
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic examination of AuIII-mediated 1,5-enyne cycloisomerization by AuBr2(N-imidate)(NHC)/AgX precatalysts – is the active catalyst AuIII or AuI?†
Jonathan P.
Reeds
a,
Mark P.
Healy‡
b and
Ian J. S.
Fairlamb
*a
aDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: ian.fairlamb@york.ac.uk
bGlaxoSmithKline, New Frontiers Science Park, Third Avenue, Harlow, Essex CM19 5AW, UK
First published on 24th July 2014
Abstract
Gold(III) catalysts mediate 1,5-enyne cycloisomerization or tandem nucleophilic substitution-1,5-enyne cycloisomerization processes in an efficient manner. This study examines the reaction kinetics of 1,5-enyne cycloisomerization, mediated by AuBr2(N-imidate)(NHC) catalysts {where N-imidate = N-tetrafluorosuccinimide (N-TFS) or N-phthalimide (N-phthal) and NHC = N,N′-di-tert-pentylimidazol-2-ylidene (ItPe)}, in the presence of AgOTf, in comparison with AuIIIBr3(NHC) and AuIBr(NHC). The nature of N-imidate anion influences catalyst efficacy. NMR spectroscopic investigations have allowed the ease of reduction of AuBr2(N-TFS)(NHC) to AuIX(NHC) (where X = N-TFS or Br) to be examined. Br2 is liberated from AuIII, which has been trapped by a sacrificial alkene. Under working catalyst conditions cationic AuIII is reduced to AuI.
Introduction
Organic transformations catalysed by Au species are of current interest within both the synthetic chemistry and catalysis communities.1 Of the reaction classes, 1,n-enyne cycloisomerization processes (n = 5 and 6) have attracted notable attention.2 While several transition metals are able to affect 1,n-enyne substrates, Au occupies a special position in this arena,2,3 as an eclectic array of structurally diverse organic products can be formed, depending on the catalyst and substrate identities, in addition to the reaction conditions used.1,6-Enyne cycloisomerization processes generally involve 6-exo-dig cyclization and are mechanistically well understood.4 1,5-Enyne cycloisomerization processes, involving 5-endo-dig cyclization, have not been examined to the same extent, despite the numerous synthetic possibilities. Nevertheless, several catalyst manifolds are known to deliver different products depending on the electronic character of the Au centre (see Scheme 1 for selected examples). This prompted Sun and Lin to explore the mechanisms of 1,5-enyne cycloisomerization, mediated by AuI, by DFT calculations (B3LYP).5
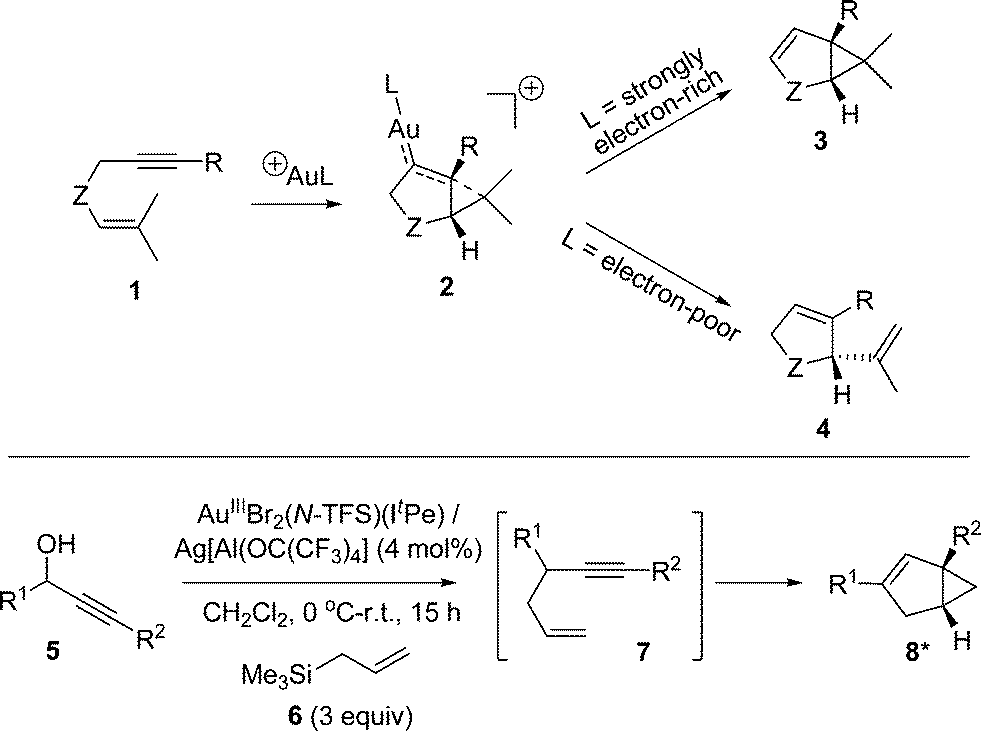 | ||
| Scheme 1 Top: 1,5-enyne cycloisomerization showing how L can direct selectivity (3 & 4 representative products; others possible). Bottom: a tandem nucleophilic substitution-1,5-enyne cycloisomerization – thermal 1,3-rearrangements are possible for 8 (R1/R2 = aryl groups). | ||
In an earlier study we developed a tandem nucleophilic substitution-1,5-enyne cycloisomerization process (5 → 8),6 starting from propargyl alcohol (5) and allyl silane (6), to deliver bicyclo[3.1.0]hexene products (8). For this process, AuIIIBr2(N-TFS)(ItPe) in combination with the non-coordinating anion, Ag[Al(OC(CF3)3)4], proved an active and selective catalyst for the tandem process. By comparison with AuIBr(ItPe) (note: near-inactive for the tandem process), it was shown that a AuIII species was required for the initial nucleophilic substitution reaction. However, a question that arose from the study was the nature of the active catalyst species in the cycloisomerization step, i.e. is AuIII or AuI responsible for cycloisomerization?
In this paper we examine the reaction kinetics of 1,5-enyne cycloisomerization, mediated by AuBr2(N-imidate)(NHC) catalysts {where N-imidate = N-tetrafluorosuccinimide (N-TFS) or N-phthalimide (N-phthal) and NHC = N,N′-di-tert-pentylimidazol-2-ylidene (ItPe)} in the presence of AgOTf. The findings allow an understanding of 1,5-enyne substrate turnover, indicating that catalyst decomposition is an issue for certain catalysts. It has been found that the N-imidate anion plays a stabilizing role for effective catalysis. NMR spectroscopic investigations have been used to examine the degradation of AuIII to AuI, which support the kinetic studies.
Results and discussion
The study is divided into two parts: (a) kinetic investigations; (b) NMR spectroscopic investigations.(a) Kinetic investigations
The 1,5-enyne cycloisomerization reaction (9 → 10) examined in these kinetic investigations is shown in Scheme 2, which has previously been validated as a useful benchmark reaction6b for testing Au catalyst efficacy (the reaction is selective for 10 and negligible side-reactions are seen).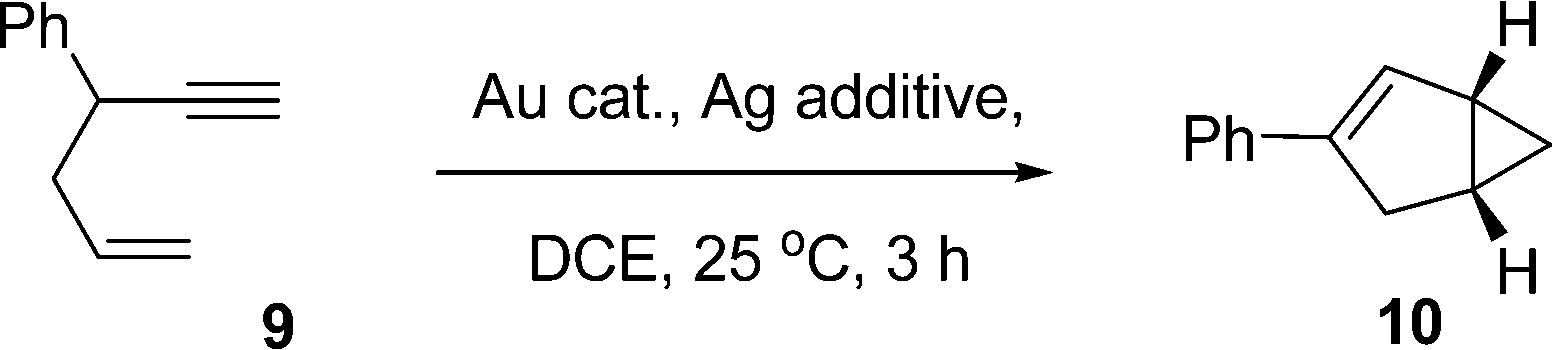 | ||
| Scheme 2 1,5-Enyne cycloisomerization reaction for the kinetic investigations. | ||
The catalysts used in this study are collated in Fig. 1, which were selected from a wider library of catalysts previously reported by our group.6a,b
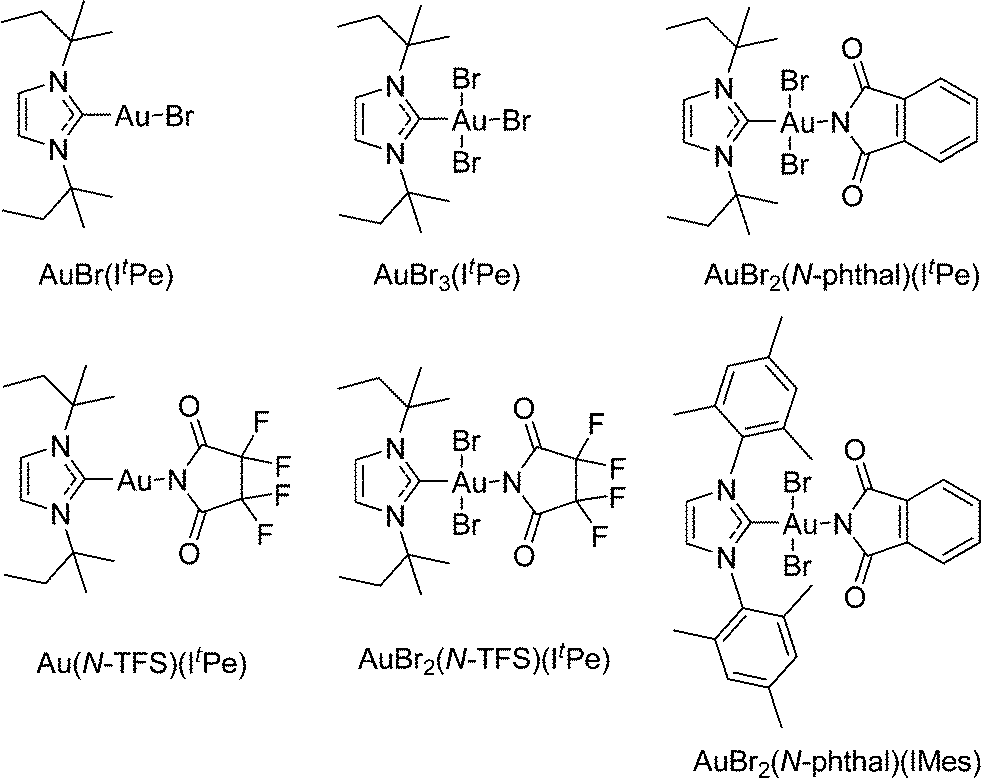 | ||
| Fig. 1 The structures of the catalysts used in this mechanistic study. | ||
The reaction of 9 → 10 was monitored by HRGC analysis. The response factors are near identical for 9 and 10 (authenticated by 1H NMR spectroscopic analysis), which is expected as they are isomers. The conversion of pure 9 (ascertained by 1H NMR spectroscopy) and associated kinetic profile is shown in Fig. 2 for the reaction mediated by 1 mol% AuBr2(N-TFS)(ItPe), in the presence of either 1 or 2 mol% AgOTf in DCE at 25 °C. The first equivalent (relative to Au cat.) serves to abstract bromide from AuBr2(N-TFS)(ItPe) forming [AuBr(N-TFS)(ItPe)]OTf and AgBr in situ. The second equivalent of AgOTf could in principle abstract another bromide.
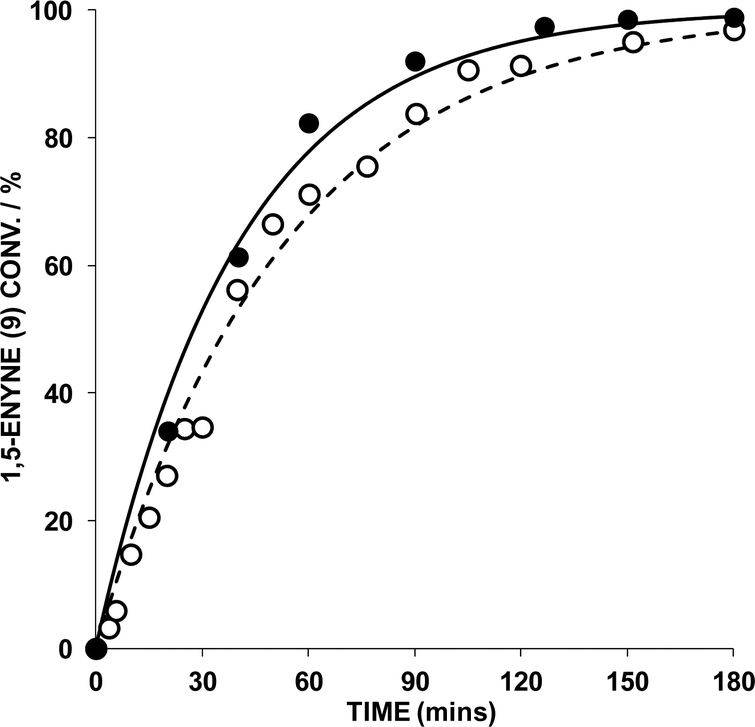 | ||
| Fig. 2 Effect of AgOTf equivalents relative to AuBr2(N-TFS)(ItPe) in 1,5-enyne cycloisomerisation of 9 (key: ● = 2 mol% AgOTf; ○ = 1 mol%. AgOTf). Other details: [9] = 0.2 M, DCE, 1 mol% Au cat., 25 °C; 1,5-enyne conversion was monitored by HRGC – each sample point was quenched by addition of N(n-Bu)4Br. The kinetic curves were fitted using Dynafit™. | ||
Examination of the kinetic profiles in Fig. 2 indicate that there is no benefit in an additional equivalent of AgOTf. Both AgOTf and AuBr2(N-TFS)(ItPe) are necessary for active catalysis (confirmed by control experiments). The data generated by HRGC could be fitted to a pseudo-first order rate equation using Dynafit™ (by curve fitting; regression analysis of ln[enyne] vs. time afforded similar data), which allowed the observed rate (kobs) and initial rate to be determined.7 For the reaction where the AuBr2(N-TFS)(ItPe)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AgOTf was 1:1, it was determined that kobs = 3.14 × 10−4 s−1 and the initial rate (T0) = 6.28 × 10−5 moldm−3 s−1 (standard error = 2.5%). A marginally faster reaction was recorded when the ratio was 1:2 (kobs = 4.16 × 10−4 s−1; T0 = 8.32 × 10−5 moldm−3 s−1; standard error = 3.3%).
:AgOTf was 1:1, it was determined that kobs = 3.14 × 10−4 s−1 and the initial rate (T0) = 6.28 × 10−5 moldm−3 s−1 (standard error = 2.5%). A marginally faster reaction was recorded when the ratio was 1:2 (kobs = 4.16 × 10−4 s−1; T0 = 8.32 × 10−5 moldm−3 s−1; standard error = 3.3%).
Based on these data the ratio of the Au catalyst to Ag additive was maintained at 1:1 for all subsequent reactions, which is in-keeping with the ratio commonly used for many related cycloisomerization processes.
The kinetic profiles for the conversion of 9 (by HRGC), catalyzed by five different Au catalysts, are shown in Fig. 3. The kinetic data is collated in Table 1. AuBr(ItPe), AuBr2(N-TFS)(ItPe), AuBr2(N-phthal)(ItPe) and AuBr2(N-phthal)(IMes) show reasonable first order behaviour in 9. The catalyst showing the highest observed rate is AuBr(ItPe) (kobs = 13.98 × 10−4 s−1), which has an initial rate commensurate with AuBr3(ItPe) (T0 = 27.96 × 10−5 moldm−3 s−1 and 21.87 × 10−5, respectively). It is important to note that AuBr3(ItPe) did not display clean first order behaviour and catalyst deactivation is apparent after ca. 60 substrate turnovers. The kinetic profile could, however be fitted to a second order rate equation by curve-fitting, in which it was assumed that the Au catalyst decomposed over time and that the rate depended on both the concentration of 9 and Au catalyst.
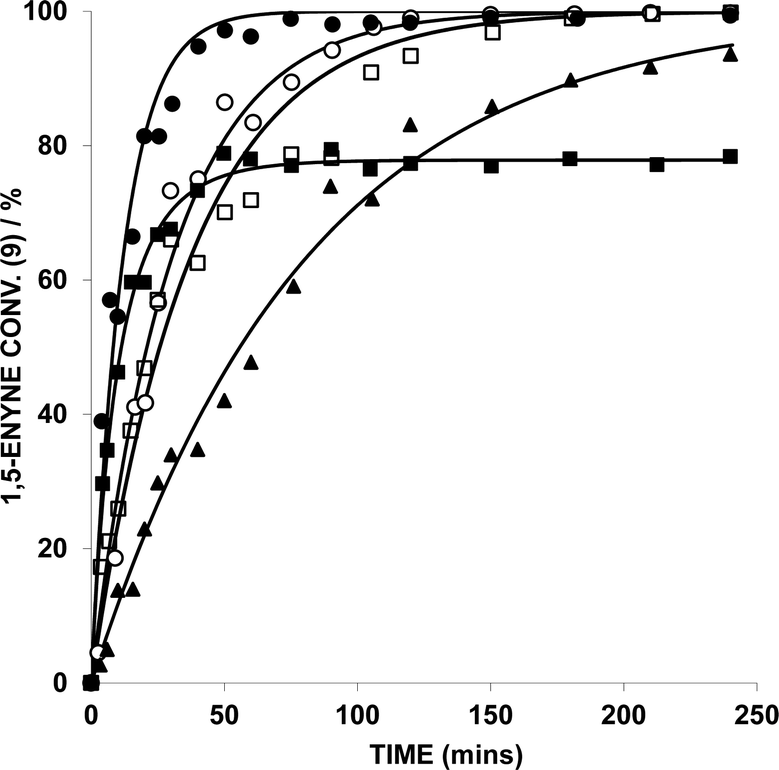 | ||
| Fig. 3 Effect of 1,5-enyne concentration [9] on the kinetics in a reaction mediated by various Au catalysts (1 mol%), AgOTf (1 mol%), in DCE at 25 °C (key: closed circles = AuBr(ItPe); open circles = AuBr2(N-TFS)(ItPe); closed squares = AuBr3(ItPe); open squares = AuBr2(N-phthal)(ItPe); closed triangles = AuBr2(N-phthal)(IMes)). 1,5-Enyne conversion monitored by HRGC analysis – each sample point was quenched by addition of (n-Bu)4NBr. The kinetic curves were fitted using Dynafit™ with standard errors < 5%. | ||
| Catalyst | k obs × 10−4 (s−1) | Initial ratea | Error (%) |
|---|---|---|---|
| a Rate at T0 × 10−5 (moldm−3 s−1). b Second order, k1obs = 0.5468 mol−1 dm3 s−1, k2obs = 9.07 × 10−4 s−1 (error 3.0%). | |||
| [AuBr(ItPe)] | 13.98 | 27.96 | 4.2 |
| [AuBr3(ItPe)] | —b | 21.87 | 2.3 |
| [AuBr2(N-TFS)(ItPe)] | 5.52 | 11.04 | 3.0 |
| [AuBr2(N-phthal)(ItPe)] | 4.49 | 8.97 | 4.2 |
| [AuBr2(N-phthal)(IMes)] | 2.07 | 4.14 | 2.0 |
The second order rate equation was deduced from initial rate = k1obs[enyne]0[Au]0 = 21.87 × 10−5 moldm−3 s−1, where k1obs (0.5468 mol−1 dm3 s−1) is the rate constant for the equation: 9 (1,5-enyne) + Au → Au + 10 (product). The rate of catalyst decomposition was determined by the initial rate of decomposition = k2obs[Au]0 = 1.81 × 10−6 s−1, where k2obs (9.07 × 10−4 s−1) is the rate constant for the decomposition of the Au active catalyst. The inactive Au is more likely colloidal, as evidenced by the kinetic data and purple colouration of the reaction mixture.
Catalyst decomposition does not appear to hinder catalysis by the other AuIII complexes tested in this series. Comparison of kobs for the AuBr2(N-phthal)(ItPe) and AuBr2(N-phthal)(IMes) catalysts, shows that the former is twice as active as the latter (Table 1). AuBr2(N-TFS)(ItPe) has a marginally higher activity than AuBr2(N-phthal)(ItPe). It is evident that the N-imidate ligand could be playing a stabilizing role, when comparing N-imidate-containing catalysts directly with AuBr3(ItPe).
In the course of these kinetic studies it was determined that ‘aged’ 1,5-enyne significantly affects the kinetic profiles recorded. Aldehyde decomposition products are formed from 9 (observed by 1H NMR, ca. 5%, after several weeks), which could interfere with the catalysis (i.e. slowing catalytic turnover). It was therefore found essential to employ either freshly prepared or purified 9 for the kinetic studies, which gave reproducible kinetic data.
Further examination of the catalytic behaviour of AuBr2(N-phthal)(ItPe) by variation of [9] reveals an interesting trend (Fig. 4, Table 2).
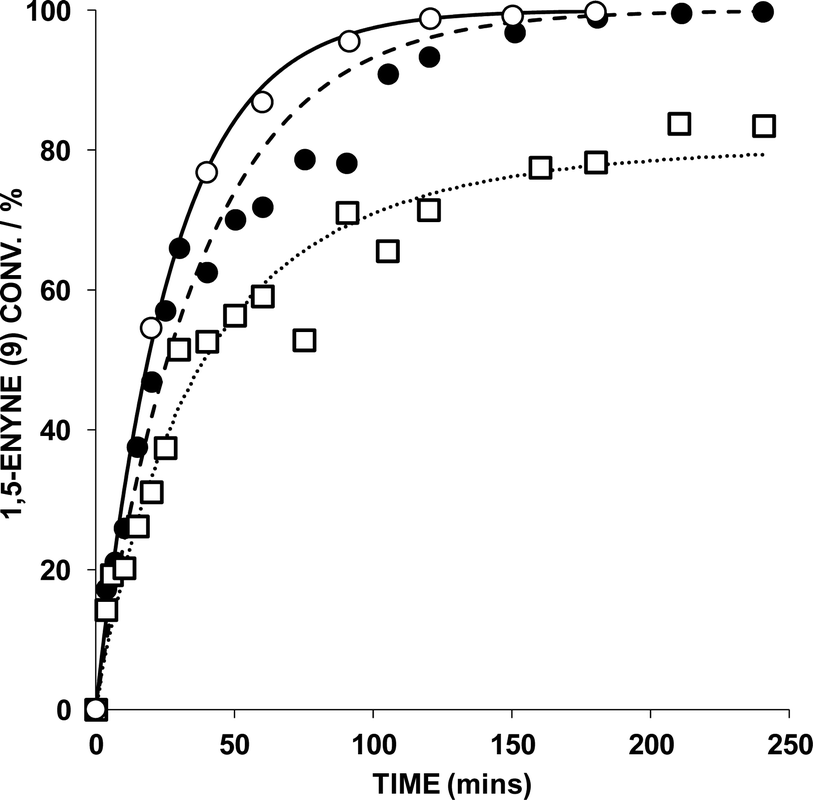 | ||
| Fig. 4 Effect of 1,5-enyne concentration [9] in a reaction mediated by AuBr2(N-phthal)(ItPe) (1 mol%), AgOTf (1 mol%), in DCE at 25 °C (key: ○ = 0.1 M 9; ● = 0.2 M 9; □ = 0.5 M 9). 1,5-Enyne conversion monitored by GC analysis – each sample point was quenched by addition of (n-Bu)4NBr. The kinetic curves were fitted using Dynafit™. | ||
Increasing [9] causes a decrease in the observed rate constants, with the initial rates increasing with concentration, relating to the change in [9] over time. At 0.5 M [9] it was necessary to fit the kinetic curve to a second order rate equation, again factoring in the Au catalyst deactivation step. The observation is consistent with increasing [Au], which is concomitant with increasing [9]. Given these data, product inhibition and Au aggregation/decomposition are likely at higher [9].
A question arising from these studies is the nature of the active catalyst species. Therefore, a competition experiment was devised to discern any subtle differences in catalyst behaviour. Dimethyl allylpropargylmalonate 11 was selected as a viable 1,6-enyne benchmark substrate, which principally affords the cyclic products 12a and 12b (Scheme 3).
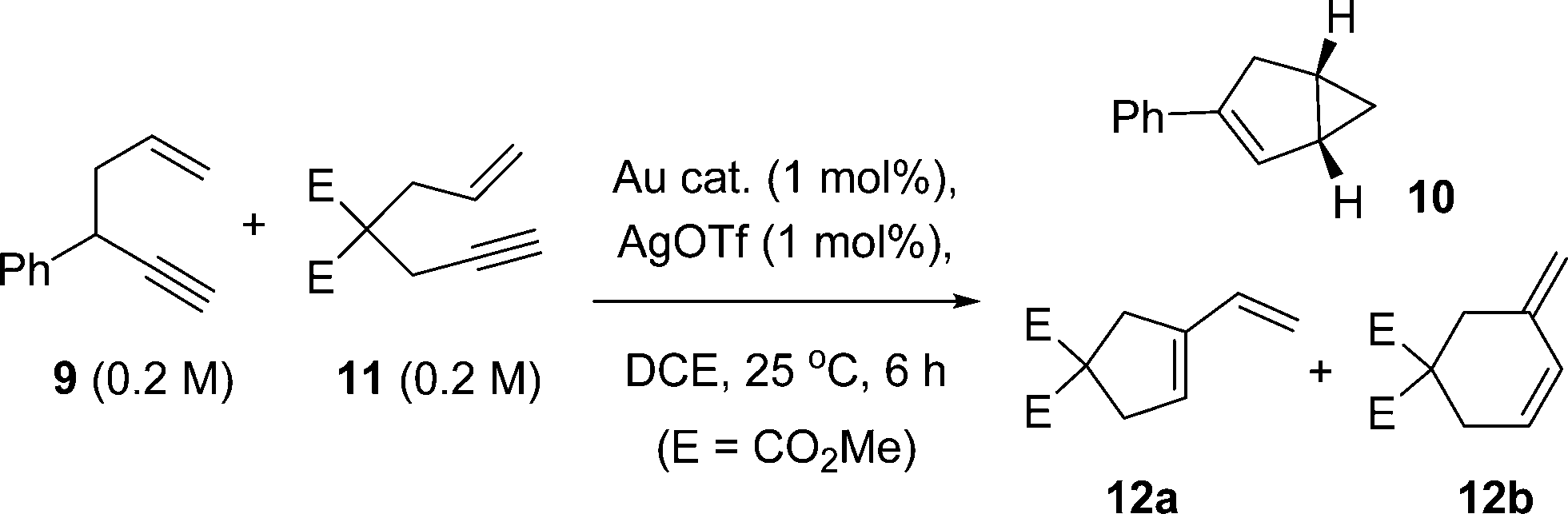 | ||
| Scheme 3 Competition experiment of a 1,5-enyne 9 and 1,6-enyne 11. Product 11 derives from 9 and products 12a and 12b from 11. The kinetic profiles are shown in Fig. 6 and product distributions of 12a:12b shown in Fig. 7. | ||
1,5-Enyne 9 (0.2 M) and 1,6-enyne 11 (0.2 M) were subjected to a reaction where the only variable was the Au catalyst {AuBr(ItPe), AuBr2(N-TFS)(ItPe) and AuBr2(N-phthal)(ItPe)} (Fig. 5 and Table 3). In addition to examining the reactivity difference between 9 and 11, the experiment allows one to probe the ratio of products formed in the latter 1,6-enyne cycloisomerization reaction (12a:12b) as the substrate is consumed over time.
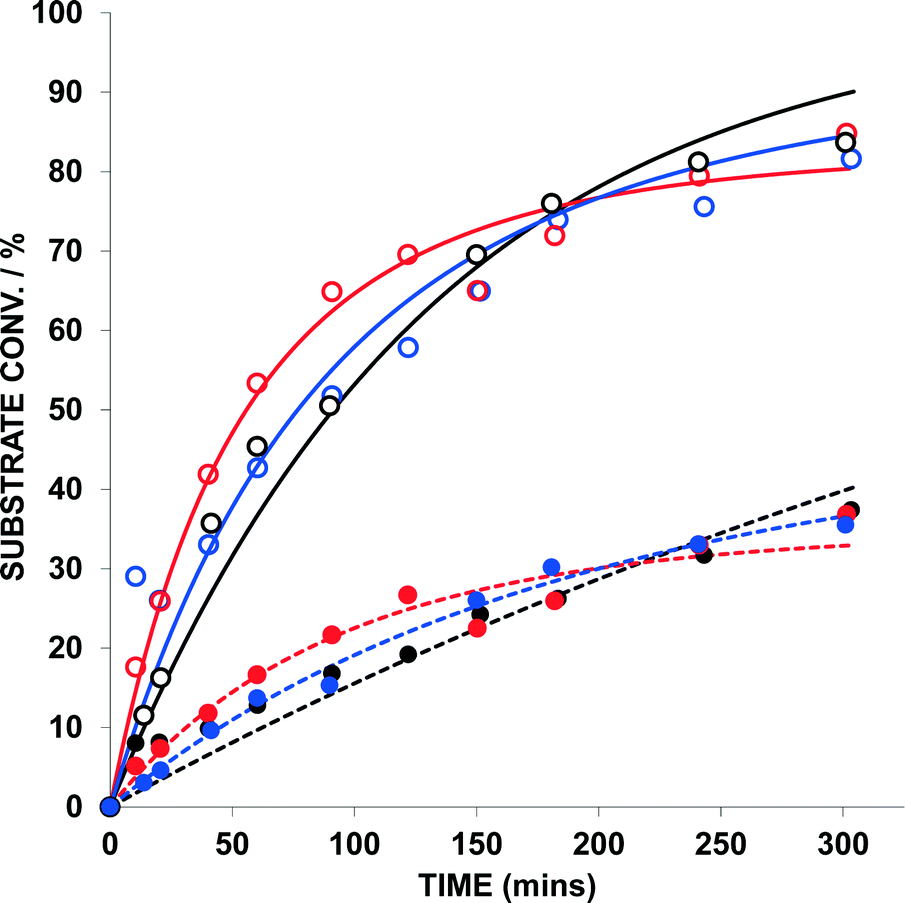 | ||
| Fig. 5 Kinetic profile for the concurrent cycloisomerisation of 4-phenyl-1-hexen-5-yne (9) and dimethyl allylpropargylmalonate (11) mediated by selected Au catalysts. Key: 1,5-enyne conversion (9, open circles) and 1,6-enyne conversion (11, closed circles) – black = AuBr(ItPe); blue = AuBr2(N-TFS)(ItPe); red = AuBr2(N-phthal)(ItPe). Conditions: 0.2 M 9, 0.2 M 11 in 1,2-dichloroethane, 1 mol% Au, 1 mol% AgOTf, 25 °C. 1,n-enyne (n = 5 or 6) conversion monitored by GC analysis – each sample point was quenched by addition of (n-Bu)4NBr. The kinetic curves were fitted using Dynafit™ with standard errors ~ 5%. | ||
| Catalyst | k obs × 10−4 (s−1) | Ratea | Error (%) | Ratio 12a:12bb |
|---|---|---|---|---|
| a Rate at T0 × 10−5 (moldm−3 s−1). b After 6 h. c Second order k1obs = 8.685 × 10−2 mol−1 dm3 s−1. d k 2obs = 6.38 × 10−5 s−1 (error 9.2%). e Second order k1obs = 0.1321 mol−1 dm3 s−1. f k 2obs = 1.52 × 10−4 s−1 (error 8.0%). g Second order k1obs = 2.128 × 10−2 mol−1 dm3 s−1. h Second order k1obs = 3.242 × 10−2 mol−1 dm3 s−1. | ||||
| 1,5-Enyne | ||||
| AuBr(ItPe) | 1.27 | 2.53 | 5.3 | — |
| AuBr2(N-TFS)(ItPe) | —c,d | 3.47 | 2.6 | — |
| AuBr2(N-phthal)(ItPe) | —e,f | 5.28 | 4.2 | — |
| 1,6-Enyne | ||||
| AuBr(ItPe) | 0.28 | 0.56 | 8.4 | 0.28 |
| AuBr(ItPe) (1,6-enyne 11 only) | 1.08 | 2.15 | 2.2 | 0.30 |
| AuBr2(N-TFS)(ItPe) | —d,g | 0.85 | 3.8 | 0.45 |
| AuBr2(N-phthal)(ItPe) | —f,h | 1.30 | 5.6 | 0.36 |
Firstly, AuBr(ItPe) is a competent catalyst for the cycloisomerization of 11 → 12 (kobs = 1.08 × 10−4 s−1; T0 = 2.15 × 10−5 moldm−3 s−1; standard error = 2.2%). The kinetic analysis of the competition experiments is complicated by the presence of two substrates and two competing catalytic cycles, which is evident in the error analysis. This complication aside, the kinetic profiles are similar in all cases (see Fig. 5). Catalyst performance is reduced for AuBr(ItPe) as compared to the AuIII catalysts – the presence of 12 slows catalytic turnover in the case of AuBr(ItPe).
A plot of the ratio of 12a:12b over the duration of the 1,6-enyne cycloisomerization reaction, reveals an interesting trend as a function of catalyst (Fig. 6).
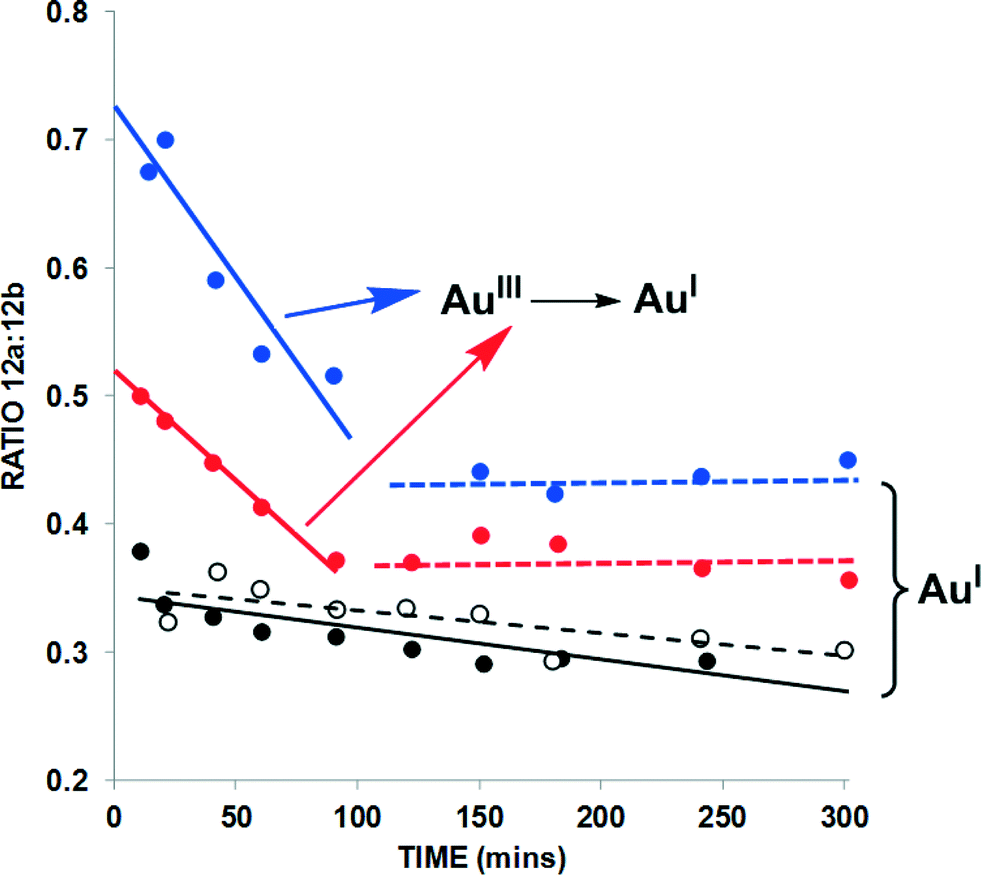 | ||
| Fig. 6 Product distribution from the cycloisomerization of 11 → 12a and 12b over time {data extracted from the kinetic profiles given in Fig. 5 – closed black circles = AuBr(ItPe); open black circles = AuBr(ItPe)/1.6-eyne 11 only; closed blue circles = AuBr2(N-TFS)(ItPe); closed red circles = AuBr2(N-phthal)(ItPe)}. The trendlines added to the graph serve as a guide to the eye. | ||
Firstly, the independent 1,6-enyne cycloisomerisation of 11 → 12a/12b mediated by AuBr(ItPe) gives a ratio of ca. 1:3, which does not alter appreciably over the 6 h reaction time. The same trend is revealed in the competition experiment in the presence of both 9 and 12, using AuBr(ItPe) as the catalyst. The product distribution is different for the AuIII catalysts, with AuBr2(N-TFS)(ItPe) showing a smaller ratio than AuBr2(N-phthal)(ItPe). Crucially, at ca. 1.5 h the ratio of 12a/12b is identical to the AuBr(ItPe) catalyst. This finding, coupled with our previous observation that dual AuIII/AuI catalyst behaviour is necessary for a successful tandem nucleophilic/1,5-enyne cycloisomerization reaction,6a indicates that AuIII to AuI degradation under the working reaction conditions, affects the ratio of products depending on distribution of AuIII/AuI species that are present at any given time. It may be conjectured that liberated N-imidate anion could affect the distribution of 12a/12b under the reaction conditions. However, in control experiments we have detected liberation of Br2 from these AuIII catalysts (see section b, below).
(b) NMR spectroscopic investigations
NMR spectroscopic analysis allows us to discern whether AuIII could be playing a role in the catalysis detailed earlier. It has been suggested previously that AuIII can be reduced to AuI under the reaction conditions.8 Nolan and co-workers8a stated that the active catalyst in the hydration of alkynes and polymerisation of styrene mediated by AuBr3(NHC) type complexes is a AuI species, formed by reductive elimination of bromide ligands. Further examination of the behaviour of the Au catalysts described herein was deemed necessary, especially whether any evidence for the formation of a cationic AuIII catalyst species, and degradation to AuI, could be gathered.1H NMR spectroscopy was used to assess whether cationic AuIII species were formed by a stoichiometric reaction of AuBr2(N-TFS)(ItPe) with AgOTf in acetone-d6 (this solvent was selected to increase solubility of all the salts). The ItPe imidazole proton signals were the most characteristic and sensitive to changes in the Au electronic configuration. It was necessary to compare NMR spectra with reference materials, namely AuBr(ItPe) and Au(N-TFS)(ItPe), as shown in Fig. 7.
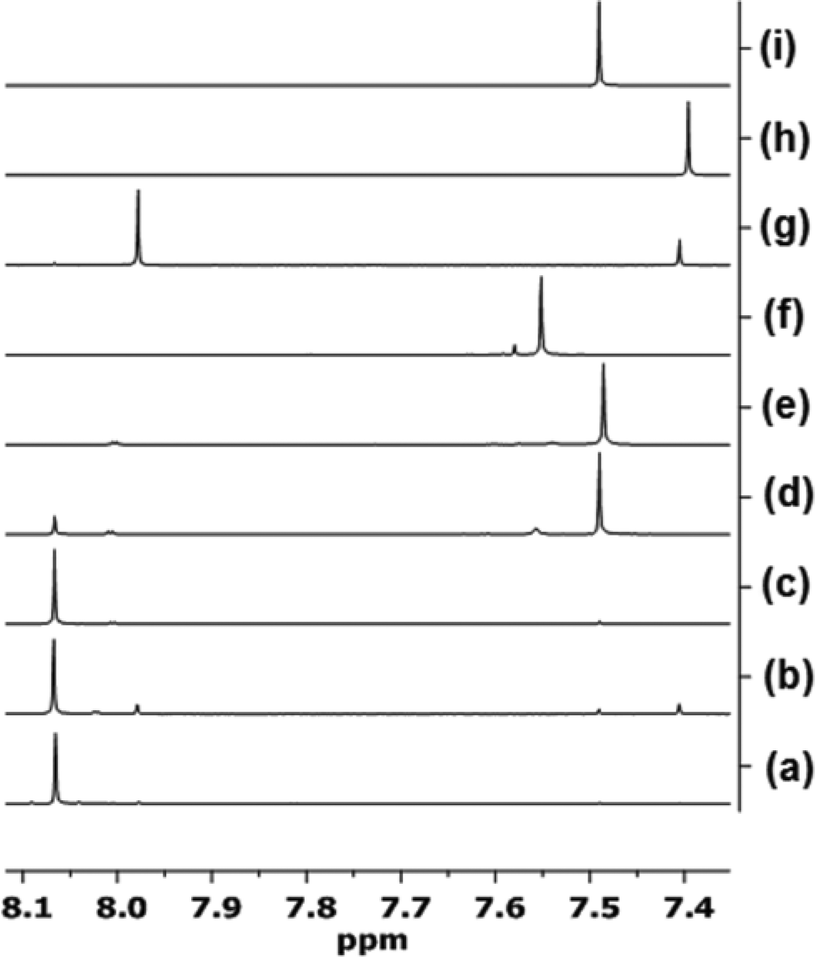 | ||
| Fig. 7
1H NMR spectroscopic analysis of the reaction of AuBr2(N-TFS)(ItPe) with AgOTf in acetone-d6 over time. (a) AuBr2(N-TFS)(ItPe) only; (b) AuBr2(N-TFS)(ItPe), aged for 48 h; (c) AuBr2(N-TFS)(ItPe)/AgOTf (1:1), 10 min; (d) as for (c), 4.5 h; (e) as for (c), 22.5 h; (f) AuBr(ItPe)/AgOTf (1:1), 22.5 h; (g) solution of AuBr3(ItPe) only, aged for 48 h; (h) AuBr(ItPe) only; (i) Au(N-TFS)(ItPe) only. | ||
The AuBr(ItPe) imidazole signal shifts 0.15 ppm downfield on treatment with AgOTf, giving Au(OTf)(ItPe) quantitatively {compare spectra (h) and (f), Fig. 7}. AuBr2(N-TFS)(ItPe) reacts with AgOTf to afford Au(N-TFS)(ItPe) and trace Au(OTf)(ItPe), with no cationic AuIII species observed after 4.5 h {compare spectra (d), (e) and (i)}. The outcome is consistent with the liberation of Br2 (as noted by a brown colouration). Furthermore, AuBr3(ItPe) decomposes in solution in the absence of AgOTf, leading to the formation of AuBr(ItPe) {compare spectra (g) and (h)}. Ageing of AuBr2(N-TFS)(ItPe) led to slow formation of trace but observable amounts of AuBr3(ItPe), AuBr(ItPe) and Au(N-TFS)(ItPe) {see spectrum (b)}. The formation of Au(N-TFS)(ItPe) is detected by 19F NMR spectroscopic analysis (see ESI†).
A similar reaction of AuBr2(N-TFS)(ItPe) with Ag[Al(OC(CF3)3)4] in acetone-d6 afforded Au(N-TFS)(ItPe) and a new broad signal at slightly higher chemical shift (δ 7.52) which we suggest is [Au(κ1-O = CMe2)(ItPe)]+[Al(OC(CF3)3)4]− (see ESI†).
The binding of the AuBr2(N-TFS)(ItPe) and AgOTf toward 1-hexene in CD2Cl2 was evaluated to reveal evidence for any alkene–AuIII interaction (Table 4), in addition to showing whether liberated Br2 could be sequestered by 1-hexene. For comparison, the reported spectroscopic data9 of [Au(μ2-H2C = CHC4H9)(IPr)][SbF6] is included in Table 4. A reduction in the alkene proton coupling constants, relative to free 1-hexene, is evidence for π-back donation, i.e. an increase in p character for the orbitals at carbon.
| Complex/salt | 1-Hexene (equiv.) | δ HA (ppm) | Coupling constant (Hz)a |
|---|---|---|---|
| a The central proton (HA) was observed as a ddt. | |||
| — | — | 5.82 | 17.0, 10.1, 6.7 |
| AgOTf | 2 | 6.21 | 17.7, 8.9, 6.6 |
| [AuBr2(N-TFS)(ItPe)] | 1 | 5.82 | 17.0, 10.1, 6.7 |
| [AuBr2(N-TFS)(ItPe)]/AgOTf | 1 | 6.26 | 18.0, 8.7, 6.5 |
| [AuBr2(N-TFS)(ItPe)]/AgOTf | 2 | 6.00 | 17.0, 10.1, 6.7 |
| [Au(μ2-H2C = CHC4H9)(IPr)][SbF6] | 1 | 6.05 | 17, 9, 4.5 |
The 1H NMR spectral data show no clear evidence for 1-hexene binding to neutral AuBr2(N-TFS)(ItPe), in the absence of AgOTf (Table 4). Upon mixing AuBr2(N-TFS)(ItPe) with 1 equiv. of AgOTf, the solution turns a cloudy colour (due to AgBr formation). Addition of 1-hexene (1 equiv.) results in a shift of Δδ = 0.44 ppm (relative to free 1-hexene). Coupling constants of 18.0 Hz (ΔJE–H = +1 Hz) for the trans-proton and 8.7 Hz (ΔJZ–H = −1.4 Hz) for the cis-coupling were recorded. Addition of 2 equiv. of 1-hexene leads to a reduction in the chemical shift (Δδ = 0.18 ppm), with little change in coupling constants observed. The excess 1-hexene is therefore in exchange with the alkene–AuIII species or alkene–AgI species. After 24 h, the 1H NMR spectrum had changed significantly, with a number of small ItPe imidazole signals corresponding to AuI and AuIII species, in addition to a large broad signal (ca. 80% of the total imidazole proton signal) in the AuI region. Another broad signal at δ 4.45–4.09 ppm was recorded, which we tentatively suggest is the alkene coordinated to AuI; the broadness in the proton signal is due to fluxionality of the alkene as either Au(μ2-H2C = CHC4H9)(N-TFS)(ItPe) or the naked cationic species [Au(μ2-H2C = CHC4H9)(ItPe)]+. Signals corresponding to free 1-hexene and a new set of proton signals corresponding to 1,2-dibromohexane, ca. 50% abundance compared to free 1-hexene, i.e. 1 equivalent. After 24 h, there was no evidence of 1-hexene binding to Ag or Au. The 19F NMR spectrum showed a very large broad signal (−182.2 ppm) suggesting the observed alkene complex is that of Au(μ2-H2C = CHC4H9)(N-TFS)(ItPe). In the absence of 1-hexene, the decomposition of AuBr2(N-TFS)(ItPe) was not observed.
It is important to note that AgOTf alone binds to 1-hexene (Δδ = 0.39 ppm; ΔJE–H = +0.7 Hz and ΔJZ–H = −1.2 Hz), which is similar to the binding of AuBr2(N-TFS)(ItPe)/1 equiv. AgOTf. Whilst AgBr is formed, we cannot rule out the involvement of chemical equilibria with AuBr2(N-TFS)(ItPe) and AgOTf. Moreover, under the catalytic conditions there is an excess of substrate and solvent relative to the catalyst system, therefore solubilisation of all the metal species is possible.
(c) Mechanistic discussion
The experimental evidence from this study indicates that AuBr(ItPe) is the most active catalyst for 1,5-enyne cycloisomerization in the series tested, showing first order behaviour. The AuIII catalyst, AuBr3(ItPe), mediates a slower reaction, which was successfully modelled kinetically as a second order process. The inclusion of a catalyst deactivation step was found necessary for the analysis. The 1H NMR spectroscopic analysis revealed that AuBr3(ItPe) degrades to AuBr(ItBu), even in the absence of AgOTf.The presence of an N-imidate anion led to higher catalyst efficacy (defined as leading to full substrate conversion) in comparison with AuBr3(ItPe).10 Of the N-imidate series, AuBr2(N-TFS)(ItPe), is the most catalytically active. The nature of the NHC ligand exhibits a significant effect. For example, AuBr2(N-phthal)(ItPe) is twice as active as AuBr2(N-phthal)(IMes).
Increasing the 1,5-enyne [9], and therefore the concentration of Au catalyst {AuBr2(N-phthal)(ItPe)}, leads to catalyst deactivation. Optimal catalyst performance was recorded at [9] between 0.1–0.2 M. At 0.5 M [9] it was necessary to model the kinetics to a second order process, again involving a catalyst deactivation step. Product inhibition is also possible at higher concentrations, and formation of Au colloids is visible by the naked eye (purple colouration, relating to the Au plasmon band).
It was possible to assess subtle differences in catalyst behaviour by a concurrent cycloisomerization competition experiment, involving 1,5-enyne 9 and 1,6-enyne 11. The former gives 10 selectively, whereas 11 afford one of two products, 12a or 12b. Three catalysts were tested, namely AuBr(ItPe), AuBr2(N-TFS)(ItPe) and AuBr2(N-phthal)(ItPe). In all cases, 1,5-enyne 9 was found to be more reactive than 1,6-enyne 11. Further analysis of the product ratios 12a:12b from the 1,6-enyne cycloisomerization reaction revealed key differences for the AuIII catalysts when compared to AuBr(ItPe). After ca. 1.5 h, the ratios of 12a:12b for AuBr2(N-TFS)(ItPe) and AuBr2(N-phthal)(ItPe) mirrored the reaction mediated by AuBr(ItPe). Therefore, while AuIII is still present, 12a is formed to a greater extent than in reactions solely mediated by AuI.
Our findings on the 1,5-enyne cycloisomerization process (9 → 11) are in-keeping with the DFT calculations reported by Sun and Lin.5 In that study, it was shown that the 1,5-enyne cycloisomerization reaction mediated by AuCl is both kinetically and thermodynamically favoured for the formation of the bicyclo[3.1.0]hexene product, with key catalytic steps (TS-Icomp carbene formation and TS-IIcomp hydrogen migration) being of similar energy (Scheme 4).
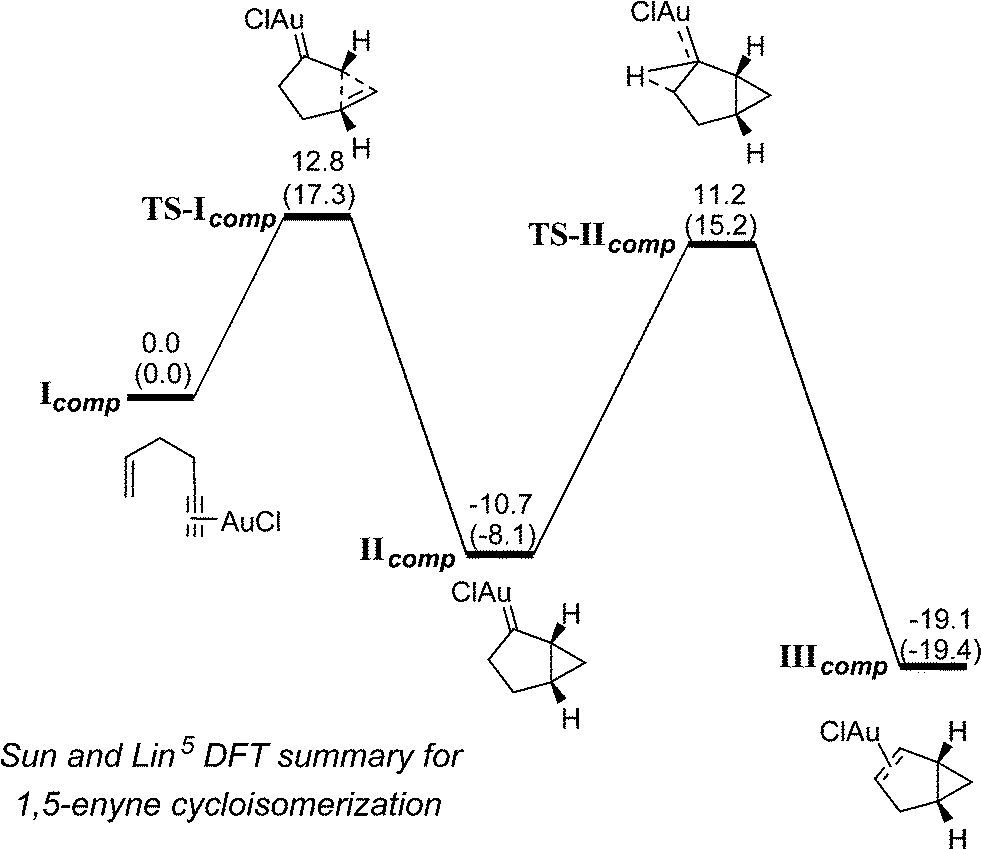 | ||
| Scheme 4 Top: proposed mechanism for the 1,5-enyne cycloisomerization 9 → 10 (a role for Ag is not shown in the mechanism, although this cannot be ruled out). Bottom: DFT computed energies for the intermediates and transition states for the a 1,5-enyne cycloisomerization mediated by AuCl, reported by Sun and Lin. | ||
From our experimental findings, we propose that Br2 is liberated from cationic AuIII–NHC catalyst species, affording AuI–NHC catalyst species in situ. The finding is supported by the liberation and trapping of the Br2 by a sacrificial alkene (1-hexene) in the NMR spectroscopic investigations. Decomposition of the AuI–NHC catalyst species affords Au colloids, which are most likely a moribund-form, especially as reduced catalyst performance is seen at higher [Au] and [9] (Scheme 5).
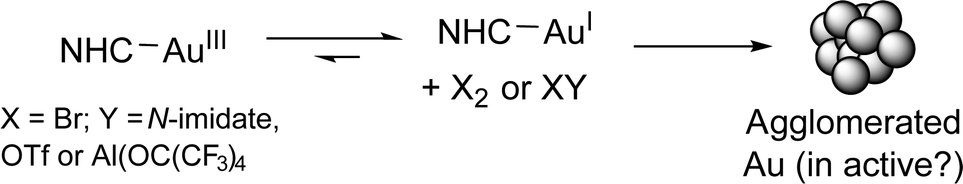 | ||
| Scheme 5 Catalyst activation and deactivation pathways. | ||
The binding experiments which explored the interaction of 1-hexene with AuBr2(N-TFS)(ItPe)/AgOTf, and AgOTf alone, demonstrates that 1-hexene binding is influenced in both cases. Crucially, 1-hexene mediates the decomposition of AuBr2(N-TFS)(ItPe) to Au(N-TFS)(ItPe), which was not observed in the absence of 1-hexene in CD2Cl2. The results taken together demonstrate that unsaturated substrates are involved in: (i) catalyst activation to give AuI, with concomitant decomposition of AuIII; (ii) solubilisation of the Ag salt, which is likely non-innocent in the catalysis. Subsequently, the nature of the coordinating ligand (and substrate and any impurities, as shown by substrate-aging experiments) is likely to affect catalytic turnover.
Finally, it is important to emphasise that other products can result from the cycloisomerization of different 1,5-enynes, catalyzed by either AuBr(ItPe) or AuBr2(N-TFS)(ItPe). In keeping with previous observations,3a,d we find that a 1,5-enyne containing a highly substituted carbon tether gives a cyclohexadiene product, both in the tandem nucleophilic substitution-1,5-enyne cycloisomerization (13 → 14 → 15) and the latter independent reaction (14 → 15) (Scheme 6). The outcome serves to highlight that the catalysts described here, and elsewhere, mirror related observations from other studies.
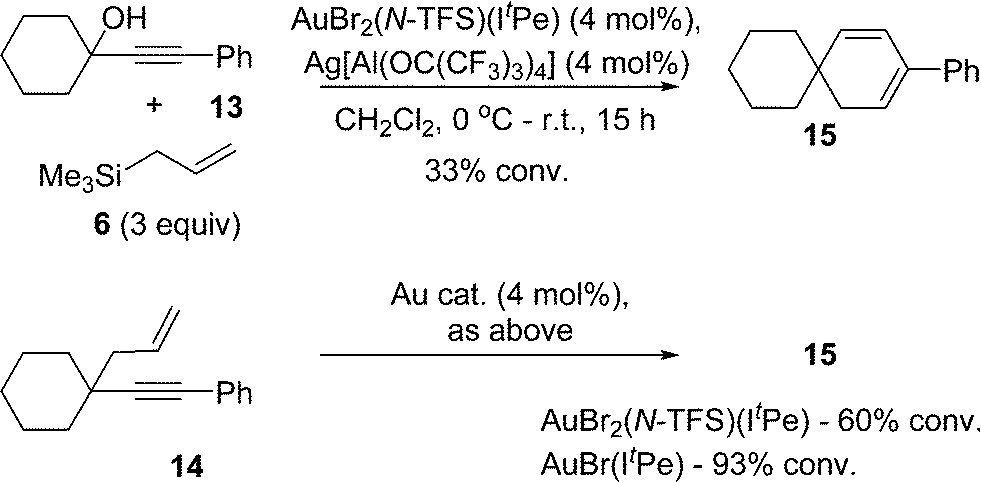 | ||
| Scheme 6 1,5-Enyne cycloisomerization leading to 1,3-cyclohexadiene formation. | ||
Conclusions
In this paper the ability of both AuIII and AuI catalysts to mediate a 1,5-enyne cycloisomerization reaction has been examined. It was necessary to understand whether AuIII was being reduced under working catalyst conditions, especially as the success of the tandem nucleophilic substitution-1,5-enyne cycloisomerization relies on AuIII for the initial nucleophilic substitution step.6aThe reaction kinetics for 1,5-enyne cycloisomerization, mediated by AuBr2(N-imidate)(NHC) catalysts {where N-imidate = N-tetrafluorosuccinimide (N-TFS) or N-phthalimide (N-phthal) and NHC = N,N′-di-tert-pentylimidazol-2-ylidene (ItPe)}, in the presence of AgOTf, revealed that the nature of the N-imidate anionic ligand influences catalyst efficacy. A direct comparison was made with AuIIIBr3(NHC), which exhibited significant catalyst deactivation in the absence of the stabilizing N-imidate ligand. In a concurrent cycloisomerization of 1,5- and 1,6-enynes (9 and 11) we were able to tease out subtle differences in the product distribution (11 → 12a:12b) during the early stages of catalytic turnover by the AuIII catalysts. NMR spectroscopic investigations have allowed the ease of reduction of AuBr2(N-TFS)(NHC) to AuIX(NHC) (where X = N-TFS or Br) to be examined. In these experiments, 1-hexene was shown to mediate AuIII decomposition. The liberation of Br2 from the AuIII centre was trapped by this sacrificial alkene.
Taking together all of the results, we conclude, that under working catalyst conditions, cationic AuIII is reduced to AuI. The role of AgI has been more difficult to understand (i.e. beyond simple bromide abstraction). The interplay of Au and Ag salts in 1,n-enyne cycloisomerization processes is currently being studied within our laboratories.
Experimental
General experimental details
All reactions involving silver salts were carried out in the absence of light. Deuterated and non-deuterated dichloromethane and acetonitrile were dried by passing through a column of activated alumina. Where necessary dichloromethane Infra-red spectra were recorded on a Unicam Research Series FT-IR spectrometer. Mass spectrometry was carried out using a Fisons Analytical (VG) Autospec instrument. 1H and 13C NMR spectra were collected on a JEOL ECX400 spectrometer operating at 400 and 101 MHz, respectively, and referenced to residual solvent signals. 13C NMR signals are singlets unless otherwise stated. All column chromatography was performed using silica-gel (mesh 220–440) purchased from Fluka Chemicals with the solvent systems specified within the text. TLC analysis was performed using Merck 5554 aluminium backed silica plates, compounds were visualised using UV light (254 nm) and a basic aqueous solution of potassium permanganate. Melting points were measured in open capillary tubes using a Stuart SMP3 Digital Melting Point Apparatus and are uncorrected. 1-Phenyl-2-propyn-1-ol and AgOTf were purchased from Alfa Aesar. All other chemicals were purchased from Sigma-Aldrich Inc. and used without further purification unless otherwise stated. All of the Au catalysts used in this study have been reported; see our previous paper for full details.6bKinetic experiments
Values for the integrated areas of the GC signals corresponding to starting materials and products were inputted into Microsoft Excel spreadsheet software. This software was used to calculate percent conversions by comparison of starting material and product signal areas. The concentration of the species in solution was determined from the percent conversions and the initial starting material concentration. Selected aliquots were analysed by 1H NMR spectroscopy to ensure consistency with conversions measured by GC.11
General procedure for the kinetic evaluation of the Au-catalysed cycloisomerization of 4-phenyl-1-hexen-5-yne (9) by gas chromatography
Au complex (3.2 μmol, 0.01 equiv.) and AgOTf (0.8 mg, 3.2 μmol, 0.01 equiv.) were mixed in CH2Cl2 (or DCE) in a screw cap vial (1 min) and the solvent removed in vacuo. A solution of 4-phenyl-1-hexen-5-yne (9) (50.0 mg, 321 μmol, 1 equiv.) in 1,2-dichloroethane (1.60 ml, 0.20 M) was added, the vial sealed with a rubber septum and a positive pressure of argon applied via an argon balloon. The mixture was stirred at 25 °C in the absence of light. Samples of 10 μl were taken via syringe and added immediately to a solution of tetra-n-butylammonium chloride (8 mM, 20 μl) in CH2Cl2. Conversion was determined via gas chromatography using a 1 μl sample. The chromatogram was run with an injector temperature of 250 °C and an initial oven temperature of 80 °C (0.5 min), heated to 160 °C, at a rate of 20 °C min−1, and maintained at 160 °C (1 min). The retention times were: 2.58 min for 4-phenyl-1-hexen-5-yne (9) and 3.89 min for 3-phenylbicyclo[3.1.0]hex-2-ene (10).NMR experiments
In CD2Cl2/CD3CN: AuBr2(N-TFS)(ItPe) (5 mg, 6.8 × 10−6 mols) was reacted with AgOTf (7 mg, 6.8 × 10−6) or Ag[Al(OC(CF3)3)4] (7.3 mg, 6.8 × 10−6 mols) in CD2Cl2 (0.6 mL) and CD3CN (0.2 mL) at 25 °C. The mixture was stirred for 1 h and then a 1H NMR spectrum was recorded (500 MHz). Characterisation details are included in the main body of the text.
Acknowledgements
We acknowledge GlaxoSmithKline, EPSRC and the Royal Society for funding. Dr J. M. Slattery is thanked for a sample of Ag[Al(OC(CF3)3)4] and discussions concerning this salt. This paper builds on work funded by a previous EPSRC grant (EP/D078776/1).Notes and references
- (a) A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS PubMed; (b) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed; (c) Zigang Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3351–3378 CrossRef PubMed; (d) E. Jiménez-Núñez and A. M. Echavarren, Chem. Commun., 2007, 333–346 RSC.
- (a) Y. Harrak, C. Blaszykowski, M. Bernard, K. Cariou, E. Mainetti, V. Mouriès, A. Dhimane, L. Fensterbank and M. Malacria, J. Am. Chem. Soc., 2004, 126, 8656–8657 CrossRef CAS PubMed; (b) M. R. Luzung, J. P. Markham and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 10858–10859 CrossRef CAS PubMed; (c) Y. Horino, T. Yamamoto, K. Ueda, S. Kuroda and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2809–2811 CrossRef CAS PubMed; (d) P. Y. Toullec and V. Michelet, Top. Curr. Chem., 2011, 302, 31–80 CrossRef CAS.
- (a) J. Sun, M. P. Conley, L. Zhang and S. A. Kozmin, J. Am. Chem. Soc., 2006, 128, 9705–9710 CrossRef CAS PubMed; (b) L. Zhang and S. A. Kozmin, J. Am. Chem. Soc., 2004, 126, 11806–11807 CrossRef CAS PubMed; (c) V. Mamane, T. Gress, H. Krause and A. Fürstner, J. Am. Chem. Soc., 2004, 126, 8654–8655 CrossRef CAS PubMed; (d) F. Gagosz, Org. Lett., 2005, 7, 4129–4132 CrossRef CAS PubMed.
- (a) N. Marion, G. Lemière, A. Correa, C. Costabile, R. Ramón, X. Moreau, P. D. Frémont, R. Dahmane, A. Hours, A. Lesage, J. C. Tablet, J. P. Goddard, V. Gandon, L. Cavallo, L. Fensterbank, M. Malacira and S. P. Nolan, Chem. – Eur. J., 2009, 15, 3243–3260 CrossRef CAS PubMed; (b) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed; (c) T. Godet and P. Belmont, Synlett, 2008, 2513–2517 CAS; (d) S. M. Ma, S. C. Yu and Z. H. Gu, Angew. Chem., Int. Ed., 2006, 45, 200–203 CrossRef CAS PubMed; (e) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef CAS; (f) C. Nieto-Oberhuber, M. P. Muñoz, E. Buñuel, C. Nevado, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 2402–2406 CrossRef CAS PubMed.
- T. Fan, X. Chen, J. Sun and Z. Lin, Organometallics, 2012, 31, 4221–4227 CrossRef CAS.
- (a) J. P. Reeds, A. C. Whitwood, M. P. Healy and I. J. S. Fairlamb, Organometallics, 2013, 32, 3108–3120 CrossRef CAS; (b) J. P. Reeds, A. C. Whitwood, M. P. Healy and I. J. S. Fairlamb, Chem. Commun., 2010, 46, 2046–2049 RSC.
- Dynafit program published by Biokin, see: P. Kuzmic, Anal. Biochem., 1996, 237, 260–273 CrossRef CAS PubMed.
- (a) S. Gaillard, A. M. Z. Slawin, A. T. Bonura, E. D. Stevens and S. P. Nolan, Organometallics, 2010, 29, 394–402 CrossRef CAS; (b) A. S. K. Hashmi, M. C. Blanco, D. Fischer and J. W. Bats, Eur. J. Org. Chem., 2006, 1387–1389 CrossRef CAS; (c) N. Morita and N. Krause, Eur. J. Org. Chem., 2006, 4634–4641 CrossRef CAS; (d) S. Komiya and J. K. Kochi, J. Am. Chem. Soc., 1976, 98, 7599–7607 CrossRef CAS; (e) S. Komiya, T. A. Albright, R. Hoffmann and J. K. Kochi, J. Am. Chem. Soc., 1976, 98, 7255–7265 CrossRef CAS.
- T. J. Brown, M. G. Dickens and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 6350–6351 CrossRef CAS PubMed.
- (a) We have seen N-imidate ligand effects in catalytic cross-coupling chemistry, see: J. L. Serrano, L. García, J. Pérez, E. Pérez, J. García, G. Sánchez, P. E. Sehnal, S. De Ornellas, T. J. Williams and I. J. S. Fairlamb, Organometallics, 2011, 30, 5095–5109 CrossRef CAS; (b) I. J. S. Fairlamb, P. Sehnal and R. J. K. Taylor, Synthesis, 2009, 508–510 CrossRef CAS PubMed; (c) M. J. Burns, I. J. S. Fairlamb, A. R. Kapdi, P. Sehnal and R. J. K. Taylor, Org. Lett., 2007, 9, 5397–5400 CrossRef CAS PubMed; (d) C. M. Crawforth, I. J. S. Fairlamb, A. R. Kapdi, J. L. Serrano, R. J. K. Taylor and G. Sanchez, Adv. Synth. Catal., 2006, 348, 405–412 CrossRef CAS; (e) S. Burling, C. M. Crawforth, I. J. S. Fairlamb, A. R. Kapdi, R. J. K. Taylor and A. C. Whitwood, Tetrahedron, 2005, 61, 9736–9751 CrossRef PubMed; (f) C. M. Crawforth, I. J. S. Fairlamb and R. J. K. Taylor, Tetrahedron Lett., 2004, 45, 461–465 CrossRef CAS PubMed; (g) I. J. S. Fairlamb, A. R. Kapdi, A. F. Lee, G. Sánchez, G. López, J. L. Serrano, L. García, J. Pérez and E. Pérez, Dalton Trans., 2004, 3970–3981 RSC; (h) J. L. Serrano, I. J. S. Fairlamb, G. Sanchez, L. Garcia, J. Perez, J. Vives, G. Lopez, C. M. Crawforth and R. J. K. Taylor, Eur. J. Inorg. Chem., 2004, 2706–2715 CrossRef CAS; (i) I. J. S. Fairlamb, A. R. Kapdi, J. M. Lynam, R. J. K. Taylor and A. C. Whitwood, Tetrahedron, 2004, 60, 5711–5718 CrossRef CAS PubMed; (j) C. M. Crawforth, S. Burling, I. J. S. Fairlamb, R. J. K. Taylor and A. C. Whitwood, Chem. Commun., 2003, 2194–2195 RSC.
- E. H. Niemelä, A. F. Lee and I. J. S. Fairlamb, Tetrahedron Lett., 2004, 45, 3593–3595 CrossRef PubMed.
- Z. Zhan, J. Yu, H. Liu, Y. Cui, R. Yang, W. Yang and J. Li, J. Org. Chem., 2006, 71, 8298–8301 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Representative NMR spectra and other characterization data. See DOI: 10.1039/c4cy00617h |
| ‡ Novartis Pharmaceuticals UK Limited, Horsham Research Centre, Wimblehurst Road, GB-Horsham, West Sussex RH12 5AB, U.K. |
| This journal is © The Royal Society of Chemistry 2014 |