 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper based TiO2 honeycomb monoliths for CO2 photoreduction
Oluwafunmilola
Ola
* and
M.
Mercedes Maroto-Valer
Centre for Innovation in Carbon Capture and Storage (CICCS), School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: O.O.Ola@hw.ac.uk; Fax: +44 (0)131 451 3180; Tel: +44 (0)131 451 4737
First published on 21st February 2014
Abstract
The direct photoreduction of CO2via catalytic conversion of copper supported on TiO2 based monolithic structures is a means by which solar fuels can be produced. Copper based monolithic structures with varying loadings were synthesized through a sol–gel dip coating procedure and tested for CO2 reduction with H2O as a reductant in the gaseous phase. Results established that increased copper concentration can decrease crystalline size and promote anatase to rutile phase transformation. The coated monolithic structures were dominated by mainly Cu1+ species, as confirmed by XPS while bulk characterization suggests that these species are present in the crystal lattice via substitution of Ti4+ ions with Cu1+ ions. The catalytic performance of the Cu doped TiO2 monoliths for hydrocarbon formation was found to be considerably higher when compared to pure TiO2 under UVA or visible light irradiation.
1. Introduction
The utilization of carbon dioxide (CO2) for photocatalytic reduction driven by solar energy is a promising strategy for producing sustainable fuels that are suitable for use in existing energy infrastructure. Although the feasibility of using titanium dioxide (TiO2) based materials for UV induced photocatalysis has been demonstrated,1,2 its visible light applications are limited.3 The tailoring of the properties of titanium dioxide (TiO2) by the addition of metals that are relatively inexpensive and readily available for CO2 photoreduction systems is highly desirable. In this regard, the use of copper species has been increasingly investigated.4Several researchers have reported that Cu2+ species are the active sites on Cu based TiO2 catalysts for the degradation of rhodamine B,5 photocatalytic water splitting6 and CO2 reduction.7,8 For CO2 reduction studies, it is generally accepted that CuO can trap photoexcited electrons from the conduction band of TiO2 and these trapped elections can participate in reduction reactions with the surface adsorbed species thus preventing electron–hole recombination.1 Furthermore, previous studies have established that the addition of copper can improve visible light absorption and efficiency of TiO2; however, little is known about the effect of these materials on supports i.e. monoliths for the photocatalytic reduction of CO2.
Many researchers have focused on ways of anchoring photocatalysts onto supports since high photoconversion efficiencies and improved light harvesting can only be achieved through the combined use of an optimized photoreactor and photocatalyst configurations. Nishimura et al. dip coated TiO2 on a silica–alumina gas separation membrane to obtain 3.5 ppmV h−1 of CO after 336 hours,9 while Pathak et al. used the hydrophilic structural cavities in Nafion-117 membrane films to host TiO2 coated with nanoscale silver and obtained methanol as the major product and formic acid as the minor product.10 Their results were reproducible even when these films were reused. Cybula et al. employed a flat perforated steel or plastic tray as a support for the dispersion of TiO2 in a tubular reactor designed for CO2 photoreduction studies.11 They observed that the type of support used not only played a critical role in determining the amount of immobilized catalyst, but also influenced the photoconversion rate when the same coating procedure was used. A decrease in catalyst loading and methane production (from 90 ppm to 34 ppm) was observed when the support was switched from steel to plastic due to weaker adhesive properties of plastic compared to those of steel.
The interconnected three-dimensional structures like the honeycomb monolith containing parallel straight channels have been exploited for industrial processes due to its potentially high surface to volume ratio, ease of scale-up through an increase of its dimensions and channels, and control of structural parameters (i.e. pore volume, pore size and surface area) etc.12,13 Photocatalytic studies conducted using a monolith as a support have identified low light utilization efficiency, due to little or no light absorption in the pores or channels of the honeycomb monolith.14 Not all immobilised photocatalysts may be activated due to limited light distribution arising from the catalyst coated on the outer surface absorbing most of the light.15 The light intensity also decays along the opaque channels of the monolith.16 More recently, it has been reported that the drawbacks of limited light penetration and efficiency of CO2 reduction can be improved by threading channels of monolithic structures with optical fibres.2,17,18 Comparison of the slurry reactor system with the monolith system demonstrated that higher conversion and quantum efficiency can be achieved when the monolith was employed as a catalyst carrier.18 This was attributed to the combined advantages of the higher geometrical internal surface area of the monolith and the elimination of uneven light distribution via the optical fibres. Accordingly, experimental analyses using copper based nanomaterials immobilized onto monolithic structures threaded with optical fibres for CO2 reduction were conducted. Detailed characterization techniques were employed in order to investigate the effect of copper doping on the physicochemical properties of TiO2 based monoliths and correlate these properties to CO2 photoconversion.
2. Experimental
2.1 Preparation of copper based TiO2 monoliths
A series of Cu doped TiO2 monoliths within the range of 0.2–2 wt% were prepared by the sol–gel method (Fig. 1). The monoliths were pre-coated with the SiO2 sol prior to dip-coating in the Cu–TiO2 sol. As shown in Fig. 1, the Cu–TiO2 sol was synthesized by adding a mixture of titanium(IV) butoxide and n-butanol to calculated amounts of copper(II) chloride dihydrate (CuCl2·2H2O) dissolved in 14 ml of acetic acid. Subsequently, polyethylene glycol (PEG) solution was added to the metal loaded sol and stirred for 6 hours. The pre-coated SiO2 monoliths were then dip-coated in the resulting Cu–TiO2 sol for 30 minutes. The Cu–TiO2 coated monoliths and the remaining sol were dried and calcined in a furnace at 150 °C and 500 °C, respectively. This procedure is detailed in previous work.18 | ||
| Fig. 1 Sol–gel procedure for Cu–TiO2 monoliths. | ||
2.2 Photocatalyst characterization
Detailed information about the crystallographic structure of the sample showing the integrated intensity, peak positions, planes and unit cell parameters was obtained using a Hiltonbrooks X-ray powder diffractometer with a Philips PW 1050 goniometer and a proportional detector. Nickel filtered Cu Kα radiation was used, operating at 20 mA and 40 kV with a scan range of 5–65 (2θ), a scan speed of 2 degrees (2θ) per minute and a step size of 0.05. The morphology of the nanoparticles was studied by transmission electron microscopy (TEM) using a JEOL 2100F instrument at an acceleration voltage of 200 kV. A Quanta 600 model equipped with an energy dispersive X-ray (EDX) system was used to perform quantitative analysis and observe the morphology of the catalysts at the voltages of 25 kV and 30 kV, respectively. Specific surface area measurements were estimated from N2 adsorption–desorption isotherms at 77 K that were measured using a ChemBET TPR/TPD analyzer connected to a linear mass flow controller/gas blender. The porosity and pore size distributions of the monoliths were characterized using a mercury (Hg) porosimetry analyzer (Micromeritics Autopore IV 9520 V1.05) with Hg pressure in the range of 0.7–275![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 790 kPa.
790 kPa.
The elemental ratios of the metals contained within the nanoparticles were quantified using the Varian Vista MPX ICP-OES (inductively coupled plasma optical emission spectroscopy) system that used an echelle polychromator with a mega-pixel CCD detector. Prior to sample injection, approximately 25 mg of Cu–TiO2 samples were digested in a mixture of 5 ml of H2SO4 and 0.5 ml of HClO4. The solution was then made up to 100 ml in deionised water. X-ray photoelectron spectroscopy (XPS) was performed on the nanoparticles using a Kratos AXIS ULTRA instrument with a mono-chromated Al Kα X-ray source (1486.69 eV) operated at a 15 mA emission current and a 12 kV anode potential (180 W). High resolution scans were taken for 5 or 10 minutes each over the appropriate regions for the photoelectron peaks with a step of 0.1 eV and a pass energy of 20 eV. Wide/survey scans over the full energy range B.E. of 1400–5 eV were performed on each sample at a pass energy of 80 eV. The wide scans were used to estimate quantification of each element present based on the peak areas using the CASAXPS software with Kratos sensitivity factors. The high resolution scans were charge corrected to the main C 1s peak = 285 eV and used to determine the chemical states of the elements detected. Spectral fitting was performed using the CasaXPS software with a line shape based on a Gaussian/Lorentzian mix of 70:30 (GL30). The band gap, threshold wavelength and the absorbance of ultraviolet light as a function of the transmittance were measured using the diffusive reflective ultraviolet-visible spectrophotometer (Varian Cary 300). The band gap energy of the samples were calculated using Eg = hc/λ where h, c and λ represent the Planck's constant, velocity of light and wavelength, respectively.
2.3 Photoreduction of CO2
The photocatalytic reduction of CO2 under UVA or visible light was conducted in a cylindrical Pyrex glass reactor with a volume of 216 cm3. The catalyst coated ceramic honeycomb monoliths with 177 channels were threaded with optical fibres to ensure light distribution within the internal channels of the monolith. The humidifier was connected before the gas inlet, while the temperature and pressure were monitored via a type T thermocouple and a pressure gauge, respectively, connected by 1/8′′ fittings after the product outlet. Light was irradiated into the side of the reactor by a light guide, with the illumination system being either a 200 W mercury lamp or a 500 W halogen lamp, with light intensities of 33.42 and 68.35 mW cm−2 respectively.After performing a leak test with helium (He) gas, ultra pure CO2 (Air Products, 99.9995%) gas saturated with water vapour was bubbled into the reactor for 1 hour at a flow rate of 4 ml min−1. Subsequently, the light source was turned on and readings were taken after 4 hours. The flow of CO2 saturated with water vapour was continuous throughout the reaction. The H2O content in the feed was 50 ml and the pressure was maintained at 1 bar for every experimental run. Products extracted from the outlet of the gas-phase photoreactor were analyzed using a mass spectrometer (MS, Hiden Analytical) equipped with a capillary, quadrupole mass analyser (HAL 201-RC) and Faraday/Secondary electron multiplier (SEM) detectors. Prior to every photocatalytic experiment, blank reactions were performed to confirm product formation was due to CO2 photoreduction.
3. Results and discussion
3.1 Textural properties of supported Cu catalysts
As shown in Fig. 2, the XRD diffraction patterns of TiO2 monoliths doped with different concentrations of copper consist mainly of two diffraction phases of anatase (A) and rutile (R). The rutile phase was detected in these samples at peak positions of 27.4° and 36.1° after calcination at 500 °C. The crystallite size of all doped Cu–TiO2 based monoliths calculated from the Scherrer equation was within the range of 16.37–19.12 nm (Table 1). As the crystallite size of anatase decreased, an increase in rutile content was observed with increased metal concentration, with the 2 wt% Cu–TiO2 sample showing the maximum growth of rutile nuclei. This is due to the ability of Cu in enhancing the particle sintering process i.e. accelerating densification and grain growth, and thus promoting mineral phase transformation.19 | ||
| Fig. 2 XRD pattern of TiO2 monoliths with different Cu loadings (1: TiO2, 2: 0.2 wt% Cu–TiO2; 3: 0.5 wt% Cu–TiO2, 4: 1 wt% Cu–TiO2, 5: 1.5 wt% Cu–TiO2, 6: 2 wt% Cu–TiO2, anatase (A) and rutile (R)). | ||
| Photocatalysts | Crystallite size (nm)/phase content (%) | Lattice parametersa | S BET (m2 g−1) | ICP-OES | Band gap (eV) | ||
|---|---|---|---|---|---|---|---|
| Anatase | Rutile | a (Å) | c (Å) | Cu (wt%) | |||
| a Estimated using the Scherrer equation on the (101) diffraction peak of anatase TiO2. b BET surface area. | |||||||
| TiO2 | 12.99 (96.50) | 4.91 (3.50) | 3.7892 | 9.4803 | 52.50 | 0.00 | 3.08 |
| 0.2 wt% Cu–TiO2 | 23.12 (96.70) | 4.79 (3.30) | 3.8039 | 9.4899 | 34.77 | 0.25 | 3.02 |
| 0.5 wt% Cu–TiO2 | 19.12 (96.00) | 5.97 (4.00) | 3.8166 | 9.4943 | 37.52 | 0.53 | 2.96 |
| 1.0 wt% Cu–TiO2 | 18.69 (94.60) | 8.51 (5.40) | 3.8186 | 9.4981 | 48.22 | 0.99 | 2.82 |
| 1.5 wt% Cu–TiO2 | 18.29 (90.80) | 8.77 (9.20) | 3.8206 | 9.507 | 71.34 | 1.60 | 2.74 |
| 2.0 wt% Cu–TiO2 | 16.37 (89.10) | 25.51 (10.90) | 3.8295 | 9.5242 | 88.96 | 2.03 | 2.61 |
These results suggest that the addition of Cu causes the gradual transformation of anatase to rutile with increasing metal concentration. The lattice constants (a & c) of Cu–TiO2 monoliths calculated based on the anatase (101) diffraction peaks, as listed in Table 1, increase with a higher doping amount, when compared to the lattice parameters of TiO2 (a = 3.7892 Å, c = 9.4803 Å). Lattice parameter measurements were repeated thrice for verifying reproducibility. The standard error of the lattice parameter measurement via XRD is within the range of ±0.05–0.28%. The lattice parameter of these Cu doped TiO2 monoliths increases as the crystallite size of anatase decreases.
The high resolution (HR) TEM images of 1 wt% Cu–TiO2 using different magnifications illustrated in Fig. 3a show aggregates of spherical nanocrystals with varying sizes from 5–27 nm. The SEM-EDS (energy dispersive spectroscopy) micrograph of the 1 wt% Cu–TiO2 monolith presented in Fig. 3b confirms the presence of Cu, with the morphological features of the samples remaining unchanged by doping. The thickness of the 1 wt% Cu–TiO2 film measured by SEM was up to 0.32 μm on the surface of the monolith. The pore size distribution of the 1 wt% Cu–TiO2 monolith measured by mercury porosimetry is illustrated in Fig. 4. The porosity and total intrusion volume for the 1 wt% Cu–TiO2 sample were 35.04% and 0.17 mL g−1 while the pore size distribution was within the macropore range with the average pore diameter being 250 Å.
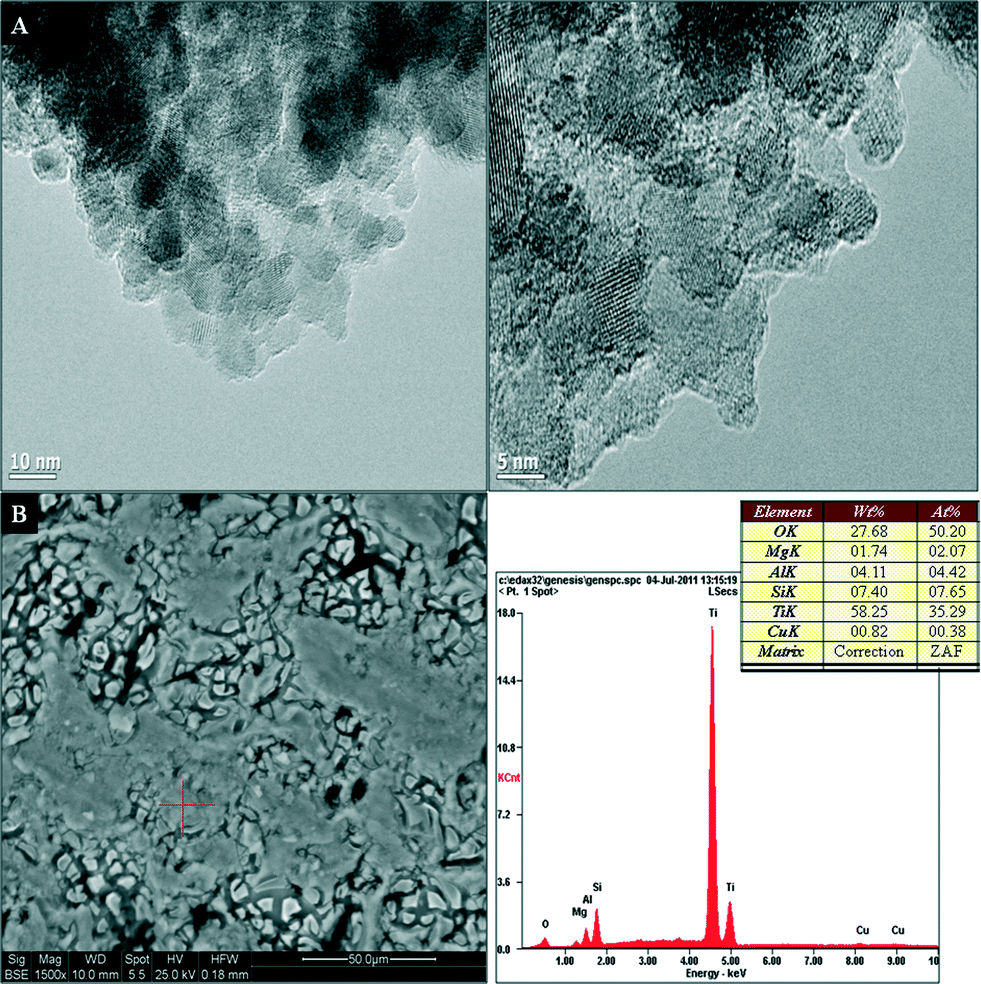 | ||
| Fig. 3 TEM (A) and SEM-EDS (B) micrographs of 1 wt% Cu–TiO2. | ||
 | ||
| Fig. 4 Pore size distribution measured by mercury porosimetry of the 1 wt% Cu–TiO2 monolith showing the cumulative intrusion (A) and differential intrusion volume (B). | ||
The BET specific surface area of the Cu–TiO2 based monoliths was within the range of 34.77–88.96 m2 g−1 (Table 1), and the standard error of these measurements is within the range of +0.02–0.5%. An increase in specific surface area of TiO2 occurs with an increase in the Cu loading.
3.2 ICP-OES and XPS analyses
Table 1 lists the quantitative analysis results calculated from ICP-OES. The ICP-OES analysis of Cu–TiO2 based monoliths demonstrated that Cu was present in the TiO2 matrix. The bulk elemental ratios of the samples are in agreement with the elemental concentration present in the precursor and show an increasing trend with increased metal concentration. This suggests that the added metals were primarily located in the crystal lattice via substitution of the Ti4+ ions with Cu2+ ions.The high resolution XPS spectra of Cu 2p of Cu–TiO2 monoliths are presented in Fig. 5. The Cu 2p3/2 and 2p1/2 peaks formed doublets by peak fitting suggesting that the chemical state is mainly Cu1+ with small amounts of Cu2+.20,21 It has been reported that Cu may be reduced under the X-ray beam during XPS analysis.22 In the work presented here, three sets of scans were collected for each sample on three different areas. The experiment times were about 1 hour per area analysed with a monochromated source which has lower X-ray flux at the sample than at a conventional ‘flood’ source. Nevertheless, it is possible that the Cu oxidation state started out before analysis proceeded, but analysis of auger peaks of Cu was not possible as scans were not recorded over sufficiently long exposure time to confirm reduction.
 | ||
| Fig. 5 XPS spectra of Cu 2p of Cu–TiO2 monoliths A) 0.5 wt% Cu–TiO2, B) 1 wt% Cu–TiO2 C) 1.5 wt% Cu–TiO2 D) 2 wt% Cu–TiO2. | ||
The intensity of the characteristic satellite peak for Cu2+ observed at 942.3 eV increased with the increasing Cu concentration (Fig. 5).4 Colon et al. reported that the key difference between Cu1+ and Cu2+ species was the prominent satellite peak present on the high binding energy sides.23 These satellite peaks which have been reported to be responsible for the shakeup transitions by ligand to metal 3d charge transfer cannot be found in metallic Cu and Cu1+ species, due to their completely filled 3d shells.23 The satellite peaks were observed at 941.7 eV and 942.5 eV for 1 wt% Cu/N–TiO2 and 1 wt% Cu–TiO2 samples calcined at 600 °C. Liu et al. also observed satellite peaks at 942.2 eV and 942.4 eV for 1 wt% and 5 wt% Cu–TiO2 samples prepared by simple precipitation, respectively.4 The XPS spectra of binding energies for Ti 2p were observed at 458.8 eV and 464.6 eV which correspond to Ti4+ in TiO2.24,25 These results are in agreement with the literature, where Ti4+ peaks were observed at 457.7 eV and 463.4 eV for the 1 wt% Cu–10 wt% I–TiO2 sample.24
The XPS spectra of the O 1s region suggest that oxygen exists in three forms on the sample surface with the binding energies of 529.5, 530.1 and 531.7 eV. The main peak appears at 529.9 eV and can be assigned to the bulk oxygen bound on TiO2. This value is consistent with the value of 530.1 eV reported in the literature for anatase TiO2.21 The peak at 529.5 eV probably corresponds to the O 1s peak of CuO26 while the other peak at 531.7 eV can be attributed to surface adsorbed components of the hydroxyl (OH−) group.25,27
3.3 Diffuse reflectance UV-Vis spectra of the Cu–TiO2 monoliths
The UV-Vis diffuse reflectance spectra of pure TiO2 and Cu-monoliths at various loading ratios are shown in Fig. 6. The absorption spectra of the resulting Cu-based TiO2 photocatalysts showed an increased shift in the visible light with increased Cu loading concentration in comparison with pure TiO2. The band gap energies of these catalysts were within the range of 2.61–3.02 eV. The lowest band gap energy was observed with the 2 wt% Cu–TiO2 sample which is consistent with the literature, where increasing metal loadings results in a shift in the absorption edges of the TiO2 based samples.4,5,7,23 The defects created in the TiO2 network and crystalline structure is responsible for change in band gap energy.7 The absorption edge between 400–600 nm can be attributed to the presence of surface defects created during annealing along with the crystallization of the rutile phase.28 Sahu and Biswas29 also observed increased absorption with the increasing Cu2+ concentration. The change in light absorption was attributed to the incorporation of Cu1+ ions into TiO2 crystal lattice via the substitution of Ti4+ by Cu2+ atoms. The increased Cu2+ concentration was also reported to increase oxygen vacancies due to the charge compensation effect. | ||
| Fig. 6 UV-Vis diffuse reflectance spectra of Cu–TiO2 monoliths. | ||
3.4 Photocatalytic reduction of CO2
The photocatalytic activities of the Cu-based TiO2 monoliths threaded with optical fibres were evaluated for CO2 photoreduction under UVA and visible light irradiation (Fig. 6). As shown in Fig. 7, several products such as hydrogen, methanol, acetaldehyde and ethanol were formed after 4 hours of light irradiation. The CO2 reduction experiments were repeated thrice, with the production rates averaged and the standard deviations reported in Fig. 7. The product rates steadily increase with an increase in the metal concentration to give the optimal ratio of 0.5 wt% Cu–TiO2 for the internally illuminated monolith photoreactor systems under either UVA or visible light irradiation, after which reduced product rates were observed for the subsequent higher doping ratios. Hydrogen and methanol were favourably produced; with maximum product rates of 12.55 μmol gcat−1 h−1 and 3.92 μmol gcat−1 h−1, respectively under UVA (Fig. 7(I)) and 3.73 μmol gcat−1 h−1 and 0.23 μmol gcat−1 h−1, respectively under visible light irradiation (Fig. 7(II)). The higher hydrocarbon evolution rate observed when the monolith was used as a catalyst carrier was due to the improved light distribution in the internally illuminated monolith photoreactor system. | ||
| Fig. 7 Effect of Cu doping on the product rate using the monolith as a catalyst carrier under UVA (I) and visible (II) irradiation A) TiO2, B) 0.2 wt% Cu–TiO2, C) 0.5 wt% Cu–TiO2, D) 1 wt% Cu–TiO2 E) 1.5 wt% Cu–TiO2 F) 2 wt% Cu–TiO2. | ||
3.5 Correlation between catalyst characterization and yields of photoconversion
The improved photoreduction activity demonstrated by the 0.5 wt% Cu–TiO2 coated monolith in UV and visible light regions compared to pure TiO2 can be attributed to the incorporation of Cu1+ ions into the TiO2 matrix and the good bi-crystallized TiO2 structure (i.e. crystallite phase of anatase with a small percentage of rutile). Phase transformation can be facilitated by substitutional dopants when cations enter the anatase lattice and cause an increase in the level of oxygen vacancies through valence or reduction/oxidation effects.30Since the ionic radius of Cu1+ is similar to Ti4+, results from XRD confirm that the probability of substitutional doping occuring is high i.e. these metal ions occupying the lattice points of Ti. The decrease in the crystalline size with increased Cu loading and lattice expansion observed in the diffraction patterns of the Cu doped samples explain the peak broadening observed which is associated with substitutional doping.
Nair et al. reported that cations with oxidation states of 3+ or lower tend to increase the oxygen vacancies in the lattice of TiO2 if placed within the lattice points.19 This increased concentration causes the subsequent rearrangement of atoms and reorganization of the structure for the rutile phase in the lattice of TiO2 through the substitution of Ti4+ with cations.5,30 Based on this, an increase in the concentration of oxygen vacancies will occur, which will enhance the nucleation process (i.e. anatase to rutile transformation) as also observed in this study.19
The phase transformation of Cu based TiO2 samples with increased metal concentration observed in this study were probably enhanced due to increased concentration of oxygen vacancies which simultaneously increased atomic mobility. Sahu and Biswas29 reported that the addition of metal dopants can alter the crystal phase of TiO2, with the degree of mineral phase transition being dependent on the metal type and the concentration. This same phenomenon was observed by Nair et al.,19 where increased enhancement was observed over CuO doped TiO2 samples compared to NiO doped TiO2. Colon et al.23 also observed lower anatase content with increased Cu concentration due to the higher amounts of dopants favouring the rutilization process. The influence of these substitutional ions is further confirmed by the change in light absorption properties and the electronic structure of the metal loaded TiO2 samples observed in the UV-Vis spectra when compared to pure TiO2. According to Li et al.,31 electronic states introduced by substitutional metal ions on the bottom of the conduction band edge of TiO2 cause the formation of a new higher unoccupied molecular orbital. This molecular orbital narrows the band gap; as also found in this study (Fig. 6) and thus influences photon absorption.
The synergistic effect between the two crystalline phases in the Cu based samples could also be another plausible reason for improved activity. Improved charge separation and high reactivity at the anatase to rutile interface occur during electron transfer from rutile to anatase at this interface where defect sites with unique charge trapping and adsorption properties can be created.32,33 Bouras et al.33 and Zhang et al.34 reported that electron hole recombination can be retarded through the creation of energy wells and surface anatase/rutile phase junctions which serve as electron traps formed from the lower band gap of rutile thus facilitating charge separation and increasing the lifetime of photogenerated electrons and holes. The presence of mixed crystalline phases of titania (i.e. anatase and rutile) has also been reported to show improved photocatalytic activity due to the synergistic effect derived from better charge separation and high surface area.35
After the optimal doping ratio of Cu1+ was exceeded within the series of synthesized catalysts (0.5 wt%), reduced photoactivity was observed. This result could be due to the coverage of the surface of TiO2 with increased metal ions which inhibited interfacial charge transfer due to the insufficient amount of light energy available for activation of all of the catalyst particles. These results are in agreement with Li et al.36 where the copper dopant below or above the optimum value of 0.5 wt% resulted in reduced production rates. The decrease in production rates at lower doping ratios below the optimum value was attributed to low Cu concentration while reduced catalytic activity at higher loadings was attributed to excess Cu species acting as recombination centres for photogenerated electrons and holes. When the doping content of Cu2+ exceeded 5 wt%, Tian et al.37 recorded a decrease in photocatalytic activity due to electron hole recombination. According to Schiavello,38 photoreactivity can be negatively influenced by either a high concentration of metallic islands on the semiconductor surface or an enhancement of their size. When this occurs, reduced surface illumination of catalysts and increased recombination rate are observed.
4. Conclusions
The photocatalytic reduction of CO2 over Cu–TiO2 coated monolithic structures threaded with optical fibres was conducted under UV and visible light irradiation. The copper species present in the substitutional sites of the TiO2 matrix were found to modify the crystalline and optical properties of TiO2. Cu1+ was identified as the primary Cu species which facilitated multi electron reactions and thus improved the efficiency of CO2 photoreduction. The increase in the Cu1+ concentration facilitated the anatase to rutile transformation due to the substitution of Cu1+ by Ti4+ in the TiO2 structure. Upon UV and visible light irradiation, the Cu doped photocatalysts exhibited improved activity compared to pure TiO2 at optimal doping ratios. The decline in the production rate observed upon increased Cu1+ concentration was probably due to the coverage of the surface of TiO2 with excess metal particles. This inhibited interfacial charge transfer was due to the insufficient amount of light energy available for activation of the catalyst particles. More importantly, the improved conversion efficiency was probably due to improved charge separation at the anatase to rutile interface and the presence of Cu1+ species serving as electron traps which suppressed electron–hole recombination.Acknowledgements
We acknowledge the financial support from the School of Engineering and Physical Sciences and the CICCS (EPSRC grant EP/F012098/2) at Heriot-Watt University. M. Maroto-Valer is grateful for the support from the Leverhulme Trust (Philip Leverhulme Prize). We thank Emily Smith, University of Nottingham, for XPS analysis.Notes and references
- D. Liu, Y. Fernandez, O. Ola, S. Mackintosh, M. Maroto-Valer, C. Parlett, A. Lee and J. Wu, Catal. Commun., 2012, 25, 78–82 CrossRef CAS
.
- O. Ola and M. Maroto-Valer, J. Catal., 2014, 309, 300–308 CrossRef CAS
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS
- L. Liu, F. Gao, H. Zhao and Y. Li, Appl. Catal., B, 2013, 134–135, 349–358 CrossRef CAS
- B. Xin, P. Wang, D. Ding, J. Liu, Z. Ren and H. Fu, Appl. Surf. Sci., 2008, 254, 2569–2574 CrossRef CAS
- J. Li, J. Zeng, L. Jia and W. Fang, Int. J. Hydrogen Energy, 2010, 35, 12733–12740 CrossRef CAS
- H. Slamet, E. Purnama, K. Riyan and J. Gunlazuardi, Catal. Commun., 2005, 6, 313–319 CrossRef CAS
- H. Slamet, E. Purnama, K. Riyan and J. Gunlazuardi, World Appl. Sci. J., 2009, 6, 112–122 CAS
- A. Nishimura, N. Komatsu, G. Mitsui, M. Hirota and E. Hu, Catal. Today, 2009, 148, 341–349 CrossRef CAS
- P. Pathak, M. Meziani, L. Castillo and Y. Sun, Green Chem., 2005, 7, 667–670 RSC
- A. Cybula, M. Klein, A. Zielińska-Jurek, M. Janczarek and A. Zaleska, Physicochem. Probl. Miner. Process., 2012, 48, 159–167 CAS
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS
- H. Lin and K. Valsaraj, J. Appl. Electrochem., 2005, 35, 699–708 CrossRef CAS
- Y. Yu, Y. Pan, Y. Wu, J. Lasek and J. Wu, Catal. Today, 2011, 174, 141–147 CrossRef CAS
- M. Dijkstra, H. Buwalda, A. W. F. de Jong, A. Michorius, J. G. M. Winkelman and A. A. C. Beenackers, Chem. Eng. Sci., 2001, 56, 547–555 CrossRef CAS
- M. Singh, I. Salvadó-Estivill and G. Puma, AIChE J., 2007, 53, 678–686 CrossRef CAS
- P. Liou, S. Chen, J. C. Wu, D. Liu, S. Mackintosh, M. Maroto-Valer and R. Linforth, Energy Environ. Sci., 2011, 4, 1487–1494 CAS
- O. Ola, M. Maroto-Valer, D. Liu, S. Mackintosh, C. Lee and J. Wu, Appl. Catal., B, 2012, 126, 172–179 CrossRef CAS
- J. Nair, P. Nair, F. Mizukami, Y. Oosawa and T. Okubo, Mater. Res. Bull., 1999, 34, 1275–1290 CrossRef CAS
-
C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble, NIST X-ray Photoelectron Spectroscopy Database, 20 Version 3.5, 2007 Search PubMed
- M. Biesinger, L. Lau, A. Gerson and R. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS
- C. Chusuei, M. Brookshier and D. Goodman, Langmuir, 1999, 15, 2806–2808 CrossRef CAS
- G. Colon, M. Maicu, M. Hidalgo and J. Navio, Appl. Catal., B, 2006, 67, 41–51 CrossRef CAS
- Q. Zhang, T. Gao, J. Andino and Y. Li, Appl. Catal., B, 2012, 13, 257–264 CrossRef
- Q. Zhang, Y. Li, E. Ackerman, M. Gajdardziska-Josifovska and H. Li, Appl. Catal., A, 2011, 400, 195–202 CrossRef CAS
- E. Z. Kurmaev, V. R. Galakhov, V. V. Fedorenko, L. V. Elokhina, S. Bartkowski, M. Neumann, C. Greaves, P. Edwards, M. Al-Mamouri and D. L. Novikov, Phys. Rev. B, 1995, 52, 2390–2394 CrossRef CAS
- Y. Wang, B. Li, C. Zhang, L. Cui, S. Kang, X. Li and L. Zhou, Appl. Catal., B, 2013, 130–131, 277–284 CrossRef CAS
- A. Heciak, A. Morawski, B. Grzmil and S. Mozia, Appl. Catal., B, 2013, 140–141, 108–114 CrossRef CAS
- M. Sahu and P. Biswas, Nanoscale Res. Lett., 2011, 6, 441–454 CrossRef PubMed
- D. Hanaor and C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS
- W. Li, A. Frenkel, J. Woicik, C. Ni and S. Shah, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 155315–155321 CrossRef
- J. Carneiro, T. Savenije, J. Moulijn and G. Mul, J. Phys. Chem. C, 2011, 115, 2211–2217 CAS
- P. Bouras, E. Stathatos and P. Lianos, Appl. Catal., B, 2007, 73, 51–59 CrossRef CAS
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed
- K. Schulte, P. DeSario and K. Gray, Appl. Catal., B, 2010, 97, 354–360 CrossRef CAS
- Y. Li, W. Wang, Z. Zhan, M. Woo, C. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS
-
C. Tian, Y. Zhao, J. Zhang and C. Zheng, in Cleaner Combustion and Sustainable World, Springer-Verlag, Berlin, 2013 Search PubMed
-
M. Schiavello, Heterogeneous Photocatalysis, John Wiley and Sons, Chicester, 1997 Search PubMed
| This journal is © The Royal Society of Chemistry 2014 |