 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A single centre aluminium(III) catalyst and TBAB as an ionic organo-catalyst for the homogeneous catalytic synthesis of styrene carbonate†
Somsak
Supasitmongkol
and
Peter
Styring
*
UK Centre for Carbon Dioxide Utilization, Department of Chemical & Biological Engineering, The University of Sheffield, Sir Robert Hadfield Building, Sheffield S1 3JD, UK. E-mail: p.styring@sheffield.ac.uk
First published on 3rd February 2014
Abstract
A non-symmetrical aluminium complex containing a single metal centre has been synthesised and used as a catalyst in the cycloaddition reaction of carbon dioxide to styrene oxide to yield a cyclic carbonate. The reaction proceeds to around 70% conversion using the catalyst alone; however, it can be increased to 90% conversion with the addition of tetrabutylammonium bromide (TBAB) as a co-catalyst. Interestingly, the reaction proceeds as well with the organo-catalyst TBAB alone as it does with the aluminium catalyst alone. This offers the potential for reduced cost processing and scale-up of the reaction through elimination of the aluminium catalyst due to reduced catalyst costs and ease of separation of TBAB as a salt from the reaction mixture. The rate constants for the aluminium catalysed reactions were determined as a function of process temperature in the range 80–150 °C, as well as the activation energies for the reactions determined from the Arrhenius plots. The activation energy for the aluminium catalyst alone was determined to be 34 kJ mol−1, while adding TBAB as a co-catalyst reduced the activation energy by 11 kJ mol−1.
1. Introduction
Carbon dioxide utilization (CDU) from waste gas streams or even the atmosphere has received growing attention over the recent years. Indeed the World Economic Forum1 has identified CDU as one of the top ten emerging technologies for 2012 and 2013. There has been much debate as to whether CDU can play a significant role in climate change mitigation because of the vast quantities of carbon dioxide emitted globally each year. The amount of CO2 that could be captured and reused has been estimated at various levels up to approximately 10% of emissions.2 Carbon dioxide is already used widely in the food industries and increasingly in the field of CO2-enhanced oil recovery (CO2-EOR). However, with the exception of a recent paper by Jaramillo et al.,3 accurate life cycle analyses over the latter have yet to be demonstrated as there are no defined and agreed boundary conditions for the process.2 Regardless of climate mitigation, CDU can provide an alternative chemical feedstock with decreased reliance on petrochemical derivatives.4–6Carbon dioxide is relatively stable and so unreactive having a standard heat of formation (ΔfH°) of −394 kJ mol−1. Therefore, catalysts are needed to reduce the activation energy of any reaction in which it is involved. The formation of cyclic carbonates is a key area that has attracted considerable interest. Cyclic carbonates are valuable products as they can be used as solvents in chemical reactions and as constituents of batteries. Furthermore, ring opening polymerisation reactions give rise to polycarbonates which are valuable structural and functional materials. Their high strength and transparency have made them useful as replacement materials for security glass applications and in the packaging of foodstuffs. CO2 insertion is also beneficial as it eliminates the use of highly toxic phosgene which is used in conventional carbonate and polycarbonate syntheses.7 Furthermore, the formation of cyclic carbonates is an example of an atom efficient reaction with each atom present in the two reactants being incorporated into the final product.
Several methods have been reported for the synthesis of cyclic carbonates from terminal epoxides by homogeneous cycloaddition of carbon dioxide with a variety of catalysts. Catalyst systems based primarily on the highly active (salen)metal-based catalyst system have been reported. Among such transition-metal complexes, chromium,8 cobalt,9,10 zinc11 and magnesium12 have been reported. Examples of such complexes are given in Fig. 1. In particular, salen complexes of aluminium have received considerable interest in the synthesis of cyclic carbonates, mainly due to the low environmental impact of aluminium, its high Earth abundance and its high catalytic activity.13 Studies have reported the synthesis of cyclic carbonates using different symmetrical aluminium salen complexes and Lewis bases as co-catalysts, including quaternary ammonium salts, pyridine, imidazolium, 18-crown-6-KI and N,N′-dimethylaminopyridine (DMAP).
 | ||
| Fig. 1 The structure of (salen)metal complexes: (a) (salen)MX complex and (b) bimetallic complex [(salen)M]2O catalyst discussed in Table 1. | ||
Lu et al. have reported the effect of a co-catalyst on the catalytic activity of aluminium salen complexes.14,15 They concluded that the binary catalyst system of (salen)AlX/Bu4NBr (TBAB) showed better catalytic activity than (salen)AlX alone. Tetrabutylammonium bromide (TBAB) has been shown to enhance catalytic activity in comparison with other Lewis bases as shown in Table 1. He et al. suggested that the existence of halides in the axial (Al–X) group enhanced the catalytic activity of (salen)AlX.12 They also proposed the mechanism for the cyclic carbonate synthesis using the binary catalyst system which has since been developed by North and co-workers.16–18
| Epoxide | Catalyst | Solvent | Conditions | TONd | TOFe (h−1) | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| (salen)metal complex | Co-catalyst | T (°C) | P (bar) | Time (h) | |||||
| a R1, R2 = H, M = Al. b R1, R2 = tBu, R3, R4 = Ph, M = Cr, X = Cl. c R1, R2 = tBu, M = Al. d Moles of carbonate produced per mole of catalyst. e Moles of carbonate produced per mole of catalyst per hour. | |||||||||
| PO | (salen)AlCla | Bu4NI | — | 25 | 6 | 8 | 492 | 61.5 | 14, 15 |
| (salen)AlCl | Bu4NBr | — | 25 | 6 | 8 | 504 | 63.0 | ||
| (salen)AlCl | Bu4NCl | — | 25 | 6 | 8 | 276 | 34.5 | ||
| (salen)AlCl | 18-Crown-6-KI | — | 25 | 6 | 8 | 463 | 57.9 | ||
| (salen)AlBr | 18-Crown-6-KI | — | 25 | 6 | 8 | 498 | 62.3 | ||
| (salen)AlEt | 18-Crown-6-KI | — | 25 | 6 | 8 | 492 | 61.5 | ||
| EO | (salen)AlCla | — | CH2Cl2 | 110 | 150–160 | 1 | 174 | 174 | 12 |
| (salen)AlCl | Pyridine | — | 110 | 150–160 | 1 | 690 | 690 | ||
| (salen)AlCl | 1-MeIm | — | 110 | 150–160 | 1 | 526 | 526 | ||
| (salen)AlCl | Bu4NBr | — | 110 | 150–160 | 1 | 2220 | 2220 | ||
| PO | (salen)CrClb | DMAP | CH2Cl2 | 100 | 8 | 1 | 916 | 916 | 8 |
| PO | [(salen)Al]2Oc | Bu4NBr | — | 25 | 1 | 3 | 24.8 | 8.3 | 16, 18 |
Cyclic carbonates are synthesised by forming the hexa-coordinated complex between the oxygen atom of the epoxide and the central metal in (salen)AlCl. The formation of a hexa-coordinated complex results in a reduction of the Al–Cl bond strength. The nucleophilicity of the Cl atom in the (salen)AlCl complex favours interaction with the epoxide, leading to facile cleavage of the epoxide ring (Fig. 2). At the same time, the highly reactive anion of the quarternary ammonium salt TBAB catalyses the opening of the epoxide ring by nucleophilic attack at the epoxide, initiating the attack at the carbon atom of carbon dioxide by the bromo-alkoxide. The lone pair of electrons of one oxygen atom of the carbon dioxide weakly coordinates with the central metal ion in the (salen)AlCl complex, leading to a linear carbonate form. The linear carbonate is then transformed into the cyclic carbonate via spontaneous intramolecular cyclic elimination.
 | ||
| Fig. 2 Proposed mechanism for the (salen)AlCl–TBAB catalysed synthesis of cyclic carbonates from epoxides and carbon dioxide. | ||
As can be seen in Table 1, the symmetrical aluminium salen complexes have been reported to show catalytic activity in cyclic carbonate synthesis at high CO2 pressure and/or high temperature, whereas only the bimetallic aluminium salen complex was an effective catalyst under ambient conditions.18 Catalytic cycloaddition by a non-symmetrical aluminium salen complex has not been reported among the catalysts described to date. Non-symmetrical salen complexes are potentially useful if they can be functionalised with a motif that allows subsequent polymerisation or grafting to a pre-formed polymeric structure, as this immobilises the catalyst so it can act heterogeneously.19–21 In this study, the synthesis of styrene carbonate using the non-symmetrical aluminium salen–acac hybrid complex (salenac), TBAB and a combined catalytic system was undertaken as a function of temperature at atmospheric CO2 pressure.
2. Experimental methods
All chemicals and solvents were obtained from Aldrich and Fisher Scientific and used as received without further purification. The following reagents were used: dichloromethane (AR, Fischer), methanol (AR, Fischer), 2,4-pentanedione (99%, Aldrich), 1,2-diaminoethane (99%, Aldrich), 2,4-dihydroxybenzaldehyde (98%, Aldrich), diethylaluminium chloride (1.8 M solution in toluene, Aldrich), tetrabutylammonium bromide (99%, Aldrich), styrene oxide (98%, Aldrich) and diethyl ether (99.8%, Fisher). CO2 (99.8%) gas was supplied by BOC Gases and dried using an inline molecular sieve/silica gel drying tube when used.NMR spectra were recorded using a Bruker DRX500 spectrometer at 500.3 MHz for 1H-NMR and 125.77 MHz for 13C-NMR. CHN analysis was performed on a Perkin Elmer 2400 CHNS/O Series II Elemental Analyser. Chloride content was determined by titration by the University of Sheffield Elemental Analysis Service in the Department of Chemistry. Mass spectra were recorded on a VG Autospec mass spectrometer using Electron Ionisation (EI). Gas chromatography (GC) was performed on a Varian 3900 GC with a 15 m × 25 mm i.d. capillary column with hydrogen as the carrier gas. The conditions used for investigating styrene oxide conversion were: hold for 2 min at an initial temperature of 60 °C, then ramped at a rate of 15 °C min−1 from 60 to 270 °C and held at 270 °C for 5 min.
Synthesis of the non-symmetrical ligand (2 in Scheme 1)
A solution of 2,4-pentanedione (1.09 ml, 10.5 mmol) in dichloromethane (8 ml) was added dropwise to a solution of 1,2-diaminoethane (1.36 ml, 20.1 mmol) in dichloromethane (8 ml). The resulting mixture was heated under reflux for 1 h. The excess 1,2-diaminoethane was removed from the resulting mixture by rotary evaporation under vacuum at 60 °C for 30 min. The residue was dissolved in dichloromethane (16 ml), then a solution of 2,4-dihydroxybenzaldehyde (1.42 g, 10.1 mmol) in methanol (7 ml) and dichloromethane (7 ml) was added slowly. The resulting solution was stirred at ambient temperature for 30 min. The product was filtrated and dried overnight in a vacuum oven at 80 °C until there was no further mass loss. The product was obtained as a pale yellow powder (1.989 g, 75%). The structure of the (salenac)OH ligand is shown in Fig. 3. 1H-NMR (500.1 MHz, (CD3)2SO) δH (ppm): 1.85 (3H, –C(CH3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), 1.90 (3H, 3× H1), 3.30 (2H, 2× H6), 3.75 (2H, 2× H7), 4.90 (1H, H3), 6.15 (1H, H5′), 6.25 (1H, H3′), 7.15 (1H, H6′), 8.35 (1H, –NCH–), 9.95 (1H, Ph–OH), 10.70 (1H, –OH) and 13.65 (1H, –C–OH). Elemental analysis: calculated for C14H18N2O3: C 64.08, H 6.87 and N 10.68%; found: C 63.52, H 6.93 and N 10.81%.
N–), 1.90 (3H, 3× H1), 3.30 (2H, 2× H6), 3.75 (2H, 2× H7), 4.90 (1H, H3), 6.15 (1H, H5′), 6.25 (1H, H3′), 7.15 (1H, H6′), 8.35 (1H, –NCH–), 9.95 (1H, Ph–OH), 10.70 (1H, –OH) and 13.65 (1H, –C–OH). Elemental analysis: calculated for C14H18N2O3: C 64.08, H 6.87 and N 10.68%; found: C 63.52, H 6.93 and N 10.81%.
 | ||
| Fig. 3 Structure of the non-symmetrical Schiff-base salenac ligand. | ||
Synthesis of the AlCl(salenac)OH complex (Al1cat) (3 in Scheme 1)
The non-symmetrical aluminium salen complex was synthesised by the reaction of the non-symmetrical Schiff-base salenac ligand (2) with diethylaluminium chloride (Et2AlCl) as shown in Scheme 1 in the results and discussion section. The (salenac)OH ligand (0.08 g, 0.305 mmol) was dissolved in dried dichloromethane (20 ml) under a nitrogen atmosphere at ambient temperature. Diethylaluminium chloride (0.17 ml, 1.8 M solution in toluene, 0.305 mmol) was added slowly to the stirred solution under a nitrogen atmosphere. After stirring for 4 h at room temperature, the product was removed by filtration and dried overnight in a vacuum oven at 80 °C until the weight was constant. This yielded 0.089 g (91%) of a bright yellow powder. The structure of the AlCl(salenac)OH complex is shown in Fig. 4 and the NMR spectra in the ESI.†1H-NMR (500.1 MHz, (CD3)2SO) δH (ppm): 1.85 (3H, –C(CH3)N–), 1.90 (3H, 3× H1), 3.30 (2H, 2× H6), 3.75 (2H, 2× H7), 4.90 (1H, H3), 6.15 (1H, H5′), 6.25 (1H, H3′), 7.15 (1H, H6′), 8.35 (1H, –NCH–) and 9.95 (1H, –OH). 13C-NMR (125.7 MHz, CDCl3) δC (ppm): 21.52 (C1), 25.59 (–C(CH3)N–), 52.27 (C6), 55.99 (C7), 98.56 (C3), 105.00 (C5′), 105.86 (C3′), 112.90 (C1′), 135.36 (C6′), 164.18 (C9), 165.05 (C4′), 166.89 (C4), 170.66 (C2′) and 175.79 (C2). Elemental analysis: calculated for C14H16N2O3AlCl: C 52.06, H 4.96, N 8.68 and Cl 10.99%; found: C 51.95, H 5.03, N 8.73 and Cl 11.07%.
 | ||
| Scheme 1 Synthesis of the non-symmetric (2) ligand and the Al1cat catalyst (3) used in the studies. | ||
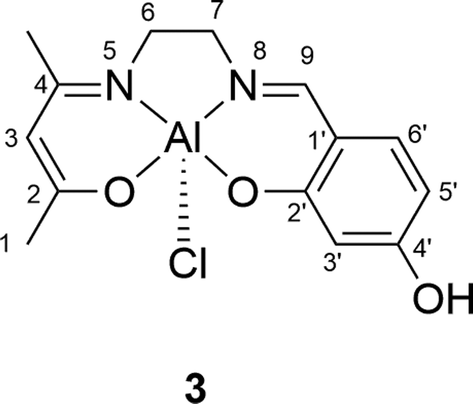 | ||
| Fig. 4 Structure of the AlCl(salenac)OH complex (Al1cat). | ||
Synthesis of styrene carbonate (4) using the Al1cat catalyst
The procedure for the homogeneously catalysed insertion reaction of carbon dioxide into styrene oxide was adapted from the literature.18 The catalytic reaction was carried out in reaction tubes using a Radleys Carousel parallel synthesiser. The reaction temperature was controlled to ±0.1 °C using an IKA fuzzy logic controller unit, while the reflux was controlled using a water cooled heat exchanger that was integrated into the synthesiser. Carbon dioxide was dried by passing it through a dryer column (molecular sieve/self-indicating silica gel) before being introduced into a Radleys Carousel reaction tube at 1 bar pressure using a PTFE sparging tube directly into the stirred solution. Agitation was achieved using a magnetic stirrer bar.A mixture of styrene oxide (2 mmol), tetrabutylammonium bromide (0–2.5 mol%), Al1cat catalyst (AlCl(salenac)OH complex, 0–2.5 mol%) and dichloromethane (5 ml) as a co-solvent was loaded into a Radleys reaction tube. This was tightly sealed and the reaction was stirred vigorously until complete dissolution of the reagents was achieved. The reaction was heated to the desired temperature (80–150 °C) and carbon dioxide was introduced to the reaction at atmospheric pressure by bubbling it through the solution. The reaction was monitored using gas chromatography until it approached steady state conversion, typically after 48 hours. The reaction solution was extracted into methanol in order to determine the styrene oxide conversion by GC analysis with tetradecane as an internal standard. To obtain pure styrene carbonate, the reaction solution was first extracted into diethyl ether and separated by column chromatography on silica gel using hexane–ethyl acetate (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as an eluent. Evaporation of the combined product fractions gave styrene oxide in 41% isolated yield as a white solid. The melting point of 50–52 °C agreed well with the literature values (50–54 °C).22,231H-NMR (500.1 MHz, CDCl3) δH (ppm): 4.35 (1H, OCH), 4.81 (1H, OCH), 5.68 (1H, PhCHO) and 7.2–7.5 (5H, ArH); 13C-NMR (125.7 MHz, CDCl3) δC (ppm): 71.1 (CH2), 77.9 (CPh), 125.81, 128.55, 129.16 and 135.8 (Ph), 154.78 (CO). Electron ionisation MS analysis gave the molecular ion of styrene carbonate at m/z 164.
:3) as an eluent. Evaporation of the combined product fractions gave styrene oxide in 41% isolated yield as a white solid. The melting point of 50–52 °C agreed well with the literature values (50–54 °C).22,231H-NMR (500.1 MHz, CDCl3) δH (ppm): 4.35 (1H, OCH), 4.81 (1H, OCH), 5.68 (1H, PhCHO) and 7.2–7.5 (5H, ArH); 13C-NMR (125.7 MHz, CDCl3) δC (ppm): 71.1 (CH2), 77.9 (CPh), 125.81, 128.55, 129.16 and 135.8 (Ph), 154.78 (CO). Electron ionisation MS analysis gave the molecular ion of styrene carbonate at m/z 164.
Kinetic parameters of styrene carbonate synthesis
The procedure for investigating the kinetic parameters for the cycloaddition of carbon dioxide to styrene oxide was adapted from the literature.18 Styrene oxide (0.23 ml, 2 mmol), tetrabutylammonium bromide (6.45 mg, 1 mol), Al1cat (6.45 mg, 1 mol) and dichloromethane (5 ml) were loaded into a Radleys reaction tube. The solution was stirred vigorously using a magnetic stirrer and heated to the desired temperature (80–150 °C). Carbon dioxide was introduced to the reaction at 1 bar by bubbling through the solution. The reaction was monitored every 3 hours using gas chromatography as described previously. The rate constant was determined from the experimental data assuming pseudo-first order reaction kinetics and the activation energy for the process was determined using the Arrhenius equation based on the calculated rate constants at different temperatures.3. Results and discussion
The ligand used in this study was prepared by adapting the synthetic procedure described by Styring et al.19–21 which was used in the synthesis of other non-symmetrical metal complexes. The ligand was dried thoroughly before forming the coordinated complex with diethylaluminium chloride. The general procedure is shown in Scheme 1.Styrene carbonate was synthesised via the catalytic cycloaddition of carbon dioxide to styrene oxide in a homogeneous reaction (Scheme 2). The mixture was stirred at elevated temperatures (80–150 °C). After reaction for 48 h, the mixture was worked up to give a yield of 41% of styrene carbonate as a white solid. The melting point (mp 50–52 °C) agreed well with the range of melting points (50–54 °C) of styrene carbonate reported in the literature.22,23 The NMR analysis was consistent with that for styrene carbonate reported in the literature.16,22,23
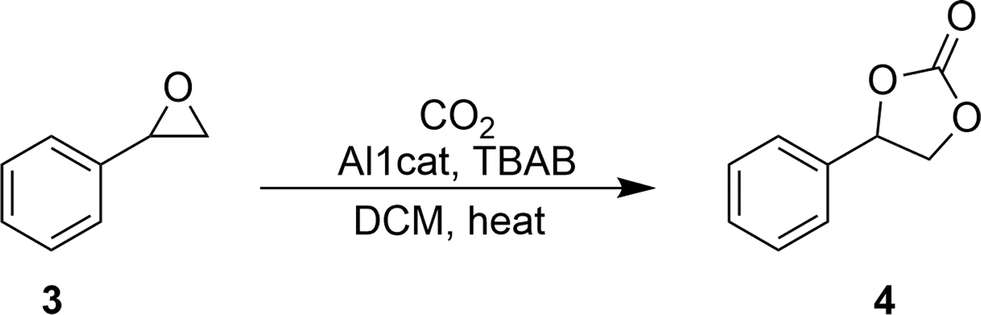 | ||
| Scheme 2 Cyclic styrene carbonate (4) synthesis through CO2 insertion into styrene oxide (3). | ||
Catalytic activity of the non-symmetrical aluminium salenac complex
A summary of the catalytic behaviour of the Al1cat complex in the styrene carbonate synthesis from styrene oxide and CO2 is reported in Table 2. The symmetrical (salen)AlCl complex was also studied for comparison. Previous studies at 100 bar CO2 pressure and 80 °C showed 19% conversion to the cyclic carbonate with a TON of 59.13 However, at 1 bar pressure we observed no conversion at temperatures up to 110 °C.| Entry | Catalyst (mol%) | Co-catalyst (mol%) | Temp (°C) | Conversionb (%) |
|---|---|---|---|---|
| a Reaction conditions: styrene oxide (2 mmol), CO2 1 bar and co-solvent (DCM) 5 ml for 48 h. b Conversions were calculated from GC analysis data based on styrene oxide by using tetradecane as internal standard. | ||||
| 1 | Al1cat (2.5) | TBAB (2.5) | 25 | 0 |
| 2 | Al1cat (1.0) | TBAB (1.0) | 25 | 0 |
| 3 | Al1cat (1.0) | TBAB (1.0) | 80 | 77 |
| 4 | Al1cat (1.0) | — | 80 | 48 |
| 5 | Al1cat (2.0) | TBAB (2.0) | 110 | 92 |
| 6 | Al1cat (1.0) | TBAB (1.0) | 110 | 90 |
| 7 | Al1cat (1.0) | — | 110 | 73 |
| 8 | — | TBAB (1.0) | 110 | 70 |
The catalytic cycloaddition was conducted initially at three different temperatures: 25, 80 and 110 °C. There was no activity at 25 °C, either with Al1cat alone or as a mixed catalyst system with TBAB (entries 1 & 2 in Table 2). The reaction was carried out in dichloromethane as the solvent. While this may seem a curious choice of solvent for a reaction at elevated temperatures and 1 bar pressure, it was used because it solubilized both the catalyst complex and the organic substrate. CO2 was sparged with constant flow directly into the solution, with the reaction tube being pressure equalised by placing an identical diameter syringe needle to the sparger inlet as the outlet in the silicone septum. Dichloromethane vapour condensed back into the reactor using the Radleys Carousel reflux unit and there was no noticeable loss of solvent at the end of the reaction. When the temperature was increased to 80 °C, a conversion of 77% was observed with added TBAB (entry 3) or 48% for Al1cat alone (entry 4). Good conversion was achieved at 110 °C under a number of conditions. For the reaction with 2 mol% each of Al1cat and TBAB (entry 5), 92% conversion was achieved, while for 1 mol% of each (entry 6) the conversion was comparable at 90%. When 1 mol% Al1cat complex was used without added TBAB at 110 °C, reactivity was decreased with lower styrene oxide conversion of 73% (entry 7), relative to that of the Al1cat and TBAB binary system (90% conversion) at the same temperature. This result followed a similar trend to those reported by Lu et al.,14,15 although the symmetrical aluminium salen complex and TBAB were reported in the literature. Interestingly, the catalytic reactivity of TBAB alone was comparable to the Al1cat system, with 70% conversion (entry 8). This shows that the aluminium catalyst may well be unnecessary for cyclic carbonate synthesis which will not only reduce costs but also simplify the post-reaction separation process, especially if supported TBAB-type reagents can be used.
All of the results of both the binary catalyst and catalyst alone showed increased styrene oxide conversion with increasing temperature, with no conversion at room temperature. Furthermore, the catalytic activity of the Al1cat complex can be compared with a symmetrical aluminium salen complex ((salen)AlCl) and a dimetallic aluminium salen complex ([(salen)Al]2O) as shown in Table 3.
| Entry | Catalyst | Solvent | Condition | TONa | TOFb (h−1) | Ref | |||
|---|---|---|---|---|---|---|---|---|---|
| Metal cat | Co-cat | T (°C) | P (bar) | Time (h) | |||||
| a Moles of carbonate produced per mole of catalyst. b Moles of carbonate produced per mole of catalyst per hour. | |||||||||
| 1 | (salen)AlCl | 1-MeIm | — | 80 | 100 | 6 | 59 | 9.83 | 13 |
| 80 | 20 | 6 | 10 | 1.67 | |||||
| 2 | [(salen)Al]2O | — | — | 25 | 1 | 6 | 35.6 | 6 | 18 |
| 3 | Al1cat | Bu4NBr | CH2Cl2 | 110 | 1 | 48 | 41 | 0.85 | This work |
| Al1cat | — | CH2Cl2 | 110 | 1 | 48 | 30 | 0.63 | ||
Although styrene carbonate can be produced in a shorter reaction time using (salen)AlCl in comparison with Al1cat reported here, (salen)AlCl suffered from low catalyst activity due to the need for a co-catalyst and the requirement for high temperature and/or high CO2 pressure (80 °C, 100 bar). The TON value of 30 for Al1cat alone as the catalyst (entry 3) was 3 times larger than that of (salen)AlCl along with methylimidazole at 20 bar of CO2 pressure (entry 1) as shown in Table 3. However, at the same low pressure, the dimetallic aluminium salen complex ([(salen)Al]2O) displayed better catalytic activity than the Al1cat complex. Higher temperatures and longer reaction times (110 °C, 48 h) were required for the catalytic reaction using Al1cat, although a TBAB co-catalyst was also used. This could be due to an increase in the inherent oxophilicity of the Al atom in the dimetallic salen complex. Both aluminium ions of the bimetallic complex dominate in activating the coordination of CO2 to the aluminium ion and then forming styrene carbonate through intramolecular substitution and cyclic elimination.18 While the bimetallic complexes show higher catalytic activity than Al1cat, they are symmetrical so any functional group is mirrored in the molecule. By contrast, the non-symmetrical Al1cat complex can be formed bearing a single, discrete functional group, in this case a phenolic OH group. This allows further derivativisation of the complex to form, for example, an ether or ester group with a support material such as a Merrifield resin. This important feature allows the initial homogeneous catalyst to be supported on a solid phase, which in turn allows the catalyst to be included in continuous flow reactors that are essential if large volumes of CO2 are to be processed.
The fact that the Al1cat complex functions well at ambient pressure is, we believe, not a consequence of any change in the mechanism proposed in Fig. 2, which would be equally valid for any salen-based aluminium(III) chloride complex. The enhanced activity is related to the experimental setup which intensifies the contact between the CO2 gas and the liquid solution through active sparging of the gas continuously through the solution.
The activation energy for styrene carbonate synthesis
Although the best results for the homogeneous cycloaddition reaction was achieved for the binary catalyst (Table 2, entries 5 & 6), it is possible that a higher temperature promotes the reactivity of the catalyst alone. To investigate the kinetics of the reaction, gas chromatography was used to analyse styrene oxide consumption relative to the initial concentration over the course of the reaction.18 It was noted that standard styrene carbonate is not available commercially, so styrene oxide was used as a reference standard in analysing the reaction kinetics. Tetradecane was used as an internal standard, and the area ratios of GC peaks at different concentration ratios of styrene oxide/tetradecane were used in the calibration. The reaction profile was determined by fitting the data to a plot of the concentration ratios against peak area ratios. The kinetic parameters were studied over a wide temperature range from 80 to 150 °C. The data were fitted to the following rate equations:| Rate = k[epoxide]a[CO2]b[Al1cat]c[TBAB]d | (1) |
| Rate = kobs[epoxide]a | (2) |
 | (3) |
| − ln[epoxide] = kobst | (4) |
Using the Arrhenius equation, the activation energy for the reaction can be calculated from the relationship between the observed rate constant and temperature:
| kobs = A exp(−Ea/RT) | (5) |
As can be seen in Fig. 5, the styrene oxide concentration decreased with reaction time, when CO2 was introduced in the reaction mixture containing Al1cat alone or the Al1cat/TBAB binary system at different temperatures. An induction period can be observed in the reaction catalysed by Al1cat alone or the Al1cat/TBAB binary system. For both catalyst systems the induction period was reduced with increasing temperature. The Al1cat/TBAB binary system showed a shorter induction period than Al1cat alone at the same temperature. A similar behaviour has been described previously in a work reported by North and Pasquale18 who studied the cyclic carbonate synthesis by using a combination of the dimetallic aluminium salen complex [(salen)Al]2O and TBAB. They reported that the induction period disappeared when a high concentration of TBAB was used in the reaction mixture. The shorter induction period and faster approach to steady state in the binary catalyst system indicate that TBAB is acting as a catalytic enhancer for styrene carbonate synthesis in this study. This can be clearly observed in the activation energy values of both catalytic systems reported in Table 4.
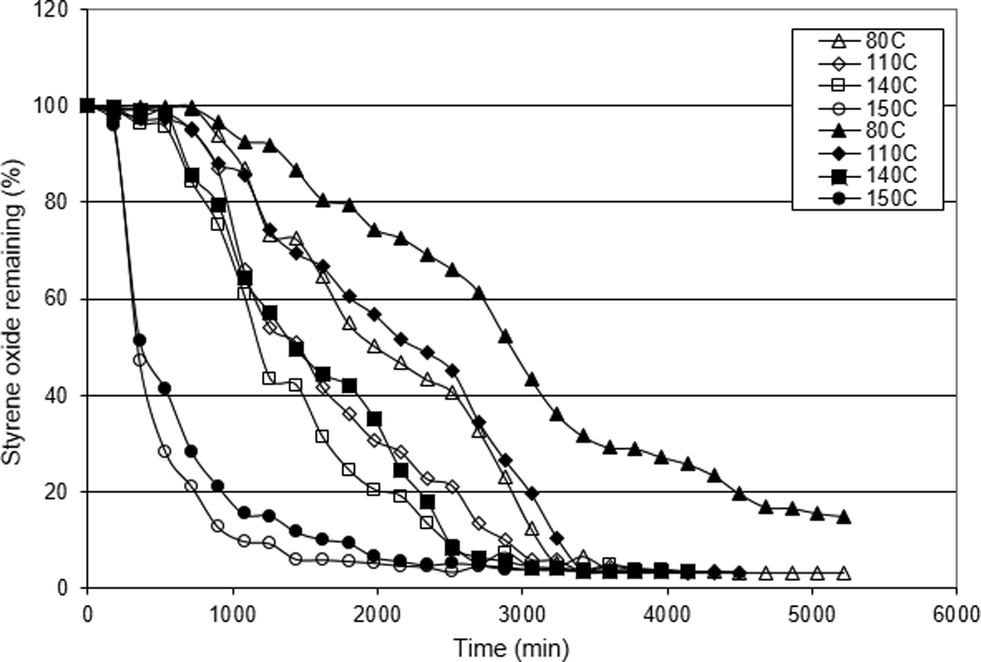 | ||
| Fig. 5 Styrene oxide conversion as a function of reaction temperature for the Al1cat catalyst alone (solid markers) and with added TBAB co-catalyst (open markers). | ||
| Catalyst | T (°C) | The observed first-order rate constant,akobs (×10−3) (min−1) | Kinetic parametersb | ||
|---|---|---|---|---|---|
| E a (kJ mol−1) | A (min−1) | R 2 | |||
| a The average and standard deviation of the observed first-order kinetic rate constants of all of the samples obtained from the three experiments at each temperature. b Kinetic parameters and the R2 values of the Al1cat catalyst alone obtained from the linear Arrhenius plot of ln kobs against 1/T. | |||||
| Al1cat/TBAB | 80 | 0.53 ± 0.06 | 23 | 1.3 | 0.91 |
| 110 | 0.87 ± 0.15 | ||||
| 140 | 1.20 ± 0.10 | ||||
| 150 | 2.20 ± 0.20 | ||||
| Al1cat | 80 | 0.23 ± 0.10 | 34 | 18.0 | 0.90 |
| 110 | 0.40 ± 0.10 | ||||
| 140 | 0.73 ± 0.15 | ||||
| 150 | 1.73 ± 0.25 | ||||
The observed rate constant was determined from the slope of a linear plot of the natural logarithm of the changing sample concentration with time at different temperatures based on eqn (4). Fig. 6 shows the determination of the observed rate constant for styrene oxide conversion by the Al1cat and TBAB catalysts at 150 °C. This was repeated for other reaction temperatures and catalyst compositions. The results indicate the reaction to be a pseudo-first-order overall under each of the different conditions.
 | ||
| Fig. 6 Pseudo-first order kinetic plot of styrene oxide concentration against time for the Al1cat/TBAB binary catalyst system at 150 °C. | ||
The activation energies for the CO2 insertion reaction into styrene oxide catalysed by Al1cat and the Al1cat/TBAB binary catalyst system were determined over the range 80–150 °C by fitting the data from a plot of the natural logarithm of the observed pseudo-first order rate constant (ln kobs) against the reciprocal absolute temperature (K/T). Fig. 7 shows the Arrhenius plots for both catalytic systems and the data derived from the studies are summarised in Table 4.
 | ||
| Fig. 7 Arrhenius plots for the cycloaddition of carbon dioxide to styrene oxide using Al1cat alone (■) and the AL1cat/TBAB co-catalyst system (♦). | ||
For the homogeneous catalyst Al1cat alone, the activation energy was calculated to be 34 kJ mol−1. When the Al1cat/TBAB binary catalyst system was used this value decreased to 23 kJ mol−1. The data show that TBAB acts by reducing the activation energy value by 11 kJ mol−1 in the catalysed reaction. From the observation of the induction period and activation energy data, it can be concluded that styrene oxide cyclic carbonation can be catalysed by Al1cat, while the TBAB acts as a catalytic promoter or a co-catalyst in this system. This is consistent with many reports, although the symmetrical aluminium salen complexes were used in the binary catalyst system studied by He et al.,12 Lu et al.14,15 and North and Pasquale.18 The mechanism of cyclic carbonate catalysis has been well established in the presence of a binary catalyst using TBAB as a co-catalyst and this is shown in Fig. 2. This mechanism correlates well with the behaviour of the catalysed reaction observed in this study. The central metal in Al1cat coordinates with an oxygen atom of epoxide, activating it towards a nucleophilic attack by the bromide ion from TBAB. This results in elimination of the chloride ion from the aluminium atom. The cleavage of the epoxide ring results in the formation of a linear bromo-alkoxide coordinated with the aluminium centre. The oxygen coordinated to the aluminium is activated and can attack the carbon dioxide molecule, which is essentially a nucleophilic attack of the oxygen lone pair of electrons at a carbonyl group. The carbonate anion formed then undergoes an intramolecular cyclisation reaction, eliminating the bromide ion back into the catalytic cycle. The displaced chloride ion then reattaches at the aluminium centre to eliminate the cyclic carbonate product.
For the same reaction of CO2 and styrene oxide catalysed by calcined Mg–Al mixed oxides, the activation energy was reported to be 78 kJ mol−1.24 It has been reported that the calcined Mg–Al mixed oxides exhibit good catalytic activity due to the presence of both acid and strong basic sites on their surfaces. However, the styrene carbonate formation in this case required a larger activation energy than Al1cat (34 kJ mol−1), especially the binary Al1cat/TBAB catalyst (23 kJ mol−1). This may be due to the fact that the homogeneous catalyst complex, Al1cat alone or the binary catalyst system, is uniformly distributed in the reaction medium and offers good mass transfer. By contrast, only the surface atoms of the calcined heterogeneous Mg–Al mixed oxides are active in the reaction medium and so the catalytic activity of the mixed oxides depends on surface coverage and reaction of the solid. The diffusion of reactants and products through internal pores of the solid therefore dominates the reaction kinetics.
One important feature of the catalytic studies was the observation that a reaction was observed with TBAB alone at 110 °C, with a conversion comparable with Al1cat alone. This is potentially important as it means that the aluminium catalyst can be eliminated from processes making the scale-up of the process significantly less expensive in terms of both materials and process/separation costs, whilst also offering the opportunity to form immobilized analogues as heterogeneous catalysts. Similar results have also been reported by Caló et al.25 for a variety of alkenes, using TBAB as both the catalyst and solvent. Cyclic carbonate formation directly from alkenes has been reported previously; however, the process is a multi-phase system using CO2 at elevated pressures.26 The proposed mechanism is shown in Fig. 8. The mechanism is remarkably similar to the Al1cat catalyst process and offers an explanation as to why the metal catalyst combined with TBAB has lower activation energy compared to Al1cat alone. The aluminium catalyst activates the epoxide towards attack of the bromide ion. However, in the metal-free system, the nucleophilic bromide ion attack at the epoxide can still occur without prior activation of the epoxide. Cycloaddition of carbon dioxide with concurrent bromide ion elimination can then occur to yield the cyclic carbonate, with the bromide ion re-joining the catalytic cycle as TBAB.
 | ||
| Fig. 8 Mechanism for the TBAB catalysed cycloaddition of CO2 to a terminal epoxide. | ||
Conclusions
The novel non-symmetrical aluminium(III) salenac complex (Al1cat) exhibits catalytic activity toward the cycloaddition of CO2 into styrene oxide at atmospheric pressure. It displayed better catalytic activity than the symmetrical aluminium salen complex reported by Alvaro et al.13 at low CO2 pressure. However, a high temperature was required in order to achieve high yields. The activation energy for Al1cat was found to be 34 kJ mol−1, which was reduced to 23 kJ mol−1 for the binary catalyst system (Al1cat/TBAB) at reaction temperatures in the range 80–150 °C. Importantly, catalysis using TBAB alone was found to be as effective as using Al1cat alone at 110 °C. This has significant implications in the industrialisation of the process as it allows the aluminium complex to be eliminated from the reaction, relying only on an organic salt to carry out the catalysis which can be readily separated post-reaction. Future studies will investigate the use of immobilised TBAB analogues, solvent-less systems and scaled up processes to further improve the technology readiness of the reaction and process.Acknowledgements
We thank the Royal Thai Government and the National Metal and Materials Technology Center (MTEC) for funding the PhD studentship to SS, and the Engineering and Physical Sciences Research Council (EPSRC) for its funding of the C-Cycle Research Programme (EP/E010318/1) and the CO2Chem Network (EP/H035702/1 and EP/K007947/1).References
- D. King, World Economic Forum, The Top 10 Emerging Technologies for 2013, http://forumblog.org/2013/02/top-10-emerging-technologies-for-2013/, (last accessed 29/11/13).
- N. von der Essen, J. Jung and A. Bardow, Energy Environ. Sci., 2013, 6, 2721–2734 Search PubMed.
- P. Jaramillo, W. M. Griffin and S. T. McCoy, Environ. Sci. Technol., 2009, 43, 8027–8032 CrossRef CAS PubMed.
- Carbon Dioxide as a Chemical Feedstock, ed. M. Aresta, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- P. Styring, D. Jansen, H. de Coninck, H. Reith and K. Armstrong, Carbon Capture and Utilisation in the Green Economy, CO2Chem Media and Publishing, UK, 2011, ISBN: 978-0-9572588-1-5 Search PubMed.
- R. Martín and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1263 CrossRef PubMed.
- R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498–11499 CrossRef CAS.
- X.-B. Lu, B. Liang, Y.-J. Zhang, Y.-Z. Tian, Y.-M. Wang, C.-X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732–3733 CrossRef CAS PubMed.
- W.-M. Ren, G.-P. Wu, F. Lin, J.-Y. Jiang, C. Liu, Y. Luo and X.-B. Lu, Chem. Sci., 2012, 3, 2094–2102 RSC.
- W. M. Shen, W.-L. Duan and M. Shi, J. Org. Chem., 2003, 68, 1559–1562 CrossRef PubMed.
- R. He, X.-B. Lu and X.-J. Feng, Appl. Catal., A, 2002, 234, 25–33 CrossRef.
- M. Alvaro, C. Baleizao, E. Carbonell, M. El Ghoul, H. GarcÍa and B. Gigante, Tetrahedron, 2005, 61, 12131–12139 CrossRef CAS PubMed.
- X.-B. Lu, Y.-J. Zhang, K. Jin, L.-M. Luo and H. Wang, J. Catal., 2004, 227(2), 537–541 CrossRef CAS PubMed.
- X.-B. Lu, Y.-J. Zhang, B. Liang, X. Li and H. Wang, J. Mol. Catal. A: Chem., 2004, 210, 31–34 CrossRef CAS PubMed.
- M. North, J. Melendez and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323–3327 Search PubMed.
- M. North, J. Melendez and P. Villuendas, Chem. Commun., 2009, 2577–2579 Search PubMed.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- P. Styring, C. Grindon and C. M. Fisher, Catal. Lett., 2001, 4, 219–225 CrossRef.
- N. T. S. Phan, D. H. Brown, H. Adams, S. Spey and P. Styring, Dalton Trans., 2004, 1348–1357 RSC.
- N. T. S. Phan and P. Styring, Green Chem., 2008, 10, 1055–1060 RSC.
- T. Iida and T. Itaya, Tetrahedron, 1993, 49, 10511–10530 CrossRef CAS.
- J.-L. Wang, J.-Q. Wang, L.-N. He, X.-Y. Dou and F. Wu, Green Chem., 2008, 10, 1218–1223 RSC.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef PubMed.
- J. Sun, S.-I. Fujita, B. M. Bhange and M. Arai, Catal. Today, 2004, 93–95, 383–388 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy01015e |
| This journal is © The Royal Society of Chemistry 2014 |