Sorbitol dehydration in a ZnCl2 molten salt hydrate medium: molecular modeling
Jianrong
Li
a,
Wim
Buijs
a,
Rob J.
Berger
b,
Jacob A.
Moulijn
a and
Michiel
Makkee
*a
aCatalysis Engineering, Department of Chemical Engineering (ChemE), Delft University of Technology, Julianalaan 136, 2628 BL, Delft, The Netherlands. E-mail: m.makkee@tudelft.nl; Fax: +31 15 278 5006; Tel: +31 15 278 1391
bAnaproc c/o Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands
First published on 22nd October 2013
Abstract
A molecular modelling study, using standard DFT B3LYP/6-31G*, was carried out to develop a better understanding of sorbitol dehydration into isosorbide in ZnCl2 molten salt hydrate medium. Catalysis of sorbitol dehydration by ZnCl2 most likely starts with complexation of the sugar alcohol functions to Zn, followed by an internal SN2 mechanism of a secondary alcohol function attacking a primary alcohol function with the Zn-complex acting as a favourable leaving group. The dehydration reactions to 1,4- and 3,6-anhydrosorbitol show a very similar activation barrier in good accordance with experimental results. The same holds for the formation of isosorbide from 1,4- and 3,6-anhydrosorbitol, albeit with a slightly higher activation barrier. The relative level of the activation barriers reflects the increased strain in the sorbitol skeleton in the corresponding transition states. ZnCl2 turns the dehydration reaction from an endothermic one to an exothermic one by forming a strong complex with the released water. Finally, the ZnCl2–H2O system has been compared with HCl–H2O, which could have been an alternative; it, however, turned out not to be the case.
Introduction
Due to the high potential value of isosorbide and the wide interest in sugar chemistry, sorbitol dehydration has been studied and reviewed extensively in the past by Flèche and Huchette1 and recently by Rose and Palkovits.2 Several catalytic systems have been discovered, such as strong mineral acid catalysts (H2SO4,1–3 HCl,4,5etc.), solid catalysts,2,6–12 ionic liquids and molten salt hydrates.7,8,13,14 Quantitative data on the reaction kinetics, however, are hardly available in the literature. Important conformational aspects of the starting sorbitol in the catalytic mechanism using a mineral acid as catalyst have been proposed by Cekovic5 and by Dosen-Micovic and Cekovic,6 using molecular mechanics. Basically, the reaction can be described as an SN2 reaction mechanism beginning with protonation of the primary hydroxyl group of sorbitol to improve its leaving group ability, followed by attack of a secondary alcohol in the sorbitol skeleton.1,2,5,6In another study, we described sorbitol dehydration in a molten salt hydrate medium8 (ZnCl2·xH2O, x = 3–4) and developed a simplified overall kinetic model using Athena9 software.
A molecular modeling study was undertaken to elucidate the reaction mechanism and to understand the results of the kinetic modeling study. In this kinetic study, the reaction scheme presented in Fig. 1 was used with the following simplification: 1,4- and 3,6-anhydrosorbitol and, in addition, 1,5-anhydrosorbitol and 2,5-anhydromannitol were each lumped together into one stable pseudo-component as one stable side product, although both components could be separated in the HPLC analysis.
 | ||
| Fig. 1 Scheme of sorbitol dehydration in a molten salt hydrate medium,9 A, sorbitol; B, 1,4-anhydrosorbitol + 3,6-anhydrosorbitol; C, 1,4:3,6-dianhydrosorbitol (isosorbide); and D, 2,5-anhydromannitol + 1,5-anhydrosorbitol. | ||
In the molecular modeling study presented here, this kinetic model was used, but the lumping was avoided. In this model, the formation of side products has not been taken into account.
Under practical conditions, the amounts of side products and unknown products are limited up to 3–5% at full sorbitol and 1,4- and 3,6-anhydrosorbitol conversion.
The kinetic model is given in Table 1. The relevant kinetic parameters obtained are shown in Table 2.
| Equations | |
|---|---|
| a Note: Tref = reference temperature. | |
| Reactions | |
1)  |
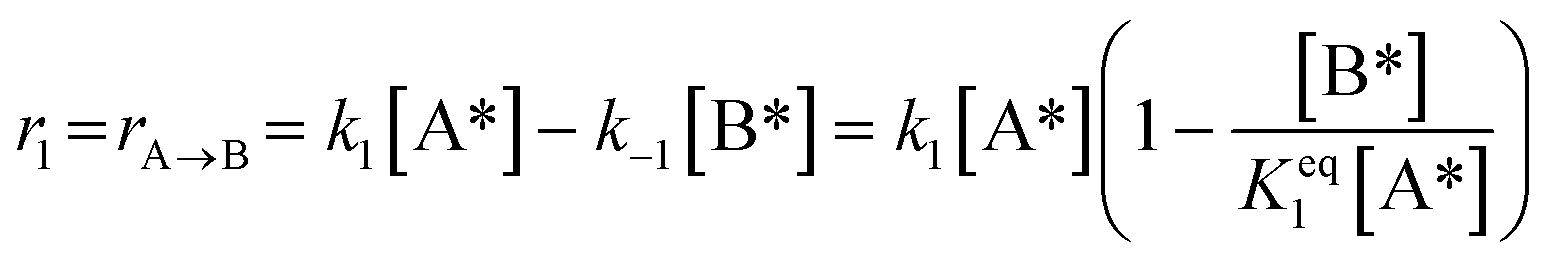
|
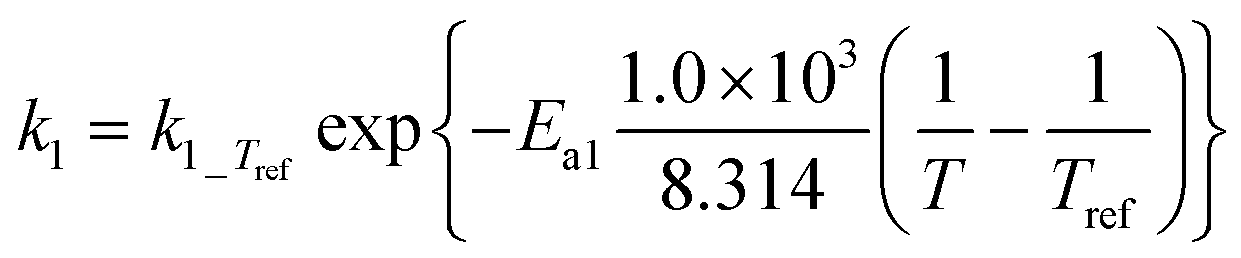
|
|
2)  |
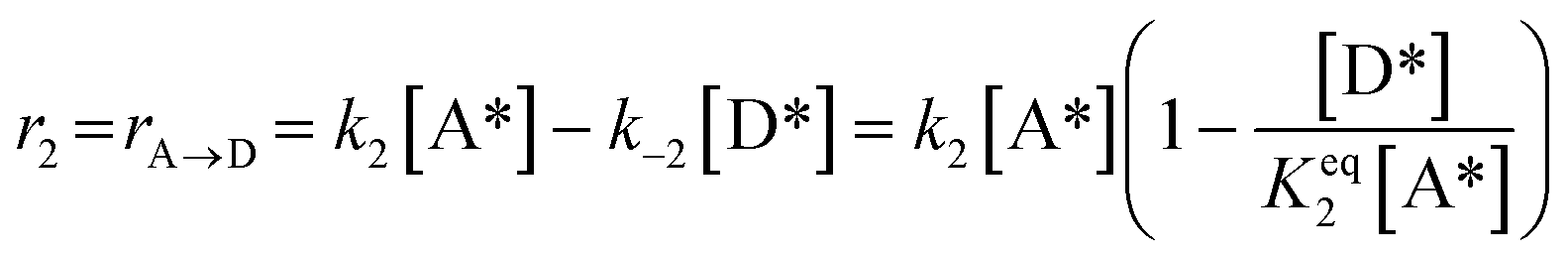
|
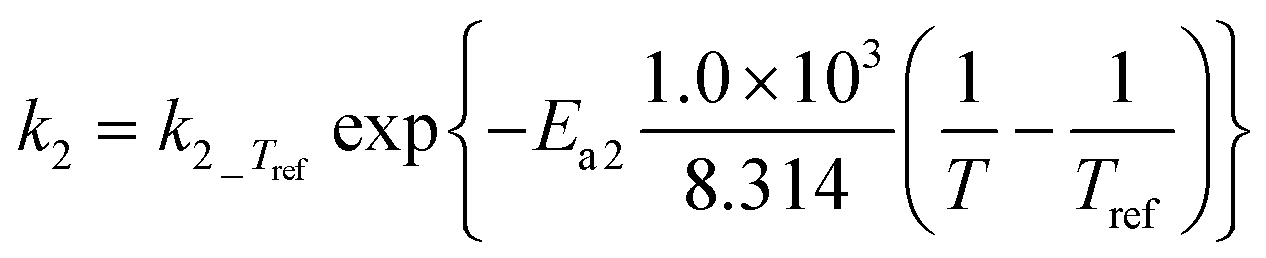
|
|
3)  |
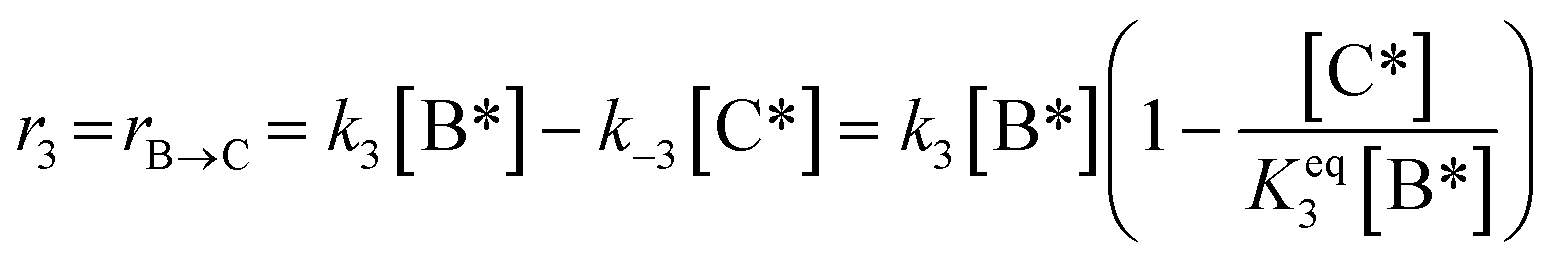
|
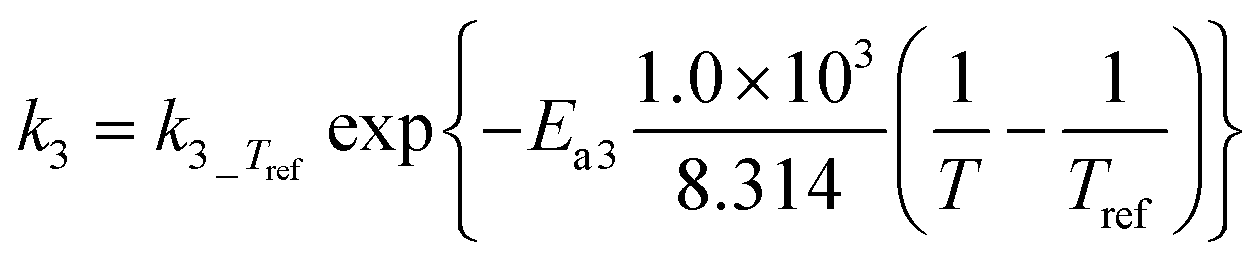
|
|
| Species balance | [*] + [A*] + [B*] + [D*] + [C*] = [*tot] |
| Thermodynamic equilibria |
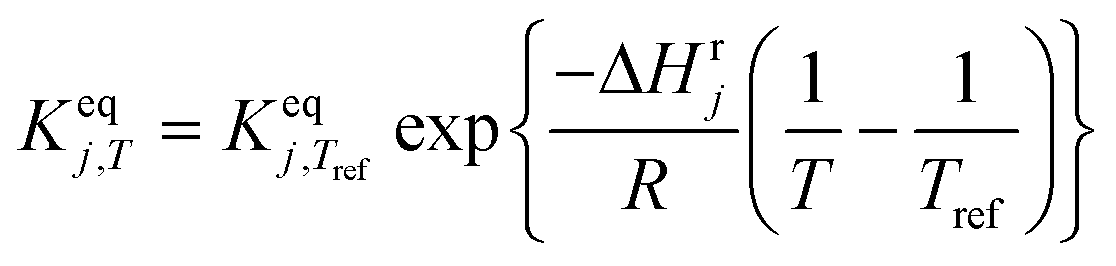
|
| Parameter | Prediction | 95% confidence range | Units |
|---|---|---|---|
a
T
ref = reference temperature.
b Equilibrium constant fixed at infinity by neglecting the backward reaction since it is too large to allow a statistically independent estimation. An analysis of the final fit showed that this corresponds with 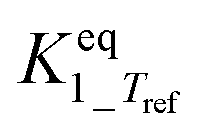 ≥ 10 and also ≥ 10 and also 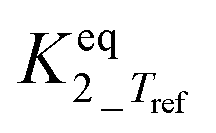 ≥ 10. ≥ 10.
|
|||
| k 1_Tref | 0.0447 | ±0.00004 | s−1 |
| k 2_Tref | 0.002939 | ±0.000001 | s−1 |
| k 3_Tref | 0.0319 | ±0.0025 | s−1 |
| E a1 | 176.76 | ±0.08 | kJ mol−1 |
| E a2 | 206.5 | ±4.6 | kJ mol−1 |
| E a3 | 187.0 | ±11.3 | kJ mol−1 |
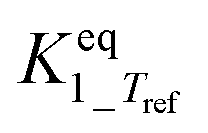
|
Infinityb | — | — |
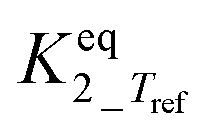
|
Infinityb | — | — |
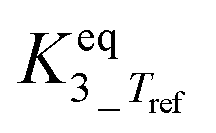
|
5.84 | ±1.85 | — |
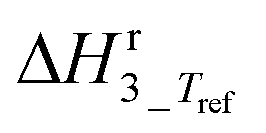
|
31 | ±48 | kJ mol−1 |
The main reaction pathway consists of a first dehydration step of sorbitol (A) into a mixture (B) of 1,4-anhydrosorbitol and 3,6-anhydrosorbitol (r1) followed by a second dehydration step into 1,4:3,6-dianhydrosorbitol, called isosorbide (C) (r3). The second sorbitol dehydration step requires a slightly higher activation energy (187.0 ± 11.3 kJ mol−1) than that of the first step of sorbitol dehydration (176.76 ± 0.08 kJ mol−1). Additionally, small amounts of two other monoanhydrohexitols were produced: 1,5-anhydrosorbitol and 2,5-anhydromannitol (D), with a relatively high activation barrier of 206.5 ± 4.6 kJ mol−1. Because they do not react further to a dianhydrosorbitol, they are referred to as “stable intermediates”. The second step seems slightly endothermic, but it should be noted that the 95% confidence range is larger than the absolute value of the reaction enthalpy.
Methods
The Spartan'10 software package10 was used for all calculations. Starting structures for the flexible sorbitol and anhydrosorbitol isomers were obtained using the Conformer Distribution option combined with a molecular mechanics calculation with the (Merck) Molecular Mechanics Force Field 94 (MMFF). Using this option, a series containing the first 100 sorbitol conformers with the lowest energy was kept out of an initial series of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The underlying assumption is that the molten salt medium does not behave like an aqueous solvent. Next, all MMFF structures, within a range of 10 kJ mol−1 of strain energy compared to the lowest one and with a geometry suitable for the desired reaction, were optimized on the DFT B3LYP/6-31G* level. Zn-complexes without sugar ligands were optimised on the B3LYP/6-31G* level starting from the corresponding MMFF-structures. Zn-complexes with sugar ligands were optimised on the B3LYP/6-31G* level starting from a manual ensemble of the B3LYP/6-31G* optimised Zn-complex and the optimised sugar.
000. The underlying assumption is that the molten salt medium does not behave like an aqueous solvent. Next, all MMFF structures, within a range of 10 kJ mol−1 of strain energy compared to the lowest one and with a geometry suitable for the desired reaction, were optimized on the DFT B3LYP/6-31G* level. Zn-complexes without sugar ligands were optimised on the B3LYP/6-31G* level starting from the corresponding MMFF-structures. Zn-complexes with sugar ligands were optimised on the B3LYP/6-31G* level starting from a manual ensemble of the B3LYP/6-31G* optimised Zn-complex and the optimised sugar.
Furthermore, transition states were optimised similarly on the B3LYP/6-31G* level. They were characterised using the unique imaginary vibration frequency and internal reaction coordinate (IRC) procedure.
All energy comparisons and calculations are based on total energies, corrected with zero-point energies (ZPE) of the relevant species.
The use of B3LYP/6-31G* is justified by the fact that it is in the most validated DFT-code at the lowest computational cost; as for structure and energy calculations, the errors are very similar to those obtained with codes like MP2/6-31G*.11 Finally, in several figures, electron isodensity maps (0.08 e au−3) are listed, with the electrostatic potential projected on its surface to provide insight into the nature of the chemical bonds and electrostatic interactions involved.
Results
ZnCl2 molten salt hydrate medium
As the experimental system is rather unusual, it is worthwhile to pay attention to its composition and structure. ZnCl2 hydrates exist in several different forms, including ZnCl2·3H2O and ZnCl2·4H2O. In this study, at the start of the reaction, the experimental system consists of 600 g of 70% ZnCl2 in water medium with 50 g of sorbitol. Thus, the molar ratio of ZnCl2:H2O is 1:3.17 and the molar ratio of ZnCl2:sorbitol is 11.48:1.
During the course of the reaction, two molecules of water per sorbitol molecule are produced (based on 100% sorbitol conversion with 100% selectivity to isosorbide), thus leading to a final molar ratio of ZnCl2:H2O ~ 1:3.34. Thus, the system should not be considered as an aqueous solution, as Zn will take up to 6 H2O molecules in its coordination sphere.
ZnCl2·3H2O and ZnCl2·4H2O seem to be obvious starting points for the calculations. Fig. 2 gives an overview of the two series of structures considered, with relevant angles, distances, and total B3LYP energy values. Table 3 lists the geometric data of these structures together with the total energies.
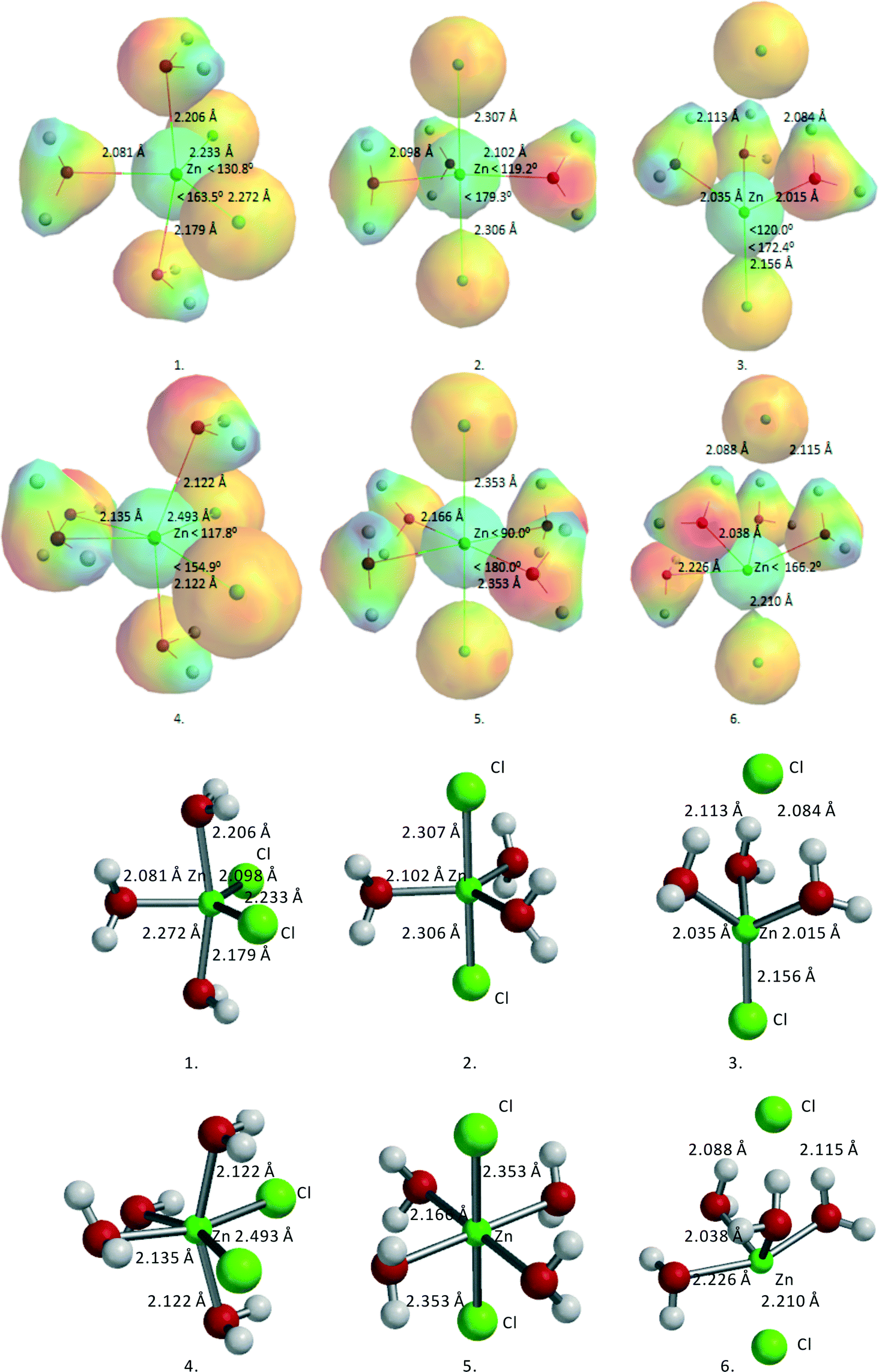 | ||
| Fig. 2 Overview of different ZnCl2·nH2O structures (n = 3, 4). For the convenience of the reader, ball-and-stick models are added. | ||
| Structures | Symmetry | E-total (kJ mol−1) | Zn–On (Å) | Zn–Cln (Å) | ∠Cl–Zn–Cl (degree) |
|---|---|---|---|---|---|
| 1. ZnCl2·3H2O cis | tbp | −7689937.7 |
2.080 | 2.233 | 130.8 |
| 2.179 | 2.272 | ||||
| 2.205 | |||||
| 2. ZnCl2·3H2O trans | tbp | −7689925.5 |
2.095 | 2.303 | 179.4 |
| 2.100 | 2.311 | ||||
| 2.100 | |||||
| 3. ZnCl2·3H2O, 1 Cl out | thd | −7689939.3 |
2.015 | 2.156 | 172.4 |
| 2.017 | 3.489 | ||||
| 2.035 | |||||
| 4. ZnCl2·4H2O cis | oh | −7890601.7 |
2.122 | 2.493 | 117.8 |
| 2.122 | 2.493 | ||||
| 2.135 | |||||
| 2.135 | |||||
| 5. ZnCl2·4H2O trans | oh | −7890589.6 |
2.166 | 2.353 | 180.0 |
| 2.166 | 2.353 | ||||
| 2.166 | |||||
| 2.166 | |||||
| 6. ZnCl2·4H2O, 1 Cl out | tbp | −7890621.3 |
2.038 | 2.210 | 166.2 |
| 2.088 | 3.543 | ||||
| 2.088 | |||||
| 2.226 |
For ZnCl2·3H2O, three structures were considered, two with trigonal bipyramidal (tbp) geometry and one with tetrahedral (thd) geometry. The trans-tbp structure 2, with two Cl's in apical position, turned out to be the highest in energy. The cis-tbp structure 1, with two Cl's in equatorial position, was found to be ~12 kJ mol−1 lower in energy than the trans-tbp structure 2, while the tetrahedral structure 3, with one Cl out of direct coordination to Zn but with three hydrogen bridges to the coordinating water molecules, is ~2 kJ mol−1 lower in energy than the cis-tbp structure 1. For ZnCl2·4H2O, it was found that the cis-structure 4 is ~12 kJ mol−1 lower in energy than the corresponding trans-structure 5, similar to ZnCl2·3H2O. Finally, the ZnCl2·4H2O tbp structure 6, with one Cl out of direct coordination to Zn but with three hydrogen bridges to the coordinating water molecules, is even ~20 kJ mol−1 lower in energy than the cis-structure 4.
Thus, the different Zn complexes turn out to be very flexible, showing a great coordination variety around Zn, ranging from tetrahedral (thd) to octahedral (oh). Hydrogen bridge formation of coordinating water molecules with Cl− also plays a very important role in determining the overall stability. This observation is important for understanding the interactions with the different sugar moieties as they have multiple possibilities to form hydrogen bridges via their various alcohol functions.
The series have been extended from ZnCl2·0H2O to ZnCl2·6H2O to obtain general conclusions on the effect of an increasing amount of water in the ZnCl2·nH2O system. Going from ZnCl2·0H2O to ZnCl2·6H2O, approximately −70 kJ mol−1 is gained for each added H2O molecule in a fairly constant way. The most stable structure of ZnCl2·6H2O is shown in Fig. 3, consistent with the most stable structures for ZnCl2·3H2O and ZnCl2·4H2O.
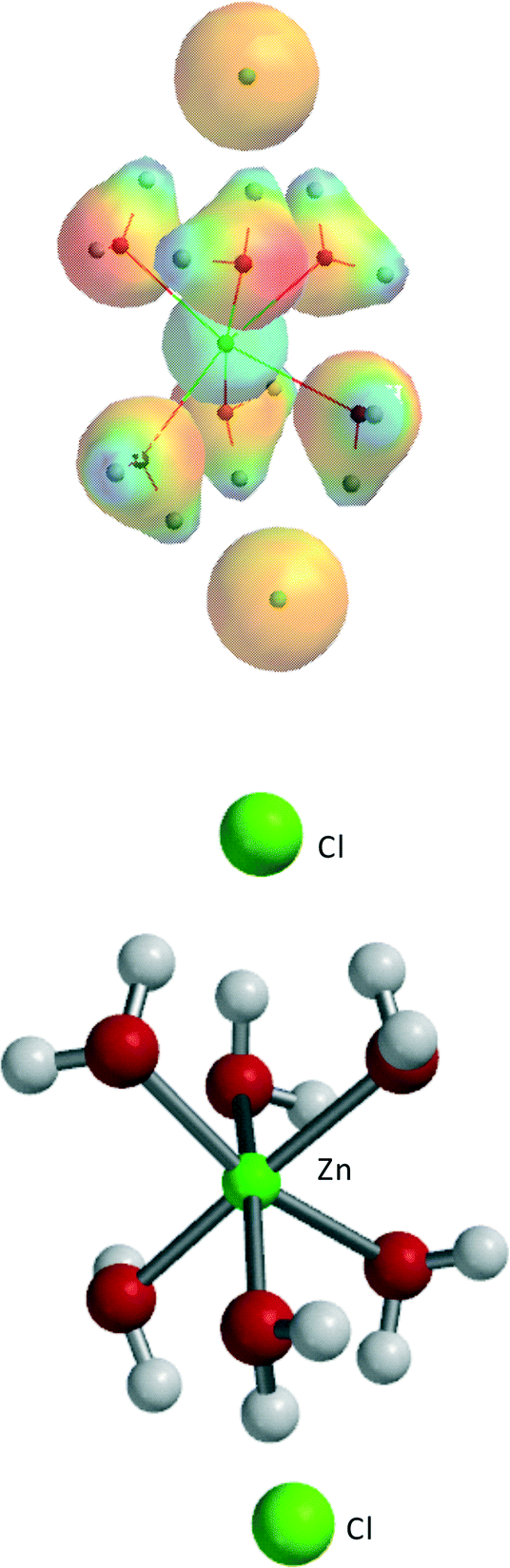 | ||
| Fig. 3 The most stable structure of ZnCl2·6H2O. | ||
ZnCl2(H2O)2–sorbitol complexes
It is presumed that prior to reaction, ZnCl2·nH2O–sorbitol complexes will be formed. The most stable sorbitol (stereochemistry, 2S,3R,4R,5R,D-glucitol) structure, obtained from a conformer distribution calculation, is shown in Fig. 4.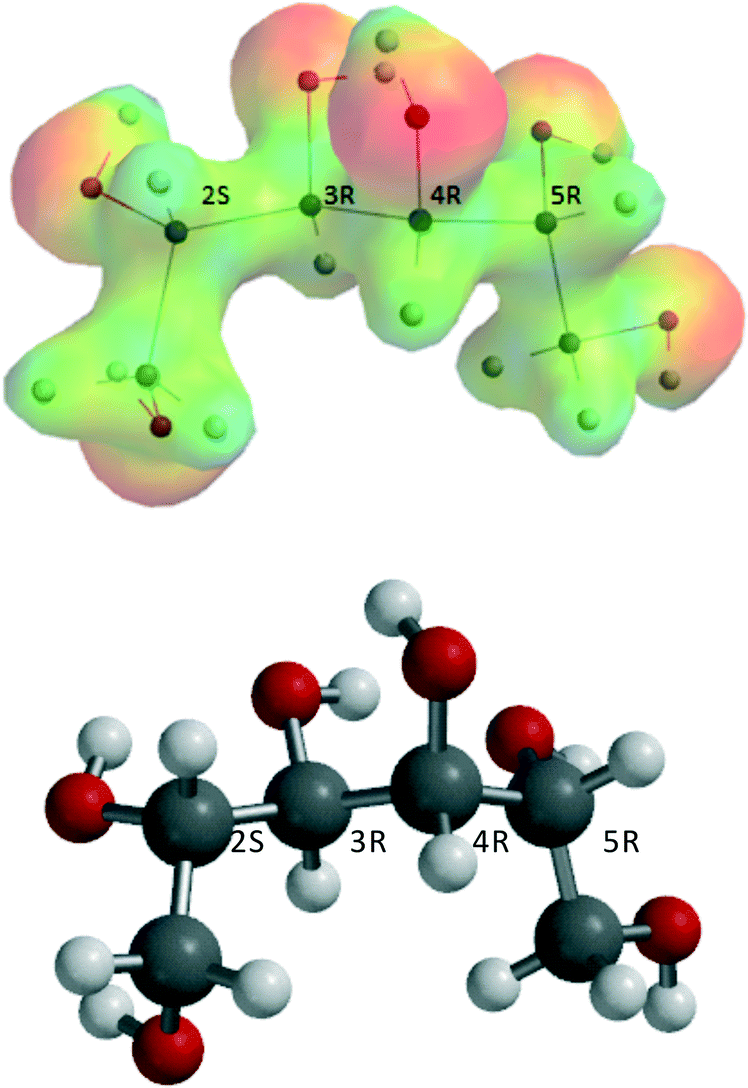 | ||
| Fig. 4 Sorbitol structure. | ||
Two types of complexes can be distinguished:
a) complexes wherein a terminal primary alcohol function is involved, and
b) complexes wherein only internal secondary alcohol functions are involved.
Though complexation of sorbitol with more than one Zn-center is likely to occur, the minimum requirement is complexation with just one Zn-center. Theoretically, complexation of sorbitol up to six Zn-centers might be possible. Most of these complexes, however, do not lead to the chemical reaction investigated and, therefore, were not considered. Starting from ZnCl2·3H2O, it was assumed that sorbitol will act as a bidentate ligand with two vicinal –OH groups coordinated and that one molecule of water will be released from the parent complex to yield in principle a six-coordinated Zn-complex.
The released water is assumed to be taken up by another ZnCl2·3H2O to yield ZnCl2·4H2O, anticipating the loss of H2O from sorbitol and anhydrosorbitols in the individual reaction steps. Thus, a consistent thermodynamic system for the dehydration was developed, while the calculation of the activation barriers of the different reaction steps became straightforward. Five complexes were considered, i.e., ZnCl2(H2O)2–1,2-dihydroxy-sorbitol, ZnCl2(H2O)2–2,3-dihydroxy-sorbitol, ZnCl2(H2O)2–3,4-dihydroxy-sorbitol, ZnCl2(H2O)2–4,5-dihydroxy-sorbitol, and ZnCl2(H2O)2–5,6-dihydroxy-sorbitol. Table 4 lists the relative energies and amounts of these five complexes.
| ZnCl2(H2O)2–sorbitol complexes | Relative energy (kJ mol−1) | Boltzmann partition (%, at 298 K) |
|---|---|---|
| a Not considered in the Boltzmann partition, see the discussion below. | ||
| ZnCl2(H2O)2–1,2-dihydroxy-sorbitol | 0.0 | 56.1 |
| ZnCl2(H2O)2–2,3-dihydroxy-sorbitol | 6.9 | 3.5 |
| ZnCl2(H2O)2–3,4-dihydroxy-sorbitol | −19.7a | — |
| ZnCl2(H2O)2–4,5-dihydroxy-sorbitol | 7.7 | 2.5 |
| ZnCl2(H2O)2–5,6-dihydroxy-sorbitol | 1.0 | 37.9 |
Fig. 5 shows a typical structure example, ZnCl2·(H2O)2–1,2-dihydroxy-sorbitol.
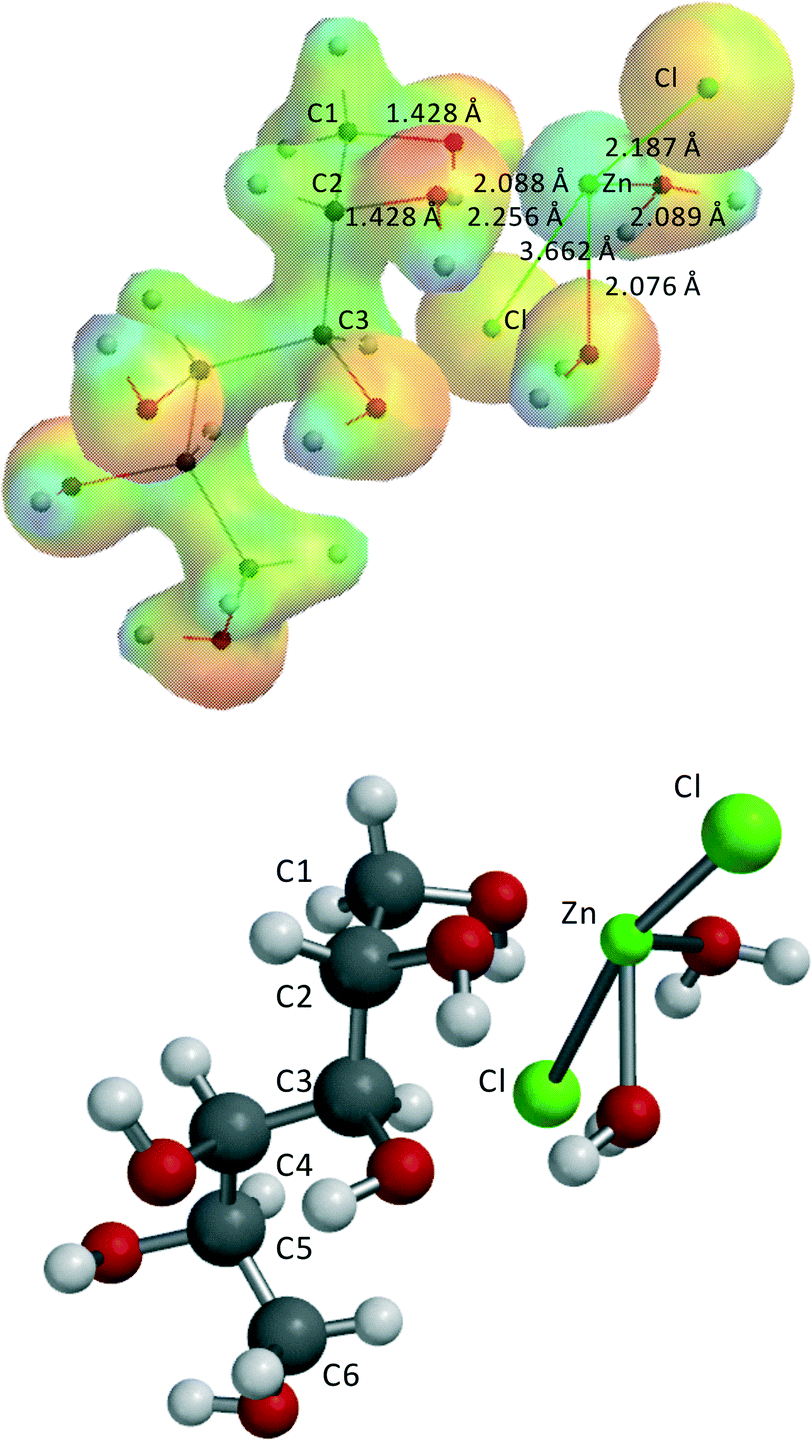 | ||
| Fig. 5 ZnCl2(H2O)2–1,2-dihydroxy-sorbitol. This complex can be described as a distorted square pyramid: Zn–(H2O)1 = 2.076 Å, Zn–(H2O)2 = 2.089 Å, Zn–(OH)1 = 2.088 Å, Zn–(OH)2 = 2.256 Å, Zn–Cl1 = 2.187 Å, Zn–Cl2 = 3.662 Å. The second Cl− is not directly coordinated to Zn(II) but forms three hydrogen bridges with the two H2O-ligands and C2–OH of sorbitol, with lengths of 2.055, 2.146, and 2.191 Å. | ||
Similar to ZnCl2·3H2O and ZnCl2·4H2O in the ZnCl2(H2O)2–1,2-dihydroxy-sorbitol complex, as well as in the other complexes, the second Cl− is not directly coordinated to Zn(II) but rather forms hydrogen bridges, in this case with two H2O-ligands and one –OH group of sorbitol. No additional internal hydrogen bridges were allowed as this will harm the comparison of the relative stabilities of the 5 complexes. The results of these complexes indicate that the two ZnCl2(H2O)n–sorbitol complexes which have a primary alcohol coordinated to Zn(II), i.e., ZnCl2(H2O)2–1,2-dihydroxy-sorbitol and ZnCl2(H2O)2–5,6-dihydroxy-sorbitol, are slightly more stable and, as a consequence, more abundant than the two complexes with only secondary alcohol groups coordinated to Zn(II), i.e., ZnCl2(H2O)2–2,3-dihydroxy-sorbitol and ZnCl2(H2O)2–4,5-dihydroxy-sorbitol. The ZnCl2(H2O)2–3,4-dihydroxy-sorbitol complex with two secondary alcohol groups coordinated to Zn(II) shows the lowest energy: −19.7 kJ mol−1. However, this is not due to any kind of strong coordinative bonding to Zn(II), but due to the presence of additional H bridges in the complex, which cannot be avoided in a computationally correct manner. Therefore, this ZnCl2(H2O)2–3,4-dihydroxy-sorbitol complex is not further considered in the calculation of the different energy levels and the reaction mechanism.
It is important to note that the abundant ZnCl2(H2O)2–1,2-dihydroxy-sorbitol and ZnCl2(H2O)2–5,6-dihydroxy-sorbitol complexes are required for the formation of 1,4- and 3,6-anhydrosorbitol, respectively, via a pathway involving a secondary position (C4–, C3–) alcohol attack on the primary position carbon (C1, C6). The ZnCl2(H2O)2–2,3-dihydroxy-sorbitol and ZnCl2(H2O)2–4,5-dihydroxy-sorbitol complexes are needed for a pathway involving a primary position alcohol attack on a secondary position carbon. As coordination of sorbitol to Zn(II) might not be limited to 1/1 complexes, it also indicates that decomplexation of secondary alcohol complexes of sorbitol to Zn(II) is thermodynamically favored over primary alcohol complexes of sorbitol to Zn(II).
Transition state of ZnCl2(H2O)2–sorbitol/anhydrosorbitol complexes
Fig. 6 shows the transition state for the ZnCl2(H2O)n-catalyzed reaction of sorbitol to 1,4-anhydrosorbitol. As stated above for this specific reaction, the complex ZnCl2(H2O)2–1,2-dihydroxy-sorbitol is the reactant.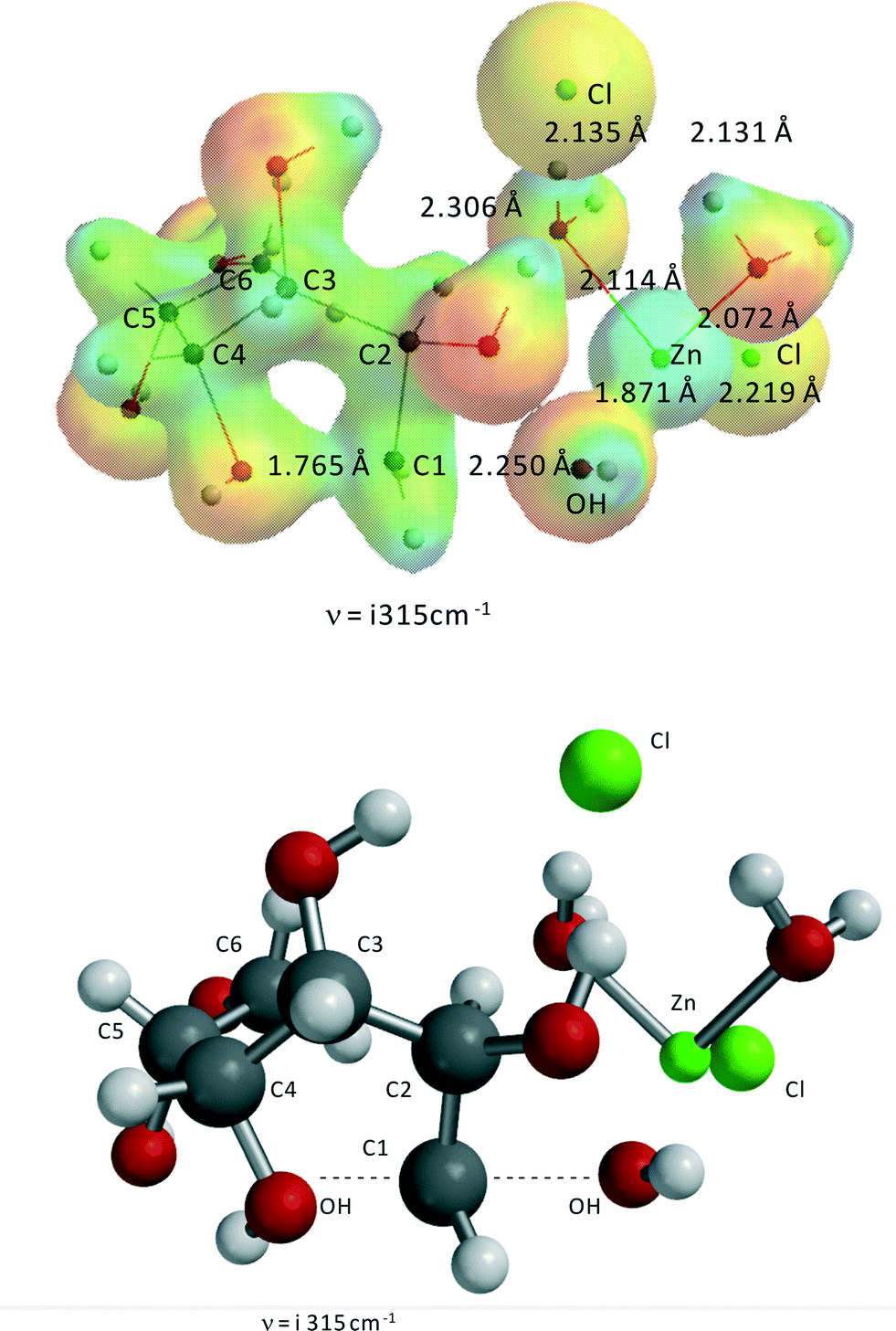 | ||
| Fig. 6 Transition state of Zn(II)Cl2(H2O)2-catalyzed formation of 1,4-anhydrosorbitol from sorbitol. ν = i315 cm−1, C4O–C1H2 = 1.765 Å, C1H2–OH = 2.250 Å, Zn–(H2O)1 = 2.072 Å, Zn–(H2O)2 = 2.114 Å, Zn–(OH)1 = 1.871 Å, Zn–(OH)2 = 3.673 Å, Zn–Cl1 = 2.219 Å, Zn–Cl2 = 3.892 Å. The second Cl− is not directly coordinated to Zn(II) but forms three hydrogen bridges with the two H2O-ligands and OH2 of sorbitol, with lengths of 2.131, 2.135, and 2.060 Å. The OH-group at C2 is also no longer coordinated to Zn(II). | ||
Clearly, the reaction can be described as an internal SN2-reaction with a late transition state. Bond formation of the C4–OH and C1 is almost complete, and the OH− leaving group (originally at C1) is close to Zn(II). The activation barrier is 177.1 kJ mol−1, almost identical to the experimental value from kinetic modeling, i.e., 176.8 kJ mol−1 (Table 2).
The alternative transition state with primary alcohol (C1–OH) attack on C4 (ZnCl2(H2O)2–4,5-dihydroxy sorbitol complex) to form 1,4-anhydrosorbitol shows an activation barrier of ~207 kJ mol−1, significantly deviating from the experimental value; thus, it is concluded that this is not a viable alternative reaction step.
The transition state for the ZnCl2(H2O)n-catalyzed reaction of sorbitol to 3,6-anhydrosorbitol is very similar to the one leading to 1,4-anhydrosorbitol. In this case, the ZnCl2(H2O)2–5,6-dihydroxy-sorbitol complex is the reactant. It shows an activation barrier of 175.5 kJ mol−1, which is again close to the experimentally derived value of 176.8 kJ mol−1 (see Table 2.) Fig. 7 shows the corresponding transition state.
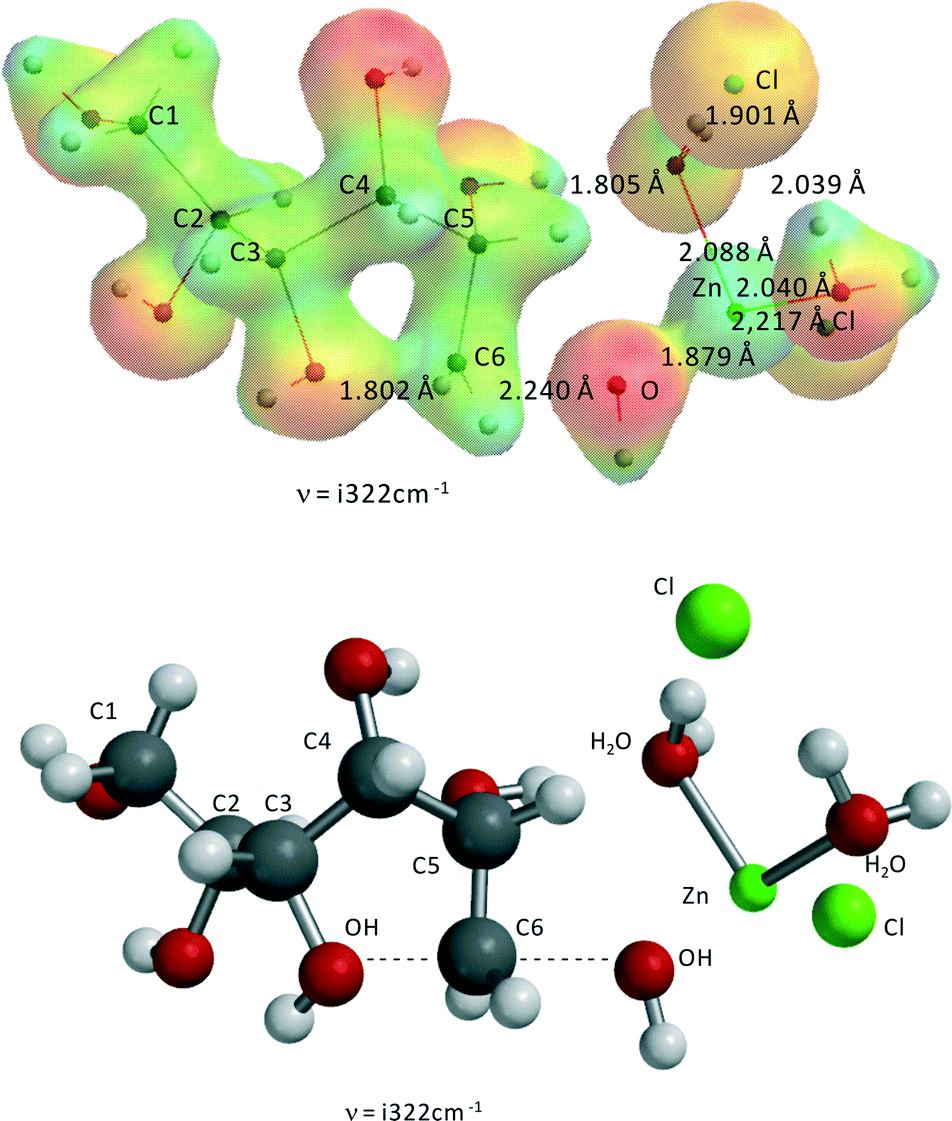 | ||
| Fig. 7 Transition state of Zn(II)Cl2(H2O)2-catalyzed formation of 3,6-anhydrosorbitol from sorbitol. ν = i322 cm−1, C3O–C6H2 = 1.802 Å, C6H2–OH = 2.240 Å, Zn–(H2O)1 = 2.040 Å, Zn–(H2O)2 = 2.088 Å, Zn–(OH)1 = 1.879 Å, Zn–(OH)2 = 3.839 Å, Zn–Cl1 = 2.217 Å, Zn–Cl2 = 3.752 Å. The second Cl− is not directly coordinated to Zn(II) but forms two hydrogen bridges with the two H2O-ligands, with lengths of 1.901 and 2.039 Å. The OH-group at C5 is also no longer coordinated to Zn(II). | ||
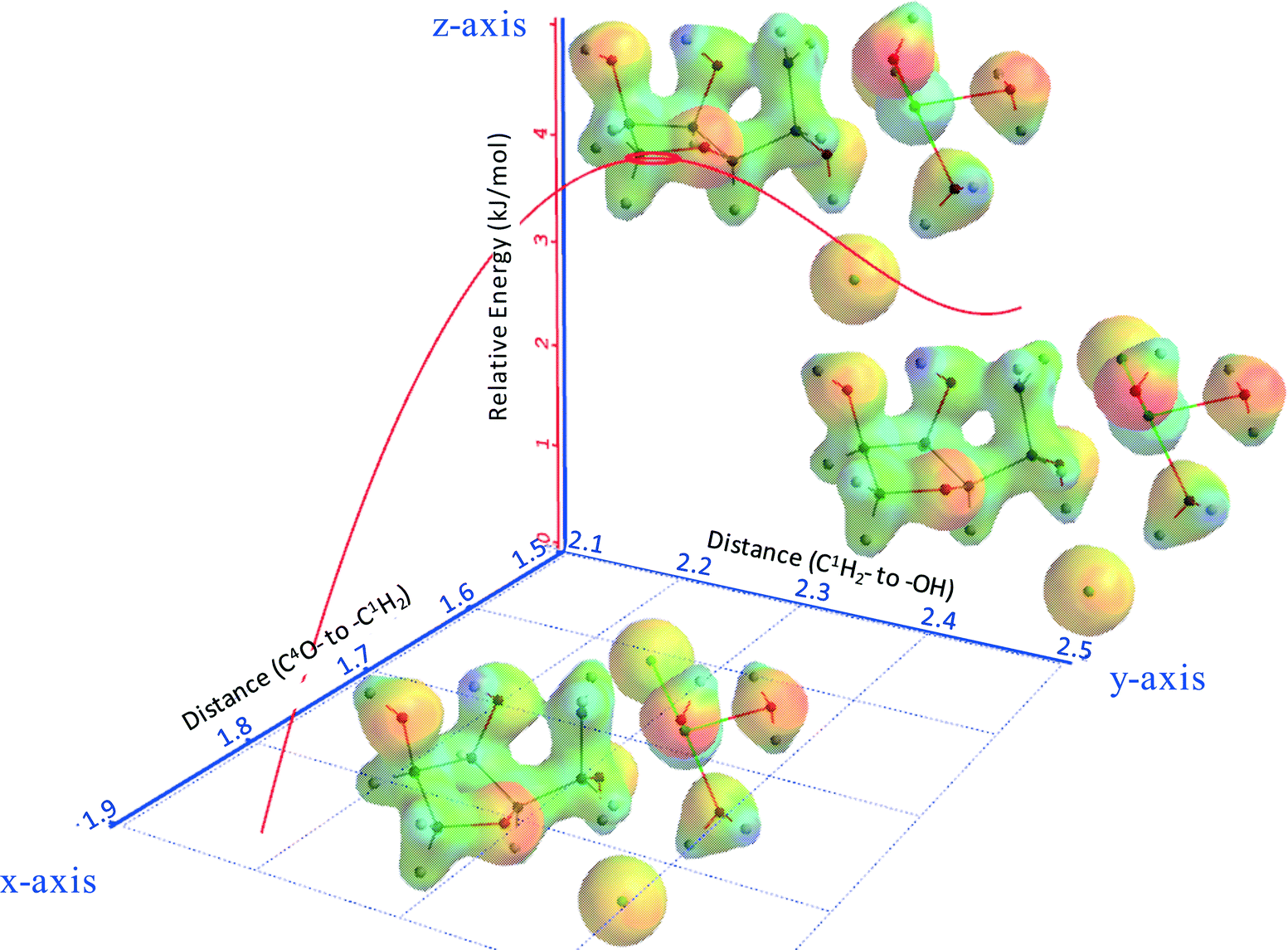
Fig. 8 shows part of the internal reaction coordinate (IRC) of 3,6-anhydrosorbitol formation from sorbitol, wherein the relative energy (z-axis) was plotted against the distance of C3O– to –C6H2 (x-axis) and the distance of C6H2– to –OH (y-axis). The x-axis represents the distance of the attacking secondary alcohol to C6, and the y-axis represents the distance of C6 to the primary alcohol leaving group. The IRC thus fully supports the view of an intramolecular SN2 reaction, as the change in the x-direction is paralleled by the change in the y-direction. Because this sequence ends with a formal ion pair consisting of 3,6-anhydrosorbitol-OH+ and ZnCl2(H2O)2(OH)−, of course, the overall reaction enthalpy is not in agreement with Fig. 13 (see below).
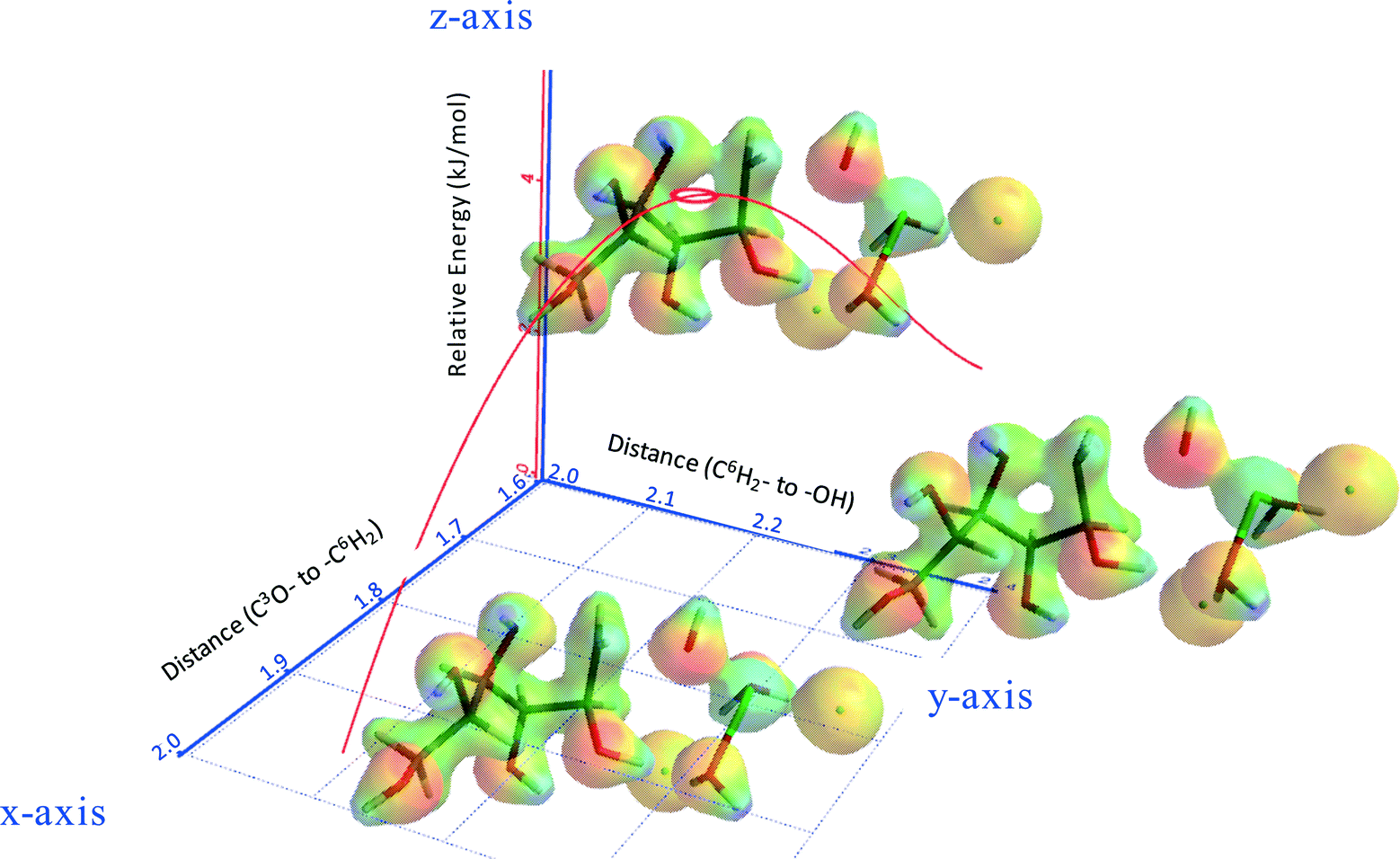 | ||
| Fig. 8 Internal reaction coordinate (IRC) of 3,6-anhydrosorbitol formation from sorbitol; x-axis: the distance of C3O– to –C6H2; y-axis: the distance of C6H2– to –OH; and z-axis: relative energy (kJ mol−1). | ||
However, it is expected that final proton transfer to an OH− group in this polar system is an almost barrier-free process.
Both 1,4-anhydrosorbitol and 3,6-anhydrosorbitol can undergo a second ring closure to isosorbide. Fig. 9 shows the transition state for the second ring closure of 1,4-anhydrosorbitol resulting in isosorbide. The activation barrier is 191.9 kJ mol−1, which is approximately 5 kJ mol−1 above the experimentally derived value and thus within the error margin of the kinetic modeling study. Fig. 10 shows the transition state for the ring closure and dehydration of 3,6-anhydrosorbitol to isosorbide. The activation barrier is 194.1 kJ mol−1, which is approximately 7 kJ mol−1 above the experimentally derived value and thus also within the error margin of the kinetic model.
 | ||
| Fig. 9 Transition state of Zn(II)Cl2(H2O)2-catalysed formation of isosorbide from 1,4-anhydrosorbitol. ν = i293 cm−1, C3O–C6H2 = 1.732 Å, C6H2–OH = 2.304 Å, Zn–(H2O)1 = 2.069 Å, Zn–(H2O)2 = 2.077 Å, Zn–(OH)1 = 1.882 Å, Zn–Cl1 = 2.205 Å, Zn–Cl2 = 4.280 Å. The second Cl− is not directly coordinated to Zn(II) but forms two hydrogen bridges with the two H2O-ligands, with lengths of 2.038, and 2.105 Å. The OH-group at C5 is also no longer coordinated to Zn(II) but already shows the more favorable endo-interaction with the oxygen of the 1,4-anhydrosorbitol ring. | ||
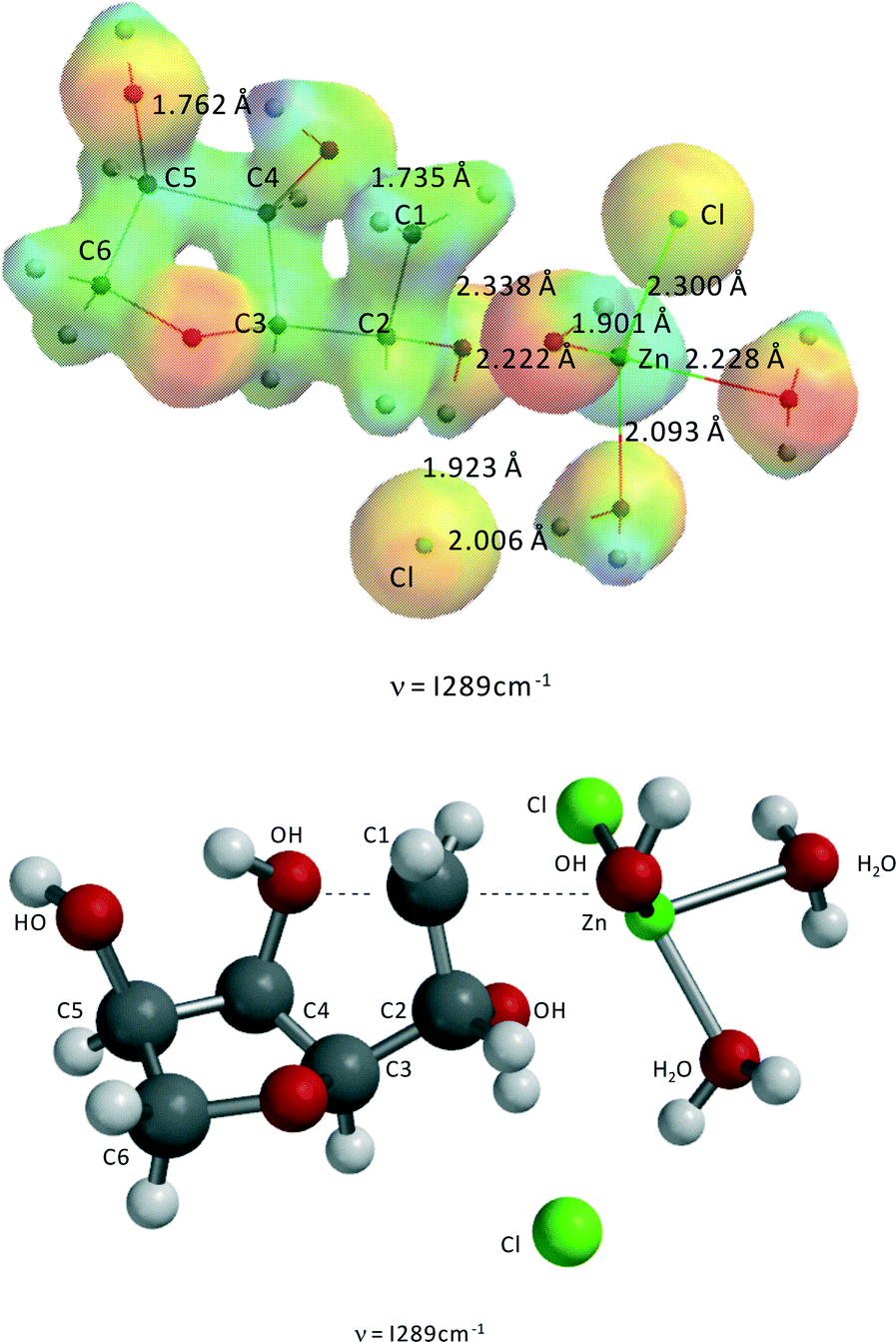 | ||
| Fig. 10 Transition state of Zn(II)Cl2(H2O)2-catalyzed formation of isosorbide from 3,6-anhydrosorbitol. ν = i289 cm−1, C4O–CH21 = 1.735 Å, CH21–OH = 2.338 Å, Zn–(H2O)1 = 2.093 Å, Zn–(H2O)2 = 2.228 Å, Zn–(OH)1 = 1.901 Å, Zn–(OH)2 = 2.222 Å, Zn–Cl1 = 2.300 Å, Zn–Cl2 = 4.139 Å. The second Cl− is not directly coordinated to Zn(II) but forms two hydrogen bridges with a H2O-ligand and a –OH at C2 of sorbitol, with lengths of 2.006 and 1.923 Å, respectively. | ||
Fig. 11 shows part of the IRC of isosorbide formation from 3,6-anhydrosorbitol. The relative energy (z-axis) was plotted against the distance of C4O– to –C1H2 (x-axis) and the distance of C1H2– to –OH (y-axis). The result is fully analogous to that of the first ring closure in the formation of 3,6-anhydrosorbitol. Again, it can be concluded that the reaction pathway is an intramolecular SN2 reaction.
 | ||
| Fig. 11 Internal reaction coordinate (IRC) of isosorbide formation from 3,6-anhydrosorbitol; x-axis: the distance of C4O– to –C1H2; y-axis: the distance of C1H2– to –OH; and z-axis: relative energy (kJ mol−1). | ||
Fig. 12 shows the transition states for the formation of the “stable intermediates” 2,5-anhydromannitol (a) and 1,5-anhydrosorbitol (b). The activation barriers for these two processes are 204.5 kJ mol−1 and 210.8 kJ mol−1, respectively. These values are in good agreement with the experimentally derived value (206.5 kJ mol−1). The slightly higher activation barrier for 1,5-anhydrosorbitol compared to 2,5-anhydromannitol is consistent with the experimental observation that 2,5-anhydromannitol seems to be formed faster than 1,5-anhydrosorbitol. In agreement with these data, the amounts of both byproducts (1,5-anhydrosorbitol and 2,5-anhydromannitol) are low compared to 1,4- and 3,6-anhydrosorbitol.
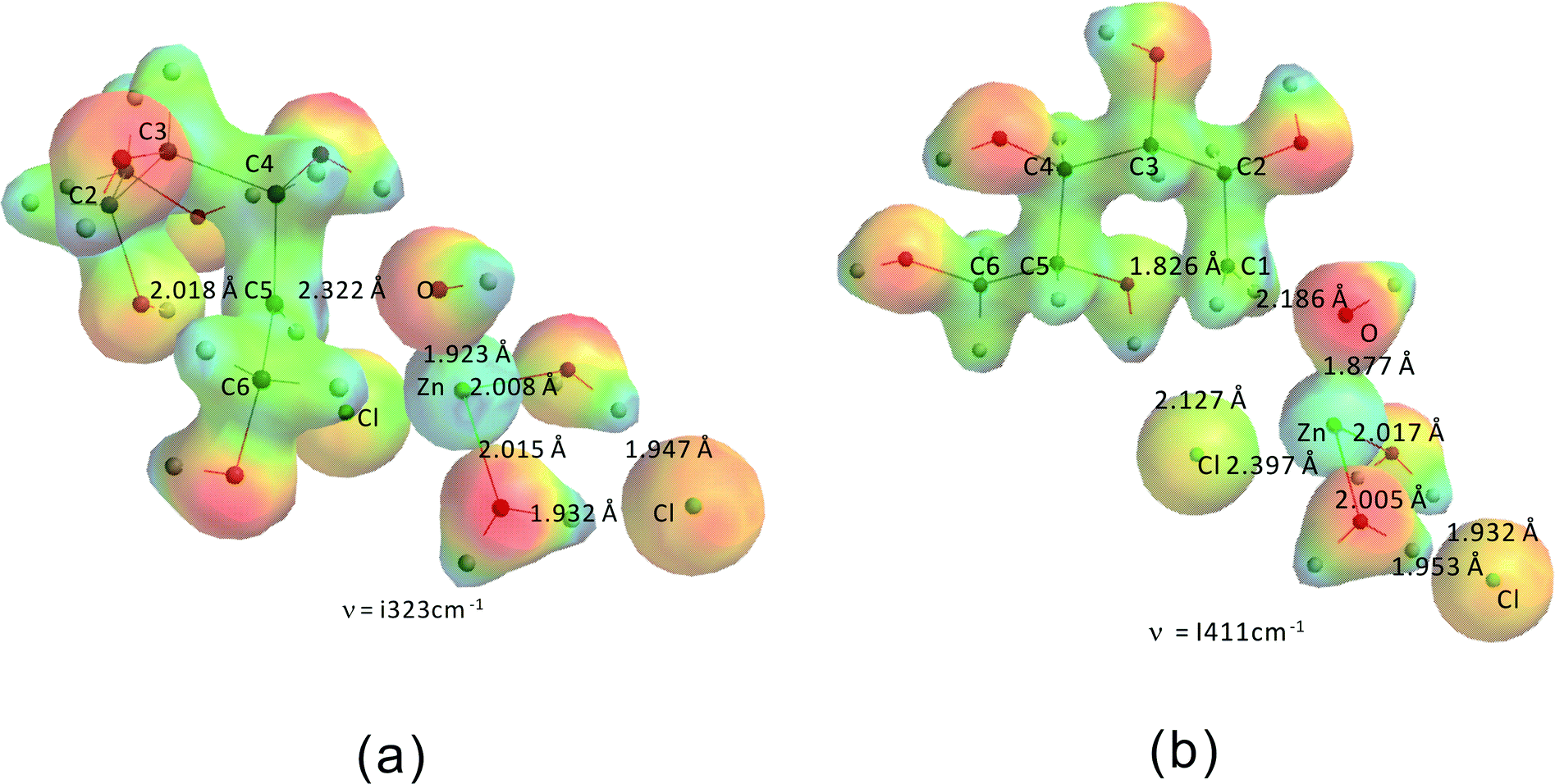 | ||
| Fig. 12 Transition states for the formation of 2,5-anhydromannitol (a) and 1,5-anhydrosorbitol (b). | ||
Energy diagram of the dehydration reactions of sorbitol to isosorbide
The molecular modeling results give good insight in the thermodynamic path of the sorbitol dehydration system. From Fig. 13, it can be seen that all reactions of sorbitol begin with an exothermic complexation to Zn(II). This is in good accordance with experimental experience that the molten salt generates heat upon adding water. Included in the modeling, the exothermic complexation of sorbitol and anhydrosorbitols to Zn(II) is essential to arrive at quantitatively correct activation barriers consistent with the results of the kinetic study. Furthermore, the intermediate 3,6-anhydrosorbitol seems slightly favored over 1,4-anhydrosorbitol. Table 5 shows the calculated and experimental activation barriers.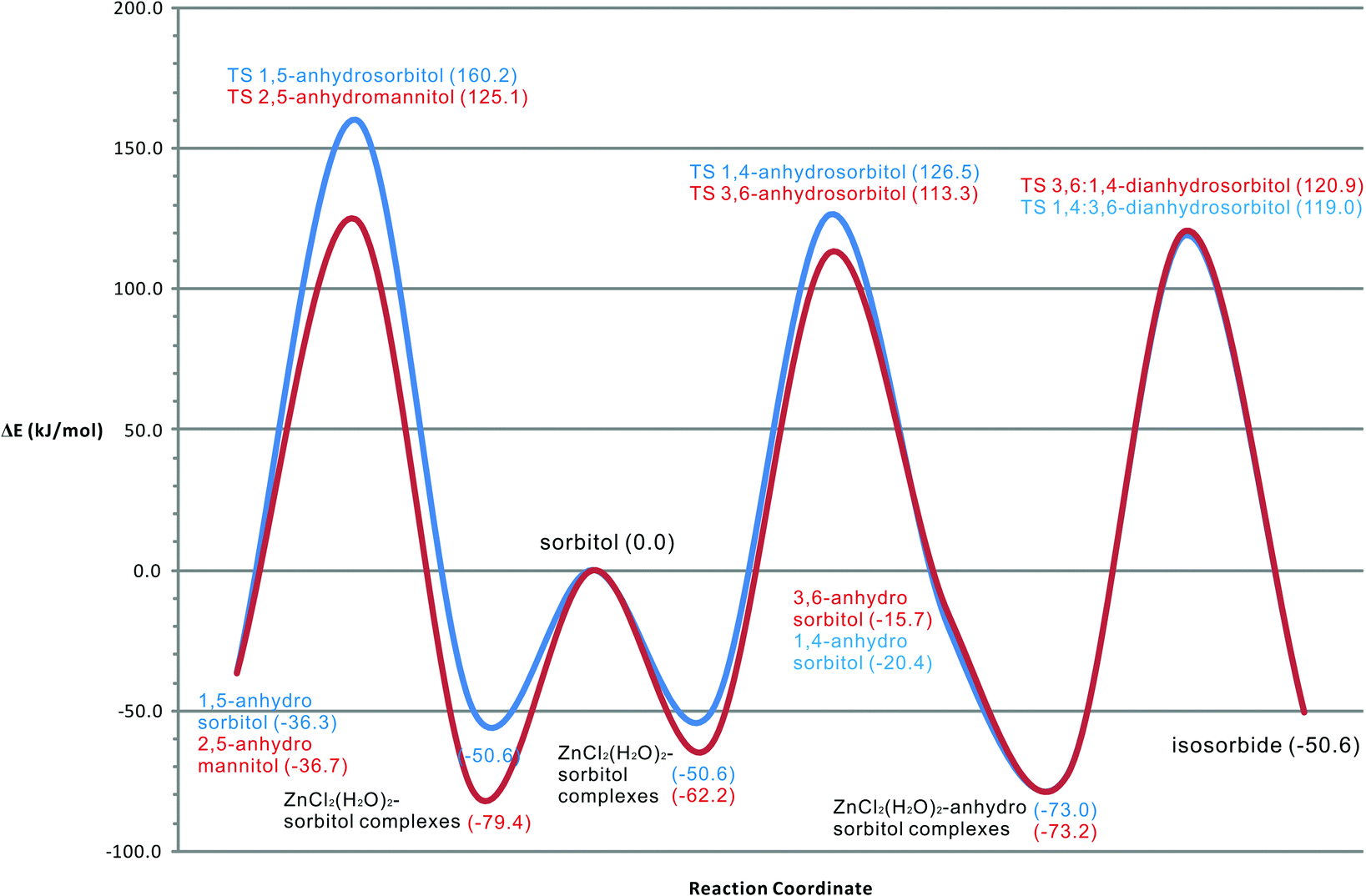 | ||
| Fig. 13 Reaction energy diagram of sorbitol dehydration reactions in ZnCl2·xH2O. | ||
| Ring closure to: | E a-calc. (kJ mol−1) | Ring closure to: | E a-calc. (kJ mol−1) | Ring closure to: | E a-exp. (kJ mol−1) |
|---|---|---|---|---|---|
| 1,4-Anhydrosorbitol | 177.1 | 3,6-Anhydrosorbitol | 175.5 | 1,4-Anhydrosorbitol | 176.8 |
| 3,6-Anhydrosorbitol | |||||
| Isosorbide (1,4:3,6-) | 191.9 | Isosorbide (3,6:1,4-) | 194.1 | Isosorbide | 187.0 |
| 2,5-Anhydromannitol | 204.5 | 1,5-Anhydrosorbitol | 210.8 | 2,5-Anhydromannitol | 206.5 |
| 1,5-Anhydrosorbitol |
Compared to 1,4-anhydrosorbitol, the activation barrier for the formation of 3,6-anhydrosorbitol is somewhat lower, but the difference is small and perhaps not significant. These results coincide nicely with the experimental results and with kinetic modeling, but the differences might be too small to draw definite conclusions. The difference between the computational (molecular modeling) derived activation barriers and the one from kinetic modeling for the second dehydration step leading to isosorbide is somewhat larger; however, it is still within the kinetic modeling error margin (±11 kJ mol−1). It might well be a simple consequence of the kinetic model with a second consecutive step, leading inherently to a larger error margin in that step. The activation barriers for the two known byproducts, 1,5-anhydrosorbitol and 2,5-anhydromannitol, are in agreement with experimental data, showing that the latter is formed to a larger extent. All dehydration steps are exothermic. The formation of the intermediates 1,4- and 3,6-anhydrosorbitol is only slightly exothermic, the formation of 1,5-anhydrosorbitol and 2,5-anhydromannitol is more exothermic and the formation of isosorbide is the most exothermic process.
Catalysis by HCl–H2O
To assess the potential contribution of catalysis by HCl–H2O that might be present in the ZnCl2(H2O)n (n = 3, 4) molten salt system, HCl–H2O in the absence of ZnCl2 catalysis was modeled as well. Fig. 14 shows the transition state for the formation of 1,4-anhydrosorbitol in HCl–H2O.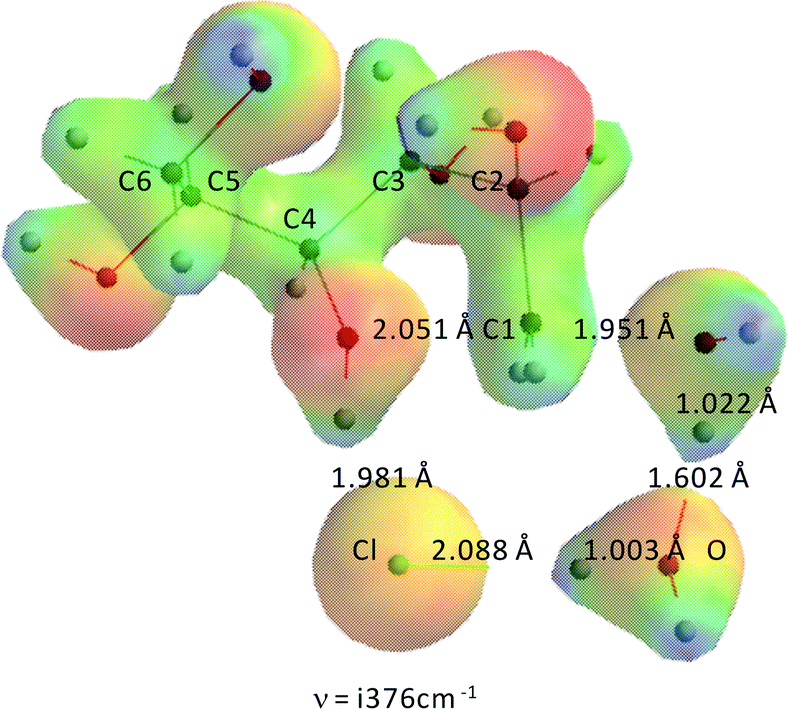 | ||
| Fig. 14 Transition state of formation of 1,4-anhydrosorbitol from sorbitol, catalyzed by HCl–H2O. ν = i376 cm−1, C4O–CH21 = 2.051 Å, CH21–OH = 1.951 Å, C4OH–Cl = 1.981 Å, Cl–H2O = 2.088 Å, H–O (in H2O) = 1.003 Å, H2O–H = 1.602 Å. | ||
As can be seen from Fig. 14, and it might have been expected from experimental chemistry, the proton of HCl has moved entirely to the original H2O molecule, which in turn has transferred the proton almost completely to the basic OH− leaving group. This process can be described as a solvent assisted/acid catalysed SN2-reaction with an earlier transition state as in the case of Zn(II) catalysis. The activation barriers are 59.7 kJ mol−1 and 53.6 kJ mol−1 for the system wherein the positions of HCl and water have been exchanged, both quite similar to the value of concentrated sulfuric acid in the water-catalyzed sorbitol dehydration system,15 and very far from our experimental values for the ZnCl2·xH2O system (due to the complexation between sorbitol and ZnCl2·xH2O).
Discussion
Although the ZnCl2(H2O)2–3,4-dihydroxy-sorbitol complex is the strongest complex in our calculation due to the additional hydrogen bridges, we do not consider this complex in the discussion. An important question is, however, how the ZnCl2(H2O)n (n = 3, 4) molten salt hydrate catalytic system relates to other systems described in the literature for sorbitol dehydration reactions. From the results, it seems clear that the chosen molecular modeling approach leads to a good explanation for the experimentally observed activation energies. The largest deviation is noted for the ring closure and dehydration of the anhydrosorbitol intermediates to isosorbide, although still within the error margin (±11 kJ mol−1) of the kinetic modeling derived value. The quantitative differences between the activation barrier for sorbitol ring closure to 1,4- and 3,6-anhydrosorbitol (177.1 and 175.5 kJ mol−1), the ring closure of the latter two intermediates to isosorbide (191.9 and 194.1 kJ mol−1), and the ring closure to 2,5-anhydromannitol and 1,5-anhydrosorbitol (204.5 and 210.8 kJ mol−1) nicely resemble the experimental findings. They explain why the desired intermediates (leading to isosorbide) are favored with respect to the usually undesired “stable intermediates”. The differences found between the 1,4- and 3,6-isomers and between the 2,5- and 1,5-isomers cannot be compared with experimental values. It is extremely difficult to measure them with the high level of accuracy needed.Our computational results are in full accordance with the literature published by Dosen-Micovic et al.,6 who reported that ring closure and subsequent elimination of water occur with retention of configuration at C3 and C4 under protic acidic conditions despite the clear differences between the two systems. Furthermore, it was observed that the relative stability of the required sorbitol conformer is reflected in the corresponding transition state, again in line with the findings of Dosen-Micovic.6
So the question arises if the ZnCl2(H2O)n (n = 3, 4) molten salt system for dehydration of sorbitol actually might lead to the formation of HCl in an aqueous environment, similarly as indicated by Montassier13 (CuCl2 catalysis) and Kuchkarev14 (Zn halides system). In the literature, no activation barrier data were found for HCl in water-catalyzed dehydration of sorbitol, but for concentrated H2SO42,15 and water itself at high temperature,16 data are reported. Table 6 lists an overview of the different systems.
We assume that for concentrated mineral acids in pure sorbitol or aqueous sorbitol, the difference between H2SO4 and HCl will be small as in both cases H3O+ will be the actual acid catalyst, albeit the role of concentration and the specific role of the anion cannot be excluded completely. The reported activation barriers of 47.9 and 51.0 kJ mol−1 in the H2SO4-catalyzed system for the ring closure to the 1,4- and 3,6-anhydrosorbitol and isosorbide,15 respectively, are much lower than the activation barriers observed in the ZnCl2(H2O)n (n = 3, 4) system. It should be noted that in the work of Xie et al.,15 the activity is measured with concentrated H2SO4 with pure sorbitol in a melt under vacuum (3 kPa in order to remove water for the enhancement of the reaction rate), whereas for the reported activation energy, sulfuric acid in aqueous sorbitol was applied. It should also be noted that our computational results for just HCl–H2O, as the simplest model for aqueous catalysis, showed an activation barrier of 53.6 kJ mol−1, very similar to that of the H2SO4-catalyzed system.
The pH of water is highly dependent on temperature,17 and the experimental activation barrier of ca. 127 kJ mol−1 can be well explained by this. The experimental data support the conclusion that the ZnCl2(H2O)n (n = 3, 4) system should not be treated as an aqueous system with or without mineral acid catalysis.
The reaction temperature of the ZnCl2(H2O)n (n = 3, 4) molten salt system for the dehydration of sorbitol (~200 °C) is much higher than the temperature in the mineral acid catalysis case (~130 °C), which is directly reflected in the activation barriers, as in both cases, intramolecular SN2-reactions of a secondary alcohol attacking a primary alcohol function provide a good explanation of the experimental data. In the case of the ZnCl2(H2O)n (n = 3, 4) system, complexation of the primary alcohol to Zn(II) enhances the leaving group ability of the alcohol. Molecular modeling fully supports this view. Furthermore, due to the zinc complexation, the required reaction temperature is around 70 °C higher than in the case of a concentrated inorganic acid homogeneous catalyzed dehydration reaction. Molecular modeling fully supports the view of the complexation between zinc and sugar alcohol. Also, the simultaneous removal of water from the equilibrium reaction enhanced the reaction rate by applying a vacuum.
Finally, another important function of the ZnCl2(H2O)n (n = 3, 4) system is the absorption of the water (or complexation with water) formed in the dehydration processes, as indicated by molecular modeling. There is a distinct order in the stability of the different Zn-complexes. Water is the strongest coordinating ligand to Zn(II), alcohols are in between water and Cl−, while OH− is a stronger coordinating anion than Cl−. In addition, Cl− is strongly solvated by water, leading to the formation of hydrogen bridges. All this turns the overall endothermic reaction into an exothermic one. As a net reaction, sorbitol loses two molecules of water. This is also consistent with the experimental observation during preparation of ZnCl2 molten salt hydrate medium that upon addition of water to ZnCl2, the reaction mixture becomes hot, clearly an indication of an exothermic process.
Thus, ZnCl2(H2O)n (n = 3, 4) should be considered as a catalytic reaction system on its own and definitely not as an aqueous system with mineral acid.
The ZnCl2(H2O)n (n = 3, 4) system might also play a role in stabilizing isosorbide, inhibiting its isomerization into isoidide and isomannide, while in another reaction system, extensive isosorbide isomerization into isoidide and isomannide is reported.18
Conclusions
Molecular modeling offers a detailed insight into the reaction pathways and kinetics aspects of the dehydration of sorbitol in the ZnCl2(H2O)n (n = 3, 4) molten salt hydrate system. All product and byproduct forming reactions can be described as internal SN2-reactions enabled by the ZnCl2(H2O)n (n = 3, 4) system. All computational activation barriers are quantitatively in line with the results derived from kinetic modeling of experimental data. The role of the ZnCl2(H2O)n (n = 3, 4) system is at least twofold:1. It acts like a selective Lewis acid catalyst by preferential complexation, enhancing the leaving group ability of the primary OH-group in the sugar moieties in intramolecular SN2 reactions with a late transition state.
2. It turns an endothermic reaction into an exothermic one by absorbing/complexing/coordinating water released from the consecutive dehydration reactions, thus making the reactions thermodynamically more favorable.
In HCl–H2O systems, the reaction pathway is similar, except for the complexation ability of ZnCl2 resulting in higher stabilities due to complex formation of reactants, intermediates, and products.
Acknowledgements
The authors would like to thank BIOeCON for financial support.Notes and references
- G. Flèche and M. Huchette, Starch/Staerke, 1986, 38, 26–30 CrossRef.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass, Vol. I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, 2004 Search PubMed.
- C. A. Ramirez-Lopez, J. R. Ochoa-Gomez, S. Gil-Rio, O. Gomez-Jimenez-Aberasturi and J. Torrecilla-Soria, J. Chem. Technol. Biotechnol., 2011, 86, 867–874 CrossRef CAS.
- Z. Cekovic, J. Serb. Chem. Soc., 1986, 51, 205–211 CAS.
- L. Dosen-Micovic and Z. Cekovic, J. Phys. Org. Chem., 1998, 11, 887–894 CrossRef CAS.
- R. Menegassi de Almeida, J. Li, C. Nederlof, P. O'Connor, M. Makkee and J. A. Moulijn, ChemSusChem, 2010, 3, 325–328 CrossRef CAS PubMed.
- J. Li, A. Spina, J. A. Moulijn and M. Makkee, Catal. Sci. Technol., 2013, 3, 1540–1546 CAS.
- J. Li, PhD thesis, Lignocellulosic biomass conversion into platform chemicals in molten salt hydrate media, ch. 5 (in English), TU Delft, 2012, ISBN 978-90-9027073-9 Search PubMed.
- Spartan'10 software package Search PubMed.
- W. J. Hehre, A Guide to Molecular Mechanics and Quantum Chemical Calculations, 2003 Search PubMed.
- B. Op de Beeck, J. Geboers, S. Van de Vyver, J. Van Lishout, J. Snelders, W. J. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199–208 CrossRef CAS PubMed.
- C. Montassier, J. C. Menezo, J. Naja, P. Granger, J. Barbier, P. Sarrazin and B. Didillon, J. Mol. Catal., 1994, 91, 119–128 CrossRef CAS.
- A. B. Kuchkarev and N. I. Shuikin, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1956, 1511–1519 CAS.
- Y. Xie, D. Yu, P. Sun, H. Li and Z. Li, Shiyou Huagong, 2010, 39, 285–290 CAS.
- A. Yamaguchi, N. Hiyoshi, O. Sato and M. Shirai, Green Chem., 2011, 13, 873–881 RSC.
- http://www.lsbu.ac.uk/water/ionis.html .
- L. W. Wright and J. D. Brandner, US Pat., 3023223, 1962 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |