Vapour phase dehydration of glycerol over VPO catalyst supported on zirconium phosphate†
N.
Pethan Rajan
,
Ginjupalli Srinivasa
Rao
,
Vanama
Pavankumar
and
Komandur V. R.
Chary
*
Catalysis Division, Indian Institute of Chemical Technology, Hyderabad 500-007, India. E-mail: kvrchary@iict.res.in; Fax: +91 40 27160921; Tel: +91 40 27193162
First published on 9th October 2013
Abstract
A series of VPO catalysts supported on porous zirconium phosphate with varying VPO loadings ranging from 5–30 wt% were prepared by solid–solid wetting method. The calcined catalysts are well characterized by BJH for pore size distribution, X-ray diffraction (XRD), FTIR, scanning electron microscopy (SEM), temperature programmed reduction (TPR) and UV diffuse reflectance spectroscopy. The acidic properties of the catalysts were investigated by ex-situ FTIR analysis of adsorbed pyridine and temperature programmed desorption (TPD) of NH3. The catalytic properties were evaluated for the gas phase dehydration of glycerol to acrolein. The results of pore size distribution by the BJH method suggest that the VPO loadings considerably affect the textural properties. XRD and FTIR analysis suggest that the formation of vanadyl pyrophosphate phase on the zirconium phosphate support. SEM image of unsupported VPO catalyst clearly exhibits the formation of a rosette like structure. UV-DRS study reveals the formation of vanadyl pyrophosphate phase along with some extent of vanadium orthophosphate phase. TPR analysis suggests that the reducibility of the active VPO phase depends on the VPO loading and also the active phase strongly interacts with the support. The results of vapour phase glycerol dehydration reaction are well correlated with the acidity of the catalysts measured by the ex-situ adsorbed pyridine by FTIR and TPD of ammonia. The findings derived from several characterization techniques clearly suggest that VPO species is highly dispersed on the ZrP support.
1. Introduction
In the recent past glycerol received particular attention as by-product from biodiesel production. The extracted triglycerides can undergo transesterification with methanol, yielding the methyl ester (biodiesel) and glycerol as by-product on a stoichiometric basis. The increasing production of biodiesel has resulted in a price decline of crude glycerol. This makes aqueous glycerol as an attractive compound for the synthesis of fine and crude chemicals. Catalytic conversion of glycerol to acrolein by a double dehydration reaction could be an important route for using glycerol resources. However this process would provide a cost effective and sustainable alternative to the current commercial catalytic petrochemical process based on the oxidation of propylene over a Bi/Mo mixed oxide catalyst. Acrolein is an important and versatile chemical intermediate and raw material for the production of acrylic acid esters, super absorber polymers, fiber treatment, detergents and production of other value-added derivatives.1,2Various types of solid acid catalysts such as zeolites,3 metal oxides,4,5 metal phosphates,6 heteropoly acids on different supports,7,8 WO3–ZrO2,9,10 have been investigated for the glycerol dehydration reaction. Among the various metal phosphate catalysts used for this reaction, vanadium phosphorus oxide (hereinafter VPO) catalyst was also studied by Wang et al.11 because it possesses strong acidic sites. Moreover, they found that the addition of molecular oxygen with glycerol feed greatly reduces the side product formation and maintains the glycerol conversion and acrolein yield. The same group also investigated the effect of calcination temperature on pure VPO catalyst and found that calcination temperature affects the Lewis acidity of the catalyst.12
Vanadium phosphorus oxides (VPO), is well known as catalyst for the industrially important selective oxidation of n-butane to maleic anhydride.13 The VPO catalyst contains several VPO phases with vanadium in 5+ oxidation state (α-, β-, γ-, δ-VOPO4), 4+ oxidation state ((VO)2P2O7) and 3+ oxidation state (VPO4). It is generally accepted that well crystallized vanadyl pyrophosphate is the major phase in industrial VPO catalyst for oxidation of n-butane to maleic anhydride. The activity of the VPO catalyst generally depends on the type of reagents used in the preparation, the ratio of phosphorous to vanadium, the nature of the solvent and the conditions of the activation step. Vanadyl pyrophosphate is prepared from the thermal treatment of another crystalline compound, the precursor VOHPO4·0.5H2O. The most recent and common commercial formulations for the preparation of the precursor prefer organic compounds as solvents.14 The crystalline structure and the morphology have been determined with a combination of X-ray diffraction and electron microscopy techniques. The acidity of the VPO catalysts can be attributed to the presence of surface P–OH groups and to coordinatively unsaturated vanadium ions exposed on the surface coupled to V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds.15 An IR study of the acidic sites using ammonia, pyridine and acetonitrile as probe molecules showed the existence of Lewis and Brønsted acid sites.16–21 Kamiya et al.22 studied the acidic character of the vanadyl pyrophosphate phase and reported that this phase possesses both Brønsted and Lewis acidic sites.
O double bonds.15 An IR study of the acidic sites using ammonia, pyridine and acetonitrile as probe molecules showed the existence of Lewis and Brønsted acid sites.16–21 Kamiya et al.22 studied the acidic character of the vanadyl pyrophosphate phase and reported that this phase possesses both Brønsted and Lewis acidic sites.
One of the extensively investigated methods to improve the catalytic performance of vanadium phosphorous oxide catalyst is to support the active phase on an oxide surface. In the case of supported catalyst, the support should have some features including better heat transfer character, larger surface area to volume ratio of active component, better mechanical strength, and controllable catalyst textures. The first employed supports for VPO were the classical oxides such as SiO2, TiO2, Al2O3 and also new kind of materials such as SiC.23–28 Au et al.29 studied the performance of SBA-15 supported VPO catalyst for oxidation of n-butane to maleic anhydride and they showed that the supported VPO species dispersed well on SBA-15 and mainly exists as crystalline (VO)2P2O7. There are other supports such as MCM-41, Al–MCM-41, fumed silica, silica, alumina, titania, phosphoric acid treated zirconia which are also used as the supports for VPO catalyst.30–33 It is well documented that the use of a support for metal oxide catalysts favours enhancement of active phase dispersion and also imparts stability to the catalysts. The use of supported catalyst during glycerol dehydration reaction have several advantages such as the enhancement of dispersion of the active phase on the support, the distribution of the pore size of the support and the acidity of the catalyst.34
Porous zirconium phosphate (ZrP) has been used as a solid acid catalyst in many reactions such as dehydration, isomerisation of olefins due to its high acidic sites.35–37 Zirconium phosphate support also exhibit high thermal stability and high surface area due to the porous nature. Hence, these properties make ZrP an interesting support material for other catalytically active metal species.38 However the VPO supported on zirconium phosphate have not been investigated so far. Hence our interest is to focus on the effect of zirconium phosphate support on VPO catalyst and the interaction between VPO and zirconium phosphate support and to investigate the acidic nature of the catalyst.
In the present investigation we report a systematic study on the characterization of VPO catalysts supported on zirconium phosphate by N2 adsorption desorption analysis, XRD, FTIR, SEM, UV-DRS analysis, ex-situ adsorbed pyridine FTIR analysis, temperature programmed reduction (TPR) and temperature programmed desorption of NH3. The catalytic properties were evaluated for the dehydration of glycerol over a series of VPO supported on zirconium phosphate catalyst. The purpose of this work is to examine the catalytic properties of VPO supported on zirconium phosphate and also to find the relation between the surface structural properties of the vanadium phosphorous oxide species as a function of an active component loading and catalytic functionalities during glycerol dehydration reaction.
2. Experimental section
Porous zirconium phosphate support was prepared from zirconium n-propoxide precursor and 85% phosphoric acid following the procedure described elsewhere.39 About 0.01 mol of zirconium n-propoxide, (70 wt% solution in 1-propanol, Aldrich) was added drop wise to a 60 mL solution of H3PO4 (0.1 mol L−1) under stirring. After 2 h of stirring at room temperature, the obtained mixture was transferred into a Teflon lined autoclave and aged statically at 80 °C for 24 h. The final material was filtered, dried and calcined at 400 °C for 5 hours.The preparation of dihydrate VOPO4·2H2O has been reported elsewhere.40 Briefly, the procedure involves the addition of a mixture of V2O5 (24 g), H3PO4 (85%, 133 mL), and H2O (577 mL) was refluxed at 115 °C for 16 h. The reaction produced a bright yellow solid, which was collected by filtration, washed with 100 mL acetone, and dried for 10 h under ambient conditions. VOHPO4·0.5H2O was prepared by reduction of VOPO4·2H2O in organic alcohol. A suspension of VOPO4·2H2O (5 g) powder in 2-butanol (50 mL) was stirred under reflux for 23 h (oil bath temperature: 101 °C). The resulting light blue solid was collected by filtration, washed with 100 mL of acetone, and dried for 10 h under ambient conditions.
Desired amount of VPO precursor and zirconium phosphate powders were mixed together in a mortar by physical mixing for about 15 min, i.e. until the colour of solid mixture was perfectly uniform. Using the same procedure, various catalysts over a wide range of VPO contents were prepared. Finally the catalysts were calcined in presence of nitrogen at 550 °C, for 4 h at the heating rate of 1 °C min−1. The VPO loadings were varied from 5 wt% to 30 wt%.
X-ray diffraction (XRD) patterns were obtained on Rigaku miniflex diffractometer using graphite filtered Cu Kα (K = 0.15406 nm) radiation. FTIR spectra of the catalysts were taken on the IR (Model: GC-FT-IR Nicolet 670) spectrometer by KBr disc method under ambient condition. The SEM analysis samples were obtained by mounting the sample on an aluminum support using a double adhesive tape coated with gold and observed in Hitachi S-520 SEM unit. The specific surface area of the calcined catalysts were analysed using N2 adsorption at −196 °C by the multipoint BET method taking 0.0162 nm2 as its cross-sectional area using Autosorb 1 (Quantachrome instruments). The Pore size distribution of the samples were analysed by BJH method using N2 adsorption desorption isotherm obtained on Autosorb 1 instrument. The UV–Vis diffused reflectance spectra were recorded on a GBC UV-Visible Cintra 10e spectrometer with an integrating sphere reflectance accessory. The spectra were recorded in air at room temperature and the data was transformed according the Kubelka–Munk equation f(R) = (1 − Rα) 2/2rα.
The ex-situ experiments of FTIR spectra of pyridine adsorbed samples were carried out to find the Brønsted and Lewis acid sites. Pyridine was adsorbed on the activated catalysts at 120 °C until saturation. Prior to adsorption experiments the catalysts were activated in N2 flow at 300 °C for 1 h to remove moisture from the samples. After such activation the samples were cooled to room temperature. The IR spectra were recorded using a IR (model: GC-FT-IR Nicolet 670) spectrometer by KBr disc method under ambient conditions.
Temperature programmed reduction studies were conducted on AutoChem 2910 (Micromeritics, USA) instrument. In TPR experiment, 100 mg of oven dried sample was taken in a U shaped quartz sample tube. The catalyst was mounted on a quartz wool plug. Prior to TPR studies, the catalyst sample was pretreated by passing helium gas in a flow of 50 mL min−1 at 200 °C for 1 h. After pretreatment the sample was cooled to ambient temperature and TPR analysis was carried out in a flow of 5% H2–Ar (50 mL min−1) from ambient temperature to 850 °C at a heating rate of 10 °C min−1. The H2 consumption & Tmax positions are calculated using GRAMS/32 software.
TPD experiments were also conducted on AutoChem 2910 (Micromeritics, USA) instrument. In a typical experiment for TPD studies, 100 mg of oven dried sample was taken in a U shaped quartz sample tube. The catalyst was mounted on a quartz wool plug. Prior to TPD studies, the sample was pretreated by passage of high purity (99.995%) helium (50 mL min−1) at 200 °C for 1 h. After pretreatment, the sample was saturated with highly pure anhydrous ammonia (50 mL min−1) with a mixture of 10% NH3–He at 80 °C for 1 h and subsequently flushed with He flow (50 mL min−1) at 80 °C for 30 min to remove physisorbed ammonia. TPD analysis was carried out from ambient temperature to 800 °C at a heating rate of 10 °C min−1. The amount of NH3 desorbed was calculated using GRAMS/32 software.
The gas phase dehydration of glycerol was carried out under atmospheric pressure in a vertical fixed bed quartz reactor (40 cm length, 9 mm i.d.) using 0.2 g of catalyst. Prior to the reaction, the catalysts were pretreated at 300 °C for 1 h in flowing dry N2 (16 mL min−1). An aqueous solution containing 20 wt% glycerol was fed into the reactor by a micro syringe pump at 0.5 mL h−1 (WHSV-2.6 h−1). The reaction products were condensed in an ice–water trap and collected hourly for analysis on a gas chromatograph GC-2014 (Shimadzu) equipped with a DB-wax 123-7033 (Agilent) capillary column (0.32 mm i.d., 30 m long) and equipped with flame ionization detector.
3. Results and discussion
In order to understand the textural properties of the catalyst, the N2 adsorption desorption measurements have been carried out to measure the surface area and pore size distribution and the results are presented in Table 1. As can be seen the results of Table 1 that VPO loadings have shown a clear impact on the surface area of the zirconium phosphate support. The surface area of the pure zirconium phosphate support was found to be 334 m2 g−1 and decreases as a function of VPO content (Table 1). This decrease of surface area with increasing VPO loading might be due to blocking the pores of the support by crystalline vanadium phosphorous oxide. The surface area of pure VPO catalyst (unsupported) was found to be 16.5 m2 g−1. The N2 adsorption desorption isotherm of the pure zirconium phosphate support and VPO supported on zirconium phosphate have shown in Fig. S1.† The isotherm of pure zirconium phosphate support exhibits a high uptake of nitrogen at low relative pressures (P/Po) of 0.1–0.4, indicating that the pore sizes are present in the ranges of micropore and mesopore region.41 BJH analysis of the pore size distribution on the adsorption isotherm reveals that a narrow pore size distribution (Fig. 1) centred around 2.0 nm. When VPO is supported on the zirconium phosphate the pore volume decreases because of the added VPO components are occupied in the pores of the zirconium phosphate support. In order to gain more information about the calcination effect on the pure zirconium phosphate, the BJH pore size distribution measurements (Fig. 1) was also carried out on pure zirconium phosphate calcined at 550 °C (P-ZrP-550 °C) and the results are reported in Table 1. As can be seen from Fig. 1 that there is a considerable change was observed in the surface area and pore volume of the pure P-ZrP-550 °C. Hence the calcination of pure ZrP at 550 °C affects the textural properties of the pure zirconium phosphate support. However, the decrease in the pore volume and surface area for supported VPO catalyst was noticed when VPO is loaded on the ZrP support indicates that the addition of VPO influences the pore size of the resulting catalyst.42 As VPO loading increases on the zirconium phosphate support, the mean pore diameter of the sample is also increasing. This might be due to filling of the micropores of zirconium phosphate support by VPO catalyst42 and these were not included in the measurement. These findings are further confirmed by the pore size distribution profiles shown in Fig. 1.| Sample No. | VPO loadings (wt%) | BET SA (m2 g−1) | Total pore volume (cc g−1) | Mean pore diameter (Å) |
|---|---|---|---|---|
| 1 | Pure ZrP-400 | 334 | 0.36 | 32 |
| 2 | Pure ZrP-550 | 271 | 0.32 | 32 |
| 3 | 5 | 214 | 0.28 | 47 |
| 4 | 10 | 179 | 0.27 | 60 |
| 5 | 15 | 161 | 0.26 | 65 |
| 6 | 20 | 152 | 0.28 | 74 |
| 7 | 25 | 148 | 0.29 | 78 |
| 8 | 30 | 145 | 0.15 | 39 |
| 9 | Pure VPO | 17 | 0.20 | 47 |
 | ||
| Fig. 1 BJH isotherm of pure ZrP and different wt% loadings of VPO. | ||
X-ray diffraction patterns of pure zirconium phosphate support and various VPO/ZrP catalysts with VPO loadings ranging from 5 to 30 wt% are shown in Fig. 2. XRD results suggest that the synthesized zirconium phosphate is found to be X-ray amorphous. As the VPO loadings are increasing on the zirconium phosphate support the diffraction peak intensity is also increasing. At lower loadings, the supported VPO catalyst (up to 20 wt% VPO loadings) does not exhibit any peaks corresponding to VPO species suggesting that the active phase is present in highly dispersed form. However it cannot be ruled out for the presence of VPO crystalline species having the size with less than 4 nm which is beyond the detection limit of powder X-ray diffraction techniques. The higher loadings of supported VPO catalyst (25 & 30 wt%) shows the main reflection peak at 2θ in the range of 23.00°, 28.45°, and 29.95° confirms the formation of vanadyl pyrophosphate on the zirconium phosphate support.43,44 The XRD results suggested that VPO catalyst is present in the vanadyl pyrophosphate phase on the zirconium phosphate support. Moreover the obtained vanadyl pyrophosphate sample showed a poor crystalline in nature because the sample was calcined in the nitrogen atmosphere. The XRD results also indicates that there is no other phase formation or compound formation taking place in the pure VPO catalyst and supported VPO catalyst.
 | ||
| Fig. 2 XRD pattern of pure ZrP and various VPO/ZrP. | ||
Fig. 3 shows the FTIR spectra of pure zirconium phosphate support and different wt% of VPO loading catalyst supported on zirconium phosphate. All the IR spectra show that the bands in the region 3400–2400 cm−1 are due to surface OH groups and the IR band at 1630 cm−1 is assigned to bending adsorbed mode of water molecule by the catalyst. Pure zirconium phosphate shows a broad band at 1020 cm−1 for Zr–O–P vibration.45 While increasing the VPO loading on zirconium phosphate support, the intensity of the peak is also increasing. At lower loadings, only one broad peak was observed at 1062 cm−1 and it does not provide any information about VPO species. This does not suggest that VPO active phase is not detected by FTIR. It might be due to the active VPO phase present in the highly dispersed state in the pores of the zirconium phosphate support or the detected peaks are over shaded by the most intense bands of the pure support. At higher loadings of VPO (25 wt% and 30 wt%) show characteristic clear IR peaks. At higher loadings (25 wt% and 30 wt%) of the sample there is also formation of a new peak at 968 cm−1 which can be attributed to stretching frequency of V4+O species and further confirms the formation of vanadyl pyrophosphate phase.46 Hence FTIR spectra is a good tool for supporting the evidence of results obtained from XRD on the formation of active vanadyl pyrophosphate phase.
 | ||
| Fig. 3 FTIR spectra of pure ZrP and various VPO/ZrP. | ||
The surface morphology of the samples were examined by SEM and Fig. 4 shows the representative SEM micrographs of pure zirconium phosphate, supported VPO catalyst with different loadings and pure VPO catalyst. The pure VPO sample has rosette like morphologies with uniform particle size distribution. The structure of the pure VPO catalyst consists of plate like crystals, which are arranged to the characteristic rosette-shape clusters. The rosette type agglomerates are made up of agglomerates of (VO)2P2O7 platelets that preferentially expose (100) crystal planes.47,48 Pure zirconium phosphate shows plate like morphology. From the SEM (Fig. 4) results it is observed that at lower loadings the VPO catalyst is well dispersed on the support and at higher loadings the dispersion decreases on the support as the particles are agglomerated.
 | ||
| Fig. 4 SEM images of pure ZrP and various VPO/ZrP. | ||
UV–Vis-DRS investigations were carried out to obtain information of the vanadium oxidation state in VPO and supported VPO catalyst. From the literature it is clear that V5+ exhibits the charge transfer absorption band at around 500 nm whereas V4+ shows the charge transfer absorption band at around 220 nm.49Fig. 5 shows the UV-DRS spectra of pure uncalcined and calcined VPO catalyst. The uncalcined VPO shows a broad charge transfer transition band at 200 to 350 nm due to the charge transfer transition from O2− to V4+ and another transition band from 600 nm due to d–d transition of V4+. After calcination in nitrogen atmosphere, the d–d transition band of V4+ disappears and the intensity of charge transfer band of calcined sample remains unchanged. In general the intensity of charge transfer transition band is much higher than the d–d transition of vanadium ions. These results are well in agreement with the previously reported findings of Martin et al.50Fig. 5 shows the UV-DRS spectra of pure zirconium phosphate and VPO catalyst supported on zirconium phosphate. The spectra are dominated by the charge transfer (CT) transition of the type O2− to V4+ in the range of 200–400 nm with multiplicity. The parent zirconium phosphate showed a band at 300 nm, which might be due to Zr(IV) cations interacting with the phosphate counter anions in the framework.51 As the VPO loadings on zirconium phosphate increases, the intensity of the charge transfer transition band of V4+ also increased. From Fig. 5 it is observed that, similar to pure VPO, the charge transfer band of V4+ and V5+ are clearly visible at the higher loading of supported VPO catalyst. This result confirms the formation of vanadyl pyrophosphate phase on the zirconium phosphate support along with some of the vanadium orthophosphate phase. However, XRD results of pure VPO and supported VPO catalyst shows the presence of only vanadyl pyrophosphate phase in the calcined sample (Fig. 2). This result reflects that various types of vanadium orthophosphate phases might be present in the amorphous form.
 | ||
| Fig. 5 UV DRS profiles of pure VPO and various VPO/ZrP. | ||
The reduction behaviour of the pure VPO and supported VPO catalysts were examined by the temperature programmed reduction (TPR) by using 5% H2/Ar. Fig. 6 shows the TPR profiles of pure VPO catalyst and different wt% VPO loadings supported on zirconium phosphate catalyst. It is interesting to see that the VPO dispersion on zirconium phosphate support strongly changes the reduction behaviour of vanadium present in the VPO catalyst. In the TPR profiles, the pure VPO catalyst showing only one sharp reduction peak at 750 °C indicates that there is only one type of reducible vanadium present in the pure VPO catalyst and it attributes to the reduction of V4+ to V3+.25 Similarly, in the case of different wt% VPO supported on zirconium phosphate catalyst, only one peak appeared in the TPR profiles at around 700 °C due to the reduction of V4+ to V3+. The reduction temperature and the volume of hydrogen consumption are shown in Table 2. As the VPO loadings are increasing the amount of hydrogen consumption by the VPO also increasing. It is very interesting to note that the reduction temperature of the supported VPO catalyst is less than the pure VPO catalyst suggesting that there is a significant interaction between VPO and zirconium phosphate support. It reflects that supported VPO catalysts are more reducible than the pure VPO catalyst. The zirconium phosphate support itself was not reduced in the applied temperature range, the observed reducibility should be attributed to the supported VPO species. At lower VPO loadings the reducibility of the catalyst is easier than the higher loadings. The reduction temperature increased with increase in VPO loadings. This clearly suggests that the reducibility decreasing with increasing of VPO loadings. At lower loadings the reducibility of VPO species is much easier because of the VPO species occupied the pores of the zirconium phosphate support leading to the well dispersion of active phase on the support. The portion of the oxygen atoms of the VPO are shared with zirconium species present in the zirconium phosphate leads to the enhanced stability of vanadium atoms and it increases the reducibility of the catalyst. At higher loadings the remarkable shift in the reduction temperature of supported VPO catalyst (>20 wt% loadings) due to the VPO species are located on the surface of the support leads to the formation of bulk type VPO catalyst and it decreases the reducibility of VPO catalyst.
 | ||
| Fig. 6 TPR profiles of various VPO/ZrP. | ||
| Sample No. | VPO loadings (wt%) | Temperature (°C) | Amount of hydrogen consumption (μmol g−1) |
|---|---|---|---|
| 1 | 5 | 674 | 296 |
| 2 | 10 | 677 | 753 |
| 3 | 15 | 680 | 1025 |
| 4 | 20 | 688 | 1273 |
| 5 | 25 | 708 | 2019 |
| 6 | 30 | 720 | 3051 |
| 7 | Pure VPO | 750 | 4536 |
The surface acidity measurements of all the samples have been carried out by ammonia TPD method. Temperature programmed desorption (TPD) of probe molecules like ammonia and pyridine is a popular method for the determination of acidity of solid catalysts as well as acid strength distribution because it is an easy and reproducible method. Ammonia is used frequently as a probe molecule because of its small molecular size, stability and strong basic strength. The ammonia TPD profiles of the pure VPO, pure zirconium phosphate support and different wt% loadings of VPO/ZrP catalysts are presented in Fig. 7. This figure clearly demonstrated the effect of acidic properties of different wt% of VPO loadings on zirconium phosphate support. The pure zirconium phosphate support has shown one broad peak in the temperature range of 100–450 °C. According to Tanabe et al.52 the strength of solid acid sites within TPD profiles can be classified by the temperature of NH3 desorption as weak (120–300 °C), moderate (300–450 °C) and strong (above 450 °C). Thus, the observed acidity in the TPD profile of zirconium phosphate is found to be in the weak moderate acidic region (120–450 °C). It can be seen from Fig. 7 that as VPO loadings are increasing the intensity of the weak moderate acidic region peak is increasing. The pure VPO catalyst has one broad peak at the weak moderate acidic region and one sharp peak at strong acidic region. The acidity of the VPO catalysts can be attributed to the presence of surface P–OH groups and to coordinatively unsaturated vanadium ions exposed in the surface coupled to VO double bonds. The ammonia uptake values and the peak temperatures are summarised in Table 3. It is interesting to note that the total acidity value of pure VPO is higher than the supported VPO catalyst. The higher acidity of pure VPO is mainly due to the presence of strong acidic sites as shown in Table 3. These strong acidic sites are absent in the case supported VPO catalyst. From Table 3 we can observe that the weak moderate acid sites are increasing with the increase of VPO loading up to 20 wt% VPO/ZrP and beyond this loading (25 & 30 wt% VPO/ZrP) it is decreasing. It is interesting to note that the total acidity values of supported VPO catalysts were also increasing up to 20 wt% VPO/ZrP and decreasing at higher loadings (25 & 30 wt% VPO/ZrP).
 | ||
| Fig. 7 TPD of ammonia profiles of Pure ZrP and various VPO/ZrP. | ||
| Sample No. | VPO loadings (wt%) | Physisorbed ammoniaa (μmol g−1) | Temperature (Tmax1) (°C) | Desorbed ammonia (μmol g−1) | Temperature (Tmax2) (°C) | Desorbed ammonia (μmol g−1) | Total ammonia desorption (μmol g−1) |
|---|---|---|---|---|---|---|---|
| a Physisorbed ammonia is not included in the total acidity value. | |||||||
| 1 | Pure ZrP | 2104 | 358 | 1123 | — | — | 1123 |
| 2 | 5 | 973 | 247 | 583 | — | — | 583 |
| 3 | 10 | 1109 | 244 | 637 | — | — | 637 |
| 4 | 15 | 583 | 260 | 998 | — | — | 998 |
| 5 | 20 | 643 | 268 | 1291 | — | — | 1291 |
| 6 | 25 | 393 | 361 | 740 | — | — | 740 |
| 7 | 30 | 377 | 358 | 685 | — | — | 685 |
| 8 | Pure VPO | — | 228 | 789 | 720 | 1162 | 1951 |
| 9 | Spent 20 wt% | 81 | 320 | 650 | — | — | 650 |
We have employed FT-IR analysis by using pyridine as a probe molecule which was saturated at 120 °C to find the nature of acid sites on VPO catalysts. Usually, the IR bands appeared at 1540–1548 cm−1 and 1445–1460 cm−1 are characteristic bands of Brønsted (B) and Lewis (L) acid sites, respectively. Furthermore, the bands correspond to combination of both Brønsted and Lewis (B + L) acid sites are appeared at 1490–1500 cm−1. It should be noted that the intensity of the IR band is proportional to the concentration of acid sites. The FTIR spectra of pure zirconium phosphate, supported VPO and Pure VPO obtained after pyridine adsorptions are illustrated in Fig. 8. All the catalysts have shown bands at 1444 cm−1 corresponding to Lewis sites and the other band appeared at 1550 cm−1 is attributed to Brønsted sites. It can also be seen from Fig. 8 that all the catalysts contain both Lewis and Brønsted sites in different proportions depending upon the VPO loadings on the support. The pure zirconium phosphate shows the sharp band at 1550 cm−1 due to Brønsted acidic sites. The VPO is supported on the zirconium phosphate and the Brønsted acidity decreases initially after that it increases continuously as the VPO loadings increases until 20 wt% VPO/ZrP. At higher loadings the Brønsted acidity did not change appreciably. The Lewis acidity also increasing with VPO loadings until 20 wt% VPO/ZrP after that it decreases. The pure VPO as well as 20 wt% VPO/ZrP showed the maximum intensity of Brønsted and Lewis acidic sites.
 | ||
| Fig. 8 Ex-situ pyridine adsorbed FTIR of pure VPO and various VPO/ZrP. | ||
The results of vapour phase glycerol dehydration to acrolein are shown in Table 4. It is important to emphasize that the comparison of conversion and selectivity between different catalysts is possible only at low time on stream since with increasing time of reaction, the deactivation process becomes predominant. The catalytic results of dehydration of glycerol over various different wt% VPO loadings on zirconium phosphate by passing nitrogen are shown in Table 4. Pure zirconium phosphate support exhibited complete glycerol conversion with 42% selectivity towards acrolein. The major by-products of this catalyst were acetaldehyde and acetol. When the VPO loading increases on the zirconium phosphate support, the acrolein selectivity decreases to 30% and then it increases with the VPO loadings and reaches a maximum of 60% and again decreases at higher loadings. Whereas the conversion of glycerol always maximum irrespective of the VPO loadings. The pure VPO shows only 90% conversion towards glycerol and 47% selectivity towards acrolein. The major by-products were acetol, acetaldehyde, acetic acid, allyl alcohol and some minor unidentified products. From this result, it was observed that the supported VPO catalyst showed better result compared to the pure VPO catalyst and pure zirconium phosphate support. The Pure zirconium phosphate support gave acetol and acetaldehyde as the major by-products. At lower loadings these by-products are formed in higher amount because the zirconium phosphate support is not covered fully by the VPO and when the VPO loadings increases these by-products decreases as a result acrolein selectivity increases. At lower loadings the reducibility of the catalyst also plays a key role in the conversion and selectivity towards acrolein.
| Sample No. | VPO loadings (wt%) | Conversion (mol%) | Selectivity (mol%) | ||||
|---|---|---|---|---|---|---|---|
| Acrolein | Hydroxyacetone | Acetaldehyde | Acetic acid | Others | |||
Reaction conditions: 0.2 g catalyst was used and diluted with 1.0 g of quartz. The aqueous glycerol (20% wt/wt) was fed at the speed of 0.5 g h−1 by syringe pump under 16 mL min−1 N2 atmosphere i.e. the reactant fed composition gly![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:N2 was 1.37:50.0:42.83 in molar ratio and reaction temperature was 300 °C. In parentheses, the same reaction condition except the reactant fed composition gly:H2O:N2:air was 1.37:50.0:32.12:10.7 in molar ratio. :H2O:N2 was 1.37:50.0:42.83 in molar ratio and reaction temperature was 300 °C. In parentheses, the same reaction condition except the reactant fed composition gly:H2O:N2:air was 1.37:50.0:32.12:10.7 in molar ratio. |
|||||||
| 1 | Pure ZrP | 100 (100) | 42 (42) | 16 (16) | 9 (9) | 3 (3) | 30 (30) |
| 2 | 5 | 100 (100) | 30 (52) | 23 (—) | 5 (10) | 14 (21) | 28 (17) |
| 3 | 10 | 100 (100) | 46 (58) | 10 (—) | 10 (9) | 18 (17) | 16 (16) |
| 4 | 15 | 100 (100) | 52 (62) | 8 (—) | 13 (8) | 9(18) | 18 (12) |
| 5 | 20 | 100 (100) | 60 (66) | 4 (—) | 16 (11) | 3 (14) | 17 (9) |
| 6 | 25 | 100 (100) | 55 (63) | 12 (—) | 18 (14) | 3 (12) | 20 (11) |
| 7 | 30 | 100 (100) | 48 (62) | 14 (—) | 8 (12) | 2 (17) | 28 (9) |
| 8 | Pure VPO | 90 (90) | 47 (50) | 2 (—) | 8 (4) | 13 (18) | 30 (27) |
Wang et al.11 studied the glycerol dehydration reaction over pure VPO catalyst by passing molecular oxygen with nitrogen gas to maintain the glycerol conversion, acrolein yield and greatly reduces the side product formation. In the present investigation the dehydration of glycerol was carried out over different wt% loadings of VPO on zirconium phosphate by passing air along with nitrogen gas and the results are shown in Table 4. Passing air along with nitrogen stopped the deactivation of the catalyst as well as reduces the formation of hydroxyacetone and allyl alcohol and as a result acrolein selectivity increases. When the VPO loading increases, the acrolein selectivity also increases until 20 wt% VPO on zirconium phosphate and decreases gradually at higher loadings. However, there is no significant decrease in the acrolein selectivity is noticed. From Table 4 it is observed that conversion of glycerol is always found to be 100%. It is interesting to note that the supported VPO at lower loadings gave better acrolein selectivity in presence of air and nitrogen when compared to passing nitrogen gas only, due to reduction of other side product formation. The results of the product distribution of glycerol dehydration reaction at constant reaction conditions except the gas flow conditions are shown in Table 4. The major products of the glycerol conversion over pure zirconium phosphate support, pure VPO, different weight percentage loadings of VPO on zirconium phosphate were acrolein, acetol, allyl alcohol, acetic acid and acetaldehyde. Products grouped under the label of other included, acrylic acid, acetone, and some unidentified products. By passing only nitrogen as a carrier gas, the lower loadings of VPO catalyst gave lower acrolein selectivity which might be due to the increase in the reducibility of the catalyst and the major side products are hydroxyacetone, allyl alcohol and acetic acid. When the air was introduced in the reaction medium, the formation of allyl alcohol and hydroxyacetone is reduced and acetic acid formation is increased. The allyl alcohol is formed by the hydrogenation of acrolein in the reaction medium. The hydrogen is mainly produced from the degradation of glycerol into the coke. The hydroxyacetone is produced through the dehydration at the terminal hydroxyl group of glycerol. The oxygen presents in the air restricts the hydrogenation of acrolein and favours the dehydration at the central hydroxyl group of the glycerol.
The effect of reaction temperature (280–340 °C) on the dehydration of glycerol was examined over 20 wt% VPO/ZrP and the results are shown in Fig. 9. At 280 °C the conversion of glycerol is 96% and it reaches maximum at higher temperatures. At 300 °C this catalyst showed maximum conversion and selectivity with the increase of temperature, there is no effect on glycerol conversion. However, at higher temperatures the acrolein selectivity decreased due to increase in the formation of acetaldehyde, acetic acid and trace amounts of acrylic acid (8%) and few unidentified products. This result suggests that at higher temperature (340 °C) this catalyst favours the C–C bond cleavage and oxidation instead of dehydration. The major side product obtained is pure acetaldehyde formed due to C–C bond cleavage of glycerol and oxidation of acetaldehyde leads to the acetic acid formation. A small amount of acrylic acid is also formed due to oxidation of acrolein. Thus the higher reaction temperature only favours the acrolein oxidation by VPO supported catalyst due to the increase of reducibility of vanadium in the catalyst.
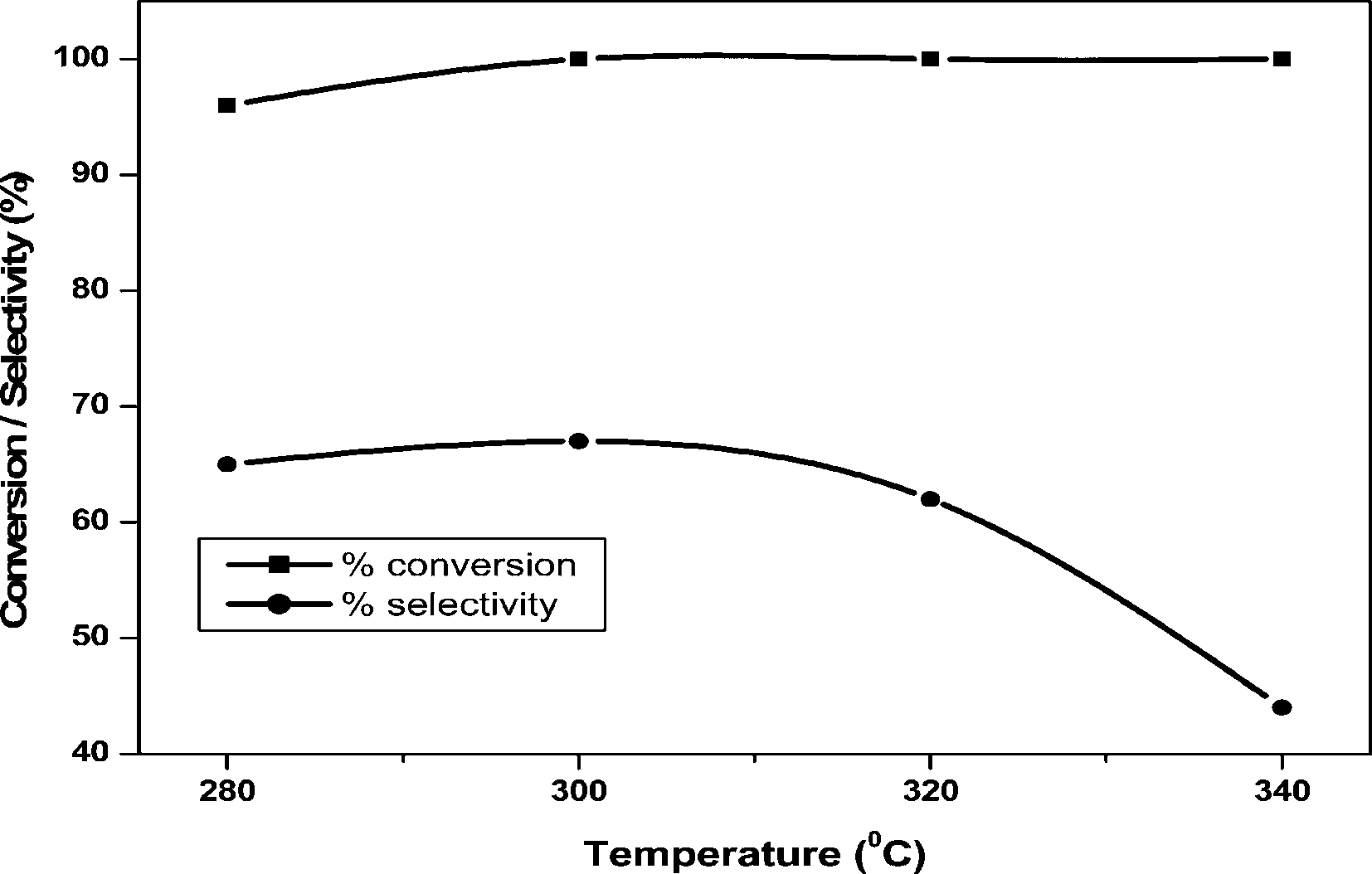 | ||
| Fig. 9 Effect of temperature over 20 wt% VPO/ZrP catalyst. | ||
The influence of the VPO loadings in the supported catalyst on the glycerol dehydration in nitrogen flow was examined with the time on stream and the results are shown in Fig. 10. As can be seen from Fig. 10, the decrease in conversion with the reaction time is noticed over all the catalysts in the catalytic dehydration of glycerol. This deactivation of the catalyst is mainly due to the formation of coke over the active sites of the catalyst.11,12 In the case of pure VPO catalyst, deactivation was more pronounced than the supported VPO catalyst because of its strong acidic character (from TPD of ammonia graph). The TPD profile of supported VPO catalyst shows the absence of strong acidic sites. In the case of supported VPO catalyst, 20 & 30 wt% VPO/ZrP showed lower rate of deactivation and higher acrolein selectivity when compared to the other tested catalysts which might be due to presence of larger pores as evidenced from the results of pore size distribution (Table 1). When these active phases are deposited on a support, it is also possible to adjust an additional parameter i.e. the pore size of the catalyst, and thus reduce diffusion limitations. In general the pore size has a direct influence on the selectivity of the catalysts. If pores are too narrow then it may hinder the rapid desorption and diffusion of the reactants and of the products in the porous network of the catalyst. Thus instead of dehydration, condensation is more likely to occur, which results in lower selectivity for acrolein and fast deactivation of the catalyst due to carbon deposition (coke). The best results were obtained for supported VPO catalysts rather than pure VPO catalyst.
 | ||
| Fig. 10 Time on stream over VPO on zirconium phosphate by passing N2 flow. | ||
The results with the time on stream of catalytic dehydration of glycerol over pure VPO and 20 wt% VPO/ZrP by passing air and nitrogen are shown in Fig. 11. By passing air along with nitrogen, the pure VPO catalyst exhibited maximum conversion of 90% and maximum selectivity of 50% at the initial hour and further the conversion decreases to 30% at 20 h and the acrolein selectivity reach almost 36% at the same reaction time. Under similar reaction conditions, the 20 wt% VPO supported on zirconium phosphate has shown almost 100% conversion and acrolein selectivity increased to 65% at 6 h of the reaction and it shows 52% conversion and 48% selectivity of acrolein after 40 h. It is interesting to that a faster deactivation was observed after 30 h with the supported VPO catalysts whereas the pure VPO catalyst exhibited the deactivation after 10 h. In the initial hours of the supported VPO catalyst, higher formation of acetaldehyde and acetic acid was observed thereby decreases the acrolein selectivity. A comparison activity of 20 wt% VPO/ZrP catalyst with the pure catalysts has been reported in Table 5. The present catalyst 20% VPO/ZrP showed better catalytic activity with respect to the pure VPO catalyst.
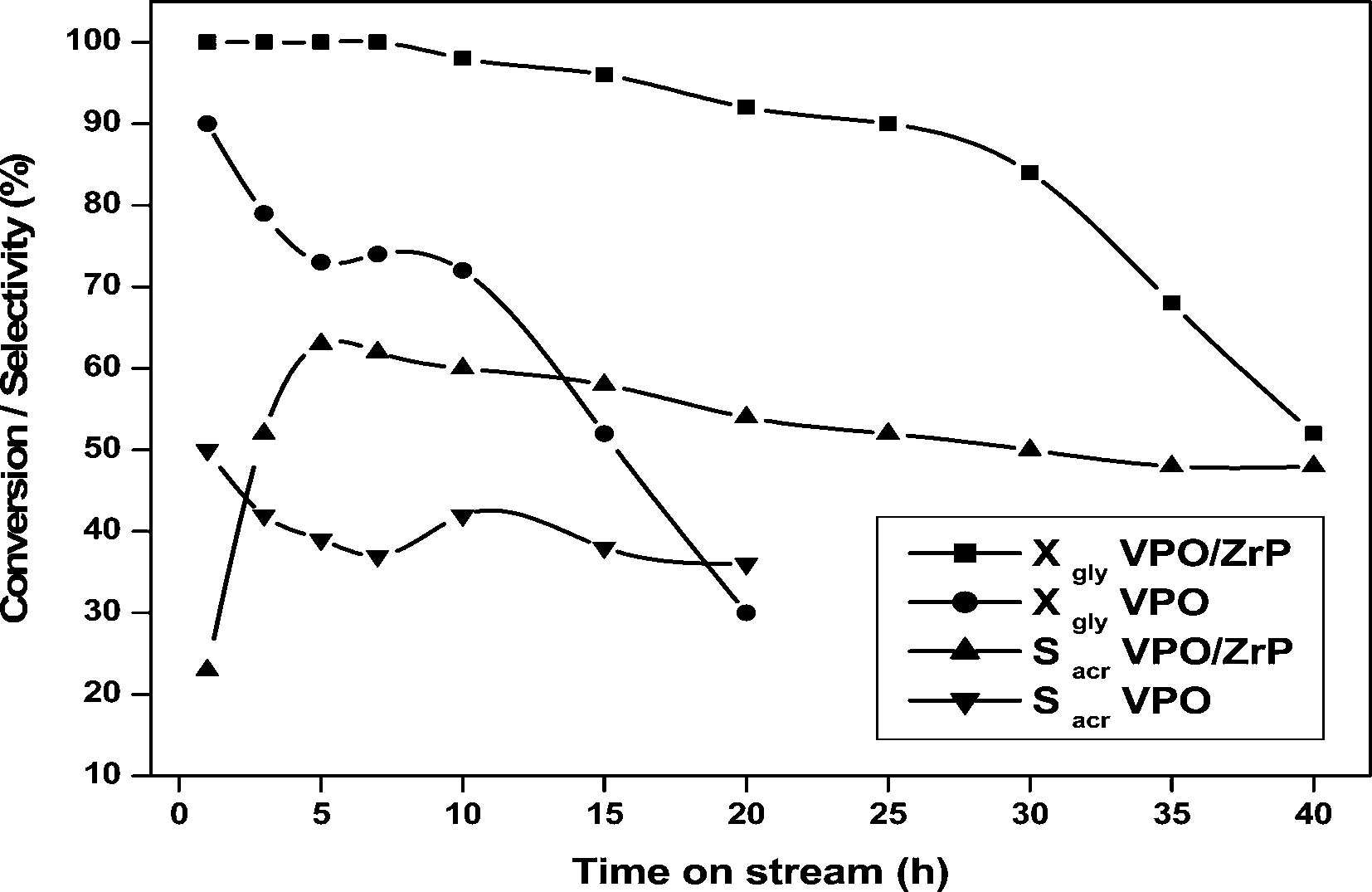 | ||
| Fig. 11 Time on stream over Pure VPO and 20 wt% VPO/ZrP by passing N2 and air flow. | ||
| Sample No. | Catalyst | Calcination temperature (°C) | Composition of fed gases (molar ratio) | Glycerol conversion (mol%) | Acrolein selectivity (mol%) | References |
|---|---|---|---|---|---|---|
| 1. | VPO | 550 | N2:O2:H2O:glycerol 57.2:12.7:28.7:1.4 |
100 | 41 | 11 |
| 2. | VPO | 500 | N2:O2:H2O:glycerol 66.6:1.7:30.3:1.5 |
47 | 39 | 12 |
| 3. | VPO | 600 | N2:O2:H2O:glycerol 66.6:1.7:30.3:1.5 |
61 | 33 | 12 |
| 4. | 20% VPO/ZrP | 550 | gly:H2O:N2:air 1.37:50.0:32.12:10.7 |
100 | 65 | — |
The acid strength and acid amount of the catalyst were significant for determining the catalytic activity. Inappropriate acidity of the catalyst may cause poor results, such as lower selectivity accompanied with severe coke deposition, and short catalyst life time. From Fig. 12 the weak moderate acidic sites or total acidity are increasing up to 20 wt% VPO/ZrP after that it decreases significantly. The selectivity of the acrolein also follows the same order. This result suggests that the weak moderate acidic sites are necessary for the formation of acrolein. Though pure zirconium phosphate shows more acidity than the supported catalyst, the acrolein selectivity is low because of the presence of Lewis acidic sites. These Lewis acidic sites can lead to single dehydration to form larger amount of hydroxyl acetone. In the case of pure VPO catalyst the maximum conversion and selectivity is obtained in the early hour (TOS 1 h) and conversion and selectivity sharply decreases with time (Fig. 11) due to the formation of hydroxyl acetone, allyl alcohol and unidentified products. This could be due to the strong acid sites favours side reactions among the reactant and the products such as acrolein etc. Because of these side reactions coke is deposited on the active acidic sites leading to the decrease of the conversion of the glycerol.
 | ||
| Fig. 12 Weak moderate acidic strength, acrolein selectivity vs. VPO loadings. | ||
To understand the role of acidic sites on the glycerol dehydration reaction, the ex-situ pyridine adsorbed FTIR analysis measurements were carried out and the results are shown in Fig. 9. From the literature it is observed that the Brønsted acidic sites are responsible for the acrolein selectivity.53 In fact, Brønsted acid sites directly protonate the secondary hydroxyl group of glycerol, which finally leads to the formation of acrolein. The increase of acrolein selectivity with VPO loadings is due to the increase of Brønsted acidic sites until 20% VPO/ZrP after that there is no significant change in the acrolein selectivity.
The spent sample of 20 wt% VPO/ZrP after 40 h reaction time was characterised by XRD, TPD of ammonia and the ex-situ pyridine FTIR analysis to find the information about the changes of the active sites during the reaction. The XRD results of 20 wt% VPO on zirconium phosphate after reaction of 40 h shows the X-ray diffraction lines at 2θ = 23.00°, 28.45°, and 29.95° corresponding to the vanadyl pyrophosphate. This result shows that the crystallite size of VPO catalyst increases after 40 h reaction time. It might be due to the severe reaction conditions employed which results rapid deactivation of the catalyst. The results of TPD of ammonia of fresh and spent samples are shown in Fig. 13. This result suggests that the acidity of the catalyst decreases when compared to the fresh samples as a result the conversion and selectivity decreases considerably. However, the peak position during TPD analysis also changed abruptly and the temperature maximum of moderate acidic site peak also shifted to higher position. The ex-situ pyridine FTIR analysis of the spent and fresh samples is shown in Fig. 14. As can be seen from Fig. 14 the Brønsted acidic sites of the spent catalyst decreases significantly. However, a marginal decrease of Lewis acidic sites is noticed. The intensity of both acidic sites (B + L) also decreases as a result the conversion and selectivity decreases after 40 h TOS.
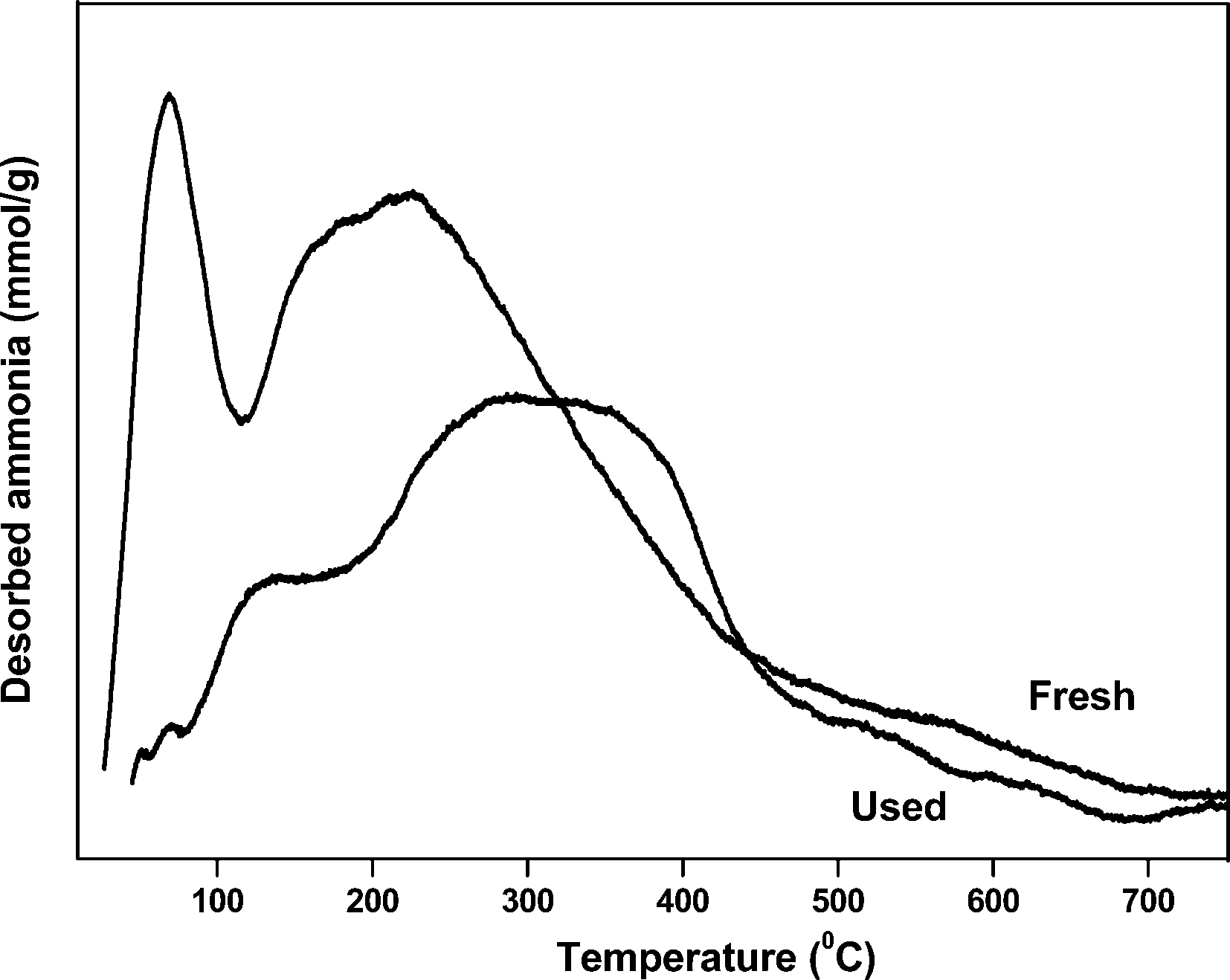 | ||
| Fig. 13 TPD of ammonia profiles of fresh and spent samples of 20 wt% VPO/ZrP. | ||
 | ||
| Fig. 14 Ex-situ pyridine adsorbed FTIR of fresh and spent samples of 20 wt% VPO/ZrP. | ||
4. Conclusion
In summary, a series of porous zirconium phosphate supported VPO catalysts were prepared by the solid–solid wetting method for acid catalysed glycerol dehydration reaction and these catalysts were characterised by various spectroscopic and adsorption techniques. The characterization results showed that the VPO species is well dispersed on the zirconium phosphate and presents mainly in the form of vanadyl pyrophosphate phase along with small amount of vanadium orthophosphate phase. The acidity of the catalyst plays a detrimental role in the formation of acrolein during glycerol dehydration reaction. The supported VPO catalysts showed better conversion and selectivity towards acrolein than the pure VPO catalyst. This is due to the appropriate acidic properties, higher surface area and higher pore diameter in the supported VPO catalysts. Among the different catalysts examined for vapour phase glycerol dehydration, 20 wt% VPO/ZrP showed the best activity towards glycerol conversion (100%) and acrolein selectivity (65%) compared to other catalysts. The spent catalyst was also characterised by TPD of ammonia and ex- situ pyridine FTIR analysis. These results show that there is a significant decrease in the acidity and Brønsted acidic sites of the spent samples leading to a decrease in the conversion and selectivity of the catalyst after 40 h reaction.Acknowledgements
N. P. R., G. S. R. and V. P. K. thank CSIR, New Delhi for the award of Senior Research Fellowships.References
- Y. Zheng, X. Chen and Y. Shen, Chem. Rev., 2008, 108, 5253 CrossRef CAS PubMed.
- B. Katryniok, S. Paul, V. Belliere Baca, P. Rey and F. Dumeignil, Green Chem., 2010, 12, 2079 RSC.
- Y. Gua, N. Cuia, Q. Yua, C. Li and Q. Cuib, Appl. Catal., A, 2012, 429–430, 9 CrossRef PubMed.
- M. Massa, A. Andersson, E. Finocchio, G. Busca, F. Lenrick and L. R. Wallenberg, J. Catal., 2013, 297, 93 CrossRef CAS PubMed.
- K. Omata, S. Izumi, T. Murayama and W. Ueda, Catal. Today, 2013, 201, 7 CrossRef CAS PubMed.
- W. Suprun, M. Lutecki, T. Haber and H. Papp, J. Mol. Catal. A: Chem., 2009, 309, 71 CrossRef CAS PubMed.
- S. H. Chai, H. P. Wang, Y. Liang and B. Q. Xu, Green Chem., 2008, 10, 1087 RSC.
- S. H. Chai, H. P. Wang, Y. Liang and B. Q. Xu, Appl. Catal., A, 2009, 353, 213 CrossRef CAS PubMed.
- J. L. Dubois, C. Duquenne and W. Holderich, FR2884818, 2006, to Arkema (FR).
- J. L. Dubois, C. Duquenne and W. Holderich, FR2882052, 2006, to Arkema (FR).
- F. Wang, J. L. Dubois and W. Ueda, J. Catal., 2009, 268, 260 CrossRef CAS PubMed.
- F. Wang, J. L. Dubois and W. Ueda, Appl. Catal., A, 2010, 376, 25 CrossRef CAS PubMed.
- Catal. Today, ed. G. Centi, 1993, vol. 16, l Search PubMed.
- G. Centi, F. Trifiro, J. R. Ebner and V. M. Franchetti, Chem. Rev., 1988, 88, 55 CrossRef CAS.
- G. Busca, G. Centi, F. Trifiro and V. Lorenzelli, J. Phys. Chem., 1986, 90, 1337 CrossRef CAS.
- F. Cavani and F. Trfiro, Appl. Catal., A, 1997, 157, 195 CrossRef CAS.
- G. Busca, G. Ramis and V. Lorenzelli, J. Mol. Catal., 1989, 50, 231 CrossRef CAS.
- L. M. Cornaglia, E. A. Lombardo, J. A. Anderson and J. L. Garcia Fierro, Appl. Catal., A, 1993, 100, 37 CrossRef CAS.
- L. M. Cornaglia and E. A. Lombardo, Stud. Surf. Sci. Catal., 90, 429 CrossRef CAS.
- S. Ben Abdelmalek, H. Batis and A. Ghorbel, J. Soc. Alger. Chim., 1993, 3, 265 Search PubMed.
- M. Abon and J. C. Volta, Appl. Catal., A, 1997, 157, 173 CrossRef CAS.
- Y. Kamiya, J. Jpn. Pet. Inst., 2003, 46, 62 CrossRef.
- K. E. Birkeland, S. M. Babitz, O. K. Bethke, H. H. Kung, G. W. Coulston and S. R. Bare, J. Phys. Chem. B, 1997, 101, 6895 CrossRef CAS.
- R. A. Overbeek, A. R. C. J. Pekelharing, A. J. Van Dillen and J. W. Geus, Appl. Catal., A, 1996, 135, 231 CrossRef CAS.
- R. A. Overbeek, P. A. Warringa, M. J. D. Combag, L. M. Visser, A. J. Van Dillen and J. W. Geus, Appl. Catal., A, 1996, 135, 209 CrossRef CAS.
- J. Marc, C. C. Ledoux, P. H. Cuong, T. Vincent, K. Kostantinos, M. Patrick and L. Jan, J. Catal., 2001, 203, 495 CrossRef.
- J. M. C. Buenoa, G. K. Bethkeb, M. C. Kung and H. H. Kung, Catal. Today, 1998, 43, 101 CrossRef.
- Z. Q. Zhou, H. Y. Xu, W. J. Ji and Y. Chen, Catal. Lett., 2004, 96, 221 CrossRef CAS.
- X. K. Li, W. J. Ji, J. Zhao, Z. B. Zhang and C. T. Au, J. Catal., 2006, 238, 232 CrossRef CAS PubMed.
- X. K. Li, W. J. Ji, J. Zhao, Z. B. Zhang and C. T. Au, Appl. Catal., A, 2006, 306, 8 CrossRef CAS PubMed.
- W. Nie, Z. Wang, W. Ji, Y. Chen and C. T. Au, Appl. Catal., A, 2003, 244, 265 CrossRef CAS.
- C. Y. Xiao, X. Chen, Z. Y. Wang, W. J. Ji, Y. Chen and C. T. Au, Catal. Today, 2004, 93–95, 223 CrossRef CAS PubMed.
- R. M. Feng, X. J. Yang, W. J. Ji, Y. Chen and C. T. Au, J. Catal., 2007, 246, 166 CrossRef CAS PubMed.
- H. Atia, U. Armbruster and A. Martin, J. Catal., 2008, 258, 71 CrossRef CAS PubMed.
- A. Clearfield and D. S. Takur, J. Catal., 1980, 65, 185 CrossRef CAS.
- K. Segawa, Y. Kurusu, Y. Nakajima and M. Kinoshita, J. Catal., 1985, 94, 491 CrossRef CAS.
- A. La Ginestra, P. Partono, M. L. Berardelli, P. Galli, C. Ferragina and M. A. Massucci, J. Catal., 1987, 103, 346 CrossRef CAS.
- Ch. Srilakshmi, K. Ramesh, P. Nagaraju, N. Lingaiah and P. S. Sai Prasad, Catal. Lett., 2006, 106, 115 CrossRef CAS.
- T. Z. Ren, Z. Y. Yuan and B. L. Su, Chem. Commun., 2004, 2730 RSC.
- N. Yamamoto, N. Hiyoshi and T. Okuhara, Chem. Mater., 2002, 14, 3882 CrossRef CAS.
- K. Narasimha Rao, A. Sridhar, A. F. Lee, S. J. Tavener, N. A. Young and K. Wilson, Green Chem., 2006, 8, 790 RSC.
- V. N. Kalevaru, N. Madaan and A. Martin, Appl. Catal., A, 2011, 391, 52 CrossRef CAS PubMed.
- G. J. Hutchings, J. Mater. Chem., 2009, 19, 1222 RSC.
- G. Koyano, T. Okuhara and M. Misono, J. Am. Chem. Soc., 1998, 120, 767 CrossRef CAS.
- Z. Y. Yuan, A. Vantomme, A. Leonard and B. L. Su, Chem. Commun., 2003, 1558 RSC.
- B. H. Sakakini, Y. H. Taufiq Yap and K. C. Waugh, J. Catal., 2000, 189, 253 CrossRef CAS.
- C. J. Kiely, C. J. Sajip, I. J. Ellison, M. T. Sananes, G. J. Hutchings and J. Volta, Catal. Lett., 1995, 33, 357 CrossRef CAS.
- Y. H. Taufiq Yap, M. H. Looi, K. C. Waugh, M. Z. Hussein, Z. Zainal and R. Samsuddin, Catal. Lett., 2001, 74, 99 CrossRef CAS.
- F. Cavani, S. Ligi, T. Monti, F. Pierelli, F. Trifiro, S. Albonetti and G. Mazzoni, Catal. Today, 2000, 61, 203 CrossRef CAS.
- A. Martin, C. Janke and V. N. Kalevaru, Appl. Catal., A, 2010, 376, 13 CrossRef CAS PubMed.
- M. Ziyada, M. Rouimia and J. L. Portefaixb, Appl. Catal., A, 1999, 183, 93 CrossRef.
- K. Tanabe, M. Misono, Y. Ono and H. Hattori, Stud. Surf. Sci. Catal., 51, 5 Search PubMed.
- A. Alhanash, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., A, 2010, 378, 11 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c3cy00430a |
| This journal is © The Royal Society of Chemistry 2014 |