-tetrametaphosphate complexes are summarized in Table S1.† The IR spectrum of complex [TBA]4[(P4O12)Mo(CO)3]·2H2O (2) features three carbonyl bands at 1874, 1708, and 1702 cm−1 consistent with C1 symmetry. The compound [TBA]3[(P4O12)Mo(CO)2(η3-C3H5)] (3) features two stretching bands at 1898 and 1709 cm−1 for the carbonyl groups which are higher in energy compared to those observed in the case of salt 2, as would be expected as a function of the higher oxidation state of the molybdenum center.31,32 The salt [TBA]3[(P4O12)MoO2Cl] (4) shows two stretches for the MoO2 moiety (896 and 880 cm−1), consistent with the presence of a pair of cis oxo ligands.33 [TBA]3[(P4O12)VOF2]·Et2O (5) shows only one stretch (912 cm−1) for the V![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond. The value observed for this stretch is in the expected range.34–3751V-NMR shows a broad peak at −594.5 ppm similar to the observations reported by Roesky et al. for the complex Ph3NVOF2.34
O bond. The value observed for this stretch is in the expected range.34–3751V-NMR shows a broad peak at −594.5 ppm similar to the observations reported by Roesky et al. for the complex Ph3NVOF2.34
Complex [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)] (7) has two characteristic CO stretches at 1918 and 1812 cm−1 which, as expected, appear at higher energy compared to those of the already reported salt [PPN]3[(P3O9)Mo(CO)3] (6).38 Complex [PPN]2[(P3O9)MoO2Cl] evinces two IR bands at 923 and 898 cm−1, characteristic of a molybdenum dioxo moiety, while complex [PPN]2[(P3O9)MoOCl2] (9) has a single stretch for the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond at 932 cm−1. The FT-IR spectrum of compound [PPN]2[(P3O9)VOF2]·2CH2Cl2 (10) manifests one signal at 930 cm−1, characteristic of a VO stretch (Table S2†). To the best of our knowledge, there is only one previous report of a fully characterized and stabilized monomeric oxo fluoride vanadium compound, reported by Roesky et al. The complex Ph3PNVOF2 was prepared by treating the ligand (Ph3PNSiMe3) with the precursor (VOF3) at low temperature (−73 °C) and stirring for twelve hours.34 The salt [PPN]2[(P3O9)WO2Cl] (11) exhibits two absorption bands for the WO2 moiety at 938 and 907 cm−1.
O bond at 932 cm−1. The FT-IR spectrum of compound [PPN]2[(P3O9)VOF2]·2CH2Cl2 (10) manifests one signal at 930 cm−1, characteristic of a VO stretch (Table S2†). To the best of our knowledge, there is only one previous report of a fully characterized and stabilized monomeric oxo fluoride vanadium compound, reported by Roesky et al. The complex Ph3PNVOF2 was prepared by treating the ligand (Ph3PNSiMe3) with the precursor (VOF3) at low temperature (−73 °C) and stirring for twelve hours.34 The salt [PPN]2[(P3O9)WO2Cl] (11) exhibits two absorption bands for the WO2 moiety at 938 and 907 cm−1.
Table 1 summarizes the carbonyl stretches of the compounds reported in this work (i.e.2, 3, 6, and 7) and compounds that contain a tripodal ligand (Kläui, (pyrazolyl)borate and 1,3,5-triazacyclohexane ligands) bearing the same metal–carbonyl moiety. From the values reported, we may conclude that the cyclo-tetrametaphosphate ligand is a better donor than the cyclo-trimetaphosphate ligand and the other tridentate ligands listed in Table 1. This is reflected in decreased νCO stretching frequencies corresponding to weaker CO bonds.43,44
In addition to IR spectroscopy, 31P-NMR spectroscopy is a helpful tool for determining the connectivity of the metaphosphate ligand with respect to the metal center. All the cyclo-tetrametaphosphate-based complexes reported herein exhibit three signals in their 31P-NMR spectra in an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1. This supports that the polyanionic ligand is binding in a tripodal fashion, leading to κ3 connectivity (similar to a previous report from our group).8 On the other hand, [TBA]3[(P4O12)VOF2]·Et2O exhibits only two peaks in a ratio of 1:3, possibly due to a fluxional process or to a change to κ2 coordination in solution (this could possibly be attributed to the lability of the oxygen trans to the oxo ligand), leading to broad peaks instead of the expected triplets. Variable temperature NMR experiments confirmed the behavior described above. At lower temperatures (Fig. 4) the spectrum for complex 4 shows a new pattern with a spin system AM2X represented by three triplets in a ratio of 1:2:1 while at room temperature the spectrum consists of a singlet.
:2:1. This supports that the polyanionic ligand is binding in a tripodal fashion, leading to κ3 connectivity (similar to a previous report from our group).8 On the other hand, [TBA]3[(P4O12)VOF2]·Et2O exhibits only two peaks in a ratio of 1:3, possibly due to a fluxional process or to a change to κ2 coordination in solution (this could possibly be attributed to the lability of the oxygen trans to the oxo ligand), leading to broad peaks instead of the expected triplets. Variable temperature NMR experiments confirmed the behavior described above. At lower temperatures (Fig. 4) the spectrum for complex 4 shows a new pattern with a spin system AM2X represented by three triplets in a ratio of 1:2:1 while at room temperature the spectrum consists of a singlet.
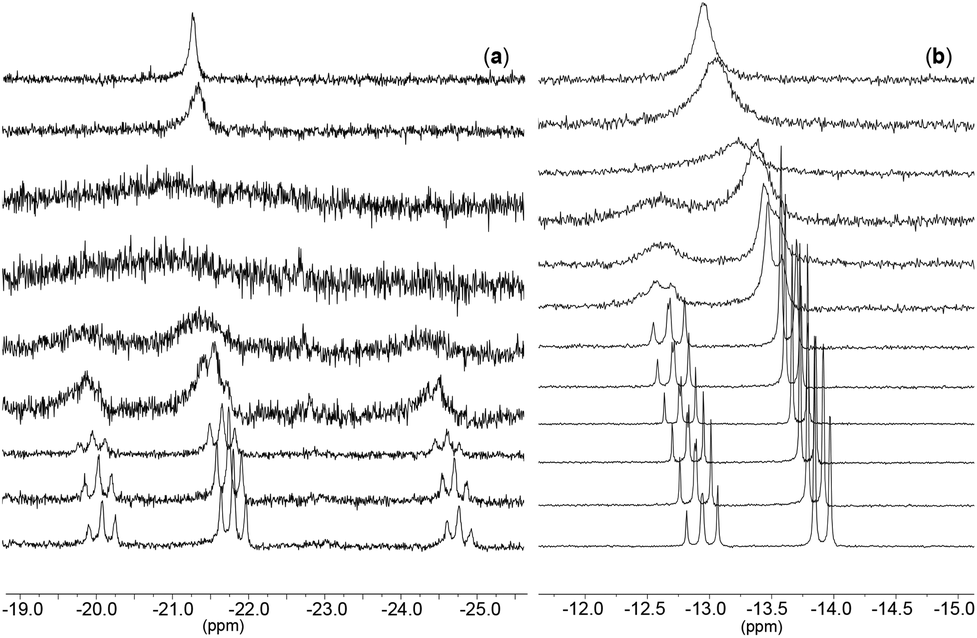 | ||
| Fig. 4 VT-31P-NMR of (a): [TBA]3[(P4O12)MoO2Cl] and (b): [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)]. The spectra were taken with an increase of 10 °C from +20 °C (top) to −60 °C (bottom) in case of spectra a and from +70 °C (top) to −40 °C (bottom) in case of spectrum b. | ||
Fig. 5 shows the spectrum observed for complex [TBA]3[(P4O12)MoO2Cl] at −60 °C (a) along with the corresponding simulation (b) which was computed using the program gNMR.45 The simulation reinforces the proposed four spin system AM2X in line with the existence of two phosphate moieties which are similar to each other and strongly coupled. These coupled phosphate units have two different phosphate neighbors which do not show any coupling between them. This suggests, for example, a κ3 connectivity of the phosphate to the metal in a fashion similar to that observed by Montag et al.8
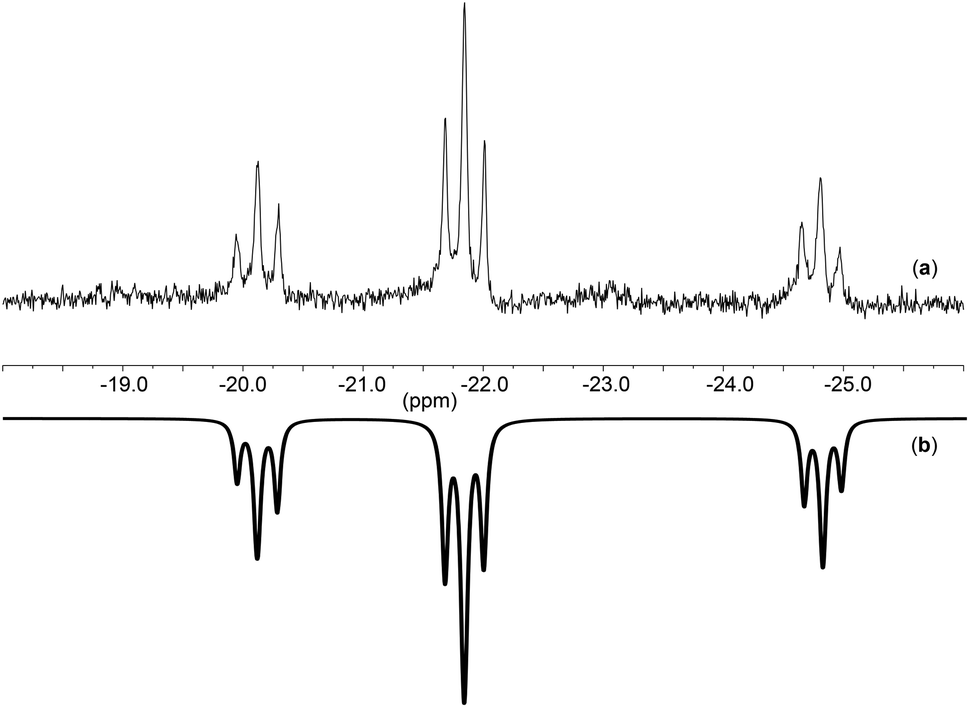 | ||
| Fig. 5 31P-NMR of (a): [TBA]3[(P4O12)MoO2Cl] at −60 °C and (b): simulated spectrum using gNMR.45 | ||
31P{1H}-NMR spectra of compounds 7, 8 and 10 show three signals in a ratio of 4:1:2 corresponding to [PPN]+ and the respective anion. The 31P-NMR spectrum of complex 7 was recorded at different temperatures (Fig. 4) showing that at high temperatures it consists of a singlet and at low temperatures it evolves into a triplet and a doublet. This behavior could be related to fluctuation of the ligand around the metal. The 1H-NMR spectrum of salt 7 shows the presence of four signals corresponding to the hydrogens of the allyl group and [PPN]+, wherein the apical and syn protons appear at very similar chemical shifts. The 31P{1H}-NMR spectrum of [(P3O9)MoO2Cl]2− is characteristic of an A2B spin system indicating a κ3 connectivity of the metaphosphate ligand. The 31P{1H}-NMR spectrum of salts 9 and 11 features only two signals in a 4:3 ratio, corresponding to [PPN]+ and the cyclo-trimetaphosphate-metal complex respectively.
The simulated spectrum for the complex [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)] at −30 °C is shown in Fig. 6. The simulation demonstrates that the spin system of this molecule is as proposed A2B giving rise to a doublet coupled to an apparent triplet. This spin system suggests that one out of the three metaphosphate moieties has a unique chemical environment.
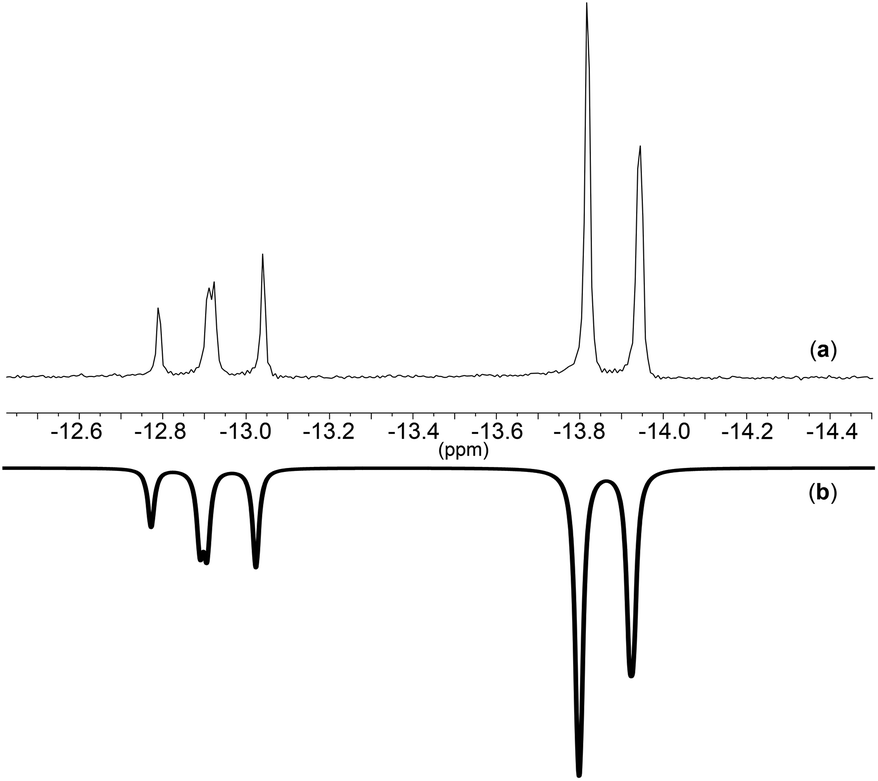 | ||
| Fig. 6 31P-NMR of (a): [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)] at −30 °C and (b): spectrum simulated using gNMR.45 | ||
2.2. Crystallographic studies
X-ray diffraction quality crystals of [TBA]3[(P4O12)Mo(CO)2(η3-C3H5)] (Fig. 7) were grown from a mixture of diethyl ether and dichloromethane. The symmetry of the complex is C1 and the connectivity of the metaphosphate is κ3. This result is consistent with the conclusions drawn from 31P-NMR and FT-IR spectroscopies studies. As the NMR simulations suggested, there are three different phosphorous environments and two of these are not coupled (the dangling metaphosphate and the farthest counterpart). The configuration of the allyl group is endo, and the bond length between the metal and the central carbon is, as expected, shorter than those between the metal and the terminal carbons.46 The distances between molybdenum and the oxygen atoms are all similar and only differ by 0.035 Å. The crystal structure of this salt also contains a dichloromethane molecule which is hydrogen bonded to the dangling metaphosphate, partially stabilizing the negative charge on the metaphosphate. The D⋯A distance is 3.065 Å (Table S3†) suggesting that this is a weak hydrogen bond.47 This structure suggests that it may be possible to pre-organize a substrate by interaction with the dangling phosphate residue.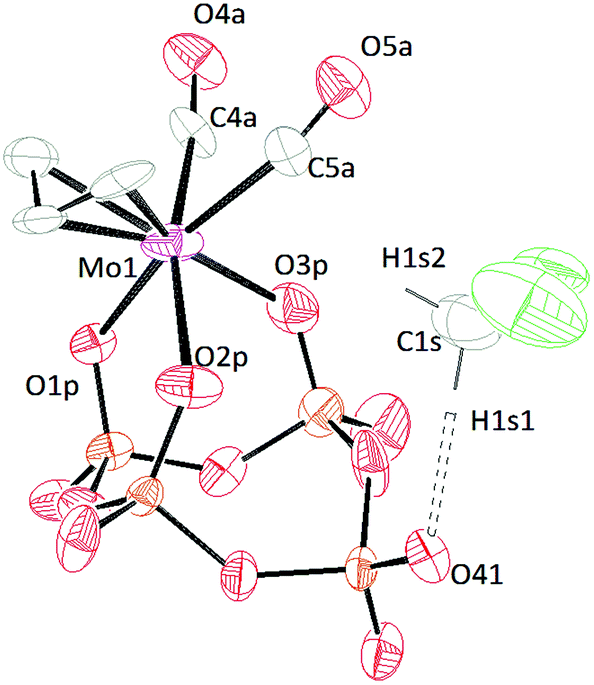 | ||
| Fig. 7 Solid-state molecular structure of anion [(P4O12)Mo(CO)2(η3-C3H5)]3− rendered using ORTEP48 with ellipsoids at the 50% probability level; the [TBA]+ cations and minor disorder are omitted for clarity. | ||
Crystals of [TBA]3[(P4O12)MoO2Cl] were obtained from a mixture of dichloromethane and diethyl ether, and were subjected to study via X-ray crystallography. The conformation of the oxo atoms is cis and the connectivity of the metaphosphate is κ3, which is again in complete agreement with FT-IR and NMR spectroscopy studies (Fig. 8). The values observed for the interatomic distances of compound [TBA]3[(P4O12)MoO2Cl] are very similar to the values for MoO2Cl2(OSMe2)2.49 As was the case for 3, this derivative (complex 4) possesses C1 symmetry. The crystal structure of this compound also contains a dichloromethane molecule that is hydrogen bonded with one of the oxygen atoms in the metaphosphate ligand. The D⋯A distance is 3.253 Å (Table S3†), indicative of a weak hydrogen bond.47 Selected interatomic distances for salts 3 and 4 are summarized in Table 2.
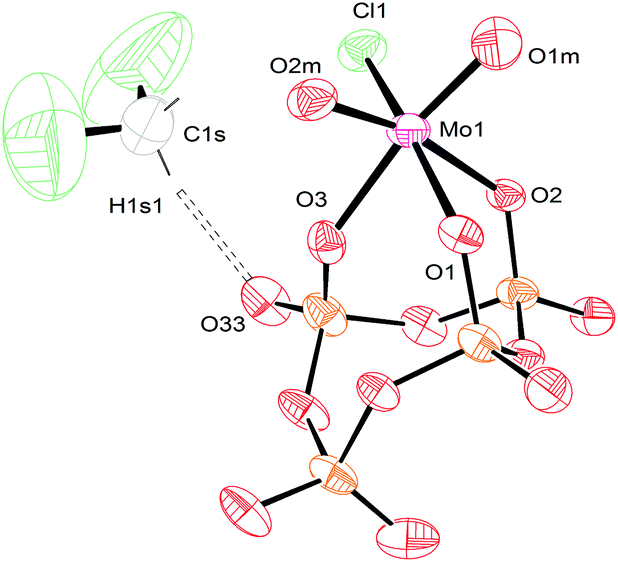 | ||
| Fig. 8 Solid-state molecular structure of anion [(P4O12)MoO2Cl]3− rendered using ORTEP48 with ellipsoids at the 50% probability level; the [TBA]+ cations and minor disorder are omitted for clarity. | ||
| Bond | 3 | 4 |
|---|---|---|
| 3: [TBA]3[(P4O12)Mo(CO)2(η3-C3H5)], 4: [TBA]3[(P4O12)MoO2Cl]. | ||
| Mo–O1 | 2.181(4) | 2.0764(4) |
| Mo–O2 | 2.216(4) | 2.163(4) |
| Mo–O3 | 2.200(5) | 2.172(4) |
| Mo–CO | 1.979(11)–2.038(13) | |
| Mo–Cl | 2.396(2) | |
| Mo–Om | 1.691(4)–1.794(8) | |
The decreased Mo–OP distance in 4versus that of 3 correlates with the increased oxidation state in the former. Even though there was no crystal structure reported for the starting material (CH3CN)2Mo(CO)2(η3-C3H5)Cl, the crystal structure of (CH3CN)2Mo(CO)2(η3-2-methyl-allyl)Cl was reported by Baker et al. The data from the solid-state structure of this complex show that the values of the interatomic distances of MoO and Mo–Cl are very similar to those observed in our complexes.50
Crystal structures were obtained for all the cyclo-trimetaphosphate salts reported herein. In general, all the complexes have Cs symmetry. Crystals of [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)] were grown by vapor diffusion of diethyl ether into a dilute dichloromethane solution (Fig. 9). The solid-state structure of [(P3O9)Mo(CO)2(η3-C3H5)]2− exhibits κ3 connectivity (consistent with the 31P{1H}-NMR spectrum) wherein all of the Mo–OP bonds differ by 0.085 Å. As suggested by the NMR simulations, one of the Mo–O distances is shorter than the other two (Mo–O1) leading to a slightly different phosphorus environment. The Mo–CO interatomic distances are similar to those reported for [(P3O9)Mo(CO)3]3−.38 The configuration of the allyl group in the solid state is endo, and the bond length between the metal and the central carbon is shorter than the bond between the metal and the external carbons by 0.14 Å.
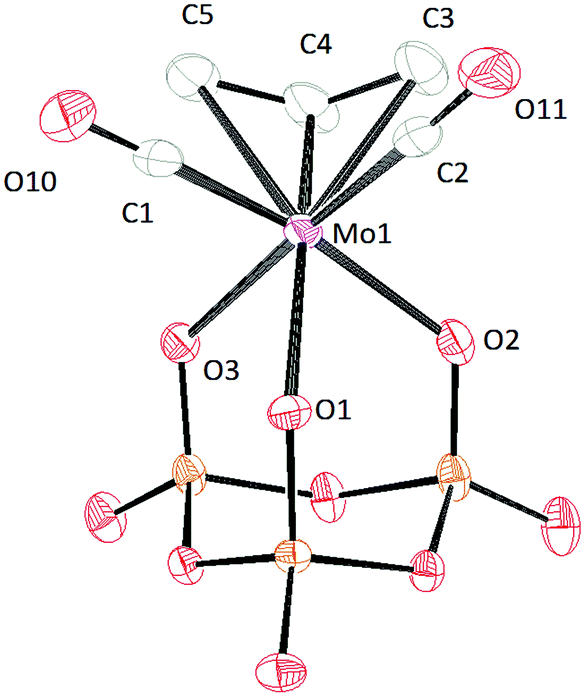 | ||
| Fig. 9 Solid-state molecular structure of anion [(P3O9)Mo(CO)2(η3-C3H5)]2− rendered using ORTEP48 with ellipsoids at the 50% probability level; the [PPN]+ cations are omitted for clarity. | ||
Crystals of [PPN]2[(P3O9)MoO2Cl] and [PPN]2[(P3O9)WO2Cl] were grown by vapor diffusion of diethyl ether into a saturated acetonitrile solution affording colorless crystals (Fig. 10 and 11 respectively). The crystal structure of the compound [PPN]2[(P3O9)WO2Cl] shows that the configuration of the two oxo atoms is cis which is consistent with the result obtained by IR spectroscopy. Crystals of [PPN]2[(P3O9)MoOCl2] were grown from a mixture of dichloromethane–toluene (2:1) affording green crystals (Fig. 12).
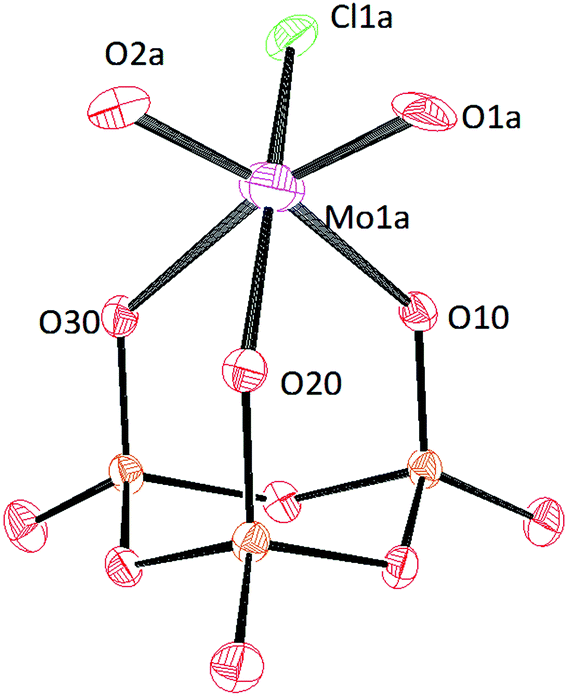 | ||
| Fig. 10 Solid-state molecular structure of anion [(P3O9)MoO2Cl]2− rendered using ORTEP48 with ellipsoids at the 50% probability level; the [PPN]+ cations and minor disorder are omitted for clarity. | ||
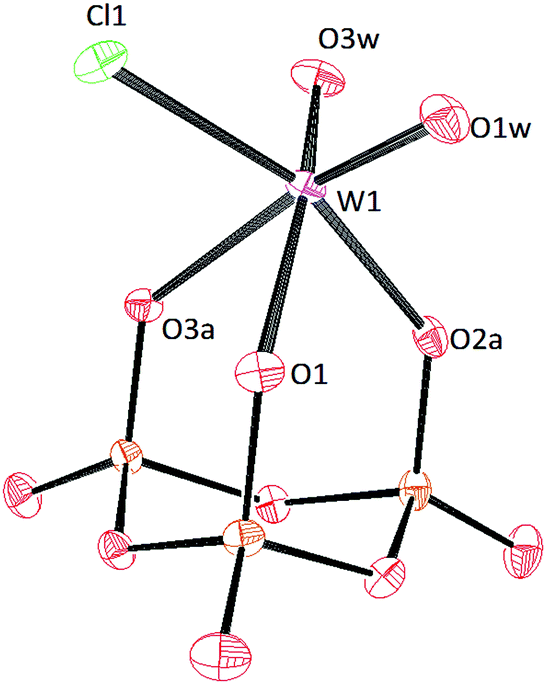 | ||
| Fig. 11 Solid-state molecular structure of the anion [(P3O9)WO2Cl]2− rendered using ORTEP48 with ellipsoids at the 50% probability level; the [PPN]+ cations and solvent are omitted for clarity. | ||
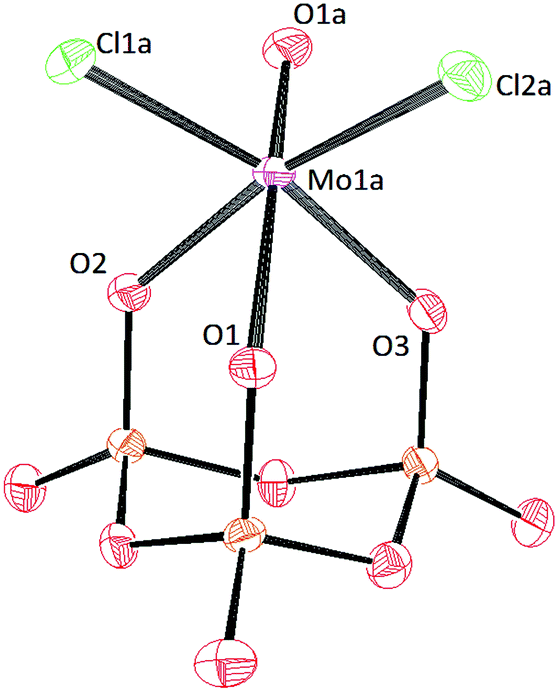 | ||
| Fig. 12 Solid-state molecular structure of anion [(P3O9)MoOCl2]2− rendered using ORTEP48 with ellipsoids at the 50% probability level; the [PPN]+ cations and minor disorder are omitted for clarity. | ||
Crystals of [PPN]2[(P3O9)VOF2]·2CH2Cl2 were grown from a mixture of dichloromethane and diethyl ether (Fig. 13). The VO distance is 1.6242(19) Å indicating a bond order of three.51 In the solid-state structure of salt 10, two solvent molecules are present. One dichloromethane solvent molecule engages in a weak hydrogen bond (D⋯A distance 3.317 Å) to one of the fluoride ions attached to the vanadium center (Table S3†).47
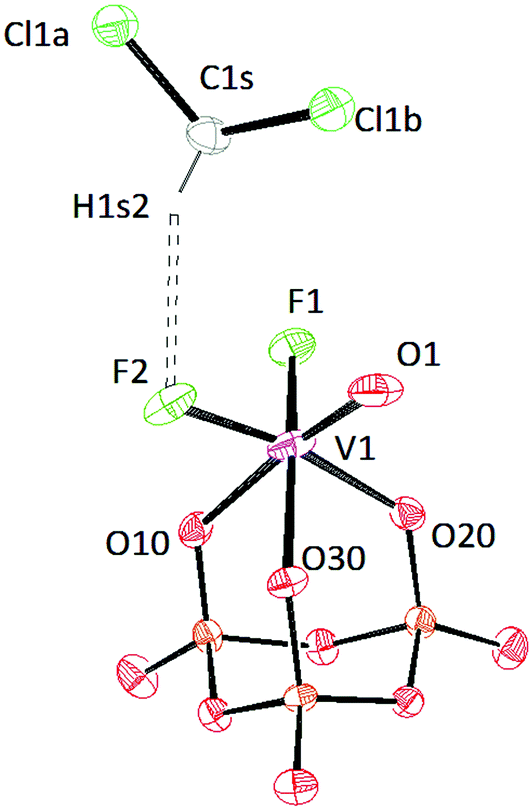 | ||
| Fig. 13 Solid-state molecular structure of the anion [(P3O9)VOF2]2− rendered using ORTEP48 with ellipsoids at the 50% probability level; the [PPN]+ cation and solvent are omitted for clarity. | ||
Selected interatomic distances for all the crystal structures of cyclo-trimetaphosphate complexes are summarized in Table 3. In general, the higher the oxidation state of the metal, the shorter the Mo–OP bond. The M–Om bond (Om = terminal oxo atom) becomes shorter as the oxidation state of the metal increases. The bond length of VO is shorter than the other M–Om bonds, lending credence to its interpretation as a triple bond.51
| Bond | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|
| 6: [PPN]3[(P3O9)Mo(CO)3]; 7: [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)]; 8: [PPN]2[(P3O9)MoO2Cl]; 9: [PPN]2[(P3O9)MoOCl2]; 10: [PPN]2[(P3O9)VOF2]·2CH2Cl2; 11: [PPN]2[(P3O9)WO2Cl]; X = CO, Cl, F; Om = terminal oxo atom. | ||||||
| M–O1 | 2.278(3) | 2.1467(11) | 2.1966(14) | 2.204(7) | 2.1725(16) | 2.1904(11) |
| M–O2 | 2.272(3) | 2.2204(13) | 2.1938(14) | 2.068(7) | 2.0395(17) | 2.0453(17) |
| M–O3 | 2.300(3) | 2.2325(14) | 2.0587(15) | 2.083(8) | 1.9692(16) | 2.1790(18) |
| M–X | 1.940(4)–1.961(2) | 1.942(2)–1.945(2) | 2.3628(11) | 2.352(10)–2.360(18) | 2.347(18)–2.35(4) | 2.3562(6) |
| 2.3562(6) | ||||||
| M–Om | 1.687(4)–1.694(2) | 1.73(5) | 1.6242(19) | 1.7152(13)–1.711(3) | ||
3. Conclusions
In summary, the facile synthesis of a series of early transition-metal-cyclophosphate complexes (bearing labile ligands) is reported. The complexes (2–5 and 7–11) were isolated in good yield and were fully characterized by means of IR and NMR spectroscopy. The obtained crystal structures and NMR spectroscopy data for these complexes showed that both cyclo-triphosphate and cyclo-tetraphosphate behave as tridentate ligands connected in κ3 mode: in the case of [P4O12]4−, one of the metaphosphate units is uncoordinated.4. Experimental
General
Unless stated otherwise, all the operations were performed in a Vacuum Atmospheres drybox under an atmosphere of purified nitrogen. Mo(CO)6 was purchased from Strem and used without further purification. Tetrabutylammonium hydroxide 1 M and DOWEX® 50 WX4-200 ion-exchange resin were purchased from Sigma-Aldrich. [Na]4[P4O12]·5H2O,30 [PPN]3[P3O9]·H2O,52 (CH3CN)3Mo(CO)3,53 (CH3CN)2Mo(CO)2(η3-C3H5)Cl,54 MoO2Cl2(OSMe2)2,49 MoOCl3,55 WO2Cl2(CH3CN)2,55 WOCl455 and [PPN]3[(P3O9)Mo(CO)3] (6)38 were prepared according to published procedures. Aqueous solutions were prepared using reagent grade deionized water (r ≥ 18 MΩ cm; Rica Chemical Company, USA). Diethyl ether, acetonitrile, dichloromethane and tetrahydrofuran were dried and deoxygenated by the method of Grubbs using a system built by SG Water USA, LLC and stored over 4 Å molecular sieves. 4 Å molecular sieves were dried under reduced pressure at a temperature above 200 °C over the course of one week. 1H, 13C{1H} and 31P{1H}-NMR spectra were recorded on Varian Mercury-300 or Bruker AVANCE-400 spectrometers. NMR solvents were obtained from Cambridge Isotope Laboratories. 1H and 13C chemical shifts are reported with respect to internal solvent resonances (DMSO-d6, δ: 2.50 and 39.52 ppm respectively). 31P-NMR chemical shifts are reported with respect to an external reference (85% H3PO4, δ: 0.0 ppm). 51V-NMR chemical shifts are reported with respect to an external reference (VOCl3, δ: 0.0 ppm). J values are given in Hz. Infrared spectra were recorded on a Bruker TENSOR37 FT-IR spectrometer. X-ray data collections were carried out on a Siemens Platform three-circle goniometer with a CCD detector using Mo-Kα radiation, λ = 0.71073 Å. An Agilent Technologies 5975C Mass Selective Detector operating in electron impact ionization mode was used to collect mass spectrometric data. Mass Spectrometry was performed on a Micromass Q-TOF ESI spectrometer in anionic mode. Samples were prepared in a glovebox and placed into a 1.5 mL GC vial with a septum to prevent exposure to moisture and oxygen and immediately analyzed by ESI mass spectrometry. All samples were prepared at a concentration of 0.1 mg mL−1 in acetonitrile. Prior to injection of the samples, the capillary tubing was flushed with 3 mL of dry acetonitrile (dried over 4 Å molecular sieves for 3 days). The anions of interest were observed to be sensitive to instrument voltages. Optimum instrument settings were obtained with a capillary voltage of 3200 V, a MCP detector voltage of 2500 V, source temperature of 100 °C and a desolvation gas temperature of 150 °C. EPR measurements were performed using a Bruker EMX EPR spectrometer, an ER 4199HS cavity and a Gunn diode microwave source producing X-band (8–10 GHz) radiation.
O) and 896m (MO). δH (400 MHz; DMSO-d6) 0.87 (t, 3JHH = 7.2, CH3–CH2, 36H), 1.27 (m, CH2–CH2, 24H), 1.52 (m, CH2–CH2, 24H), 3.14 (t, 3JHH = 7.2, CH2–N, 24H) ppm. δC (100 MHz; DMSO-d6) 57.2 (CH2, 12C), 23.2 (CH2, 12C), 19.1 (CH3, 12C), 13.4 (CH3, 12C) ppm. δP (162 MHz; CH3CN) −21.2 (b, 4P) ppm.
O). δH (400 MHz; DMSO-d6) 0.88 (t, 3JHH = 7.2, CH3–CH2, 36H), 1.28 (m, CH2–CH2, 24H), 1.53 (m, CH2–CH2, 24H), 3.15 (t, 3JHH = 7.1, CH2–N, 24H) ppm. δC (100 MHz; DMSO-d6) 57.4 (CH2, 12C), 23.0 (CH2, 12C), 19.1 (CH2, 12C), 13.4 (CH3, 12C) ppm. δV (104 MHz; DMSO-d6) −594.5 ppm.
O) and 923m (MO). δH (400 MHz; DMSO-d6) 7.57 (m, HAr, 48H), 7.72 (m, HAr, 12H) ppm. δC (100 MHz; DMSO-d6) 133.7 (m, para, 12C), 132.3 (m, ortho, 24C), 129.9 (m, meta, 24C), 126.33 (dd, 1JPC = 107.1, 3JPC = 1.5, ipso, 12C). δP (121.5 MHz; CH3CN) +22.3 (s, 4P), −18.2 (t, 2JPP = 18.1, 1P), −19.0 (d, 2JPP = 18.1, 2P) ppm. ESI-MS (m/z, CH2Cl2): 939.9281 (M − PPN)−, 200.8703 (M)2−.
:1 CH2Cl2–toluene mixture to provide the desired product as green crystals which were collected by filtration and washed with a mixture 1:3 acetonitrile–diethyl ether (3 × 2 mL) (0.30 g, 75%). Elem. Anal. Found: C, 57.81; H, 4.20; N, 1.75%. Calc for C72H60Cl2MoN2O10P7: C, 57.77; H, 4.04; N, 1.87%. FT-IR (ATR) νmax/cm−1 932m (MO). δH (300 MHz; DMSO-d6) 7.56 (m, HAr, 48H), 7.71 (m, HAr, 12H) ppm. δC (100 MHz; DMSO-d6): 133.6 (m, para, 12C), 131.8 (m, ortho, 24C), 129.7 (m, meta, 24C), 126.8 (dd, 1JPC = 107.1, 3JPC = 1.5, ipso, 6C) ppm. δP (121.5 MHz, CH3CN): +21.54 (s, 4P) ppm. EPR (CH2Cl2, g-value): 1.934. ESI-MS (m/z, CH2Cl2): 421.72 (MH)−, 385.74 (M − Cl)− and 341.75 (M − PO3)−.
O). δH (400 MHz; DMSO-d6) 7.59 (m, HAr, 60H) ppm. δC (100 MHz; DMSO-d6): 133.6 (m, para, 12C), 131.9 (m, ortho, 24C), 129.5 (m, meta, 24C), 126.8 (dd, 1JPC = 107.1, 3JPC = 1.5, ipso, 6C) ppm. δP (121.5 MHz; CH3CN) +22.16 (s, 4P), −18.19 (s, 1P), −22.15 (s, 2P). δV (104 MHz; DMSO-d6): −593.4 ppm.
:1), solution of the residue to give colorless crystals which were collected by filtration and washed with a mixture 1:3 acetonitrile–diethyl ether (3 × 2 mL) (0.32 g, 77%). Elem. Anal. Found: C, 55.38; H, 3.90; N, 1.75%. Calc. for C72H60ClN2O11P7W: C, 55.24; H, 3.86; N, 1.79%. FT-IR (ATR) νmax/cm−1 907m (WO), 938m (WO). δH (300 MHz; DMSO-d6) 7.56 (m, HAr, 48H), 7.71 (m, HAr, 12H) ppm. δC (100 MHz; DMSO-d6) 133.6 (m, para, 12C), 131.8 (m, ortho, 24C), 129.7 (m, meta, 24C), 126.8 (dd, 1JPC = 107.1, 3JPC = 1.5, ipso, 6C) ppm. δP (121.5 MHz; CH3CN) +22.20 (s, 4P), −17.79 (m, 3P) ppm. ESI-MS (m/z, CH2Cl2) 488.80 (MH)−, 452.81 (M − Cl)−, 408.83 (M − PO3)−, 243.89 (M)2−.
Acknowledgements
The authors thank Eni SpA under the Eni-MIT Alliance Solar Frontiers Program for financial support.References
- S. Kamimura, T. Matsunaga, S. Kuwata, M. Iwasaki and Y. Ishii, Inorg. Chem., 2004, 43, 6127–6129 CrossRef CAS PubMed.
- B. Boonchom, J. Optoelectron. Adv. Mater., 2009, 1, 103–114 Search PubMed.
- M. Trojan and P. Sulcová, Dyes Pigm., 2000, 47, 291–294 CrossRef CAS.
- H. Onoda, K. Okumoto, A. Nakahira and I. Tanaka, Materials, 2009, 2, 1–9 CrossRef CAS.
- K. Kanao, Y. Ikeda, K. Kimura, S. Kamimura, Y. Tanabe, Y. Mutoh, M. Iwasaki and Y. Ishii, Organometallics, 2013, 32, 527–537 CrossRef CAS.
- Y. Ikeda, T. Yamaguchi, K. Kanao, K. Kimura, S. Kamimura, Y. Mutoh, Y. Tanabe and Y. Ishii, J. Am. Chem. Soc., 2008, 130, 16856–16857 CrossRef CAS PubMed.
- S. Kamimura, S. Kuwata, M. Iwasaki and Y. Ishii, Inorg. Chem., 2004, 43, 399–401 CrossRef CAS PubMed.
- M. Montag, C. R. Clough, P. Müller and C. C. Cummins, Chem. Commun., 2011, 47, 662–664 RSC.
- S. Mohamady and S. D. Taylor, Org. Lett., 2013, 15(11), 2612–2615 CrossRef CAS PubMed.
- P. M. Laügt, J. Guitel and I. T. E. G. Bassi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 201–208 CrossRef.
- A. G. Nord, Acta Chem., Scand. Ser. A, 1983, 37, 539–543 CrossRef PubMed.
- M. Trojan, D. Brandová and Z. Solc, Thermochim. Acta, 1987, 110, 343–358 CrossRef CAS.
- M. Trojan, Mater. Lett., 1989, 8, 247–252 CrossRef CAS.
- W. Gunsser, D. Fruehauf, K. Rohwer and A. Zimmermann, J. Solid State Chem., 1989, 82, 43–51 CrossRef CAS.
- M. Trojan and J. Palme, Thermochim. Acta, 1993, 224, 165–175 CrossRef CAS.
- M. Trojan, J. Palme and P. Mazan, Thermochim. Acta, 1993, 224, 177–182 CrossRef CAS.
- B. Boonchoma, M. Thongkamc, S. Kongtaweelert and N. Vittayakorn, J. Alloys Compd., 2009, 486, 689–692 CrossRef PubMed.
- B. Boonchom and N. Vittayakorn, J. Mater. Sci., 2010, 45, 1459–1463 CrossRef CAS PubMed.
- H. Hemissi, M. Rzaigui and Z. A. Al Othman, Acta Crystallogr., Sect E: Struct. Rep. Online, 2010, 66, m186–m187 CAS.
- C. J. Besecker and W. G. Klemperer, J. Organomet. Chem., 1981, 205, C31–C32 CrossRef CAS.
- C. J. Besecker, V. W. Day and W. G. Klemperer, Organometallics, 1985, 4, 564–570 CrossRef CAS.
- V. W. Day, W. G. Klemperer and D. J. Main, Inorg. Chem., 1990, 29, 2345–2355 CrossRef CAS.
- V. W. Day, W. G. Klemperer, S. P. Lockledge and D. J. Main, J. Am. Chem. Soc., 1990, 112, 2031–2033 CrossRef CAS.
- V. W. Day, T. A. Eberspacher, W. G. Klemperer, R. P. Planalp, P. W. Schiller, A. Yagasaki and B. Zhong, Inorg. Chem., 1993, 32, 1629–1637 CrossRef CAS.
- W. G. Klemperer and B. Zhong, Inorg. Chem., 1993, 32, 5821–5826 CrossRef CAS.
- K. N. Han, D. Whang, H. J. Lee, Y. Do and K. Kim, Inorg. Chem., 1993, 32, 2597–2599 CrossRef CAS.
- V. W. Day, T. A. Eberspacher, W. G. Klemperer and B. Zhong, J. Am. Chem. Soc., 1994, 116, 3119–3120 CrossRef CAS.
- S. Ryu, D. Whang, J. Kim, W. Yeo and K. Kim, J. Chem. Soc., Dalton Trans., 1993, 205–209 RSC.
- D. Attanasio, F. Bachechi and L. Suber, J. Chem. Soc., Dalton Trans., 1993, 2373–2378 RSC.
- S. Greenfield and M. Clift, Analytical chemistry of the condensed phosphates, Pergamon Press, 1975, vol. 57, p. 190 Search PubMed.
- J. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds – Part A, John Wiley & Sons, Inc., 6th edn, 2009, vol. B, pp. 222–223 Search PubMed.
- H. H. Kung, Transition Metal Oxides: Surface Chemistry and Catalysis, Elsevier Science Publishers B.V., 1st edn, 1989, p. 67 Search PubMed.
- J. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, Inc., 5th edn, 1997, vol. B, pp. 168–169 Search PubMed.
- H. W. Roesky, I. Leichtweis and M. Noltemeyer, Inorg. Chem., 1993, 32, 5102–5104 CrossRef CAS.
- J. Harrelda, H. Wonga, B. Daveb, B. Dunna and L. Nazar, J. Non-Cryst. Solids, 1995, 225, 319–324 CrossRef.
- K. Kanamoria, K. Nishidaa, N. Miyataa, K. Okamotob, Y. Miyoshic, A. Tamura and H. Sakurai, J. Inorg. Biochem., 2001, 86, 649–656 CrossRef.
- M. D. Hoops and B. S. Ault, J. Mol. Struct., 2002, 616, 91–101 CrossRef CAS.
- C. R. Clough, J. S. Silvia, P. Müller and C. C. Cummins, Inorg. Chim. Acta, 2012, 382, 195–198 CrossRef CAS PubMed.
- W. Kläui, A. Müller, W. Eberspach, R. Boese and I. Goldberg, J. Am. Chem. Soc., 1987, 109, 164–169 CrossRef.
- M. D. Curtis and K.-B. Shiu, Inorg. Chem., 1985, 24, 1213–1218 CrossRef CAS.
- N. L. Armanasco, M. V. Baker, M. R. North, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1998, 1145–1149 RSC.
- Y. D. Ward, L. A. Villanueva, G. D. Allred, S. C. Payne, M. A. Semones and L. S. Liebeskind, Organometallics, 1995, 14, 4132–4156 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 2010, pp. 27–32 Search PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley-Interscience, New York, 6th edn, 1999, pp. 637–638 Search PubMed.
- gNMR V5, Adept Scientific plc, Letchworth Herts, UK, 2003 Search PubMed.
- R. H. Crabteree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Inc., 4th edn, 2005, pp. 131–133 Search PubMed.
- G. Desiraju and T. Steiner, The Weak Hydrogen Bond: In Structural Chemistry and Biology, Oxford University Press, 2001, pp. 12–16 Search PubMed.
- M. N. Burnett and C. K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Ridge National Laboratory Report ORNL-6895, 1996 Search PubMed.
- F. J. Arnaiz, R. Aguado, M. R. Pedrosa and A. D. Cian, Inorg. Chim. Acta, 2003, 347, 33–40 CrossRef CAS.
- P. Baker, M. Drew, A. Johans and M. Meehan, J. Chem. Crystallogr., 1998, 28, 839–841 CrossRef CAS.
- D. Shriver and P. Atkins, Inorganic Chemistry, W. H. Freeman and Company, 5th edn, 2010, pp. 461–462 Search PubMed.
- W. G. Klemperer and D. J. Main, Inorg. Chem., 1990, 29, 2355–2360 CrossRef CAS.
- D. P. Tate, W. R. Knipple and J. M. Augl, Inorg. Chem., 1962, 1, 433–434 CrossRef CAS.
- D. A. Clark, D. L. Jones and R. J. Mawby, J. Chem. Soc., Dalton Trans., 1980, 4, 565–569 RSC.
- V. C. Gibson, T. P. Kee and A. Shaw, Polyhedron, 1990, 9, 2293–2298 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 898619–898621, 898623, 898624, 928307 and 928308. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52526k |
| This journal is © The Royal Society of Chemistry 2014 |