 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond post-synthesis modification: evolution of metal–organic frameworks via building block replacement
Pravas
Deria
a,
Joseph E.
Mondloch
a,
Olga
Karagiaridi
a,
Wojciech
Bury
*ab,
Joseph T.
Hupp
*a and
Omar K.
Farha
*ac
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: j-hupp@northwestern.edu; o-farha@northwestern.edu
bDepartment of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: wojbur@ch.pw.edu.pl
cDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 11th April 2014
Abstract
Metal–organic frameworks (MOFs) are hybrid porous materials with many potential applications, which intimately depend on the presence of chemical functionality either at the organic linkers and/or at the metal nodes. Functionality that cannot be introduced into MOFs directly via de novo syntheses can be accessed through post-synthesis modification (PSM) on the reactive moieties of the linkers and/or nodes without disrupting the metal–linker bonds. Even more intriguing methods that go beyond PSM are herein termed building block replacement (BBR) which encompasses (i) solvent-assisted linker exchange (SALE), (ii) non-bridging ligand replacement, and (iii) transmetalation. These one-step or tandem BBR processes involve exchanging key structural components of the MOF, which in turn should allow for the evolution of protoMOF structures (i.e., the utilization of a parent MOF as a template) to design MOFs composed of completely new components, presumably via single crystal to single crystal transformations. The influence of building block replacement on the stability and properties of MOFs will be discussed, and some insights into their mechanistic aspects are provided. Future perspectives providing a glimpse into how these techniques can lead to various unexplored areas of MOF chemistry are also presented.
1. Introduction
Metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), define a class of hybrid solids consisting of periodic coordination networks containing multitopic organic linkers and metal-ion-containing nodes or secondary building units (SBUs).1–5 These crystalline, highly porous solids can possess ultra-high Brunauer–Emmett–Teller (BET) surface areas (∼7000 m2 g−1)6–9 and thus have garnered much interest for gas capture, separation and storage.10–17 However, the modular nature of the MOF structures, stemming from the tunability of the linker and metal SBUs, has also sparked interest in applications involving catalysis,18–20 light harvesting,21–25 sensing,26 optical luminescence,27,28 ionic conductivity,29–32 and non-linear optical behavior.33 Access to a wide range of potential applications is intimately tied to the ability to present a similarly wide range of chemical functionality within MOFs. Incorporation of functionality is straightforward if appropriately derivatized organic linkers can be synthesized and then directly employed in solvothermal materials syntheses. In some cases, however, challenges due to limited linker solubility, chemical stability, thermal stability, or functional group compatibility can preclude direct synthesis. Similarly, undesired reactions between linker functional groups and metal ions can lead to undesired products, to the exclusion of desired materials. Overly strong metal-ion–linker bonding can lead to formation of amorphous kinetic products to the exclusion of crystalline compounds.34 Finally, undesired polymorphs of crystalline MOFs may be obtained to the exclusion of desired, and typically higher energy (i.e., thermodynamically less stable), polymorphs.35 These issues have been a limiting factor in MOF synthesis, which in turn has truncated the range of accessible chemical functionality that can be incorporated into MOFs.36–38Post-synthesis modification (PSM), extensively reviewed by Cohen38,39 and by Burrows,40 effectively removes the potential for functional-group interference during MOF assembly. PSM involves heterogeneous chemical reactions to functionalize preassembled MOF structures38,39via:
(i) modification of linkers including (a) covalent modification (or elaboration), (b) deprotection of linker functionality (including demetalation),38,39,41–44 and (c) electron addition (reduction) and concomitant incorporation of charge compensating ions.45
(ii) modification of metal-containing nodes including (a) incorporation of non-framework or pendant ligands via dative bonding to coordinatively unsaturated metal sites,38,39,46–50 (b) alkyl or silyl grafting to oxygen atoms in metal-oxide nodes,51,52 and (c) attachment of metal ions or complexes at node oxygen sites via atomic layer deposition (ALD)53 or via reaction with organometallic species in dry solutions.53,54
Beyond these extensively studied PSM approaches, various conceptually different post-synthesis routes are now emerging. Building block replacement (BBR) involves replacement of key structural components of the MOF including (i) solvent-assisted linker exchange (SALE), (ii) non-bridging ligand replacement, and (iii) transmetalation at nodes or within linkers. (Fig. 1 shows some examples of MOFs used in the BBR processes discussed below.) BBR is intriguing given that it opens up a very broad strategy for the synthesis of isostructural MOFs that in turn should enable functional MOF chemistry. Both one-component and tandem building block replacement processes can provide access to otherwise unattainable MOF structures and functionality. They also may provide access to MOFs featuring gradient compositions and chemical properties.
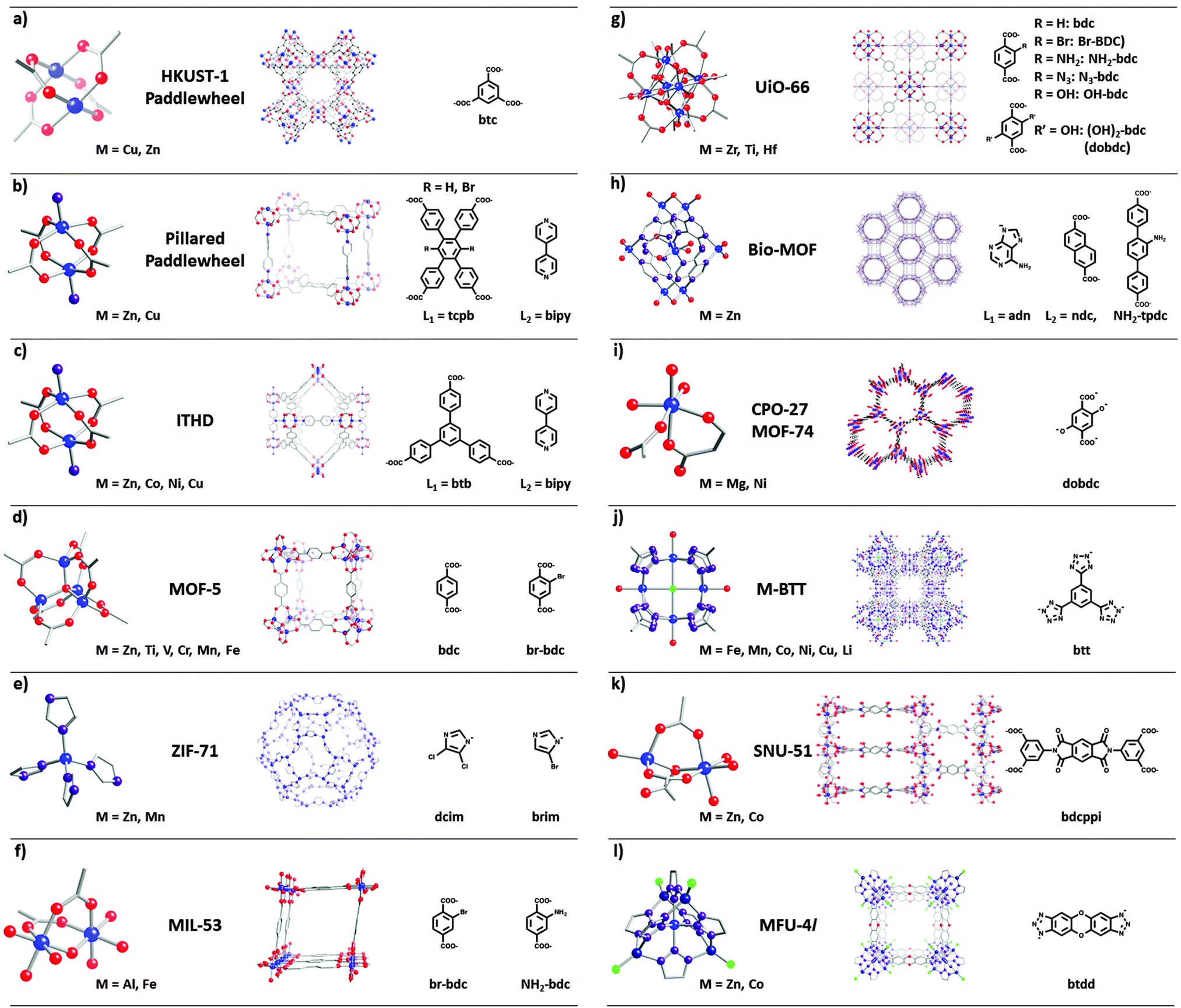 | ||
| Fig. 1 Lattice structures (middle) and corresponding SBUs (metal nodes (left), and organic linkers (right)) of some of the MOFs discussed in this review. (Atom definition: blue – metal, red – oxygen, purple – nitrogen, grey – carbon, green – chlorine.) | ||
Solvent-assisted linker exchange, (SALE),55–57 often offers complete exchange of one organic linker for another possessing new functionality. Likewise, while transmetalation58 provides a route to incorporate desired metal ions that are difficult to incorporate via de novo synthesis, replacement of non-bridging charge-balancing ligands represents an efficient way to engender new functional chemistry. In general, BBR reactions involve heterogeneous, linker, ligand, or metal-ion exchange, via breaking and remaking chemical bonds within the parent MOF.55–59 Although it is difficult to be definitive, most examples of BBR to date appear to involve single crystal-to-single crystal transformations, rather than dissolution and recrystallization.
Finally, in many cases it is relatively easy to find more mild conditions for BBR than the solvothermal conditions typically required for direct syntheses of MOFs. In our experience, BBR reactions also often work well with reactant concentrations that are too low for effective direct synthesis. Thus BBR can be especially useful when desired linkers or non-bridging ligands are expensive, delicate, or inherently incompatible with typical conditions for direct MOF synthesis.
BBR has been used to take on some recurring challenges in MOF synthesis, including: (i) preparing MOFs that appear to be inaccessible by direct methods; (ii) engineering the pores or nodes within MOFs (including controlling the pore volume, environment, aperture size, and chemical stability); and (iii) imparting or enhancing desired functional behaviour, including catalysis, selective gas sorption, ionic conductivity, redox conductivity, and mediating site-isolated chemistry. In addition, we discuss some of the key chemical factors controlling the extent, or even the feasibility, of BBR, both from the perspective of gaining a fundamental understanding and of discovering useful and transferable design or reaction rules. BBR reactions occurring only at or near the external surface of a MOF crystal60–63 point to an intriguing further level of potential structural and functional complexity; however those studies are beyond the scope of the review. Finally, we briefly speculate on possible new ways in which BBR may prove synthetically or functionally useful.
2. Solvent-assisted linker exchange
While PSM is often effective for circumventing problems associated with direct MOF preparation (vide supra),38,39 replacement of linkers is another viable strategy. It has variously been termed “stepwise synthesis”,64 “bridging linker replacement”,64 “post-synthetic exchange”,59 isomorphous ligand replacement,65 and stepwise ligand exchange.64 We prefer the term “SALE”55–57,64–74 as it highlights the importance of the solvent during linker exchange. Conceptually SALE occurs at the solid–solution interface; a parent MOF is placed in a solution containing a second linker and a daughter MOF retaining the parent MOF topology is obtained. Similar linker exchange approaches have been utilized in metal diphosphonate-pillared zirconium phosphate,75 cross-linked organophosphazene polymers,76 and supramolecular coordination complexes, including metal–organic polyhedra,77,78 but these are beyond the scope of this review.2.1. Pore engineering
One might suspect smaller pore volumes to be readily obtainable via SALE—the diffusion of smaller linkers should be facile into pores of larger dimension (vide infra). In contrast, SALE of longer linkers might be significantly more difficult given presumably slower diffusion as well as the necessity for significant structural changes within the framework upon exchange. Recently however, Karagiaridi et al. demonstrated that linkers of increasing length could be introduced into the pillared-paddlewheel MOF, SALEM-5 [Zn2(Br-tcpb)(dped)], via SALE (Fig. 2).56SALEM-5 contains layers of Br-tcpb linker and the 9 Å dped pillars which could be replaced by dipyridyl pillars of 11 Å tmbbp (SALEM-6), 14 Å nbp (SALEM-7) and 17 Å tmbebp (SALEM-8) (Fig. 2). In the most striking example (i.e., SALEM-5 with dped pillar to SALEM-8 with tmbebp pillar), the c-axis of the resultant unit cell expands from 17.75 to 23.50 Å. Likewise, Rosi and co-workers demonstrated that SALE of longer linkers is possible in MOFs containing carboxylate linkers, specifically during the transformation of Bio-MOF-101 (Fig. 1h), which consists of the zinc-adeninate clusters (Zn8(adn)4O28+ periodically linked with ndc linker), to Bio-MOF-103 (Zn8(adn)4O28+ periodically linked with NH2-tpdc linker).79 In this series the unit cell was increased from 62.04 to 82.25 Å. Notably, most MOFs with increased pore volume could not be prepared de novo.
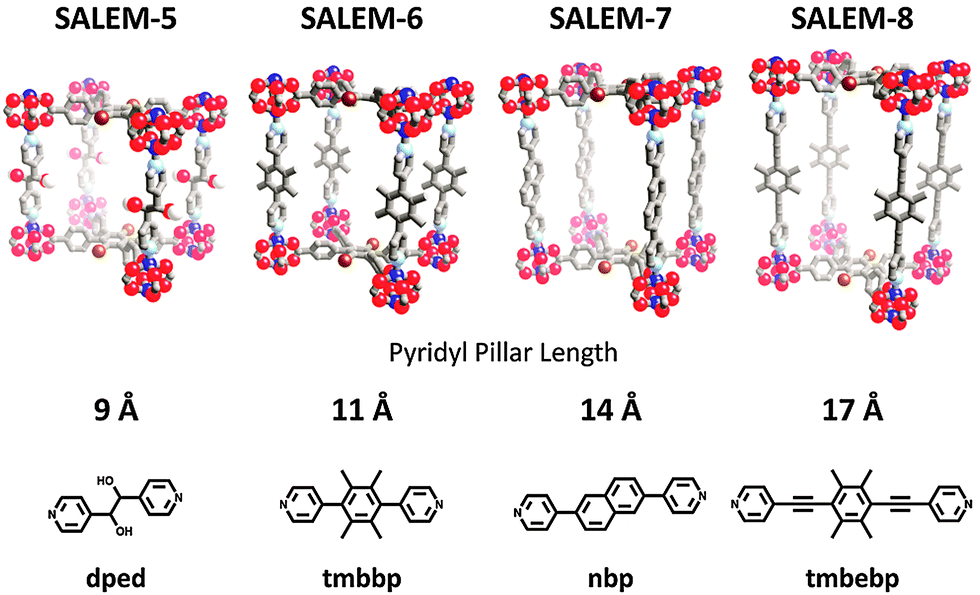 | ||
| Fig. 2 Increasing pore sizes in an isoreticular series of pillared-paddlewheel MOFs synthesized via SALE. The value in Å represents the length of the dipyridyl linkers utilized for SALE on the parent MOF SALEM-5. | ||
Framework catenation can also drastically change (diminish) MOF pore volumes. Bury et al. found that SALE could be used to access the non-catenated version of an otherwise catenated material. The SALE starting point was the non-catenated pillared-paddlewheel compound DO-MOF [Zn2(tcpb)(dped); see Fig. 1b for a related structure].67DO-MOF contains 2-D sheets of the zinc-coordinated tetracarboxylate linker, tcpb, separated by the dipyridyl pillar, dped. In contrast, the tcpb linker with dipyridyl bipy and abp pillars yields two-fold catenated structures. However, DO-MOF, under SALE conditions with bipy and abp replaces the dped pillars and yields the non-catenated MOFs containing bipy and abp ligands. Control over catenation was confirmed via thermogravimetric analysis as well as structural modelling via powder X-ray diffraction (PXRD). This study delineates an interesting alternative avenue to prepare non-catenated MOFs from those that exhibit catenation.
Karagiaridi et al. also demonstrated that pore functionality could be readily controlled in another highly sought after class of MOFs, namely zeolitic imidazolate frameworks (ZIFs).55 The eim linkers in {Cd(eim)2} (CdIF-4), a ZIF possessing RHO topology and Cd2+ nodes, could be exchanged for nim and mim linkers to form {Cd(nim)2} (CdIF-9), and {Cd(mim)2} (SALEM-1) respectively. The SALE reactions that occur between CdIF-4, CdIF-9 and SALEM-1 are summarized in Fig. 3. Importantly the studies from Kim et al., as well as Karagiaridi et al., demonstrate that some of the most robust MOF structures known are indeed amenable to SALE.
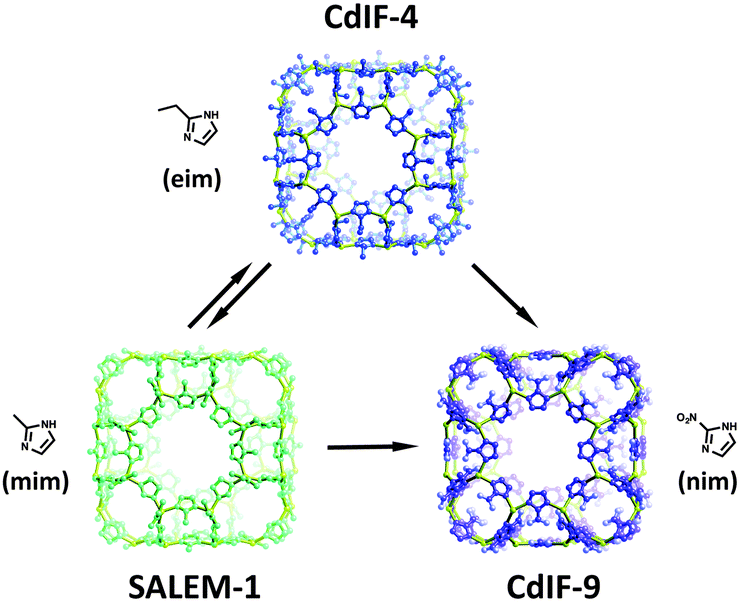 | ||
| Fig. 3 Pore environment modification through SALE in ZIFs. | ||
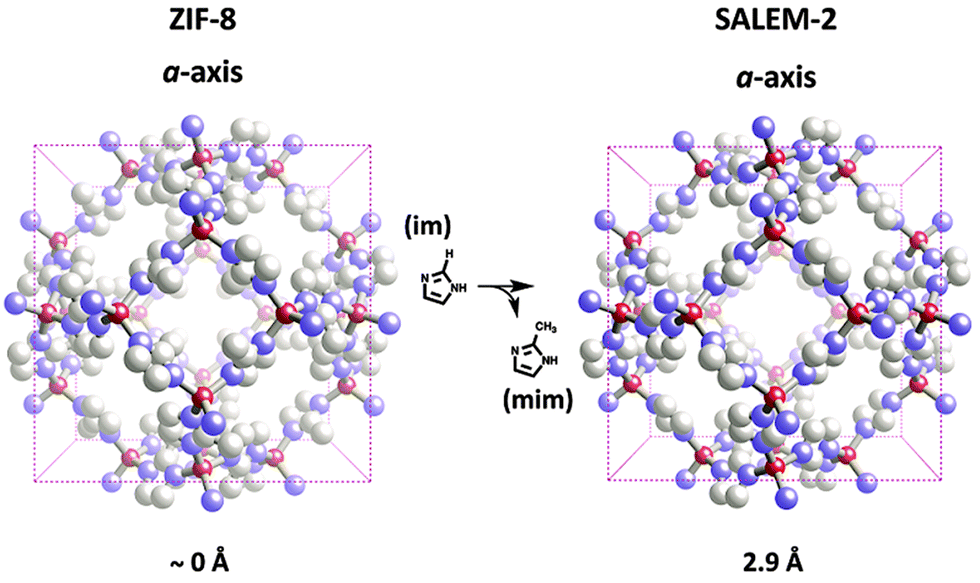 | ||
| Fig. 4 Aperture modulation in ZIF-8 through SALE. | ||
2.2. Enabling functional MOF chemistry through SALE
Returning back to UiO-66 (Fig. 1g), Pullen et al. utilized SALE to incorporate the linker FeFe(dcbdt)(CO)6 into UiO-66.73 The FeFe(dcbdt)(CO)6 linker resembles the active site of known FeFe hydrogenase enzymes which are highly active H+ reduction catalysts.81,82 In combination with a photosensitizer ([Ru(bpy)3]2+) and a sacrificial electron donor (ascorbate) all under illumination, the FeFe(dcbdt)(CO)6 containing UiO-66 derivative is an active H2 evolution catalyst (albeit largely at the surface of the MOF; Fig. 5). Pullen et al.'s results demonstrate the use of SALE to incorporate catalytically active moieties for the conversion of solar energy into useable chemical fuels within MOFs.
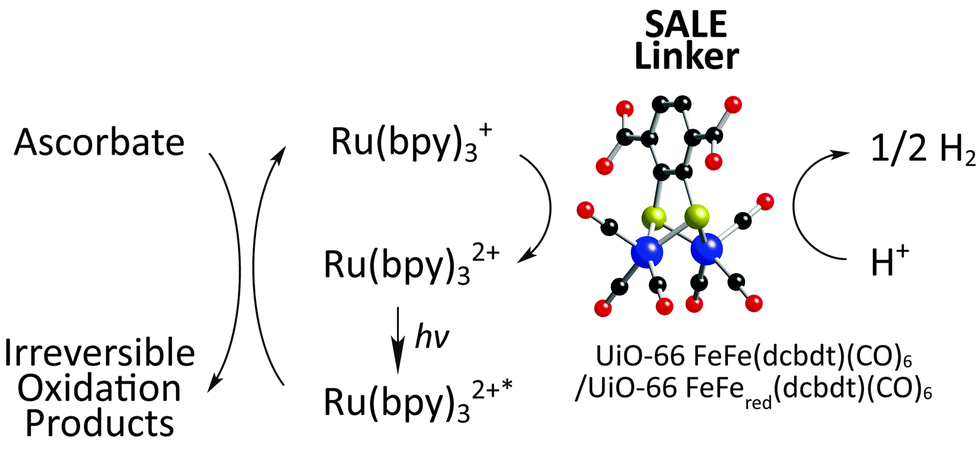 | ||
| Fig. 5 Schematic representation of photocatalytic proton reduction process. The atoms in the linker utilized for SALE are defined as: red – oxygen, black – carbon, yellow – sulfur, and purple – Fe. Ru(bpy)32+ and ascorbate were employed as photosensitizer and sacrificial electron donor, respectively. | ||
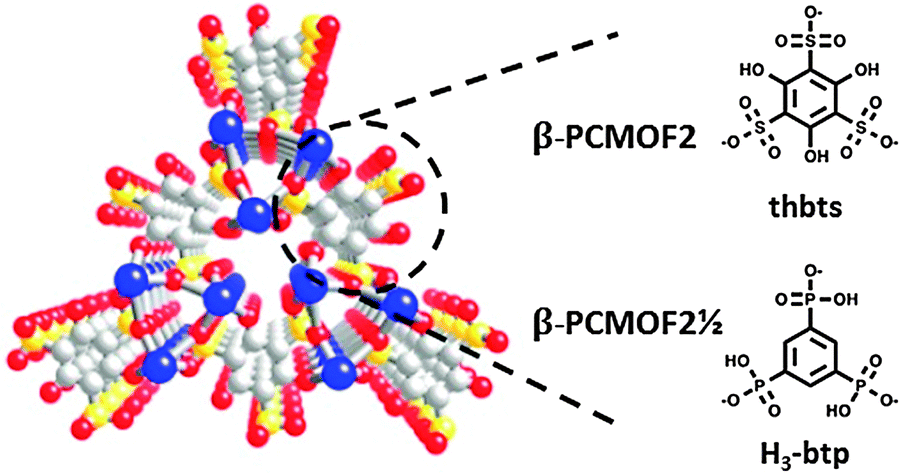 | ||
| Fig. 6 Structure of β-PCMOF2 and β-PCMOF2½. The atoms are defined as: blue – sodium, red – oxygen, grey – carbon, and yellow – sulphur/phosphorus. | ||
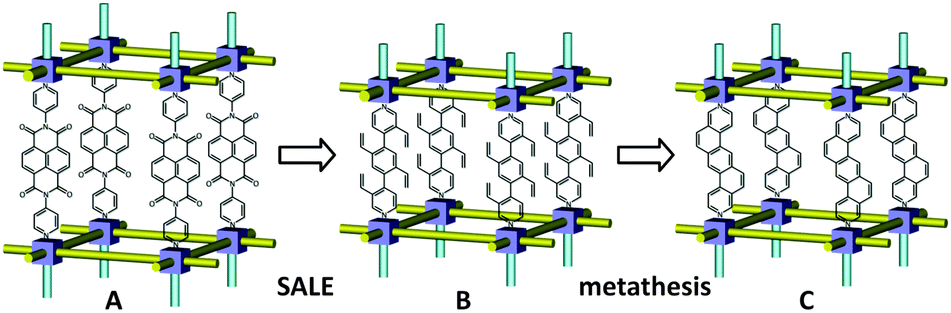 | ||
| Fig. 7 Isolating reaction sites through SALE. SALE in Br-YOMOF (A) resulting in SALEM-14 (B), where the isolated vinyl groups, followed by post-synthesis Grubbs catalysis reaction, provide PAH-MOF-1 (C). | ||
2.3. Feasibility of SALE: chemical principles
Recent experiments have given insight into some fundamental aspects of SALE. For most of the cases discussed above researchers have concluded that SALE is occurring in a single-crystal-to-single-crystal (rather than dissolution-reassembly) manner; however, more work is required to realize a complete mechanistic picture of SALE. As expected a fundamental understanding will take both thermodynamics (e.g., pKa of incoming linkers) and kinetics (e.g., diffusion of linkers) into consideration and as expected the role of the solvent merits considerable attention.Another kinetic aspect that deserves evaluation is the role of crystallite size on the rate of SALE. In their work on RPM systems, Takaishi et al. experimented with varying the crystal size of the MOF by grinding and monitoring the rate of the reaction.70 They discovered that the ground crystals reached SALE equilibrium more rapidly than the larger crystals indicating the salient role of diffusion in the SALE process. This finding was further corroborated by scanning electron microscopy – energy dispersive X-ray spectroscopy (SEM-EDX), which established that the concentration of Sn (the metal present in the incoming metallo-porphyrin based dipyridyl pillars) was much higher around the crystal surface than in the crystal core.
3. Non-bridging ligand replacement
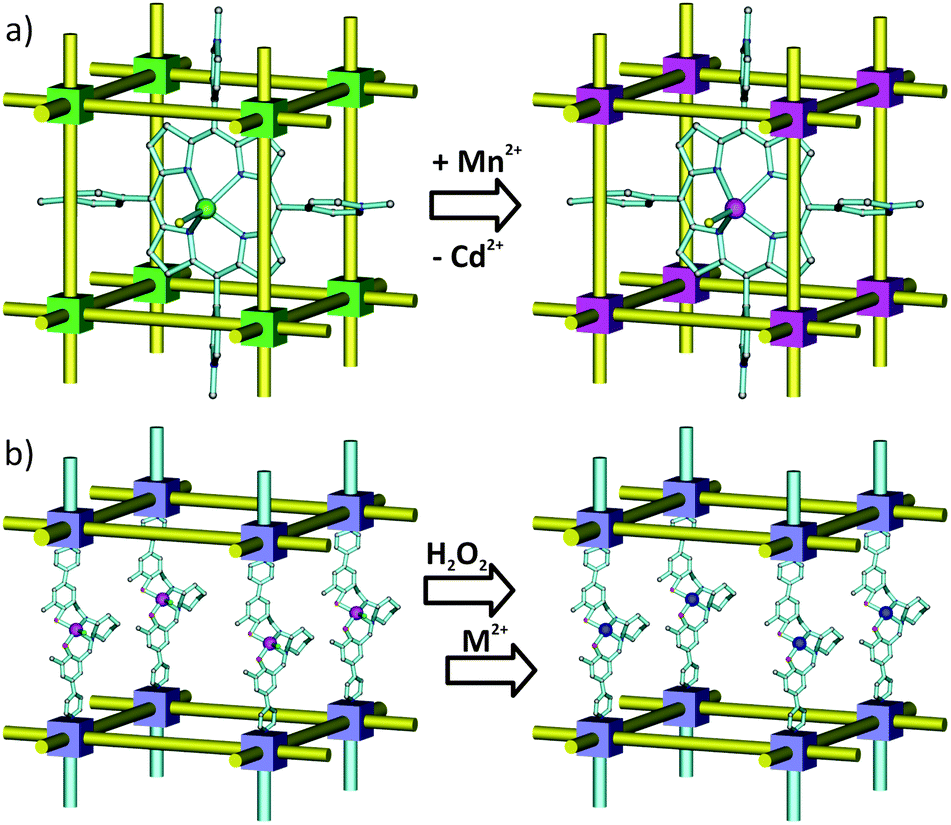
While MOF nodes can be composed of single metal ions, they can also be comprised of discrete metal-containing clusters (see Fig. 1 for examples). These metal-cluster containing nodes offer an opportunity to introduce a battery of new functionality through the replacement of charge balancing non-bridging ligands. While typical metal node functionalization utilizes dative bonding to coordinatively unsaturated metal sites,46–50 non-bridging ligand replacement relies on either acid–base or ligand exchange chemistry between the existing anionic ligand on the MOF node and the incoming anionic ligand. The result is the introduction of functional groups as charge compensating and strongly bound moieties to the MOF node via ionic bonding. Like SALE, non-bridging ligand replacement is expected to be governed by many of the factors that govern SALE including solvent selection, diffusion, and pKa of the conjugate acids (vide infra).This idea was recently realized when Deria et al. reported a new BBR technique called solvent-assisted ligand incorporation (SALI) for Zr-MOFs. The authors reported the efficient incorporation of carboxylate-based organic functional groups (CFGs) into the mesoporous MOF, NU-1000 (Fig. 8a), consisting of [Zr6(μ3-OH)8(–OH)8]8+ nodes and TBAPy linkers.84 The incorporation of new carboxylate based functionalities (CFGs) via SALI is encompassed by two boundary compositions of the Zr6-node – [Zr6(OH)16(RCOO)8] – before functionalization, and [Zr6(O)4(OH)4(RCOO)8(R′COO)4] – after the SALI process, which is governed by an acid–base equilibrium between the carboxylic acid moiety of the incoming ligand (R′COOH) and a pair of hydroxyl ligands of the node (forming a water molecule as a by-product). Notably, carboxylates (R′COO−) of weaker conjugate acids were found to be efficiently supplanted by those of stronger conjugate acids. Thus the distinctive feature of SALI, from a SALE reaction perspective, is that it does not involve replacement of a bridging linker, but instead a charge compensating and non-bridging ligand (e.g. hydroxide or a carboxylate ligand). The authors incorporated perfluoroalkane functionality within the mesoporous channels of NU-1000 by utilizing perfluoroalkyl carboxylic acids of varying chain length (–CF3, –C3F7, –C7F15 and –C9F19). The resulting materials showed increased Qst values for CO2 and slightly less affinity for water vapor.84 Notably, this technique not only proved to be versatile, tolerates many functionalities, but is also shown to yield new materials that are robust and effective for secondary functionalization at the SALI-incorporated moiety.85
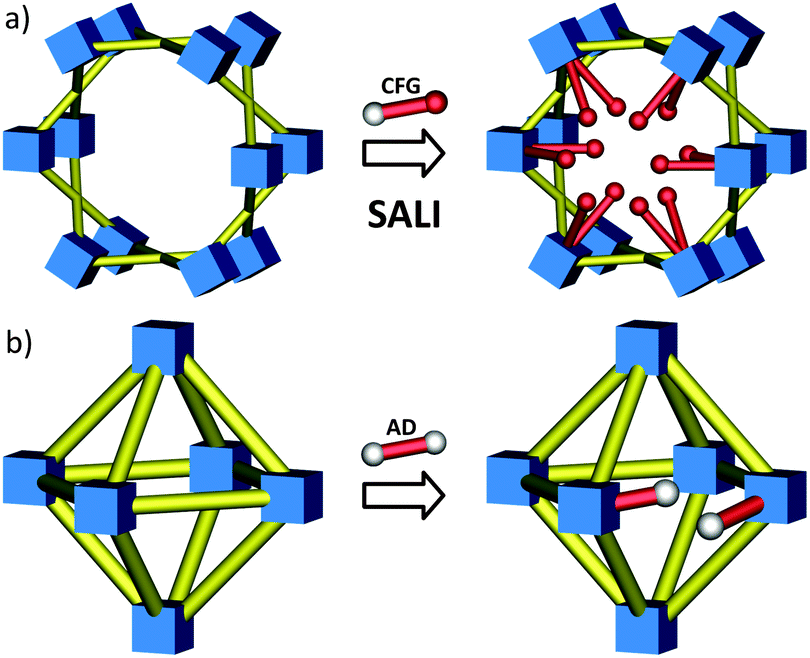 | ||
| Fig. 8 Schematic representation of (a) solvent-assisted linker incorporation (SALI) with CFGs and (b) linker replacement with alkanedioic acids in various Zr-MOFs. | ||
In view of their high thermal, mechanical, and chemical stability,86UiO-66-type materials constitute a similarly intriguing class of MOFs compounds, considering a comprehensive picture regarding the stability and reactivity of this group of MOF materials. For instance, it was demonstrated by Wu et al.87 and subsequently Katz et al.88 that depending on the synthetic conditions the UiO-66 framework (see Fig. 1g) may lack one or more structural elements54 (i.e., they may exhibit the missing linker phenomenon) without losing its integrity and topology; such systems can facilitate non-bridging ligand exchange. In a recent study, Suh and coworkers employed a slightly modified SALE approach to exchange the bdc linkers in UiO-66 material for various flexible alkanedioic acids (AD).89 As a result the authors obtained a series of modified UiO-66-ADn derivatives (Fig. 8b; ADn is defined as HOOC–(CH2)n–COOH, where n = 4, 6, 8, and 10). In contrast to a conventional SALE process, during the post-synthetic linker exchange, a single ditopic terephthalate linker (bdc) was replaced by two flexible ditopic alkanedioate ligands, leading to UiO-66 derivatives functionalized with pendant carboxylic groups. Given that UiO-66 materials can possess “missing linker”-defects (for UiO-66 consisting 12 linker nodes, a geometric surface area of 800 m2 g−1 was estimated88), it is quite possible that a part of the AD-functionalization could proceed via a non-bridging hydroxyl ligand replacement (similar to that involved in SALI; vide supra) in addition to the bdc linker replacement process as described by the authors. Using ideal adsorption solution theory (IAST), they demonstrated that for landfill gas separation (a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of CO2 and CH4), all of the UiO-66-ADns showed higher selectivity for CO2 over CH4 relative to UiO-66.
:50 mixture of CO2 and CH4), all of the UiO-66-ADns showed higher selectivity for CO2 over CH4 relative to UiO-66.
4. Transmetalation
In this section we will discuss reactions that involve post-synthesis replacement of MOF building blocks via transmetalation.58 Also known as “metal metathesis”90 or “metal-ion exchange”,91 transmetalation entails partial or complete replacement of the metal nodes (or sometimes extra-framework metal moieties) of a MOF by a reaction with a metal precursor (in most cases a solution of a metal salt) in a single crystal-to-single crystal fashion. The emergence of this synthetic pathway in MOFs has been relatively recent, with around thirty reports of successful cases;41,62,63,66,68,90–112 (including a few examples of complete replacement). Besides discussing how transmetalation engenders new MOF evolution and enables new functional chemistry, we analyze the current literature in order to provide a set of fundamental principles that govern successful metal replacement reactions. These principles appear to offer insight into why only a handful of transmetalation experiments lead to full metal exchange, whereas partial transmetalation is more common.4.1. Metal node engineering
Similarly, attempts via conventional solvothermal syntheses to create Cu2+, Co2+ and Ni2+ analogues of {[Zn7(btt)3(H2O)7]n–[Zn5(ppbotctc)3(H2O)5]n}, a unique interpenetrated cationic/anionic polyhedron-based double framework with square pyramidal, triangular-pyramidal, octahedral and triangular-paddlewheel Zn2+ nodes were unsuccessful;105 so were the efforts of Denysenko et al. to synthesize a Co2+-based MOF-5-like structure {Zn5Cl4(btdd)3} (MFU-4l; Fig. 1l) with large btdd linkers that they had obtained for Zn2+.94 To avoid tedious optimization, all the above research teams utilized transmetalation to successfully achieve their goals.
Certain metal ions are particularly challenging to incorporate into MOFs under standard solvothermal synthesis conditions. One set of examples consists of various elements in low-oxidation states – for example, Ti3+, V2+ and Cr2+. Standard solvothermal synthesis conditions (at elevated temperatures in the presence of air) often induce their oxidation.93 Small metal cations such as Ti4+ pose difficulties when it comes to incorporating them into structures that demand high coordination numbers, such as the {M6O4(OH)4(CO2)12} cluster of UiO-66 (see Fig. 1g).66 Conversely, it is not trivial to introduce metals that favor unusually high coordination numbers, e.g., lanthanides, into the low-density sodalite topology of {M3[(M4X)3L8]} (where metals are found in an octahedrally coordinated geometry; see Fig. 1j, and X = Cl or O and L = btt or hett).90 As discussed in more detail below, transmetalation has in several instances proven to be an effective way to meet these challenges and has helped expand the synthetic possibilities in MOF chemistry.
4.2. Introducing new chemical functionality in MOFs via transmetalation
 | ||
| Fig. 9 Partial transmetalation in Zn4O node within MOF-5. | ||
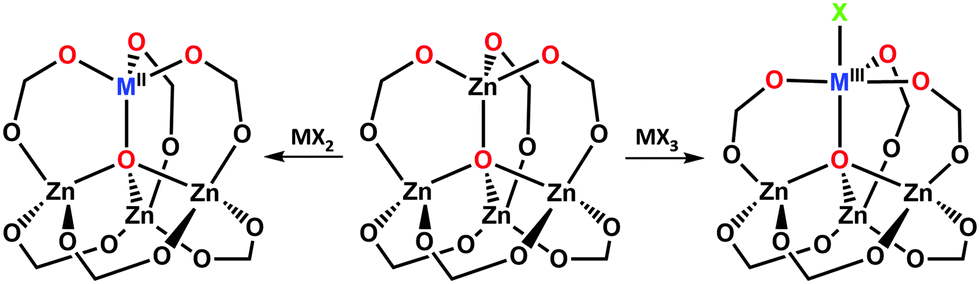
Transmetalation offers an attractive strategy for the incorporation of catalytically active metal centers in MOF linkers. The synthesis of catalytic MOFs featuring salen-41 or porphyrin-based104 linkers has particularly benefitted from this approach. Shultz et al. were able to expand a family of M-salen-containing pillared-paddlewheel MSO-MOF materials [Zn2(tcpb)(salen)] by a stepwise replacement of Mn3+ in the salen linker with Co2+, Zn2+, Cr2+, Cu2+, Ni2+ or Mn2+ (Fig. 10b) resulting in a series of potential catalysts.41 Similarly, Zhang et al. applied transmetalation to their “ship-in-a-bottle” material porph@MOM-10, a [meso-tetra(N-methyl-4-pyridyl)porphine tetratosylate] encapsulated in the {Cd6(bpt)4Cl4(H2O)4} framework, which contains catalytically inactive Cd2+ ions both in its nodes and in the porphyrin moiety.104 Replacement of Cd2+ throughout the material with Mn2+ afforded a product (Fig. 10a) that could catalyze the epoxidation of trans-stilbene with 75% conversion (compared to only 7% conversion provided by the original Cd2+ containing porph@MOM-10). Notably, the Mn2+ analogue proved inaccessible de novo.
 | ||
| Fig. 10 Transmetalation in (a) porph@MOM-10, and (b) MSO-MOF. Note that for the panel (b), the metalation was carried out after the demetalation of the parent MOF in a sequential manner. | ||
:1.000, whereas for the Cu+ analogue, a higher Qst value of 9.9 kJ mol−1 was measured even with an extremely low Cu/Mn ratio of 0.028:1.000 (note that for the parent material with Mn2+, a Qst of 10.1 kJ mol−1 was measured). This transmetalation study enabled the authors to measure, for the first time, the Qst values associated with hydrogen binding to Fe2+ and Ni2+ ions. The work of Dincǎ et al. demonstrates the wealth of information that can be obtained from studying isostructural MOFs functionalized by different metal ions – a goal that can be readily achieved by transmetalation, given its single crystal-to-single crystal nature.
Transmetalation has also been implemented for the improvement of CO2 sorption properties. It has been recently shown that the Zr4+ nodes of the highly robust UiO-66 (Fig. 1g) can be partially transmetalated with the significantly smaller cation Ti4+.66 Lau et al. demonstrated that modification of the UiO-66 framework leads to pore constriction that promotes better accommodation of CO2 molecules, and which is ultimately manifested in an enhancement of the CO2 uptake.96 In fact, replacing only 56% of the heavy Zr4+ nodes with Ti4+ led to an 80% improvement of the framework's ability to take up CO2 (from 2.3 mmol g−1 to 4 mmol g−1 at 120 kPa). Notably, this enhancement of the gravimetric CO2 uptake is much higher than that expected from reduction of the molecular weight of the MOF (calculated to be 19% for a model of a 100% Ti4+-exchanged UiO-66). Botas et al. also observed a similar improvement in CO2 uptake – in partially Co2+-doped MOF-5.112 They attributed this effect to an increase in charge–quadrupole interactions between the doped framework and CO2.
4.3. Feasibility of transmetalation: fundamental principles
Numerous examples of transmetalation in MOFs allow us to analyze its scope and feasibility to draw a more detailed picture regarding the fundamental processes involved. Below we provide an analysis of the most important factors that determine the success of transmetalation.The first aspect to consider is the thermodynamic stability of the daughter MOF relative to the parent framework. To probe the stability of MOF nodes, we can resort to a criterion developed for transition metal complexes – the Irving–Williams series. The Irving–Williams series predicts increasingly stronger metal–ligand interactions in high-spin octahedral complexes of 3d M2+ species. The stability of the respective complex peaks for Cu2+, then drops for Zn2+ since its fully occupied d orbitals do not experience additional crystal field stabilization. The Irving–Williams series nicely correlates with the stability of MOFs featuring octahedral metal centers (e.g., MOFs with paddlewheel-based structural building units). Many transmetalation studies report the feasibility of performing a transmetalation to Cu2+. For example, Prasad et al. successfully replaced the Zn2+ centers in the previously described material SNU-51 with Cu2+.91 Similarly, Zhang et al. inserted Cu2+ into the 2D material {Na0.25[(CH3)2NH2]1.75[Cd(hmcem)2]} via replacement of the Cd2+ nodes.102 Interestingly, Cu2+ tends to be the only ion capable of replacing the otherwise difficult intra-framework metal nodes in the case of the aforementioned {Mn3[(Mn4Cl)3(btt)8(CH3OH)10]2} and MSO-MOF systems.41,95
The applicability of the Irving–Williams series to assess MOF stability is especially evident in studies that focus on comparing the feasibility of transmetalation by different metal species. Zhang et al. observed that their attempts to replace zinc ions in the double polyhedron [Zn7(btt)3(H2O)7]n–[Zn5(ppbotctc)3(H2O)5]n with Cu2+, Co2+ or Ni2+ were only successful for Cu2+.105 Mukherjee et al. synthesized a 2D network [{[Zn(btin)2(H2O)2]·2PF6 pyrene·2(H2O)}n] of Kagomé dual topology with octahedral Zn2+ centers and performed an extended transmetalation study on it: the results of the study indicated that the compound could be rapidly transmetalated with Cu2+ or Cd2+, with the Cd2+ nodes subsequently able to be transmetalated to Cu2+; transmetalation of Cu2+ to Cd2+, on the other hand, proceeded very slowly, whereas the transmetalation of Cu2+ to Zn2+ proved not feasible.99 Reverse and serial transmetalation experiments conducted independently by Yao et al. and Song et al. on the paddlewheel-based MOFs SUMOF-1 with the general formula of {M6(btb)4(bipy)3} (where M = Zn2+) and the ITHD series (Fig. 1c), respectively, establish the thermodynamic stability trend in MOF metal nodes to be in the order Cu2+ ≫ Ni2+ > Co2+ ≈ Zn2+, which is in good agreement with the Irving–Williams series (Fig. 11).63,101 These results, along with the recorded data indicating strong Ni–O bonds and large formation constants for Ni–ammine complexes,120 can explain the choice of Ni2+ in transmetalation experiments geared to enhance the framework stability (vide supra).
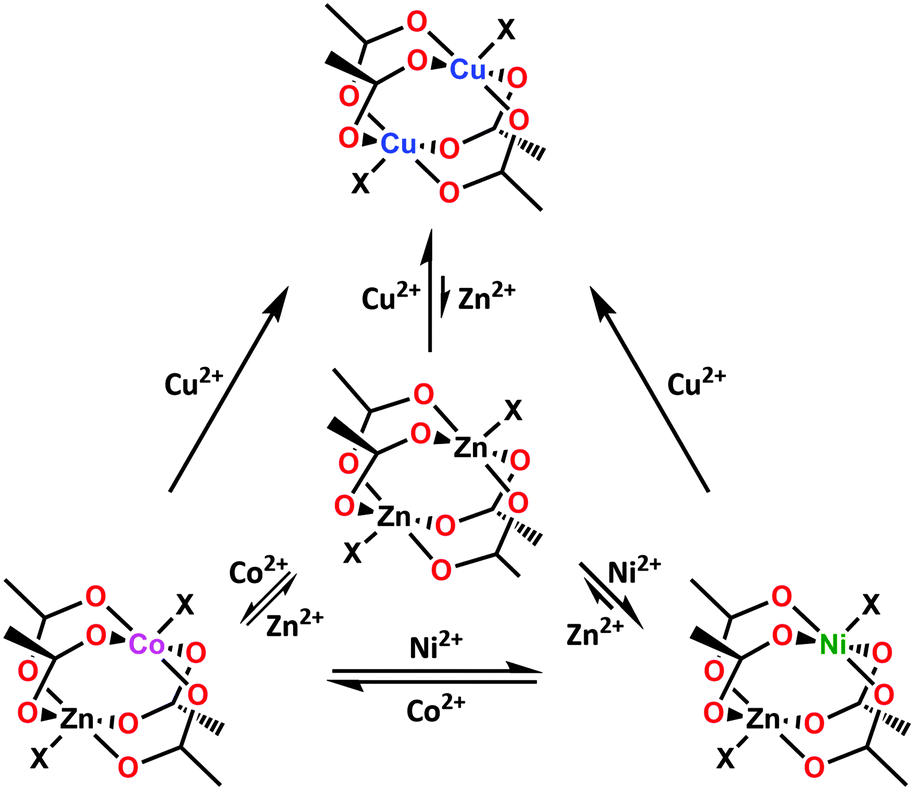 | ||
| Fig. 11 Feasibility of partial vs. complete transmetalation for the first row transition metal ions, M2+, in paddlewheel nodes showing a trend parallel to the stability of Irving–Williams series. | ||
Another aspect governing transmetalation is the preferable coordination state that a given metal assumes. For example, it is well known that Ni2+ favors octahedral coordination, unless it is coerced into a tetrahedral geometry by sterically demanding linkers. Consistent with these ideas, incorporation of Ni2+ into the Zn4O cluster of MOF-5 (which requires tetrahedral coordination of metal centers) has proven challenging. Brozek et al. observed that even after prolonged soaking of MOF-5 in a Ni2+ solution, the extent of transmetalation was no more than 25%, meaning that, on average, only one Zn2+ per cluster was replaced uniformly by Ni2+ (Fig. 9).92 Furthermore, inside the MOF-5 cluster the Ni2+ ion assumes an octahedral geometry by coordinating to two DMF molecules, as evidenced by UV-Vis spectroscopy – becoming four-coordinated only following heating. Similar results were earlier reported for Co2+ doped MOF-5, again with only one Zn2+ ion per node being substituted by Co2+.112 Partial metal exchange has also been observed for MFU-4l, a material topologically related to MOF-5.94 The pentanuclear node of MFU-4l consists of two types of Zn2+ metal centers, four outer tetrahedral metals and one inner octahedral metal. Although outer zinc ions can be replaced, even under forcing conditions Co2+ does not supplant the central, six-coordinated zinc ion, which results in the Co:Zn molar ratio of 4:1 in the exchanged material.
Finally, the success of transmetalation depends on the ability of the metal ion to access the framework interior. There is good evidence that rates of transmetalation can be limited by diffusion. For example, in some cases of partial transmetalation higher degrees of transmetalation have been measured at and near the MOF crystal exterior surface than in the interior.62 Thus, a MOF must possess channels that enable diffusion of the incoming metal species to the crystal core. MFU-4 [Zn5Cl4(bbta)3], which possesses very narrow (2.5 Å) pores was found to be impermeable to Co2+, unlike its larger counterpart, MFU-4l [i.e., Zn5Cl4(btdd)3], which features 9.2 Å pores and successfully undergoes transmetalation as discussed above.94 Additionally, pore flexibility may also facilitate the entry of metal ions. When transmetalation experiments with Cu2+ were performed on Zn-HKUST-1 (Fig. 1a), a paddlewheel-based MOF with relatively small and rigid btc linkers, replacement was only 56% complete even after 3 months. In contrast, Zn-PMOF-2 [Zn4(tdpeb)8(H2O)12], another paddlewheel-based framework possessing more flexible tdpeb linkers, underwent complete transmetalation in three days.62
5. Outlook and future directions
In previous sections we discussed numerous examples of BBR, where only one structural element, namely the metal ions, charge-balancing ligands in the node or the organic linkers had been replaced. These examples nicely demonstrate that MOF structures are highly modular and can be altered via BBR. Recently presented strategies for tuning the physical and chemical properties of porous crystalline materials emphasize a deliberate introduction of framework heterogeneity by increasing the number of constituents. Introducing a variety of building units into a single crystalline solid containing gradients of functionality may open up new opportunities for application-based MOF chemistry. This goal can be achieved, to some extent, by using de novo approaches – for example, incorporating multiple linkers into the same crystal containing different functional groups.37,121Importantly, some recent examples demonstrate a high potential for the construction of multiple building block (nodes and the organic linkers) MOFs by utilizing the BBR strategy. An important exploration of BBR reactions towards multiple building block MOF structures is a stepwise replacement of linkers and metal ions in nodes within the parent framework. Cohen and co-workers selected two ZIF materials, zinc 4,5-dichloro-imidazolate {Zn(dcim)2} (ZIF-71) with RHO topology (Fig. 1e) and zinc 2-methylimidazolate {Zn(mim)2} (ZIF-8) with SOD topology (Fig. 4) for tandem building block replacement experiments (Fig. 12).68 The exchange process for ZIF-71 was conducted in two ways; in the first, the compound was exposed to a solution of 4-bromoimidazole (brim) for linker exchange, and then the metal cation was exchanged by soaking the brominated ZIF in a methanolic solution of Mn(acac)2. For the reverse process, the exchange of a metal ion (Zn2+ to Mn2+) was followed by the linker exchange (dcim to brim). After 3 days of incubation of ZIF-71 in a methanolic solution of brim, ∼35% of the brim had been incorporated into ZIF-71. Consecutive treatment of the obtained ZIF-71(dcim/brim) powder with Mn(acac)2 in MeOH, resulted in bimetallic and multifunctional ZIF-71(dcim/brim)-(Zn/Mn), where ∼12% Mn2+ could be observed as a result of the metal exchange. The uniform distribution of linkers and metal centers in ZIF-71(dcim/brim)-(Zn/Mn) was further confirmed by SEM-EDX mapping. Additionally, ATOFMS indicated that the microcrystalline particles of the resulting material indeed contain the multiple building block ZIF-71(dcim/brim)-(Zn/Mn). The tandem BBR experiment performed in the reverse order gave a nearly identical result, yielding the analogous ZIF-71(Zn/Mn)-(dcim/brim), which contained ∼12% Mn2+ and 30% brim. These results nicely demonstrate that tandem building block replacement is capable of incorporating a variety of building blocks into a single protoMOF.
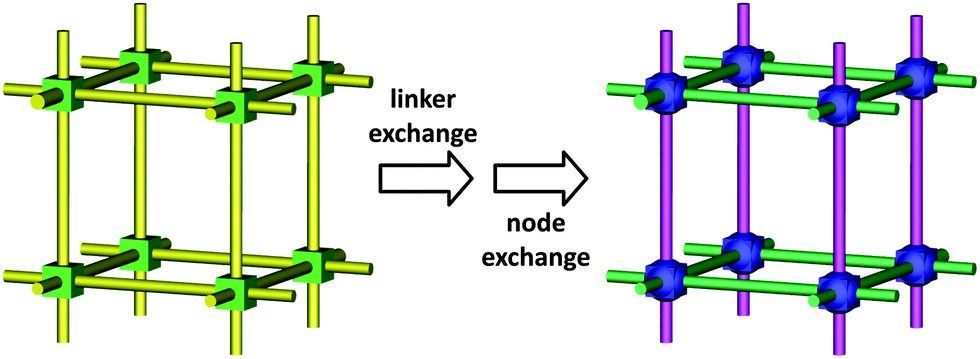 | ||
| Fig. 12 Accessing multiple building block MOF via sequential building block replacements. | ||
While sequential transmetalation63,101 and SALE56,79 are known separately, Cohen's work provides the first example of sequential metal and linker replacement in the same system, which nicely illustrates the synthetic potential of BBR for the introduction of functional variety into MOF scaffolds. Looking forward, another interesting possibility for BBR would be the complete conversion of a protoMOF structure into a new material with new nodes and linkers by tandem building block replacement.
Like selective transmetalation, facilitated by the difference in metal–linker interactions, selective replacement of specific linkers via SALE in MOFs with multiple but similar linkers (e.g., bipyridines, imidazolates, or carboxylates) can be viable due to the difference in their basicity. Such selective SALE, though not realized yet, can potentially open up new possibilities in preparing isoreticular MOFs with spatially controllable pore functionality.
Similarly, we speculate the future utility of BBR can conceptually encompass the following areas:
(i) BBR in MOF membranes: MOF membranes are regarded as interesting materials for gas separations.14,122–128 However, the formation of MOF membranes is limited by synthetic obstacles (related to solvothermal MOF synthesis and preparation of a good quality membrane)122,124,126,127,129 and the incorporation of desired functionality is also challenging. In this view, BBR in MOF membranes, especially ZIF-containing systems,128–132 could offer new possibilities for enhancing the stability and versatility of separations.
(ii) BBR in Layer-by-Layer (LbL)-grown MOF films; the LbL technique has become a promising methodology for growing MOF films, with precise control over thickness, orientation, and porosity, on various substrates.25,122 BBR in combination with LbL could significantly widen the scope of the LbL approach by application of post-LbL SALE or transmetalation, leading to multifunctional MOF films that are difficult to synthesize de novo.
(iii) Different attachment chemistry for non-bridging, charge compensating ligand exchange (SALI): exploration of different attachment chemistry for SALI is another way forward for the synthesis of highly robust functional MOFs (vide supra). This new concept could be further expanded in several directions by incorporation of various types of charged anchoring moieties (not limited to carboxylic acids) resulting in a broader scope of SALI. Utilizing this approach might lead to interesting directions such as incorporation of multiple light harvesters or redox active moieties for the construction of functional arrays or photoredox antenna systems.
While it is clear that SALE, transmetalation and non-bridging ligand exchange stand to become essential components in the MOF synthesis toolbox, it is also clear that much work is needed to fully understand many of the fundamental aspects governing these unique chemical transformations. Looking forward we anticipate that BBR will become a key player in the conversion of protoMOF structures that contain entirely new linkers and nodes and that those materials will exhibit unique properties that are rarely attainable utilizing traditional synthetic methods.
List of abbreviations
| abp | 4,4′-Azobis(pyridine) |
| AD | Alkanedioic acid |
| adn | Adeninate |
| bbta | Benzobistriazolate |
| bdc | Benzene dicarboxylate |
| bdcppi | N,N′-Bis(3,5-dicarboxylphenyl)pyromellitic diimide |
| bodipy | Boron dipyrromethene |
| brim | 4-Bromoimidazolate |
| Br-tcpb | 1,4-Dibromo-2,3,5,6-tetrakis(4-carboxyphenyl)benzene |
| btb | 4,4′,4′′-Benzene-1,3,5-triyl-tribenzoate |
| btc | Benzene-1,3,5-tricarboxylate |
| btdd | Bis(1,2,3-triazolate-[4,5-b],[4′,5′-i])dibenzo-[1,4]-dioxin |
| btin | Benzene-1,3,5-triyltriisonicotinate |
| btp | 1,3,5-Benzenetriphosphonate |
| btt | 1,3,5-Benzenetristetrazolate |
| bipy | 4,4′-Bipyridine |
| bpdc | 4,4′-Diphenyldicarboxylate |
| bpt | Biphenyl-3,4′,5-tricarboxylate |
| bpy | 2,2′-Bipyridine |
| CdIF | Cadmium imidazolate framework |
| dabco | Triethylenediamine |
| dcbdt | 1,4-Dicarboxylbenzene-2,3-dithiolate |
| dcim | 4,5-Dichloroimidazolate |
| DEF | N,N′-Diethylformamide |
| DMF | N,N′-Dimethylformamide |
| dobdc | 2,5-Dioxido-1,4-benzenedicarboxylate |
| dped | meso-1,2-Di(4-pyridyl)-1,2ethanediol |
| dpni | N,N′-Di-4-pyridylnaphthalene tetracarboxydiimide |
| eim | 2-Ethylimidazolate |
| hbtc | Monoprotonated benzene-1,3,5-tricarboxylate |
| hett | 5,5′,10,10′,15,15′-Hexaethayltruxene-2,7,12-tricarboxylate |
| H4BDCPPI | N,N′-Bis(3,5-dicarboxyphenyl) pyromellitic diimide |
| H2-hmcem | 2-Hydroxymethyl-4,6-bi(2′-methoxyl-4′-(2′′-1′′-carboxyl)-ethylene)-1,3,5-mesitylene |
| HKUST | Hong Kong University of Science and Technology |
| ICP | Inductively coupled plasma |
| im | Imidazolate |
| LMCT | Ligand-to-metal charge transfer |
| MeOH | Methanol |
| MFU | Metal–organic framework Ulm University |
| MIL | Materials Institute Lavoisier |
| mim | 2-Methylimidazolate |
| MOM | Metal–organic material |
| MSO | Metal salen octopus |
| ndc | 2,6-Naphthalenedicarboxylate |
| NH2-tpdc | 2′-Amino-1,1′:4,1′′-terphenyl-4,4′′-dicarboxylate |
| nim | Nitroimidazole |
| NMR | Nuclear magnetic resonance |
| NU | Northwestern University |
| PAH | Polyaromatic hydrocarbon |
| PCMOF | Proton conducting metal–organic framework |
| PMOF | Polyhedron-based metal–organic framework |
| ppbotctc | N-Phenyl-N′-phenyl bicyclo[2,2,2]oct-7-ene-2,3,5,6-tetracarboxydiimide tetracarboxylic acid |
| PPF | Porphyrin paddlewheel framework |
| PXRD | Powder X-ray diffraction |
| RPM | Robust porphyrinic material |
| SALE | Solvent-assisted linker exchange |
| SALEM | Solvent-assisted linker exchanged material |
| salen | (R,R)-(−)-1,2-Cyclohexanediamino-N,N′-bis(3-tert-butyl-5-(4-pyridyl)salicylidene)MnIIICl |
| SALI | Solvent-assisted ligand insertion |
| SBU | Structural building unit |
| SNU | Seoul National University |
| SUMOF | Stockholm University metal–organic framework |
| TBAPy | 1,3,6,8-Tetrakis(p-benzoate)pyrene |
| tcpb | 2,3,5,6-Tetrakis(4-carboxyphenyl)benzene |
| tdpeb | 1,3,5-Tris(3,5-dicarboxylphenylethynyl)benzene |
| thbts | 2,4,6-Trihydroxy-1,3,5-benzenetrisulfonate |
| tmbbp | 2,3,5,6-Tetramethyl-1,4-bis(4-pyridyl)benzene |
| tmbebp | 2,3,5,6-Tetramethyl-1,4-bis-ethynyl-(4-pyridyl)benzene |
| Zn-tcpp | Tetrakis(4-carboxyphenyl)porphyrinatozinc(II) |
| UiO | Universitetet i Oslo |
| ZIF | Zeolitic imidazolate framework |
Acknowledgements
For support of our own work on building-block replacement chemistry we gratefully acknowledge U.S. Dept. of Energy, Office of Science, Basic Energy Sciences Program (chemical separations and synthesis methods development), the Stanford Global Climate and Energy Project (carbon capture), the Defense Threat Reduction Agency (degradation and capture of chemical threats), the U.S. Army Research Office (capture of toxic industrial chemicals), the U.S. Dept. of Energy, ARPA-E (methane storage and release); the Northwestern University Institute for Chemistry in Energy Processes (energy-related chemical catalysis), and the Institute for Sustainability and Energy at Northwestern (equipment acquisition). WB acknowledges support from the Foundation for Polish Science through the “Kolumb” Program.Notes and references
- G. Férey, Hybrid Porous Solids: Past, Present, Future, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- O. M. Yaghi, M. O'Keefe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Reticular Synthesis and the Design of New Materials, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Soft Porous Crystals, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Functional Porous Coordination Polymers, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341, 1230444 CrossRef PubMed.
- O. K. Farha, A. Ö. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, De Novo Synthesis of a Metal–Organic Framework Material Featuring Ultrahigh Surface Area and Gas Storage Capacities, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydin and J. T. Hupp, Metal–Organic Framework Materials with Ultrahigh Surface Areas: Is the Sky the Limit?, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- D. Yuan, D. Zhao, D. Sun and H.-C. Zhou, An Isoreticular Series of Metal–Organic Frameworks with Dendritic Hexacarboxylate Ligands and Exceptionally High Gas-Uptake Capacity, Angew. Chem., Int. Ed., 2010, 49, 5357–5361 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Ultrahigh Porosity in Metal-Organic Frameworks, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- C. E. Wilmer, O. K. Farha, T. Yildirim, I. Eryazici, V. Krungleviciute, A. A. Sarjeant, R. Q. Snurr and J. T. Hupp, Gram-Scale, High-Yield Synthesis of a Robust Metal–Organic Framework for Storing Methane and other Gases, Energy Environ. Sci., 2013, 6, 1158–1163 CAS.
- C. E. Wilmer, O. K. Farha, Y.-S. Bae, J. T. Hupp and R. Q. Snurr, Structure–Property Relationships of Porous Materials for Carbon Dioxide Separation and Capture, Energy Environ. Sci., 2012, 5, 9849–9856 CAS.
- L. J. Murray, M. Dincă and J. R. Long, Hydrogen Storage in Metal–Organic Frameworks, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Selective Gas Adsorption and Separation in Metal–Organic Frameworks, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Carbon Dioxide Capture in Metal–Organic Frameworks, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Hydrogen Storage in Metal–Organic Frameworks, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Metal–Organic Frameworks for Separations, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, Methane Storage in Metal–Organic Frameworks: Current Records, Surprise Findings, and Challenges, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Metal-Organic Framework Materials as Catalysts, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- L. Ma, C. Abney and W. Lin, Enantioselective Catalysis with Homochiral Metal–Organic Frameworks, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- M. Yoon, R. Srirambalaji and K. Kim, Homochiral Metal–Organic Frameworks for Asymmetric Heterogeneous Catalysis, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- C. A. Kent, D. Liu, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, Light Harvesting in Microscale Metal–Organic Frameworks by Energy Migration and Interfacial Electron Transfer Quenching, J. Am. Chem. Soc., 2011, 133, 12940–12943 CrossRef CAS PubMed.
- C. A. Kent, B. P. Mehl, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, Energy Transfer Dynamics in Metal-Organic Frameworks, J. Am. Chem. Soc., 2010, 132, 12767–12769 CrossRef CAS PubMed.
- S. Jin, H.-J. Son, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, Energy Transfer from Quantum Dots to Metal–Organic Frameworks for Enhanced Light Harvesting, J. Am. Chem. Soc., 2013, 135, 955–958 CrossRef CAS PubMed.
- H.-J. Son, S. Jin, S. Patwardhan, S. J. Wezenberg, N. C. Jeong, M. So, C. E. Wilmer, A. A. Sarjeant, G. C. Schatz, R. Q. Snurr, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, Light-Harvesting and Ultrafast Energy Migration in Porphyrin-Based Metal–Organic Frameworks, J. Am. Chem. Soc., 2013, 135, 862–869 CrossRef CAS PubMed.
- M. So, S. Jin, H.-J. Son, G. P. Wiederrecht, O. K. Farha and J. T. Hupp, Layer-by-Layer Fabrication of Oriented Porous Thin Films Based on Porphyrin-Containing Metal–Organic Frameworks, J. Am. Chem. Soc., 2013, 135, 15698–15701 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal–Organic Framework Materials as Chemical Sensors, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Luminescent Functional Metal–Organic Frameworks, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- C.-Y. Sun, X.-L. Wang, X. Zhang, C. Qin, P. Li, Z.-M. Su, D.-X. Zhu, G.-G. Shan, K.-Z. Shao, H. Wu and J. Li, Efficient and tunable white-light emission of metal–organic frameworks by iridium-complex encapsulation, Nat. Commun., 2013, 4, 2717 Search PubMed.
- S. Horike, D. Umeyama and S. Kitagawa, Ion Conductivity and Transport by Porous Coordination Polymers and Metal-Organic Frameworks, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed.
- A. Shigematsu, T. Yamada and H. Kitagawa, Wide Control of Proton Conductivity in Porous Coordination Polymers, J. Am. Chem. Soc., 2011, 133, 2034–2036 CrossRef CAS PubMed.
- M. L. Aubrey, R. Ameloot, B. M. Wiers and J. R. Long, Metal–Organic Frameworks as Solid Magnesium Electrolytes, Energy Environ. Sci., 2014, 7, 667–671 CAS.
- G. K. H. Shimizu, J. M. Taylor and S. Kim, Proton Conduction with Metal-Organic Frameworks, Science, 2013, 341, 354–355 CrossRef CAS PubMed.
- C. Wang, T. Zhang and W. Lin, Rational Synthesis of Noncentrosymmetric Metal–Organic Frameworks for Second-Order Nonlinear Optics, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed.
- A. Hu, H. L. Ngo and W. Lin, Chiral Porous Hybrid Solids for Practical Heterogeneous Asymmetric Hydrogenation of Aromatic Ketones, J. Am. Chem. Soc., 2003, 125, 11490–11491 CrossRef CAS PubMed.
- D. W. Lewis, A. R. Ruiz-Salvador, A. Gomez, L. M. Rodriguez-Albelo, F.-X. Coudert, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, Zeolitic Imidazole Frameworks: Structural and Energetics Trends Compared with their Zeolite Analogues, CrystEngComm, 2009, 11, 2272–2276 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keefe and O. M. Yaghi, Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Multiple Functional Groups of Varying Ratios in Metal-Organic Frameworks, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- S. M. Cohen, Postsynthetic Methods for the Functionalization of Metal–Organic Frameworks, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- K. K. Tanabe and S. M. Cohen, Postsynthetic Modification of Metal–Organic Frameworks—a Progress Report, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- A. D. Burrows, in Metal Organic Frameworks as Heterogeneous Catalysts, ed. F. L. i. Xamena and J. Gascon, The Royal Society of Chemistry, 2013, pp. 31–75 Search PubMed.
- A. M. Shultz, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, Post-Synthesis Modification of a Metal–Organic Framework To Form Metallosalen-Containing MOF Materials, J. Am. Chem. Soc., 2011, 133, 13252–13255 CrossRef CAS PubMed.
- T. Gadzikwa, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, Selective Bifunctional Modification of a Non-catenated Metal-Organic Framework Material via “Click” Chemistry, J. Am. Chem. Soc., 2009, 131, 13613–13615 CrossRef CAS PubMed.
- T. Gadzikwa, G. Lu, C. L. Stern, S. R. Wilson, J. T. Hupp and S. T. Nguyen, Covalent Surface Modification of a Metal–Organic Framework: Selective Surface Engineering via CuI-Catalyzed Huisgen Cycloaddition, Chem. Commun., 2008, 5493–5495 RSC.
- H. Sato, R. Matsuda, K. Sugimoto, M. Takata and S. Kitagawa, Photoactivation of a Nanoporous Crystal for On-Demand Guest Trapping and Conversion, Nat. Mater., 2010, 9, 661–666 CrossRef CAS PubMed.
- K. L. Mulfort and J. T. Hupp, Alkali Metal Cation Effects on Hydrogen Uptake and Binding in Metal-Organic Frameworks, Inorg. Chem., 2008, 47, 7936–7938 CrossRef CAS PubMed.
- O. K. Farha, K. L. Mulfort and J. T. Hupp, An Example of Node-Based Postassembly Elaboration of a Hydrogen-Sorbing, Metal–Organic Framework Material, Inorg. Chem., 2008, 47, 10223–10225 CrossRef CAS PubMed.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Amine Grafting on Coordinatively Unsaturated Metal Centers of MOFs: Consequences for Catalysis and Metal Encapsulation, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Enhanced Carbon Dioxide Capture upon Incorporation of N,N′-Dimethylethylenediamine in the Metal–Organic Framework CuBTTri, Chem. Sci., 2011, 2, 2022–2028 RSC.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, A Homochiral Metal-Organic Porous Material for Enantioselective Separation and Catalysis, Nature, 2000, 404, 982–986 CrossRef CAS PubMed.
- S.-T. Zheng, X. Zhao, S. Lau, A. Fuhr, P. Feng and X. Bu, Entrapment of Metal Clusters in Metal–Organic Framework Channels by Extended Hooks Anchored at Open Metal Sites, J. Am. Chem. Soc., 2013, 135, 10270–10273 CrossRef CAS PubMed.
- R. Ameloot, M. Aubrey, B. M. Wiers, A. P. Gómora-Figueroa, S. N. Patel, N. P. Balsara and J. R. Long, Ionic Conductivity in the Metal–Organic Framework UiO-66 by Dehydration and Insertion of Lithium tert-Butoxide, Chem. – Eur. J., 2013, 19, 5533–5536 CrossRef CAS PubMed.
- M. Meilikhov, K. Yusenko and R. A. Fischer, Turning MIL-53(Al) Redox-Active by Functionalization of the Bridging OH-Group with 1,1′-Ferrocenediyl-Dimethylsilane, J. Am. Chem. Soc., 2009, 131, 9644–9645 CrossRef CAS PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, Vapor-Phase Metalation by Atomic Layer Deposition in a Metal–Organic Framework, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- C. Larabi and E. A. Quadrelli, Titration of Zr3(μ-OH) Hydroxy Groups at the Cornerstones of Bulk MOF UiO-67, [Zr6O4(OH)4(biphenyldicarboxylate)6], and Their Reaction with [AuMe(PMe3)], Eur. J. Inorg. Chem., 2012, 3014–3022 CrossRef CAS.
- O. Karagiaridi, W. Bury, A. A. Sarjeant, C. L. Stern, O. K. Farha and J. T. Hupp, Synthesis and Characterization of Isostructural Cadmium Zeolitic Imidazolate Frameworks via Solvent-Assisted Linker Exchange, Chem. Sci., 2012, 3, 3256–3260 RSC.
- O. Karagiaridi, W. Bury, E. Tylianakis, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Opening Metal–Organic Frameworks Vol. 2: Inserting Longer Pillars into Pillared-Paddlewheel Structures through Solvent-Assisted Linker Exchange, Chem. Mater., 2013, 25, 3499–3503 CrossRef CAS.
- O. Karagiaridi, M. B. Lalonde, W. Bury, A. A. Sarjeant, O. K. Farha and J. T. Hupp, Opening ZIF-8: A Catalytically Active Zeolitic Imidazolate Framework of Sodalite Topology with Unsubstituted Linkers, J. Am. Chem. Soc., 2012, 134, 18790–18796 CrossRef CAS PubMed.
- M. Lalonde, W. Bury, O. Karagiaridi, Z. Brown, J. T. Hupp and O. K. Farha, Transmetalation: Routes to Metal Exchange within Metal–Organic Frameworks, J. Mater. Chem. A, 2013, 1, 5453–5468 CAS.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Postsynthetic Ligand Exchange as a Route to Functionalization of ‘Inert’ Metal–Organic Frameworks, Chem. Sci., 2012, 3, 126–130 RSC.
- M. Kondo, S. Furukawa, K. Hirai and S. Kitagawa, Coordinatively Immobilized Monolayers on Porous Coordination Polymer Crystals, Angew. Chem., Int. Ed., 2010, 49, 5327–5330 CrossRef CAS PubMed.
- N. Yanai and S. Granick, Directional Self-Assembly of a Colloidal Metal–Organic Framework, Angew. Chem., Int. Ed., 2012, 51, 5638–5641 CrossRef CAS PubMed.
- X. Song, S. Jeong, D. Kim and M. S. Lah, Transmetalations in Two Metal–Organic Frameworks with Different Framework Flexibilities: Kinetics and Core–Shell Heterostructure, CrystEngComm, 2012, 14, 5753–5756 RSC.
- X. Song, T. K. Kim, H. Kim, D. Kim, S. Jeong, H. R. Moon and M. S. Lah, Post-Synthetic Modifications of Framework Metal Ions in Isostructural Metal–Organic Frameworks: Core–Shell Heterostructures via Selective Transmetalations, Chem. Mater., 2012, 24, 3065–3073 CrossRef CAS.
- B. J. Burnett, P. M. Barron, C. Hu and W. Choe, Stepwise Synthesis of Metal–Organic Frameworks: Replacement of Structural Organic Linkers, J. Am. Chem. Soc., 2011, 133, 9984–9987 CrossRef CAS PubMed.
- S. Kim, K. W. Dawson, B. S. Gelfand, J. M. Taylor and G. K. H. Shimizu, Enhancing Proton Conduction in a Metal–Organic Framework by Isomorphous Ligand Replacement, J. Am. Chem. Soc., 2013, 135, 963–966 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, Postsynthetic Ligand and Cation Exchange in Robust Metal–Organic Frameworks, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- W. Bury, D. Fairen-Jimenez, M. B. Lalonde, R. Q. Snurr, O. K. Farha and J. T. Hupp, Control over Catenation in Pillared Paddlewheel Metal–Organic Framework Materials via Solvent-Assisted Linker Exchange, Chem. Mater., 2013, 25, 739–744 CrossRef CAS.
- H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Tandem Postsynthetic Metal Ion and Ligand Exchange in Zeolitic Imidazolate Frameworks, Inorg. Chem., 2013, 52, 4011–4016 CrossRef CAS PubMed.
- A. F. Gross, E. Sherman, S. L. Mahoney and J. J. Vajo, Reversible Ligand Exchange in a Metal–Organic Framework (MOF): Toward MOF-Based Dynamic Combinatorial Chemical Systems, J. Phys. Chem. A, 2013, 117, 3771–3776 CrossRef CAS PubMed.
- S. Takaishi, E. J. DeMarco, M. J. Pellin, O. K. Farha and J. T. Hupp, Solvent-Assisted Linker Exchange (SALE) and Post-Assembly Metallation in Porphyrinic Metal–Organic Framework Materials, Chem. Sci., 2013, 4, 1509–1513 RSC.
- L.-H. Wang, R. Shang, Z. Zheng, C.-L. Liu and Z.-M. Wang, Two Systems of [DabcoH2]2+/[PipH2]2+–Uranyl–Oxalate Showing Reversible Crystal-to-Crystal Transformations Controlled by the Diammonium/Uranyl/Oxalate Ratios in Aqueous Solutions ([DabcoH2]2+ = 1,4-Diazabicyclo-[2.2.2]-octaneH2 and [PipH2]2+ = PiperazineH2), Cryst. Growth Des., 2013, 13, 2597–2606 CAS.
- N. A. Vermeulen, O. Karagiaridi, A. A. Sarjeant, C. L. Stern, J. T. Hupp, O. K. Farha and J. F. Stoddart, Aromatizing Olefin Metathesis by Ligand Isolation inside a Metal–Organic Framework, J. Am. Chem. Soc., 2013, 135, 14916–14919 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, Enhanced Photochemical Hydrogen Production by a Molecular Diiron Catalyst Incorporated into a Metal–Organic Framework, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- S. Jeong, D. Kim, X. Song, M. Choi, N. Park and M. S. Lah, Postsynthetic Exchanges of the Pillaring Ligand in Three-Dimensional Metal–Organic Frameworks, Chem. Mater., 2013, 25, 1047–1054 CrossRef CAS.
- G. Alberti, E. Giontella and S. Murcia-Mascarós, Mechanism of the Formation of Organic Derivatives of γ-Zirconium Phosphate by Topotactic Reactions with Phosphonic Acids in Water and Water–Acetone Media, Inorg. Chem., 1997, 36, 2844–2849 CrossRef CAS PubMed.
- H. R. Allcock and G. Y. Moore, Synthesis of Poly(organophosphazene) Copolymers and Cross-Linked Polymers by Ligand Exchange, Macromolecules, 1972, 5, 231–232 CrossRef CAS.
- J.-R. Li and H.-C. Zhou, Bridging-Ligand-Substitution Strategy for the Preparation of Metal–Organic Polyhedra, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed.
- M. Ziegler, A. V. Davis, D. W. Johnson and K. N. Raymond, Supramolecular Chirality: A Reporter of Structural Memory, Angew. Chem., Int. Ed., 2003, 42, 665–668 CrossRef CAS PubMed.
- T. Li, M. T. Kozlowski, E. A. Doud, M. N. Blakely and N. L. Rosi, Stepwise Ligand Exchange for the Preparation of a Family of Mesoporous MOFs, J. Am. Chem. Soc., 2013, 135, 11688–11691 CrossRef CAS PubMed.
- D. Peralta, G. Chaplais, A. Simon-Masseron, K. Barthelet, C. Chizallet, A.-A. Quoineaud and G. D. Pirngruber, Comparison of the Behavior of Metal–Organic Frameworks and Zeolites for Hydrocarbon Separations, J. Am. Chem. Soc., 2012, 134, 8115–8126 CrossRef CAS PubMed.
- G. A. N. Felton, A. K. Vannucci, J. Chen, L. T. Lockett, N. Okumura, B. J. Petro, U. I. Zakai, D. H. Evans, R. S. Glass and D. L. Lichtenberger, Hydrogen Generation from Weak Acids: Electrochemical and Computational Studies of a Diiron Hydrogenase Mimic, J. Am. Chem. Soc., 2007, 129, 12521–12530 CrossRef CAS PubMed.
- D. Streich, Y. Astuti, M. Orlandi, L. Schwartz, R. Lomoth, L. Hammarström and S. Ott, High-Turnover Photochemical Hydrogen Production Catalyzed by a Model Complex of the [FeFe]-Hydrogenase Active Site, Chem. – Eur. J., 2010, 16, 60–63 CrossRef CAS PubMed.
- Y. Inokuma, M. Kawano and M. Fujita, Crystalline Molecular Flasks, Nat. Chem., 2011, 3, 349–358 CrossRef CAS PubMed.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, Perfluoroalkane Functionalization of NU-1000 via Solvent-Assisted Ligand Incorporation: Synthesis and CO2 Adsorption Studies, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed.
- P. Deria, W. Bury, J. T. Hupp and O. K. Farha, Versatile Functionalization of the NU-1000 Platform by Solvent-Assisted Ligand Incorporation, Chem. Commun., 2014, 50, 1965–1968 RSC.
- H. Wu, T. Yildirim and W. Zhou, Exceptional Mechanical Stability of Highly Porous Zirconium Metal–Organic Framework UiO-66 and Its Important Implications, J. Phys. Chem. Lett., 2013, 4, 925–930 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, Unusual and Highly Tunable Missing-Linker Defects in Zirconium Metal-Organic Framework UiO-66 and Their Important Effects on Gas Adsorption, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, A Facile Synthesis of UiO-66, UiO-67 and their Derivatives, Chem. Commun., 2013, 49, 9449–9451 RSC.
- D. H. Hong and M. P. Suh, Enhancing CO2 Separation Ability of a Metal-Organic Framework by Post-Synthetic Ligand Exchange with Flexible Aliphatic Carboxylates, Chem. – Eur. J., 2014, 20, 426–434 CrossRef CAS PubMed.
- S. Das, H. Kim and K. Kim, Metathesis in Single Crystal: Complete and Reversible Exchange of Metal Ions Constituting the Frameworks of Metal–Organic Frameworks, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS PubMed.
- T. K. Prasad, D. H. Hong and M. P. Suh, High Gas Sorption and Metal-Ion Exchange of Microporous Metal–Organic Frameworks with Incorporated Imide Groups, Chem. – Eur. J., 2010, 16, 14043–14050 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, Lattice-Imposed Geometry in Metal-Organic Frameworks: Lacunary Zn4O Clusters in MOF-5 Serve as Tripodal Chelating Ligands for Ni2+, Chem. Sci., 2012, 3, 2110–2113 RSC.
- C. K. Brozek and M. Dincă, Ti3+-, V2+/3+-, Cr2+/3+-, Mn2+-, and Fe2+-Substituted MOF-5 and Redox Reactivity in Cr- and Fe-MOF-5, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- D. Denysenko, T. Werner, M. Grzywa, A. Puls, V. Hagen, G. Eickerling, J. Jelic, K. Reuter and D. Volkmer, Reversible Gas-Phase Redox Processes Catalyzed by Co-Exchanged MFU-4l(arge), Chem. Commun., 2012, 48, 1236–1238 RSC.
- M. Dincǎ and J. R. Long, High-Enthalpy Hydrogen Adsorption in Cation-Exchanged Variants of the Microporous Metal–Organic Framework Mn3[(Mn4Cl)3(BTT)8(CH3OH)10]2, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef PubMed.
- C. H. Lau, R. Babarao and M. R. Hill, A Route to Drastic Increase of CO2 Uptake in Zr Metal Organic Framework UiO-66, Chem. Commun., 2013, 49, 3634–3636 RSC.
- J. Kahr, R. E. Morris and P. A. Wright, Post-synthetic incorporation of nickel into CPO-27(Mg) to give materials with enhanced permanent porosity, CrystEngComm, 2013, 15, 9779–9786 RSC.
- J.-H. Liao, W.-T. Chen, C.-S. Tsai and C.-C. Wang, Characterization, Adsorption Properties, Metal Ion-Exchange and Crystal-to-Crystal Transformation of Cd3[(Cd4Cl)3(BTT)8(H2O)12]2 Framework, where BTT3− = 1,3,5-Benzenetristetrazolate, CrystEngComm, 2013, 15, 3377–3384 RSC.
- G. Mukherjee and K. Biradha, Post-Synthetic Modification of Isomorphic Coordination Layers: Exchange Dynamics of Metal Ions in a Single Crystal to Single Crystal Fashion, Chem. Commun., 2012, 48, 4293–4295 RSC.
- H.-M. Zhang, J. Yang, Y.-C. He and J.-F. Ma, An Unusual Porous Metal–Organic Framework that Demonstrates the Highly Efficient Exchange of Metal Ions and the Reversible Adsorption of Iodine, Chem. – Asian J., 2013, 8, 2787–2791 CrossRef CAS PubMed.
- Q. Yao, J. Sun, K. Li, J. Su, M. V. Peskov and X. Zou, A Series of Isostructural Mesoporous Metal–Organic Frameworks Obtained by Ion-Exchange Induced Single-Crystal to Single-Crystal Transformation, Dalton Trans., 2012, 41, 3953–3955 RSC.
- T.-T. Lian, S.-M. Chen, F. Wangb and J. Zhang, Metal–Organic Framework Architecture with Polyhedron-in-Polyhedron and Further Polyhedral Assembly, CrystEngComm, 2013, 15, 1036–1038 RSC.
- Z. Zhang, W.-Y. Gao, L. Wojtas, S. Ma, M. Eddaoudi and M. J. Zaworotko, Post-Synthetic Modification of Porphyrin-Encapsulating Metal–Organic Materials by Cooperative Addition of Inorganic Salts to Enhance CO2/CH4 Selectivity, Angew. Chem., Int. Ed., 2012, 51, 9330–9334 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, L. Wojtas, P. Nugent, M. Eddaoudi and M. J. Zaworotko, Templated Synthesis, Postsynthetic Metal Exchange, and Properties of a Porphyrin-Encapsulating Metal–Organic Material, J. Am. Chem. Soc., 2011, 134, 924–927 CrossRef PubMed.
- Z.-J. Zhang, W. Shi, Z. Niu, H.-H. Li, B. Zhao, P. Cheng, D.-Z. Liao and S.-P. Yan, A New Type of Polyhedron-Based Metal–Organic Frameworks with Interpenetrating Cationic and Anionic Nets Demonstrating Ion Exchange, Adsorption and Luminescent Properties, Chem. Commun., 2011, 47, 6425–6427 RSC.
- J. Li, L. Li, H. Hou and Y. Fan, Study on the Reaction of Polymeric Zinc Ferrocenyl Carboxylate with Pb(II) or Cd(II), Cryst. Growth Des., 2009, 9, 4504–4513 CAS.
- L. Mi, H. Hou, Z. Song, H. Han and Y. Fan, Polymeric Zinc Ferrocenyl Sulfonate as a Molecular Aspirator for the Removal of Toxic Metal Ions, Chem. – Eur. J., 2008, 14, 1814–1821 CrossRef CAS PubMed.
- L. Mi, H. Hou, Z. Song, H. Han, H. Xu, Y. Fan and S.-W. Ng, Rational Construction of Porous Polymeric Cadmium Ferrocene-1,1′-disulfonates for Transition Metal Ion Exchange and Sorption, Cryst. Growth Des., 2007, 7, 2553–2561 CAS.
- S. Huang, X. Li, X. Shi, H. Hou and Y. Fan, Structure Extending and Cation Exchange of Cd(II) and Co(II) Materials Compounds Inducing Fluorescence Signal Mutation, J. Mater. Chem., 2010, 20, 5695–5699 RSC.
- Z. Wei, W. Lu, H.-L. Jiang and H.-C. Zhou, A Route to Metal–Organic Frameworks through Framework Templating, Inorg. Chem., 2013, 52, 1164–1166 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Zirconium-Metalloporphyrin PCN-222: Mesoporous Metal–Organic Frameworks with Ultrahigh Stability as Biomimetic Catalysts, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- J. A. Botas, G. Calleja, M. Sánchez-Sánchez and M. G. Orcajo, Cobalt Doping of the MOF-5 Framework and Its Effect on Gas-Adsorption Properties, Langmuir, 2010, 26, 5300–5303 CrossRef CAS PubMed.
- C. J. Kepert and M. J. Rosseinsky, Zeolite-Like Crystal Structure of an Empty Microporous Molecular Framework, Chem. Commun., 1999, 375–376 RSC.
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Synthetic Strategies, Structure Patterns, and Emerging Properties in the Chemistry of Modular Porous Solids, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS.
- A. P. Nelson, O. K. Farha, K. L. Mulfort and J. T. Hupp, Supercritical Processing as a Route to High Internal Surface Areas and Permanent Microporosity in Metal-Organic Framework Materials, J. Am. Chem. Soc., 2009, 131, 458–460 CrossRef CAS PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Evaluating Metal–Organic Frameworks for Natural Gas Storage, Chem. Sci., 2014, 5, 32–51 RSC.
- C.-X. Yang, H.-B. Ren and X.-P. Yan, Fluorescent Metal–Organic Framework MIL-53(Al) for Highly Selective and Sensitive Detection of Fe3+ in Aqueous Solution, Anal. Chem., 2013, 85, 7441–7446 CrossRef CAS PubMed.
- In a MIL-53 coordination environment, Fe(III) should be relatively easy to reduce. If so, then rapid luminescence quenching by electron transfer from photo-excited BDC to Fe(III) would account qualitatively for the observations. If the photo-prepared molecular exciton can migrate from linker to linker (for example, via Förster energy transfer) only a comparatively small fraction of the Al(III) ions would need to be displaced by Fe(III) in order to engender fluorescence quenching. Although no evidence for or against such amplified quenching is presented in the original report, it's occurrence might account for the observed low Fe(III) detection threshold.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Stability and Hydrolyzation of Metal Organic Frameworks with Paddle-Wheel SBUs upon Hydration, Chem. Mater., 2012, 24, 3153–3167 CrossRef CAS.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Mapping of Functional Groups in Metal-Organic Frameworks, Science, 2013, 341, 882–885 CrossRef CAS PubMed.
- A. Bétard and R. A. Fischer, Metal–Organic Framework Thin Films: From Fundamentals to Applications., Chem. Rev., 2012, 112, 1055–1083 CrossRef PubMed.
- J. Gascon and F. Kapteijn, Metal-Organic Framework Membranes—High Potential, Bright Future?, Angew. Chem., Int. Ed., 2010, 49, 1530–1532 CrossRef CAS PubMed.
- H. Guo, G. Zhu, I. J. Hewitt and S. Qiu, “Twin Copper Source” Growth of Metal–Organic Framework Membrane: Cu3(BTC)2 with High Permeability and Selectivity for Recycling H2, J. Am. Chem. Soc., 2009, 131, 1646–1647 CrossRef CAS PubMed.
- M. Arnold, P. Kortunov, D. J. Jones, Y. Nedellec, J. Kärger and J. Caro, Oriented Crystallisation on Supports and Anisotropic Mass Transport of the Metal-Organic Framework Manganese Formate, Eur. J. Inorg. Chem., 2007, 60–64 CrossRef CAS.
- D. Zacher, O. Shekhah, C. Wöll and R. A. Fischer, Thin Films of Metal–Organic Frameworks, Chem. Soc. Rev., 2009, 38, 1418–1429 RSC.
- O. Shekhah, J. Liu, R. A. Fischer and C. Wöll, MOF Thin Films: Existing and Future Applications, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC.
- S. Aguado, C.-H. Nicolas, V. Moizan-Baslé, C. Nieto, H. Amrouche, N. Bats, N. Audebrand and D. Farrusseng, Facile Synthesis of an Ultramicroporous MOF Tubular Membrane With Selectivity Towards CO2, New J. Chem., 2011, 35, 41–44 RSC.
- O. Shekhah, R. Swaidan, Y. Belmabkhout, M. d. Plessis, T. Jacobs, L. J. Barbour, I. Pinnau and M. Eddaoudi, The Liquid Phase Epitaxy Approach for the Successful Construction of Ultra-Thin and Defect-Free ZIF-8 Membranes: Pure and Mixed Gas Transport Study, Chem. Commun., 2014, 50, 2089–2092 RSC.
- H. Bux, F. Liang, Y. Li, J. Cravillon, M. Wiebcke and J. Caro, Zeolitic Imidazolate Framework Membrane with Molecular Sieving Properties by Microwave-Assisted Solvothermal Synthesis, J. Am. Chem. Soc., 2009, 131, 16000–16001 CrossRef CAS PubMed.
- Y.-S. Li, H. Bux, A. Feldhoff, G.-L. Li, W.-S. Yang and J. Caro, Controllable Synthesis of Metal–Organic Frameworks: From MOF Nanorods to Oriented MOF Membranes, Adv. Mater., 2010, 22, 3322–3326 CrossRef CAS PubMed.
- A. Huang, H. Bux, F. Steinbach and J. Caro, Molecular-Sieve Membrane with Hydrogen Permselectivity: ZIF-22 in LTA Topology Prepared with 3-Aminopropyltriethoxysilane as Covalent Linker, Angew. Chem., Int. Ed., 2010, 49, 4958–4961 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |