
Stable and selective electrochemical reduction of carbon dioxide to ethylene on copper mesocrystals†
Chung Shou
Chen
,
Albertus D.
Handoko
,
Jane Hui
Wan
,
Liang
Ma
,
Dan
Ren
and
Boon Siang
Yeo
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: chmyeos@nus.edu.sg; Fax: +65 67791691; Tel: +65 65162836
First published on 1st September 2014
Abstract
Stable and selective electrochemical reduction of carbon dioxide to ethylene was achieved using copper mesocrystal catalysts in 0.1 M KHCO3. The Cu mesocrystal catalysts were facilely derived by the in situ reduction of a thin CuCl film during the first 200 seconds of the CO2 electroreduction process. At −0.99 V vs. RHE, the Faradaic efficiency of ethylene formation using these Cu mesocrystals was ~18× larger than that of methane and forms up to 81% of the total carbonaceous products. Control CO2 reduction experiments show that this selectivity towards C2H4 formation could not be replicated by using regular copper nanoparticles formed by pulse electrodeposition. High resolution transmission electron microscopy reveals the presence of both (100)Cu facets and atomic steps in the Cu mesocrystals which we assign as active sites in catalyzing the reduction of CO2 to C2H4. CO adsorption measurements suggest that the remarkable C2H4 selectivity could be attributed to the greater propensity of CO adsorption on Cu mesocrystals than on other types of Cu surfaces. The Cu mesocrystals remained active and selective towards C2H4 formation for longer than six hours. This is an important and industrially relevant feature missing from many reported Cu-based CO2 reduction catalysts.
Introduction
Carbon dioxide reduction to hydrocarbons and alcohols has the potential of generating a sustainable supply of valuable feedstock for our chemical industries and fuels for our energy needs.1 This process also mitigates excessive CO2 buildup in the atmosphere which contributes to global climate warming. CO2 can be electrochemically reduced to hydrocarbons such as ethylene and methane via 2CO2 + 12e− + 8H2O → C2H4 + 12OH− and CO2 + 8e− + 6H2O → CH4 + 8OH−, respectively. C2H4 is a particularly valuable product as it has widespread applications in many industries including agriculture and polymer manufacturing. To date, the most promising catalyst that can electroreduce CO2 to C2H4 is copper metal.2,3 However, alongside C2H4, many carbonaceous side-products including methane (CH4), carbon monoxide (CO) and formate (HCOO−) are also simultaneously formed.4–7 Furthermore, the Cu catalysts are highly susceptible to poisoning and deactivation, commonly within 30 minutes from the start of the CO2 reduction process.8,9 For the above reasons, considerable effort has been dedicated to understand the structure and composition of materials with the aim of developing catalysts that can selectively reduce CO2 to C2H4 over a long period of time.7,10,11Polycrystalline Cu surfaces do not show significant preference towards ethylene formation, with a C2H4/CH4 product ratio of around 1 to 2.3–5,12,13 It is thought that their lack of selectivity originates from the great heterogeneity of sites present on the polycrystalline surface, each with different catalytic activities. This is underscored by the work of Hori et al. which investigated the effect of different Cu facets on the selectivity of CO2 electroreduction.7,14 Single crystal (100)Cu surfaces were found to favor the formation of C2H4 more than (111)Cu, as indicated by their C2H4/CH4 ratios of 1.3 and 0.2, respectively.7 Interestingly, when the high index (711)Cu, (911)Cu, and (810)Cu planes, formed by cleaving (100)Cu, were examined, they displayed even higher selectivity towards C2H4 with the C2H4/CH4 ratio value increasing to 10 for (711)Cu. Whether the selectivity is due to increased population of atomic steps exposed in such high index facets or a certain periodic spacing in the exposed copper terraces, it is clear that certain Cu facets do exhibit preference towards different hydrocarbons and it is possible to tune them.
Besides single crystal Cu surfaces, enhancements in C2H4 selectivity during CO2 reduction were also observed on CuO and CuI-halide coated electrodes.15,16 Sustaining stable catalytic activities on these catalysts is however more challenging because CuII or CuI will inevitably be reduced to Cu metal during the CO2 reduction process. Thus, in the case of Cu oxide, intermittent anodic pulses of +1 to +2 V had to be applied during the CO2 reduction process to maintain the Cu oxide in its catalytically active oxidation state.15 Large cathodic potentials of up to −3 V (vs. Ag/AgCl) were also required for the CO2 reduction process, which significantly increased the energy input of the system. High surface area Cu nanoparticles have been shown to offer good selectivity towards hydrocarbon formation, especially C2H4.6 It was proposed that the numerous steps and edges formed on the surfaces of the Cu nanoparticles could be crucial for selective C2H4 formation. In support of this, quantum chemical simulations indicated that reaction intermediates like CHO* are more stable on the (211)Cu surface steps than on (100)Cu terraces. This could lead to their concentration build up and eventual dimerization to C2H4.17 More recently, thick Cu2O films have also shown promising selectivity towards C2H4 formation.18 Local pH changes associated with the thickness of the films were proposed to induce this selectivity, as pH has been shown to alter the production rates of various CO2 reduction products.10
These preceding studies have inspired us to develop a stable C2H4-selective electrocatalyst based on copper and understand how this selectivity came to fruition. Herein, we report the activity and characteristics of novel copper mesocrystals for the selective electroreduction of CO2 to C2H4. These mesocrystals were facilely prepared by electrochemically roughening a Cu electrode in KCl electrolyte to produce a thin overlayer of CuCl. CuCl was then reduced in situ during the CO2 electroreduction process to yield Cu mesocrystals. These catalysts are highly active for CO2 reduction to C2H4, which was demonstrated by the high C2H4/CH4 Faradaic efficiency (FE) ratio of ~18. The FE of C2H4 results in up to ~81% of the total carbonaceous products. Thorough materials analysis reveals that the morphology of these mesostructurally arranged 30–50 nm copper particles displayed both (100)Cu facets and numerous steps/edges. Cyclic voltammetry studies indicate that the C2H4 selectivity of these mesocrystals can be correlated with the propensity and stronger adsorption of CO intermediates on their surfaces. Control experiments performed on copper nanoparticles prepared by pulse electrodeposition or electropolished Cu surfaces show that this selectivity could not be simply replicated by ordinary nanoparticulate or bulk copper surfaces.
Our results demonstrate that both (100)Cu facets and steps formed on copper mesocrystals are essential for the selective reduction of CO2 to C2H4. These copper mesocrystals remain very active and selective for C2H4 production for over six hours. They are also robust enough to be taken out mid-reaction, exposed to the environment and reintroduced to fresh electrolyte for a full round of CO2 electroreduction without significant loss in activity. Addition of Cl− to the electrolyte did not affect the activity of the copper mesocrystals significantly. Our discovery of a facilely prepared, robust and selective catalyst based on an earth-abundant metal such as Cu represents a major step forward towards the realization of industrial-scale reduction of CO2 to C2H4.
Experimental
Catalyst preparation
Only deionized Type I water (18.2 MΩ cm, Barnstead, Thermo Scientific) was used for washing and for preparing solutions. 10 mm diameter Cu metal discs (99.99%, Goodfellow Inc.) were used as the base to prepare all catalysts. These discs were mechanically polished with SiC paper and alumina slurries, resulting in a mirror-like finish.19 Between each step, the copper discs were ultrasonicated in deionized water and 0.1 M KOH to remove any alumina particles left on their surface.The following catalysts were prepared.
1. Catalyst A: Cu mesocrystals. Polished copper discs were electrochemically roughened in aqueous 0.1 M KCl using five triangular potential scans ranging from 0.24 V to 1.74 V (vs. RHE) at a rate of 500 mV s−1. During each cycle, the potential was held at the positive and negative limits for 10 and 5 seconds, respectively. They were then rinsed and washed with copious amounts of deionized water several times. Cu mesocrystals were then formed in situ during the CO2 electroreduction process in CO2-saturated 0.1 M KHCO3 (99.99%, Sigma Aldrich). These parameters were the optimum for obtaining a mechanically stable layer of Cu mesocrystals. This sample shall be addressed in this article as catalyst A or Cu mesocrystals.
2. Catalyst B: Cu nanoparticles. Cu nanoparticles were electrodeposited on polished Cu discs using 3000 cycles of galvanostatic pulse deposition. A square wave pulsed current setting was used, alternated between −4.94 and +2.49 mA cm−2, with a 100 ms duration for both anodic and cathodic pulses. The electrolyte consisted of 0.01 M CuSO4 (extra pure, GCE Laboratory Chemicals), 0.1 M Na2SO4 (≥99.0%, Sigma Aldrich) and 0.1 M H2SO4 (98%, RCI Labscan). This sample shall be addressed as catalyst B or Cu nanoparticles.
3. Catalyst C: electropolished Cu. Mechanically-polished Cu disks were electropolished in phosphoric acid (85%, RCI Labscan) at 259.7 mA cm−2 anodic current for 60 seconds and then rinsed with deionized water.6 This sample shall be addressed as catalyst C or electropolished Cu.
Online electrochemical Gas Chromatography (GC)
Electrochemical measurements were performed using a Gamry 600 galvanostat/potentiostat in a three-electrode cell configuration with an Ag/AgCl reference electrode (ET072, eDAQ) and a Pt mesh counter electrode. The potential of the Ag/AgCl reference electrode was checked daily against a reversible hydrogen electrode (HydroFlex®, Gaskatel).A custom-made, gas-tight two compartment polytetrafluoroethylene cell was used for CO2 reduction experiments.4 The anodic and cathodic compartments were separated by an anion-exchange membrane (Selemion AMV, AGC Asahi Glass). A holder gripped the working electrode firmly in place and exposed only a circular geometric surface area of 0.385 cm2. The cathodic compartment was filled with 32 ml of electrolyte, leaving approximately 3 ml of headspace. Before each measurement, the aqueous 0.1 M KHCO3 (99.99%, Sigma Aldrich) electrolyte was saturated with CO2 gas (99.999%, Linde Gas) for at least 30 minutes. All experiments were performed at 298 K.
CO2 was continuously pumped into both the catholyte and anolyte during the CO2 reduction process at a rate of 20 sccm using calibrated mass flow controllers (MC-100SCCM-D, Alicat Scientific). The gas outlet of the cathodic compartment was connected to a gas chromatograph (GC-7890A, Agilent Technologies) for periodical sampling. The GC is equipped with a thermal conductivity detector (TCD) for detecting H2 and two flame ionization detectors (FID, one fitted with a methanizer) for detecting hydrocarbons and CO. The carrier gases for the TCD and FIDs are nitrogen and helium, respectively. The GC was calibrated regularly using standard gas mixtures (Singapore Oxygen Air Liquide and National Oxygen Pte Ltd) under standard conditions (1 atm, 298 K).
A typical CO2 electroreduction experiment at a constant applied voltage spanned over 4200 seconds. A total of six gas aliquots (2.5 cm3 each) were measured. They were injected into the GC every 10 minutes by an automated sampler. The first injection is performed 230 seconds after the start of the CO2 reduction reaction; this ensures adequate flushing of the transfer line of atmospheric contaminants. The GC data collected in this work are translated to Faradaic efficiencies (FE) (ESI† section S1). The Faradaic efficiencies of the products in all measurements amount to ~90% or better, demonstrating that all of the major electroreduction products have been detected.
NMR detection of formate
After each CO2 electroreduction experiment, 2 mL of the catholyte was mixed with 0.1 mL of an internal standard consisting of 25 mM phenol (99.5%, Scharlau) and 5 mM dimethyl sulfoxide (99.9%, Quality Reagent Chemical). 0.5 mL of this aqueous mixture was then added to 0.7 mL of D2O (99.96% deuterium, Cambridge Isotope Laboratories). Solvent suppression was applied to decrease the intensity of the water peak. The results presented here are the average of 52 1D 1H NMR scans (Avance 300, Bruker) and processed using the WIN-NMR software (Bruker). Formate (chemical shift 8.3 ppm) was the only CO2 reduction product detected in the electrolyte.4Characterization of copper catalysts
The surface morphologies of the Cu catalysts were analyzed by scanning electron microscopy (SEM, JEOL JSM-6710F) operated in secondary electron mode (5 keV, 10 mA probe current). High resolution transmission electron micrographs of selected catalysts were obtained using a JEOL TEM-3010. For the TEM measurements, the top layer of the Cu samples was scraped off the substrate and suspended in isopropanol using ultrasonication. A drop of the homogenized solution was then drop-casted on a 300 mesh nickel grid coated with lacey carbon (LC325-Ni, Electron Microscopy Sciences). Only smaller particles, with sizes ≤50 nm, could be analyzed by TEM as the electron beam cannot penetrate through larger particles.X-ray photoelectron spectroscopy (XPS) was performed using a Kratos AXIS Ultra (Al Kα emission source). An ultrathin Pt film was sputter-coated onto the catalyst surface just before XPS measurement as the internal standard for binding energy calibration. X-ray diffraction (XRD) of the films was performed using a Bruker D8 (Cu Kα, 40 kV, 40 mA). The incoming X-ray angle was kept at 0.1° to minimize the diffraction signal from the underlying copper disc.
iR drop compensation and reporting of working electrode potentials and currents
iR drop compensation was performed during the electrochemical measurements using the current interrupt technique. All of the potentials measured in this work are referenced to the RHE using the following conversion:| ERHE(V) = EAg/AgCl(V) + 0.205 V + (0.059 V × pH) |
The pH values of the electrolytes are listed in the ESI† S2. The current density values reported in this work were normalized to the geometric surface area.
Results and discussion
GC results: stable ethylene selectivity of copper mesocrystals
Representative chronoamperograms of catalysts A, B and C (A = Cu mesocrystals, B = Cu nanoparticles and C = electropolished Cu) during CO2 electroreduction at −0.99 V are presented in Fig. 1A. Catalyst A exhibited significantly higher reduction current during the first ~200 seconds that peaked at −65 mA cm−2 before stabilizing to ca. −25 mA cm−2.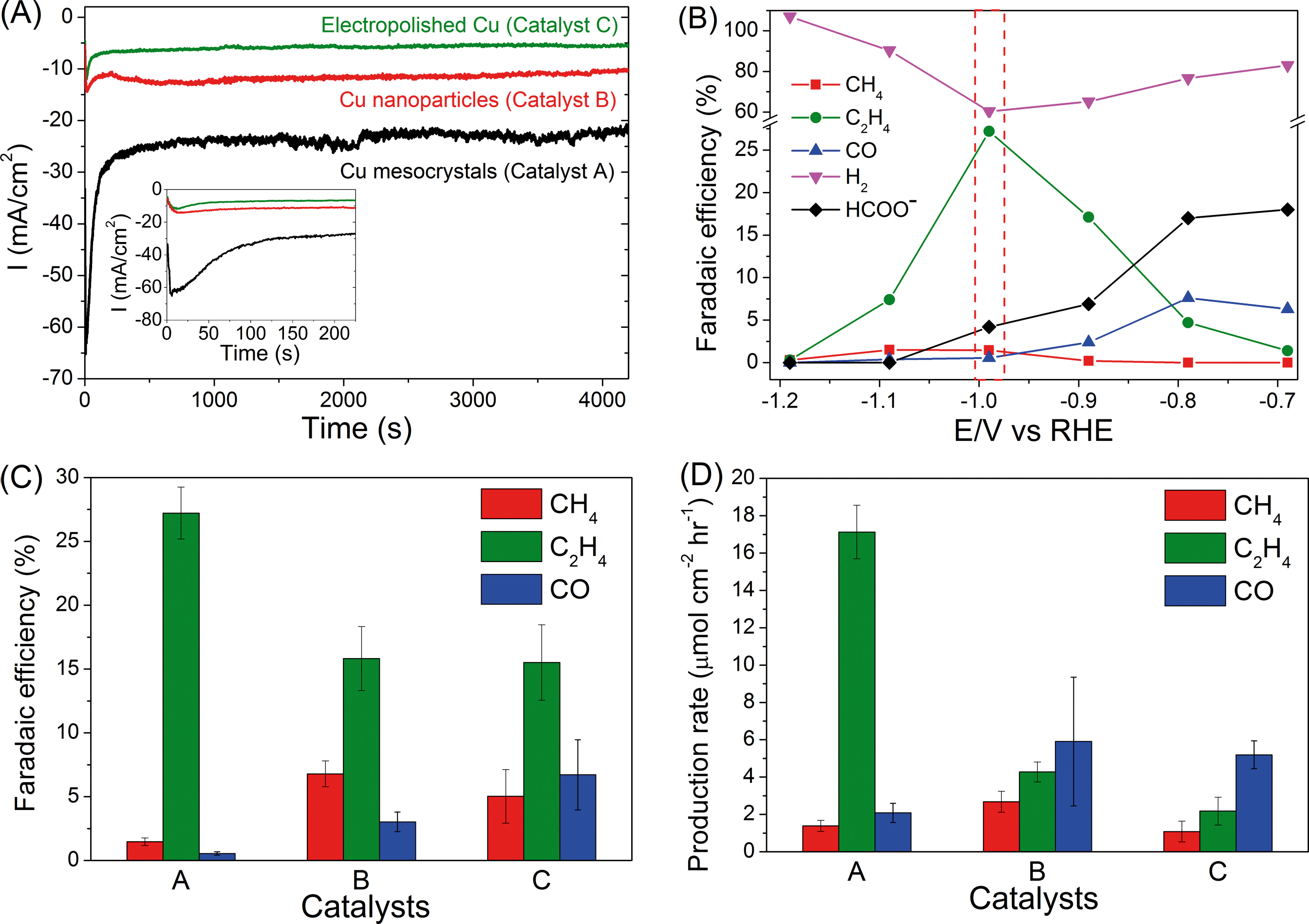 | ||
| Fig. 1 (A) CO2 reduction current as a function of time for catalysts A (Cu mesocrystals), B (Cu nanoparticles) and C (electropolished Cu). Potential applied: −0.99 V. The inset is a zoomed-in picture of the reduction currents at the start of the CO2 reduction process. (B) Faradaic efficiencies of the CO2 electroreduction products of catalyst A as a function of potential. A comparison of the (C) Faradaic efficiencies and (D) production rates of CO2 electroreduction products on catalysts A, B and C at −0.99 V. | ||
On the basis of ex situ characterization of catalyst A at different times during the electrochemical CO2 reduction (see the following section), this temporal reduction current was attributed to the reduction of CuCl to Cu mesocrystals. When steady state currents were compared, catalyst A always displayed the highest current density, which is approximately twice and three times those of catalysts B and C, respectively (ESI† section S3). We have determined, from their cyclic voltammograms, that the ratio of the electroactive surface areas of catalysts A, B and C is approximately 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1. Hence, the difference in the measured currents can be attributed to differences in the surface areas of the catalysts.
:2:1. Hence, the difference in the measured currents can be attributed to differences in the surface areas of the catalysts.
The detected products in catalysts A, B and C are methane, ethylene, carbon monoxide, formate and hydrogen, and their Faradaic efficiencies vary with the applied working potentials (Fig. 1B, ESI† section S4). Strikingly, only catalyst A exhibited significant preference towards C2H4 formation. At −0.99 V, the FE values of C2H4 and CH4 are 27.2% and 1.47%, respectively, which gives a C2H4/CH4 ratio of ~18 (Fig. 1B–C). This ratio is greater than the C2H4/CH4 selectivity of ~10 found for high index (711)Cu single crystal copper surfaces.7 As a comparison, catalysts B and C exhibited C2H4/CH4 ratios of only 2.3 and 3.1, respectively, consistent with the ratios reported previously for polycrystalline and sputtered Cu electrodes (Fig. 1C).4–6,20 C2H6 was also detected on Cu mesocrystals, with a Faradaic efficiency of ~0.3%, (ESI† section S1) but not on the other two catalysts. The Faradaic efficiency of C2H4 formation using catalyst A is ~81% of the total carbonaceous product yield (Fig. 1C). This is significantly higher than the C2H4 yield of 45% and 40% found for catalysts B and C. This figure is also higher than the 61% C2H4 yield found for (711)Cu.7 In terms of the production rate, C2H4 is produced at a rate of ~17 μmol cm−2 h−1 using catalyst A (Fig. 1D). This is approximately an order of magnitude higher than that observed for catalyst C. These figures of merit demonstrate that there is a significant improvement in the selectivity of CO2 reduction to C2H4 when the Cu mesocrystal catalyst is used.
The rapid deactivation of the Cu catalysts is a major obstacle that will hinder any efforts to industrially scale up the CO2 electroreduction process to produce C2H4.8,9 It is therefore important to assess the stability of our copper electrodes over longer CO2 electroreduction periods. A bias of −0.99 V was chosen as it showed the optimum selectivity towards C2H4, especially for copper mesocrystals (catalyst A, Fig. 1B). Results presented in Fig. 2 demonstrate that the copper mesocrystals displayed superior and stable catalytic activity towards C2H4 formation for >6 hours (Fig. 2A). In contrast, catalysts B and C showed rapid catalytic deactivation. The Faradaic efficiencies of their CH4 and C2H4 products declined over the course of the electrolysis, with the FE of C2H4 using catalyst C decreasing by 75%. H2 production has also increased for these two catalysts. Deactivation of Cu catalysts has been previously attributed to contamination of trace amounts of heavy metals present in the electrolyte.8 The subsequent sections of this work will be devoted to in-depth materials and electrochemical investigation of the origin of the higher ethylene selectivity and stability of our Cu mesocrystal catalyst.
 | ||
| Fig. 2 Composite plot of current density vs. time (black trace) at −0.99 V and Faradaic efficiencies of CO (blue trace), C2H4 (green), CH4 (red) and H2 (inset, magenta) over six hours of CO2 reduction on (A) catalyst A, (B) catalyst B, and (C) catalyst C. | ||
Chemical and structural analyses of the Cu mesocrystals
Cu mesocrystals (catalyst A) were analyzed before, during and after CO2 reduction. The initial steps of Cu mesocrystal formation involved five cycles of electrochemical roughening of the polished Cu discs in 0.1 M KCl solution. After this roughening procedure, aggregated particles of ~400–1000 nm in size were found on top of the polished Cu (Fig. 3A–B). X-ray diffraction and XPS revealed that these particles are CuCl (Fig. 3C–D).‡ The CuCl-covered Cu discs were then rinsed with deionized water and used directly for electrochemical CO2 reduction. | ||
| Fig. 3 SEM micrographs of (A) polished Cu and (B) catalyst A before CO2 reduction. (C) X-ray diffraction and (D) XPS data for catalyst A before (black trace) and after 4200 seconds of CO2 reduction (red trace). The hashes in (C) indicate trace amounts of CuCl2 while the asterisks in (D) indicate the internal Pt standard. The inset in panel (D) shows a higher resolution scan region where the Cl 2p XPS peak occurs. Time resolved ex situ SEM micrographs of catalyst A after (E) 10, (F) 100, and (G–H) 4200 seconds of CO2 electroreduction. (I–L) HRTEM micrographs of the same catalyst in (G–H) under increasing magnifications. Only lattice parameters belonging to the Cu metal were detected. The dark arrows indicate some of the steps and edges present on the Cu mesocrystals. | ||
Within 10–20 seconds from the application of −0.99 V to the electrode in CO2-saturated KHCO3, the CuCl layer was converted to cuboids approx. 400–500 nm across (Fig. 3E). A longer exposure time (~100 seconds) resulted in the growth of smaller nanoparticles (30–50 nm) over the well-defined cuboids' surface (Fig. 3F–H; see also the ESI† section S5).
These small and rough nanoparticles were determined by XRD and XPS to be crystalline copper metal (Fig. 3C–D). No CuCl, Cl, or metal contaminants such as Fe or Zn could be detected on the catalyst's XPS spectra after 4200 seconds of the CO2 electrochemical reduction process. As there should be no Cu ions present in the KHCO3 electrolyte initially, the formation of Cu mesocrystals from CuCl must have proceeded via dissolution–redeposition21 followed by oriented attachment22 or a self-limiting electrochemical aggregative growth mechanism,23 with the large Cu cuboids' surface acting as the orientation template.24
The representative high resolution transmission electron micrograph of the 30–50 nm Cu nanoparticle clusters, obtained by scraping catalyst A after 4200 seconds of CO2 electroreduction, is shown in Fig. 3I. The marked d-spacings in these nanoparticles correspond to those of the Cu metal only (Fig. 3J). The most striking observation in the HRTEM images is the clear presence of numerous atomic steps and (100)Cu terraces on the surfaces of these copper nanoparticles (Fig. 3K–L, also see the ESI† S6). This arrangement is reminiscent of the high index facet exposed by off-cutting (100)Cu single crystals, exposing numerous high index atomic steps with (100)Cu terraces in between.7 However, as our Cu mesocrystals have larger surface areas than single crystal copper surfaces, there will be significantly more high index atomic steps. The (100)Cu surface terminations of the Cu mesocrystals may have been promoted by the underlying Cu cuboid template when it was formed from CuCl (Fig. 3E). This process usually has a lower activation energy.25 Similar mesocrystals were also observed after CO2 electroreduction at different potentials between −0.69 and −0.89 V (ESI† section S7). We thus conclude that the selective reductions of CO2 to ethylene at other potentials, while showing variable product ratios, were catalyzed by the same Cu mesocrystals.
Catalyst B (Cu nanoparticles) was introduced to assess the importance of the well-defined cuboid surface (in Fig. 3E) as the template for the mesocrystals' nucleation and mesostructural arrangement. These electrodeposited copper nanoparticles are relatively small and possess high surface areas (Fig. 4A–B). However, as they were grown directly on polycrystalline copper discs, which do not have a significant number of exposed (100)Cu growth templates like catalyst A, rounded particles are formed without any clear (100)Cu termination. This morphology was retained throughout the CO2 reduction at −0.99 V (Fig. 4C). High resolution TEM revealed that the surfaces of these rounded nanoparticles were smoothly gradated, possibly with many steps, but no terraces could be found (Fig. 4D inset, see also the ESI† S8). This surface can thus be classified as an extremely high index plane with many steps but with no terraces. Although Hori et al. have shown that high index stepped surfaces are important for C2H4 selectivity, exposing an even higher index plane than (711)Cu appears to be detrimental to C2H4 selectivity.7 This suggests that (100)Cu terraces are important and high C2H4 selectivity can only be achieved by balancing the step and terrace population. Thus unsurprisingly, catalyst B only displayed a C2H4/CH4 ratio of 2.3, not very different from regular polycrystalline copper.4
 | ||
| Fig. 4 SEM micrographs of electrodeposited copper nanoparticles (A) before and (B) after CO2 reduction. (C) A representative HRTEM micrograph of electrodeposited Cu nanoparticles after CO2 reduction. (D) Increased magnification of the marked area in (C). SEM micrographs of the electropolished Cu surface (E) before and (F) after CO2 reduction. | ||
Catalyst C was introduced to represent smooth polycrystalline Cu surfaces. As seen in Fig. 4E, electropolishing strips the Cu surface of protrusions by dissolving them in phosphoric acid, resulting in a very smooth surface. As a result, catalyst C was not found to be selective towards C2H4 formation, which is in agreement with previous reports on polycrystalline copper.4,5 After CO2 reduction, the surface becomes slightly rougher with newly formed protrusions (Fig. 4F). The roughening could be attributed to the prolonged application of cathodic potentials. However, this roughened surface does not exhibit any selectivity towards C2H4 (Fig. 2C).
CO adsorption studies and robustness of Cu mesocrystals
Thus far, we have demonstrated that copper mesocrystals are highly selective towards the electrocatalytic reduction of CO2 to C2H4. We have also established, by comparing different copper surfaces, that the presence of both (100)Cu facets and atomic steps/edges is essential for their selectivity.The dimerization of CO (or CHO*) on copper has been postulated to be the key step for C2H4 formation during CO2 reduction.26–28 This suggests that the population of active sites responsible for C2H4 formation can be probed by CO adsorption.
Baseline measurements of these three catalysts in Ar-saturated 0.1 M KHCO3 were first taken. The oxidation and reduction potentials of the Cu catalysts in all three catalysts agree well with previous reports (Fig. 5A).6 Cu mesocrystals (catalyst A) show the highest current density and a more complex set of oxidation/reduction peaks. These observations can be respectively attributed to their larger surface area and to the greater number of exposed steps/facets on their surface.6,29 The CV measurements in CO- and CO2-saturated electrolytes were very similar (Fig. 5B–C), which suggests that these two compounds can react similarly on Cu surfaces.20 The shoulders at −0.7 to −0.9 V in the CVs shown in Fig. 5B–C are usually attributed to the formation of adsorbed CO.30 However, more recent work by Kortlever et al. indicates that these peaks are instead related to the direct reduction of bicarbonate to formate.31
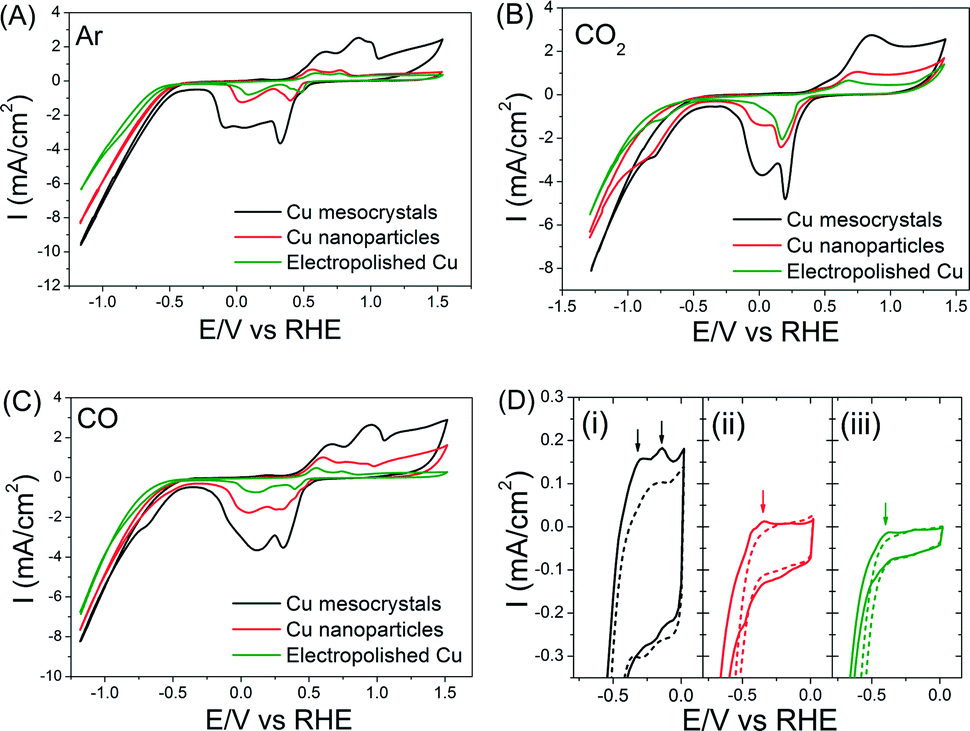 | ||
| Fig. 5 Cyclic voltammograms of the three Cu catalysts in (A) Ar-, (B) CO2- and (C) CO-saturated 0.1 M KHCO3. (D) Close comparison of the CVs of (i) Cu mesocrystals, (ii) Cu nanoparticles and (iii) electropolished Cu in Ar- (broken line) and CO- (solid line) saturated 0.1 M KHCO3. The arrows indicate CO adsorption/desorption features around −0.14 V (only on Cu mesocrystals) and −0.35 V (on all samples). These CV measurements were performed after 4200 seconds of CO2 electroreduction. | ||
When the CVs of the electrodes in the CO-saturated electrolyte were examined between 0 to −0.5 V, an anodic peak feature around −0.14 V was observed exclusively on the Cu mesocrystals (Fig. 5D(i)). In contrast, both the Cu nanoparticles and the electropolished Cu showed only a faint peak at −0.35 V. These peaks are reminiscent of the CO adsorption/desorption features observed in single crystal copper surfaces.7 The more positive position and greater peak intensity in the CV of the Cu mesocrystals indicate that they adsorb CO more readily as compared to the other two catalysts.32
Since dimerization of CO (or CHO*) is a key step in ethylene production, the fact that CO can be better adsorbed on the Cu mesocrystals can explain their preferred selectivity for ethylene production. The likely consequence is that CO and CH4 production rates would be correspondingly lower on the Cu mesocrystals. This was indeed what we observed (Fig. 1D). The Faradaic efficiency of CO during CO2 electroreduction on different Cu single crystal surfaces has also been shown to be inversely proportional to that of C2H4 in the literature.7
Unlike catalysts B and C, catalyst A's C2H4 production rate is constant for six hours (Fig. 2). A reason for this enhanced stability could be the minimum exposure of the Cu mesocrystals to the atmosphere as they were formed in situ from CuCl during the CO2 reduction process. However, we found that catalyst A remained very active even when it was taken out mid-reaction, exposed to ambient atmosphere for several minutes, and then reintroduced back to the cell for further CO2 reduction (ESI† section S9). The presence of Cl− has also been reported to alter product selectivity in CO2 electroreduction.3,16 However, we found that the activity and product distribution of CO2 reduction on the Cu mesocrystals were not affected by Cl− added to the KHCO3 electrolyte (ESI† S9). These experiments demonstrate that the Cu mesocrystals are inherently more stable and resistant to atmospheric and trace contaminants than ordinary Cu surfaces or nanoparticles.
Conclusion
In this work, we report the highly selective and stable electroreduction of CO2 to C2H4 on Cu mesocrystals. A C2H4/CH4 FE ratio of ~18 was achieved. The in situ formation of this novel electrocatalyst from CuCl resulted in the mesostructurally arranged 30–50 nm (100)Cu terminated particles bearing many steps and edges. These specific surface arrangements lead to higher availability of active sites on Cu mesocrystals, indicated by higher and stronger CO adsorption, a crucial factor that leads to high C2H4 selectivity.Control CO2 electroreduction experiments on electrodeposited Cu nanoparticles and electropolished Cu could not replicate the high C2H4 selectivity exhibited by Cu mesocrystals. Despite having large surface areas, electrodeposited Cu nanoparticles appeared more rounded and did not exhibit clear (100)Cu facets or terraces. They displayed a similar product distribution as the electropolished Cu surface. These control experiments affirmed that the presence of (100)Cu with steps as imaged in the Cu mesocrystals is indeed essential for the selective reduction of CO2 to C2H4.
Most remarkably, our Cu mesocrystals are stable and selective towards C2H4 for over six hours. Exposure to chloride anions during reaction or atmospheric contaminants also does not significantly change the catalytic activity of the Cu mesocrystals. We believe that the development of facilely prepared, stable and selective Cu mesocrystals is a significant step towards the realization of industrial-scale CO2 reduction to ethylene.
Acknowledgements
This work is supported by a start-up grant (R143-000-515-133) from the National University of Singapore. A. D. H. is grateful for a research fellowship from Singapore-Peking-Oxford Research Enterprise (R-706-000-100-414).The authors thank Dr. Yuan Ze Liang (NUS Chemical and Biomolecular Engineering) for XPS measurements and Mr. Lee Ka Yau (NUS Chemistry) for TEM measurements.
Notes and references
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 Search PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. Vayenas, R. White and M. Gamboa-Aldeco, Springer New York, 2008, ch. 3, vol. 42, pp. 89–189 Search PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 CAS.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 Search PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Norskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 Search PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 Search PubMed.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 Search PubMed.
- D. W. DeWulf, T. Jin and A. J. Bard, J. Electrochem. Soc., 1989, 136, 1686–1691 Search PubMed.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53–57 Search PubMed.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 Search PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 Search PubMed.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2001, 106, 15–17 Search PubMed.
- J. Yano, T. Morita, K. Shimano, Y. Nagami and S. Yamasaki, J. Solid State Electrochem., 2007, 11, 554–557 Search PubMed.
- K. Ogura, H. Yano and F. Shirai, J. Electrochem. Soc., 2003, 150, D163–D168 Search PubMed.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pedersen and J. K. Nørskov, Surf. Sci., 2011, 605, 1354–1359 Search PubMed.
- R. Kas, R. Kortlever, A. Milbrat, M. T. M. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 Search PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 Search PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Surf. Sci., 1995, 335, 258–263 Search PubMed.
- L. Yu, H. Sun, J. He, D. Wang, X. Jin, X. Hu and G. Z. Chen, Electrochem. Commun., 2007, 9, 1374–1381 Search PubMed.
- E. J. H. Lee, C. Ribeiro, E. Longo and E. R. Leite, J. Phys. Chem. B, 2005, 109, 20842–20846 Search PubMed.
- J. Ustarroz, J. A. Hammons, T. Altantzis, A. Hubin, S. Bals and H. Terryn, J. Am. Chem. Soc., 2013, 135, 11550–11561 Search PubMed.
- M. Tian, J. Wang, J. Kurtz, T. E. Mallouk and M. H. W. Chan, Nano Lett., 2003, 3, 919–923 Search PubMed.
- K. R. Sarma, P. J. Shlichta, W. R. Wilcox and R. A. Lefever, J. Cryst. Growth, 1997, 174, 487–494 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 Search PubMed.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 Search PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
- A. Hamelin and A. M. Martins, J. Electroanal. Chem., 1996, 407, 13–21 Search PubMed.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1988, 17–19 Search PubMed.
- R. Kortlever, K. H. Tan, Y. Kwon and M. T. M. Koper, J. Solid State Electrochem., 2013, 17, 1843–1849 Search PubMed.
- S. K. Shaw, A. Berna, J. M. Feliu, R. J. Nichols, T. Jacob and D. J. Schiffrin, Phys. Chem. Chem. Phys., 2011, 13, 5242–5251 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Calculation of Faradaic efficiencies, representative raw data, electron micrographs and electrochemical data. See DOI: 10.1039/c4cy00906a |
| ‡ Trace amounts of CuCl2 were also detected on the CuCl film. Its reflexions in the XRD data were marked with hashes in Fig. 3C. |
| This journal is © The Royal Society of Chemistry 2015 |