 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mechanism of gas-phase alcohol oxidation at the interface of Au nanoparticles and a MgCuCr2O4 spinel support†
Weiyu
Song
,
Peng
Liu
and
Emiel J. M.
Hensen
*
Inorganic Materials Chemistry, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
First published on 23rd May 2014
Abstract
The catalytic oxidation of bio-ethanol to acetaldehyde entails a promising route for valorization of biomass into many important chemicals that are currently mainly being produced from fossil-based ethylene feedstock. We employ here DFT calculations to understand the unprecedented synergy between gold clusters and a MgCuCr2O4 spinel support, which shows excellent catalytic performance for the oxidation of ethanol to acetaldehyde (space-time yield of 311 gacetaldehyde ggold−1 h−1 at 250 °C). The investigations support a mechanism involving catalytic reactions at the gold–support interface. Dissociative adsorption of ethanol is facilitated by cooperative action of a gold atom at the metal cluster–support interface and a basic oxygen atom of the support. The most difficult step is the recombinative desorption of water from the surface. The oxygen vacancy formation energy is found to be a good performance descriptor for ethanol oxidation of Au/MgMeCr2O4 (Me = Cu, Ni, Co) catalysts. The high selectivity towards acetaldehyde stems from the facile desorption of acetaldehyde as compared to the cleavage of the remaining α-C–H bond in the product. The opposite holds for methanol oxidation, explaining why experimentally we observe complete methanol oxidation over Au/MgCuCr2O4 under conditions where ethanol is selectively converted to acetaldehyde.
1. Introduction
Selective oxidation of alcohols over recyclable heterogeneous catalysts is an area of growing importance in green organic chemistry with the aim to replace stoichiometric reagents.1 Many efforts have been geared towards the use of molecular oxygen as the oxidant generating water as the only by-product. The most promising results have been obtained using metals such as Ru,2 Ag,3 Cu,4 Au5 and Pd.6 Au–Cu alloy catalysts have also been explored for this purpose.7,8 Among these, the use of supported gold nanoparticles has been preferred with many appealing examples of aerobic oxidation in the gas and liquid phases.9,10 Acetaldehyde is an important bulk chemical for the production of peracetic acid, pentaerythritol, pyridine bases, butylene glycol and chloral, with a worldwide production of over 106 tons per year.11 Currently, the main industrial process for production of acetaldehyde is the Wacker process, which uses fossil ethylene feedstock.12 On a smaller scale, the Veba-Chemie process may be employed, which involves gas-phase oxidation of ethanol over a silver catalyst at temperatures in the range of 500–650 °C.13 Traditionally, ethanol dehydrogenation processes were employed, for instance, using copper chromite as the catalyst, requiring frequent catalyst regeneration.14 Recently, we have reported on a highly efficient and robust heterogeneous system consisting of Au nanoparticles supported on a MgCuCr2O4 spinel-phase support material for aerobic ethanol oxidation to acetaldehyde. This catalyst is able to convert ethanol to acetaldehyde with a space-time-yield of 311 gethanol gAu−1 h−1 at 250 °C with over 95% acetaldehyde yield and no sign of deactivation after 500 h on stream.15 The unprecedented performance of this catalyst is hypothesized to derive from the synergy between metallic Au nanoparticles and surface Cu species stabilized in the spinel support. The topic of catalysis at the interface between metallic nanoparticles and the subjacent support material is gaining in importance, especially in the field of heterogeneous gold catalysis.16–19 In this contribution, we used the state-of-the art density functional theory method to investigate the reaction mechanism of the catalytic oxidation of ethanol to acetaldehyde on a small Au cluster adsorbed on a model MgCuCr2O4 support. Specifically, we focus on the interface region between the Au cluster and the support and attempt to identify the crucial role of Cu substitutions for Mg in the spinel support. As the results appeared to suggest that this catalyst would also be able to oxidize methanol to formaldehyde, another important aldehyde currently produced by gas-phase oxidation with silver catalysts above 600 °C or with Fe–Mo–V oxides at 250–400 °C,20 we evaluated the potential of the Au/MgCuCr2O4 catalyst in this reaction. Further calculations were carried out to explain the results.2. Methods
Density functional theory (DFT) with the PBE (Perdew–Burke–Ernzerhof) functional21 as implemented in the Vienna ab initio Simulation Package (VASP)22–24 was employed. A Hubbard U term was added to the PBE functional (DFT + U) employing the rotationally invariant formalism by Dudarev et al.,25 in which only the difference (Ueff = U − J) between the Coulomb U and the exchange J parameters enters. Spin-polarized calculations were performed. The projector augmented wave (PAW) method26–28 was used to describe the interaction between the ions and the electrons with the frozen-core approximation.27 Computations were done on the MgCr2O4 spinel in which one out of four Mg cations was replaced by Cu. The formation of the MgCuCr2O4 spinel was earlier confirmed.15 A bilayer cluster of 10 gold atoms was placed on the (111) surface of the MgCr2O4 and MgCuCr2O4 spinels. The energy cut-off was set to 400 eV, similar to the value used in a previous study of MgCr2O4 as a bulk material.29 For Cr, we used the on-site repulsion U = 3 eV and the exchange parameter J = 0.9 eV.29 For all the surface calculations, the model was a periodic slab with a 2 × 2 surface unit cell with lattice constants of 11.78 × 11.78 Å. We verified that energies computed using 2 × 2 and 3 × 2 unit cells gave similar results. The larger unit cell size was chosen such as to exclude that interactions between the organic adsorbate with the gold cluster in the neighboring periodic images influenced the energy. A Monkhorst–Pack 1 × 1 × 1 mesh was used for the Brillouin zone integration. To verify that this mesh size is adequate, we computed the vacancy formation energy of MgCuCr2O4 at a higher k-point setting of 2 × 2 × 1. The difference with the standard setting was less than 2 kJ mol−1. Based on our earlier experimental study, we chose the (311) and (111) surface terminations of MgCr2O4 to build the surface model.15 The large unit cell size required to construct the (311) surface model is computationally prohibitive. Accordingly, we used the (111) surface termination of MgCr2O4. This surface contains Cu and Mg cations in the second layer and O atoms binding to Cu cations in the first layer. Although other cuts will lead to different surface geometries, the Cu cations in the spinel structure are all four-fold coordinated, so that we do not expect a significant effect of the geometry of the surface on the Cu–O binding, which will turn out to be the main parameter to explain catalytic reactivity.The details of the model are shown in Fig. 1. It includes two stoichiometric layers (Fig. 1a) with the bottom layer kept frozen. The vacuum spacing between two slabs is 15 Å. For the Au cluster model, we first optimized a bilayer Au10 cluster on the MgCr2O4(111) surface and froze a selection of the Au atoms at their optimized positions during subsequent calculations (Fig. 1b). This choice is made to have a suitable model for the Au nanoparticle–support interface. Inclusion of a rigid nanoparticle or other suitable models thereof is not possible given the required size of the unit cell. The gold atoms in the cluster close to the surface are fully relaxed during the computations. The approach involving a partially frozen model for the Au cluster has been used before in theoretical studies to study reactions at the interface between metal clusters and oxidic supports.30
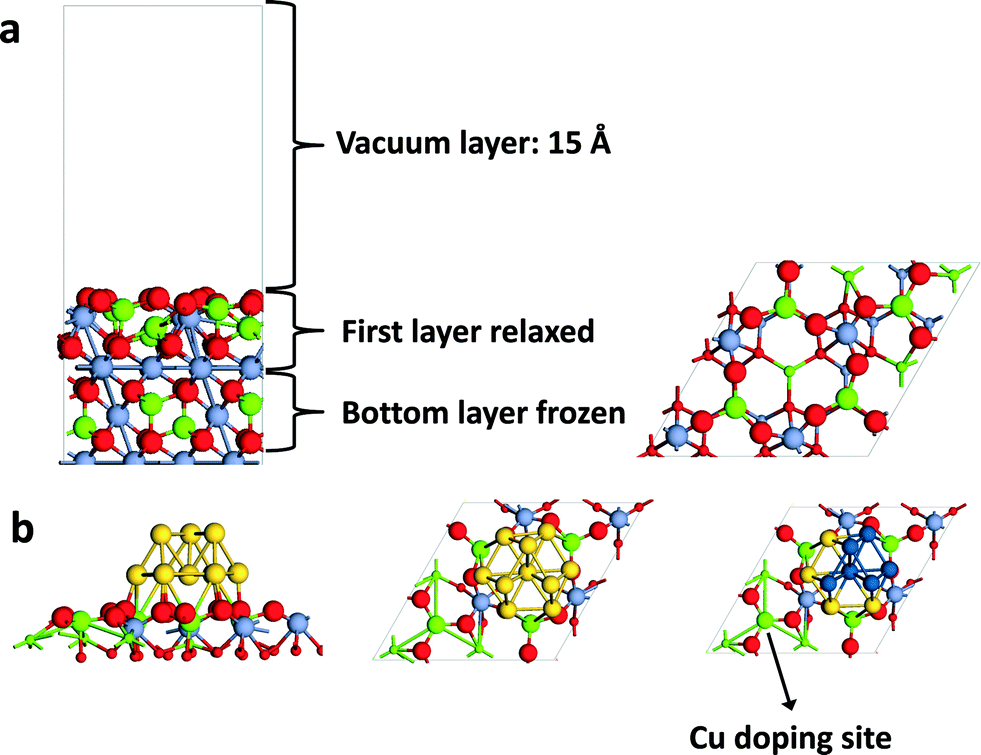 | ||
| Fig. 1 (a) Surface model of the (111)-cut of the MgCr2O4 spinel used; (b) Au cluster (left: top view; middle: side-on view) consisting of a Au10 cluster adsorbed on the (111)-cut of the MgCr2O4 spinel structure (one out of four Mg cations in the surface indicated by the arrow is replaced by a Cu substituent; color scheme – red: O; yellow: Au; green: Mg; blue: Cr; orange: Cu). | ||
Additional calculations were done for Cu(111) and Au(111) surfaces. To model these surfaces, 3 × 3 unit cells with dimensions of 7.67 × 7.67 Å for Cu and 8.87 × 8.87 Å for Au containing four metal layers were employed. The top two layers were relaxed and the bottom two layers were frozen. The thickness of the vacuum between the slabs was 10 Å. A 3 × 3 × 1 k-point mesh was employed for these calculations. The most stable hcp site was considered for O adsorption on Cu(111). For the H migration on the Au(111), the most stable three-fold hollow sites, i.e., fcc and hcp, were considered.
In all calculations, atoms were relaxed until forces were smaller than 0.05 eV Å−1. The location and energy of the transition states were calculated with the climbing-image nudged elastic band method.31
3. Results and discussion
The reaction energy diagram and key intermediates towards acetaldehyde formation are shown in Fig. 2 (the complete set of structures is shown in Fig. S1†). The reaction cycle for ethanol oxidation starts with the adsorption of ethanol on one of the Au atoms located at the interface between the gold cluster and the MgCuCr2O4 support. During geometry optimization, it was found that the hydroxyl group of the ethanol adsorbed with the O atom coordinating to an interfacial Au atom spontaneously dissociates into an ethoxy fragment coordinated to the gold cluster and a H atom coordinated to an O atom of the support bridging between Cu2+ and Mg2+ (state ii, Fig. 2). The final structure is more stable by 117 kJ mol−1 compared to ethanol in the gas phase. It has been argued that the low activity of Au/SiO2 catalysts lies in the difficulty to activate ethanol on gold itself.32 Indeed, the spontaneous dissociative adsorption of ethanol at the interface between the gold cluster and MgCuCr2O4 compares favorably to the high barriers reported for O–H bond cleavage of adsorbed ethanol, i.e. 204 kJ mol−1 for Au(111),9 80 kJ mol−1 for a Au38 cluster,33 101 kJ mol−1 for a Au nanorod33 and 117 kJ mol−1 for Au(511).33 Molecular adsorption of ethanol on Au surfaces is weak with typical values of 12–50 kJ mol–1.33 The spontaneous dissociation of ethanol on Au/MgCuCr2O4 is due to the basicity of the surface O atoms of the support and the presence of the Au cluster, which provide adsorption sites for the ethoxy group. The oxidation reaction proceeds after rotation of the adsorbed ethoxy group such that one of the α-C–H atoms points to the Au cluster (state iii, Fig. 2). The dissociation of the α-C–H bond proceeds with a barrier of 44 kJ mol−1. This reaction, which results in adsorbed acetaldehyde (state iv, Fig. 2), is thermoneutral. In the transition state, the C–H bond distance is elongated to 1.8 Å. The activation barrier for the C–H bond cleavage step of 44 kJ mol−1 is very close to previously reported values of 46 kJ mol−1 for Au(111),9 42 kJ mol−1 for Au(511),33 57 kJ mol−1 for the Au-rod model33 and 40 kJ mol−1 for a Au38 cluster.33 It is illustrative of the structure insensitivity of the α-C–H bond cleavage step.33 Acetaldehyde is only weakly bound to the surface with an adsorption energy of 17 kJ mol−1.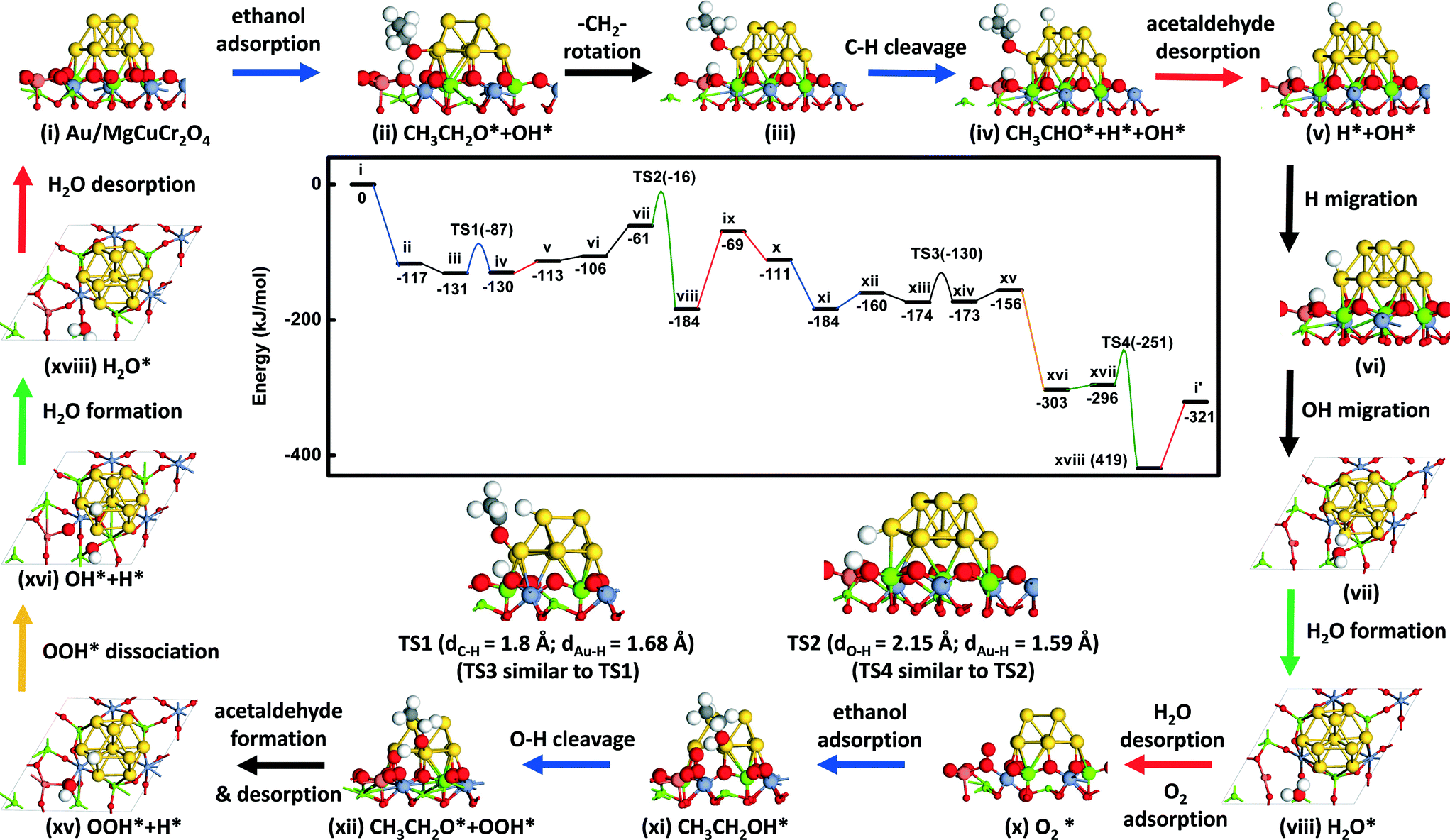 | ||
| Fig. 2 Mechanism of oxidation of ethanol to acetaldehyde for the Au10/MgCuCr2O4 model: (center top) reaction energy diagram, (center bottom) most important transition state structures and (periphery) reaction mechanism (DFT calculations for the Au10 cluster on the (111) surface of MgCuCr2O4; cut-off energy set at 400 eV). | ||
To close the first part of the catalytic cycle, the H atoms need to be removed as water. For water formation, the H atom on the Au cluster first migrates close to the interface with the support (state vi, Fig. 2) with a nearly negligible reaction energy (ΔE = 7 kJ mol−1). The barrier for this process is rather high because some of the Au atoms in the cluster were frozen. To obtain a reasonable value for the activation barrier, we studied H migration from an fcc to an hcp site on the Au(111) surface, representative of the stable surfaces on Au nanoparticles. The barrier for this H migration step is 10 kJ mol−1 (Fig. 3). Before its reaction with this H atom, the OH group on the support migrates from the initial site connected to the Cu cation (state vi, Fig. 2) to an adjacent site connected to a neighboring Mg cation (state vii, Fig. 2). This reaction is endothermic by 45 kJ mol−1 and it does not involve a notable activation barrier. Formation of water adsorbed to the support requires overcoming a barrier of 45 kJ mol−1. The reaction is thermodynamically very favorable and releases 123 kJ mol−1. Desorption of water into the gas phase takes 115 kJ mol−1. Thus, the overall water formation process from the H atom on the Au cluster with the support OH group is thermodynamically favorable.
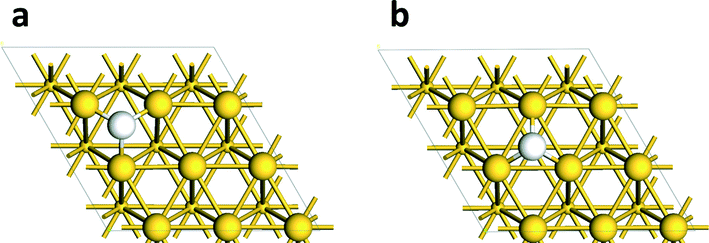 | ||
| Fig. 3 Adsorption sites for H on the Au(111) surface: (a) fcc hollow site, (b) hcp hollow site (the top two layers are shown with the second layer in smaller spheres; color scheme – yellow: Au; white: H). | ||
These reaction events generate one oxygen vacancy on the support surface close to the Au cluster and suggest that ethanol can be oxidized by the support oxygen atoms. This is consistent with the experimental finding that ethanol can also be converted to acetaldehyde and water in the absence of oxygen.15 It results, however, in catalyst deactivation, which should at least in part be due to the depletion of the O atoms of the support. Under aerobic reaction conditions, one O2 molecule will reduce two ethanol molecules to two acetaldehyde molecules. We found that O2 adsorbs on the oxygen vacancy site with an energy of 42 kJ mol−1 (state x, Fig. 2). The O–O bond distance in adsorbed O2 is elongated to 1.33 Å compared to the gas-phase value of 1.23 Å. It points to the formation of O2−. In contrast, we did not find any other adsorption sites for O2 close to the interface between the Au cluster and the stoichiometric MgCuCr2O4 support. The reason is that Cu is fully coordinated by O atoms.
The adsorption energy of ethanol on the Au atom located close to the interface is 73 kJ mol−1. Ethanol reacts with the O2− species to dissociate its hydroxyl group, forming an OOH* group on the support and an ethoxy group on the Au cluster (state xii, Fig. 2). This reaction has again a negligible barrier and is endothermic by 24 kJ mol−1. The bond distance of O–O after formation of OOH* is further elongated to 1.47 Å. The enthalpy change starting from ethanol in the gas phase to dissociated ethoxy and OOH* (from state x to state xii, Fig. 2) is −49 kJ mol−1, which is not as favorable as for the ethanol activation process by surface O (from state i to state ii, Fig. 2) is −117 kJ mol−1. Bader charge analysis34 of the O atoms on the pristine surface and of the top O atom of adsorbing O2 molecule on the defect surface predicts charges of −0.99 e for the former and −0.21 e for the latter. The difference underpins the higher basicity of the pristine surface, explaining the more exothermic activation step of gas-phase ethanol. Similar to the earlier explored pathway, the ethoxy group dissociates one of its α-C–H atoms to the Au cluster and, subsequently, desorbs as acetaldehyde.
Two different reaction pathways exist for the OOH* group adsorbed on the support. The first one involves its migration to the Au cluster and further recombination with H adsorbed to Au, whereas the second one involves O–OH dissociation and OH recombination with the H atom adsorbed to Au to close the reaction cycle. The migration of OOH from the support to the Au cluster is highly endothermic (ΔE > 100 kJ mol−1) and can therefore be excluded. During geometry optimization, the OOH group adsorbed on Au migrates spontaneously to the support vacancy. The preference for adsorption on the support is the much stronger Cu–O bond compared to the Au–O bond. In contrast, the alternative dissociation of OOH is spontaneous and exothermic by 147 kJ mol−1. The resulting OH group bridges between two Mg ions of the support (state xvi, Fig. 2) and recombines with the H atom from the Au cluster with a barrier of 45 kJ mol−1. This water formation reaction is again strongly exothermic by 123 kJ mol−1 and it takes 98 kJ mol−1 to desorb water into the gas phase, closing the whole reaction cycle.
As it was experimentally observed that substituting Mg with Cu in the spinel support is essential to obtain highly active catalysts (Au/MgCuCr2O4 exhibits an order of magnitude higher activity than Au/MgCr2O4 (ref. 15)), we investigated in more detail the role of Cu for the reaction mechanism of ethanol oxidation. For Au/MgCr2O4, we found that the dissociation of ethanol to ethoxy and OH surface species is spontaneous and exothermic by −118 kJ mol−1. This value is the same as the one computed for Au/MgCuCr2O4. It implies similar basicity of the O atoms in both support materials. The α-C–H bond cleavage to form acetaldehyde takes place on the Au cluster and we therefore do not expect any influence of Cu substitution in the support. We then found that the water formation step from H on the Au cluster with OH on the support is strongly dependent on the presence of substitutions in the support surface. Whereas the overall reaction energy for water formation is +37 kJ mol−1 for Au/MgCuCr2O4, it amounts to +136 kJ mol−1 for Au/MgCr2O4. This substantial energy difference suggests that regeneration of the active site by removal of water is facilitated by the presence of Cu. It can be understood by the stronger binding of OH to Mg2+ than to Cu2+. We should like to note here that the Au cluster is close to the oxygen vacancy site. This may influence the binding energy of surface O atoms of the support, as elegantly shown by Wahlström et al.35 As we will show below, the substitution of Mg for Cu is the dominant factor for the decrease in the oxygen vacancy formation energy.
On this basis, we speculated that the oxygen vacancy formation energy might be a useful descriptor for the reactivity in ethanol oxidation. To explore this, we defined the oxygen vacancy formation energy as E(vacancy formation) = E(defective surface) + 0.5 × E(O2) − E(pristine surface). This provides values for MgCr2O4 and MgCuCr2O4 of 160 and 102 kJ mol−1, respectively, correlating well with the difference in the energy for water formation. Substitutions of Mg in the MgCr2O4 support with Ni or Co only had a very minor effect on the ethanol oxidation activity, much less than that observed for the Cu-substituted support.15 Consistent with this, we compute oxygen vacancy formation energies of 170 kJ mol−1 for MgNiCr2O4 and 152 kJ mol−1 for MgCoCr2O4, respectively. Table 1 lists the oxygen vacancy formation energies of Au nanoparticles on substituted MgMeCr2O4 with Me = Co, Ni, Cu, Mg and experimentally measured catalytic activities of gold nanoparticles on such supports for oxidation of ethanol to acetaldehyde. When the energy needed to vacate the support surface is high, the catalytic activity is low. It relates to the removal of water as a difficult step in the overall reaction mechanism. We then attempted to apply this concept to understand the synergetic effect in Au–Cu alloy catalysts for selective alcohol oxidation.7,8 The synergy is attributed to the presence of reduced Au and Cu, the latter being possibly present as a thin CuOx film.7 Such Au–Cu catalyst systems typically deactivate, which has been correlated to the deep oxidation of Cu to bulk CuO. Our simple concept is able to explain the Au–Cu synergy and the deactivation. The binding energy for an O atom on the Cu(111) surface amounts to 120 kJ mol−1 (Fig. 4), which is only marginally higher than the energy for MgCuCr2O4. We speculate that O atoms adsorbed on the Cu metal surface will act as basic sites for ethanol activation, followed by further reaction on the Au cluster and OH removal as water. It is likely that O2 adsorption on the vacated Cu surface will be dissociative. However, the oxidative reaction conditions will lead to formation of CuO. Formation of CuO leads to a very high vacancy formation energy. For oxygen vacancy formation energy on the CuO(111) surface, we turn to the very recent results reported by Maimaiti et al.36 The value is in the range of 258–340 kJ mol−1 based on the same functional (GGA-PBE). Although the combination between Au and CuO will be able to activate ethanol into an ethoxy group and OH, the recombinative removal of OH with H as water will be prohibited by the strong binding of O in CuO. This is consistent with the experimentally observed deactivation of Au–Cu alloys due to formation of CuO.7
| Catalyst | d Au (nm) | Ratea (10−7 mol s−1) | TOFb (s−1) | E vacancy (kJ mol−1) |
|---|---|---|---|---|
a Reaction conditions: 0.1 g of catalyst, ethanol/O2/He = 1/3/63, T = 250 °C, GHSV = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL gcat−1 h−1.
b Turnover frequency (TOF) based on acetaldehyde yield and gold dispersion (D = 1.3/dAu)13 and given in molaldehyde molsurface Au−1 s−1. 000 mL gcat−1 h−1.
b Turnover frequency (TOF) based on acetaldehyde yield and gold dispersion (D = 1.3/dAu)13 and given in molaldehyde molsurface Au−1 s−1.
|
||||
| Au/MgCr2O4 | 3.3 | 3.8 | 0.20 | 160 |
| Au/MgCoCr2O4 | 3.2 | 4.2 | 0.21 | 153 |
| Au/MgNiCr2O4 | 3.3 | 3.4 | 0.18 | 170 |
| Au/MgCuCr2O4 | 3.1 | 17.2 | 0.89 | 102 |
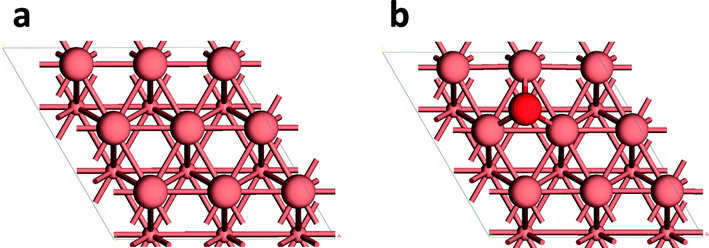 | ||
| Fig. 4 (a) Surface model of the Cu(111) surface; (b) adsorption of an O atom on the fcc hollow site of the Cu(111) surface (the top two layers are shown with the second layer in smaller spheres; color scheme – pink: Cu; red: O). | ||
Given the industrial importance of the oxidation of methanol to formaldehyde, we explored the reaction pathway from methanol to formaldehyde on Au/MgCuCr2O4 by computational modeling. We found that a similar pathway as found for ethanol oxidation to acetaldehyde is viable. Dissociative adsorption of methanol on the Au atoms located at the interface with the support (state ii, Fig. 5a) is strongly exothermic (−118 kJ mol−1) and barrierless. The reaction energy is similar to the dissociative adsorption of ethanol on this surface model. After rotation, the methoxy group dissociates one of its C–H bonds on the Au cluster (state iv, Fig. 5a) with a barrier of 38 kJ mol−1 (ΔE = 31 kJ mol−1). The adsorption energy of formaldehyde is 28 kJ mol−1, which is higher than that of acetaldehyde (17 kJ mol−1). The further reaction sequence leading to closure of the reaction cycle by water formation and desorption is similar to that of the ethanol oxidation case. Thus, we tested the optimized Au/MgCuCr2O4 catalyst for methanol oxidation but found that, instead of formaldehyde, CO2 is the main product at 250 °C. Fig. 6 shows methanol conversion and product selectivities as a function of the reaction temperature. At very low conversion at low reaction temperatures, the main products formed are formaldehyde and methyl formate. Formation of the latter product indicates that further oxidation of formaldehyde to formic acid occurs, consistent with the formation of formic acid at intermediate reaction temperatures. With increasing temperature, the formaldehyde selectivity decreases and the methyl formate selectivity goes through a maximum. The decreasing selectivities of desirable products are caused by complete oxidation of methanol to CO2. These results are very different from what was observed for ethanol oxidation, where acetaldehyde was the main product, even at a high temperature of 250 °C.
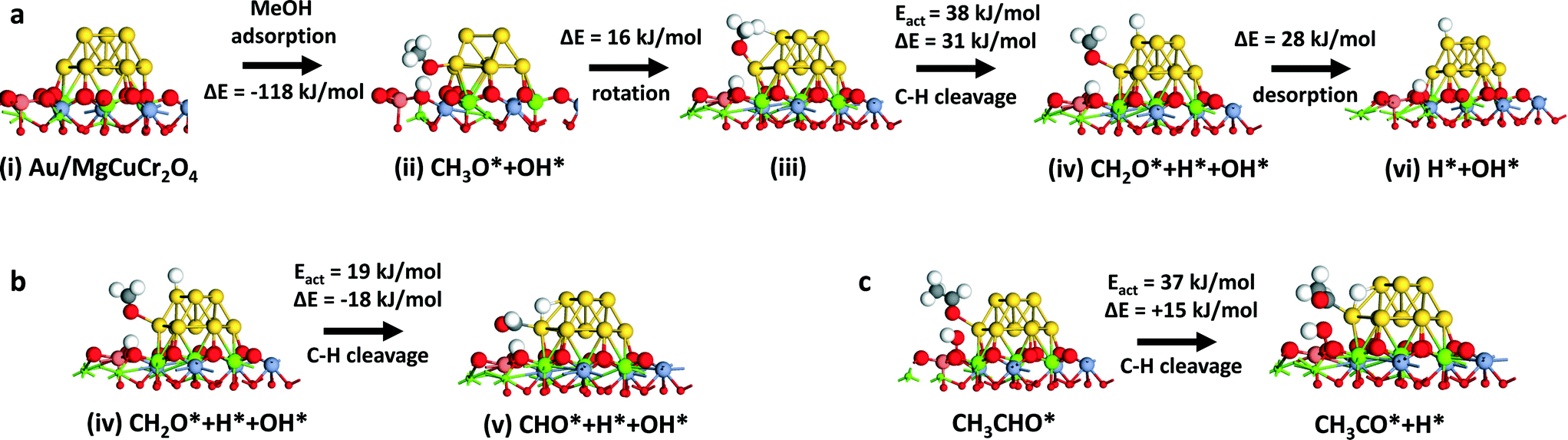 | ||
| Fig. 5 (a) Mechanism of oxidation of methanol to formaldehyde for the Au10/MgCuCr2O4 model. From state v, the recombinative desorption of water is similar to the mechanism found for ethanol oxidation (states v–ix in Fig. 2); (b) α-C–H bond cleavage in adsorbed formaldehyde and (c) α-C–H bond cleavage in adsorbed acetaldehyde. | ||
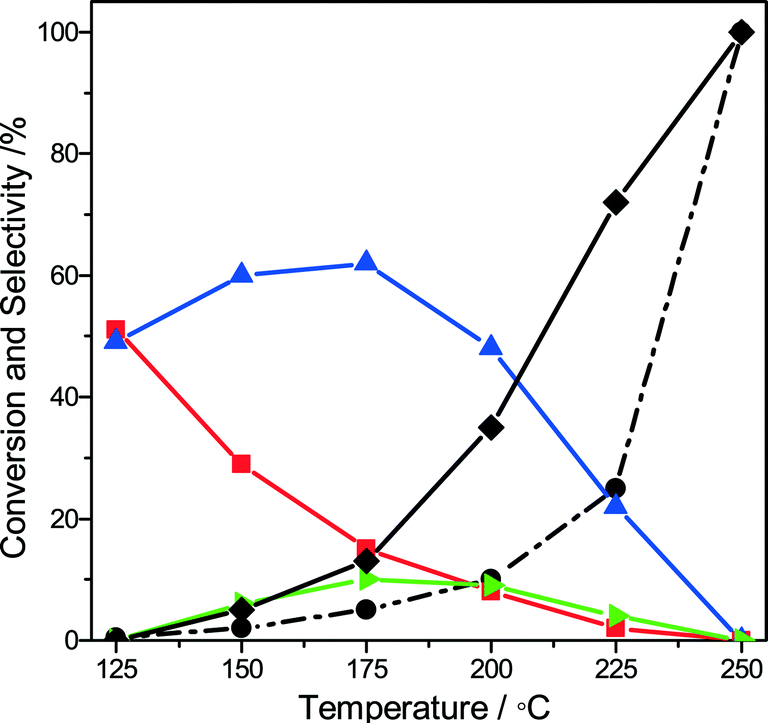 | ||
Fig. 6 Conversion and selectivity of methanol oxidation (0.1 g of catalyst, 5 vol% methanol, methanol/O2/He ratio = 1/3/16, GHSV = 60000 mL gcat−1 h−1) as a function of the reaction temperature (●: methanol,  : formaldehyde, : formaldehyde,  : methyl formate, : methyl formate,  : formic acid, ♦: carbon dioxide). : formic acid, ♦: carbon dioxide). | ||
To better understand the significant difference between methanol and ethanol oxidation, we explored further decomposition of adsorbed formaldehyde (Fig. 5b) and acetaldehyde (Fig. 5c) by quantum-chemical modeling. The cleavage of the remaining α-C–H bond in adsorbed formaldehyde has a barrier of 19 kJ mol−1 and is exothermic by 18 kJ mol−1. These energetics are more favorable than desorption of formaldehyde, explaining the preference for its further oxidation. Comparatively, the cleavage of the α-C–H bond in adsorbed acetaldehyde is much more difficult. The activation energy is 37 kJ mol−1. Consistent with the higher activation energy, the reaction energy is endothermic by 15 kJ mol−1. Thus, desorption of acetaldehyde is more favorable than further α-C–H bond cleavage to products that can lead to complete oxidation.
4. Conclusions
To summarize, we have explored for the first time the mechanism of catalytic alcohol oxidation for the highly active and selective Au/MgCuCr2O4 catalyst. Our findings underpin the importance of the interface between gold and the subjacent support, which has earlier only been investigated for CO oxidation. Dissociative adsorption of ethanol is facile, taking place through the cooperative action of gold and the basic oxygen atoms of the support. The cleavage of the C–H bond in the adsorbed ethoxy group takes place over the gold cluster followed by desorption of acetaldehyde and recombinative H2O desorption. A second ethanol molecule is oxidized in the same manner with O2 adsorbed on the oxygen vacancy acting as the basic site. The resulting OOH intermediate dissociates. The catalytic cycle is closed by recombination of the OH intermediate with the hydride atom adsorbed on gold followed by water desorption. The oxygen vacancy formation energy is found to be a good descriptor for the performance in ethanol oxidation of Au/MgMeCr2O4 (Me = Cu, Ni, Co) catalysts. It also provides a satisfactory explanation for the Au–Cu alloy synergy and deactivation in alcohol oxidation reported in the literature. Finally, we found that the high selectivity towards acetaldehyde stems from facile desorption of acetaldehyde as compared to cleavage of its remaining α-C–H bond. The opposite holds for methanol oxidation, that is formaldehyde decomposition is favorable compared to its desorption, explaining why experimentally we observe complete methanol combustion over Au/MgCuCr2O4 under conditions where ethanol is selectively converted to acetaldehyde.Acknowledgements
We acknowledge financial support for this research from ADEM, A green Deal in Energy Materials of the Ministry of Economic Affairs of The Netherlands (www.adem-innovationlab.nl). Supercomputing facilities were funded by the Netherlands Organization for Scientific Research.References
- T. Taker, N. Iguchi and M. Haruta, Catal. Surv. Asia, 2011, 15, 80 CrossRef.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538 CrossRef CAS; K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2003, 9, 4353 CrossRef PubMed.
- K. Shimizu, K. Sugino, K. Sawabe and A. Satsuma, Chem. – Eur. J., 2009, 15, 2341 CrossRef CAS PubMed.
- (a) F. Zaccheria, N. Ravasio, R. Psaro and A. Fusi, Chem. – Eur. J., 2006, 12, 6426 CrossRef CAS PubMed; (b) L. Huang, Y. Zhu, C. Huo, H. Zheng, G. Feng, C. Zhang and Y. Li, J. Mol. Catal. A: Chem., 2008, 288, 109 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed; (b) C. D. Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC; (c) A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC; (d) Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467 CrossRef CAS PubMed.
- (a) K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS PubMed; (b) J. D. Grunwaldt, M. Caravati and A. Baiker, J. Phys. Chem. B, 2006, 110, 25586 CrossRef CAS PubMed; (c) D. Ferri, C. Mondelli, F. Krumeich and A. Baiker, J. Phys. Chem. B, 2006, 110, 22982 CrossRef CAS PubMed.
- J. C. Bauer, G. M. Veith, L. F. Allard, Y. Oyola, S. H. Overbury and S. Dai, ACS Catal., 2012, 2, 2537 CrossRef CAS.
- C. D. Pina, E. Falletta and M. Rossi, J. Catal., 2008, 260, 384 CrossRef PubMed.
- B. N. Zope, D. H. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74 CrossRef CAS PubMed.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC.
- C. Caro, K. Thirunavukkarasu, M. Anilkumar, N. R. Shiju and G. Rothenberg, Adv. Synth. Catal., 2012, 354, 1327 CrossRef CAS PubMed.
- (a) R. Jira, Angew. Chem., Int. Ed., 2009, 48, 9034 CrossRef CAS PubMed; (b) J. A. Keith and P. M. Henry, Angew. Chem., Int. Ed., 2009, 48, 9038 CrossRef CAS PubMed.
- M. Eckert, G. Fleischmann, R. Jira, M. B. Hermann and K. Golka, Acetaldehyde, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- W. Opitz and W. Urbanski, Knapsack-Griesheim, DE 1 097 969, 1954; W. Opitz and W. Urbanski, DE 1 108 200, 1955.
- P. Liu and E. J. M. Hensen, J. Am. Chem. Soc., 2013, 135, 14032 CrossRef CAS PubMed.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736 CrossRef CAS PubMed.
- M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771 CrossRef CAS PubMed.
- C. Liu, Y. Tan, S. Lin, H. Li, X. Wu, L. Li, Y. Pei and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 2583 CrossRef CAS PubMed.
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134, 1560 CrossRef CAS PubMed.
- A. P. Vieria Soares, M. Farinha Portela and A. Kiennemann, Catal. Rev.: Sci. Eng., 2005, 47, 125 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Jouber, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- O. Bengone, M. Alouani, P. Blöchl and J. Hugel, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 16392–16401 CrossRef CAS.
- H. J. Xiang, E. J. Kan, S. Wei, M. H. Whangbo and X. G. Gong, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 224429 CrossRef.
- (a) L. M. Molina, M. D. Rasmussen and B. Hammer, J. Chem. Phys., 2004, 120, 7673 CrossRef CAS PubMed; (b) I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS PubMed.
- Y. Guan and E. J. M. Hensen, Appl. Catal., A, 2009, 361, 49 CrossRef CAS PubMed.
- M. Boronat, A. Corma, F. Illas, J. Radilla, T. Ródenas and M. J. Sabater, J. Catal., 2011, 278, 50 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- E. Wahlström, N. Lopez, R. Schaub, P. Thostrup, A. Rønnau, C. Africh, E. Lægsgaard, J. K. Nørskov and F. Besenbacher, Phys. Rev. Lett., 2003, 90, 026101 CrossRef.
- Y. Maimaiti, M. Nolan and S. D. Elliott, Phys. Chem. Chem. Phys., 2014, 16, 3036 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00462k |
| This journal is © The Royal Society of Chemistry 2014 |