 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of new derivatives of copper complexes of Josiphos family ligands for applications in asymmetric catalysis†
R.
Oost
,
J.
Rong
,
A. J.
Minnaard
* and
S. R.
Harutyunyan
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborg 4, 9747 AG, Groningen, The Netherlands. E-mail: S.Harutyunyan@rug.nl; A.J.Minnaard@rug.nl
First published on 24th March 2014
Abstract
A series of new copper complexes containing chiral ferrocenyl diphosphine ligands of the Josiphos family have been prepared. These complexes have been studied in the catalytic asymmetric 1,2-addition of Grignard reagents to enones and aromatic ketones. Variation of the electronic and steric properties of the ligand resulted in a positive effect in the regio- and enantioselectivity of Grignard reagents to α-H-substituted enones using the ligand in which tert-butyl substituents were introduced in the diarylphosphine moiety. The copper complexes were also successfully applied in the catalytic asymmetric conjugate addition of Grignard reagents to enoates. No increase of enantioselectivity was observed in the catalytic asymmetric addition of linear Grignard reagents, compared to that of the commercially available ligand rev-Josiphos.
Introduction
Ferrocenyl based ligands, in particular diphosphines, are amongst the most important of chiral ligands in transition metal catalysis.1 Josiphos, the primus inter pares of the ferrocenyl diphosphines, provides high activity and enantioselectivity in a wide range of catalytic transformations, including hydrogenations,2 Buchwald–Hartwig aminations,3 and Cu-catalyzed conjugate reductions4 as well as additions.5 However, fine-tuning of the steric and electronic properties of these ligands is often needed to achieve the desired excellent catalytic performance in a specific reaction or for a specific substrate.Recently, we demonstrated that rev-Josiphos, in combination with a Cu(I)-salt, catalyzes the 1,2-addition of Grignard reagents to enones and aromatic ketones.6–8 This strategy provides direct access to chiral tertiary alcohols with excellent yields and enantioselectivities (>95%). During this research we found that in the addition to enones, the α-substituent on the olefin plays an important role in the 1,2- versus 1,4-regioselectivity, with excellent 1,2-selectivity observed only with α-bromo and α-methyl substituted enones. For non-substituted enones, a mixture of 1,2- and 1,4-addition products is obtained, both with low enantioselectivities.
The most direct approach to improve catalyst performance is through fine-tuning of the ligand structure. We envisioned that changing the steric properties and tuning the electronic properties of the ligand will improve the regioselectivity of the reaction in favour of the 1,2-addition to non-substituted enones by affecting the equilibrium between the intermediate π- and σ-complexes.9
Here, we report the synthesis of five new Cu-complexes of Josiphos-type ligands, Cu-L1–L5, and their application in the Cu-catalyzed 1,2-addition of Grignard reagents to enones and aromatic ketones as well as in the 1,4-addition to α,β-unsaturated esters (Fig. 1).
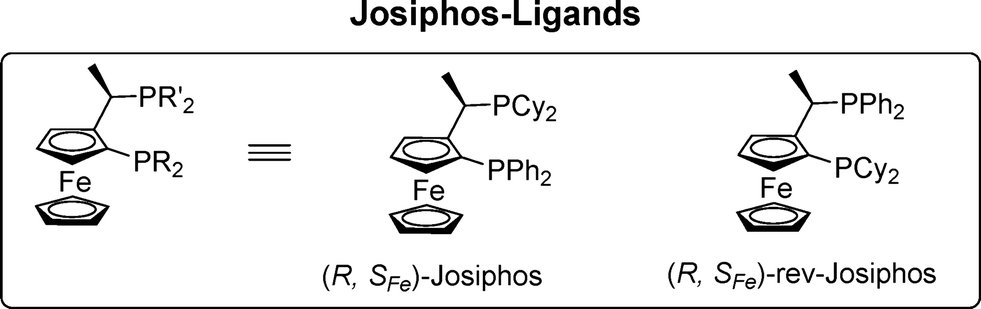 | ||
| Fig. 1 Representative ligands of Josiphos family. | ||
Results
Synthesis of Cu-L1–L5
Josiphos-type ligands are readily accessible via a two-step synthesis starting from enantiomerically pure Ugi's amine.10 Due to the stepwise introduction of the phosphine substituents, fine tuning of the ligands is achieved by variation in the substituents on each of the phosphorus atoms. An alternative approach is to modify Josiphos-type ligands by increasing the steric bulk of the ferrocenyl side-chain.11The copper(I) complexes of the Josiphos-type ligands were synthesized using a modification of the procedure reported by Togni et al. for the synthesis of ferrocenyl ligands (Scheme 1).12 The phosphines were introduced in two steps starting from (S)-Ugi's amine, in turn prepared in four steps from ferrocene.13 In the first step, (S)-Ugi's amine was diastereoselectively ortho-lithiated followed by reaction with chlorodicyclohexylphosphine to provide aminophosphine 1 in 78% yield.14 The dimethylamino moiety was then substituted with the corresponding secondary phosphane, with retention of configuration,15 to give the desired Josiphos-type ligand. As for Josiphos, these ferrocenyl diphosphines were bench-stable, but oxidized slowly in air. Therefore all ligands, without further isolation/purification, were reacted with CuBr·SMe2 in CH2Cl2 at room temperature to form their corresponding copper(I) complexes (Scheme 1). The complexes were stable in air and moisture for several months at room temperature. If needed, the copper could be removed quantitatively by treatment with ethylenediamine to recover the corresponding free ligand.16
 | ||
| Scheme 1 General procedure for the synthesis of copper complexes of rev-Josiphos derivatives. | ||
In the synthesis of Cu-L2, substitution of the dimethylamino moiety for bis(3,5-di(trifluoromethyl)phenyl)phosphine did not proceed with full retention of configuration, which is in good agreement with earlier observations,17 with a diastereomeric ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 determined by 31P-NMR spectroscopy. However, the diastereomers of the copper complexes could be separated by flash-chromatography over silica gel.
:1 determined by 31P-NMR spectroscopy. However, the diastereomers of the copper complexes could be separated by flash-chromatography over silica gel.
We attempted to introduce a sterically more demanding phosphine, i.e. diadamantylphosphine, in place of a dicyclohexylphosphine moiety to understand the influence of steric encumbrance at the other phosphine group on the reactivity/selectivity of the copper complexes in addition reactions.19 However, the reaction of ortho-lithiated Ugi's amine with diadamantylphosphine chloride did not yield the desired product (Scheme 2). Despite several efforts, we were not able to introduce this phosphine on the ortho-position, presumably due to steric bulk.
 | ||
| Scheme 2 Introduction of a diadamantylphosphine group in the ligand structure. | ||
The copper complex of the isobutyl analogue of rev-Josiphos Cu-L5 was prepared to study the effect of an increase in steric interactions in the backbone of the ligand (Scheme 3). Ketone 2 was obtained in 68% yield by Friedel–Crafts acylation of ferrocene with isovaleryl chloride. Chiral alcohol 3 was obtained by enantioselective reduction with borane–dimethyl sulfide at 0 °C in THF in the presence of 30 mol% of (S)-2-methyl-5,5-diphenyl-3,4-propano-1,3,2-oxazaborolidine. The enantiomeric excess of 3, determined by HPLC, was 98% after recrystallization. Acetylation of 3 with Ac2O in pyridine afforded acetate 4. The conversion of acetate 4 to amine 5 was accomplished in good yield by treatment with aqueous dimethylamine in methanol following the procedure of Ugi.9
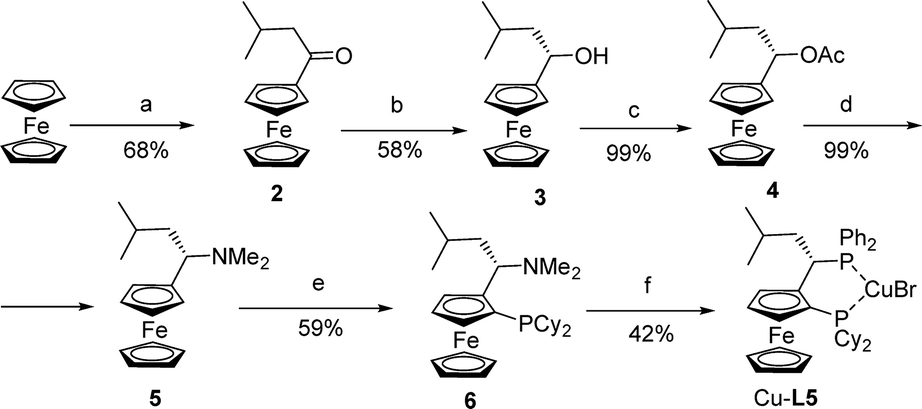 | ||
| Scheme 3 Synthesis of Cu-L5. a) 1 eq. isovaleryl chloride, 1 eq. AlCl3, DCM, rt, 3 h; b) 1.2 eq. BH3·SMe2, 30 mol% (S)-2-methyl-5,5-diphenyl-3,4-propano-1,3,2-oxazaborolidine, THF, 0 °C, 2 h; c) Ac2O, pyridine, rt, 30 h; d) aq. HNMe2, MeOH, rt, 30 h; e) first 1.2 eq. sec-BuLi, Et2O, 0 °C, then 2 eq. Cy2PCl, rt, overnight; f) first 1 eq. Ph2PH, AcOH, 90 °C, 5 h, then 1 eq. CuBr·SMe2, CH2Cl2, rt, 2 h. | ||
As with Ugi's amine, ferrocenyl amine 5 can undergo diastereoselective ortho-lithiation. This reaction was carried out with sec-butyllithium in ether at 0 °C. The use of an excess of chlorodicyclohexylphosphine was necessary to obtain 6 in acceptable yields and high diastereoselectivities (>95%). The dimethylamino group was then substituted with diphenylphosphine in acetic acid according to Togni's procedure,12 and Cu-L5 was formed in 42% yield (Scheme 3).
Application of Cu-L1–L5 in catalysis
The Cu-catalyzed 1,2-additions of both iBuMgBr and EtMgBr to enone 7 were chosen as model reactions in the evaluation of the catalytic activity/selectivity of Cu-L1–L5. The 1,2-addition product of iBuMgBr was obtained with 90% ee using Cu-rev-Josiphos (Table 1, entry 1). Similar results were obtained with Cu-L1 that contains electron donating substituents in the diarylphosphine moiety (entry 2). However, with electron-withdrawing substituents, e.g., in Cu-L2, the reaction yielded the same result as in the blank reaction (entry 3). A considerable decrease of enantioselectivity in the reactions catalysed by Cu-L3–L5 compared to that by Cu-rev-Josiphos (entries 4–6) coincides with an increase in the steric encumbrance of the ligands. Interestingly, with Cu-L3, the product obtained has the opposite configuration to that obtained with Cu-rev-Josiphos (entry 4).|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | RMgBr |
8:9:10b |
8 (ee)c,d |
|
a Reaction conditions: addition of 1.2 equiv. RMgBr to a 0.15 M solution of 7 in tBuOMe at −78 °C in the presence of 5 mol% Cu-L1–L5.
b The ratio of 8:9:10 was determined by GC analysis.
c Enantioselectivity of 8 was determined by HPLC analysis.
d The absolute configuration of 8 is R (ref. 20).
e The opposite enantiomer was obtained.
|
||||
| 1 | Cu-rev-Josiphos | iBuMgBr | 97:1:2 |
90 |
| 2 | Cu-L1 | iBuMgBr | 99:1:0 |
87 |
| 3 | Cu-L2 | iBuMgBr | 25:25:50 |
Racemic |
| 4 | Cu-L3 | iBuMgBr | 71:26:3 |
27e |
| 5 | Cu-L4 | iBuMgBr | 97:2:1 |
60 |
| 6 | Cu-L5 | iBuMgBr | 78:4:18 |
34 |
| 7 | Cu-rev-Josiphos | EtMgBr | 98:1:1 |
23 |
| 8 | Cu-L1 | EtMgBr | 96:4:0 |
11 |
| 9 | Cu-L2 | EtMgBr | 87:12:1 |
17e |
| 10 | Cu-L3 | EtMgBr | 90:0:10 |
6e |
| 11 | Cu-L4 | EtMgBr | 86:13:1 |
10 |
| 12 | Cu-L5 | EtMgBr | 99:1:0 |
14 |

In the addition of EtMgBr to α-Br-substituted enones, where the Cu-rev-Josiphos only gives 23% ee (Table 1, entry 7), we found that all complexes, Cu-L1–L5, provide a low ee as well (entries 8–12). Surprisingly, Cu-L2 catalyses this reaction with good 1,2-selectivity and 17% ee (entry 9). These data indicate that neither an increase in steric bulk on the diarylphosphine nor the introduction of an isobutyl substituent in the backbone increases the enantioselectivity in the copper-catalyzed 1,2-addition to enones. Changing the electronic properties of the copper complexes also leads to a small decrease in enantioselectivity compared to Cu-rev-Josiphos. Despite the low enantioselectivity, it is interesting to note that once again both Cu-L2 and Cu-L3 provide the product with the opposite configuration compared with Cu-rev-Josiphos.
In the addition of Grignard reagents to α-H-substituted enones, regioselectivity is an important issue. Research over the last 80 years has established that copper(I) based reagents and catalysts are the primary synthetic tool to obtain 1,4-selectivity. The use of Cu-rev-Josiphos and EtMgBr led to only 16% 1,2-addition product (Table 2, entry 1). Changing the Grignard reagent to the β-branched iBuMgBr increased the 1,2-selectivity to 29% (entry 2). This indicates that not only the α-substituent determines the regioselectivity but also the steric hindrance of the Grignard reagent is important. Therefore we chose to use iBuMgBr for our screening.
|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | RMgBr |
12:13:14b |
12 (ee)c,d |
|
a Reaction conditions: addition of 1.2 equiv. RMgBr to a 0.15 M solution of 11 in tBuOMe at −78 °C in the presence of 5 mol% Cu-L1–L5.
b The ratio of 12:13:14 was determined by GC analysis.
c Enantioselectivity of 12 was determined by HPLC analysis.
d The opposite enantiomer was obtained.
|
||||
| 1 | Cu-rev-Josiphos | EtMgBr | 16:84:0 |
14 |
| 2 | Cu-rev-Josiphos | iBuMgBr | 29:64:6 |
28 |
| 3 | Cu-L1 | iBuMgBr | 19:70:11 |
12 |
| 4 | Cu-L2 | iBuMgBr | 5:95:0 |
n.d. |
| 5 | Cu-L3 | iBuMgBr | 8:75:17 |
31d |
| 6 | Cu-L4 | iBuMgBr | 43:56:1 |
54 |
| 7 | Cu-L5 | iBuMgBr | 28:48:24 |
9 |

We found that an electron donating substituent in Cu-L1 does not change the regioselectivity considerably. In contrast, the use of electron withdrawing groups in Cu-L2 (entry 4) affects the regioselectivity drastically, with almost full 1,4-selectivity albeit as a racemate. As in the previous case, Cu-L3 led to the 1,2-addition product with the opposite configuration in 31% ee (entry 5). Importantly an increase in 1,2-selectivity as well as in enantioselectivity was observed with Cu-L4 (43% and 54%, respectively, entry 6). Comparison of the results of entries 3 and 6 (Table 2) indicates that the steric hindrance, rather than the change in electronic properties of the diarylphosphine moiety, plays a dominant role in the regio- and enantioselectivity.
The catalytic system was also applied in the 1,2-addition of Grignard reagents to aryl alkyl ketones. In previous studies it was found that although the yields were excellent, high enantioselectivities were obtained only with bulkier Grignard reagents.8 The addition of EtMgBr to ketone 15 catalysed by Cu-rev-Josiphos provided only 22% ee (Table 3). The complexes Cu-L1–L5 were screened in the same reaction. As in the addition to enones, enantioselectivities decreased compared to Cu-rev-Josiphos and the configuration of the product changed for Cu-L2 and Cu-L3. With Cu-L4 and Cu-L5, only racemic products were obtained with lower regioselectivities (entries 5 and 6).
|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | RMgBr |
16:17b |
16 (ee)c |
|
a Reaction conditions: addition of 1.2 equiv. EtMgBr to a 0.15 M solution of 15 in tBuOMe at −78 °C in the presence of 5 mol% Cu-L1–L5.
b The ratio of 16:17 was determined by GC analysis.
c Enantioselectivity of 16 was determined by HPLC analysis.
d The opposite enantiomer was obtained.
|
||||
| 1 | Cu-rev-Josiphos | EtMgBr | 99:1 |
22 |
| 2 | Cu-L1 | EtMgBr | 87:13 |
9 |
| 3 | Cu-L2 | EtMgBr | 99:1 |
14d |
| 4 | Cu-L3 | EtMgBr | 90:10 |
16d |
| 5 | Cu-L4 | EtMgBr | 56:44 |
Racemic |
| 6 | Cu-L5 | EtMgBr | 71:29 |
Racemic |
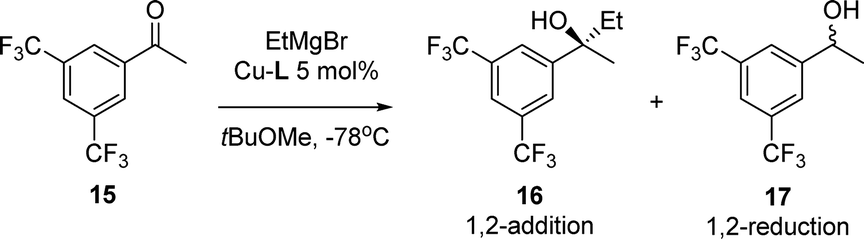
The new complexes were also studied in a reaction in which Cu-rev-Josiphos provides excellent results, the 1,4-addition of EtMgBr to α,β-unsaturated esters (Table 4).21
|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | RMgBr | Yieldb (%) | 19 (ee)c |
| a Reaction conditions: addition of 19 to a 0.3 M solution of EtMgBr in DCM at −78 °C in the presence of 5 mol% Cu-L1–L5. b Conversion determined by GC analysis. c Enantioselectivity of 19 was determined by GC analysis on a chiral phase (B-PM). | ||||
| 1 | Cu-rev-Josiphos | EtMgBr | 94 | 98 |
| 2 | Cu-L1 | EtMgBr | 95 | 96 |
| 3 | Cu-L2 | EtMgBr | 97 | 88 |
| 4 | Cu-L3 | EtMgBr | 95 | 7 |
| 5 | Cu-L4 | EtMgBr | 56 | 70 |
| 6 | Cu-L5 | EtMgBr | 96 | 81 |

All of the new complexes provided good yields, and most of them provided good enantioselectivities also. Complex Cu-L1 provided, as in the 1,2-additions, similar results to those obtained with rev-Josiphos (entry 2). The other complexes provide good enantioselectivities, but less than that obtained with the Cu-rev-Josiphos. Cu-L3 showed only 7% enantioselectivity (entry 4). Interestingly, Cu-L4 provided low 1,4-regioselectivity (entry 5). This result correlates well with relatively good 1,2-selectivity obtained using the same complex in the addition of Grignard reagents to α-H-substituted enones (Table 2, entry 6).
Discussion
In the present contribution several electronically and sterically distinct Cu-complexes have been synthesized and tested as catalysts in enantioselective 1,2- and 1,4-additions of Grignard reagents to carbonyl compounds. From the data obtained in the addition of iBuMgBr and EtMgBr to α-Br-substituted enones, one can conclude that increasing the electron donating properties of one of the phosphine moieties of the catalyst (Cu-L1) does not affect, substantially, the performance of the catalyst when compared with that observed using Cu-rev-Josiphos (Table 1, entries 1 and 2). By contrast, introducing electron-withdrawing groups at the phosphine moiety decreases the regioselectivity as well as the stereoselectivity in the 1,2-addition reaction (Table 1, compare entries 1 and 7 with entries 3 and 9). We surmise that reducing the electron density at the phosphine moiety has a negative effect on the ligands' association constant to copper(I). This in turn results in reduced catalytic activity, and therefore, the overall selectivity of the reaction decreases due to an increase in the relative contribution of the non-catalysed 1,2- and 1,4-additions as well as the 1,2-reduction.Increasing steric interactions both at the phosphine moieties (Cu-L3–L5) and at the chiral carbon center results in a pronounced decrease in enantiofacial discrimination (Table 1).
The data obtained with catalysts Cu-L1–L5 in the additions to aryl alkyl ketone 15 did not, however, show an obvious correlation. In all of the cases very low stereoselectivity was observed, and in the case of Cu-L4 and Cu-L5, lower catalytic activity was apparent from the increased extent of the 1,2-reduction to yield 17 (Table 3).
One of the goals of this study was to understand the factors that govern the regioselectivity observed in the addition of Grignard reagents to α-H-substituted enones. Research efforts over the last decades have shown that, in the case of Cu-catalysed reactions, 1,4-addition of organometallic reagents is the most common pathway. In our previous studies with chiral ferrocenyl ligands, we have observed this trend. Using Cu-rev-Josiphos as a catalyst in the addition of Grignard reagents to α-H-substituted enones provided mainly 1,4-product 13 (Table 2, entries 1 and 2). When testing our new catalysts Cu-L1–L5 in this transformation, we were pleased to find that both the regioselectivity and enantioselectivity of the 1,2-addition reaction greatly increased using catalyst Cu-L4, which bears a sterically hindered and electron rich phosphine moiety (Table 2, entry 6). From a mechanistic perspective, it is well established that transmetallated Cu-species are capable of forming π-complexes with a conjugated double bond followed by oxidative addition to form a σ-complex and reductive elimination to yield the 1,4-addition product.5 However recently it has been shown that transmetallated Cu-species are capable of π-complex formation with a conjugated carbonyl moiety also.25 Our present empirical data indicate that the presence of an α-substituent at the enone 7 destabilizes the formation of π- and σ-complexes with a conjugated double bond. This most likely drives the Cu-catalyst to form π-complexes with the carbonyl of the enone, followed by formation of the 1,2-addition product (Table 1). Therefore high 1,2-selectivity has been obtained with most of the catalysts studied. In contrast, in the case of α-H-substituted enone 11, such substrate control is absent and the reaction proceeds via π- and σ-complexes followed by 1,4-product formation. However when sterically hindered and electron rich catalyst Cu-L4 was used for the first time a clear catalyst control over the regio- and stereoselectivity of the addition was observed. This conclusion is supported by consideration of the data obtained regarding the 1,4-addition of EtMgBr to α,β-unsaturated esters. Most of the ligands were found to be excellent catalysts in regard to 1,4-selectivity, while Cu-L4 provided both lower 1,4-selectivity and lower enantioselectivity (Table 4, entry 5).
Conclusions
We report here the synthesis of five new copper complexes of rev-Josiphos-type ligands with a variation in steric and electronic properties. We successfully introduced electron donating, electron withdrawing and bulky substituents on the phosphines as well as a bulky substituent in the backbone. These complexes have been applied to the 1,2- and 1,4-additions of alkyl magnesium reagents to enones. We found that in the 1,2-addition to α-H-substituted enones, the Cu-L4 with bulky tert-butyl and electron donating methoxy groups on the aromatic ring provides higher regio- and enantioselectivities compared to Cu-rev-Josiphos. Copper complexes were also successfully applied in the 1,4-addition of EtMgBr to an α-H-substituted enone: methyl cinnamate. While all newly synthesized catalysts provided high 1,4-selectivity, Cu-L4 was found to give lower regio- and enantioselectivity. These results provide further insight into how to optimise catalyst design towards better 1,2-selectivity with α-H-substituted enones. In the 1,2-addition to aromatic ketones complexes Cu-L1–L5 did not improve the enantioselectivity of linear Grignard reagents. Further investigations to increase the enantioselectivity of linear Grignard reagents and to design better 1,2-selective catalysts are currently underway.Experimental
General
All reactions were carried out under a nitrogen atmosphere using oven-dried glassware and using standard Schlenk techniques. Dichloromethane, THF and tBuOMe were used from the solvent purification system. (S)-Ugi's amine13 and 7 (ref. 22) were prepared according to literature procedures. All other starting materials and Grignard reagents were purchased from Aldrich (iBuMgBr (2 M in Et2O) and EtMgBr (3 M in Et2O)). Chromatography was performed on silica gel (230–400 mesh). Thin-layer chromatography was performed on silica plates. Compounds were visualized by UV and cerium/molybdenum or potassium permanganate staining. Progress and conversion of the reaction were determined by GC-MS. Mass spectra were recorded on a mass spectrometer using an Orbitrap analyser. 1H, 13C, 19 F and 31P spectra were recorded on 400 and 100.59 MHz using CDCl3 as solvent. Chemical shift values are reported in ppm with the solvent resonance as the internal standard (CHCl3: δ 7.26 for 1H, δ 77.0 for 13C). Data are reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constants (Hz), and integration. Optical rotations were measured on a polarimeter with a 10 cm cell (c given in grams per 100 mL). Enantiomeric excesses were determined by chiral GC or HPLC analysis.(R)-1-Dicyclohexylphosphano-2-[α-(S)-(N,N-dimethylamino)ethyl]ferrocene 1 (ref. 12)
sec-Butyllithium (1.4 M in cyclohexane, 0.9 mL, 1.26 mmol) was added to a solution of Ugi's amine (0.3 g, 1.09 mmol) in Et2O (4 mL) at 0 °C. After 2 h, chlorodicyclohexylphosphane (0.3 mL, 1.3 mmol) was added at 0 °C and the solution was allowed to warm up to rt and stirred overnight. Saturated aqueous Na2CO3 (3 mL) was added and the layers were separated. The aqueous layer was extracted three times with CH2Cl2 (4 mL) and the combined organic layers were dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography (SiO2, pentane/Et2O 5:1) to give compound 1 as an orange solid (0.39 g, 78%). 1H-NMR (CDCl3): δ 0.9–2.0 (m, 25H), 2.08 (s, 6H), 2.35 (s, 1H), 4.03 (s, 5H), 4.08 (s, 1H), 4.24 (d, 2H). 13C-NMR (CDCl3): δ 9.0, 26.8 (d, J = 11.6 Hz), 27.6 (d, J = 8.3 Hz), 27.9 (d, J = 12.3 Hz), 28.1 (d, J = 7.1 Hz), 28.6, 28.7, 28.8, 31.0 (d, J = 9.8 Hz), 32.6 (d, J = 16.7 Hz), 32.8 (d, J = 18.5 Hz), 34.2 (d, J = 8.3 Hz), 36.7 (d, J = 12.3 Hz), 39.6, 56.8 (d, J = 8 Hz), 67.5, 68.7 (d, J = 3 Hz), 70.0, 70.3, 79.1, 95.6. 31P-NMR (CDCl3): δ – 11.3 (s).
4-Methylvalerylferrocene 2 (ref. 23)
Isovaleryl chloride (2 mL, 16.1 mmol) was added to a suspension of aluminium(III) chloride (2.3 g, 17 mmol) in CH2Cl2 (35 mL) at 0 °C. This solution was added to a solution of ferrocene (3.0 g, 16.1 mmol) in CH2Cl2 (35 mL) at 0 °C. The reaction mixture was warmed to rt and stirred for 3 h. The reaction was quenched by adding ice-cold water (50 mL) at 0 °C. The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 × 50 mL). The organic layers were washed with saturated K2CO3 solution, brine and water. The organic layer was dried over MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (SiO2, pentane/Et2O 9:1). Pure 3 was isolated as an orange solid (2.95 g, 68%). 1H-NMR (CDCl3): δ 0.98 (d, 6H, J = 6.5 Hz), 2.26 (app. nonet, 1H, J = 6.5 Hz), 2.55 (d, 2H, J = 6.5 Hz), 4.20 (s, 5H), 4.49 (s, 2H), 4.78 (s, 2H). 13C-NMR (CDCl3): 23.1 (2CH3), 25.2 (CH), 49.0 (CH2), 69.5 (2CH), 69.9 (5CH), 72.3 (2CH), 79.9 (C) 203.7 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
(S)-Ferrocenyl-3-methylbutanol 3
A stirred solution of 0.56 g of (R)-2-methyl-5,5-diphenyl-3,4-propano-1,3,2-oxazaborolidine (3 mmol, 30 mol%) in 5 mL of THF was cooled to 0 °C, and 0.6 mL of borane–Me2S solution (1.2 mmol, 2 M in THF) was added. After 15 min, a solution of 2 (1.63 g, 6 mmol) in 20 mL of THF and, separately, a borane–Me2S solution (2.4 mL, 4.8 mmol, 2 M in THF) were simultaneously added slowly over a period of 1 h at 0 °C. The reaction mixture was stirred for 1 h at 0 °C before it was quenched with 5 mL of MeOH. The solution was concentrated under reduced pressure and the crude product was purified by column chromatography (pentane/EtOAc 4:1) and recrystallized from hot hexane. Pure 3 was obtained as an orange solid (1.82 g, 58%, 98% ee). The enantiomeric ratio was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 98:2, 40 °C, detection at 254 nm, retention times (min): 16.9 (major) and 18.3 (minor). 1H-NMR (CDCl3): δ 0.91 (t, J = 5.5 Hz, 6H), 1.41 (m, 1H), 1.62 (m, 1H), 1.76 (m, 2H), 4.21 (s, 2H), 4.25 (s, 5H), 4.29 (s, 2H). 13C-NMR (CDCl3): δ 22.4 (CH3), 23.6 (CH3), 25.1 (CH), 47.5 (CH2), 61.7 (CHOH), 66.1 (2CH), 67.8 (5CH), 69.1 (2CH), 95.8 (C). HRMS (ESI+) calcd for C15H19Fe [M − OH]+ 255.08307, found 255.082.
(S)-Ferrocenylethyl-3-methylpropyl acetate 4
Ferrocenyl alcohol 3 (0.94 g, 3.5 mmol) was dissolved in 10 mL of pyridine. Acetic anhydride (5 mL, 53 mmol) was added and the solution was stirred for 30 hours at room temperature. Volatiles were removed under vacuum (0.7 mm Hg, 5 h). Without further purification, pure 4 was obtained as an orange solid (1.1 g, quantitative). 1H-NMR (CDCl3): δ 0.95 (dd, 6H), δ 1.62 (m, 2H), 1.79 (m, 1H), 2.04 (s, 3H), 4.12 (s, 7H) 4.16 (s, 1H), 4.26 (s, 1H), 5.85 (dd, 1H). 13C-NMR (CDCl3): 21.5 (CH), 22.1 (CH3), 23.7 (CH3), 24.9 (CH2), 44.0 (CH3), 66.5 (CH), 68.0 (CH), 68.2 (CH), 68.3 (CH), 68.9 (CH), 70.5 (CH), 88.45 (C). 170.8 (CO). HRMS (ESI+) calcd for C15H19Fe [M − OAc]+ 255.08307, found 255.082.
(S)-[3-(N,N-Dimethylamino)isobutyl]ferrocene 5
To a solution of 4 (1.09 g, 3.45 mmol) in MeOH (10 mL), dimethylamine (10 mL, 40% in water) was added. The reaction was stirred for 30 hour at room temperature. Then, the solution was diluted with ether (100 mL) and water (50 mL). The organic layer was separated, dried over MgSO4 and filtered. The solvent was removed in vacuo. Pure 5 was obtained as dark brown oil (1.03 g, quantitative). 1H-NMR (CDCl3): δ 1.01 (dd, J = 3.9, 6.5 Hz, 6H), 1.70 (m, 2H), 1.91 (m, 1H), 1.97 (s, 6H), 3.46 (dd, J = 3.1, 11.4 Hz, 1H), 4.0–4.1 (m, 9H). 13C-NMR (CDCl3): δ 21.9 (CH), 24.5 (CH3), 24.9 (CH3), 40.6 (CH2), 40.7 (2CH3), 60.6 (CH), 67.1 (CH), 67.3 (CH), 67.6 (CH), 68.8 (5CH), 69.5(CH), 85.4 (C). HRMS (ESI+) calcd for C15H19Fe [M − NMe2]+ 255.08307, found 255.082.(R)-1-Dicyclohexylphosphano-2-[α-(S)-(N,N-dimethylamino)isobutyl]ferrocene 6
sec-Butyllithium (1.4 M in cyclohexane, 0.86 mL, 1.2 mmol) was added to a solution of 5 (0.3 g, 1 mmol) in Et2O (2 mL) at 0 °C. After 2 h, chlorodicyclohexylphosphane (0.5 mL, 2 mmol) was added at 0 °C and the solution was allowed to warm up to rt and stirred overnight. Saturated aqueous Na2CO3 (3 mL) was added and the layers were separated. The aqueous layer was extracted three times with CH2Cl2 (4 mL) and the combined organic layers were dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography (SiO2, pentane/Et2O 10:1) to give compound 6 as an orange solid (0.29 g, 59%). 1H-NMR (CDCl3): δ 1.03 (dd, J = 5.7, 33.3 Hz, 6H), 1.1–2.0 (m, 25H), 2.19 (s, 6H), 3.96 (m, 1H), 4.04 (s, 5H), 4.08 (s, 1H), 4.21 (m, 2H). 13C-NMR (CDCl3): δ 22.6 (s, 2CH3), 24.6 (s, 1CH2), 26.3 (s, 1CH2), 26.8 (d, J = 15.4 Hz, 1CH2), 27.6 (d, J = 8 Hz, 1CH2), 28.0 (s, 1CH2), 28.1 (d, J = 4.6, 1CH2), 28.6 (d, J = 12.6, 1CH2), 29.1 (m, 1CH2), 30.8 (m, 1CH2), 32.6 (d, J = 16.9 Hz, 1CH2), 32.9 (d, J = 18.6 Hz, 1CH2), 36.3 (d, J = 12.7 Hz, 1CH2), 39.4 (s, 1CH2), 40.2 (s, 1CH), 46.3 (s, 1CH), 58.2 (s, 1CH), 67.5 (m, CH), 68.6 (d, J = 3.5 Hz, CH), 70.0 (s, 5CH), 70.6 (m, CH), 77.4 (s, 1C), 79.4 (s, 1C). 31P-NMR (CDCl3): δ −11.88 (s). HRMS (ESI+) calcd for C29H47FeNP [M + H]+ 496.27901, found 496.278.
(R)-1-[(SP)-2-(Dicyclohexylphosphino)ferrocenyl]-ethyl-(di-4-methoxyphenyl)phosphine Cu-L1
A mixture of 1 (150 mg, 0.33 mmol) and bis(4-methoxyphenyl)phosphine (82 mg, 0.33 mmol) in acetic acid (5 mL) was degassed three times and stirred for 5 h at 90 °C under nitrogen atmosphere. The solvent was removed under reduced pressure and the yellow residue was dissolved in CH2Cl2. The organic layer was washed with saturated aqueous Na2CO3, brine and H2O. The organic layer was dried on MgSO4 and the solvent was removed under reduced pressure. The crude product was dissolved in CH2Cl2 (5 mL) and CuBr·SMe2 (68 mg, 0.33 mmol, 1 equiv.) was added. The mixture was stirred at rt for 2 h. The solvent was removed in vacuo and the product was purified by column chromatography (SiO2, pentane/Et2O 5:1), and recrystallization from methanol gave Cu-L1 as an orange solid (85 mg, 32%). 1H-NMR (CDCl3): δ 1.0–2.0 (m, 25H), 3.76 (s, 3H, OMe), 3.80 (s, 3H, OMe), 4.04 (s, 5H, Cp), 4.21 (s, 1H, Cp), 4.29 (m, 1H, Cp), 4.33 (s, 1H, Cp) 6.83 (dd, 4H, J = 8.5, 57.3 Hz), 7.52 (dt, J = 8.8, 97.7 Hz, 4H). 13C-NMR (CDCl3): δ 17.7 (s, CH3), 26.2 (d, J = 29.1 Hz), 26.9 (d, J = 12.1), 27.4 (d, J = 12.3), 28.2, 28.3, 30.5 (d, J = 8.7 Hz), 30.7 (d, J = 5.5 Hz), 31.9 (d, J = 10.0 Hz), 34.5 (d, J = 10.8 Hz), 35.4 (d, J = 6.0 Hz), 35.6 (d, J = 6.3), 39.5 (d, J = 6.6 Hz), 39.6 (d, J = 6.7), 55.4 (d, J = 8.7 Hz), 69.0, 69.6, 70.1, 70.3, 73.6, 74.8 (d, J = 18.2 Hz), 77.4, 93.3 (dd, J = 17.4, 7.1 Hz), 114.4 (dd, J = 37.2, 9.6 Hz), 121.7 (dd, J = 22.3, 6.9 Hz), 124.1 (dd, J = 20.5, 10.5 Hz), 135.7 (dt, J = 13.5, 5.3 Hz), 161.1 (d, J = 30 Hz). 31P-NMR (CDCl3): δ −11.66. [α]D20 = −2.8 (c = 1, CH2Cl2). HRMS (ESI+) calcd for C38H48BrCuFeO2P2 [M]+, 796.09529, found 796.094.
(R)-1-[(SP)-2-(Dicyclohexylphosphino)ferrocenyl]-ethyl-di[3,5-bis-(trifluoromethyl)phenyl]phosphine Cu-L2
Prepared according to the procedure described above from 1 (300 mg, 0.65 mmol), bis(3,5-di(trifluoromethyl)phenyl)phosphine (300 mg, 0.65 mmol) and CuBr·SMe2 (134 mg, 0.65 mmol). Purified on column chromatography (SiO2, pentane/Et2O 5:1) to give compound Cu-L2 as an orange solid (176 mg, 25%). 1H-NMR (CDCl3): δ 1.0–2.0 (m, 25H), 3.86 (q, 1H), 4.12 (s, 1H), 4.18 (s, 5H), 4.23 (s, 1H), 4.30 (s, 1H), 7.35 (s, 1H), 7.85 (s, 1H), 7.89 (s, 2H), 8.28 (s, 2H). 13C-NMR (CDCl3): δ 16.9 (s, CH3), 25.9 (m), 26.6 (m), 27.3 (m), 28.3 (d, J = 15.7 Hz), 29.5 (m), 31.2 (s), 34.5 (s), 35.9 (m), 41.2 (m), 68.8 (s), 69.9 (s), 70.2 (s, 5CH), 71.4 (s, 1CH), 74.6 (s, 1CH), 77.4 (s, 1C), 91.3 (d, 1C), 122.5 (s), 123.0 (dq, J = 273, 30.4 Hz, 4CF3), 124.4 (s), 131.5 (dq, J = 33.9, 7.2 Hz), 132.2 (m), 136.5 (br. s). 31P-NMR (CDCl3): δ −9.53 (br. d, J = 149.9 Hz), −14.31(br. d, J = 155.3 Hz). 19F-NMR (CDCl3): δ −63.1 (d, J = 16.0 Hz). [α]D20 = −40.4 (c = 1, CH2Cl2). HRMS (ESI+) calcd for C40H40F12FeP2Cu [M]+, 929.1054, found 929.104.
(R)-1-[(Sp)-2-(Dicyclohexylphosphino)ferrocenyl]-ethyl-(di-o-tolyl)phosphine Cu-L3
Prepared according to the procedure described above from 1 (218 mg, 0.48 mmol), di(o-tolyl)phosphine (18 mg, 0.53 mmol) and CuBr·SMe2 (100 mg, 0.49 mmol). Purified on column chromatography (SiO2, pentane/Et2O 5:1) to give compound Cu-L3 as an orange solid (275 mg, 75%). 1H-NMR (CDCl3): δ 1–2.6 (m, 31H), 3.65 (q, 1H), 4.21 (s, 5H), 4.29 (s, 1H), 4.34 (s, 2H), 6.9–7.3 (m, 8H). 13C-NMR (CDCl3): δ 15.5 (d, J = 4.5 Hz, CH3), 23.2 (d, J = 15.2 Hz, CH2), 23.4 (d, J = 11.5 Hz, CH2), 26.0 (d, J = 8.2 Hz, CH2), 27.0 (d, J = 12.3 Hz, CH2), 27.3 (t, J = CH), 27.8 (d, J = 13.9 Hz, CH2), 28.2 (s), 29.8 (m), 30.9 (d, J = 8.6 Hz, CH2), 31.7 (d, J = 11.6 Hz, CH2), 34.5 (d, J = 15.5 Hz, CH2), 38.4 (d, J = 11.6 Hz, CH2), 68.8 (d, J = 3.8 Hz, 1CH), 70.1 (s, 5CH), 70.5 (d, J = 7.7 Hz, 1CH), 74.1 (s, 1CH), 74.8 (d, J = 17.9 Hz, 1CH), 77.4 (s, 1CH), 91.5 (d, Cp), 125.2 (d, J = 4.2, Hz, Ar), 125.9 (d, J = 5.0 Hz, Ar), 127.5 (dd, J = 3.9, 20.0 Hz, 1C), 129.6 (s, Ar), 129.8 (s, Ar), 130.3 (dd, J = 7.5, 17.1 Hz, 1C), 131.1 (d, J = 7.7 Hz, Ar), 131.5 (m, Ar), 131.8 (d, J = 6.2 Hz, Ar), 133.5 (s, 1CH), 142.5 (d, J = 16.9 Hz, 1C), 143.6 (d, J = 19.5 Hz, 1C). 31P-NMR (CDCl3): δ −10.2 (d, J = 181.7 Hz), −19.9 (br. d, J = 184.8 Hz). [α]D20 = +17.0 (c = 1, CH2Cl2). HRMS (ESI+) calcd for C38H48BrCuFeP2 [M]+, 764.1054, found 764.101.
(R)-1-[(SP)-2-(Dicyclohexylphosphino)ferrocenyl]-ethyl-[di(3,5-di-tert-butyl-4-methoxyphenyl)phosphine Cu-L4
Prepared according to the procedure described above from 1 (150 mg, 0.33 mmol), bis(3,5-di-tert-butyl-4-methoxyphenyl)chlorophosphine (82 mg, 0.33 mmol) and CuBr·SMe2 (68 mg, 0.33 mmol). Purified on column chromatography (SiO2, pentane/Et2O 5:1) to give compound Cu-L4 as an orange solid (202 mg, 60%). 1H-NMR (CDCl3): δ 1.0–2.0 (m, 25H), 1.29 (s, 18H, tBu), 1.41 (s, 18H, tBu), 3.42 (m, 1H, CH), 3.59 (s, 3H, OMe), 3.66 (s, 3H, OMe), 4.07 (s, 5H, Cp), 4.26 (s, 1H, Cp), 4.31 (m, 2H, Cp), 7.23 (d, 2H, J = 12.5, Ar), 7.57 (d, 2H, J = 19 Hz, Ar). 13C-NMR (CDCl3): δ 16.4, 23.2, 31.7 (d, J = 19.1 Hz), 32.6 (d, J = 11.5 Hz), 32.9, 33.6 (d, J = 14.3 Hz), 34.0, 36.0 (dd, J = 22.1, 6.2 Hz), 37.3 (d, J = 11.2 Hz), 37.9 (d, J = 6.36 Hz), 40.3 (d, J = 10.3 Hz), 41.0 (dd, J = 14.0, 5.5 Hz), 41.7 (d, J = 14 Hz), 44.5 (dd, J = 11.1, 5.6 Hz), 51.9, 70.0 (d, J = 5.3 Hz), 74.3 (d, J = 3.3 Hz), 75.6, 76.1 (t, J = 5.9 Hz), 77.0, 79.1, 80.1, 80.2, 83.0, 99.0, 110.77, 138.2 (dd, J = 60.7, 16.6 Hz), 149.5 (dd, J = 34.2 ,9.2 Hz), 166.6 (d, J = 40 Hz). 31P-NMR (CDCl3): δ −9.53, −14.31. [α]D20 = −40.8 (c = 1, CH2Cl2). HRMS (ESI+) calcd for C54H80BrCuFeO2P2 [M]+, 1020.3457, found 1020.346.
(R)-1-[(SP)-2-(Dicyclohexylphosphino)ferrocenyl]isobutyl-diphenylphosphine Cu-L5
Prepared according to the procedure described above from 6 (220 mg, 0.44 mmol), diphenylphosphine (0.08 mL, 0.44 mmol) and CuBr·SMe2 (90 mg, 1 eq.) . Purified on column chromatography (SiO2, pentane/Et2O 5:1) to give compound Cu-L5 as an orange solid (144 mg, 42%). 1H-NMR (CDCl3): δ 0.62 (d, 3H, CH3), 0.75 (d, 3H, CH3), 1.0–2.0 (m, 25H), 3.75 (s, 5H, Cp), 4.18 (s, 1H, Cp), 4.25 (s, 1H, Cp), 4.34 (s, 1H, Cp), 7.2–7.5 (m, 6H, Ar), 7.69 (bs, 2H, Ar), 7.95 (bs, 2H, Ar). 13C-NMR (CDCl3): δ 21.4, 23.4, 25.5, 26.2 (d, J = 16.6 Hz), 27.2 (d, J = 11.6 Hz), 27.7 (m), 28.4 (d, J = 16.9 Hz), 29.3 (s), 29.9 (s), 30.5, 30.7, 33.3 (d, J = 14.3 Hz), 34.2 (d, J = 12.1 Hz), 36.5 (m), 39.3 (t, J = 9.0 Hz), 43.7 (m), 46.1 (s), 68.7 (s, CH), 70.1 (s, 5CH), 72.1 (s, CH), 72.6 (s, CH), 77.4 (s, C), 93.8 (d, J = 21.5 Hz, C), 125.7 (s, C), 128.5 (m, 2CH), 128.7 (d, J = 9.0 Hz, 2CH), 129.2 (d, J = 8.1 Hz, 2CH), 130.3 (d, J = 45.8 Hz, 2CH), 134.1 (m, 2CH). 31P-NMR (CDCl3): δ −9.53 (d), −14.31 (br). [α]D20 = +59.2 (c = 0.25, CH2Cl2). HRMS (ESI+) calcd for C40H40F12FeP2Cu [M]+, 778.1211, found 778.117.
General procedure for the copper-catalyzed 1,2-addition of Grignard reagents to ketones6
A Schlenk tube equipped with septum and stirring bar was charged with Cu-L (7.5 μmol, 5 mol%). Dry tBuOMe (1.5 mL) was added and the solution was stirred under nitrogen at rt for 15 min. Then, the corresponding ketone (0.15 mmol in 0.5 mL of tBuOMe) was added and the resulting solution was cooled to −78 °C. In a separate Schlenk flask, the corresponding Grignard reagent (0.18 mmol, 1.2 eq., in Et2O) was diluted with tBuOMe (to a combined volume of 1 mL) under nitrogen and added dropwise to the reaction mixture over 3 h using a syringe pump. Once the addition was complete, the reaction mixture was monitored by TLC and GC-MS. The reaction was quenched by the addition of MeOH (1 mL) and saturated aqueous NH4Cl (2 mL), the mixture was warmed to rt and diluted with Et2O, and the layers were separated. The aqueous layer was extracted with Et2O (3 × 5 mL), the combined organic layers were dried with anhydrous Na2SO4 and filtered, and the solvent was evaporated in vacuo. Gas chromatography analysis was carried out to determine the 1,2-addition, 1,4-addition and 1,2-reduction ratio on a sample which was passed through a short plug of silica gel to remove copper residues. The crude product was purified by flash chromatography on silica gel using mixtures of n-pentane and Et2O as the eluent.(Z)-2-Bromo-3,5-dimethyl-1-phenylhex-1-en-3-ol 8a6
Using the general procedure: reaction was performed with iBuMgBr and 7. 1H-NMR (CDCl3): δ 1.0 (dd, J = 6.8 Hz, 6H), 1.60 (s, 3H), 1.66 (dd, J = 6.1, 13.9 Hz, 1H), 1.82 (app. nonet, J = 6.5 Hz, 1H), 1.90 (dd, J = 5.5, 14.1 Hz, 1H), 1.99 (br. s, 1H) 7.25 (s, 1H), 7.31 (m, 1H), 7.38 (t, J = 7.3 Hz, 2H), 7.56 (d, J = 7.9 Hz, 2H). 13C-NMR (CDCl3): δ 24.46 (CH3), 24.54 (CH3), 24.71 (CH), 29.0 (CH3), 49.3 (CH2), 77.8 (C), 126.4 (CH), 127.9 (CH), 128.3(2CH), 129.2(2CH), 134.9 (C), 136.5 (C). Enantiomeric excess was determined by chiral HPLC analysis, Chiralcel AD-H column, n-heptane/i-PrOH 98:2, 40 °C, detection at 240 nm, retention times (min): 17.9 (minor) and 21.3 (major).
(Z)-2-Bromo-3-methyl-1-phenylpent-1-en-3-ol 8b6
Using the general procedure: the reaction was performed with EtMgBr and 7. 1H-NMR (CDCl3): δ 0.92 (t, J = 7.4 Hz, 3H), 1.56 (s, 3H), 1.86 (m, 3H), 7.18 (s, 1H), 7.29 (m, 1H), 7.36 (t, J = 7.5 Hz, 2H), 7.55 (d, J = 7.8 Hz, 2H). 13C-NMR (CDCl3): δ 8.3, 27.3, 33.9, 77.7, 127.0, 127.9, 128.3, 129.3, 134.3, 136.4. Enantiomeric excess was determined by chiral HPLC analysis, Chiralcel AD-H column, n-heptane/i-PrOH 98:2, 40 °C, detection at 240 nm, retention times (min): 25.7 (minor) and 27.5 (major).
(E)-3,5-Dimethyl-1-phenylhex-1-en-3-ol 12 (ref. 23)
Using the general procedure: the reaction was performed with iBuMgBr and 11. 1H-NMR (CDCl3): δ 0.91 (t, J = 6.8 Hz, 6H), 1.37 (s, 3H), 1.56 (m, 2H), 1.78 (app. nonet, J = 6.5 Hz, 1H) 6.24 (d, J = 16.4 Hz, 1H), 6.54 (d, J = 16.2 Hz, 1H), 7.2–7.4 (m, 5H). 13C-NMR (CDCl3): δ 24.4 (CH3), 24.5 (CH3), 24.6 (CH), 29.2 (CH3), 51.5 (CH2), 73.7 (C), 126.3 (2CH), 126.4 (CH), 127.2 (2CH), 128.5 (CH), 137.1 (CH), 137.2 (CH). Enantiomeric excess was determined by chiral HPLC analysis, Chiralcel AD-H column, n-heptane/i-PrOH 98:2, 40 °C, detection at 240 nm, retention times (min): 21.2 (minor) and 22.2 (major).
2-(3,5-Bis(trifluoromethyl)phenyl)butan-2-ol 16 (ref. 8)
Using the general procedure: the reaction was performed with EtMgBr and 15. 1H-NMR (CDCl3): δ 0.80 (t, J = 7.5 Hz, 3H), 1.58 (s, 3H), 1.85 (m, 3H), 7.74 (s, 1H), 7.88 (s, 2H). 13C-NMR (CDCl3): δ 8.2 (CH3), 30.1 (CH3), 36.9 (CH2), 74.9 (C), 120.8 (q, J = 3.9 Hz, 2C), 122.4 (CH), 125.1 (CH), 125.6 (CH), 131.6 (q, J = 33.3 Hz, 2CF3), 150.7 (C). Enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane/i-PrOH 99.5:0.5, 40 °C, detection at 240 nm, retention times (min): 17.8 (minor) and 18.4 (major).
General procedure for copper-catalyzed 1,4-addition to α,β-unsaturated esters21
A Schlenk tube equipped with septum and a stirring bar was charged with Cu-L (5 μmol, 5 mol%). Dry CH2Cl2 (1 mL) was added and the solution was stirred under nitrogen at rt for 5 min. The mixture was cooled to −78 °C and EtMgBr (0.3 mmol, 3.0 M solution in Et2O) was added. After stirring for 5 min, a solution of 18 (19 mg, 0.10 mmol) in CH2Cl2 (0.25 mL) was added dropwise to the reaction mixture over 1 h using a syringe pump. Once the addition was complete, the reaction mixture was monitored by TLC and GC-MS. The reaction was quenched by the addition of MeOH (1 mL) and saturated aqueous NH4Cl (2 mL), the mixture was warmed to rt and diluted with CH2Cl2, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), the combined organic layers were dried with anhydrous MgSO4 and filtered, and the solvent was evaporated in vacuo. Gas chromatography analysis was carried out to determine the 1,2-addition and 1,4-addition ratio on a sample which was passed through a short plug of silica gel to remove copper residues. The crude product was purified by flash chromatography on silica gel using mixtures of n-pentane and Et2O as the eluent.Methyl-3-phenylpentanoate 19 (ref. 24)
Using the general procedure: the reaction was performed with EtMgBr and 18. 1H-NMR (CDCl3): δ 0.74 (t, J = 7.5 Hz, 3H), 1.63 (m, 2H), 2.58 (m, 2H), 2.95 (q, J = 7.1 Hz, 1H), 3.50 (s, 3H), 7.1 (m, 3H), 7.20 (m, 2H). 13C-NMR (CDCl3): δ 12.1 (CH3), 29.3 (CH2), 41.5 (CH2), 44.1 (CH), 51.7 (CH3), 126.6 (CH), 127.7 (CH), 128.6 (CH), 144.1 (C), 173.2 (C). Enantiomeric excess was determined by chiral GC analysis, Chiraldex B-PM column (30 m × 0.25 mm), 90 °C, retention times (min): 37.7 (minor) and 38.6 (major).Acknowledgements
Financial support from the Netherlands Organization for Scientific Research (NWO-Vidi, S.R.H.) is acknowledged.Notes and references
- H.-U. Blaser, B. Pugin, F. Spindler, E. Mejía and A. Togni, in Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley_VCH, Weinheim, Germany, 1st edn, 2011, pp. 93–136 Search PubMed.
- N. C. Zanetti, F. Spindler, J. Spencer, A. Togni and G. Rihs, Organometallics, 1996, 15, 860–866 CrossRef CAS.
- Q. Shen, S. Shekar, J. P. Stambuli and J. F. Hartwig, Angew. Chem., Int. Ed., 2005, 44, 1371–1375 CrossRef CAS PubMed; A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 8704–8704 CrossRef PubMed.
- B. H. Lipshutz and J. M. Servesko, Angew. Chem., Int. Ed., 2003, 42, 4789–4792 CrossRef CAS PubMed; B. H. Lipshutz, J. M. Servesko and B. R. Taft, J. Am. Chem. Soc., 2004, 126, 8352–8353 CrossRef PubMed; B. H. Lipshutz, N. Tanaka, B. R. Taft and C.-T. Lee, Org. Lett., 2006, 8, 1963–1966 CrossRef PubMed.
- S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed; F. Bilčík, M. Drusan, J. Marák and R. Šebesta, J. Org. Chem., 2012, 77, 760–765 CrossRef PubMed.
- A. V. R. Madduri, A. J. Minnaard and S. R. Harutyunyan, Org. Biomol. Chem., 2012, 10, 2878–2884 CAS.
- A. V. R. Madduri, A. J. Minnaard and S. R. Harutyunyan, Chem. Commun., 2012, 48, 1478–1480 RSC.
- A. V. R. Madduri, S. R. Harutyunyan and A. J. Minnaard, Angew. Chem., Int. Ed., 2012, 51, 3164–3167 CrossRef CAS PubMed.
- S. R. Harutyunyan, F. López, W. R. Browne, A. Correa, D. Peña, R. Badorrey, A. Meetsma, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 9103–9118 CrossRef CAS PubMed.
- G. Gokel, P. Hoffmann, H. Klusacek, D. Marquarding and I. Ugi, Angew. Chem., 1970, 82, 77–78 CrossRef.
- S. Yasuike, C. C. Kofink, R. J. Kloetzing, N. Gommermann, K. Tappe, A. Gavryushin and P. Knochel, Tetrahedron: Asymmetry, 2005, 16, 3385–3393 CrossRef CAS PubMed.
- U. Burckhardt, L. Hintermann, A. Schnyder and A. Togni, Organometallics, 1995, 14, 5415–5425 CrossRef CAS.
- M. Woltersdorf, R. Kranich and H.-G. Schmalz, Tetrahedron, 1997, 53, 7219–7230 CrossRef CAS.
- T. Hayashi, K. Yamamoto and M. Kumada, Tetrahedron Lett., 1974, 49–50, 4405–4408 CrossRef; T. Hayashi, T. Mise, S. Mitachi, K. Yamamoto and M. Kumada, Tetrahedron Lett., 1976, 17, 1133 CrossRef.
- G. W. Gokel, D. Marquarding and I. K. Ugi, J. Org. Chem., 1972, 37, 3052–3058 CrossRef CAS.
- F. Caprioli, A. V. R. Madduri, A. J. Minnaard and S. R. Harutyunyan, Chem. Commun., 2013, 49, 5450–5452 RSC.
- The ligand synthesis has been reported in: E. Mejía, R. Aardoom and A. Togni, Eur. J. Inorg. Chem., 2012, 31, 5021–5032 CrossRef.
- Ligand was used in: D. Hook, B. Riss, D. Kaufmann, M. Napp, E. Bappert, P. Polleux, J. Medlock and A. Zanotti-Gerosa, PCT Int. Appl., WO 2009090251, 2009 Search PubMed.
- The rev-Josiphos ligand in which cyclohexyl groups are substituted with tert-butyl groups is commercially available. However our preliminary screening of the copper complex of this ligand in 1,2-addition reactions to enones and aromatic ketones resulted in a drastic loss of enantioselectivity.
- Z. Wu, A. V. R. Madduri, S. R. Harutyunyan and A. J. Minnaard, Eur. J. Org. Chem., 2014, 575–582 CrossRef CAS.
- F. López, S. R. Harutyunyan, A. Meetsma, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 2752–2756 CrossRef PubMed.
- K.-M. Kim and I.-H. Park, Synthesis, 2004, 2641–2644 CrossRef CAS PubMed.
- J. J. McDonnell and D. J. Pochopien, J. Org. Chem., 1971, 36, 2092–2098 CrossRef CAS.
- B. D. West and K. P. Link, J. Heterocycl. Chem., 1965, 93–94 CrossRef CAS.
- S. H. Bertz, R. A. Hardin, T. J. Heavey and C. A. Ogle, Angew. Chem., Int. Ed., 2013, 39, 10440–10442 CrossRef CAS; S. H. Bertz, R. A. Hardin and C. A. Ogle, J. Am. Chem. Soc., 2013, 135, 9656–9658 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00180j |
| This journal is © The Royal Society of Chemistry 2014 |