
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale metal fluorides: a new class of heterogeneous catalysts†
Erhard
Kemnitz
Department für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, D-12489 Berlin, Germany. E-mail: erhard.kemnitz@chemie.hu-berlin.de; Fax: +493020937468; Tel: +493020937277
First published on 5th December 2014
Abstract
This perspective article focuses on nanoscopic metal fluorides and hydroxide fluorides prepared via a recently explored fluorolytic sol–gel synthesis approach. Metal fluoride phases obtained via this route exhibit distinctly different properties compared with their classically prepared homologues. Thus, extremely strong solid Lewis acids are available which give access to new catalytic reactions with sometimes unexpectedly high conversion degrees and selectivity. Even more interestingly, metal hydroxide fluorides can be obtained via this synthesis route, which are not accessible via any other approach for which the hydroxide to fluoride ratio can be adjusted over a wide range. As a result, bi-acidic (Brønsted and Lewis) solids with tunable Lewis to Brønsted acidity can be obtained which show interesting results in a variety of reactions. Finally, these new nano-metal fluorides, due to their very high surface areas and distinct acidic properties, can be used as supports for many novel metal catalysed reactions, thus showing surprising synergistic effects. This overview will briefly outline the synthesis approach of the fluorolytic sol–gel route, will present characteristic bulk and surface properties and will give several examples of their novel catalytic applications in purely Lewis acid and in bi-acidic catalysed reactions, and will also exemplarily show the potential of these new materials as supports for heterogeneous catalytic reactions.
 Erhard Kemnitz |
Introduction
As compared to their respective metal oxides, metal fluorides play just a minor role in both homogeneous and heterogeneous catalysis. The probably most prominent fluoride used in catalysis is antimony pentafluoride, SbF5, which is considered to be the strongest Lewis acid. It became very popular as the so-called Swarts catalyst in the late 1920s when it was introduced as a catalyst under homogeneous conditions for the industrial production of chlorofluorocarbons (CFCs) which were used for decades as refrigerants.1,2 However, alternatively to this liquid phase catalytic process, heterogeneously catalysed processes were developed in the 1960s that mainly used fluorinated alumina and chromia, where in fact aluminium fluoride and chromium(III)fluoride were the catalytically active phases. Measured on their production volume, SbF5, AlF3, and CrF3 are the most widely used Lewis acid catalysts. Not surprisingly, fluorinated alumina and chromia and AlF3 as well as CrF3 were intensively investigated for many different fluorination reactions that are mainly related to the synthesis of CFCs and their alternatives hydrochlorofluorocarbons, HCFCs, and hydrofluorocarbons, HFCs. The characteristics of these catalysts as well as reaction mechanisms of the heterogeneously catalysed fluorination reactions were comprehensively reviewed some few years ago (see ref. 3 and 4 and references therein).In 1999, K.O. Christe et al.5 introduced the pF scale which provides for the first time a quantification of Lewis acidity strength of metal fluorides and chlorides. Their approach is based on ab initio calculations of the free formation energies of the gas phase reactions of molecular metal fluorides with a gaseous fluoride anion (eqn (1)):
| MFx(g) + F−(g) → MF−(x+1)(g) | (1) |
These formation energy values stand for the fluoride anion affinity of the respective metal halides and, thus, can directly be taken as a measure of the Lewis acidity of these molecules. Thus, the authors consider their pF scale as a Lewis acid counterpart of the pKs scale of Brønsted acids. For selected compounds, values are listed in Table 1; for further data, see ref. 6.
| Comp. | SbF5 | AlF3 | AlF2Cl | AlFCl2 | AlCl3 | InF3 | GaF3 | AsF3 | SiF4 |
|---|---|---|---|---|---|---|---|---|---|
| pF− | 12.03 | 11.50 | 11.47 | 11.50 | 11.46 | 10.75 | 10.70 | 10.59 | 7.35 |
Reflecting on these data, there are at least two surprising facts: first, it turns out that AuF5 is an even slightly stronger Lewis acid than SbF5 is, although this is chemically not relevant since it is a very unstable compound. Second, aluminium fluoride and aluminium chloride fluoride phases (note that stoichiometric compounds are unknown so far) exhibit practically the same Lewis acidity as the strong Lewis acid AlCl3 which is like iron chloride, tin chloride or zinc chloride, a widely known Lewis acidic Friedel–Crafts catalyst. In contrast, AlF3 is not at all considered as a suitable Friedel–Crafts catalyst; this means that its experienced catalytic potential is in strong contradiction to the Lewis acidity predicted by the pF-scale.
This situation has fundamentally changed since 2003 when the so-called fluorolytic sol–gel synthesis had been introduced, giving access to nanoscopic high surface aluminium fluoride (HS-AlF3),7 and was later on applied to many other nano-metal fluorides as well.8–10
2. Synthesis and properties of nanoscopic metal fluorides
2.1. The non-aqueous fluorolytic sol–gel synthesis of binary metal fluorides
The classical aqueous sol–gel synthesis is represented by the reaction of a metal alkoxide dissolved in an organic solvent with water. In a first step, the hydrolysis reaction with water leads to the formation of M–OH-groups (eqn (2a)), which in a second reaction step undergo condensation reactions either with neighbouring M–OH or with still un-reacted M–OR groups (eqn (2b) and (2c)); thus, either water or alcohol is formed.| M(OR)n + nH2O → M(OH)n + nROH | (2a) |
| M–OH + HO–M– → –M–O–M–+ H2O | (2b) |
| –M–OH + RO–M– → –M–O–M–+ ROH | (2c) |
This brief description is in fact a simplification of the complex reactions, but when properly performed this will lead to the creation of M–O–M-bridges, resulting finally in a three-dimensional nanoscale metal oxide.
Because the initial reaction step is a hydrolysis reaction, in this context it will be defined as hydrolytic sol–gel synthesis thus indicating the differences and similarities with the fluorolytic sol–gel synthesis that will be briefly outlined here.
The simplified chemical pathway of the fluorolytic sol–gel synthesis is given by eqn (3).
| M–OR + H–F → M–F + ROH | (3a) |
nM–F → (–![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) N–F–M–F)n/2 N–F–M–F)n/2 | (3b) |
As can easily be seen, this first reaction step (eqn (3a)) resembles closely the first step of the hydrolytic sol–gel route (eqn (2a)). The only but important difference is that in the fluorolytic sol–gel synthesis water as a reactant is completely replaced by hydrogen fluoride thus directly resulting in the formation of M–F bonds. The second reaction step of both synthesis routes has nothing in common although both strategies result – if properly performed – in the formation of either nanoscale metal oxides or nanoscale metal fluorides, respectively. M–F-bonds formed according to eqn (3a) cannot undergo condensation reactions, but fortunately another typical property of a fluoride anion supports the formation of a three-dimensional fluoride network. This means that fluoride ions strongly tend to bridge (eqn (3b)). There are just some very scarce examples of solid fluorides exhibiting terminal, meaning non-bridging, fluoride ions.11 Consequently, instead of condensation in case of metal oxide formation, the strong bridging tendency of fluoride ions causes the formation of nanoscopic three-dimensional particles. This new fluorolytic route was published first time for the synthesis of nanoscopic high surface area aluminium fluoride (HS-AlF3) in 2003 (ref. 7) that has been proven in the meantime to be a general synthesis approach for nanoscopic metal fluorides.12–27
For deeper insights into the mechanism of the fluorolytic sol–gel approach, see ref. 10.
The reaction conditions differ sometimes drastically; however, the general synthesis path of the fluorolytic sol–gel synthesis for all the binary metal fluorides follows the same rule that may be summarized according to Scheme 1.
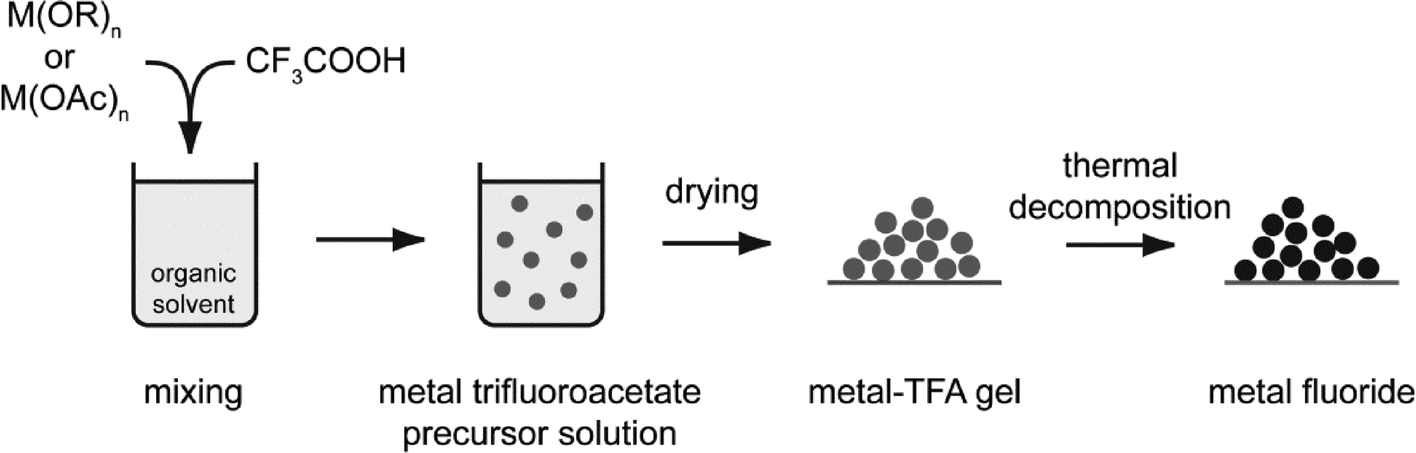 | ||
| Scheme 1 The fluorolytic sol–gel synthesis starting from a metal alkoxide reacting with anhydrous HF in a suitable organic solvent. | ||
Based on the same synthesis procedure, also ternary and quaternary metal fluorides and even complex fluorometallates are accessible.28–31
The topological properties differ inside a narrow range, e.g. the particle size of many metal fluorides prepared according to this route varies between 5 and 20 nm. Hence, for many metal fluorides optically transparent clear sols can be synthesized (Fig. 1 left) from which dry xerogels can be obtained by evaporation of the solvent (Fig. 1 right).
 | ||
| Fig. 1 Clear colloidal solution (sol) of MgF2 in ethanol on the left. MgF2 xerogel obtained from the sol in dishes on the right. | ||
Due to the characteristics of the synthesis path, highly distorted, often totally X-ray amorphous solids are formed, with BET surface areas ranging from, e.g., 250 m2 g−1 for HS-AlF3 up to 400 m2 g−1 in case of MgF2 xerogels.
Bulk and surface properties of these nano-metal fluoride phases sometimes differ drastically from their classically prepared analogues. An essential feature that is relevant for applications in catalysis is the surface topology of those phases. This will be displayed for two different AlF3 phases, namely β-AlF3 being one of the most intensively investigated crystalline AlF3-modification because it exhibits for many Lewis acid catalysed reactions the best catalytic performance (see ref. 3 and 4 and references therein). The other one is HS-AlF3 obtained according to the fluorolytic sol–gel route being almost X-ray amorphous with a surface area of >200 m2 g−1. In Fig. 2, IR difference spectra of adsorbed CO as a probe molecule on β-AlF3 are displayed.
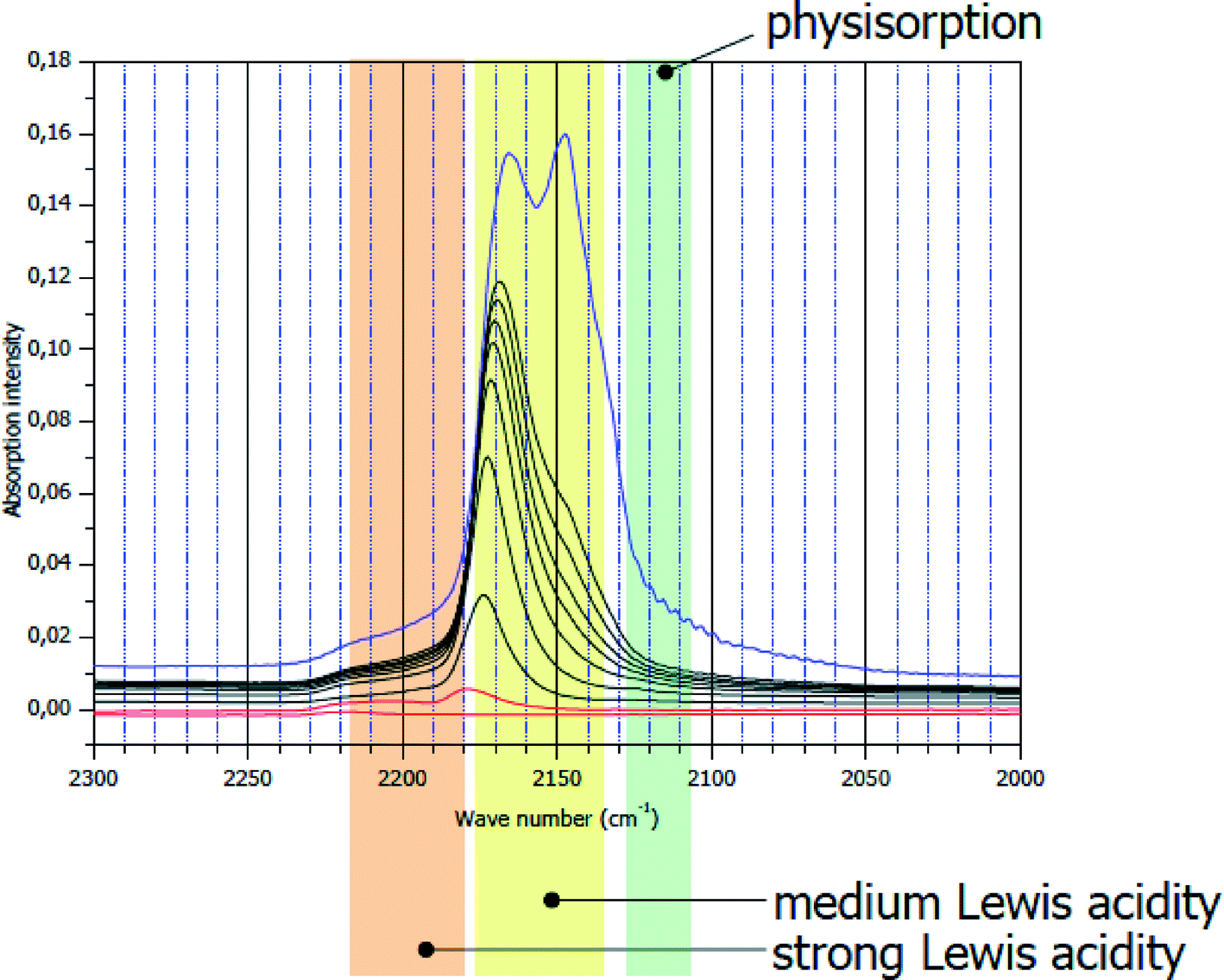 | ||
| Fig. 2 CO IR difference spectra on crystalline β-AlF3. ν(CO) = 2150 cm−1: physisorbed CO; ν(CO) = 2175 cm−1: medium strong Lewis acid sites. | ||
The bands at ν(CO) of ca. 2150 cm−1 are characteristic of weakly (physisorbed) CO, whereas those at ν(CO) of about 2175 cm−1 represent medium strong Lewis acid sites. There is a very weak shoulder appearing at ν(CO) = 2240–2220 cm−1 which can be explained by very strong Lewis acid sites.32
The same CO IR spectra of sol–gel derived HS-AlF3 are significantly different (Fig. 3). Besides the bands at ν(CO) = 2150 cm−1 and ν(CO) = 2175 cm−1, bands at ν(CO) = 2240–2220 cm−1 are the dominating feature of this sample indicating the presence of very strong Lewis acid sites.32 Note that, to the best of the author's knowledge, there is no solid Lewis acid described in the literature exhibiting such strong surface sites.
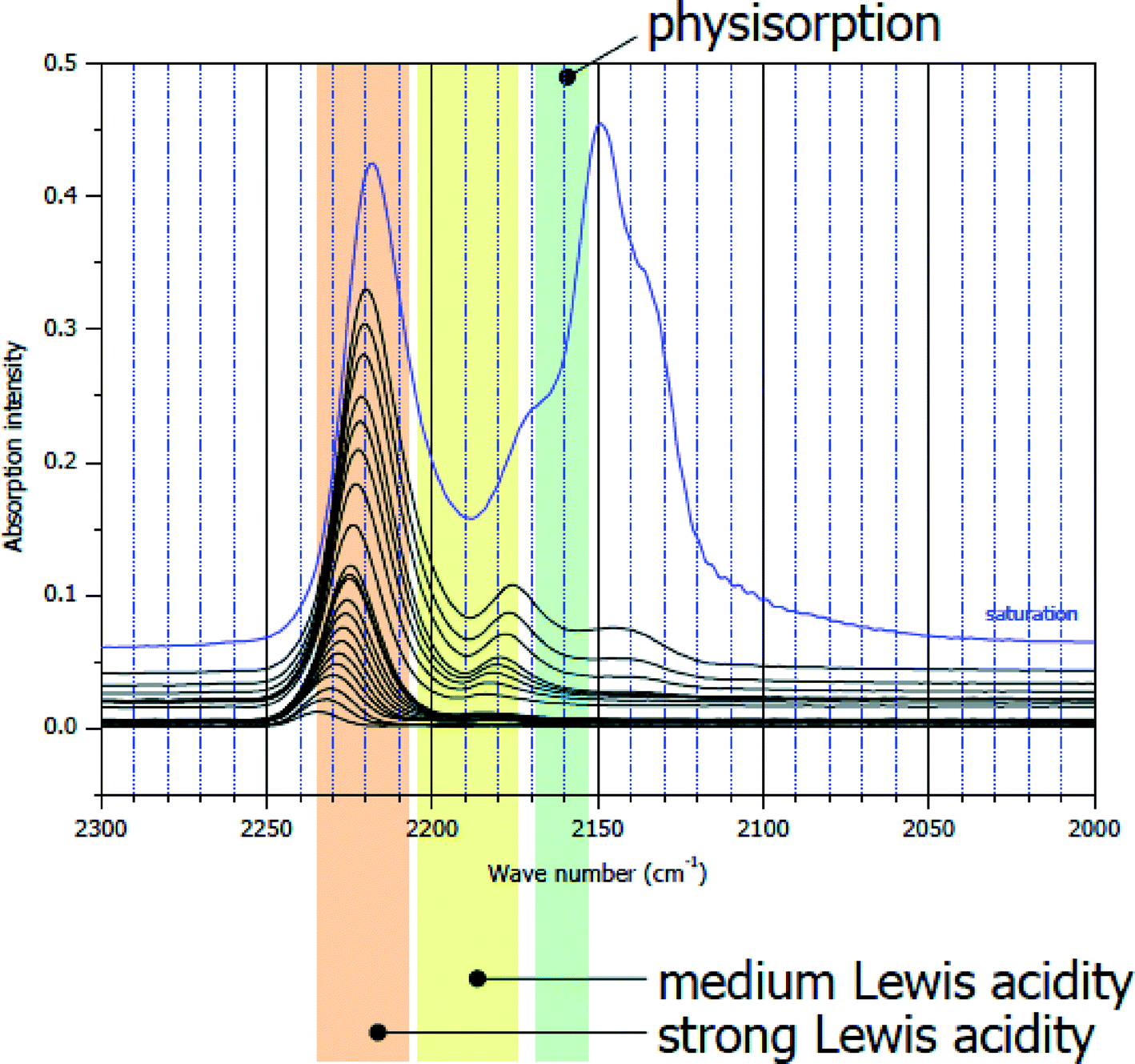 | ||
| Fig. 3 CO IR difference spectra on sol–gel derived HS-AlF3. ν(CO) = 2150 cm−1–physisorbed CO; ν(CO) = 2175 cm−1–medium strong Lewis acid sites; ν(CO) = 2240–2220 cm−1 – very strong Lewis acid sites. | ||
A rationalization of these big differences can be provided based on DFT calculation performed by N. M. Harrison et al.,33 who calculated relaxed surface structures of different crystalline AlF3 phases and predicted the presence of under-coordinated surface Al-sites which are accessible for reactant molecules. For both crystalline β-AlF3 (ref. 34) and α-AlF3 (ref. 35), F-terminated relaxed surfaces are displayed in Fig. 4 and 5, respectively.
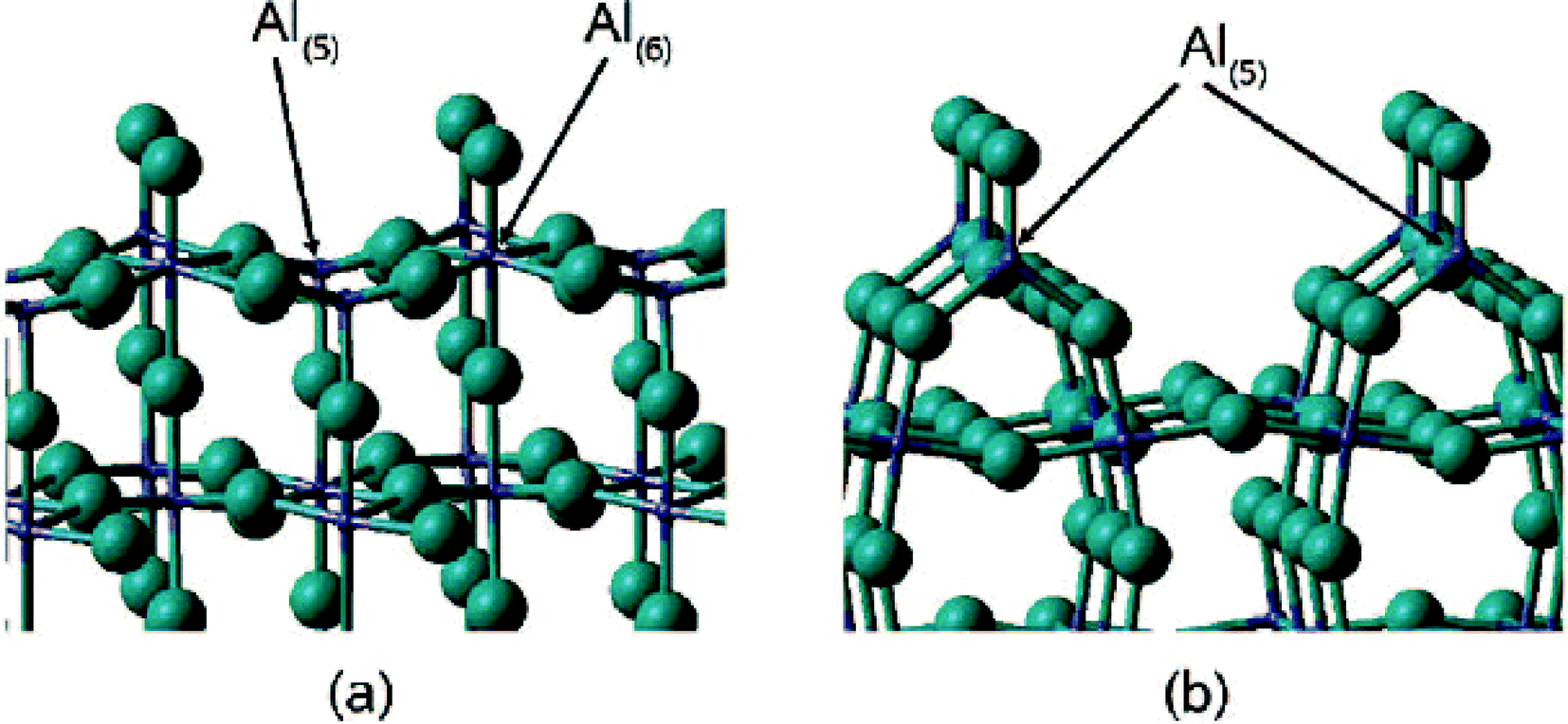 | ||
| Fig. 4 The T1 (a) and T6 (b) termination of the relaxed β-AlF3 (ref. 100) surface. The Al ions are shown in purple and the F ions in green. | ||
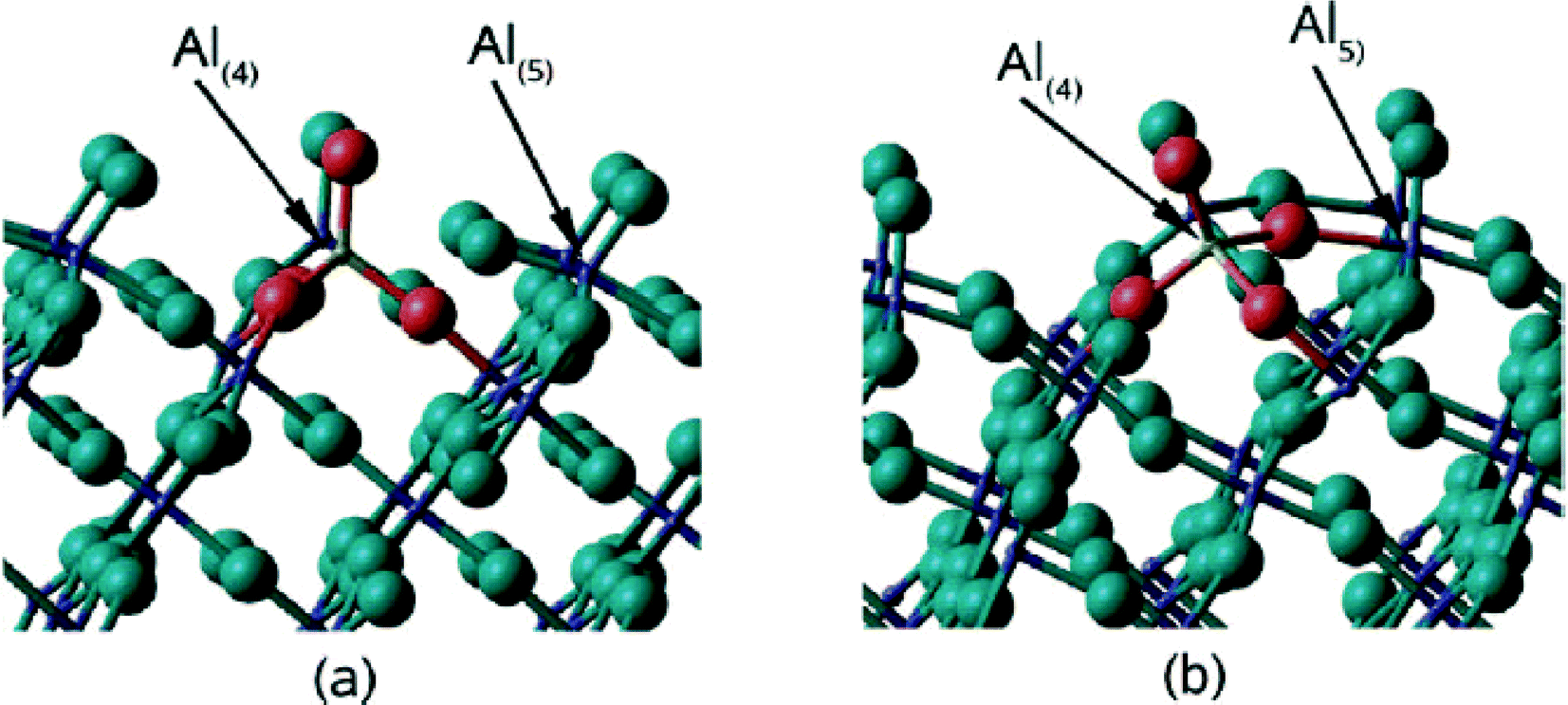 | ||
| Fig. 5 The structures of (a) the (0112) (1 × 1) termination and (b) the (0112) (√2 × √2) termination. The F ions are presented by large spheres and the Al ions by small spheres. | ||
It is evident that surfaces of both phases exhibit under-coordinated sites; thus, both should act as solid Lewis acids.36 However, because of the usually very low surface areas of these crystalline phases, just a very limited number of these surface sites are available in catalytic reactions.
Independently, Chupas et al.37 calculated two thermodynamically relaxed surfaces of α-AlF3 clusters, one formed by 1800 and the other constituted by 2400 AlF3 molecules. The major outcome was that at the surfaces of these clusters six-, five-, as well as four-fold coordinate Al sites are present. However, the percentage of five- and four-fold coordinate sites increased with a smaller cluster size! This is in full agreement with the general prediction from the surface effect of nano-science.
This effect is not restricted to AlF3. Also MgF2, which as a crystalline material has to be considered as a neutral compound,38–42 exhibits weak Lewis acid sites as evidenced by CO–IR investigations.43–45 This holds for other metal fluorides obtained via the fluorolytic sol–gel approach as well.46 Thus, ZnF2 (ref. 26) and FeF3 (ref. 27) are further examples for this new class of catalysts.
In conclusion, via the fluorolytic sol–gel synthesis, nanoscopic metal fluorides with large surface areas can be obtained exhibiting a huge number of co-ordinatively un-saturated Lewis acidic surface sites. The catalytic properties of these compounds will be described below.
| AlCl3 + (3–x) CCl3F → AlClxF3–x + (3–x)CCl4 | (4) |
Just by reacting dry AlCl3 with a suitable chlorofluorocarbon like CCl3F, an AlF3-phase is formed that under no circumstances can be obtained chloride free and was therefore designated as aluminium chloride fluoride (ACF). The latter was originally discovered by Krespan and Petrov47 who intensively investigated their catalytic potential in Lewis acid catalyzed reactions.48–54 The real structural composition as well as solid state properties were later comprehensively investigated by us.55,56 It was evidently shown that ACF can be described as a fluorine rich aluminium chloride fluoride with a variable but narrow chlorine stoichiometry corresponding to the general formula AlClxF3−x with x ≈0.05–0.25. Even a very similar aluminium bromide fluoride (ABF) has been obtained following the same general synthesis route that exhibits similar properties as ACF. The main structural features are presented in Scheme 2. The different connectivity of fluorine, on the one hand, and chloride and bromide, respectively, on the other hand, causes a highly distorted structure resulting in comparatively large surface areas (>100 m2 g−1) and co-ordinatively unsaturated surface Al-sites which evidently represent the strong Lewis acid sites.
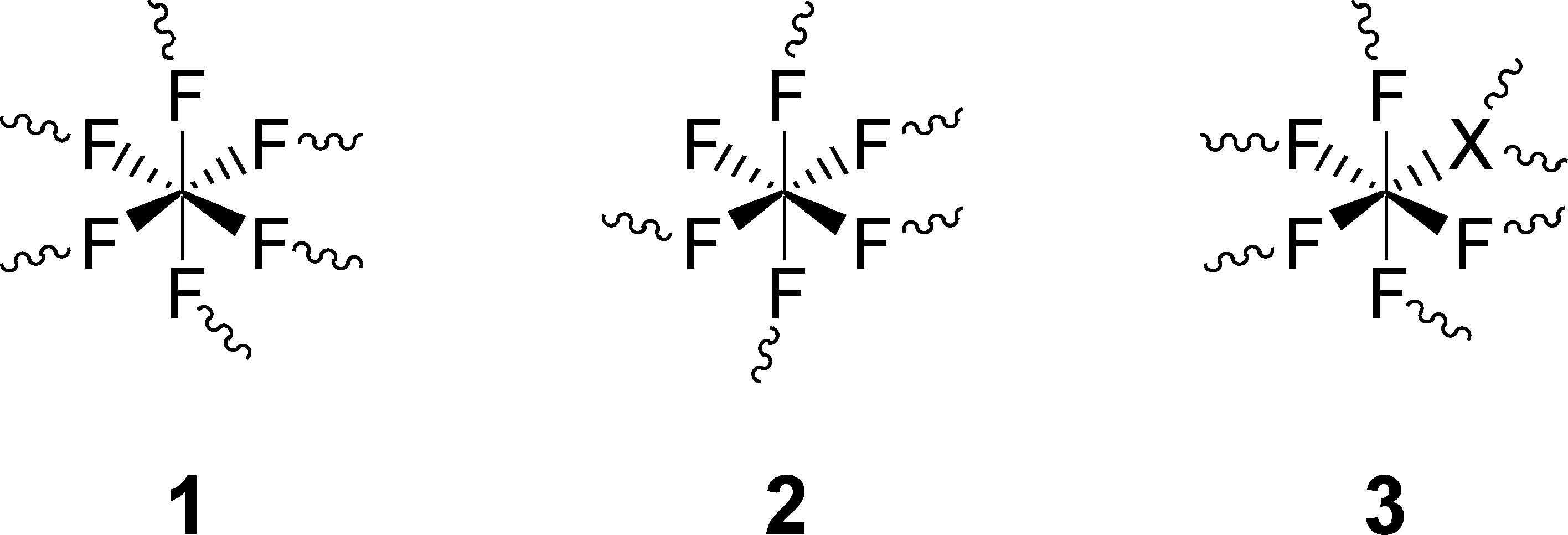 | ||
| Scheme 2 Structural octahedral units proposed in ABF and ACF (X = Cl, Br). The sinuous lines indicate the bond to the next aluminum atom. Reproduced from ref. 55 with permission from Wiley. | ||
ACF and ABF are extremely moisture sensitive compounds that lose all their novel Lewis acidic potential irreversibly in a few seconds of contact with air. Thus handling them in catalytic reactions demands working under extremely dry conditions, and therefore, their practical applications are limited because of this. A comprehensive presentation type setting of the main characteristics of ACF and ABF is reviewed in ref. 57.
2.2. Binary metal hydroxide fluorides
Metal hydroxide fluorides are scarce, but they do exist. For instance, fluorine rich phases of aluminium hydroxide fluoride in a pyrochlore structure are known and comprehensively investigated.58,59 Further examples are apatite fluoride, Ca5(PO4)3F,60 and zharchikite, AlF(OH)2.61 These are exclusively crystalline phases which did not show any catalytic activity in reactions where,e.g., crystalline phases of AlF3, CrF3 or FeF3, respectively, are catalytically active.62–65In the course of intensive mechanistic investigations that are reviewed in ref. 9 and 10, the presence of intermediately formed metal alkoxide fluorides was evidenced by both MAS-NMR22,66–72 and single crystal structure determination.73 This will be exemplarily shown for the aluminium fluoride system. 19F and 27Al MAS NMR investigations clearly show a systematic change in the chemical shift values with increasing HF to Al ratio, thus indicating the presence of mixed AlF6−x(OR)x octahedral units in the intermediately formed molecular clusters whereby the x-value decreases as the HF to Al stoichiometry increases (Fig. 6). A similar dependency was established for the MgF2-system.74
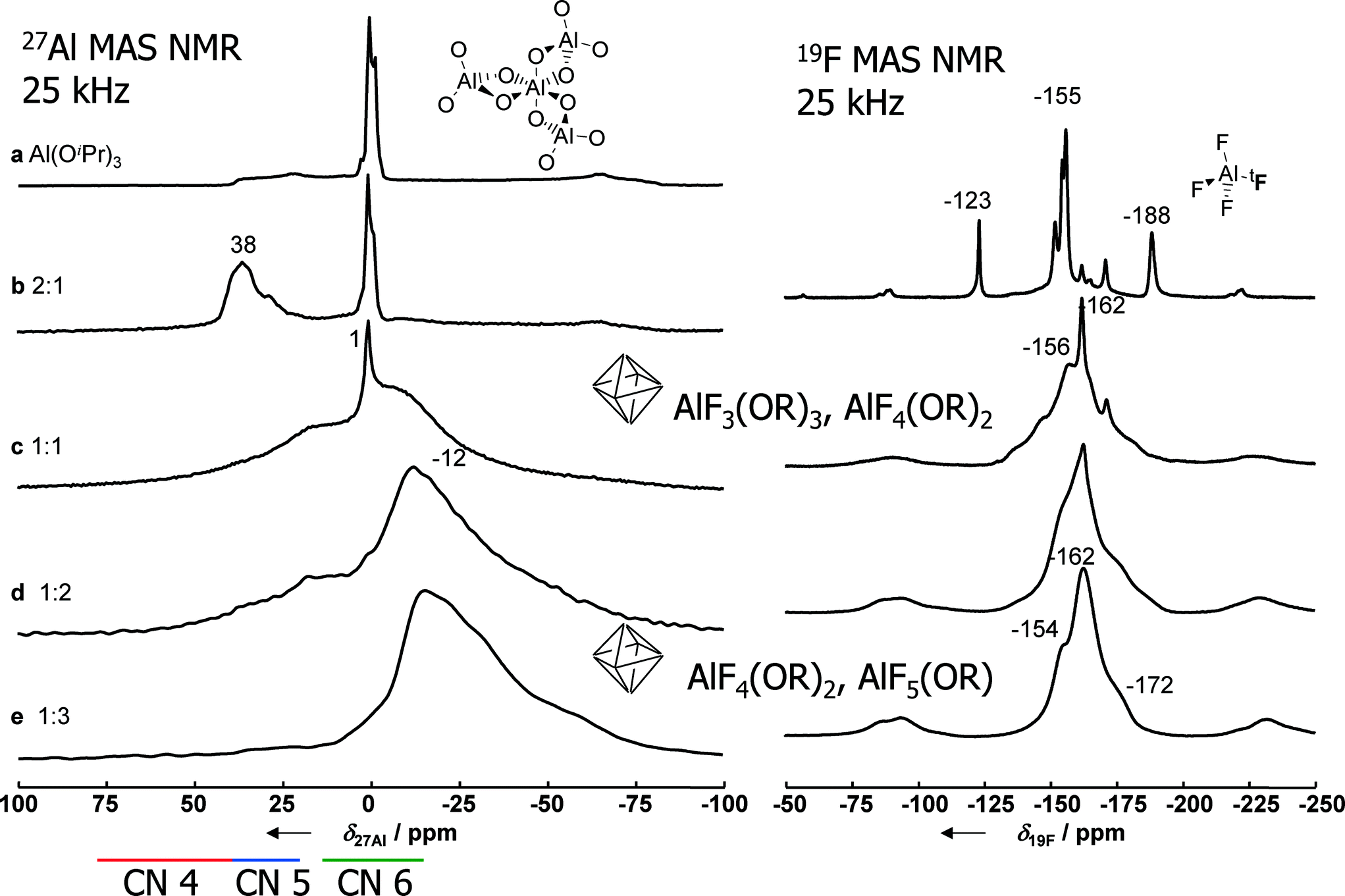 | ||
| Fig. 6
27Al and 19F MAS NMR spectra of Al(OiPr)3 and aluminium isopropoxide solids prepared with different molar ratios of Al:F as given in the figure (νrot = 25 kHz, B0 = 9.4 T). | ||
These NMR-based results were later confirmed by several few single crystals that could occasionally be isolated from synthesis batches, thus giving clear evidence for mixed F and O coordinate octahedral metal sites formed in the course of the fluorolytic sol–gel synthesis (cf.Fig. 7).
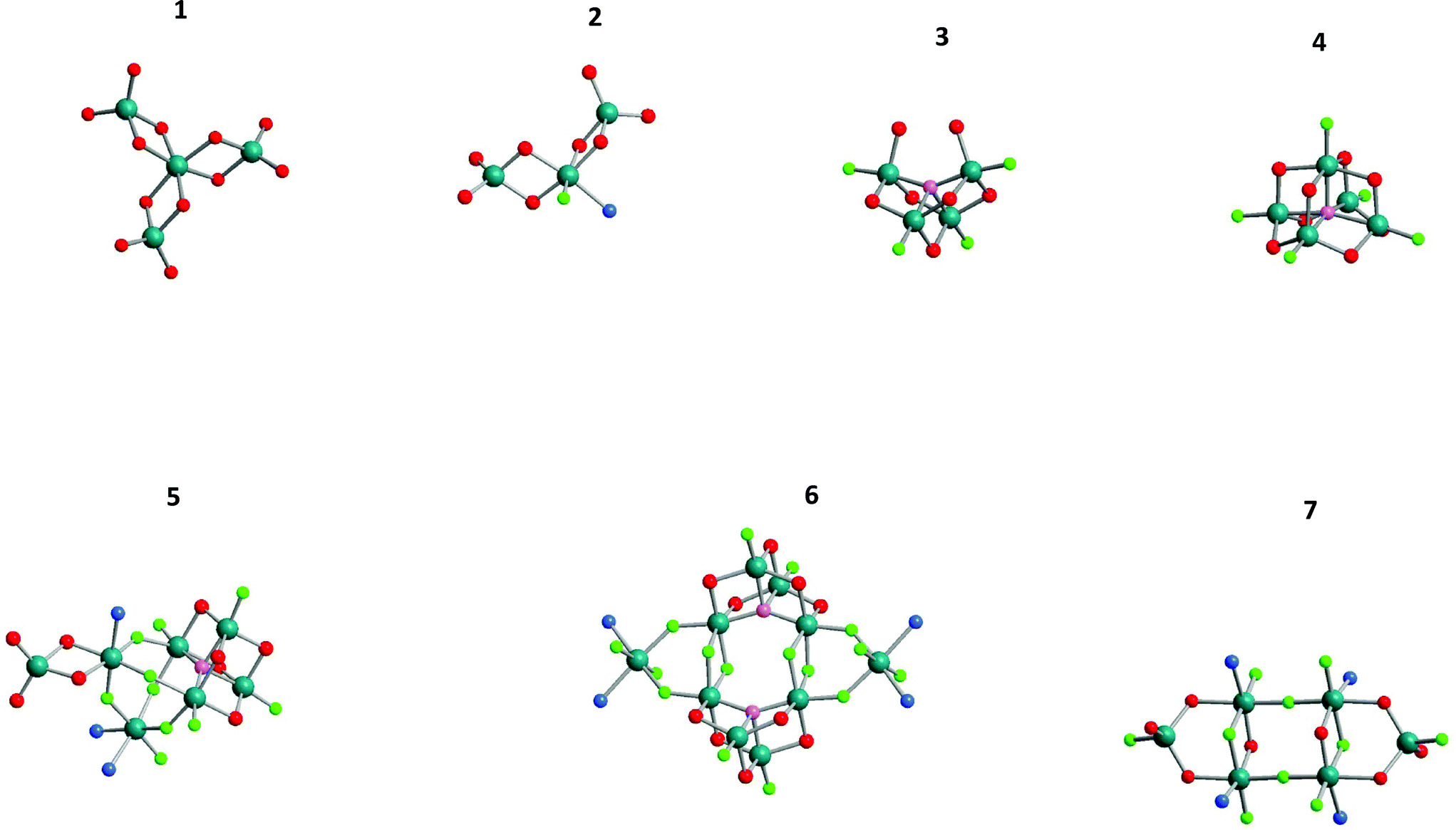 | ||
| Fig. 7 Structures of aluminium alkoxide fluorides obtained from sols of varying aluminium isopropoxide to HF ratios sorted according to their Al to F ratios within the structure. 1: Al4(OiPr)12; 2: Al3F(OiPr)8·D (D = Py, DMSO), Al:F = 3:1; 3: Al4F4(μ4O)(OiPr)5(H(iPrO)2), Al:F = 1:1; 4: Al5F5(μ5O)(OiPr)8, Al:F = 1:1; 5: Al7F10(μ4O)(OiPr)9·3Py, Al:F = 1:1.43, 6: Al10F16(μ4O)2(OiPr)10·4Py, Al:F = 1:1.6, 7: Al6F10(OiPr)8·4Py, Al:F = 1:1.67. | ||
Based on these findings, we wondered whether it might be possible to obtain metal alkoxide fluorides and hydroxide fluorides starting from such intermediately formed metal alkoxide fluorides just by introducing water into the reaction system that may transform M–OR into M–OH units. In fact, this hypothesis was worked out to a very elegant synthesis route towards metal hydroxide fluorides. Thus, the fluorolytic sol–gel synthesis is not only limited to metal fluorides but by modification also gives an excellent access to hydroxide fluorides following the reaction as displayed in eqn (5):
| M(OR)n + (n − m)HF + mHOX → MFn−m(OX)m | (5) |
Group X may be any organic group, thus organofluorides may be obtained which are of interest for the fabrication of inorganic–organic composite materials by introducing nano-metal fluorides into organic polymers.75 However, if group X is H (the reactant molecule is water that competes with HF), hydroxide fluorides will be formed.
The formation of real hydroxide fluorides (not mixtures of hydroxides and fluorides!) can doubtlessly be evidenced by MAS NMR, as is shown in Fig. 8 for the MgF2−x(OH)x-system.
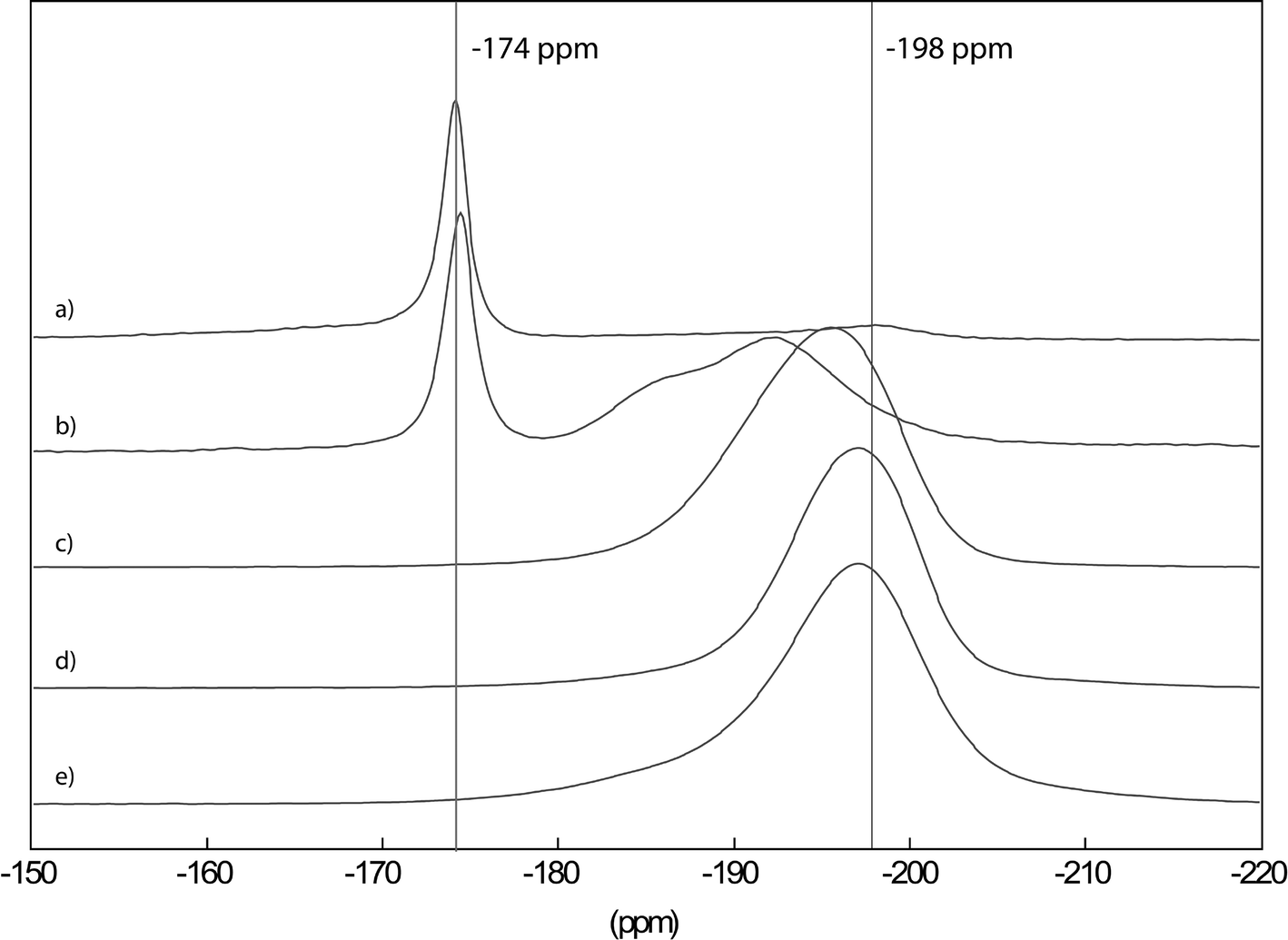 | ||
| Fig. 8 19F MAS NMR spectra of Mg(OMe)2−xFx with x = 0.3 (a), 0.4 (b), 1 (c), 1.5 (d) and 1.75 (e), recorded in a 2.5 mm probe at 25 kHz. Reproduced from ref. 74 with permission from The Royal Society of Chemistry. | ||
If for these samples a post calcination step is applied, the hydroxide fluorides can be further transformed into oxide fluorides according to eqn (6):20,24
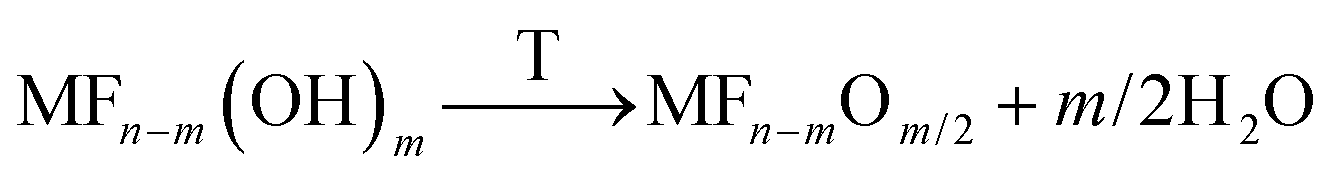 | (6) |
Surface areas up to 600 m2 g−1 were measured for these samples. Another interesting fact is the significantly increased thermal stability of metal oxide fluorides. Whereas the pure metal fluorides undergo crystallization at comparatively low temperatures (for comparison MgF2 slowly starts to crystallize above 280 °C, and AlF3 starts to crystallize above ca. 500 °C) resulting in an increased structural order and, consequently, decreased surface areas accompanied by loss of reactivity, the metal oxide fluorides are stable even up to temperatures of 700 °C. This property makes these compounds interesting candidates to be used as functional supports.
It is worth noting that this approach can be applied for multi-metal oxide fluorides as well.
In conclusion, via the fluorolytic sol–gel synthesis, nanoscopic metal fluorides with large surface areas can be obtained exhibiting a huge number of co-ordinatively un-saturated Lewis acidic surface sites. The catalytic properties of these compounds will be described below.
2.3. Partly hydroxylated metal fluorides
A closely related but distinct different synthesis approach is the competitive fluorolysis/hydrolysis reaction. This means that hydrogen fluoride will be used in almost stoichiometric portions as in the case of the original fluorolytic sol–gel synthesis (cf. eqn (3)) but small amounts of water will be added to the reaction system at the same time thus acting as a competitor for HF as shown in eqn (7):| M(OR)n + (n)HF + mH2O → MFn−x(OR)x + nROH + xHF + m − xH2O | (7) |
It was found that fluorination strongly dominates over hydrolysis, e.g. for the magnesium fluoride system a more than 10 times faster reaction with HF as compared to water was measured.74,75 Also thermodynamic considerations allow us to expect a privileged formation of the fluoride for many metals. Hence, just a very little amount of hydroxyl groups becomes incorporated when water is added to the reaction mixture as a competitor for HF, meaning the x-value may lie in the region of some few thousands of a percent only.76 Thus, the products formed this way still are nearly pure metal fluorides as can be seen from the 19F magic angle spinning (MAS) NMR spectra of, e.g., different MgF2-phases prepared at constant HF/Mg ratios of 2 but with additional varying amounts of water (see Fig. 9).
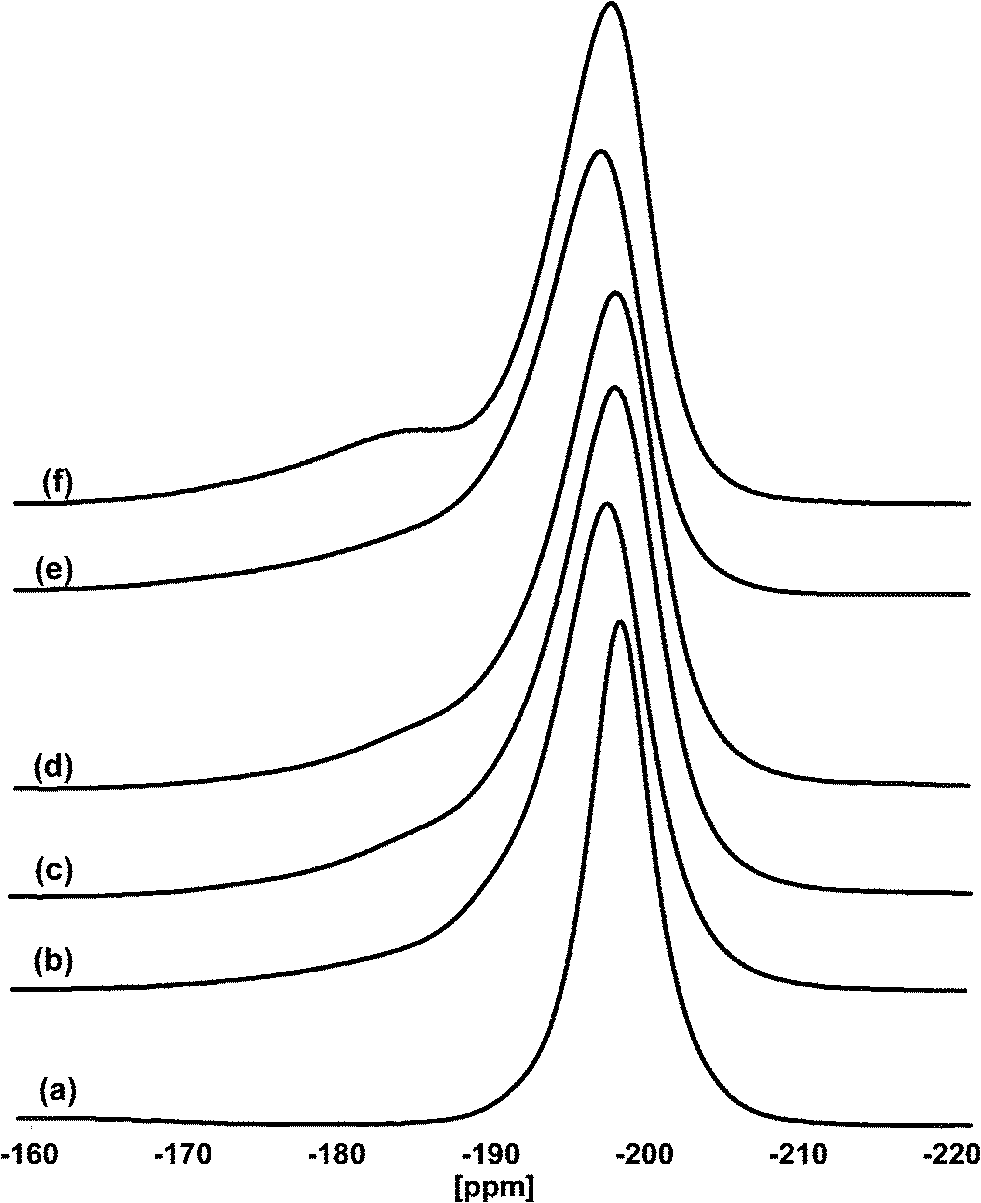 | ||
| Fig. 9 19F MAS NMR spectra of a) MgF2-crystalline, b) MgF2–100%HF, c) MgF2–87%HF, d) MgF2–71%HF, e) MgF2–57%HF, and f) MgF2–40%HF. Reproduced from ref. 44 with permission from Wiley. | ||
Therefore we may define them more appropriately as “partially hydroxylated” metal fluorides rather than as hydroxide fluorides. Just by variation of the water but maintaining the stoichiometric HF to metal ratio the extent of partial hydrolysis can be tuned at a very low OH concentration.
Thus, this general approach was successfully applied for a one-pot sol–gel synthesis of different partially hydroxylated magnesium fluorides.44 The MgF2−x(OH)x samples obtained this way exhibit almost the same bulk structure as MgF2, the absence of any carbon indicating a complete solvolysis process. 19F MAS NMR, XRD, TEM and XPS investigations show for samples obtained from aqueous HF with increasing water concentration an increase of hydroxyl groups which are mainly on the surface whereas the bulk composition remains almost unchanged. Thus, it seems that shell-like nanoparticles of MgF2−x(OH)x phases with an inner core (bulk) of mainly pure MgF2 are formed. As long as the overall OH-stoichiometry is <0.1, as a very big surprise, these hydroxyl groups are Brønsted acidic in nature as has been proven by IR difference spectra of adsorbed CO on MgF2-phases.44 The observed ν(CO) stretching vibrations at 2180–2200 cm−1 both dominant for MgF2–100%HF and MgF2–87%HF can be assigned to Lewis acidic sites of medium strength.32 The bands at 2165–2150 cm−1 can be assigned to CO coordinated to both weak Lewis and Brønsted acidic sites in this region. Therefore, lutidine as a more sensitive probe molecule for distinction between Lewis and Brønsted sites has been used. Comparing these results with those obtained from CO-adsorption measurements allows us to conclude that MgF2–100%HF and MgF2–87%HF samples exhibit exclusively Lewis centres mainly represented by 5-fold coordinate Mg surface sites whereas the samples MgF2–71%HF, MgF2–57%HF, and MgF2–40%HF are believed to carry both Lewis and Brønsted sites (Fig. 10).
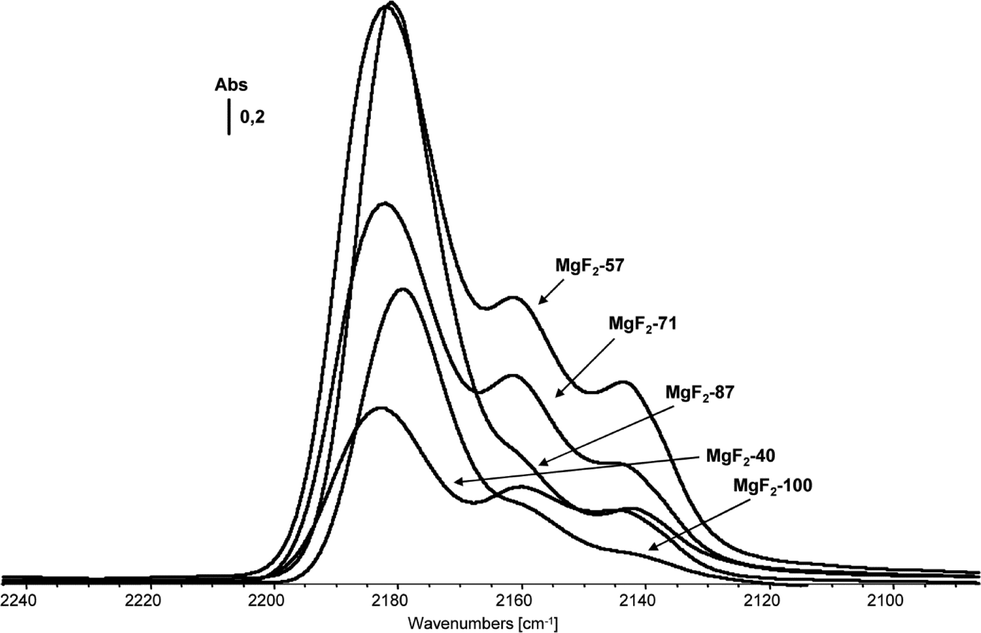 | ||
| Fig. 10 CO IR difference spectra of magnesium fluoride phases prepared using HF of different concentrations (indicated by the numbers). Note that for all samples a constant stoichiometric ratio HF/Mg = 2 was maintained. Reproduced from ref. 44 with permission from Wiley. | ||
Thus, bulk-surface techniques as well as catalytic performance with these materials confirm this unusual result, which was also found for the AlF3−x(OH)x system.77 Thus, high resolution MAS 1H NMR investigations on MgF2−x(OH)x) samples obtained from 71% aqueous HF showed an isotropic chemical shift of δ = 4.8 ppm, characteristic of remaining water on the surface and/or bulk,78 and a central signal at δ = 3.3 ppm. The latter was never observed before for MgO79 or magnesium oxide fluoride (unpublished work). Our DFT calculations showed that a 1H isotropical shift of δ = 3.3 ppm is representative for acidic OH-groups. Basic groups, in full agreement with experimental data, were calculated to be in the range of 0 < δ < −2 ppm. Hence, 1H MAS NMR strongly argues for Brønsted acidic Mg–OH groups.
The unexpected Brønsted acid character of the M–OH groups on these magnesium or aluminium fluoride surfaces, respectively, is a result of the combination of different factors as shown in Scheme 3. Mainly because of the high electronegativity the fluorine atoms induce additional electron withdrawal from Mg atoms thus strengthening the polarity of the O–H bonds (Scheme 3a). In addition, the presence of <6 coordinate magnesium surface sites further facilitates withdrawing of electron density from the O–H bond (Scheme 3b). Moreover, simultaneous fluorolysis and hydrolysis reactions facilitate formation of bridging OH-groups at the surface. Such groups are known to be stronger Brønsted acid sites than non-bridging ones (Scheme 3c).80 Even the formation of hydrogen bonds between O of an OH-group and a neighbouring F atom has to be taken into account (Scheme 3d) as was demonstrated for adsorbed water on MgF2.43
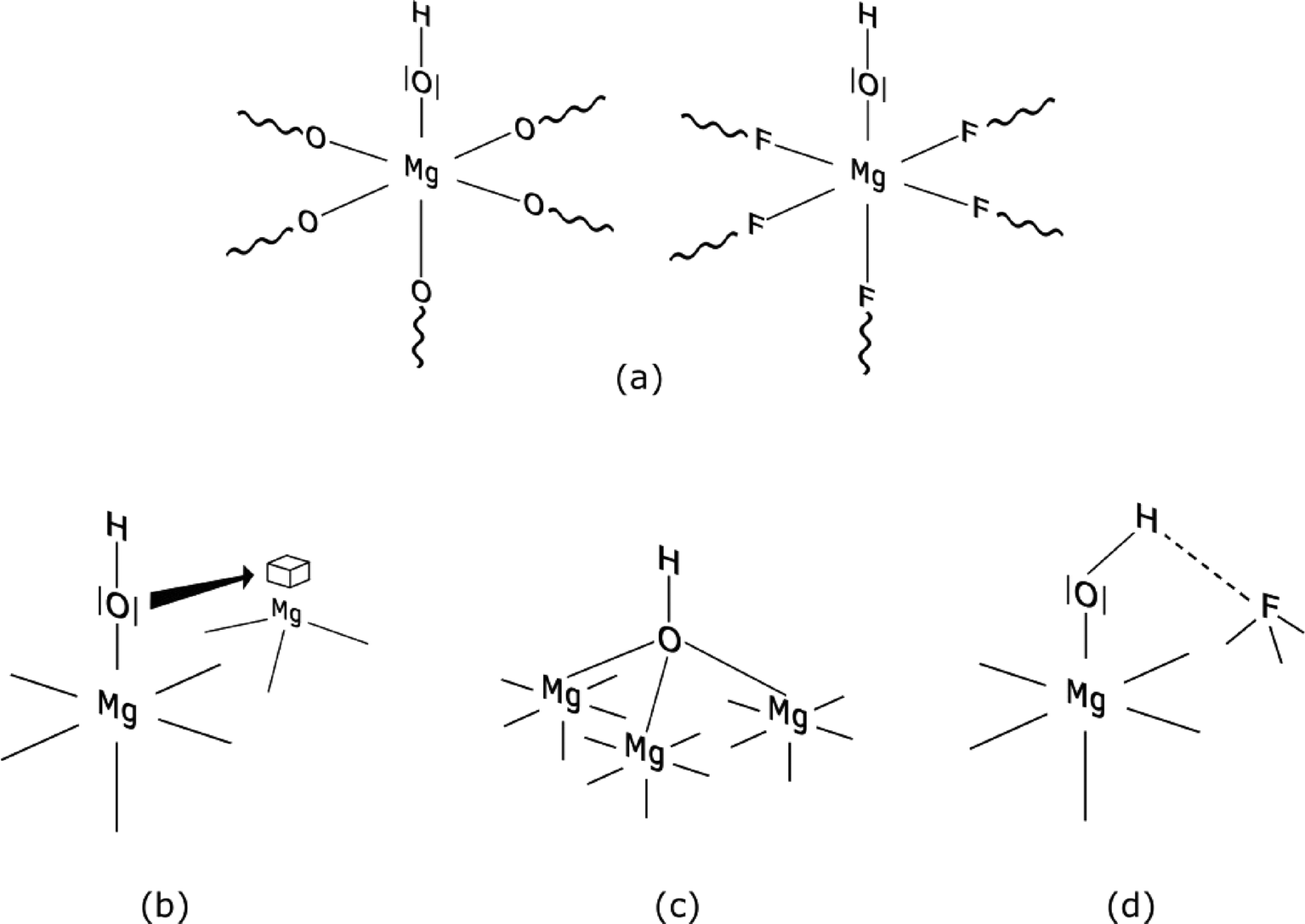 | ||
| Scheme 3 Graphical illustration of possible topological situations on the MgF2−x(OH)x surface that could explain the Brønsted acidic nature of the OH-group; (a) OH-group on a MgO or MgF2 surface, (b) interaction of an OH-group with under-coordinated magnesium, (c) branched Mg–OH group and (d) hydrogen bonding between the H of the OH-group and a F atom. Reproduced from ref. 45 with permission from Elsevier. | ||
However, all these considerations hold only as long as the OH content is low (x<0.1 for MgF2−x(OH)x). For example, by using 20 wt% aqueous HF (Mg:HF ratio is 1:2), the hydrolysis becomes significant in comparison to the fluorolysis and consequently Mg–OH groups turn from Brønsted acidic into basic sites.45 In such samples OH-groups are present not only at the surface but also are involved in the bulk structure, thus effects described in Scheme 3 become less relevant and typical OH-groups (basic) like in MgO are formed.
For hydroxylated magnesium fluoride, the range from 100 wt% to 40 wt% aqueous HF at a constant HF to Mg ratio of 2 results in Brønsted acidic samples of different strength, and for hydroxylated aluminium fluoride even higher water concentrations can be applied. These samples possess very high surface areas ranging from 200 to 450 m2 g−1. Moreover, adsorption of CO reveals the presence of five- and fourfold coordinate Mg2+ sites at the surface. This additional finding suggests that sol–gel prepared MgF2−x(OH)x samples are bi-acidic materials. The crystallisation of these samples starts at around 220 °C and limits the catalytic application to 200 °C, which is sufficient for the fine chemical production as it will be shown below.
3. Metal fluorides and hydroxide fluorides in catalytic reactions
As briefly outlined in section 2, these new nanoscopic metal fluorides are characterized by their unique acidic properties, which can be tuned from purely Lewis to bi-functional, that is Lewis and Brønsted. Moreover, due to this tunable acid character in combination with the large surface area of these new materials, they are also interesting materials as co-catalysts and/or supports for other functionalities, e.g. novel metal catalysts. Hence, the following sections will be sorted according to these different acidic properties resulting in different classes of catalytic reactions.3.1. Lewis acid catalysed reactions
The isomerization of 1,2-dibromo-hexafluoropropane into 2,2-dibromo-hexafluoropropane (eqn (8)) can be catalysed effectively only in the presence of very strong Lewis acids.| CF2Br–CFBr–CF3 → CF3–CBr2–CF3 | (8) |
Thus, with SbF5 as a catalyst >90% conversion can be obtained at around 80 °C. Surprisingly enough, both ACF and HS-AlF3 give comparable conversions (>90%) even at room temperature.57 Another example for this exciting high Lewis acidity is the dehydrofluorination reaction of 3-chloro-1,1,1,3-tetrafluorobutane (eqn (9)).81
| CF3–CH2–CFCl–CH3 → CF3–CH = CCl–CH3 + HF | (9) |
With HS-AlF3 a conversion of the starting compound >99% with a selectivity towards the dehydrofluorination product of 100% was obtained at 200 °C, which has never been achieved with any other catalyst.82 Interestingly enough, by employing nanoscopic, high surface area BaF2 as a solid catalyst, exclusively the hydrodechlorination reaction (eqn (10))
| CF3–CH2–CFCl–CH3 → CF3–CH = CF–CH3 + HCl | (10) |
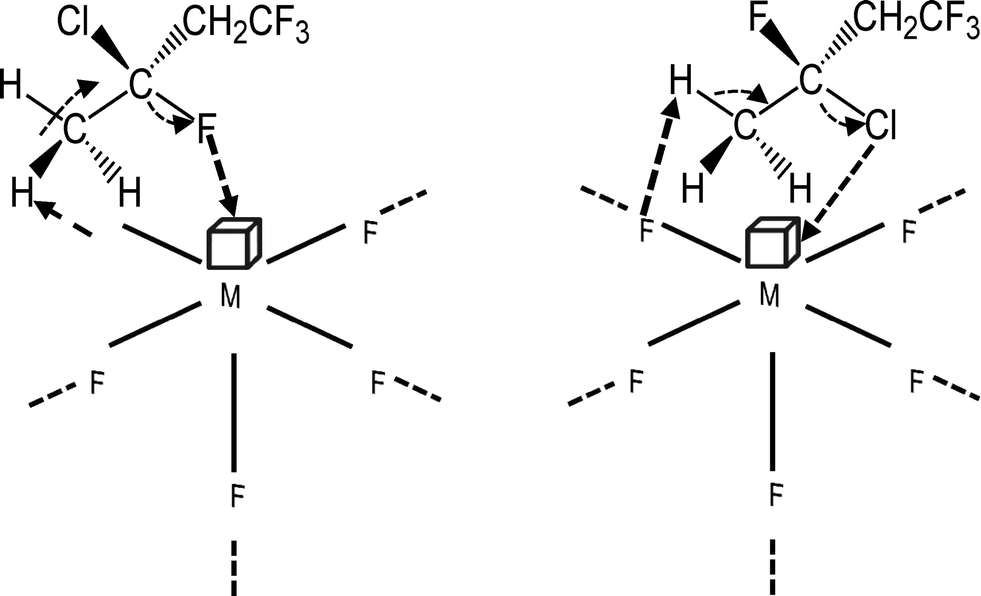 | ||
| Scheme 4 Proposed catalytic mechanism for the dehydrogenation and dehydrochlorination of 3-chloro-1,1,1,3-tetrafluorobutane. | ||
Nanoscopic MgF2, CaF2, and SrF2 gave also appreciable conversions between 83 and 96%, however with drastically different selectivities. MgF2, in crystalline form a rather “neutral” compound,38 mainly results in the dehydrofluorination product (S ≈ 90%), whereas CaF2 and SrF2 act increasingly as dehydrochlorination catalysts (50% and 70%, respectively). The behaviour of nano-MgF2 is a further good example of how catalytic activity of usually less active materials can be altered by the synthesis and the resulting high surface area. Thus it was shown by FT-IR investigations using different probe molecules that MgF2 in fact carries a considerable part of under-coordinated surface sites of medium strong Lewis acidity.44,45 This has been recently also proven by periodic calculations at the B3LYP level in combination with CO adsorption measurements which evidence the presence of five-fold, under-coordinated Mg-surface sites that are strong enough to adsorb CO.82
Whereas dehydrohalogenation reactions are thermodynamically forced by the formation of stable olefins, another type of C–F bond activation has gained a tremendous attention over the past 10 years. This means that catalytic transformation of C–F into C–H bonds is a challenging task but can provide access to otherwise inaccessible fluorinated compounds and building blocks.83,84
Deuterated compounds play a fundamental role in isotope labelling reactions in pharmaceutical, medical and spectroscopic applications.85–89 Besides the novel Shilov or Periana type precious metal catalysts90,91 and others, also solid Lewis acids like zeolites and dehydroxylated alumina are described to catalyse to some extent H/D-exchange reactions above 150 °C92–101 whereas molecular Lewis acids like AlBr3 or MoCl5 catalyse H/D exchange between arenes only.102,103 Thus, both very strong Lewis acids ACF and HS-AlF3 were tested for their catalytic activity in H/D exchange reactions between aliphatic and aromatic C–D/H bonds.104 Expectedly, H/D exchange between C6D6 and C6H12 after 22 h at 110 °C in the presence of these aluminium fluorides gave conversions between 35 and 65%, however, using instead C6D12 as deuterium source and C6H6 as proton source for H/D exchange showed almost conversions even in the range of ca. 80% at just 40 °C to 70 °C. This method is not restricted to cyclohexane. Cyclopentane, cyclooctane, and hexamethylethane were also deuterated at 40 °C with 42%, 9%, and 18% deuterium incorporation into the alkanes. A higher conversion was achieved after prolonged reaction times, that is, 45%, 64%, and 21% deuterium incorporation were obtained after 72 h at 40 °C, respectively.
Mechanistically, it was presumed that in an initial step the C–H/D bonds in organic substrates RH interact via the σ-bond with a Lewis acidic surface site at the ACF or HS-AlF3 (Scheme 5).105
 | ||
| Scheme 5 C–H-activation on bifunctional Lewis-acidic HS-AlF3 or ACF surface sites. R = alkyl, aryl. Reproduced from ref. 104 with permission from Wiley. | ||
It was shown that both aluminium fluoride phases exhibit a huge number of under-coordinated Al surface sites, which define the Lewis acidic properties of these catalysts.106 After coordination, the C–H/D bonds are further activated as a consequence of an interaction of a neighbouring fluoride with the H/D atom, which is presumably positively charged.107,108 This leads to Al bound organyl moieties that are carbanionic in nature. The presence of adjacent F⋯D–R and F⋯H–R sites results in H/D exchange via reversible C–H/C–D bond forming reactions (Scheme 5).
Catalytic C–F bond cleavage under mild reaction conditions is a fundamental challenge in synthetic chemistry.109–120 The conversions can yield otherwise inaccessible fluorinated compounds and building blocks.121–131 Despite some progress that was achieved for the intermolecular activation of highly fluorinated aromatics and olefins, C–F bond cleavage reactions of fluorinated alkanes are less developed. Since silylium or alumylium ions as homogeneous Lewis-acidic catalysts, which are stabilized by weakly coordinating anions, were successfully employed in homogeneously catalyzed reactions,132–141 we wondered whether or not this concept might be applied for heterogeneous catalysts which carry very strong Lewis acidic surface sites. In fact, by 1H MAS NMR spectroscopy, the formation of a silylium-like species at the surface was evidenced142 (cf.Fig. 11).
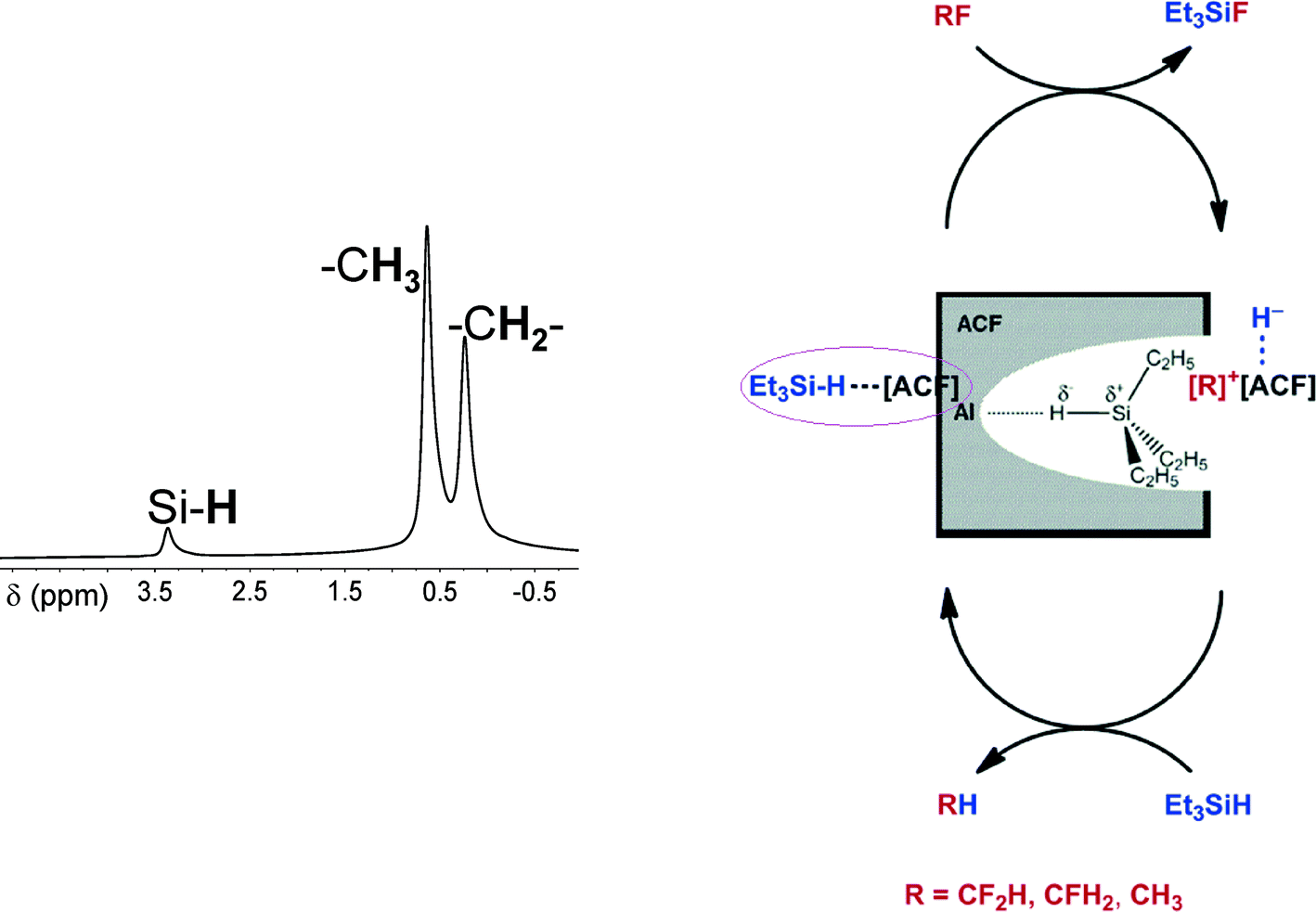 | ||
| Fig. 11 (Left) 1H MAS NMR spectrum which shows the signals for surface-bound Et3SiH (νrot = 10 kHz) with the SiH resonance at δ = 3.45 ppm. For Et3SiH in C6D6 solution, the resonance is observed at δ = 3.85 ppm. (Right side) Schematic representation of ACF⋯H–SiEt3. | ||
On treatment of CH3F, CH2F2, and CHF3 with the aluminium fluoride catalysts in C6D6 in the presence of Et3SiH at room temperature and one atmosphere, vigorous reactions and the evolution of gaseous products were observed (Fig. 12). In all these cases, formation of fully hydrogenated methane was observed for the first time under heterogeneous conditions, at low temperature and in the absence of a precious metal catalyst. Indeed, the formation of the Friedel–Crafts product [D10]diphenylmethane was observed in the C6D6 solution. For CHF3 the [D10]diphenylmethane formation involves two Friedel–Crafts reaction steps and a subsequent hydrodefluorination reaction.
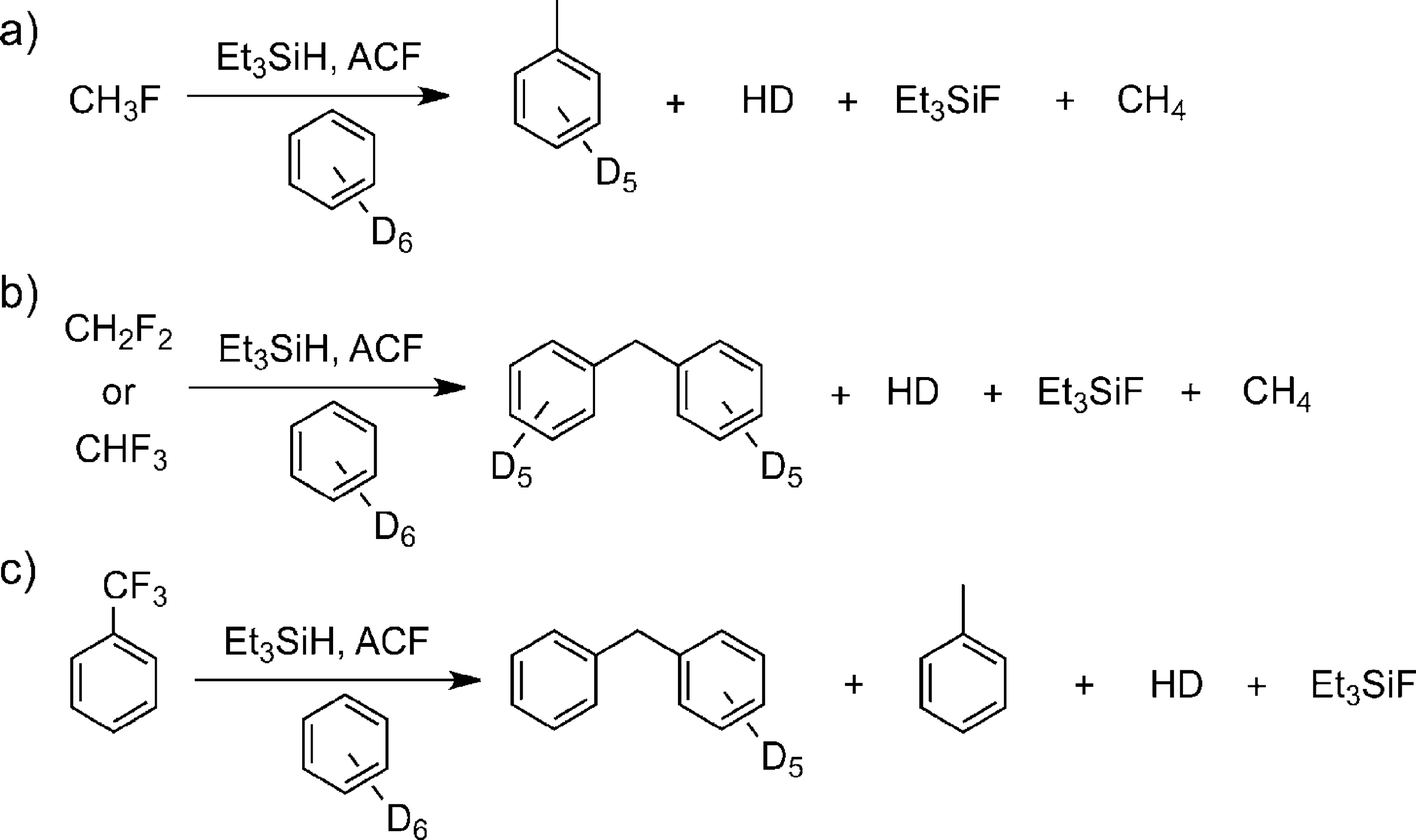 | ||
| Fig. 12 ACF-catalyzed C–F activation reactions of fluoromethanes and trifluorotoluene. Reproduced from ref. 142 with permission from Wiley. | ||
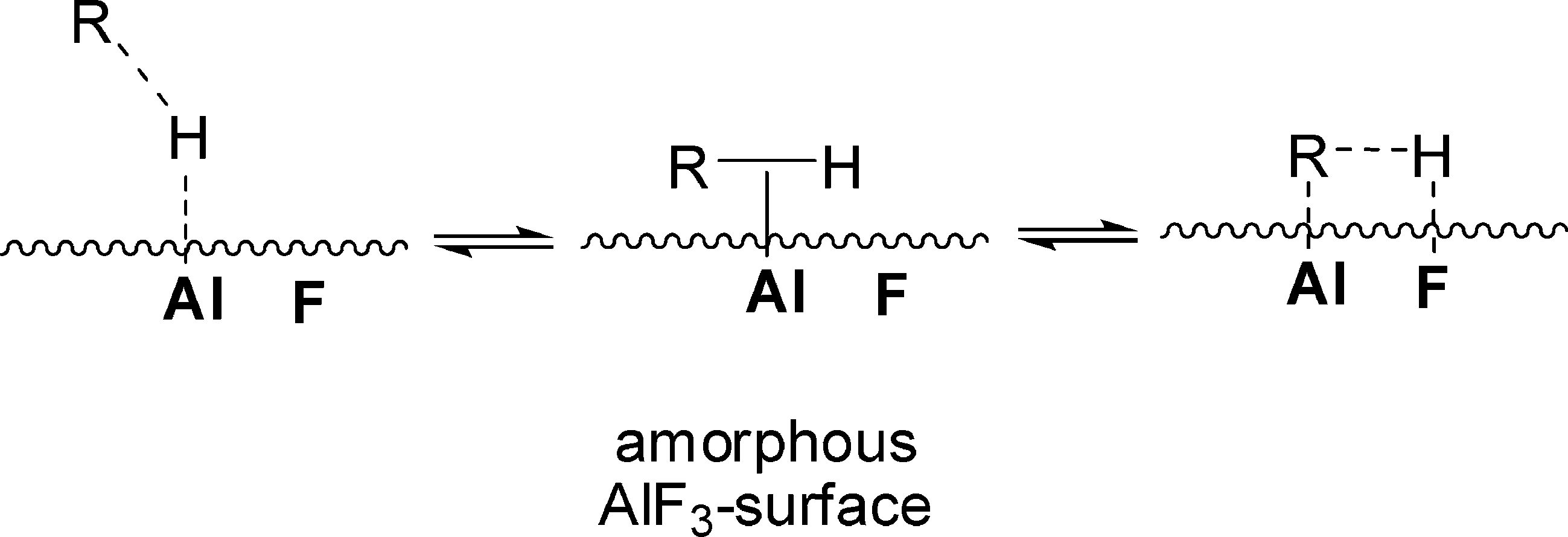
Mechanistically, it was presumed that Et3SiH initially binds at the Lewis-acidic sites of ACF, thus resulting in the surface bound Et3SiH entities ACF⋯H–SiEt3. A polarization of the Si–H bond probably leads to species which have a silylium ion character and are able to cleave a C–F bond to yield Et3SiF and carbenium-like species, which in turn attack the solvent (C6D6). The Wheland intermediates formed this way subsequently react with the remaining hydrogen atom on the ACF to release the Friedel–Crafts product as well as HD. The Lewis-acidic ACF site is thereby regenerated (Fig. 13).
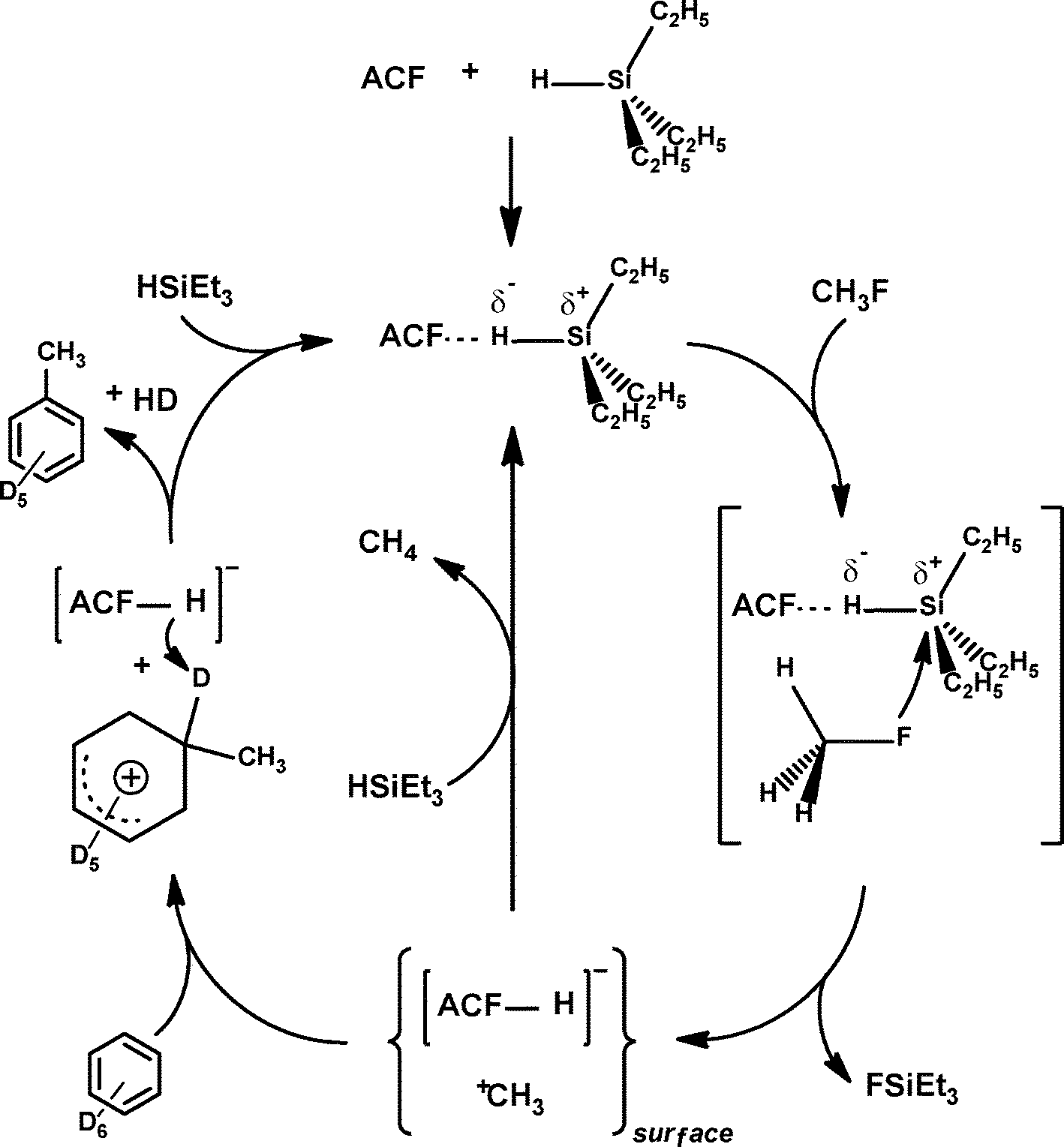 | ||
| Fig. 13 Proposed mechanisms for the ACF catalyzed C–F activation reaction including a Friedel–Crafts reaction with the solvent C6D6via a carbenium-like intermediate (left side) and the ACF-catalyzed hydrodefluorination by a concerted mechanism (right side). Both mechanisms are shown for the substrate CH3F. | ||
However, in an alternative process ACF⋯H–SiEt3 converts the C–F bonds into C–H bonds to yield the hydrodefluorination products methane and Et3SiF. This might occur by a concerted reaction pathway (Fig. 13, right cycle) or again by a two-step mechanism which also involves the carbenium-like species and the ACF-bound hydrogen (Fig. 13, left cycle). Hence, the latter entity could be regarded as a weakly coordinating anion (WCA) which stabilizes the carbenium-like species. The stabilization of fluorinated carbocations by weakly coordinating anions and the resulting Friedel–Crafts reactivity have also been described for Et3Si(carborane) in homogeneous phase.143 In the meantime, these results have been reconfirmed by employing HS-AlF3 instead, which especially in case of larger fluorinated molecules gives even higher conversions than ACF does.
These results show that the Lewis acid aluminum fluoride catalysts are superior over all (even homogeneous) catalysts reported so far and can be used in an unprecedented heterogeneous catalytic process for the C–F activation of fluoromethanes in the presence of Et3SiH. Thus, interesting new catalytic reactions will be explored based on this new finding.
3.2. Biacidic Lewis and Brønsted acid catalyzed reactions
In sections 2.1 and 2.2, it was shown that – based on the fluorolytic sol–gel synthesis – even nanoscale metal hydroxide fluorides or partly hydroxylated metal fluorides with adjustable Lewis to Brønsted surface sites can be obtained. Consequently, these new biacidic catalysts were checked for their catalytic potential, the main catalytic features of which will be briefly described below.Partly hydroxylated nanoscopic magnesium fluoride phases of different degrees of hydroxylation were used for the synthesis of vitamin E by the condensation of 2,3,6-trimethylhydroquinone (TMHQ, 1) with isophytol (IP, 2). This reaction proceeds in a first step through Friedel–Crafts alkylation–cyclization (Scheme 6,144–151) in the presence of Brønsted surface sites whereas the second ring closure step is regarded to proceed at Lewis acid sites.
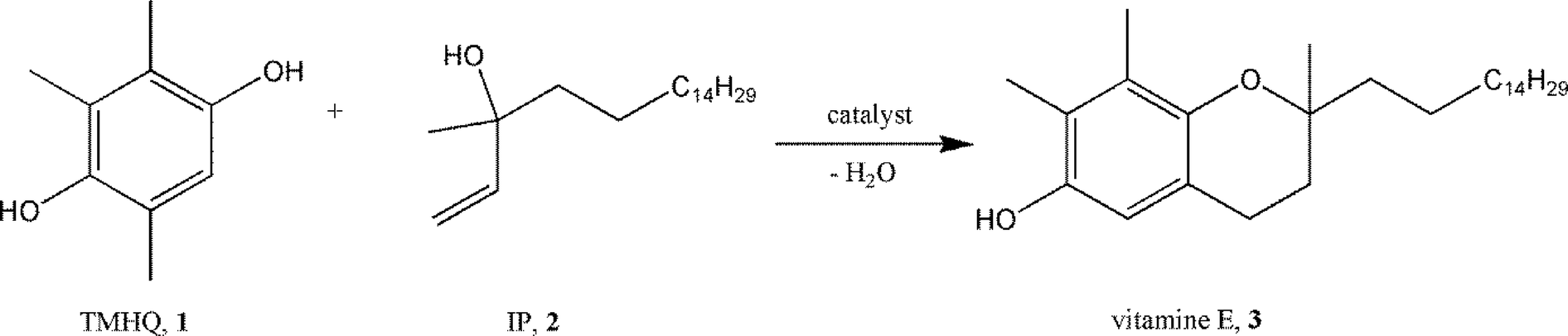 | ||
| Scheme 6 Synthesis of vitamin E by condensation of 2,3,6-trimethylhydrochinone with isophytol. | ||
Both crystalline and HS-MgF2 did not show noticeable catalytic activity. In contrast, HS-MgF2 prepared with 71% aqueous HF gave a complete conversion of isophytol and almost 100% to (all-rac)-α-tocopherol.44 Hydrofluoric acid with increased water content (higher amount of OH-groups become introduced into the MgF2-phases) resulted all in 100% conversion but with decreasing selectivity toward (all-rac)-α-tocopherol, obviously because of the decreasing strength of Lewis acidity (cf. section 2.3). Similar high conversions and selectivities were obtained for this reaction using hydroxylated HS-AlF3 (ref. 77), thus pointing out the general applicability of this approach.
Another example of multi-step Lewis–Brønsted catalysed reactions is the synthesis of vitamin K1 (Fig. 14) that involves as key step the Friedel–Crafts alkylation of menadiol acetate 12 (MDA) with isophytol 2 (IP) followed by the dihydro vitamin K1 oxidation.152
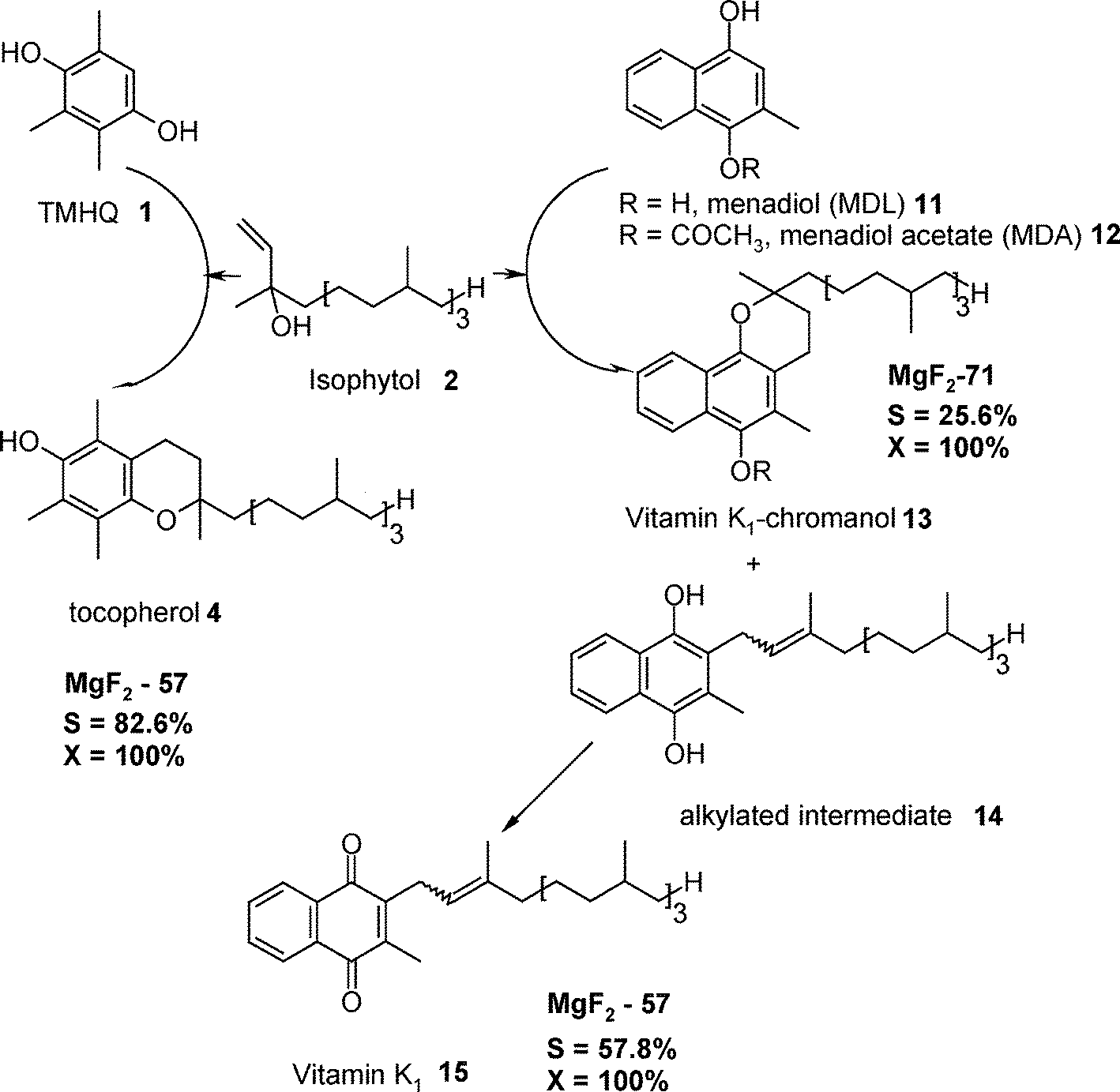 | ||
| Fig. 14 The syntheses of vitamin K115 and K1-chromanol 13 (the reaction steps are compared with the reaction steps for the synthesis of tocopherol 4). Reproduced from ref. 157 with permission from Wiley. | ||
The reaction cycles displayed in Fig. 14 summarize the main results from this heterogeneous catalytic reaction.44,153 Obviously, a combination of low amount of Lewis acid sites and medium Brønsted acid sites (e.g. MgF2-57) highly favours the formation of vitamin K115 in good yields of ca. 60% being comparable with the liquid phase catalytic process employing BF3·OEt2.
A further interesting reaction is the cyclization of citronellal to (±)-isopulegol that is an important intermediate for the (±)-menthol synthesis (Scheme 7). Here too, nanoscopic hydroxylated magnesium fluorides were successfully employed as very active and selective heterogeneous catalysts.32,106,154–156
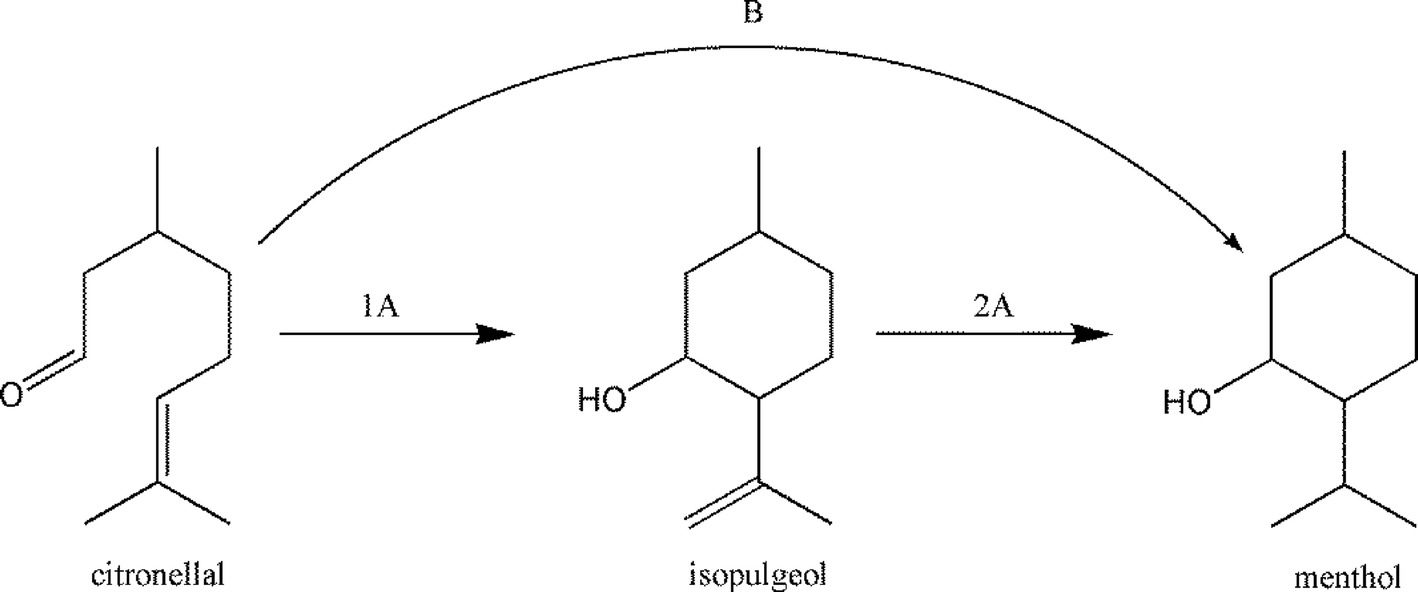 | ||
| Scheme 7 The synthesis of menthol from citronellal in two steps (route 1A + 2A) and in one-pot (route B). | ||
The hydroxylated fluoride catalysts showed unexpected diastereoselectivities (ds) (91.7%, for hydroxylated AlF3 and 84.7% for hydroxylated MgF2-71) superior to most conventional catalysts used for this reaction (71–75%).
Taking the tunable properties of these hydroxylated metal fluorides into account, it is logical to test them in benzylation reactions. Hence they were employed in alkylation reactions of benzene (Fig. 15).
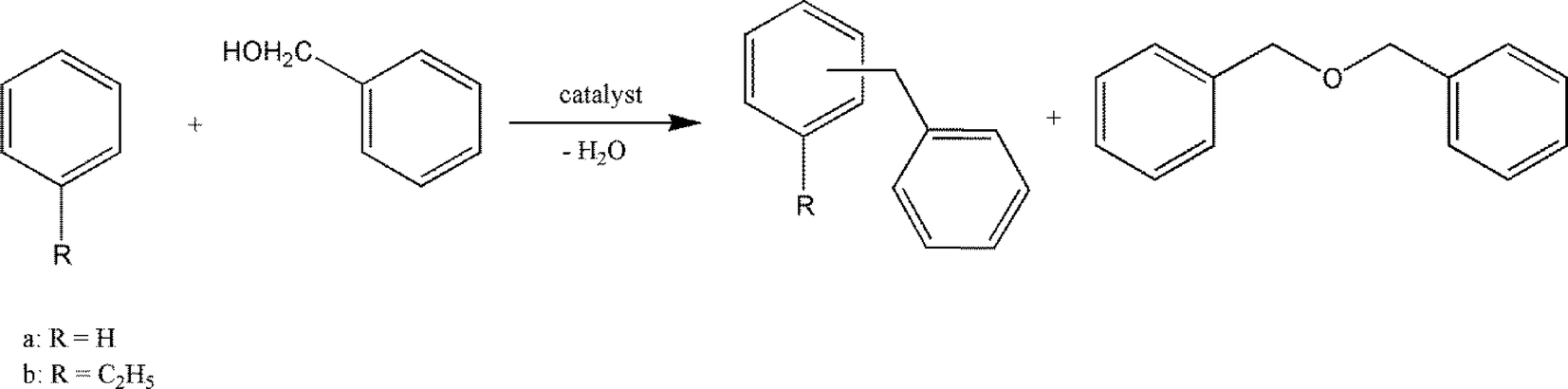 | ||
| Fig. 15 Friedel–Crafts reaction of benzene and ethylbenzene resulting in the formation of diphenylmethane or ethyldiphenylmethane, respectively, and dibenzylether. | ||
The best results were obtained with MgF2-57 and MgF2-71 catalysts which gave selectivities towards dibenzyl ether (DBE) between 85 and 97%, thus evidencing that the etherification of benzyl alcohol to DBE was much faster than the benzylation of the substrate.158
A very recent example is the dehydration of D-xylose to furfural, the mechanism of which being different in the presence of Lewis or Brønsted sites.159,160 In this case the biacidic MgF2 catalysts were further functionalized using different fluorosulfonic precursors. It was proven by TG-MS, 19F-MAS-NMR, and pyridine adsorption that surface OH groups were converted into stronger Brønsted surface sites. The catalytic data showed a significant change in the reaction kinetics and a furfural selectivity of 90% at 160 °C in water/toluene for optimized Lewis to Brønsted site ratios.
It is worth mentioning that these new biacidic catalysts are not only restricted to binary metal fluorides, but a second or even third metal may also be incorporated in the solid catalyst thus opening an even wider field of catalytically active solids. Thus, a good example for making benefit of this synthesis approach to ternary biacidic metal fluorides with tunable functionalities relates to the fast and selective saccharification of cellulose to glucose.161 Sn-doped hydroxylated MgF2 exhibited a very high catalytic activity for cellulose degradation and also high selectivity to glycose. It was concluded that the cellulose degradation proceeds via a two-step homogeneous/heterogeneous mechanism. Thus, an initial hydrolysis of cellulose starts involving hydrated tin fluoride that is partly released from the solid catalyst. Hence, the formed oligomers are small enough to enter into the pores where they can interact with the active surface sites of the heterogeneous catalyst, thus forming glucose.
3.3. Nanoscopic metal fluorides supported precious metal catalysed reactions
Anchoring of other catalytically active materials is a known approach to create even more complex catalytic functions inside not only a metal fluoride catalyst.38 Thus, impregnation of precious metals can be easily achieved by adding suitable precursors like PdCl2, H2PtCl6, or HAuCl4 to MFn or MFn−xOx/2 (M: metal, n: oxidation state of the metal) in an alcoholic solution. This “incipient wetness impregnation” method was also used for the preparation of a new cationic gold/hydroxylated magnesium fluoride catalyst, Au@MgF2−x(OH)x, that exhibits both the novel acidic properties of the nanoscopic magnesium fluoride and that of the gold metal.162 This catalyst was used for the one-pot synthesis of (±)-menthol from citronellal (Fig. 16).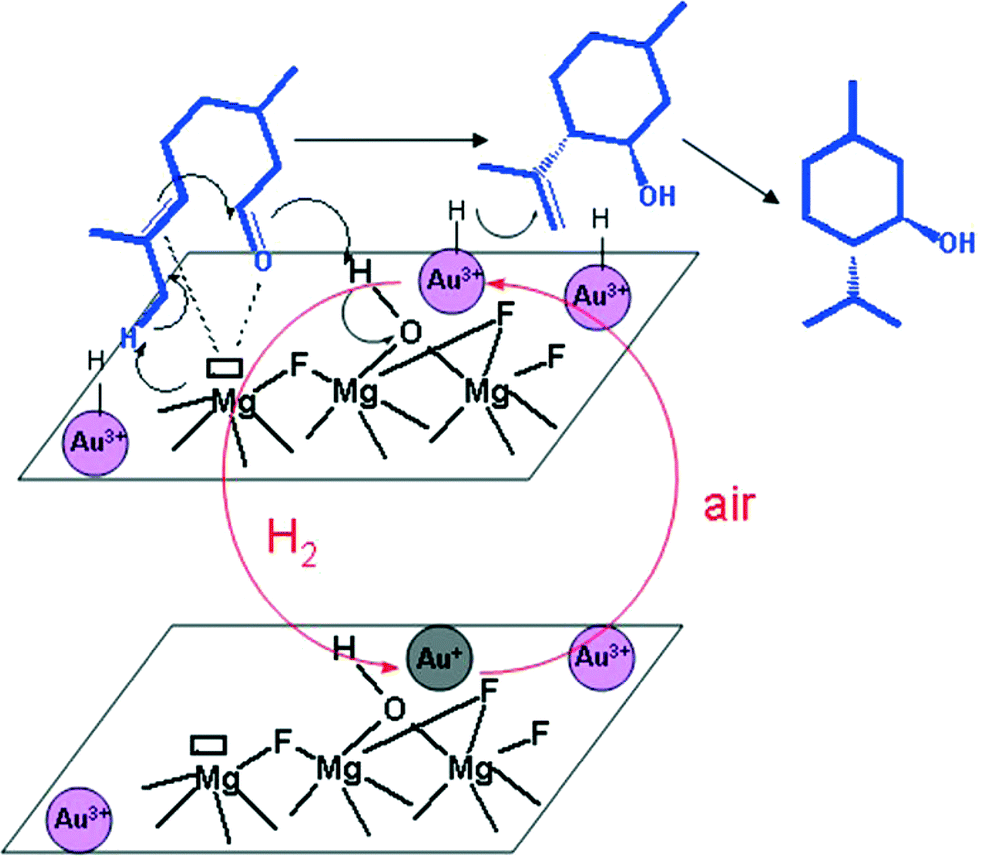 | ||
| Fig. 16 Proposed catalytic cycle in the hydrogenation step of isopulegol to (±)-menthol with ionic gold particles. | ||
This catalyst provides unexpectedly active ionic gold species for the selective hydrogenation of isopulegols to menthols. Interestingly enough, the catalytic features of the fluoride needed for the diastereoselective isomerization of citronellal to(±)-isopulegol are preserved.
By comparing the ionic gold based catalyst with Ir-based catalysts,163 it was found that the former is totally selective to menthols (no by-products such as citronellol, 3,7-dimethyloctanol, and 3,7-dimethyloctanal were observed) suggesting a different reaction mechanism (Fig. 16).
In agreement with the previous work of Comas-Vives et al.,164 who established that in the presence of an ionic gold based catalyst the heterolytic bond cleavage of H2 is more probable than its homolytic activation, we found that this effect is even more favoured by the hydroxylated MgF2 support; the proton remains on the polar surface whereas the hydride is bonded to the gold site. Thus, this novel catalyst can be used to perform such reactions in nonpolar solvents like toluene.
Nano-aluminium fluoride-supported platinum and palladium catalysts were found to be very efficient n-pentane hydroisomerization catalysts.165 By in situ preparation, very small metal nanoparticles (2 nm) finely distributed over the fluoride catalyst were created that maintained very good activity, stability, and selectivity towards isomerization (up to nearly 100%) at 350 °C. The exciting requisite for this superior catalytic behaviour was found to be the combination of very strong Lewis acidity of the HS-AlF3 and the precious metal. The performance of the 0.5 wt% Pd@HS-AlF3 catalyst appeared as good as that of the catalyst with 2 wt% Pd.
Hydrogenation reactions are another type of reactions in which M@MFn catalysts were successfully employed as very active and selective new catalysts. Thus Pd–Cu@HS-AlF3 catalysts were tested in hydroisomerization reactions of n-pentane and compared with Pd–Cu catalysts impregnated on active carbon for comparison.166
Another example of reactions in which M@MFn catalysts exhibit exiting properties is the very selective olefin hydrogenation by palladium nanoparticles supported on magnesium hydroxide fluoride. The catalysts were prepared in a one-pot synthesis procedure using palladium acetate and magnesium methoxide as precursors in the fluorolytic sol–gel synthesis as described in section 2.2. As can be seen from the TEM micrographs in Fig. 17, the majority of Pd-particles are approximately 5 nm in diameter, although the particle size distribution spreads up to 20 nm. These catalysts showed extremely high metal dispersion compared to examples reported in the literature.167
 | ||
| Fig. 17 TEM images of catalysts a) Pd–MgF2-100 and b) Pd–MgF2-71. Scale bars: 5 nm. Reproduced from ref. 166 with permission from Elsevier. | ||
First, hydrogenation of styrene was investigated as a model reaction. The reaction was performed at room temperature and atmospheric pressure by bubbling hydrogen gas through the reaction mixture (Scheme 8).
 | ||
| Scheme 8 Hydrogenation of styrene. | ||
A Pd–MgO and a commercial 5 wt% Pd/C catalyst were also used for comparison. With both catalysts, very low conversions below 1, 7, and 4%, respectively, were obtained. In contrast, on running the reaction with Pd@MgF2-40, 44% styrene conversion was obtained after 3 h, which increased to ca. 44% with Pd@MgF2-71 and as much as 100% styrene conversion was achieved with Pd@MgF2-100. It should be noted that the latter catalysts contained as little as 0.1 wt% only. Based on these encouraging results, this catalyst was screened for a large variety of substrates. For most of the even less reactive substrates up to 100% conversions were obtained. Moreover the exiting feature of this catalyst in these reactions is its surprising selectivity exclusively for olefinic double bond hydrogenation. A further valuable property is that even after 10 recycle tests, neither leaching nor a noticeable loss of activity and selectivity was observed. Hence, this magnesium fluoride based Pd catalyst represents a very efficient new catalyst for the regioselective hydrogenation of olefins in the presence of other functionalities under ambient reaction conditions which have not been reported so far for heterogeneous catalysts. Also disubstituted olefins like indene and α-methylstyrene gave almost full conversion, although a longer reaction time was necessary. Even olefins with other reducible functional groups like 2-butenal were 100% converted with 100% selectivity for olefin hydrogenation without reduction of ketone moieties. Similar exciting good conversions and selectivity data were obtained by screening 2-alkenol olefin, 3-phenylpropenol, 2-phenylpropanol, or even olefins with carboxylic group like methyl acrylate and acrylic acid.
The effect of bulkier groups and electron withdrawing reducible carbonyl groups was studied on benzalacetophenone. The conversion decreased to 65% without affecting much the selectivity to 1,5-diphenylpentanone. Obviously the presence of phenyl rings lowers the activity. The geometrical isomers cis and trans stilbene were hydrogenated. The hydrogenation needed a slightly elevated temperature (80 °C) due to higher steric hindrance of the bulkier phenyl group. Further on, the effect of the substituent on phenyl ring of styrene was examined by catalytic hydrogenation of 4-vinylanisole. The reaction showed 95% conversion with 99% selectivity for olefin hydrogenation.
The recyclability of the catalyst was tested using styrene hydrogenation at room temperature and bubbling hydrogen at ambient pressure through the reaction solution. After completion of each reaction, the catalyst was separated, washed, dried and used for the next run and the catalyst did not show noticeable loss of activity.
FTIR studies regarding adsorbed species on the catalyst surface gave no indication for adsorbed olefinic species. Hence activation of hydrogen is supposed to be the crucial step, and hence a mechanistic pathway as displayed in Fig. 18 was deduced.
 | ||
| Fig. 18 Proposed reaction mechanism for the hydrogenation of styrene in the presence of the Pd–MgF2-100 catalyst. Reproduced from ref. 166 with permission from Elsevier. | ||
4. Conclusion
The non-aqueous fluorolytic sol–gel synthesis of nanoscopic metal fluorides, developed just over the past 10 years, has given access to new metal fluoride based catalysts. Not only highly Lewis acidic solids can be obtained but also even more advantageously bi-acidic hydroxide fluorides with tunable Lewis to Brønsted acidity can be synthesized for the first time. These compounds can be obtained by modification of the synthesis procedure in two different ways. The first approach is based on the use of an under-stoichiometric amount of HF during the first synthesis step followed by the addition of the stoichiometrically requested amount of water, which completes the reaction. This way, all nominal compositions of M(OH)n−xFx are accessible. In a second step, these hydroxide fluorides can be transformed into the corresponding metal oxide, fluorides (MOn−x/2Fx). By tuning the oxide to fluoride stoichiometry inside these phases the surface side character can be altered from strongly Lewis acidic at high F-content towards weak Lewis but stronger Brønsted acidic or even basic depending on the metal M at high oxygen content. Mainly determined by the O to F ratio these oxide fluoride phases store their amorphous character (depending on the metal M) up to temperatures of about 600 °C and exhibit surface areas up to 700 m2 g−1. It is noteworthy that some of these metal fluorides or oxide fluorides, respectively, are thermally stable up to at least 1000 °C, meaning no notable hydrolysis or pyrolysis reaction, respectively, takes place.The introduction of a second or even a third metal into these new compounds allows for further functionalization resulting in unlimited new compounds with high impact on catalytic applications.
However, these nanoscale metal fluorides and hydroxide fluorides represent not only new, catalytically active classes of compounds with very high surface areas but also are excellent candidates to be used as supports especially if hydroxide fluorides are converted by calcination into oxide fluorides. Thus, other active components like, e.g., precious metals can be supported on these new fluoride based materials by incipient wetness impregnation or even more effectively by in situ incorporation during the fluorolytic sol–gel synthesis. It is noteworthy that some of such catalytically interesting new materials were successfully proven as very effective co-catalysts in combination with, e.g., precious metals like Au, Pd, Pt, etc.
As it has been exemplarily shown in this review, the new nanoscopic metal fluoride based materials and especially the large variety of metal hydroxide fluoride, MFx(OH)n−x, and metal oxide fluoride, MOn−x/2Fx, phases represent a new class of potential catalysts waiting for further exploration in heterogeneous catalysis.
Of course, limitation in applications may arise from chemical resistivity (hydrolysis) depending on the metal used. However, there are many metal fluorides being both temperature and hydrolysis resistant over a wide range.
Acknowledgements
The author would like to thank the EU for supporting a part of this work through the 6th Framework Programme (FUNFLUOS, contract no. NMP3-CT-2004-5005575) and the Deutsche Forschungsgemeinschaft, DFG (Ke-489/22, 28, and 29 as well as graduate school “Fluorine as a Key Element” GRK 1582).Special thanks goes to Prof. Simona Coman from Bucharest University who joined my group several years ago as a fellow of the Humboldt Foundation. Because of her, most of the work on metal hydroxide fluorides started. Furthermore, the impact of several former and present co-workers who contributed at least partly to the content of this paper by their results should be emphasized: Dr. Ying Guo, Dr. Katharina Teinz, Dr. Krisna Murthy Janmanchi, Dr. Sridana Shekhar, Dr. Zhijian Li, Dr. Udo Gross, Dr. Stephan Rüdiger, Dr. Pratap Patil, Dr. Thoralf Krahl, Dr. Mike Ahrens, Dr. René König, Dr. Kerstin Scheurell, PD Dr. Gudrun Scholz, Dr. Detlef Heidemann, Dr. Anton Dimitrov, Agnieszka Siweck, and Sigrid Bäßler.References
- B. C. Gates, Catalytic Chemistry, John Wiley & Sons Ltd., NY, 1991, ch. 1, p. 2 Search PubMed.
- L. E. Manzer, Catal. Today, 1992, 13, 13–22 CrossRef CAS.
- E. Kemnitz and D.-H. Menz, Prog. Solid State Chem., 1998, 26, 97–153 CrossRef CAS.
- E. Kemnitz and J. M. Winfield, in Advanced Inorganic Fluorides: Synthesis, Characterization and Applications, ed. T. Nakajima, A. Tressaud and B. Zemva, Elsevier Science S A., 2000, pp. 367–402 Search PubMed.
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and D. A. Boatz, J. Fluorine Chem., 2000, 101, 151–153 CrossRef CAS.
- K. E. Gutowski and D. A. Dixon, J. Phys. Chem. A, 2006, 110, 12044–12054 CrossRef CAS PubMed.
- E. Kemnitz, U. Groß, S. Rüdiger and S. Chandra Shekar, Angew. Chem., 2003, 115, 4383–4386 ( Angew. Chem., Int. Ed. , 2003 , 42 , 4251–4254 ) CrossRef.
- S. Rüdiger, U. Goß and E. Kemnitz, J. Fluorine Chem., 2007, 128, 353–368 CrossRef PubMed.
- S. Rüdiger and E. Kemnitz, Dalton Trans., 2008, 1117–1127 RSC.
- E. Kemnitz, G. Scholz and S. Rüdiger, in Functionalized Inorganic Fluorides, ed. A. Tressaud, Wiley, 2010, pp. 1–35 Search PubMed.
- U. Groß, D. Müller and E. Kemnitz, Angew. Chem., 2003, 115, 2739 ( Angew. Chem., Int. Ed. , 2003 , 42 , 2626–2629 ) CrossRef.
- E. Kemnitz, S. Rüdiger and U. Gross, Z. Anorg. Allg. Chem., 2004, 630, 1696–1696 CrossRef.
- S. Rüdiger, U. Groß, M. Feist, H. Prescott, S. C. Shekar, S. I. Troyanov and E. Kemnitz, J. Mater. Chem., 2005, 15, 588–597 RSC.
- J. Krishna Murthy, U. Groß, S. Rüdiger, E. Kemnitz and J. M. Winfield, J. Solid State Chem., 2006, 179, 739–746 CrossRef CAS PubMed.
- S. Rüdiger, G. Eltanany, U. Groß and E. Kemnitz, J. Sol-Gel Sci. Technol., 2007, 41, 299–311 CrossRef.
- R. König, G. Scholz, N. H. Thong and E. Kemnitz, Chem. Mater., 2007, 19, 2229–2237 CrossRef.
- S. Wuttke, G. Scholz, S. Rüdiger and E. Kemnitz, J. Mater. Chem., 2007, 17, 4980–4988 RSC.
- R. König, G. Scholz and E. Kemnitz, Solid State Nucl. Magn. Reson., 2007, 32, 78–88 CrossRef PubMed.
- G. Eltanany, S. Rüdiger and E. Kemnitz, J. Mater. Chem., 2008, 18, 2268–2275 RSC.
- C. Stosiek, G. Scholz, G. Eltanany, R. Bertram and E. Kemnitz, Chem. Mater., 2008, 20, 5687–5697 CrossRef CAS.
- S. Wuttke, A. Lehmann, G. Scholz, M. Feist, A. Dimitrov, S. I. Troyanov and E. Kemnitz, Dalton Trans., 2009, 4729–4734 RSC.
- R. König, G. Scholz, A. Pawlik, C. Jäger, B. van Rossum and E. Kemnitz, J. Phys. Chem. C, 2009, 113, 15576–15585 Search PubMed.
- A. Dimitrov, J. Koch, S. I. Troyanov and E. Kemnitz, Eur. J. Inorg. Chem., 2009, 5299–5301 CrossRef CAS.
- C. Stosiek, G. Scholz, S. L. M. Schröder and E. Kemnitz, Chem. Mater., 2010, 22, 2347–2356 CrossRef CAS.
- J. Noack, K. Teinz, C. Schaumberg, C. Fritz, S. Rüdiger and E. Kemnitz, J. Mater. Chem., 2011, 21, 334–338 RSC.
- Y. Guo, S. Wuttke, A. Vimont, M. Daturi, J.-C. Lavalley, K. Teinz and E. Kemnitz, J. Mater. Chem., 2012, 22, 14587–14593 RSC.
- Y. Guo, P. Gaczyñski, K.-D. Becker and E. Kemnitz, ChemCatChem, 2013, 5, 2223–2232 CrossRef CAS.
- J. Krishnamurthy, U. Gross, S. Rüdiger, E. Ünveren, W. Unger and E. Kemnitz, Appl. Catal., A, 2005, 282, 85–91 CrossRef CAS PubMed.
- M. Ahrens, K. Schuschke, S. Redmer and E. Kemnitz, Solid State Sci., 2007, 9, 833–837 CrossRef CAS PubMed.
- M. Ahrens, G. Scholz, M. Feist and E. Kemnitz, Solid State Sci., 2006, 8, 798–806 CrossRef CAS PubMed.
- M. Ahrens, G. Scholz and E. Kemnitz, Z. Anorg. Allg. Chem., 2008, 634, 2978–2981 CrossRef CAS.
- T. Krahl, A. Vimont, G. Eltanany, M. Daturi and E. Kemnitz, J. Phys. Chem. C, 2007, 111, 18317–18325 CAS.
- A. Wander, B. G. Searly, C. L. Bailey and N. M. Harrison, J. Phys. Chem. B, 2005, 109, 22935–22938 CrossRef CAS PubMed.
- A. Wander, C. L. Bailey, S. Mukhopadhyay, B. G. Searly and N. M. Harrison, J. Phys. Chem. C, 2008, 112, 6515–6519 CAS.
- A. Wander, B. G. Searly, C. L. Bailey and N. M. Harrison, J. Phys. Chem. B, 2005, 109, 22935–22938 CrossRef CAS PubMed.
- M. H. G. Prechtl, M. Teltewskoi, A. Dimitrov, E. Kemnitz and T. Braun, Chem. – Eur. J., 2011, 17, 14385–14388 CrossRef CAS PubMed.
- C. P. Grey and P. Chupas, et al. , Phys. Chem. Chem. Phys., 2006, 8, 5045–5055 RSC.
- M. Wojciechowska and R. Fiedorow, J. Fluorine Chem., 1980, 15, 443–452 CrossRef CAS.
- M. Wojciechowska, M. Zielinski and M. Pietrowski, J. Fluorine Chem., 2003, 120, 1–11 CrossRef CAS.
- M. Wojciechowska, M. Pietrowski and M. Zielinski, Catal. Today, 2007, 119, 338–341 CrossRef CAS PubMed.
- M. Wojciechowska, M. Zielinski, M. Przystajko and M. Pietrowski, Catal. Today, 2007, 119, 44–47 CrossRef CAS PubMed.
- M. Pietrowski, M. Zielinski and M. Wojciechowska, Catal. Lett., 2009, 128, 31–35 CrossRef CAS.
- S. Wuttke, A. Vimont, J.-C. Lavalley, M. Daturi and E. Kemnitz, J. Phys. Chem. C, 2010, 114, 5113–5120 CAS.
- S. Wuttke, S. M. Coman, G. Scholz, H. Kirmse, A. Vimont, M. Daturi, S. L. M. Schroeder and E. Kemnitz, Chem. – Eur. J., 2008, 14, 11488–11499 CrossRef CAS PubMed.
- S. Wuttke, S. M. Coman, J. Kröhnert, F. C. Jentoft and E. Kemnitz, Catal. Today, 2010, 152, 2–10 CrossRef CAS PubMed.
- E. Kemnitz and S. Rüdiger, in Functionalized Inorganic Fluorides, ed. Alain Tressaud, Wiley, 2010, pp. 69–97 Search PubMed.
- C. G. Krespan and V. A. Petrov, Chem. Rev., 1996, 96, 3269–3301 CrossRef CAS PubMed.
- C. G. Krespan, U.S. Pat. 5.162.594, (to DuPont Co.), CAN 117:69439, 1992 Search PubMed.
- A. C. Sievert, C. G. Krespan and V. A. Petrov, U.S. Pat., 5.157.171, (to DuPont Co.), CAN 115:70904, 1992 Search PubMed.
- C. G. Krespan and D. A. Dixon, J. Fluorine Chem., 1996, 77, 117–126 CrossRef CAS.
- V. A. Petrov and C. G. Krespan, J. Fluorine Chem., 2000, 102, 199–204 CrossRef CAS.
- V. A. Petrov, C. G. Krespan and B. E. Smart, J. Fluorine Chem., 1996, 77, 139–142 CrossRef CAS.
- V. A. Petrov, C. G. Krespan and B. E. Smart, J. Fluorine Chem., 1998, 89, 125–130 CrossRef CAS.
- V. A. Petrov, F. Davidson and B. E. Smart, J. Org. Chem., 1995, 60, 3419–3422 CrossRef CAS.
- T. Krahl and E. Kemnitz, Angew. Chem., Int. Ed., 2004, 43, 6653–6656 CrossRef CAS PubMed.
- T. Krahl, R. Stösser, E. Kemnitz, G. Scholz, M. Feist, G. Silly and J.-Y. Buzaré, Inorg. Chem., 2003, 42, 6474–6483 CrossRef CAS PubMed.
- T. Krahl and E. Kemnitz, J. Fluorine Chem., 2006, 127, 663–678 CrossRef CAS PubMed.
- D. Dambournet, A. Demourgues, C. Martineau, E. Durand, J. Majimel, A. Vimont, H. Leclerc, J.-C. Lavalley, M. Daturi, C. Legein, J.-Y. Buzaré, F. Fayon and A. Tressaud, J. Mater. Chem., 2008, 18, 2483–2492 RSC.
- A. Demourgues, N. Penin, D. Dambournet, R. Clarenc, A. Tressaud and E. Durand, J. Fluorine Chem., 2012, 134, 35–43 CrossRef CAS PubMed.
- C. F. Rammlesburg, Apatite, in Handbuch der Mineralchemie, Wilhelm Engelmann, Leipzig, 1860, pp. 351–355 Search PubMed.
- B. Buhn, A. H. Rankin, J. Schneider and P. Dulski, Chem. Geol., 2002, 18, 75–98 CrossRef.
- E. Kemnitz, A. Hess, G. Rother and S. Troyanov, J. Catal., 1996, 159, 332–339 CrossRef CAS.
- B. Adamczyk, A. Hess and E. Kemnitz, J. Mater. Chem., 1996, 6, 1731–1735 RSC.
- A. Heß, E. Kemnitz, A. Lippitz, W. E. S. Unger and D.-H. Menz, J. Catal., 1994, 148, 270–280 CrossRef.
- E. Kemnitz, A. Kohne, I. Grohmann, A. Lippitz and A. W. S. Unger, J. Catal., 1996, 159, 270–279 CrossRef CAS.
- M. Karg, G. Scholz, R. König and E. Kemnitz, Dalton Trans., 2012, 41, 2360–2366 RSC.
- R. König, G. Scholz, M. Veizi, C. Jäger, S. I. Troyanov and E. Kemnitz, Dalton Trans., 2011, 40, 8701–8710 RSC.
- R. König, G. Scholz and E. Kemnitz, J. Sol-Gel Sci. Technol., 2010, 56, 145–156 CrossRef.
- G. Scholz, S. Brehme, R. König, D. Heidemann and E. Kemnitz, J. Phys. Chem. C, 2010, 114, 10535–10543 CAS.
- A. Pawlik, R. König, G. Scholz, E. Kemnitz, G. Brunklaus, M. Bertmer and C. Jäger, J. Phys. Chem. C, 2009, 113, 16674–16680 CAS.
- R. König, G. Scholz, A. Pawlik, C. Jäger, B. van Rossum and E. Kemnitz, J. Phys. Chem. C, 2009, 113, 15576–15585 Search PubMed.
- R. König, G. Scholz and E. Kemnitz, J. Phys. Chem. C, 2009, 113, 6426–6438 Search PubMed.
- A. Dimitrov, J. Koch, S. I. Troyanov and E. Kemnitz, Eur. J. Inorg. Chem., 2009, 5299–5301 CrossRef CAS.
- J. Noack, F. Emmerling, H. Kirmsec and E. Kemnitz, J. Mater. Chem., 2011, 21, 15015–15021 RSC.
- J. Noack, L. Schmidt, H.-J. Gläsel, M. Bauer and E. Kemnitz, Nanoscale, 2011, 3, 4774–4779 RSC.
- C. L. Bailey, S. Mukhopadhyay, A. Wander, B. G. Searle and N. M. Harrison, J. Phys. Chem. C, 2009, 113, 4976–4983 CAS.
- S. M. Coman, S. Wuttke, A. Vimont, M. Daturi and E. Kemnitz, Adv. Synth. Catal., 2008, 350, 2517–2524 CrossRef CAS.
- H. Eckert, J. P. Yesinowsky, L. A. Silver and E. M. Stolper, J. Phys. Chem., 1988, 92, 2055–2064 CrossRef CAS.
- C. Chizallet, G. Costentin, H. L. Pernot, M. Che, C. Bonhomme, J. Maquet, F. Delbeocq and P. Sautet, J. Phys. Chem. C, 2007, 111, 18279–18287 CAS.
- H. Knözinger and P. Ratnasamy, Catal. Rev.: Sci. Eng., 1978, 17, 31–70 Search PubMed.
- K. Teinz, S. Wuttke, F. Börno, J. Eicher and E. Kemnitz, J. Catal., 2011, 282, 175–182 CrossRef CAS PubMed.
- Z. Huesges, C. Müller, B. Paulus, C. Hough, N. Harrison and E. Kemnitz, Surf. Sci., 2013, 609, 73–77 CrossRef CAS PubMed.
- T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401–406 RSC.
- J. T. Golden, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 2001, 123, 5837–5838 CrossRef CAS.
- T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401–406 RSC.
- S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092–2093 CrossRef CAS PubMed.
- H. Lowry and K. S. Richardson, Mechanism and Theory in Organic Chemistry, Harper and Row, New York, 1987 Search PubMed.
- A. F. Thomas, Deuterium Labelling in Organic Chemistry, Meredith Corporation, New York, 1971 Search PubMed.
- C. M. Yung, M. B. Skaddan and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 13033–13043 CrossRef CAS PubMed.
- T. Maegawa, Y. Fujiwara, Y. Inagaki, H. Esaki, Y. Monguchi and H. Sajiki, Angew. Chem., Int. Ed., 2008, 47, 5394–5397 CrossRef CAS PubMed.
- K. Meier, K. J. H. Young, D. H. Ess, W. J. Tenn, J. Oxgaard, W. A. Goddard and R. A. Periana, Organometallics, 2009, 28, 5293–5304 CrossRef.
- J. Engelhardt and W. K. Hall, J. Catal., 1995, 151, 1–9 CrossRef CAS.
- J. Engelhardt, G. Onyestyak and W. K. Hall, J. Catal., 1995, 157, 721–729 CrossRef CAS.
- M. Haouas, G. Fink, F. Taulelle and J. Sommer, Chem. – Eur. J., 2010, 16, 9034–9039 CrossRef CAS PubMed.
- S. G. Hindin, G. A. Mills and A. G. Oblad, J. Am. Chem. Soc., 1951, 73, 278–281 CrossRef CAS.
- B. Schoofs, J. Schuermans and R. A. Schoonheydt, Microporous Mesoporous Mater., 2000, 35–6, 99–111 CrossRef.
- J. Sommer, M. Hachoumy, F. Garin, D. Barthomeuf and J. Vedrine, J. Am. Chem. Soc., 1995, 117, 1135–1136 CrossRef CAS.
- J. Sommer, R. Jost and M. Hachoumy, Catal. Today, 1997, 38, 309–319 CrossRef CAS.
- J. G. Larson and W. K. Hall, J. Phys. Chem., 1965, 69, 3080–3089 CrossRef CAS.
- P. J. Robertson, M. S. Scurrell and C. Kemball, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 903–912 RSC.
- R. Wischert, C. Copéret, F. Delbecq and P. Sautet, Angew. Chem., Int. Ed., 2011, 50, 3202–3205 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- V. Derdau, J. Atzrodt, J. Zimmermann, C. Kroll and F. Bruckner, Chem. – Eur. J., 2009, 15, 10397–10404 CrossRef CAS PubMed.
- M. H. G. Prechtl, M. Teltewskoi, A. Dimitrov, E. Kemnitz and Th. Braun, Chem. – Eur. J., 2011, 17, 14385–14388 CrossRef CAS PubMed.
- C. Hall and R. N. Perutz, Chem. Rev., 1996, 96, 3125–3146 CrossRef CAS PubMed.
- J. K. Murthy, U. Gross, S. Rudiger, V. V. Rao, V. V. Kumar, A. Wander, C. L. Bailey, N. M. Harrison and E. Kemnitz, J. Phys. Chem. B, 2006, 110, 8314–8319 CrossRef CAS PubMed.
- V. V. Grushin, Acc. Chem. Res., 2010, 43, 160–171 CrossRef CAS PubMed.
- D. Noveski, T. Braun, B. Neumann, A. Stammler and H.-G. Stammler, Dalton Trans., 2004, 4106–4119 RSC.
- J. Burdeniuc, B. Jedlicka and R. H. Crabtree, Chem. Ber., 1997, 130, 145–154 CrossRef CAS.
- G. Meier and T. Braun, Angew. Chem., 2009, 121, 1575–1577 ( Angew. Chem., Int. Ed. , 2009 , 48 , 1546–1548 ) CrossRef.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed.
- T. Braun and F. Wehmeier, Eur. J. Inorg. Chem., 2011, 613–625 CrossRef CAS.
- E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333–348 CrossRef CAS PubMed.
- L. Keyes, A. D. Sun and J. A. Love, Eur. J. Org. Chem., 2011, 3985–3994 CrossRef CAS.
- J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS.
- A. D. Sun and J. A. Love, Dalton Trans., 2010, 39, 10362–10374 RSC.
- J.-F. Paquin, Synlett, 2011, 289–293 CrossRef CAS PubMed.
- H. Torrens, Coord. Chem. Rev., 2005, 249, 1957–1985 CrossRef CAS PubMed.
- W. D. Jones, Dalton Trans., 2003, 3991–3995 RSC.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–33481 CrossRef PubMed.
- M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., 2010, 122, 4039–4043 ( Angew. Chem., Int. Ed. , 2010 , 49 , 3947–3951 ) CrossRef.
- T. Braun, F. Wehmeier and K. Altenhçner, Angew. Chem., 2007, 119, 5415–5418 ( Angew. Chem., Int. Ed. , 2007 , 46 , 5321–5324 ) CrossRef.
- T. Braun, M. A. Salomon, K. Altenhçner, M. Teltewskoi and S. Hinze, Angew. Chem., 2009, 121, 1850–1854 ( Angew. Chem., Int. Ed. , 2009 , 48 , 1818–1822 ) CrossRef.
- M. F. Kuehnel and D. Lentz, Angew. Chem., 2010, 122, 2995–2998 ( Angew. Chem., Int. Ed. , 2010 , 49 , 2933–2936 ) CrossRef.
- G. Haufe, S. Suzuki, H. Yasui, C. Terada, T. Kitayama, M. Shiro and N. Shibata, Angew. Chem., 2012, 124, 12441–12445 ( Angew. Chem., Int. Ed. , 2012 , 51 , 12275–12279 ) CrossRef.
- M. F. Kühnel, T. Schlöder, S. Riedel, B. Nieto-Ortega, F. J. Ramirez, J. T. L. Navarette, J. Casado and D. Lentz, Angew. Chem., 2012, 124, 2261–2263 ( Angew. Chem., Int. Ed. , 2012 , 51 , 2218–2220 ) CrossRef.
- T. Schaub, M. Backes and U. Radius, J. Am. Chem. Soc., 2006, 128, 15964–15965 CrossRef CAS PubMed.
- T. Braun, R. N. Perutz and M. I. Sladek, Chem. Commun., 2001, 2254–2255 RSC.
- M. I. Sladek, T. Braun, B. Neumann and H.-G. Stammler, J. Chem. Soc., Dalton Trans., 2002, 297–299 RSC.
- A. Steffen, M. I. Sladek, T. Braun, B. Neumann and H.-G. Stammler, Organometallics, 2005, 24, 4057–4064 CrossRef CAS.
- T. Braun, V. Schorlemer, B. Neumann and H.-G. Stammler, J. Fluorine Chem., 2006, 127, 367–372 CrossRef CAS PubMed.
- O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 332, 574–577 CrossRef CAS PubMed.
- C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946–4953 CrossRef CAS PubMed.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- S. Duttwyler, C. Douvris, N. L. P. Fackler, F. S. Tham, C. A. Reed, K. K. Baldridge and J. S. Siegel, Angew. Chem., 2010, 122, 7681–7684 ( Angew. Chem., Int. Ed. , 2010 , 49 , 7519–7522 ) CrossRef.
- W. Gu, M. R. Haneline, C. Douvris and O. V. Ozerov, J. Am. Chem. Soc., 2009, 131, 11203–11212 CrossRef CAS PubMed.
- M. Klahn, C. Fischer, A. Spannenberg, U. Rosenthal and I. Krossing, Tetrahedron Lett., 2007, 48, 8900–8903 CrossRef CAS PubMed.
- R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676–9682 CrossRef CAS PubMed.
- H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176–9184 RSC.
- C. Douvris, E. S. Stoyanov, F. S. Tham and C. A. Reed, Chem. Commun., 2007, 1145–1147 RSC.
- C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27–30 CrossRef CAS.
- M. Ahrens, G. Scholz, Th. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2013, 52, 5328–5332 CrossRef CAS PubMed.
- C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27–30 CrossRef CAS.
- W. Bonrath and T. Netscher, Appl. Catal., A, 2005, 280, 55–73 CrossRef CAS PubMed.
- M. Schneider, K. Zimmermann, F. Aquino and W. Bonrath, Appl. Catal., A, 2001, 220, 51–58 CrossRef CAS.
- Y. Kokubo, A. Hasegawa, S. Kuwata, K. Ishihara, H. Yamamoto and T. Ikariya, Adv. Synth. Catal., 2005, 347, 220–224 CrossRef CAS.
- H. Wang and B.-Q. Xu, Appl. Catal., A, 2004, 275, 247–255 CrossRef CAS PubMed.
- M. C. Laufer, W. Bonrath and W. F. Hölderich, Catal. Lett., 2005, 100, 101–109 CrossRef CAS PubMed.
- A. Heidekum, M. A. Harmer and W. F. Hölderich, J. Catal., 1998, 176, 260–263 CrossRef CAS.
- S. Wang and F. Kienzle, Ind. Eng. Chem. Res., 2000, 39, 4487–4490 CrossRef CAS.
- A. Hasegawa, K. Ishihara and H. Yamamoto, Angew. Chem., Int. Ed., 2003, 42, 573190–573197 CrossRef PubMed.
- A. Rüttimann, Chimia, 1986, 40, 290–306 Search PubMed.
- S. M. Coman, V. I. Parvulescu, S. Wuttke and E. Kemnitz, ChemCatChem, 2010, 2, 92–97 CrossRef CAS.
- P. T. Patil, A. Dimitrov, H. Kirmse, W. Neumann and E. Kemnitz, Appl. Catal., B, 2007, 78, 80–91 CrossRef PubMed.
- P. T. Patil, A. Dimitrov, J. Radnik and E. Kemnitz, J. Mater. Chem., 2008, 18, 1632–1635 RSC.
- S. M. Coman, P. Patil, S. Wuttke and E. Kemnitz, Chem. Commun., 2009, 460–462 RSC.
- E. Kemnitz, S. Wuttke and S. M. Coman, Eur. J. Inorg. Chem., 2011, 4773–4794 CrossRef CAS.
- N. Candu, S. Wuttke, E. Kemnitz, S. M. Coman and V. I. Parvulescu, Appl. Catal., A, 2011, 391, 169–174 CrossRef CAS PubMed.
- I. Agirrezabal-Telleria, F. Hemmann, C. Jäger, P. L. Arias and E. Kemnitz, J. Catal., 2013, 205, 81–91 CrossRef PubMed.
- I. Agirrezabal-Telleria, Y. Guo, F. Hemmann, P. L. Arias and E. Kemnitz, Catal. Sci. Technol., 2014, 4, 1357–1368 CAS.
- S. Wuttke, A. Negoi, N. Gheorghe, V. Kuncser, E. Kemnitz, V. Parvulescu and S. M. Coman, ChemSusChem, 2012, 5, 1708–1711 CrossRef CAS PubMed.
- A. Negoi, S. Wuttke, E. Kemnitz, D. Macovei, V. I. Parvulescu, C. M. Teodorescu and S. M. Coman, Angew. Chem., Int. Ed., 2010, 49, 8134–8138 CrossRef CAS PubMed.
- F. Neatu, S. M. Coman, V. I. Parvulescu, G. Poncelet, D. de Vos and P. A. Jacobs, Top. Catal., 2009, 52, 1292–1300 CrossRef CAS PubMed.
- A. Comas-Vives, C. Gonzalez-Arellano, A. Corma, M. Iglesias, F. Sanchez and G. Ujaque, J. Am. Chem. Soc., 2006, 128, 4756–4765 CrossRef CAS PubMed.
- O. Machynskyy, E. Kemnitz and Z. Karpinski, ChemCatChem, 2014, 6, 592–602 CrossRef CAS.
- M. V. R. Acham, A. V. Biradar, M. K. Dongare, E. Kemnitz and S. B. Umbarkar, ChemCatChem, 2014, 6, 3182–3191 CrossRef.
- K. V. R. Chary, D. Naresh, V. Vishwanatan, M. Sadakane and W. Ueda, Catal. Commun., 2007, 8, 471–477 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Prof. Dr. Martin Jansen on the occasion of his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2015 |