
Peroxoniobium(V)-catalyzed selective oxidation of sulfides with hydrogen peroxide in water: a sustainable approach†
Sandhya Rani
Gogoi
,
Jeena Jyoti
Boruah
,
Gargi
Sengupta
,
Gangutri
Saikia
,
Kabirun
Ahmed
,
Kusum K.
Bania
and
Nashreen S.
Islam
*
Department of Chemical Sciences, Tezpur University, Napaam, Tezpur - 784 028, Assam, India. E-mail: nsi@tezu.ernet.in; nashreen.islam@rediffmail.com; Tel: +91 3712 267007 Tel: +91 9435380222(Off.)
First published on 19th September 2014
Abstract
An efficient and eco-compatible route for the selective oxidation of a variety of thioethers to the corresponding sulfoxide or sulfone with 30% aqueous H2O2 in water, using newly synthesized peroxoniobium (pNb) complexes as catalysts, is described. The catalysts with formulas Na2[Nb(O2)3(arg)]·2H2O (arg = arginate) (NbA) and Na2[Nb(O2)3(nic)(H2O)]·H2O (nic = nicotinate) (NbN) have been synthesized from the reaction of sodium tetraperoxoniobate with 30% H2O2 and the respective organic ligand in an aqueous medium, and these have been comprehensively characterized by elemental analysis, spectral studies (FTIR, Raman, 1H NMR, 13C NMR and UV-vis), EDX analysis and TGA-DTG analysis. The density functional theory (DFT) method has been used to investigate the structure of the synthesized pNb complexes. The catalysts are physiologically safe and can be reused for at least six reaction cycles without losing their activity or selectivity. The oxidation is chemoselective for sulfides or sulfoxides leaving the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or alcoholic moiety unaffected. The developed methodologies, apart from being high yielding and straightforward, are completely free from halogen, organic co-solvent, or co-catalysts.
C or alcoholic moiety unaffected. The developed methodologies, apart from being high yielding and straightforward, are completely free from halogen, organic co-solvent, or co-catalysts.
1 Introduction
Chemoselective oxidation of organic sulfides represents one of the most fundamentally important reactions in the domain of organic chemistry, which is fascinating from both chemical and biological perspectives.1 The practical utility of sulfoxides and sulfones as high-value commodity chemicals and their versatility as precursors for gaining access to a variety of chemically and biologically active molecules, including drugs and chiral auxiliaries, have been adequately highlighted in the literature.1a,d,e,2A number of highly promising new catalytic strategies for selective sulfoxidation using various oxidants have been developed in the past few years based on transition metal catalysts, particularly those from groups 4–7 in their highest oxidation states such as titanium,3 vanadium,4 chromium,5 iron,6 molybdenum,7 tungsten7c,8 and rhenium.9 Many of the available methods, however, rely upon the use of toxic and volatile organic solvents and harmful oxidants or require harsh reaction conditions, which lower the practical importance of otherwise efficient oxidation catalysts. Thus, notwithstanding the enormous progress in the development of catalytic protocols to achieve selective oxidation of sulfides, the important criterion of ecological sustainability is still a challenging issue to address. Sustainability of a chemical transformation is mainly governed by the solvent, reagents and catalysts used in addition to the work-up procedure employed.6b In view of the current ecological concerns, the demand for catalytic oxidation processes that use benign solvent, green oxidants and reagents which co-produce only innocuous waste seems to have intensified.7c,8,10 Out of the multitude of available organic oxidants, 30% aqueous H2O2 has been recognized as the best waste-preventing terminal oxidant because of the high oxygen content, cost, safety and easy handling.10c–e,11
Water holds great promise as an alternative to traditional organic solvents as it is inexpensive, safe and non-volatile, with unique redox stability and high heat capacity.12 Although, traditionally, water is referred to as “the universal solvent”, in organic synthesis, water has been treated as a contaminant mainly due to the concern regarding solubility.12a However, since the ‘on water’ approach pioneered by Sharpless,12b which demonstrated that solubility is not a requisite to reactivity and that many organic transformations can be performed efficiently in aqueous solvent, there has been phenomenal progress in the field of water-based organic synthesis.12b,13 This also necessitated the development of water-tolerant catalysts to support such transformations. Although very limited, there are reports on metal-catalyzed sulfoxidation reaction in an aqueous medium.6a,7a,10h,14 Very recently, Chakravarthy and co-workers reported a surfactant-based Mo catalyst for selective sulfoxidation of a variety of sulfides in an aqueous medium with 40% H2O2.7a
In the recent past, we have successfully developed a set of new recoverable heterogeneous catalysts based on polymer-immobilized peroxotungsten (pW) and peroxomolybdenum (pMo) complexes, which displayed excellent stability, selectivity and efficiency with respect to yield, TON and TOF for the oxidation of a diverse range of thioethers and dibenzothiophene by H2O2 under very mild conditions.7b,8a Few of these supported catalysts also effectively catalyzed the oxidative bromination of a variety of activated aromatics at ambient temperature and near-neutral pH.15 Furthermore, a number of dimeric and macromolecular water-soluble peroxo compounds of vanadium and tungsten that we prepared could serve as stoichiometric oxidants of organic bromides and sulfides in an aqueous organic medium.16
In our continuing endeavours devoted to developing newer catalysts and methodologies for oxidation under environmentally acceptable reaction conditions, in the present work, we have aspired to design new peroxometal-based catalytic systems for selective sulfoxidation using H2O2 as an oxidant in an aqueous medium. We selected the peroxoniobium (pNb) system as an ideal candidate for our study for a variety of reasons. Most importantly, Nb has been reported to be non-toxic to animals, with LD50 values in several thousands of milligrams per kilogram body weight in contrast to its lighter group 5 element vanadium, which is known to be moderately toxic.17 Catalysis by pNb compounds is a field of growing interest.18 The pNb species generated in situ in the presence of H2O2 have been shown to catalyze the oxidation of sulfides18c–g and alcohol18h and the epoxidation of alkene.18a,b,i For example, Egami et al. used Nb(salan) complexes to catalyze the asymmetric epoxidation of allylic alcohol with a urea–hydrogen peroxide adduct with good selectivity.18a However, the catalytic potential of discreet synthetic heteroligand pNb complexes as oxidation catalysts has rarely been investigated.
A suitable choice of the co-ligand is an important prerequisite in order to obtain stable and well-defined peroxometallates. Moreover, the coordinating ligand has been shown to have a dramatic effect on the reactivity of peroxometallates (pM) which enabled the activity of pM complexes as stoichiometric or catalytic agents to be fine-tuned with ligands.19 In the present investigation, since our focus was to develop physiologically harmless catalysts, we have chosen nicotinic acid (also known as niacin) and the amino acid arginine as potential co-ligands. These ligands possess a carboxylate functional group for easy attachment to the Nb(V) centre in addition to the N-donor site. Carboxylate anions have been known to be excellent co-ligands for stabilising pNb species.20 Consequently, a host of pNb complexes have been reported in recent years possessing ligands with a carboxylate moiety.20
On the other hand, although a number of publications, including those from our laboratory, have dealt with the synthesis and characterization of peroxometal complexes of V,16a–d,21 Mo22 and W16e,21f,23 with amino acid or nicotinic acid as a co-ligand,24 as far as we are aware, arginine and nicotinic acid have not been used so far to obtain a heteroleptic pNb complex in the solid state. Apart from being biologically relevant, these ligands are reasonably inexpensive, water soluble, and commercially available.
We report herein the catalytic activity of a pair of newly synthesised pNb complexes for the controlled oxidation of sulfides with H2O2 in an aqueous medium, in terms of selectivity, yield, reusability and sustainability, to obtain sulfoxide or sulfone.
2 Results and discussion
2.1 Synthesis and characterization
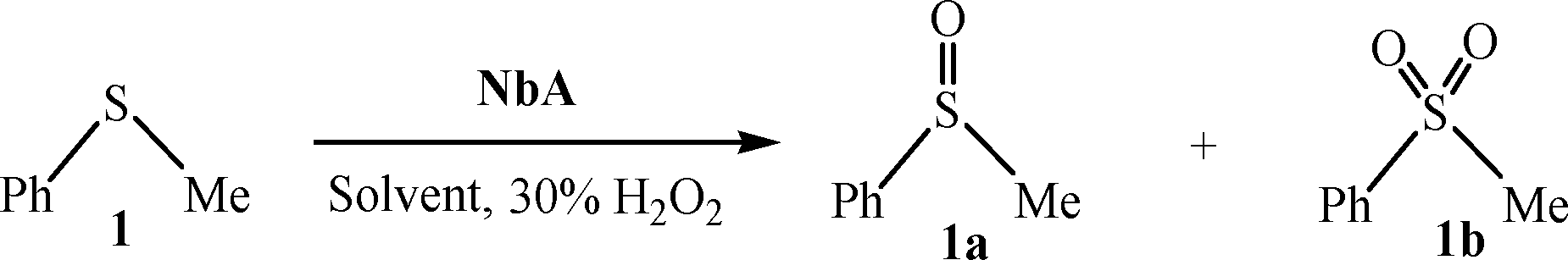
A reasonably straightforward synthetic route has been established to obtain a pair of stable and water-soluble pNb complexes with arginine or niacin as a co-ligand. The procedure is based on the reaction of sodium tetraperoxoniobate with 30% H2O2 and the respective co-ligand in an aqueous medium at near-neutral pH. Synthesis of soluble niobium(V) compounds is considered to be rather challenging because of very low solubility of the common Nb-containing starting materials.18d In the present study, water-soluble Na3[Nb(O2)4]·13H2O has been used as a precursor complex, which was prepared by the reported method.18i The maintenance of pH of ca. 6 was found to be essential for the formation of the triperoxoniobium species and its stabilisation by the chosen organic ligands, occurring in their anionic form, leading to the isolation of the NbA and NbN complexes. The procedure included other requirements such as maintenance of temperature at ≤4 °C and limiting water to that contributed by 30% H2O2. The compounds were observed to be stable in the solid state for several weeks when stored dry in a closed container at <30 °C.The elemental analysis data (Table 1) for each of the title compounds indicated the presence of three peroxide groups and one ancillary ligand, either arginine or niacin per Nb(V) centre with the formula Na2[Nb(O2)3(arg)]·2H2O or Na2[Nb(O2)3(nic)(H2O)]·H2O. Energy-dispersive X-ray (EDX) spectroscopic analysis clearly showed the presence of Nb, Na, C, N and O in the complexes (Fig. 1). The obtained data on the composition of the compounds from EDX analysis were in good agreement with elemental analysis values (Table 1). Despite many attempts, it was not possible to obtain crystals of the compounds large enough for an X-ray crystal structure. The compounds were diamagnetic in nature as was evident from the magnetic susceptibility measurements, in conformity with the presence of Nb in its +5 oxidation state.
| Complexes | % Found from elemental analysis/EDX analysis (theoretical %) | % O22− content (theoretical %) | ||||
|---|---|---|---|---|---|---|
| C | H | N | Nb | Na | ||
| a Determined by AAS. | ||||||
| Na2[Nb(O2)3(arg)]·2H2O | 16.29 | 3.76 | 12.46 | 21.01a | 11.14 | 21.71 |
| 16.15 | 12.76 | 20.84 | 10.47 | |||
| (16.21) | (3.83) | (12.61) | (20.92) | (10.36) | (21.61) | |
| Na2[Nb(O2)3(nic)(H2O)]·H2O | 18.41 | 1.99 | 3.55 | 23.52a | 11.63 | 24.54 |
| 18.37 | 3.61 | 23.57 | 11.66 | |||
| (18.32) | (2.03) | (3.56) | (23.64) | (11.70) | (24.43) | |
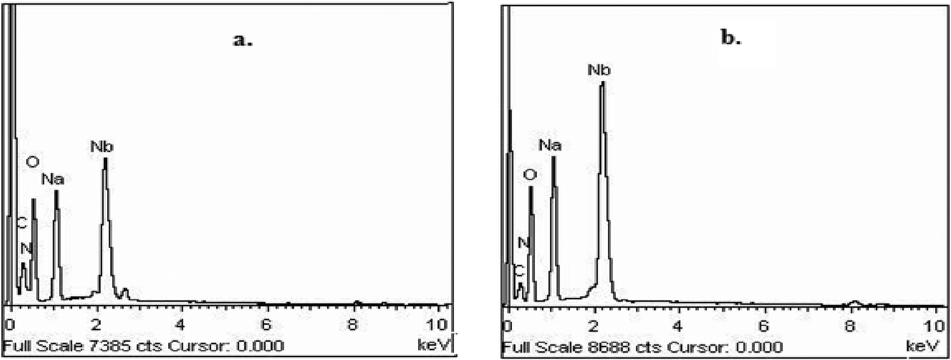 | ||
| Fig. 1 EDX spectra of (a) NbA and (b) NbN. | ||
| Assignment | NbA | NbN | ||
|---|---|---|---|---|
| a s, strong; m, medium; vw, very weak; sh, shoulder. | ||||
| ν(O–O) | IR | Exp. | 846(m), 822(sh), 813(s) | 847(m), 826(sh), 811(s) |
| Calc. | 837, 865, 889 | 807, 872 | ||
| R | Exp. | 823(sh), 847(s), 857(sh) | 817(sh), 847(s), 867(sh) | |
| Calc. | 806, 853, 879 | 813, 828, 876 | ||
| ν s(Nb–O2) | IR | Exp. | 547(s) | 547(s) |
| Calc. | 528 | 523 | ||
| R | Exp. | 538(s) | 542(s) | |
| Calc. | 528 | 554 | ||
| ν as(Nb–O2) | IR | Exp. | 592(m) | 593(sh) |
| Calc. | 604 | 605 | ||
| R | Exp. | 567(sh) | 571(sh) | |
| Calc. | 575 | 598 | ||
| ν as(COO−) | IR | Exp. | 1636(s) | 1625(s) |
| Calc. | 1615 | 1649 | ||
| R | Exp. | 1630(vw) | 1627(sh) | |
| Calc. | 1609 | 1627 | ||
| ν s(COO−) | IR | Exp. | 1406(m) | 1388(s) |
| Calc. | 1375 | 1378 | ||
| R | Exp. | 1410(vw) | 1393(sh) | |
| Calc. | 1403 | 1335 | ||
 | ||
| Fig. 2 Raman spectra of (a) NbN and (b) NbA. | ||
A triperoxo niobium species with a triangularly bonded peroxo group has been reported to exhibit a diagnostic IR pattern with three ν(O–O) bands in the 800–880 cm−1 region.18d,25 The IR and Raman spectra of each of the NbA and NbN compounds showed clear identification of three sharp absorptions representing the characteristic ν(O–O) modes of the peroxo group, in addition to the νasym(Nb–O2) and νsym(Nb–O2) vibrations, as has been expected in the 870–810 and 500–600 cm−1 regions, respectively.18d,25
On the basis of the available reported data pertaining to metal compounds with coordinated amino acid and niacin as ligands, empirical assignments could be derived for the IR and Raman bands observed for the catalysts NbA and NbN.26
The IR spectra of arginine and arginato–metal complexes have been reported previously.27 In the spectrum of free arginine, νas(COO−) and νs(COO−) modes are observed at 1606 and 1425 cm−1, respectively, with Δν = 181 [Δν = νas(COO−) − νs(COO−)], whereas in the case of the NbA complex, the corresponding absorptions appeared at 1636 cm−1 and 1406 cm−1, respectively. The shift of νas(COO−) to a higher frequency and that of νs(COO−) to a lower frequency compared to the free ligand values, with an increase in the Δν (230 cm−1), are typical of the unidentate coordination of the carboxylate group.26a In the Raman spectrum of the compound, weak intensity bands representing νas(COO−) and νs(COO−) vibrations have been located at 1630 and 1410 cm−1, respectively. The ν(C–H) occurred as an intense peak at 2934 cm−1 in the Raman spectrum in contrast to its presence as a weak band in the IR. Two bands typical of ν(NH2) are observed at 3352 and 3288 cm−1 in the free ligand spectrum.27a A new peak appeared at 3193 cm−1 in the spectrum of the NbA complex indicative of a coordinated amino group.27a However, the other absorptions representing ν(N–H) could not be assigned with certainty owing to their overlapping with ν(OH) modes of lattice water, appearing as a broad band in the 3500–3300 cm−1 region.
In the NbN catalyst, the presence of carboxylato-bonded niacin has been clearly demonstrated in its IR and Raman spectra (Table 2). A number of reports are available on IR spectral characterization of metal complexes containing coordinated niacin.26c,e,f Moreover, the IR and Raman spectra of NIA have been thoroughly investigated by Kumar and co-workers and others.26d Solid sodium nicotinate exhibits major carboxylate IR bands at 1620 and 1416 cm−1 for νas(COO−) and νs(COO−), respectively.26f The positions of νas(COO−) and νs(COO−) stretching in both IR and Raman spectra of NbN and the corresponding Δν value (237 cm−1) provide clear evidence of unidentate coordination of a non-protonated carboxylato group to the metal atom. Furthermore, a significantly less intense aromatic ν(CC) band at 1568 cm−1 depicts monodentate coordination of a carboxylate group to Nb.24 Since the ν(CC) mode, ν(CN) absorption and pyridine ring vibrations at 1476, 1016 and 940 cm−1, respectively, do not undergo any positive shift relative to the respective free ligand values, coordination via nitrogen in the pyridine ring can be ruled out safely.26c The presence of water molecules in the complex was apparent from the observed broad and intense ν(OH) bands in the 3500 to 3400 cm−1 region. Further confirmation for the presence of coordinated water in the compound was obtained from the consistent appearance of a moderate intensity signal at 769 cm−1 attributable to the rocking mode of water.28 The intense band appearing at 3077 cm−1 in the Raman spectrum has been ascribed to ν(CH) vibration.
The spectral pattern of the pNb complex with arginine as a co-ligand resembled closely the spectrum of the free ligand by exhibiting 4 major peaks at 3.52, 3.11, 1.74 and 1.59 ppm.30a The spectrum of the complex, however, showed a distinct upfield shift of all the resonances relative to the free ligand, indicating coordination of the ligand to the metal centre.30 Similar observations were made previously in the case of metal compounds containing complexed amino acid.30,31
The 13C chemical shift induced by coordination has been widely utilized as a convenient means of understanding the bonding pattern of ancillary ligands in peroxo–metal complexes.32 The 13C NMR spectrum of the free arginine in D2O displays a typical resonance for a carboxylate carbon atom at 183.17 ppm in addition to the five other well-resolved peaks corresponding to carbon atoms C(2) to C(6).30a The spectrum of NbA, on the other hand, displayed the carboxylate resonance at a lower field of 215.45 ppm, thus testifying to the existence of a complexed carboxylate group.30a,33 The substantial downfield shift relative to free carboxylate, with Δδ (δcomplex − δfree carboxylate) ≈ 32 ppm, indicated strong metal–ligand interaction as has been reported earlier in the case of some other peroxo–metal carboxylate complexes.7b,33 The guanidyl C resonance along with the resonances of alkyl groups (C-5 and C-4) showed very little shift, whereas the resonances of α-CH and β-CH2 groups shifted to a higher field by ca. 1 ppm [Fig. S3(a) (ESI†)]. These results indicated the coordination of carboxylate and amino groups of the ligand to the niobium centre and are consistent with observations made in the case of other reported arginine-containing metal compounds.30a The 1H and 13C NMR spectra of nicotinic acid in D2O have been studied and reported by Khan and co-workers under varying pH conditions.34 A 1H NMR pattern typical of a nicotinate anion was observed with four well-resolved resonances in the spectrum of the NbN compound.34 As expected for the nicotinate anion, the spectrum showed a distinct upfield shift of each of the four resonances corresponding to the aromatic protons H(2) to H(6) relative to the zwitterionic free ligand values [Fig. S2(a) (ESI†)].30b
The 13C spectrum provided further persuasive evidence in support of the presence of a nicotinate anion in the NbN compound by displaying resonances for ring carbon atoms in the region expected for the nicotinate anion.34 The peak attributable to the metal-bound carboxylate carbon, as in the case of NbA, appeared at a very low field of 210.78 ppm compared to the carboxylate resonance of the free nicotinic acid [Fig. S3(b) (ESI†)].7b,33 Occurrence of the carboxylate carbon as a singlet in the spectrum of each of the catalysts, NbA and NbN, indicated a single-carbon environment for complexed carboxylate.33 Thus, the results of the NMR analysis showed evidence of the presence of only one complex species in solution in each case. It is therefore apparent that catalysts did not hydrolyze and retained their solid-state structure in solution.
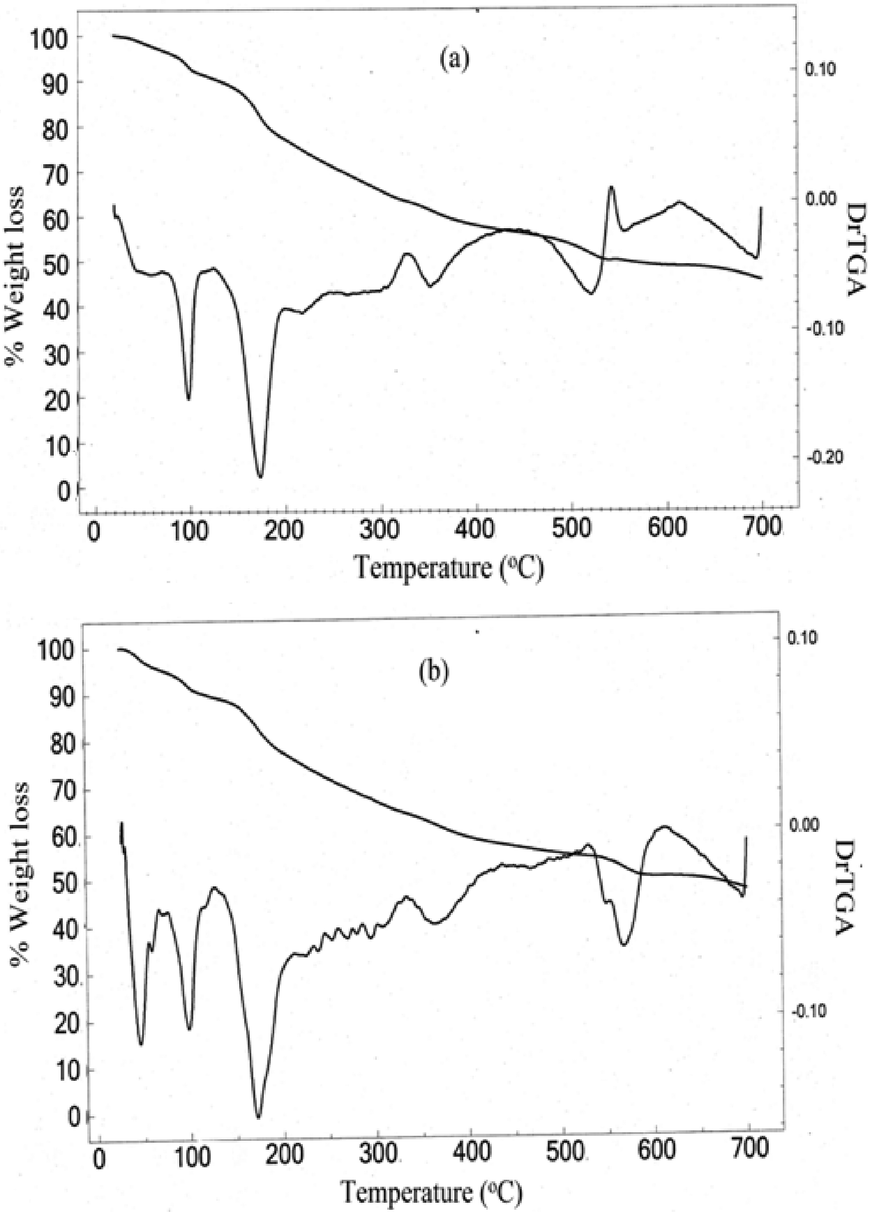 | ||
| Fig. 3 TGA-DTG plot of (a) NbA and (b) NbN. | ||
The first stage of decomposition for NbA [Fig. 3(a)] occurs in the temperature range of 78–102 °C with the liberation of the lattice water from the complex. The corresponding weight loss of 8.9% is in good agreement with the value of 8.1% calculated for two molecules of water of crystallization. The next decomposition stage is in the temperature range of 157–189 °C attributable to loss of peroxo groups from the complexes with a weight loss of 18.8%. As the observed weight loss is slightly less than the expected value, it is likely that part of the oxygen is retained with niobium to form oxoniobium species, as has been observed previously in the case of neat as well as heteroligand pNb complexes.20b,f,35 A further increase in temperature leads to continuous degradation of the arginine ligand up to a final decomposition temperature of 700 °C. The total weight loss which occurred during the overall decomposition process was evaluated to be 54.3%, which agreed well with the theoretically calculated value of 54.6% for the loss of the components, viz. water molecules, coordinated peroxide, and the co-ligand, assuming that four of the ligand oxygen atoms were being retained to form the oxoniobate species as the final degradation product.
It is notable that the thermogram for NbN [Fig. 3(b)] displayed a two-step dehydration process in the temperature range of 45–105 °C, providing conclusive evidence of the presence of outer sphere coordinated water molecules in the compound, consistent with the formula assigned. The decomposition step occurring at relatively higher temperature between 90 and 105 °C, after the initial liberation of the outer sphere water molecule in the temperature range of 45–70 °C, is attributable to the loss of the coordinated water molecule. The observed total weight loss of 9.8% corresponding to the two steps combined is close to the calculated value of 9.2% for the release of two molecules of water from the complex. After the dehydration, the thermal behaviour of the NbA and NbN catalysts is quite similar. The NbN compound undergoes continuous degradation with loss of peroxide in the temperature range of 158–189 °C analogous to the NbA compound, followed by the loss of the niacin ligand up to the final temperature of 700 °C. The residue remaining after the complete degradation of each of the pNb compounds was found to be oxoniobate species as indicated by the IR spectral analysis which showed the typical ν(NbO) stretching and was devoid of absorptions attributable to the peroxo group and the respective co-ligands of the starting complex.
The above results are consistent with the proposed structures of the compounds NbA and NbN shown schematically in Fig. 4. The structure of NbA shows the Nb atom displaying a coordination number of 8, surrounded by the three peroxo groups and the arginate ligand bonded via its unidentate carboxylate group and the amino group. The structure of NbN includes a nicotinate anion occurring as a unidentate ligand bonded to the Nb centre through the carboxylate group, the side-on bound peroxo groups and a coordinated water molecule completing eightfold coordination around the Nb.
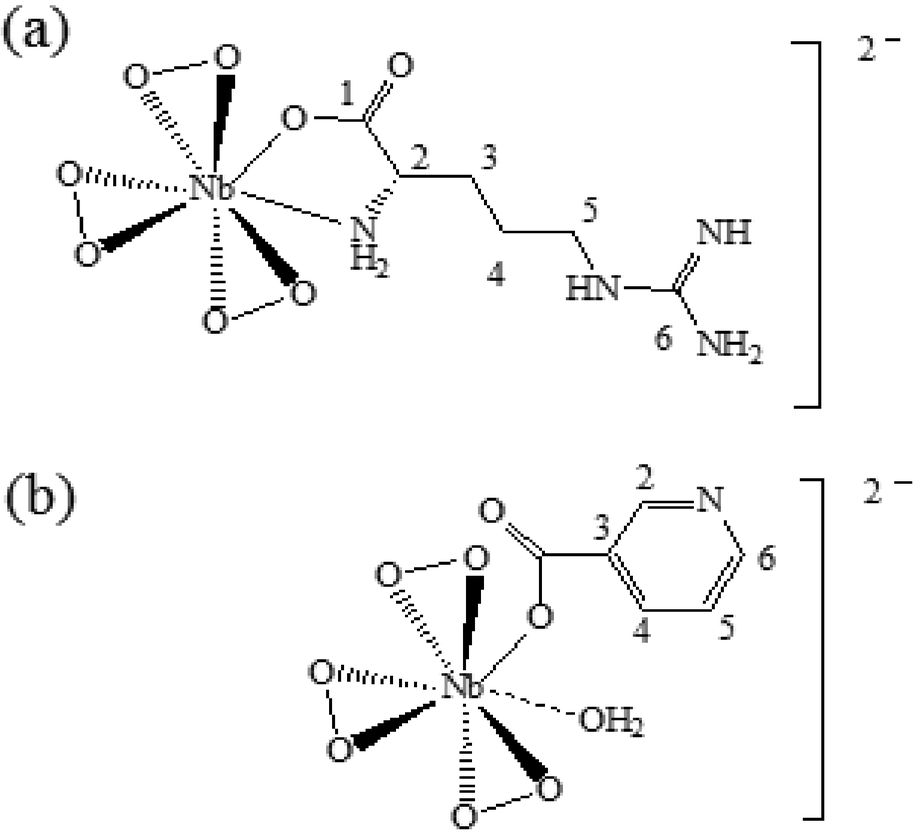 | ||
| Fig. 4 Proposed structure of (a) NbA and (b) NbN. | ||
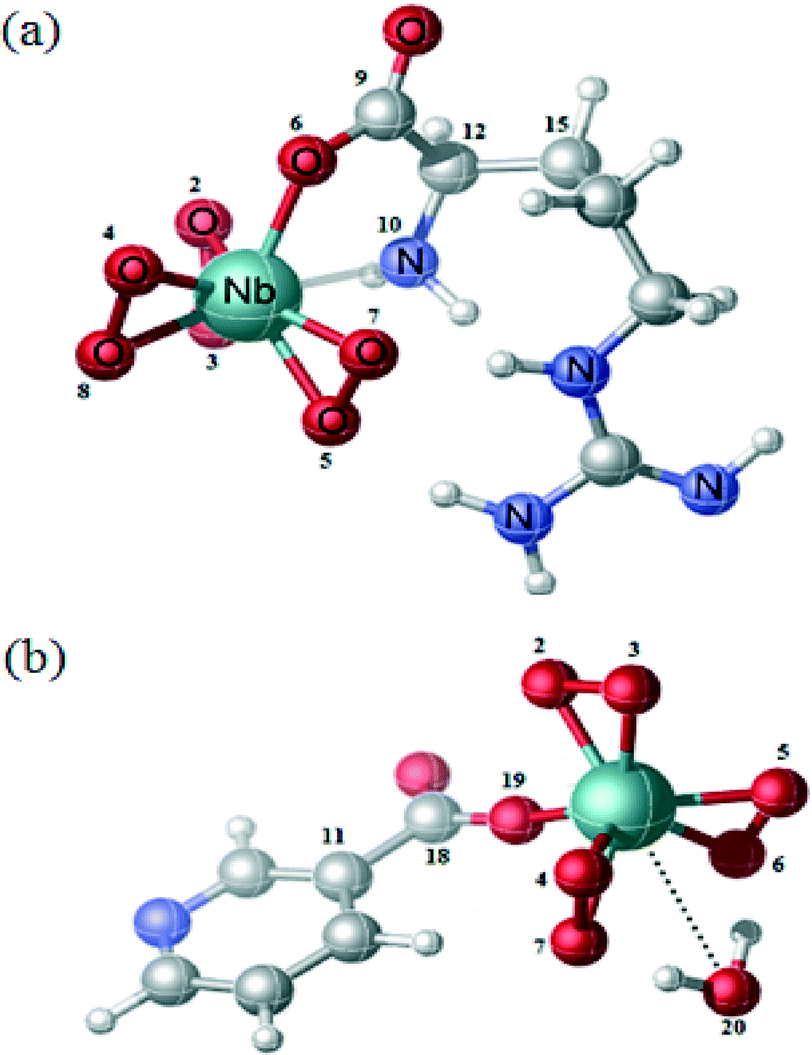 | ||
| Fig. 5 Optimized geometry of (a) NbA and (b) NbN. The numerical numbers represent the labeling of the atoms as in Table 5. | ||
In the case of NbA, the central metal atom (Nb) is coordinated to six oxygen atoms belonging to three η2-peroxo groups, in addition to one oxygen atom from the carboxylato group and a N (amino) atom from the deprotonated argininate ligand. One of the bidentate peroxo groups is in trans position to the co-ligand, whereas the other two are in cis configuration. The coordination sphere of the NbN complex comprises a niobium atom surrounded by seven oxygen atoms contributed by three η2-peroxo groups and one from the unidentate carboxylato group of the nicotinate ligand. The eighth coordination site is satisfied by a water molecule which is weakly bonded to the central atom, as has been observed previously by Djordjevic et al. in the case of nicotinate containing a peroxomolybdenum complex.24 Thus, the structure of NbN can be described as a distorted octahedron with one of the axial distances slightly longer. For both the complexes, the coordination distances are within the range characteristic of the heteroleptic peroxo complexes of niobium(V).20f The geometrical parameters (bond angle and bond length) obtained from DFT calculations are depicted in Table 5. The Nb–O(peroxo) bond lengths are within the range of 1.977 to 2.086 Å, whereas the (O–O) distances range from 1.526 to 1.533 Å. The Nb–O (carboxylate) bond distances in NbA and NbN are 2.162 Å and 2.138 Å, respectively. The Nb–N bond distance in NbA is 2.370 Å. The geometrical parameters obtained from our theoretical calculation correlated well with the reported crystallographic parameters pertaining to other heteroleptic triperoxo niobate complexes with coordination environment comprising N,O- or O-donor co-ligands.18d–i,19a–d,20a–f The small differences observed between the experimental and calculated geometrical parameters are expected as the ground-state geometries were obtained in the gas phase by full geometry optimization.
| Structural indexa | NbA | Structural indexa | NbN |
|---|---|---|---|
| a See Fig. 5 for the atomic numbering. | |||
| Nb–O2 | 2.072 | Nb–O2 | 2.047 |
| Nb–O3 | 2.008 | Nb–O3 | 1.999 |
| Nb–O4 | 1.999 | Nb–O4 | 1.977 |
| Nb–O5 | 2.066 | Nb–O5 | 2.033 |
| Nb–O6 | 2.162 | Nb–O6 | 1.985 |
| Nb–O7 | 2.086 | Nb–O7 | 2.040 |
| Nb–O8 | 2.011 | Nb–O19 | 2.138 |
| Nb–N10 | 2.370 | Nb–O20 | 2.831 |
| O2–O3 | 1.526 | O2–O3 | 1.527 |
| O4–O8 | 1.533 | O5–O6 | 1.530 |
| O5–O7 | 1.530 | O4–O7 | 1.532 |
| C12–C15 | 1.537 | C18–C11 | 1.521 |
| C9–O6 | 1.307 | ||
| ∠O2–Nb–O3 | 43.9 | ∠O2–Nb–O3 | 44.3 |
| ∠O4–Nb–O8 | 44.9 | ∠O4–Nb–O7 | 44.8 |
| ∠O5–Nb–O7 | 44.2 | ∠O5–Nb–O6 | 44.7 |
| ∠O6–Nb–N10 | 72.1 | ∠O3–Nb–O19 | 82.9 |
We have further calculated the vibrational frequencies for the optimized geometries of the compounds and compared the data obtained with the experimentally determined frequencies as illustrated in Table 2. The calculated IR and Raman spectra for the two complexes are found to simulate well with the experimental ones. The small deviations between the calculated and experimental spectral data are anticipated as the calculated spectral data are obtained for those of the gas-phase-optimized geometries. These observed discrepancies appear to be acceptable as the average error for frequencies calculated with the B3LYP functional was reported to be of the order 40–50 cm−1 for inorganic molecules.37 Moreover, such deviations in the FTIR and Raman vibrational bands obtained from DFT-based calculations on eight-coordinated peroxo complexes of niobium(V) are not unprecedented.20f Thus, the results obtained from our theoretical calculations truly support our experimental findings and completely validate the predicted geometries for the synthesized complexes.
2.2 Catalytic activity of the NbA and NbN complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate molar ratio of 1:1000, was conducted in water at ambient temperature under magnetic stirring. The reaction proceeded rapidly within a reasonably short period in the presence of each of the catalysts. The reaction under these conditions was, however, observed to be mildly exothermic, leading to the formation of a mixture of 1a and 1b with a molar ratio of 85:15, as presented in Table 6 (entry 1). Subsequently, the reaction conditions for selective sulfoxidation including substrate:oxidant stoichiometry, catalyst concentration, solvent type and reaction temperature were optimized using MPS as the model substrate and the NbA complex as the catalyst. We have attempted to control the degree of oxidation by lowering the reaction temperature to 0 °C by conducting the reaction in an ice bath.
:substrate molar ratio of 1:1000, was conducted in water at ambient temperature under magnetic stirring. The reaction proceeded rapidly within a reasonably short period in the presence of each of the catalysts. The reaction under these conditions was, however, observed to be mildly exothermic, leading to the formation of a mixture of 1a and 1b with a molar ratio of 85:15, as presented in Table 6 (entry 1). Subsequently, the reaction conditions for selective sulfoxidation including substrate:oxidant stoichiometry, catalyst concentration, solvent type and reaction temperature were optimized using MPS as the model substrate and the NbA complex as the catalyst. We have attempted to control the degree of oxidation by lowering the reaction temperature to 0 °C by conducting the reaction in an ice bath.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Molar ratio (catalyst:MPS) |
30% H2O2 (equiv.) | Solvent | Time (min) | Isolated yield (%) |
1a:1b |
TON | TOF (h−1) |
| a Reactions were carried out with 5 mmol of the substrate in 5 mL of solvent. Catalyst amount = 2.22 mg for 0.005 mmol of NbA. b Reaction at room temperature. c Reaction at 0 °C in ice bath. d Na3[Nb(O2)4]·13H2O as the catalyst. e Using H2SO4 (1 mmol). f Blank experiment (without a catalyst). | ||||||||
| 1 | 1:1000b |
1 | H2O | 75 | 76 | 85:15 |
760 | 608 |
| 2 | 1:1000c |
1 | H2O | 75 | 70 | 100:0 |
700 | 560 |
| 3 | 1:500c |
2 | H2O | 38 | 97 | 85:15 |
485 | 766 |
| 4 | 1:1000c |
2 | H2O | 40 | 95 | 100:0 |
950 | 1425 |
| 5 | 1:2000c |
2 | H2O | 43 | 96 | 100:0 |
1920 | 2679 |
| 6 | 1:2500c |
2 | H2O | 50 | 97 | 100:0 |
2425 | 2910 |
| 7 | 1:3000c |
2 | H2O | 75 | 95 | 100:0 |
2844 | 2275 |
| 8 | 1:2500b |
2 | CH3OH | 115 | 75 | 100:0 |
1875 | 978 |
| 9 | 1:2500b |
2 | CH3CN | 120 | 70 | 100:0 |
1750 | 875 |
| 10d | 1:2500c |
2 | H2O | 95 | 97 | 55:45 |
2425 | 1531 |
| 11e | 1:2500c |
2 | H2O | 20 | 96 | 100:0 |
2400 | 7200 |
| 12f | — | 2 | H2O | 50 | 9 | 90:10 |
— | — |
As has been anticipated, the reaction indeed proceeded smoothly to yield sulfoxide with 100% selectivity and nearly 70% conversion under these conditions. Complete oxidation of MPS to pure sulfoxide could be attained with excellent TOF (without affecting the selectivity) by increasing the oxidant:substrate molar ratio to 2:1 without altering the other reaction conditions.
Next, we have examined the effect of the catalyst amount on the rate and selectivity of the reaction under otherwise identical reaction conditions. As illustrated in Table 6, although the rate was faster at higher catalyst concentration and a reasonably good TOF could be attained even at a catalyst:substrate ratio of 1:3000, the optimal catalyst:substrate molar ratio was found to be 1:2500 for achieving the highest TOF along with complete selectivity. The pNb species has an important contribution in facilitating the reactions, which was confirmed by conducting a control experiment without the catalyst. The reaction was extremely slow and non-selective in the absence of the catalyst, affording a mixture of sulfoxide and sulfone in <9% yield under the optimized reaction conditions (Table 6, entry 12). We have also compared the catalytic efficiency of the newly developed catalysts with the neat tetraperoxoniobate (TPNB) complex Na3[Nb(O2)4]·13H2O under analogous reaction conditions. As shown in Table 6 (entry 10), the reaction proceeded smoothly in the presence of TPNB as well, although product selectivity could not be obtained under the maintained reaction conditions. From these observations, it is apparent that the co-ligand environment influences the catalytic activity of the pNb compounds.
In addition to water, we have screened the sulfoxidation reaction in relatively safer organic solvents such as CH3OH and CH3CN. The solvent effect has been evaluated in the oxidation of MPS. Interestingly, the catalytic protocol for sulfoxidation was found to be compatible with these organic solvents as well; however, the efficiency of the catalysts was observed to vary with the nature of the solvent. Although the catalysts are insoluble in neat organic solvents, including methanol or acetonitrile, in the presence of aqueous H2O2 used as an oxidant, each of the catalysts dissolves completely in these water-miscible solvents, leading to the homogeneity of the catalytic process. It is pertinent to note that we have strategically avoided the use of hazardous chlorinated solvents in the present work. The data presented in Table 6 (entries 8 and 9) demonstrate that although the NbA is highly potent in methanol as well as in acetonitrile, MeOH proved to be a relatively better solvent, affording both product selectivity and high yield at ambient temperature (Table 6, entry 8). Significantly, the selective sulfoxidation in the chosen organic solvents could be achieved at room temperature. It is thus remarkable that by using the same set of catalyst, it is possible to achieve selective oxidation of sulfide in water and organic solvents under mild reaction conditions. Water, however, proved to be the best solvent with respect to catalyst efficiency as demonstrated by higher TOF and product selectivity, notwithstanding the insolubility of most of the chosen organic substrates in water. This is not surprising, considering the observations made by Sharpless et al.12b that several reactions involving water-insoluble organic reactants could proceed optimally in pure water. Our results are also in agreement with earlier findings that chemoselective sulfoxidation is favoured in polar protic solvent with strong hydrogen bonding ability.38
The sulfoxidation reaction has been carried out at the natural pH attained by the reaction mixture (ca. 5). However, a substantial increase in TOF was noted on addition of acid to the reaction medium (Table 6, entry 11). The finding is in accord with the reports related to other peroxo metal systems (Ti, Mo and W),39 where it has been demonstrated that the use of acidic additives led to an improvement in catalytic activity of the peroxometallates. The role of protons in the activation of titanium peroxo complexes has been extensively investigated by Kholdeeva and co-workers.39b In the present work, however, since our goal has been to maintain a mild reaction condition, addition of acid or other additives was avoided as far as possible. Therefore no attempt has been made to adjust the pH of the reaction.
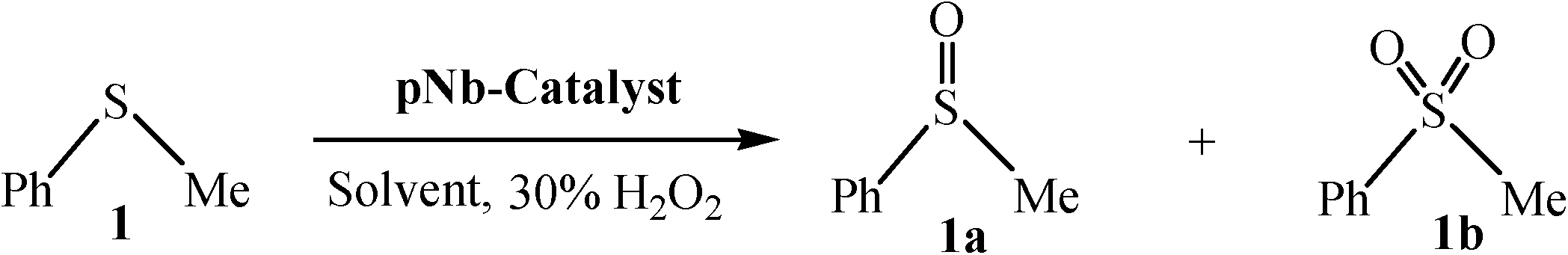
The aforementioned findings were further exploited to obtain pure sulfoxide from a series of aryl alkyl, aryl vinyl, aryl alcohol and dialkyl sulfides listed in Table 7. Evidently, both the catalysts were effective in leading to the facile and selective transformation of each of the substrates to the corresponding sulfoxide with impressive yield, although the NbN catalyst displayed relatively superior activity. The transformations worked well for both aliphatic and aromatic substrates irrespective of having electron-donating or electron-withdrawing moieties. The nature of the substrate and the attached substituent, however, appeared to influence the rates of oxidation.8g,11b The observed trend in variations in the rate of oxidation of the chosen substrates is consistent with the previous findings that with increasing nucleophilicity of the sulfide, the rate of oxidation with H2O2 increases.8g,11b It is therefore not unexpected that dialkyl sulfides were oxidized by H2O2 at a faster rate leading to the highest TOF, relative to conjugated systems such as allylic and vinylic sulfides or aromatic sulfides. It is notable that even in the case of a less nucleophilic diaromatic sulfide the corresponding sulfoxide was effectively obtained.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | NbA | NbN | ||||||
| Time (min) | Isolated yield of sulfoxide (%) | TONb | TOFc (h−1) | Time (min) | Isolated yield of sulfoxide (%) | TONb | TOFc (h−1) | ||
| a Optimized conditions: 5 mmol of substrate, 10 mmol of 30% H2O2 and 0.002 mmol of catalyst in H2O at 0 °C. b TON (turnover number) = millimoles of the product per millimole of the catalyst. c TOF (turnover frequency) = millimoles of the product per millimole of the catalyst per hour. d Yield of the 6th reaction cycle. e Scale-up data (6.24 g of MPS). | |||||||||
| 1. |
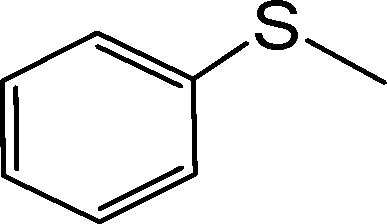
|
50 | 97 | 2425 | 2910 | 45 | 96 | 2400 | 3200 |
| 50 | 93d | 2325 | 2790 | 45 | 94d | 2350 | 3133 | ||
| 50 | 96e | 2400 | 2880 | 45 | 95e | 2375 | 3166 | ||
| 2. |
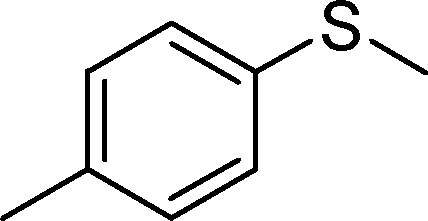
|
45 | 97 | 2425 | 3233 | 40 | 95 | 2375 | 3562 |
| 3. |
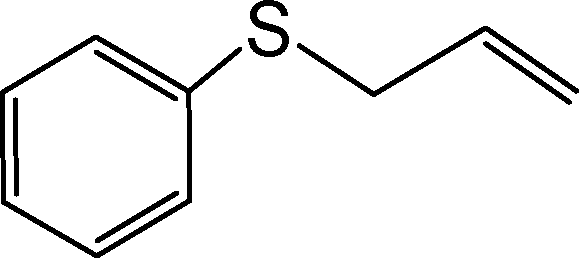
|
85 | 93 | 2325 | 1641 | 75 | 94 | 2350 | 1880 |
| 4. |
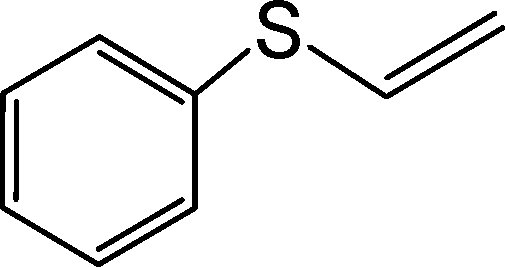
|
240 | 95 | 2375 | 593 | 225 | 93 | 2325 | 620 |
| 5. |
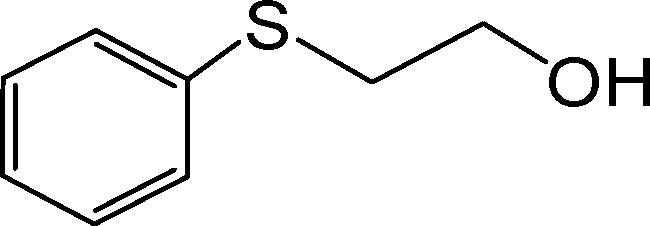
|
115 | 97 | 2425 | 1265 | 105 | 95 | 2375 | 1357 |
| 6. |

|
55 | 96 | 2400 | 2618 | 50 | 95 | 2375 | 2850 |
| 7. |
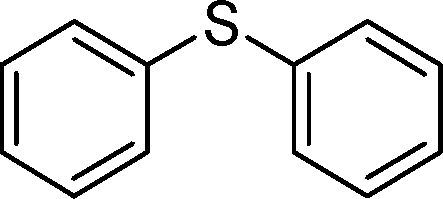
|
210 | 93 | 2325 | 664 | 195 | 94 | 2350 | 723 |
| 8. |
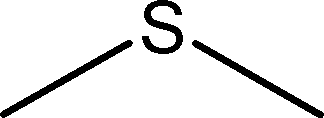
|
30 | 97 | 2425 | 4850 | 25 | 96 | 2400 | 5760 |
| 9. |

|
40 | 96 | 2400 | 3600 | 35 | 93 | 2325 | 3985 |
| 10. |

|
40 | 97 | 2425 | 3637 | 35 | 95 | 2375 | 4071 |
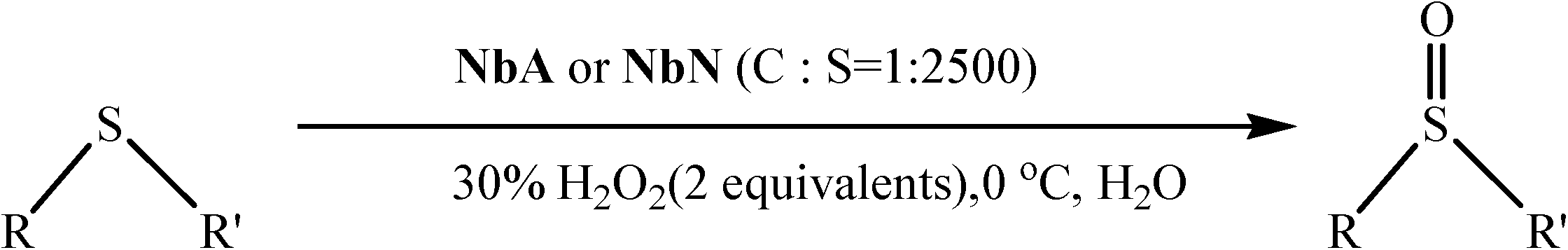
A salient feature of the methodology, which enhances the synthetic utility of the oxidation, is the excellent chemoselectivity displayed by the catalysts for the sulfur group of substituted sulfides such as allylic, vinylic and alcoholic sulfides, with co-existing sensitive functional groups (Table 7, entries 3–5). Importantly, allylic and vinylic sulfoxides were obtained without an epoxidation product. Moreover, the –OH group of alcoholic sulfides and the benzylic C–H bond remained unaffected when the benzylic and alcoholic sulfides could be oxidized to the corresponding sulfoxide under the maintained reaction conditions. The potential of the developed protocol for scaled-up synthetic application has been demonstrated by conducting the oxidation with 6.24 g of thioanisole (tenfold scale) under optimized conditions (Table 7, entry 1e). The H2O2 efficiency in the oxidation in the presence of both the catalysts NbA and NbN was found to be higher than 90%. The H2O2 efficiency, which is a measure of the effective use of H2O2, has been defined as 100 × moles of H2O2 consumed in the formation of oxyfunctionalized products per mole of H2O2 converted11a [text S4, (ESI†)].
:substrate ratio of 1:1000 using 2 equiv. of H2O2 (Table 8, entry 1).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Molar ratio (catalyst:MPS) |
H2O2 (equiv.) | Solvent | Time (min) | Isolated yield (%) |
1a:1b |
TON | TOF(h−1) |
| a Reactions are carried out with 5 mmol of the substrate in 5 mL of solvent at room temperature. | ||||||||
| 1. | 1:1000 |
2 | H2O | 80 | 96 | 0:100 |
960 | 720 |
| 2. | 1:2000 |
2 | H2O | 175 | 95 | 0:100 |
1900 | 651 |
| 3. | 1:2500 |
2 | H2O | 240 | 96 | 25:75 |
2400 | 600 |
| 4. | 1:1000 |
2 | CH3OH | 240 | 96 | 70:30 |
960 | 240 |
| 5. | 1:1000 |
2 | CH3CN | 240 | 93 | 75:25 |
930 | 232 |
Apart from MPS, the protocol could be conveniently applied to obtain pure sulfone from a variety of aromatic and aliphatic sulfides as shown in Table 9. The transformations were chemoselective (Table 9, entries 3–5) and amenable for scale-up (Table 9, entry 1e) as has been observed in the case of sulfoxidation reaction. These findings underscore the synthetic value of the methodology.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | NbA | NbN | ||||||
| Time (min) | Isolated yield of sulfone (%) | TONb | TOFc (h−1) | Time (min) | Isolated yield of sulfone (%) | TONb | TOFc (h−1) | ||
| a Optimized conditions: 5 mmol of the substrate, 10 mmol of 30% H2O2 and 0.005 mmol of the catalyst in H2O at RT. b TON (turnover number) = millimoles of the product per millimole of the catalyst. c TOF (turnover frequency) = millimoles of the product per millimole of the catalyst per hour. d Yield of the 6th reaction cycle. e Scale-up data (6.24 g of MPS). | |||||||||
| 1. |
|
80 | 96 | 960 | 720 | 70 | 97 | 970 | 831 |
| 80 | 93d | 930 | 697 | 70 | 93 | 930 | 797 | ||
| 80 | 95e | 950 | 712 | 70 | 94e | 940 | 805 | ||
| 2. |
|
75 | 95 | 950 | 760 | 65 | 97 | 970 | 895 |
| 3. |
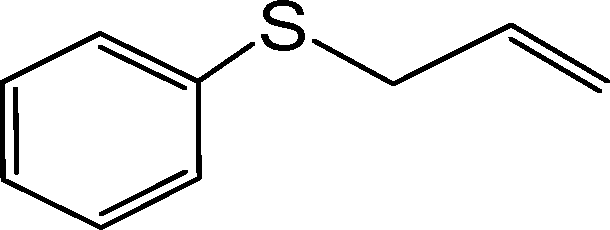
|
145 | 93 | 930 | 384 | 135 | 94 | 940 | 431 |
| 4. |
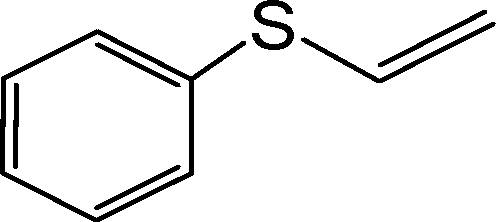
|
255 | 94 | 940 | 221 | 250 | 93 | 930 | 235 |
| 5. |
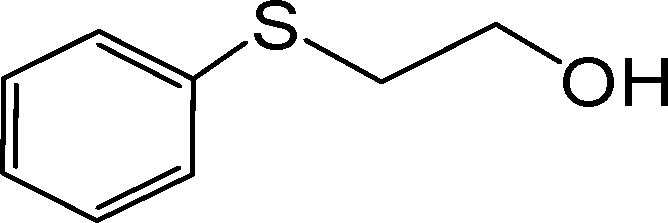
|
190 | 97 | 970 | 306 | 180 | 95 | 950 | 316 |
| 6. |
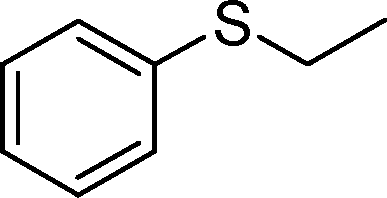
|
90 | 95 | 950 | 633 | 80 | 94 | 940 | 705 |
| 7. |
|
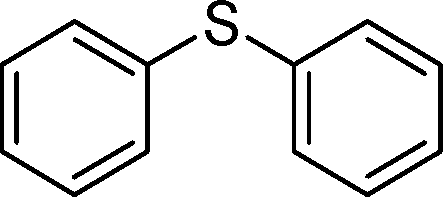
315 | 93 | 930 | 177 | 310 | 93 | 930 | 180 |
| 8. |

|
65 | 94 | 940 | 867 | 55 | 96 | 960 | 1047 |
| 9. |

|
70 | 93 | 930 | 797 | 60 | 93 | 930 | 930 |
| 10. |

|
70 | 95 | 950 | 814 | 60 | 94 | 940 | 940 |
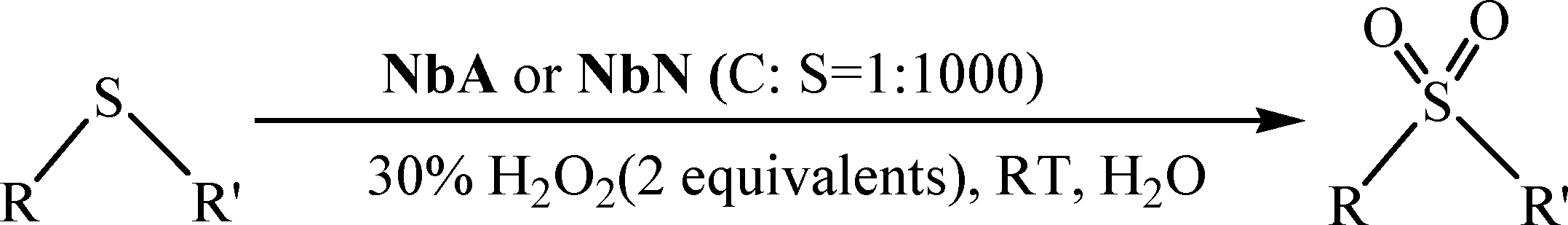
Interestingly, the procedure with NbA used as a catalyst provided an overall TOF of ca. 17130 h−1 (ca. 18999 h−1 for NbN) after 6 cycles of oxidation of MPS to sulfoxide and ca. 4259 h−1 (ca. 4884 h−1 with NbN) for conversion to sulfone. The results further demonstrate the superior activity of the catalysts under mild conditions over other reported methods for sulfide oxidation involving Nb(V) catalysts,20a–f as well as many other protocols based on Mo(VI) or W(VI)/H2O27b–e,8b–e,g,10c,g,h,38a,40 systems including the polymer-supported heterogeneous Mo(VI) catalysts that we reported recently.7b
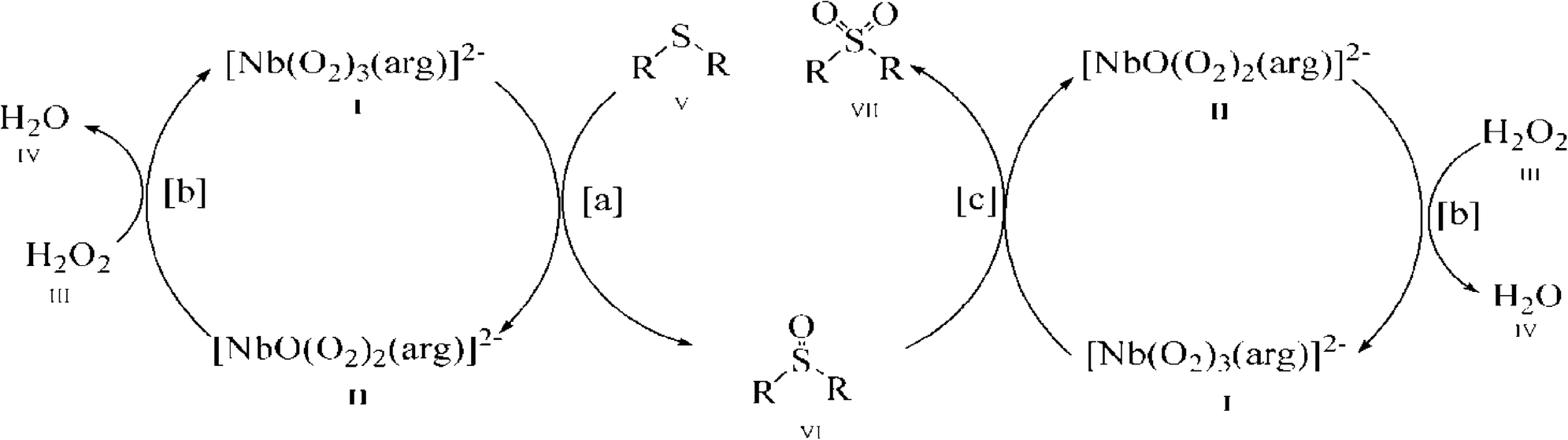 | ||
| Fig. 6 The proposed mechanism. | ||
As shown in Fig. 6, with the NbA catalyst as a representative, it is possible that the reaction proceeds through the formation of the diperoxoniobate intermediate II, subsequent to the facile transfer of electrophilic oxygen from the triperoxoniobium complex I to the substrate V to yield sulfoxide (reaction a). The intermediate II combines with the peroxide of H2O2 to regenerate the starting triperoxoniobate complex (reaction b) leading to a catalytic cycle. The resulting sulfoxide formed may undergo further oxidation in a separate cycle by reacting with a triperoxo Nb species to yield sulfone (reaction c). The sulfone formation thus seems to be a two-step process. The greater ease of oxidation of sulfide to sulfoxide compared to the second oxidation of sulfoxide to sulfone is likely to be a consequence of higher nucleophilicity of sulfide relative to sulfoxide. The proposed mechanism is in line with the reaction pathway proposed previously for peroxoniobate-catalysed oxidation of sulfide.18a,d,20g
It is worth noting that although the mechanism of action of peroxo complexes of other d0 metals such as V(V), Mo(VI) and W(VI) has been extensively investigated in organic oxidation, the chemistry of Nb(V) peroxide still remains relatively unexplored.7b,d,8a,16e,21d,24,40a,d,41 The previous studies from several laboratories including ours7b,8a,16e,f have shown that during substrate oxidation performed by active diperoxo complexes of Mo(VI) or W(VI), a more stable monoperoxo species is formed which is practically inactive in oxidation.7d,24,41k–n Taking into account this finding, it is reasonable to expect the formation of a less reactive diperoxoniobate (DPNb) intermediate from an active triperoxoniobate (TPNb) species during sulfide oxidation, as shown in the proposed mechanism (reaction intermediate II). In order to establish the involvement of such an intermediate in the reaction pathway, a separate experiment was conducted using NbA as a stoichiometric oxidant of MPS, maintaining an NbA:substrate molar ratio of 1:1 in the absence of H2O2 at 0 °C. The substrate was completely and selectively transformed into sulfoxide within a reaction time of ca. 40 minutes. The product isolated from the aqueous part of the spent reaction mixture was subsequently subjected to spectral and elemental analyses. The data obtained indicated a Nb:peroxo ratio of 1:2, clearly suggesting the formation of a DPNb species. This was further confirmed from the IR spectrum which showed two distinct bands, in addition to the coordinated amino acid ligands, characteristic of a DPNb moiety [Fig. S1(d) (ESI†)] in contrast to the three peroxo absorptions of the original triperoxoniobium catalyst. Similar reaction of MPS conducted with the isolated DPNb complex was noted to be extremely slow, as has been anticipated, remaining incomplete even after 14 h of reaction time. The aforementioned findings lent further credence to the proposed mechanism.
3. Experimental section
3.1 Materials
Acetone, hydrogen peroxide, acetonitrile, methanol, ethylacetate, petroleum ether, diethyl ether, silica gel (60–120 mesh) (RANKEM), L-arginine (CDH, New Delhi, India), nicotinic acid (HIMEDIA), sodium hydroxide, and sodium sulfate (E. Merck, India) were used in this study. Niobium pentoxide, methyl phenyl sulfide (MPS), methyl p-tolyl sulfide (MpTS), ethyl phenyl sulfide (EPS), dimethyl sulfide (DMS), dibutyl sulfide (DBS), phenylvinyl sulfide (PVS), 2-(phenylthio)ethanol (PTE), dihexyl sulfide (DHS), diphenyl sulfide (DPS) and allyl phenyl sulfide (APS) were obtained from Sigma-Aldrich Chemical Company, Milwaukee, USA. The water used for solution preparation was deionized and distilled.3.2 Synthesis of the complexes
3.3 Elemental analysis
The Perkin-Elmer 2400 series II elemental analyzer was used for C, H and N elemental analysis of the synthesized compounds. The niobium content was estimated gravimetrically as NbO(C13H10NO2)3 (ref. 42) and by atomic absorption spectroscopy (AAS). Niobium, sodium, C and N composition contents were also obtained by EDX analysis. In addition, the sodium content was quantified with an ionometer (ORION VERSASTER). The peroxide content of the compounds was determined by adding a weighed amount of the compound to a cold solution of 1.5% boric acid (w/v) in 0.7 M sulfuric acid (100 mL) and titrating with standard sodium thiosulfate solution.433.4 Physical and spectroscopic measurements
The IR spectra were recorded at ambient temperature by making pressed pellets of the compounds with KBr pellets using a Perkin-Elmer spectrum 100 FTIR spectrophotometer. Raman spectra of the compounds were recorded using a Renishaw InVia Raman microscope equipped with an argon ion laser with an excitation wavelength of 514 nm and a laser maximum output power of 20 mW. The measurement parameters were 10 s of exposure time, 1 accumulation, laser power 10% of the output power and 50× objective, and the spectral resolution was set to 0.3 cm−1. Thermogravimetric analysis was performed in a SHIMADZU TGA-50 system at a heating rate of 10 °C min−1 under an atmosphere of nitrogen using an aluminium pan. Energy-dispersive X-ray analysis was done by using a JEOL JSM-6390LV scanning electron micrograph attached to an energy-dispersive X-ray detector. Atomic absorption spectrometry was done using a Thermo iCE 3000 series atomic absorption spectrophotometer model analyst 200. The 13C NMR spectra for arginine, NbA, nicotinic acid and NbN were recorded using a JEOL JNM-ECS400 spectrometer with the following parameters: a carbon frequency of 100.5 MHz, 4096 X-resolution points, the number of scans equal to 1000–20000, 1 s of acquisition time, and 30° pulse length (D2O as solvent). 1H NMR study for the synthesised complexes along with free ligands has been carried out with a JEOL JNM-ECS400 spectrometer with 16 scans, 2 s of acquisition time and 45° pulse length (D2O as solvent). The 1H and 13C NMR spectra of organic sulfoxides and sulfones were recorded using a JEOL JNM-ECS400 spectrometer (CDCl3 as solvent and TMS as an internal standard). Melting points were determined using open capillary tubes on a Büchi melting point B-540 apparatus and were uncorrected. GC analysis was carried out using a CIC model 2010 gas chromatograph and an SE-52 packed column (length 2 m, 1/8 inch OD) with a flame ionization detector (FID) and nitrogen as the carrier gas (30 mL min−1).
3.5 Catalytic activity
:substrate molar ratio of 1:2500 and a substrate:H2O2 ratio of 1:2 in a 50 mL round-bottomed flask. The reaction was conducted at 0 °C in an ice bath under continuous stirring. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion of the reaction, the products were extracted with diethyl ether and dried over anhydrous Na2SO4 and distilled under reduced pressure to remove excess diethyl ether. The corresponding sulfoxide obtained was purified by column chromatography on silica gel using ethyl acetate and n-hexane (1:9).
The products obtained were characterized by IR, 1H NMR, and 13C NMR spectroscopy, and in the case of solid sulfoxide products, in addition to the above spectral analysis, we have also carried out melting point determination (see the ESI†).
:substrate molar ratio of 1:1000 and a substrate:H2O2 ratio of 1:2 at room temperature (RT) under continuous stirring. The reaction was monitored by thin-layer chromatography (TLC). After completion of the reaction, the products were extracted with diethyl ether and dried over anhydrous Na2SO4 and distilled under reduced pressure to remove excess diethyl ether. The corresponding sulfone obtained was purified by column chromatography on silica gel using ethyl acetate and n-hexane (1:9).
The IR, 1H NMR, and 13C NMR spectroscopy tools were used to characterize the products. In addition to the above spectral analysis, we have also carried out melting point determination for the products (see the ESI†).
3.6 Regeneration of the catalyst
Any of the following procedures could be adopted to regenerate the catalysts for reuse. The regeneration of the catalyst was carried out for the reaction using methyl phenyl sulfide (MPS). After completion of the oxidation reaction and subsequent extraction of the organic reaction product, the aqueous part of the reaction mixture was transferred to a 250 mL beaker. Keeping the solution in an ice bath, 30% H2O2 was added to it maintaining a Nb:peroxide ratio of 1:2, followed by addition of pre-cooled acetone under constant stirring until a white pasty mass separated out. From this precipitate, the catalyst was finally obtained as a microcrystalline solid by following the work-up procedure mentioned under Section 3.2.2. The regenerated pNb catalyst was then placed into a fresh reaction mixture consisting of MPS and hydrogen peroxide in water, and the reaction was allowed to proceed under optimised conditions as mentioned under Section 3.5.1 (for sulfoxide) or Section 3.5.2 (for sulfone). The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion of the reaction, the process was repeated for a total of six reaction cycles.
In an alternative procedure, recycling of the catalyst could be performed in situ after completion of the reaction cycle and extraction of the organic reaction product. Regeneration of the used reagent could be achieved by adding 30% H2O2 and a fresh batch of substrates to the aqueous portion of the spent reaction mixture, maintaining the same procedure as mentioned under Section 3.5.1 (for sulfoxide) or Section 3.5.2 (for sulfone), and conducting the reaction under optimised conditions. Each of the procedures was repeated for six reaction cycles.
3.7 Computational details
The density functional theory (DFT)36 calculations were performed using the Gaussian09 programme44 at the B3LYP/LANL2DZ level of theory. The ground-state geometry of the two niobium complexes were obtained in the gas phase, and the minima of the optimized structures were verified by the absence of imaginary frequencies.Conclusions
In summary, a pair of new heteroligand triperoxoniobium(V) complexes has been synthesized, characterized and successfully applied in the selective oxidation of variously substituted sulfides to selectively obtain sulfoxide or sulfone with 30% H2O2 in neat water. The reactions proceed under mild conditions to afford the resulting products with impressive yield and TON or TOF. The catalysts display excellent chemoselectivity toward the sulfur group of substrates with other oxidizable functional groups, including the hydroxyl group and CC bonds. The adherence of the developed protocol to the principles of green chemistry is ensured by the fact that the reactions, apart from using neat water as a standard green solvent and aqueous 30% H2O2 as an oxidant, employ a non-toxic pNb catalyst which can be reused in subsequent cycles without losing its activity. In addition, the oxidation is absolutely free from organic co-solvent, co-catalyst or any other auxiliaries, involves easy work-up procedure and is amenable for ready scalability. All of these features make the presented catalytic strategies attractive and interesting from both economic and environmental perspectives.
Acknowledgements
The authors gratefully acknowledge the Department of Science and Technology, New Delhi, India, for providing financial support. We are also grateful to the University Grants Commission, Basic Science Research Fellowship, New Delhi, India, for providing a junior research fellowship to S.R.G. We thank Dr. B. Choudhury, Department of Physics, Tezpur University, Tezpur, Assam, India, for the Raman spectra.Notes and references
- (a) H. L. Holland, Chem. Rev., 1988, 88, 473–485 CrossRef CAS; (b) M. Tanaka, H. Yamazaki, H. Hakusui, N. Nakamichi and H. Sekino, Chirality, 1997, 9, 17–21 CrossRef CAS; (c) H. Cotton, T. Elebring, M. Larsson, L. Li, H. H. Sorensen and S. Von Unge, Tetrahedron: Asymmetry, 2000, 11, 3819–3825 CrossRef CAS; (d) M. C. Carreno, Chem. Rev., 1995, 95, 1717–1760 CrossRef CAS; (e) S. Patai and Z. Rappoport, Synthesis of Sulfones, Sulfoxides and Cyclic Sulfides, J. Wiley, Chichester, 1994 Search PubMed.
- (a) I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651–3706 CrossRef CAS PubMed; (b) J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS.
- (a) V. Hulea, F. Fajula and J. Bousquet, J. Catal., 2001, 198, 179–186 CrossRef CAS; (b) M. Iwamoto, Y. Tanaka, J. Hirosumi, N. Kita and S. Triwahyono, Microporous Mesoporous Mater., 2001, 48, 271–277 CrossRef CAS.
- (a) S. Hussain, D. Talukdar, S. K. Bharadwaj and M. K. Chaudhuri, Tetrahedron Lett., 2012, 53, 6512–6515 CrossRef CAS PubMed; (b) V. Conte and B. Floris, Dalton Trans., 2011, 40, 1419–1436 RSC; (c) V. Conte, F. Fabbianesi, B. Floris, P. Galloni, D. Sordi, I. C. W. E. Arends, M. Bonchio, D. Rehder and D. Bogdal, Pure Appl. Chem., 2009, 81, 1265–1277 CrossRef CAS; (d) F. Gregori, I. Nobili, F. Bigi, F. Maggi and G. Predieri, J. Mol. Catal. A: Chem., 2008, 286, 124–127 CrossRef CAS PubMed; (e) F. Di Furia, G. Modena and R. Curci, Tetrahedron Lett., 1976, 17, 4637–4638 CrossRef.
- (a) L. Xu, J. Cheng and M. L. Trudell, J. Org. Chem., 2003, 68, 5388–5391 CrossRef CAS PubMed; (b) N. S. Venkataramanan, G. Kuppuraj and S. Rajagopal, Coord. Chem. Rev., 2005, 249, 1249–1268 CrossRef CAS PubMed.
- (a) F. Rajabi, S. Naserian, A. Primo and R. Luque, Adv. Synth. Catal., 2011, 353, 2060–2066 CrossRef; (b) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–8941 CrossRef CAS PubMed.
- (a) R. D. Chakravarthy, V. Ramkumar and D. K. Chand, Green Chem., 2014, 16, 2190–2196 RSC; (b) J. J. Boruah, S. P. Das, S. R. Ankireddy, S. R. Gogoi and N. S. Islam, Green Chem., 2013, 15, 2944–2959 RSC; (c) K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS PubMed; (d) N. Gharah, S. Chakraborty, A. K. Mukherjee and R. Bhattacharyya, Inorg. Chim. Acta, 2009, 362, 1089–1100 CrossRef CAS PubMed; (e) N. M. Gresley, W. P. Griffith, A. C. Laemmel, H. I. S. Nogueira and B. C. Parkin, J. Mol. Catal. A: Chem., 1997, 117, 185–198 CrossRef CAS.
- (a) S. P. Das, J. J. Boruah, N. Sharma and N. S. Islam, J. Mol. Catal. A: Chem., 2012, 356, 36–45 CrossRef CAS PubMed; (b) B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 CrossRef CAS PubMed; (c) X.-Y. Shi and J. F. Wei, J. Mol. Catal. A: Chem., 2008, 280, 142–147 CrossRef CAS PubMed; (d) D. H. Koo, M. Kim and S. Chang, Org. Lett., 2005, 7, 5015–5018 CrossRef CAS PubMed; (e) Y. M. A. Yamada, H. Tabata, M. Ichinohe and H. T. S. Ikegami, Tetrahedron, 2004, 60, 4087–4096 CrossRef CAS PubMed; (f) B. M. Choudary, B. Bharathi, C. V. Reddy and M. L. Kantam, J. Chem. Soc., Perkin Trans. 1, 2002, 1, 2069–2074 RSC; (g) K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS.
- K. J. Stanger, J. W. Wiench, M. Pruski, J. H. Espenson, G. A. Kraus and R. J. Angelici, J. Mol. Catal. A: Chem., 2006, 243, 158–169 CrossRef CAS PubMed.
- (a) P. S. Kulkarni and C. A. M. Afonso, Green Chem., 2010, 12, 1139–1149 RSC; (b) M. V. Gómez, R. Caballero, E. Vázquez, A. Moreno, A. Hoz and Á. Díaz-Ortiz, Green Chem., 2007, 9, 331–336 RSC; (c) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC; (d) K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef CAS; (e) C. W. Jones, Applications of Hydrogen Peroxide and Derivatives, Royal Society of Chemistry, Cambridge, 1999 Search PubMed; (f) In Catalytic Oxidations with Hydrogen Peroxide as Oxidant, ed. G. Strukul, Kluwer Academic Publishers, Dordrecht, 1992 Search PubMed; (g) P. Kowalski, K. Mitka, K. Ossowska and Z. Kolarska, Tetrahedron, 2005, 61, 1933–1953 CrossRef CAS PubMed; (h) B. M. Choudary, B. Bharathi, C. V. Reddy and M. L. Kantam, J. Chem. Soc., Perkin Trans. 1, 2002, 2069–2074 RSC; (i) G. Strukul and A. Scarso, Environmentally Benign Oxidants, in Liquid Phase Oxidation via Heterogeneous Catalysis, ed. M. G. Clerici and O. A. Kholdeeva, John Wiley & Sons, Inc., Hoboken, New Jersey, 2013, ch. 1, pp. 1–20 Search PubMed.
- (a) V. Hulea, A.-L. Maciuca, F. Fajula and E. Dumitriu, Appl. Catal., A, 2006, 313, 200–207 CrossRef CAS PubMed; (b) B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2474 CrossRef CAS PubMed.
- (a) U. M. Lindstrom, Chem. Rev., 2002, 102, 2751–2772 CrossRef PubMed; (b) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- (a) Organic Reactions in Water, ed. U. M. Lindstrom, Blackwell Publishing, Oxford, 2007 Search PubMed; (b) D. C. Rideout and R. J. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- (a) K. Surendra, N. S. Krishnaveni, V. P. Kumar, R. Sridhar and K. R. Rao, Tetrahedron Lett., 2005, 46, 4581–4583 CrossRef CAS PubMed; (b) K. L. Prasanth and H. Maheswaran, J. Mol. Catal. A: Chem., 2007, 268, 45–49 CrossRef CAS PubMed; (c) X.-M. Zeng, J.-M. Chen, A. Yoshimura, K. Middletonb and V. V. Zhdankin, RSC Adv., 2011, 1, 973–977 RSC; (d) A. Rezaeifard, M. Jafarpour, A. Naeimi and M. Salimi, Inorg. Chem. Commun., 2012, 15, 230–234 CrossRef CAS PubMed; (e) Y. Imada, T. Kitagawa, H.-K. Wanga, N. Komiya and T. Naota, Tetrahedron Lett., 2013, 54, 621–624 CrossRef CAS PubMed; (f) B. Yu, C.-X. Guo, C.-L. Zhong, Z.-F. Diao and L.-N. He, Tetrahedron Lett., 2014, 55, 1818–1821 CrossRef CAS PubMed.
- J. J. Boruah, S. P. Das, R. Borah, S. R. Gogoi and N. S. Islam, Polyhedron, 2013, 52, 246–254 CrossRef CAS PubMed.
- (a) D. Kalita, S. Sarmah, S. P. Das, D. Baishya, A. Patowary, S. Baruah and N. S. Islam, React. Funct. Polym., 2008, 68, 876–890 CrossRef CAS PubMed; (b) S. Sarmah, D. Kaita, P. Hazarika, R. Borah and N. S. Islam, Polyhedron, 2004, 50, 8046–8062 Search PubMed; (c) S. Sarmah, P. Hazarika, R. Borah, N. S. Islam, A. V. S. Rao and T. Ramasarma, Mol. Cell. Biochem., 2002, 236, 95–105 CrossRef CAS; (d) S. Sarmah and N. S. Islam, J. Chem. Res., 2001, 172–174 CrossRef CAS; (e) P. Hazarika, D. Kalita, S. Sarmah, R. Borah and N. S. Islam, Polyhedron, 2006, 25, 3501–3508 CrossRef CAS PubMed; (f) S. P. Das, J. J. Boruah, H. Chetry and N. S. Islam, Tetrahedron Lett., 2012, 53, 1163–1168 CrossRef CAS PubMed.
- K. Rydzynski and D. Pakulska, Patty's Industrial Hygiene and Toxicology, John Wiley & Sons Press, 2012 Search PubMed.
- (a) H. Egami, T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 5886–5895 CrossRef CAS PubMed; (b) H. Egami and T. Katsuki, Angew. Chem., Int. Ed., 2008, 47, 5171–5174 CrossRef CAS PubMed; (c) M. Kirihara, J. Yamamoto, T. Noguchi, A. Itou, S. Naito and Y. Hirai, Tetrahedron, 2009, 65, 10477–10484 CrossRef CAS PubMed; (d) D. Bayot and M. Devillers, Coord. Chem. Rev., 2006, 250, 2610–2626 CrossRef CAS PubMed; (e) F. V. Tkhai, A. V. Tarakanova, O. V. Kostyuchenko, B. N. Tarasevich, N. S. Kulikov and A. V. Anisimov, Theor. Found. Chem. Eng., 2008, 42, 636–642 CrossRef; (f) E. V. Rakhmanov, A. L. Maximov, A. V. Tarakanova, F. V. Tkhay and A. V. Anisimov, Moscow Univ. Chem. Bull., 2010, 65, 380–383 CrossRef; (g) E. V. Rakhmanov, Z. Sinyan, A. V. Tarakanova, A. V. Anisimov, A. V. Akopyan and N. S. Baleeva, Russ. J. Gen. Chem., 2012, 82, 1118–1121 CrossRef CAS; (h) C. M. S. Batista, S. C. S. Melo, G. Gelbard and E. R. Lachter, J. Chem. Res., Synop., 1997, 92–93 RSC; (i) L. C. Passoni, M. R. H. Siddiqui, A. Steiner and I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2000, 153, 103–108 CrossRef CAS.
- (a) V. Conte and F. Di Furia, in Catylic Oxidations with Hydrogen Peroxide as Oxidant, ed. G. Strukul, Kluwer Academic Publishers, The Netherlands, 1992, p. 223 Search PubMed; (b) V. Conte, F. Di Furia and S. Moro, J. Mol. Catal., 1997, 120, 93–99 CrossRef CAS; (c) V. Conte, F. Di Furia and S. Moro, J. Mol. Catal. A: Chem., 1997, 117, 139–149 CrossRef CAS; (d) H. Mimoun, L. Saussine, E. Daire, M. Postel, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1983, 105, 3101–3110 CrossRef CAS.
- (a) D. Bayot, B. Tinant, B. Mathieu, J.-P. Declercq and M. Devillers, Eur. J. Inorg. Chem., 2003, 737–743 CrossRef CAS; (b) D. Bayot, B. Tinant and M. Devillers, Inorg. Chem., 2004, 43, 5999–6005 CrossRef CAS PubMed; (c) D. Bayot, M. Degand and M. Devillers, J. Solid State Chem., 2005, 178, 2635–2642 CrossRef CAS PubMed; (d) D. Bayot, B. Tinant and M. Devillers, Inorg. Chem., 2005, 44, 1554–1562 CrossRef CAS PubMed; (e) D. Bayot, M. Degand, B. Tinant and M. Devillers, Inorg. Chim. Acta, 2006, 359, 1390–1394 CrossRef PubMed; (f) A. Maniatakou, C. Makedonas, C. A. Mitsopoulou, C. Raptopoulou, I. Rizopoulou, A. Terzis and A. Karaliota, Polyhedron, 2008, 27, 3398–3408 CrossRef CAS PubMed; (g) A. Maniatakou, S. Karaliota, M. Mavri, C. Raptopoulou, A. Terzis and A. Karaliota, J. Inorg. Biochem., 2009, 103, 859–868 CrossRef CAS PubMed; (h) A. C. Dengel and W. P. Griffith, Polyhedron, 1989, 8, 1371–1377 CrossRef CAS; (i) Y. Narendar and G. L. Messing, Chem. Mater., 1997, 9, 580–587 CrossRef CAS.
- (a) C. Djordjevic, N. Vuletic, M. L. Renslo, B. C. Puryear and R. Alimard, Mol. Cell. Biochem., 1995, 153, 25–29 CrossRef CAS; (b) D. Kalita, R. C. Deka and N. S. Islam, Inorg. Chem. Commun., 2007, 10, 45–48 CrossRef CAS PubMed; (c) P. Hazarika, S. Sarmah, D. Kalita and N. S. Islam, Transition Met. Chem., 2008, 33, 69–77 CrossRef CAS; (d) D. Kalita, S. P. Das and N. S. Islam, Biol. Trace Elem. Res., 2009, 128, 200–219 CrossRef CAS PubMed; (e) A. K. Haldar, S. Banerjee, K. Naskar, D. Kalita, N. S. Islam and S. Roy, Exp. Parasitol., 2009, 122, 145–154 CrossRef CAS PubMed; (f) V. Khanna, M. Jain, M. K. Barthwal, D. Kalita, J. J. Boruah, S. P. Das, N. S. Islam, T. Ramasarma and M. Dikshit, Pharmacol. Res., 2011, 64, 274–282 CrossRef CAS PubMed.
- C. Djordjevic, N. Vuletic, B. A. Jacobs, M. Lee-Renslo and E. Sinn, Inorg. Chem., 1997, 36, 1798–1805 CrossRef CAS PubMed.
- (a) P. Hazarika, D. Kalita, S. Sarmah and N. S. Islam, Mol. Cell. Biochem., 2006, 284, 39–47 CrossRef CAS PubMed; (b) P. Hazarika, D. Kalita and N. S. Islam, J. Enzyme Inhib. Med. Chem., 2008, 23, 504–513 CrossRef CAS PubMed.
- C. Djordjevic, B. C. Puryear, N. Vuletic, C. J. Abelt and S. J. Sheffield, Inorg. Chem., 1988, 27, 2926–2932 CrossRef CAS.
- D. Bayot, M. Devillers and D. Peeters, Eur. J. Inorg. Chem., 2005, 4118–4123 CrossRef CAS.
- (a) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Co-ordination Compounds, Part B, Wiley and Sons, New York, 5th edn, 1997, pp. 62–67 Search PubMed; (b) J. D. Gelder, K. D. Gussem, P. Vandenabeele and L. Moens, J. Raman Spectrosc., 2007, 38, 1133–1147 CrossRef; (c) M. Nazir1 and I. I. Naqvi, Am. J. Anal. Chem., 2013, 4, 134–140 CrossRef; (d) M. Kumar and R. A. Yadav, Spectrochim. Acta, Part A, 2011, 79, 1316–1325 CrossRef CAS PubMed; (e) C. S. Dilip, K. J. Venkatachalam, A. P. Raj and T. Ramachandramoorthy, Int. J. Life Sci. Pharma Res., 2011, 1, 80–88 Search PubMed; (f) J. C. Chang, L. E. Gerdom, N. C. Baeniger and H. M. Goff, Inorg. Chem., 1983, 22, 1739–1744 CrossRef CAS.
- (a) S. T. Chow and C. A. Mcauliffe, J. Inorg. Nucl. Chem., 1975, 37, 1059–1064 CrossRef CAS; (b) S. Kumar and S. B. Rai, Indian J. Pure Appl. Phys., 2010, 48, 251–255 CAS; (c) R. E. Marsh and J. Donahue, Adv. Protein Chem., 1967, 22, 235–256 CrossRef CAS; (d) I. Viera, M. H. Torre, O. E. Piro, E. E. Castellano and E. J. Baran, J. Inorg. Biochem., 2005, 99, 1250–1254 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Co-ordination Compounds, Part B, Wiley and Sons, New York, 5th edn, 1997, p. 55 Search PubMed.
- H. Thomadaki, A. Lymberopoulou-Karaliota, A. Maniatakou and A. Scorilas, J. Inorg. Biochem., 2011, 105, 155–163 CrossRef CAS PubMed.
- (a) M. A. Girasolo, S. Rubino, P. Portanova, G. Calvaruso, G. Ruisi and G. Stocco, J. Organomet. Chem., 2010, 695, 609–618 CrossRef PubMed; (b) M. Nath, R. Yadav, G. Eng and P. Musingarimi, Appl. Organomet. Chem., 1999, 13, 29–37 CrossRef CAS.
- E. M. A. Ratilla, B. K. Scott, M. S. Moxness and N. M. Kostie, Inorg. Chem., 1990, 29, 918–926 CrossRef CAS.
- (a) D. Bayot, B. Tinant and M. Devillers, Inorg. Chim. Acta, 2004, 357, 809–816 CrossRef CAS PubMed; (b) L. L. G. Justino, M. L. Ramos, M. M. Caldeira and V. M. S. Gil, Inorg. Chim. Acta, 2003, 356, 179–186 CrossRef CAS; (c) A. C. Dengel, W. P. Griffith, R. D. Powell and A. C. Skapski, J. Chem. Soc., Dalton Trans., 1987, 991–995 RSC; (d) S. E. Jacobson, R. Tang and F. Mares, Inorg. Chem., 1978, 17, 3055–3063 CrossRef CAS; (e) L. Pettersson, I. Andersson and A. Gorzsas, Coord. Chem. Rev., 2003, 237, 77–87 CrossRef CAS; (f) V. Conte, F. D. Furia and S. Moro, J. Mol. Catal., 1994, 94, 323–333 CrossRef CAS.
- J. J. Boruah, D. Kalita, S. P. Das, S. Paul and N. S. Islam, Inorg. Chem., 2011, 50, 8046–8062 CrossRef CAS PubMed.
- J.-C. Khan, M.-P. Halle, M.-P. Slmonnln and R. Schaal, J. Phys. Chem., 1977, 81, 587–590 CrossRef.
- (a) D. Bayot, B. Tinant and M. Devillers, Catal. Today, 2003, 78, 439–447 CrossRef CAS; (b) J. K. Ghosh and G. V. Jere, Thermochim. Acta, 1988, 136, 73–80 CrossRef CAS; (c) G. V. Jere, L. Surendra and M. K. Gupta, Thermochim. Acta, 1983, 63, 229–236 CrossRef CAS.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989 Search PubMed.
- I. Bytheway and M. W. Wong, Chem. Phys. Lett., 1998, 282, 219–226 CrossRef CAS.
- (a) S. K. Maiti, S. Banerjee, A. K. Mukherjee, K. M. A. Malik and R. Bhattacharyya, New J. Chem., 2005, 29, 554–563 RSC; (b) E. Baciocchi, M. F. Gerini and A. Lapi, J. Org. Chem., 2004, 69, 3586–3589 CrossRef CAS PubMed; (c) T. Patonay, W. Adam, A. Levai, P. Kover, M. Nemeth, E.-M. Peters and K. Peters, J. Org. Chem., 2001, 66, 2275–2280 CrossRef CAS PubMed.
- (a) O. A. Kholdeeva and R. I. Maksimovskaya, J. Mol. Catal. A: Chem., 2007, 262, 7–24 CrossRef CAS PubMed; (b) O. A. Kholdeeva, Eur. J. Inorg. Chem., 2013, 1595–1605 CrossRef CAS; (c) O. A. Kholdeeva, G. M. Maksimov, R. I. Maksimovskaya, L. A. Kovaleva and M. A. Fedotov, React. Kinet. Catal. Lett., 1999, 66, 311–317 CrossRef CAS; (d) H. S. Schultz, H. B. Freyermuth and S. R. Buc, J. Org. Chem., 1963, 28, 1140–1142 CrossRef CAS; (e) O. Bortolini, F. Di Furia, G. Modena and R. Seraglia, J. Org. Chem., 1985, 50, 2688–2690 CrossRef CAS.
- (a) K. Jeyakumar, R. D. Chakravarthy and D. K. Chand, Catal. Commun., 2009, 10, 1948–1951 CrossRef CAS PubMed; (b) C. A. Gamelas, T. Lourenço, A. P. da Costa, A. L. Simplício, B. Royo and C. C. Romão, Tetrahedron Lett., 2008, 49, 4708–4712 CrossRef CAS PubMed; (c) A. Fuerte, M. Iglesias, F. Sánchez and A. Corma, J. Mol. Catal. A: Chem., 2004, 211, 227–235 CrossRef CAS PubMed; (d) M. H. Dickman and M. T. Pope, Chem. Rev., 1994, 94, 569–584 CrossRef CAS; (e) B. Tamamiand and H. Yeganeh, Eur. Polym. J., 1999, 35, 1445–1450 CrossRef; (f) O. Bortolini, F. Di Furia, G. Modena and R. Seraglia, J. Org. Chem., 1985, 50, 2688–2690 CrossRef CAS; (g) W. Zhu, G. Zhu, H. Li, Y. Chao, Y. Chang, G. Chen and C. Han, J. Mol. Catal. A: Chem., 2011, 347, 8–14 CrossRef CAS PubMed; (h) H. S. Schultz, H. B. Freyermuth and S. R. Buc, J. Org. Chem., 1963, 28, 1140–1142 CrossRef CAS; (i) I. Sheikhshoaie, A. Rezaeifard, N. Monadi and S. Kaafi, Polyhedron, 2009, 28, 733–738 CrossRef CAS PubMed; (j) M. Bagherzadeh, L. Tahsini, R. Latifi, A. Ellern and L. K. Woo, Inorg. Chim. Acta, 2008, 361, 2019–2024 CrossRef CAS PubMed; (k) A. Basak, A. U. Barlan and H. Yamamoto, Tetrahedron: Asymmetry, 2006, 17, 508–511 CrossRef CAS PubMed; (l) K. Jeyakumar and D. K. Chand, Tetrahedron Lett., 2006, 47, 4573–4576 CrossRef CAS PubMed; (m) G. P. Romanelli, P. I. Villabrille, C. V. Cáceres, P. G. Vázquez and P. Tundo, Catal. Commun., 2011, 12, 726–730 CrossRef CAS PubMed; (n) P. Tundo, G. P. Romanelli, P. G. Vázquez and F. Aricò, Catal. Commun., 2010, 11, 1181–1184 CrossRef CAS PubMed.
- (a) A. F. Ghiron and R. C. Thompson, Inorg. Chem., 1989, 28, 3647–3650 CrossRef CAS; (b) K. S. Kirshenbaum and K. B. Sharpless, J. Org. Chem., 1985, 50, 1979–1982 CrossRef CAS; (c) H. Mimoun, Catal. Today, 1987, 1, 281–295 CrossRef CAS; (d) M. Herbert, F. Montilla and A. Galindo, J. Mol. Catal. A: Chem., 2011, 338, 111–120 CAS; (e) H. Mimoun, I. Seree de Roch and L. Sajus, Bull. Soc. Chim. Fr., 1969, 5, 1481–1492 Search PubMed; (f) K. A. Joergensen, Chem. Rev., 1989, 89, 431–458 CrossRef CAS; (g) N. Gharah, S. Chakraborty, A. K. Mukherjee and R. Bhattacharyya, Chem. Commun., 2004, 2630–2632 RSC , and references cited therein; (h) W. R. Thiel and J. Eppinger, Chem. – Eur. J., 1997, 3, 696–705 CrossRef CAS; (i) S. K. Maiti, K. M. A. Malik, S. Gupta, S. Chakraborty, A. K. Ganguly, A. K. Mukherjee and R. Bhattacharyya, Inorg. Chem., 2006, 45, 9843–9857 CrossRef CAS PubMed; (j) S. K. Maiti, S. Dinda, N. Gharah and R. Bhattacharyya, New J. Chem., 2006, 30, 479–489 RSC; (k) M. S. Reynolds, S. J. Morandi, J. W. Raebiger, S. P. Melican and S. P. E. Smith, Inorg. Chem., 1994, 33, 4977–4984 CrossRef CAS; (l) G. E. Meister and A. Butler, Inorg. Chem., 1994, 33, 3269–3275 CrossRef CAS; (m) F. R. Sensato, R. Custodio, E. Longo, V. S. Safont and J. Andres, J. Org. Chem., 2003, 68, 5870–5874 CrossRef CAS PubMed; (n) C. Djordjevic, N. Vuletic and E. Sinn, Inorg. Chim. Acta, 1985, 104, L7–L9 CrossRef CAS.
- A. K. Majumdar and A. K. Mukherjee, Anal. Chim. Acta, 1959, 21, 245–247 CrossRef CAS.
- M. K. Chaudhuri, S. K. Ghosh and N. S. Islam, Inorg. Chem., 1985, 24, 2706–2707 CrossRef CAS.
- M. C. Frisch, et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed; Gaussian 09, Revision A.1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, 13C NMR and 1H NMR spectra of complexes, hydrogen peroxide efficiency, characterization of sulfoxides and sulfones, and bar diagram for recyclability of the catalyst. See DOI: 10.1039/c4cy00864b |
| This journal is © The Royal Society of Chemistry 2015 |