 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic Lewis base functionalisation of carboxylic acids, esters and anhydrides via C1-ammonium or azolium enolates
Louis C.
Morrill
and
Andrew D.
Smith
*
EastCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife, UK KY16 9ST. E-mail: ads10@st-andrews.ac.uk
First published on 28th May 2014
Abstract
This tutorial review highlights the organocatalytic Lewis base functionalisation of carboxylic acids, esters and anhydrides via C1-ammonium/azolium enolates. The generation and synthetic utility of these powerful intermediates is highlighted through their application in various methodologies including aldol-lactonisations, Michael-lactonisations/lactamisations and [2,3]-rearrangements.
 Louis C. Morrill | Louis was born in Wick (UK) and obtained his MChem degree from the University of St Andrews, including a one-year placement at Astrazeneca in Loughborough (UK). He recently completed his PhD under the supervision of Prof. Andrew Smith at the University of St Andrews, funded by The Carnegie Trust for the Universities of Scotland. His research focused on the organocatalytic functionalisation of carboxylic acids using isothioureas. |
 Andrew D. Smith | Andy was born in Middlesbrough (UK) and gained a DPhil (supervised by Prof. Steve Davies) in 2000 from Jesus College, University of Oxford. Awarded the cross-disciplinary Weston Junior Research Fellowship for post-doctoral studies (New College, Oxford, 2000) a further research collaboration with Prof. Davies was established. In October 2005, ADS was appointed as a Royal Society URF within the School of Chemistry at the University of St Andrews, was promoted to Reader in 2010, and Professor in 2012. Research within the ADS group is currently directed towards the development of novel catalytic methods, with a particular interest in the development of Lewis base promoted catalytic processes, including the use of NHCs and isothioureas in organocatalysis. |
Key learning pointsThis tutorial review will:(1) Define C1-ammonium and azolium enolates and place them in context with other related methodologies. (2) Outline how these enolates can be accessed from carboxylic acids, esters and anhydrides and why this is desirable. (3) Showcase the utility of these enolates in organic synthesis. (4) Provide clear explanations of the mechanisms of reactions and insights into the origin of stereocontrol. (5) Show the current scope and limitations of this area and define challenges for the future. |
1. Introduction
The development of new synthetic methods that allow rapid access to valuable chemical motifs from readily accessible starting materials is an enduring goal of organic synthesis. Organocatalysis is now a thoroughly established member of the organic chemists' toolbox, with a range of reaction modes defined and applied to diverse synthetic transformations.1 Lewis bases,2 most simply defined by their ability to donate a lone pair of electrons, have found widespread utility in organocatalysis, particularly via enamine,3 iminium4 and SOMO5 activation. Lewis base organocatalysis also encompasses both ammonium6 and azolium7 enolate reaction modes that utilise tertiary amine or N-heterocyclic carbene (NHC) catalysts respectively. Both have become powerful synthetic methods for challenging catalytic and asymmetric bond forming reactions and useful synthetic transformations. Ammonium and azolium enolates are commonly classified into three subtypes, C1-, C2- and C3-ammonium/azolium enolates based upon the number of atoms between the corresponding Lewis basic catalyst (LB, Fig. 1) and the oxygen atom of the enolate.6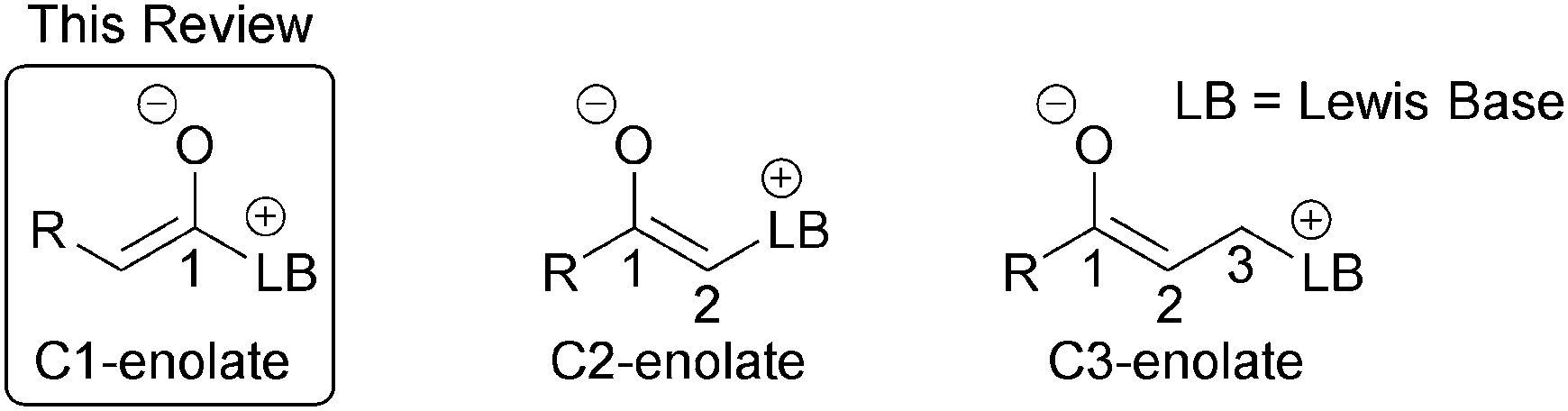 | ||
| Fig. 1 Classification of ammonium/azolium enolates. | ||
C1-ammonium enolates have traditionally been accessed via the nucleophilic attack of a tertiary amine catalyst on either a pre-formed or in situ generated ketene formed from an acid chloride (Fig. 2).6,8 C1-azolium enolates can be formed in a similar manner using NHCs and ketenes. Alternatively, α-functionalised aldehydes or enals can be employed as precursors in redox catalysed transformations,7 while aliphatic aldehydes are useful C1-azolium enolate precursors in the presence of a stoichiometric oxidant.9 However, the development of alternative routes to C1-ammonium or azolium intermediates that do not employ unstable ketenes, their precursors or a redox transformation is an active field of research. To this end, this review exclusively concentrates on strategies for the generation of C1-ammonium or azolium intermediates from alternative, bench-stable starting materials at the carboxylic acid oxidation level, namely carboxylic acids, esters or anhydrides.
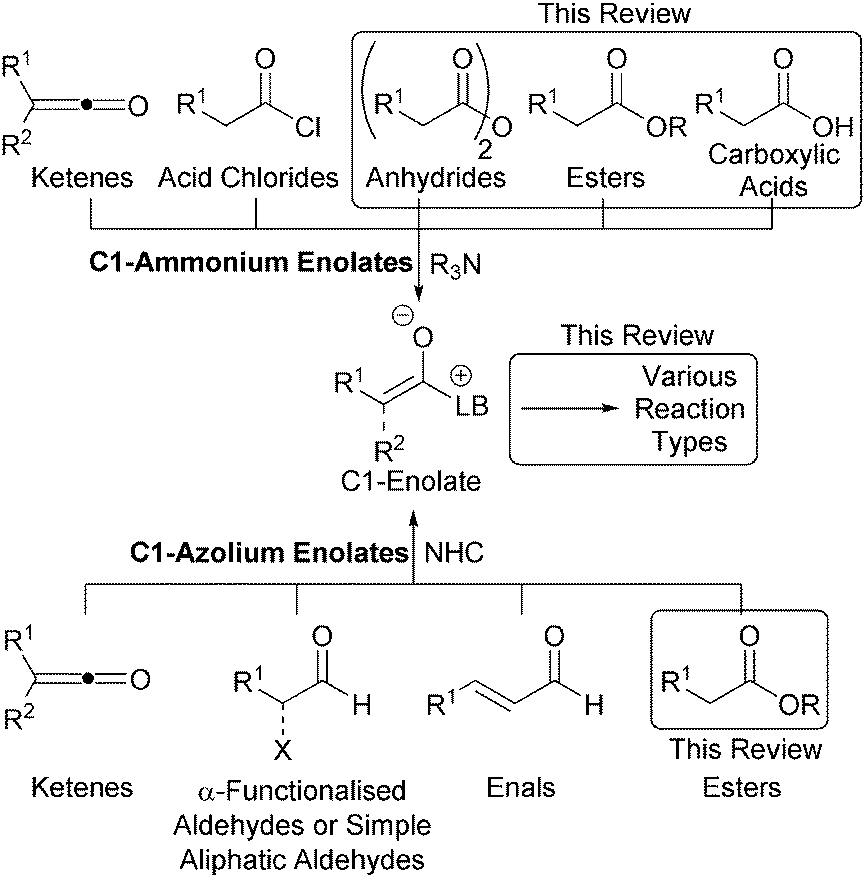 | ||
| Fig. 2 Overview of organocatalytic C1-enolate generation. | ||
These strategies all rely upon the initial acylation of a tertiary amine or NHC Lewis base catalyst to form a transient acylammonium or acylazolium intermediate from a carboxylic acid, anhydride or ester starting material that can subsequently undergo deprotonation to afford the corresponding C(1)-enolate (Fig. 3). Despite the commercial availability, high stability and low cost of a variety of carboxylic acids, they have been sparsely used as starting materials in Lewis base organocatalysis. Direct reaction of an acid with a Lewis base results in non-productive salt formation, so acid substrates must firstly be derivatised in situ to generate an “activated” electrophilic species (such as a mixed anhydride) that can be readily intercepted by the Lewis basic tertiary amine or NHC, generating an acylammonium or acylazolium intermediate en route to the corresponding enolate. Alternatively, bench stable acid anhydrides or “activated” phenolic esters are directly susceptible to nucleophilic attack by a Lewis base to generate the desired acylammonium or acylazolium intermediate.
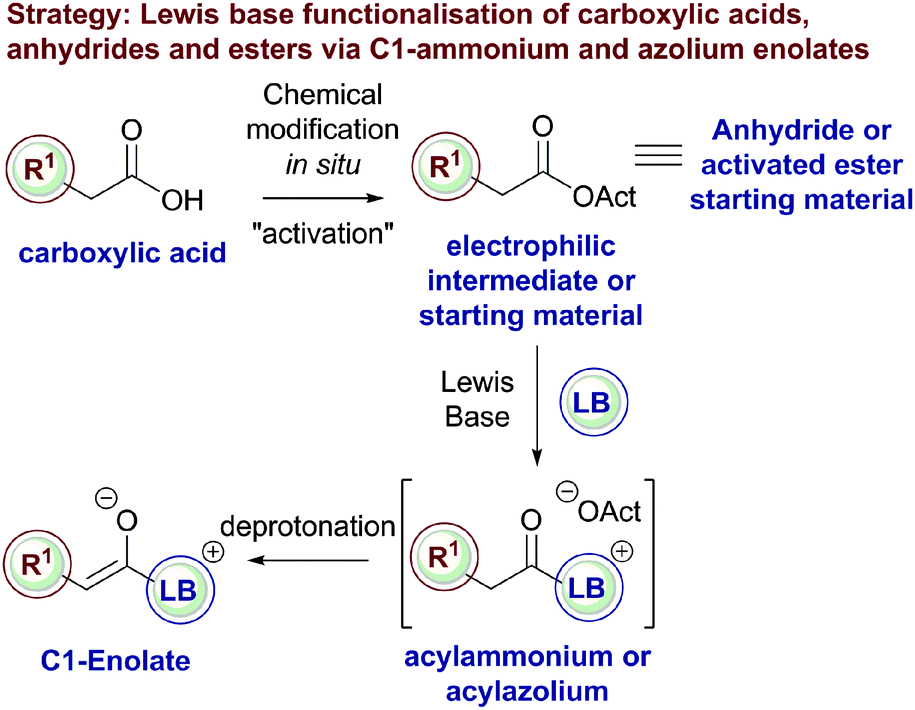 | ||
| Fig. 3 Overview of strategy of C1-enolate generation from carboxylic acids, anhydrides and esters. | ||
In this context, this review will describe the generation of C1-ammonium enolates from carboxylic acid, ester and anhydride starting materials. The use of various tertiary amine Lewis basic catalysts will be demonstrated, including Cinchona alkaloids, pyridine-derived nucleophiles and isothioureas. The subsequent utility of these enolates towards both intra- and intermolecular processes will be discussed and placed in context with their utility in natural product synthesis. The generation of C1-azolium enolates from carboxylic ester starting materials using NHC catalysts will also be described; to date the use of carboxylic acids and anhydrides to prepare azolium anolates has not been described but this clearly offers new opportunities for exploitation.
2. Carboxylic acids as C1-ammonium enolate precursors
2.1. Intramolecular processes
Taking inspiration from the early work of Wynberg in generating β-lactones in a catalytic and asymmetric fashion,10 Romo and co-workers developed the first example of a catalytic and asymmetric nucleophile-catalysed aldol-lactonisation (NCAL) reaction using a carboxylic acid starting material.11 While the Wynberg methodology generates enantiomerically enriched β-lactones, the Romo methodology addressed limitations of this approach – such as the use of pre-generated ketene and the necessity for using highly reactive aldehyde substrates such as chloral. Treatment of a range of aldehyde-acids 2 with Mukaiyama reagent 112 and Et3N promotes the intramolecular NCAL reaction, generating a variety of cis-bicyclic β-lactones 3 in good yields (Scheme 1). Syringe pump addition of substrate over 10 h and using CH3CN as solvent were identified as important factors for obtaining high yields.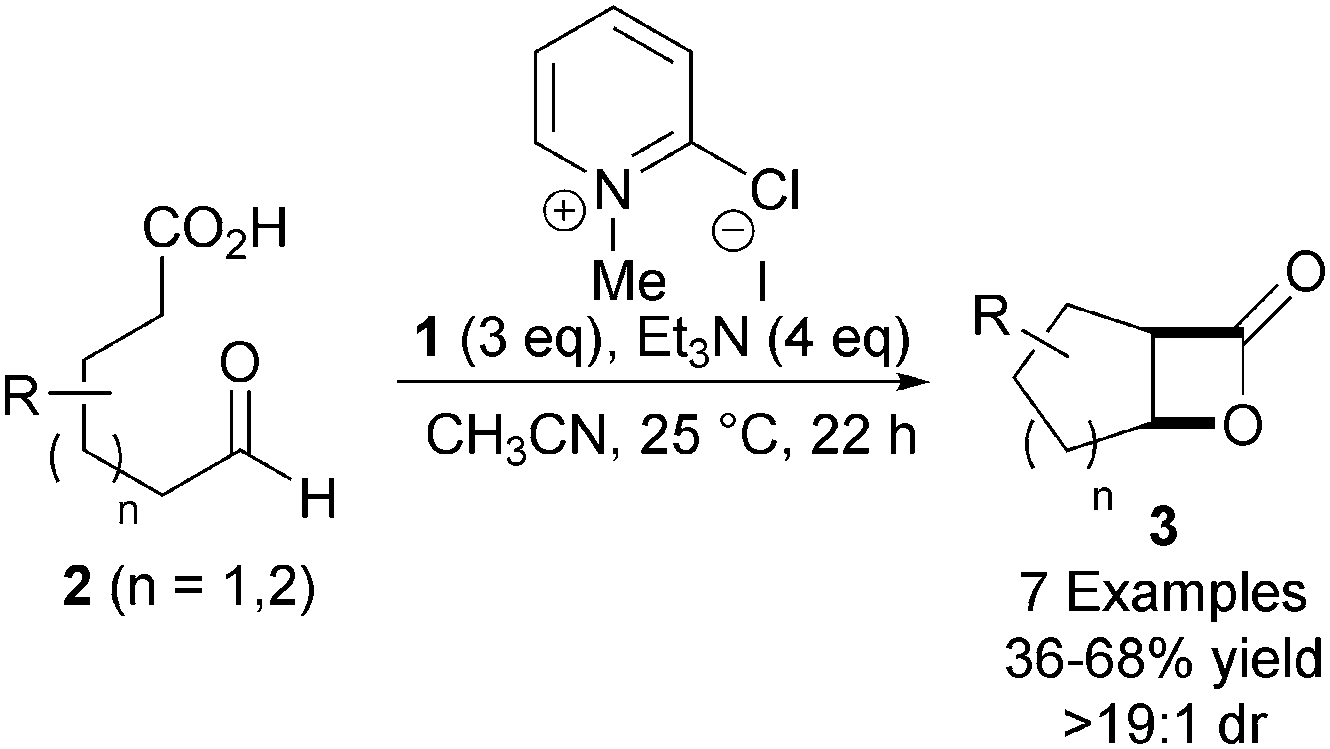 | ||
| Scheme 1 Nucleophile-catalysed aldol-lactonisation (NCAL) reaction. | ||
This process was rendered asymmetric through use of chiral tertiary amines. For example, using O-acetyl quinidine 4 as the Lewis base generates bicyclic β-lactone (1R,2S)-5 from 6 in high ee, providing evidence for a NCAL reaction pathway over a possible thermal [2+2] cycloaddition (Scheme 2).
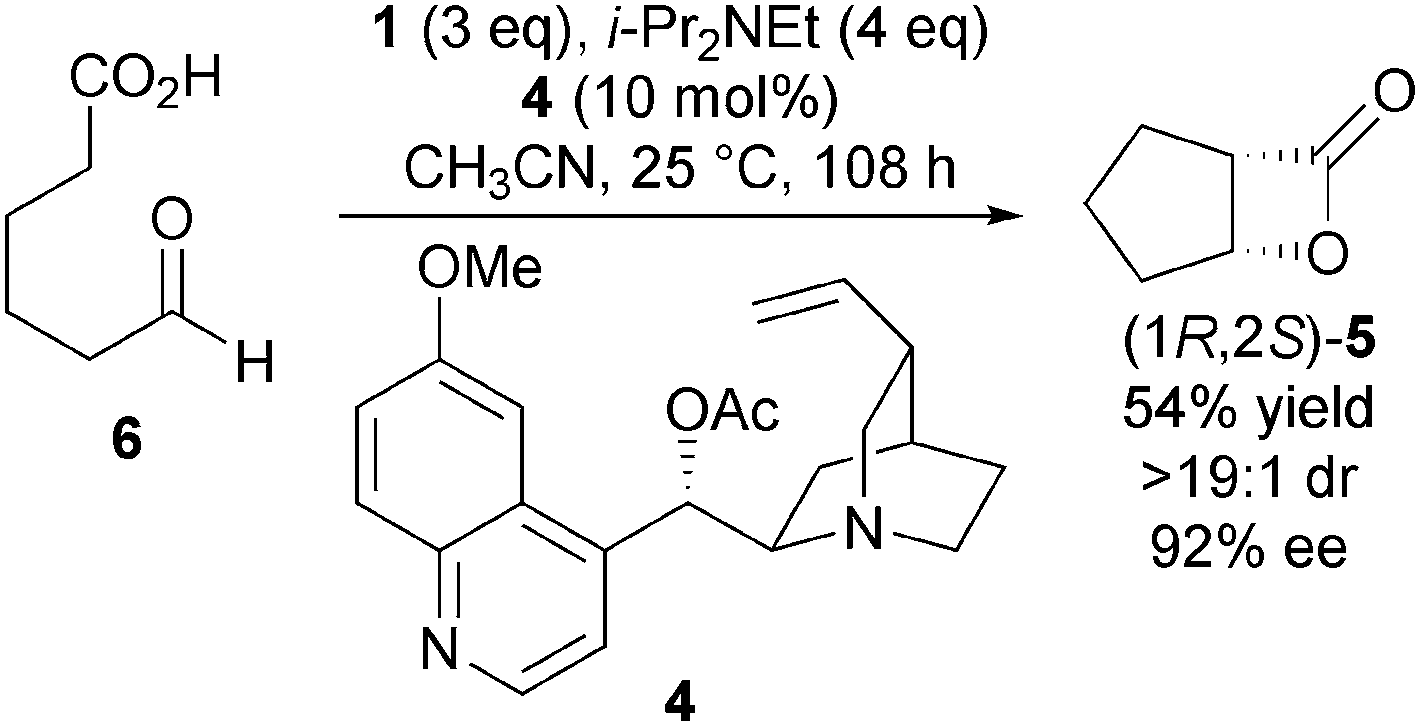 | ||
| Scheme 2 Chiral amine catalysed asymmetric NCAL reaction. | ||
The proposed mechanism involves initial derivatisation of carboxylic acid 6 with Mukaiyama's reagent and base to form activated ester 7in situ (Scheme 3). Two possible pathways are then proposed: (i) ketene 8 may form via elimination, which can be intercepted by the cinchona catalyst to generate ammonium enolate 9; or (ii) the catalyst may undergo acylation with the activated ester to form acyl ammonium 10 that gives ammonium enolate 9 upon deprotonation. It is likely that the acyl ammonium pathway (path ii) is more prevalent due to ketene 8 being present at very low concentrations at room temperature.
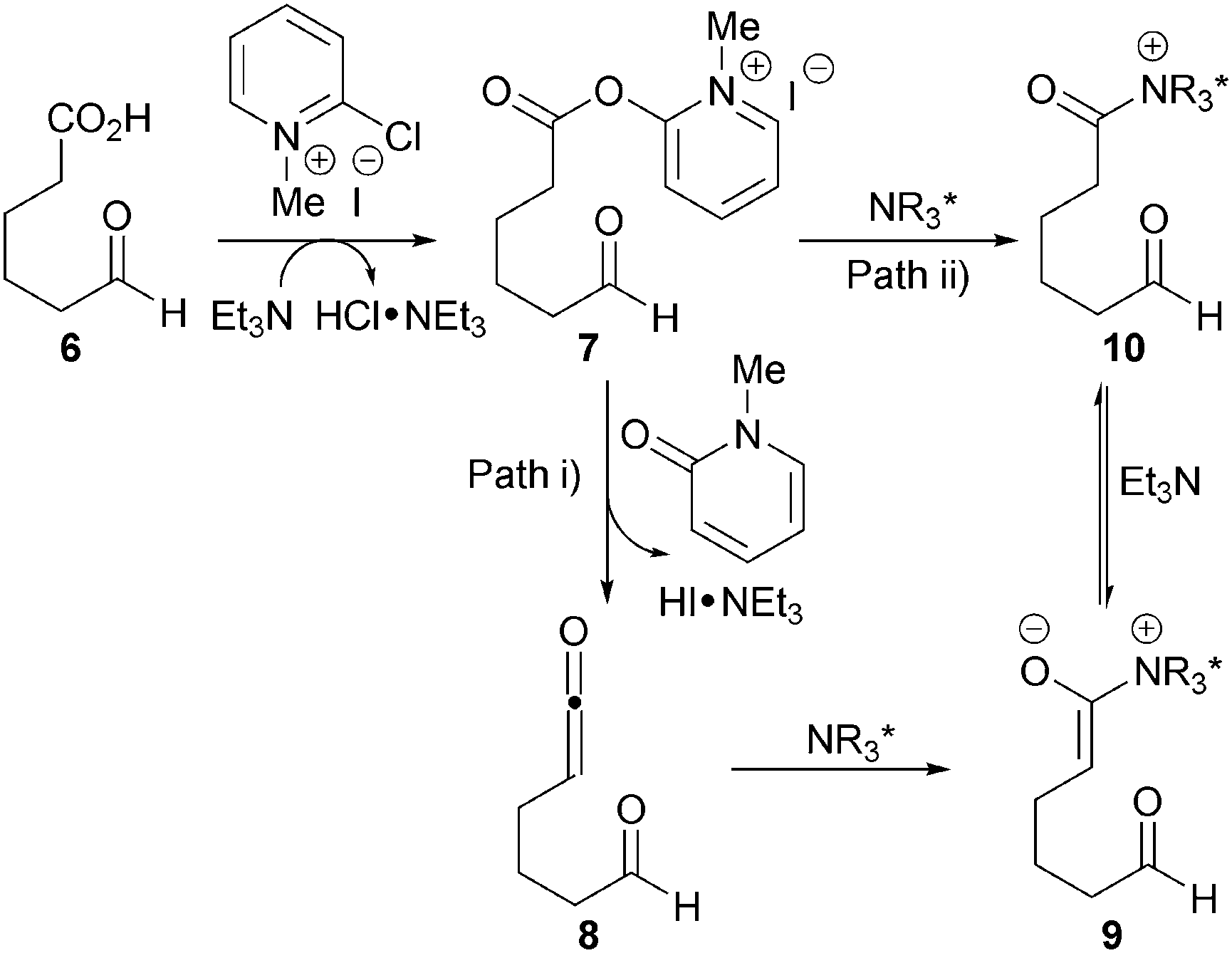 | ||
| Scheme 3 Generation of ammonium enolate 9 from aldehyde-acids. | ||
The origin of the stereoselectivity in this process can be explained by the aldehyde approaching the Re face of ammonium enolate 11, opposite to the steric bulk of the quinoline ring, generating 12. Nucleophilic attack on the Si face of the aldehyde gives cis-aldolate 13, which undergoes lactonisation to generate the cis-β-lactone 5 (Fig. 4).
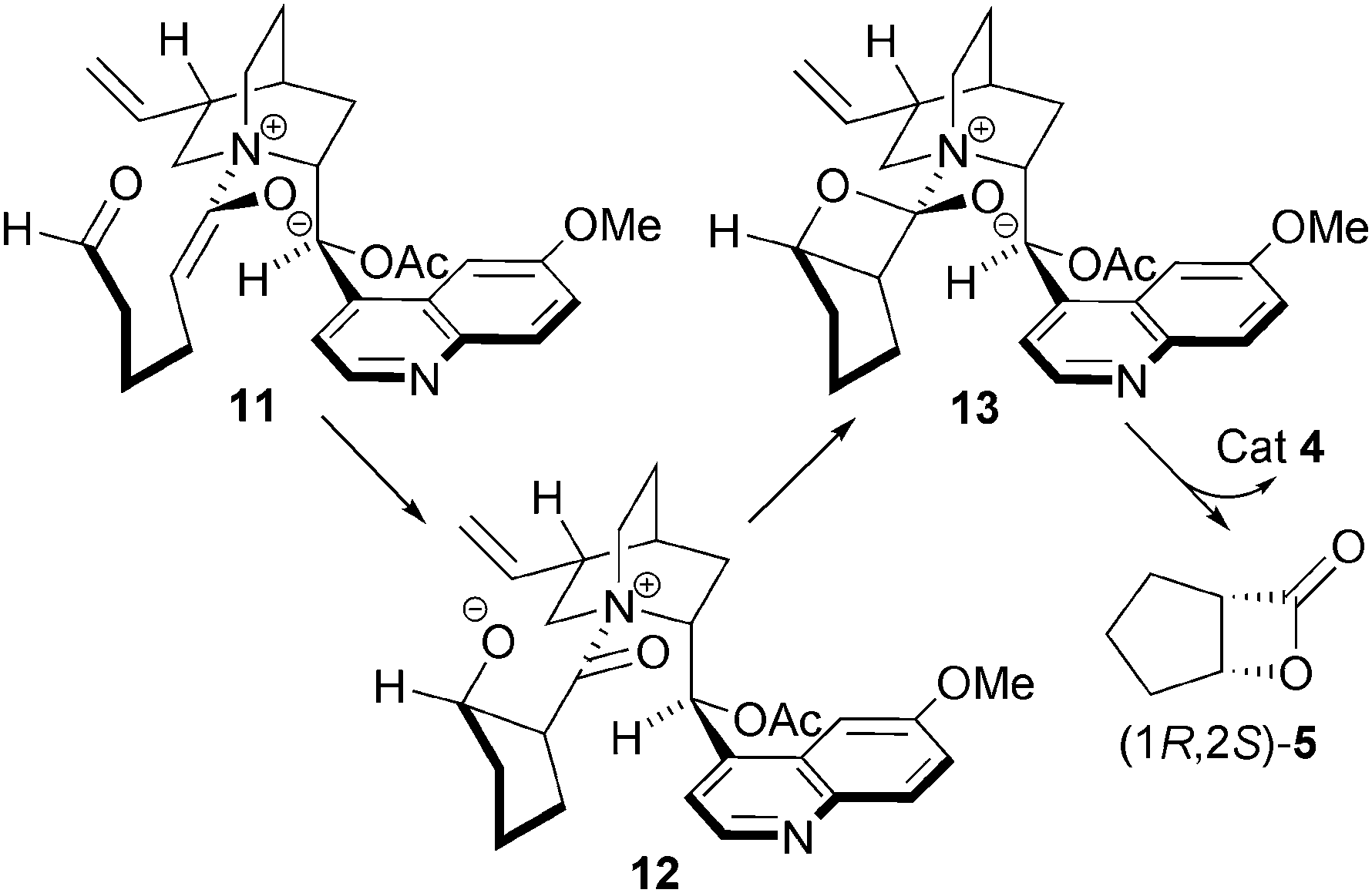 | ||
| Fig. 4 Stereochemical rationale for the asymmetric NCAL reaction. | ||
Various quinidine derivatives were tested as catalysts in this NCAL protocol, with changing the nature of the functional group at C9 (a variety of esters, a carbamate and a carbonate) having minimal influence upon observed enantioselectivities (Scheme 4).13 Significantly, through the use of either pseudo enantiomeric O-acetyl quinine 14, or rigidified catalyst 15, the enantiomeric β-lactone (1S,2R)-5 can be prepared in high ee.
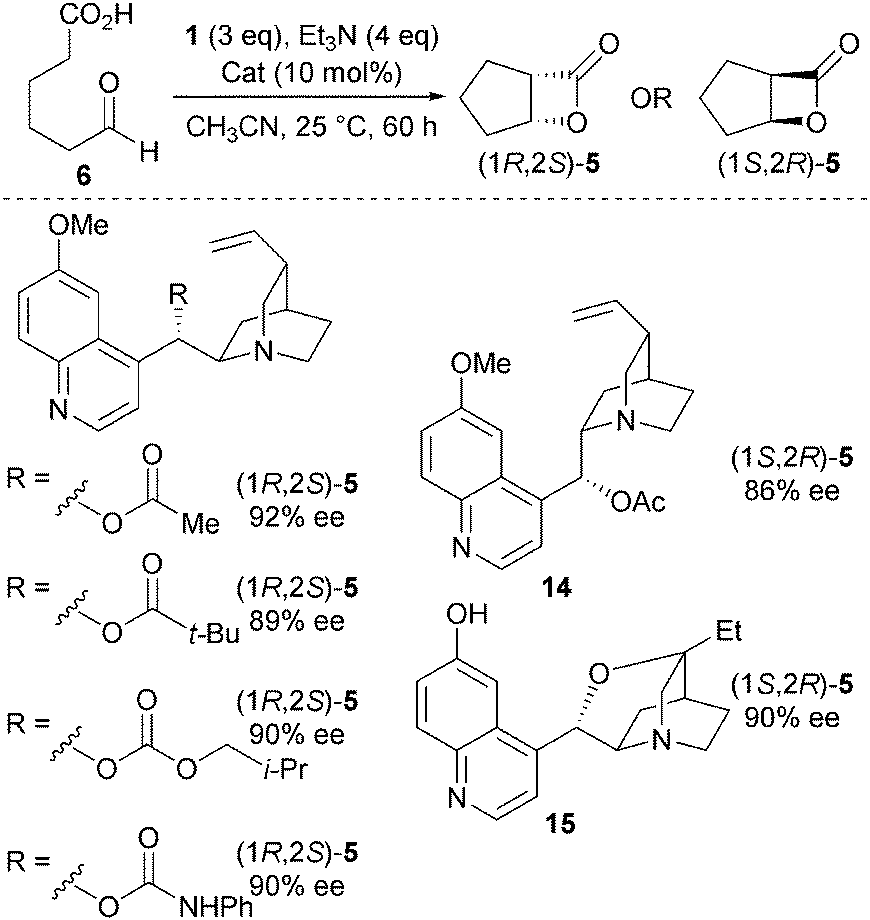 | ||
| Scheme 4 Various chiral amine catalysts for the NCAL reaction. | ||
The ability of modified Mukaiyama reagents 16–18 to increase reaction yields for the catalytic asymmetric NCAL reaction whilst maintaining high levels of enantioselectivity have also been investigated (Fig. 5).14,15 This was primarily attributed to: (i) improved reagent solubility, allowing reactions to be carried out in less polar solvents such as THF and CH2Cl2 and minimising potential ring-opening reactions promoted by polar media; (ii) less nucleophilic counterions – triflate and tetrafluoroborate – reducing β-lactone decomposition.
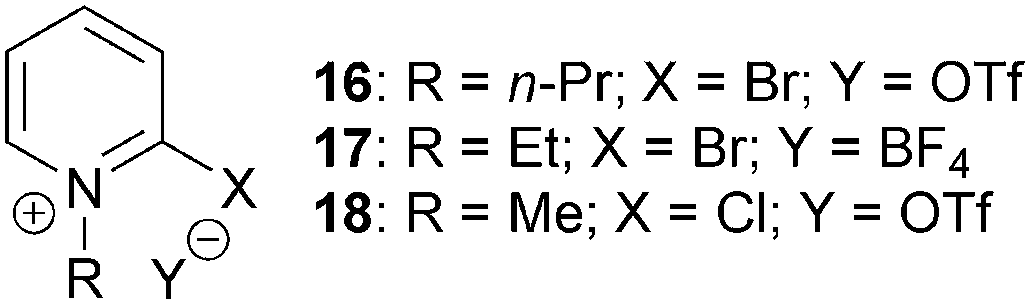 | ||
| Fig. 5 Modified Mukaiyama reagents for the NCAL reaction. | ||
In 2011, the Dikshit group demonstrated a catalytic asymmetric aza variant of the NCAL reaction (Scheme 5).16 Using modified Mukaiyama's reagent 19, amino acid derived aldehyde-acids 20 could be converted into either enantiomer of the corresponding bicyclic β-lactone 21 through use of either O-acetyl quinine 14 or O-acetyl quinidine 4, in good yields (51–67%) and high enantioselectivities (83–97% ee). This strategy provides evidence for the wider application of Romo's NCAL strategy towards alternative heterocyclic systems.
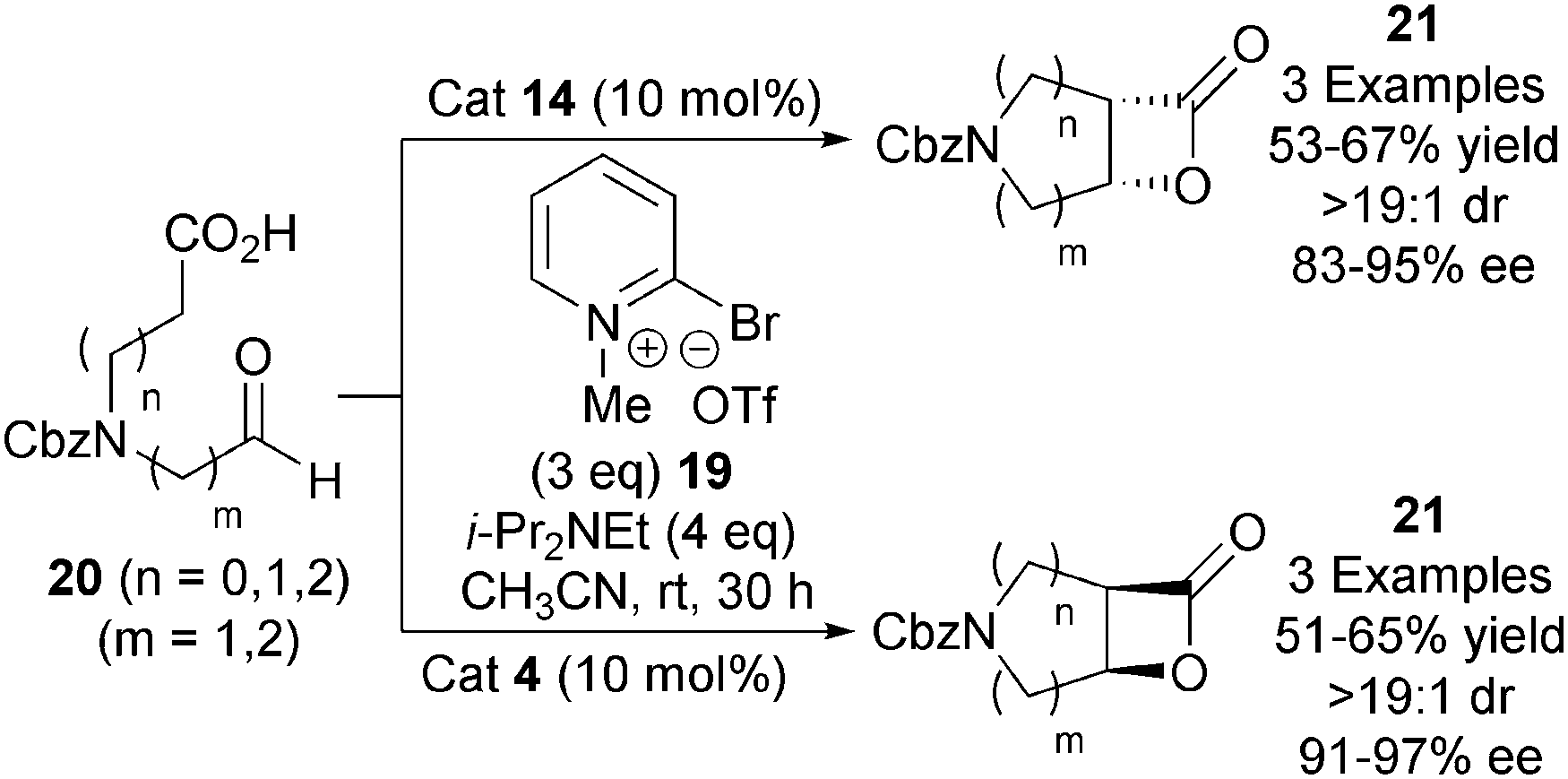 | ||
| Scheme 5 Aza variant of the intramolecular NCAL reaction. | ||
Building upon this work, Romo developed a diastereoselective NCAL reaction that allowed the conversion of enantiomerically enriched aldehyde-acid substrates bearing γ- and δ-substituents into bicyclic β-lactones in high diastereoselectivity.17 Amongst several examples, treatment of aldehyde-acid 22 (87% ee) under the standard NCAL conditions using Et3N as the nucleophilic promoter, gives a substrate controlled 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomeric anti- and syn-β-lactones 23 and 24 respectively (Table 1). Chiral catalysts were next examined to test if catalyst control could override the inherent substrate bias. Use of O-TMSQD results in a reversal of diastereoselectivity (1:7 anti:syn) representative of the mismatched case, while employing O-TMSQN gives exclusively the anti-β-lactone (>19:1 anti:syn) indicative of a matched situation.
:1 mixture of diastereomeric anti- and syn-β-lactones 23 and 24 respectively (Table 1). Chiral catalysts were next examined to test if catalyst control could override the inherent substrate bias. Use of O-TMSQD results in a reversal of diastereoselectivity (1:7 anti:syn) representative of the mismatched case, while employing O-TMSQN gives exclusively the anti-β-lactone (>19:1 anti:syn) indicative of a matched situation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Base | Time (h) | Yield (%) | dr (23:24) |
| 1 | Et3N (100) | Et3N | 48 | 58 | 2:1 |
| 2 | O-TMSQD (10) | i-Pr2EtN | 72 | 73 | 1:7 |
| 3 | O-TMSQN (10) | i-Pr2EtN | 72 | 60 | >19:1 |
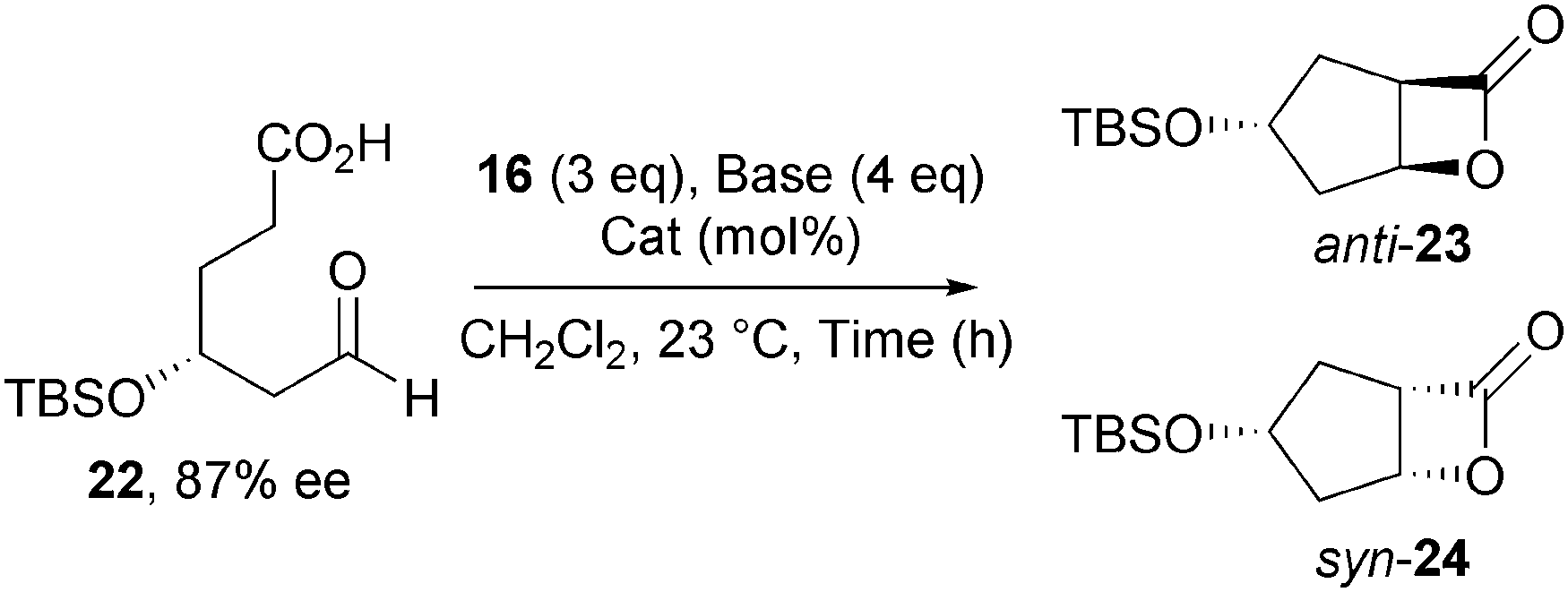
The potential to access both tetrahydrofuran- and tetrahydropyran-fused β-lactones via the same NCAL process was also investigated, however this generally resulted in both poor diastereoselectivities and isolated yields.
In a significant advance, the NCAL methodology was extended towards less reactive keto-acid substrates 25.18 Using super-stoichiometric quantities of 4-pyrrolidino pyridine (PPY) 26, the ammonium enolate formed is sufficiently reactive to undergo bis-cyclisation, affording a variety of bicyclic and tricyclic β-lactones 27 in high yield and diastereoselectivity (Scheme 6).
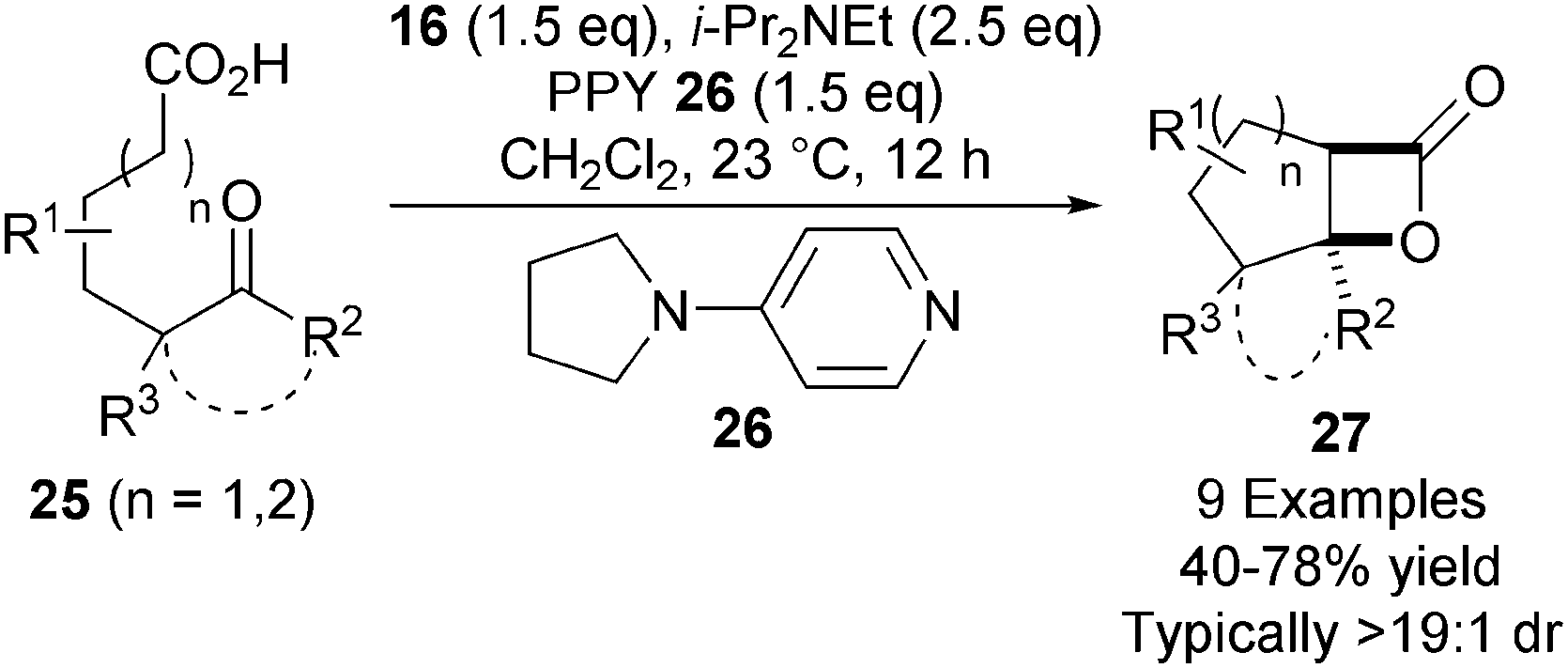 | ||
| Scheme 6 Bis-cyclisation of keto-acids. | ||
To showcase the utility of this methodology, it was used as the key step in an enantioselective synthesis of (+)-dihydroplakevulin A 28, known to be a precursor to DNA polymerase inhibitor plakeuvulin A 29 (Scheme 7). Under optimised conditions, enantioenriched keto-acid 30 undergoes bis-cyclisation to give bicyclic β-lactone 31 in moderate yield and excellent dr, which is easily converted over two steps into (+)-dihydroplakevulin A 28 through ring-opening with MeOH followed by silyl deprotection.
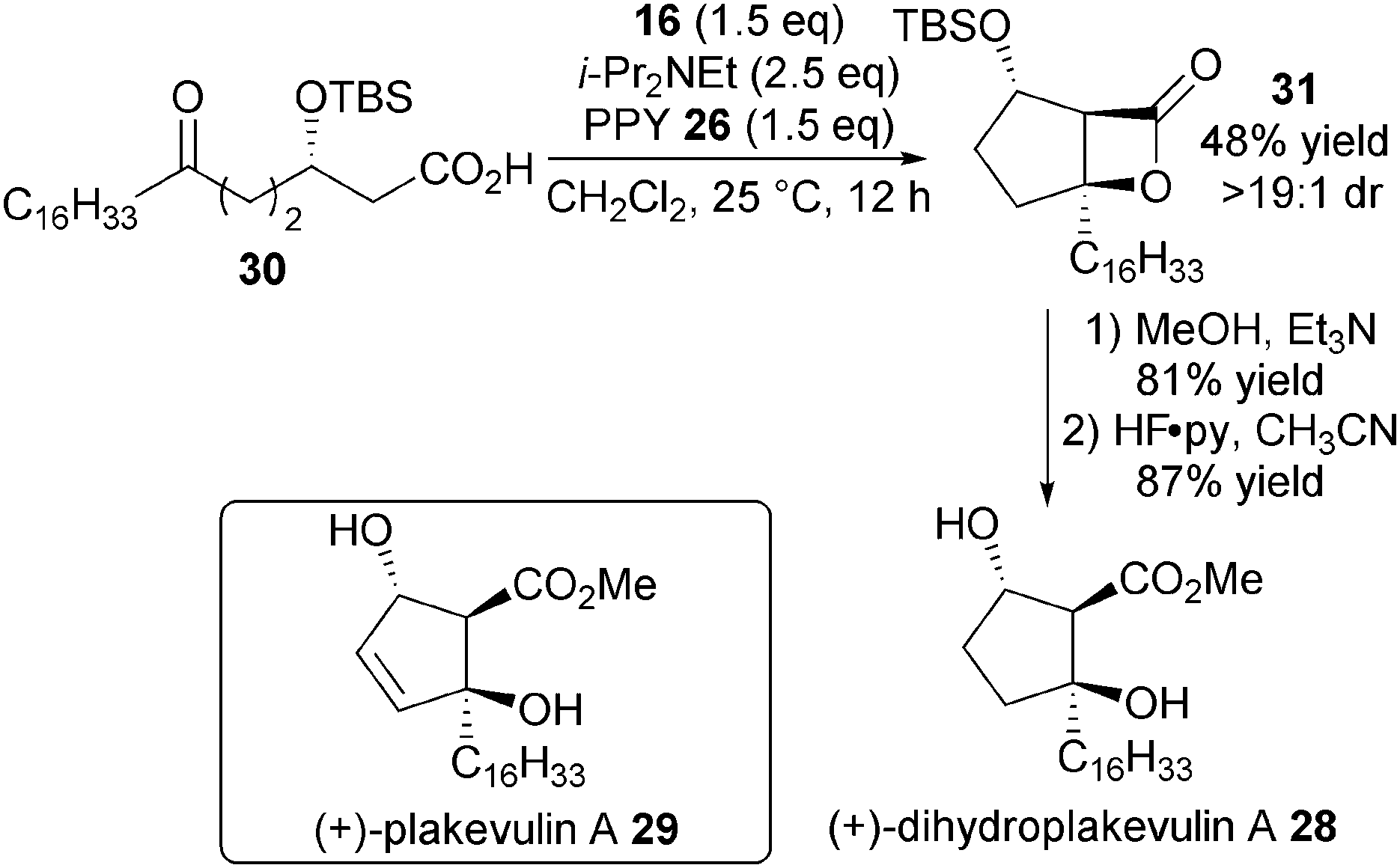 | ||
| Scheme 7 Synthesis of (+)-dihydroplakevulin A. | ||
Further applications of this methodology were disclosed in the synthesis of both (±)-cinnabaramide A and (±)-salinosporamide A 32 (Scheme 8).19 For example, bis-cyclisation of keto-acid 33 (1:1 dr), catalysed by PPY, gives β-lactone 34 in 34% yield and 2:1 dr with the major diastereoisomer subsequently elaborated to 32. The moderate yield of the key bis-cyclisation step is likely due to the difficult functionalisation of a sterically congested α,α-disubstituted carboxylic acid group and again highlights some of the current limitations of this methodology.
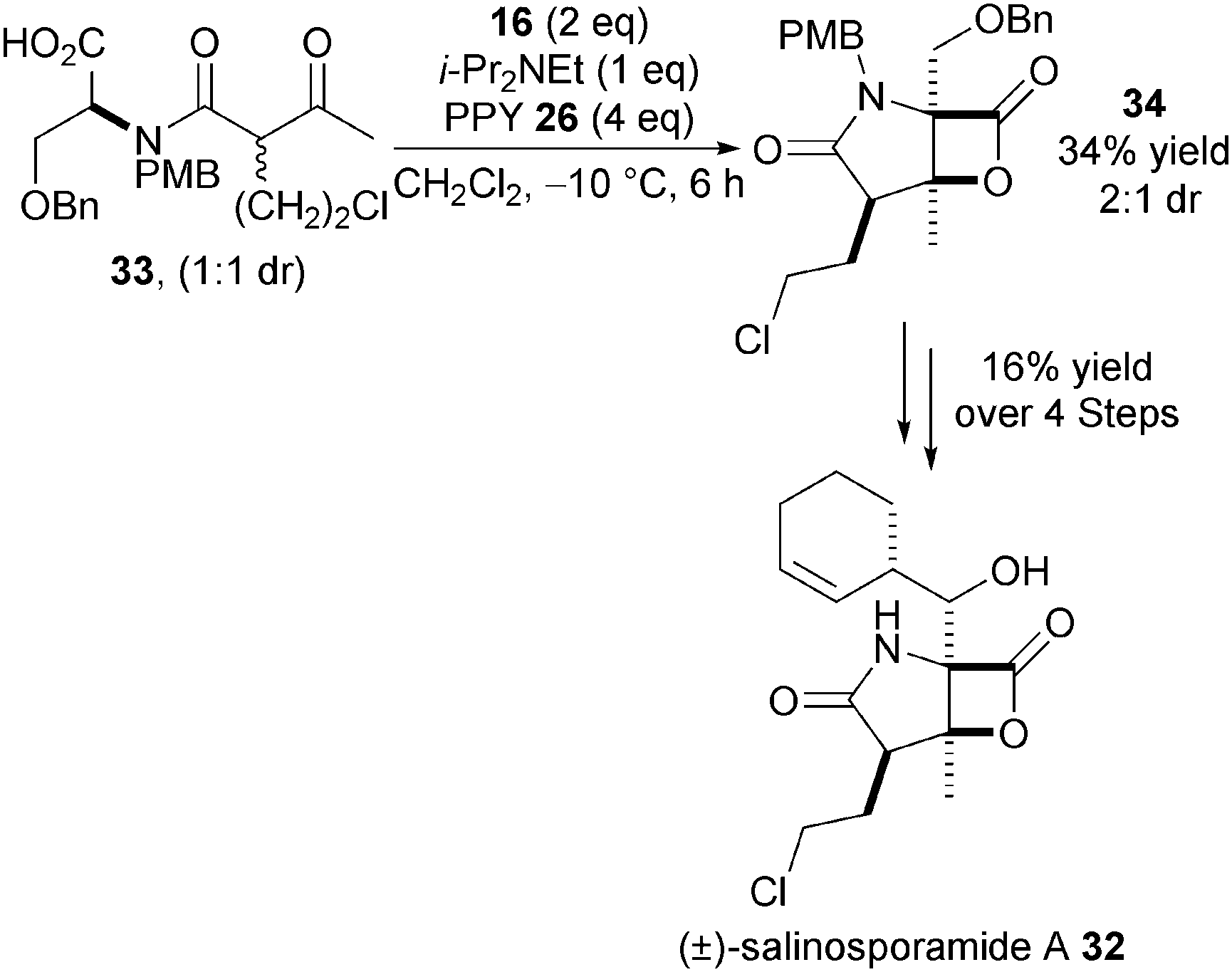 | ||
| Scheme 8 Synthesis of (±)-salinosporamide A. | ||
Romo subsequently demonstrated that (−)-salinosporamide A 32 could be obtained in enantiomerically enriched form (Scheme 9).20,21 This was achieved by synthesising enantio- and diastereomerically pure keto-acid 33 and utilising modified bis-cyclisation conditions that minimise racemisation of the starting material. Employing mesyl chloride as activating agent at −5 to −10 °C in toluene gave bicyclic β-lactone 35 in 53% yield, 5:1 dr and 90% ee on gram scale. Elaboration of 35 into (−)-salinosporamide A 33 was analogous to the previous synthesis.
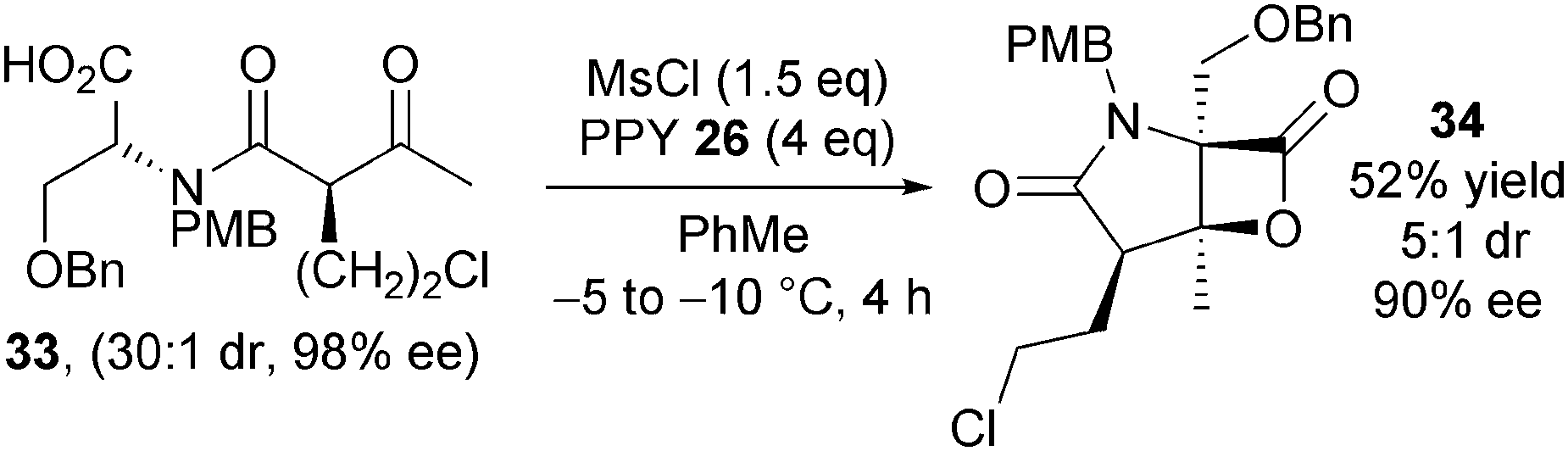 | ||
| Scheme 9 Modified route towards (−)-salinosporamide A. | ||
In 2011, Romo and co-workers again demonstrated the utility of the NCAL reaction towards natural product synthesis. Bis-cyclisation of (R)-carvone derived keto-acid 35, using TsCl as activating agent and 4-PPY as the nucleophilic Lewis base catalyst, gave the desired β-lactone product 36 in 83% yield and >19:1 dr (Scheme 10).22 Importantly, in the context of natural product synthesis, this NCAL reaction can be carried out on greater than 10 g scale. β-Lactone 36 was elegantly elaborated into molecularly complex (+)-omphadiol 37 over 7 steps in an impressive 33% overall yield, representing the first total synthesis of this product.
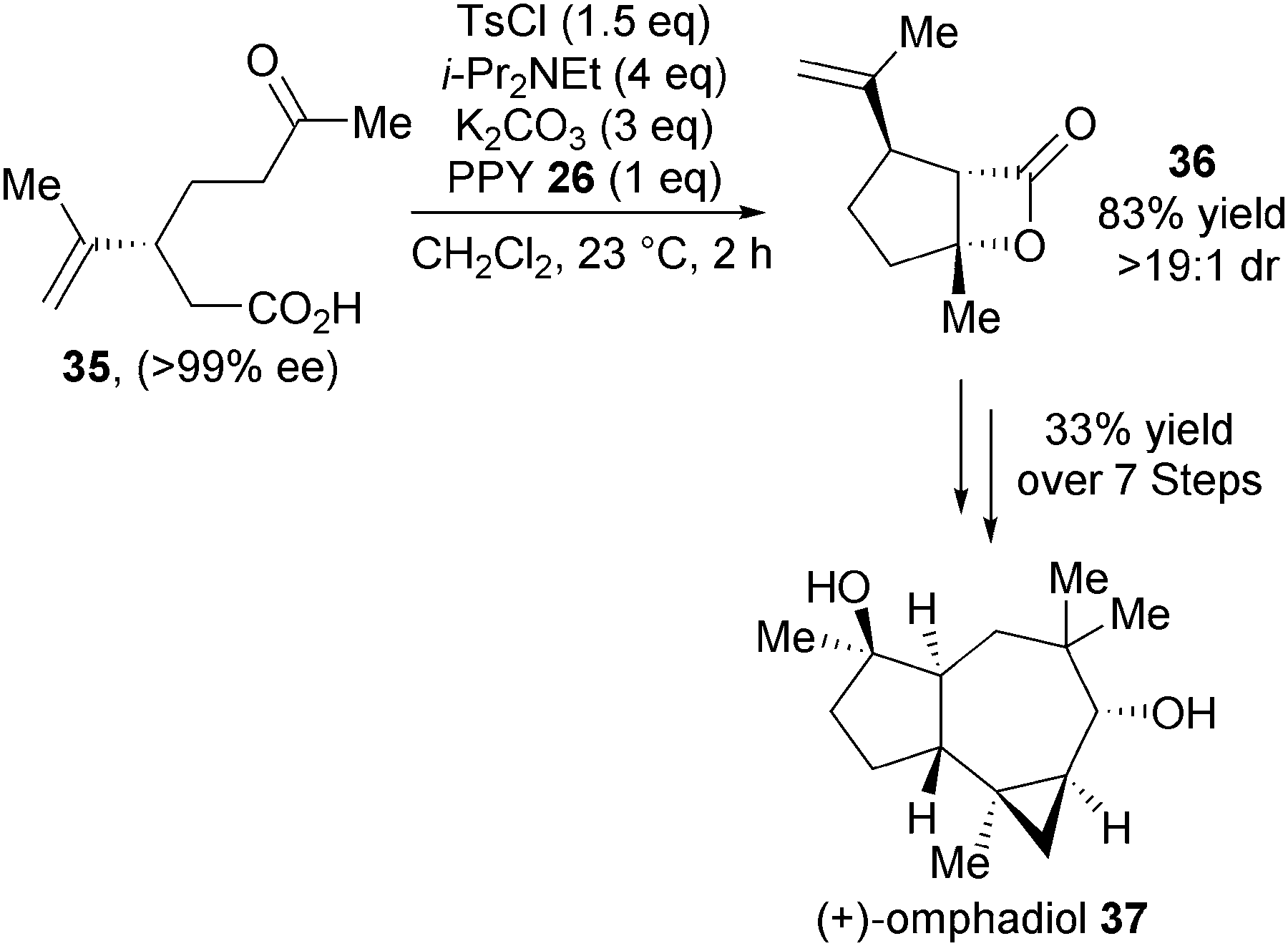 | ||
| Scheme 10 Application of NCAL towards synthesis of (+)-omphadiol. | ||
In the key bis-cyclisation step towards the synthesis of (+)-omphadiol, Romo and co-workers noted that K2CO3 as base in combination with i-Pr2NEt as “shuttle base” was optimal for achieving a high yield of 36 in a significantly reduced reaction time of 2 h (typically 24 h). This led to the development of a more practical and scalable general procedure for the highly diastereoselective NCAL reaction of keto-acids 38 into bicyclic β-lactones 39 using commercially available p-TsCl as activating agent and DMAP 40 as nucleophilic catalyst (Scheme 11).23
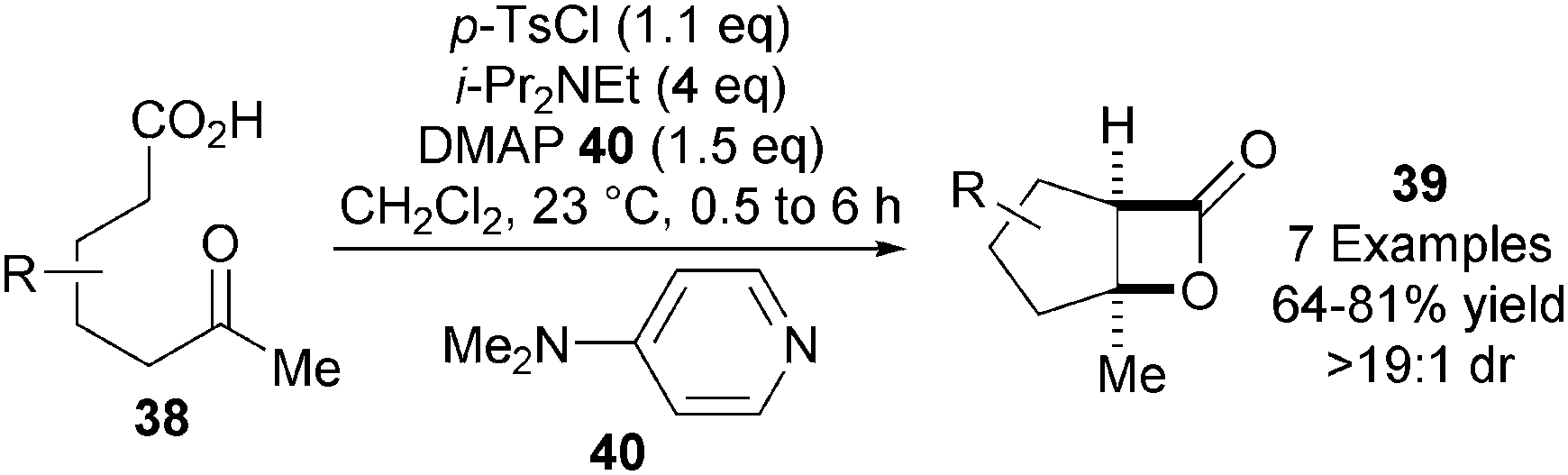 | ||
| Scheme 11 Improved procedure for NCAL of keto-acids. | ||
In 2008, Romo and co-workers utilised the NCAL reaction to access a range of tricyclic β-lactones 41 from keto-acids 42 using their standard reaction conditions (Scheme 12).24 Interestingly, these products undergo an unusual dyotropic rearrangement via 1,2-acyl migration upon treatment with Lewis acidic Zn(OTf)2, giving bridged γ-lactones 43 with high stereospecificity.
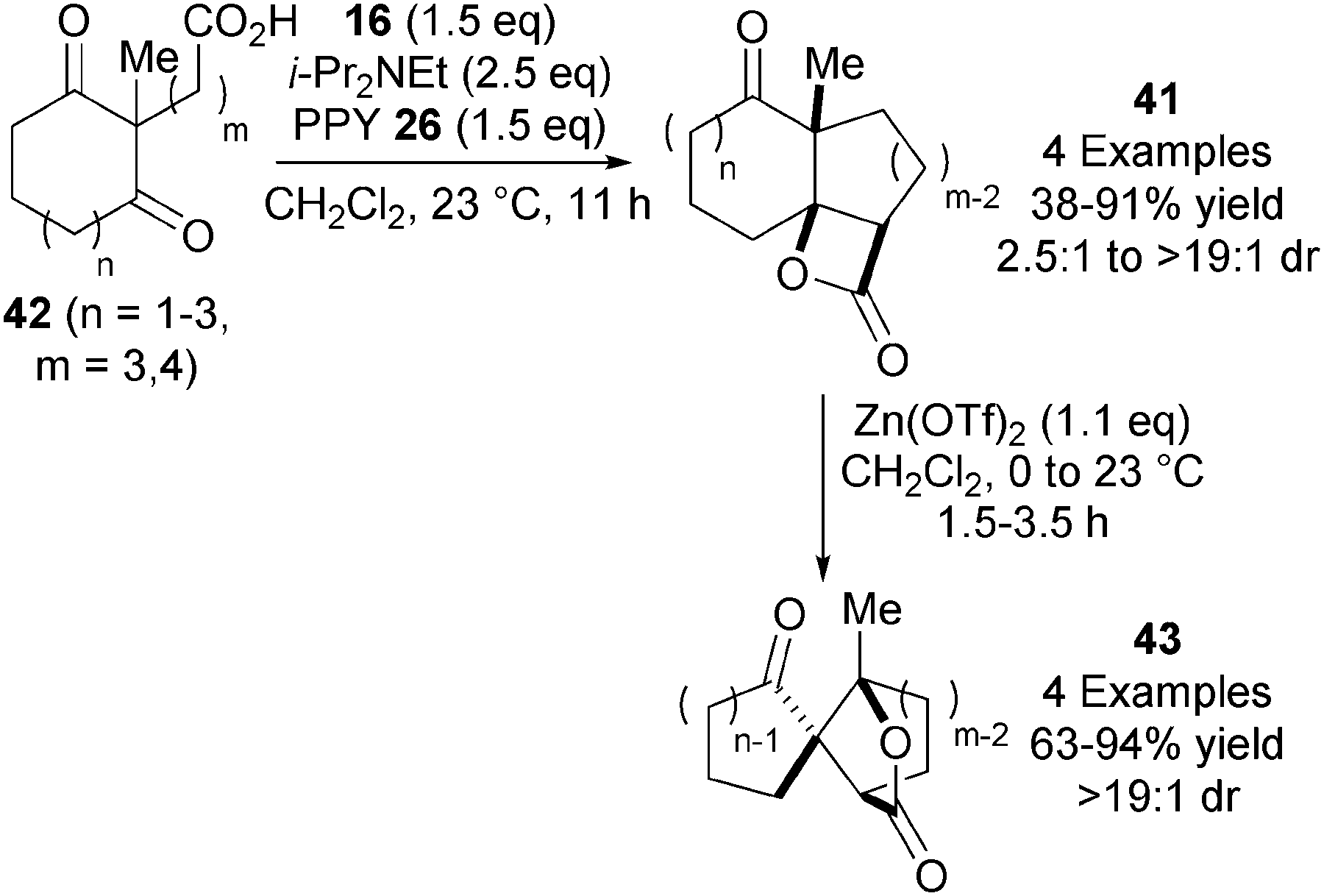 | ||
| Scheme 12 Tricyclic β-lactone synthesis and dyotropic rearrangements. | ||
As a proof of principle, Romo reported the first asymmetric bis-cyclisation of keto-acid 44 using stoichiometric quantities of commercially available (S)-tetramisole hydrochloride 45, forming 46 in 97% ee (Scheme 13). This experiment provided both the first direct evidence for nucleophile involvement in the stereodefining step of biscyclisations with keto-acid substrates and the first demonstration of isothioureas in ammonium enolate chemistry.
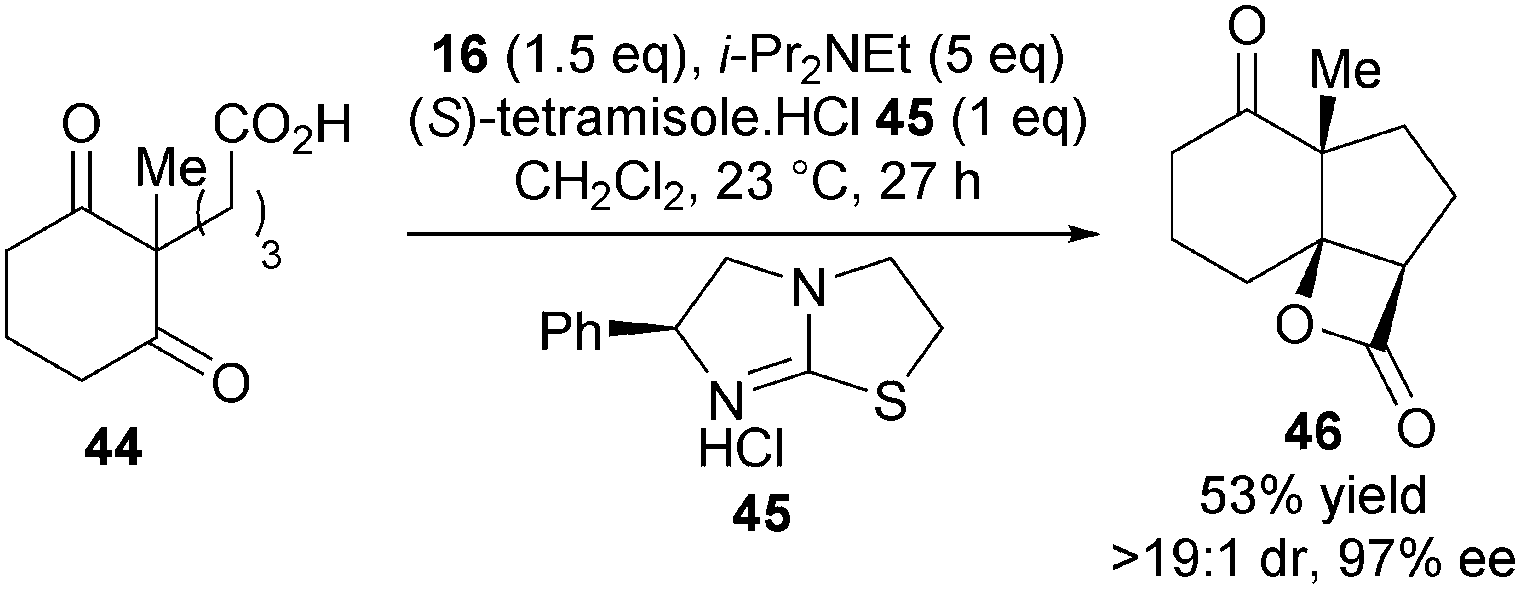 | ||
| Scheme 13 Stoichiometric asymmetric synthesis of tricyclic β-lactone. | ||
In a significant breakthrough, Romo extended this approach to the first catalytic asymmetric NCAL reaction of keto-acids.25 Isothiourea (S)-HBTM 47, developed by Birman as a highly efficient O-acyl transfer agent,26 catalyses the transformation of a range of keto-acids 48 into bi- and tricyclic β-lactones 49 (Scheme 14). Careful optimisation showed that that p-TsCl was the optimal carboxylic acid activating agent and that use of both 0.1 M concentrations and 1 eq. of LiCl Lewis acid co-catalyst were optimal for obtaining 49 in high isolated yields (71–93%) and in excellent enantioselectivities (84 to >98% ee).
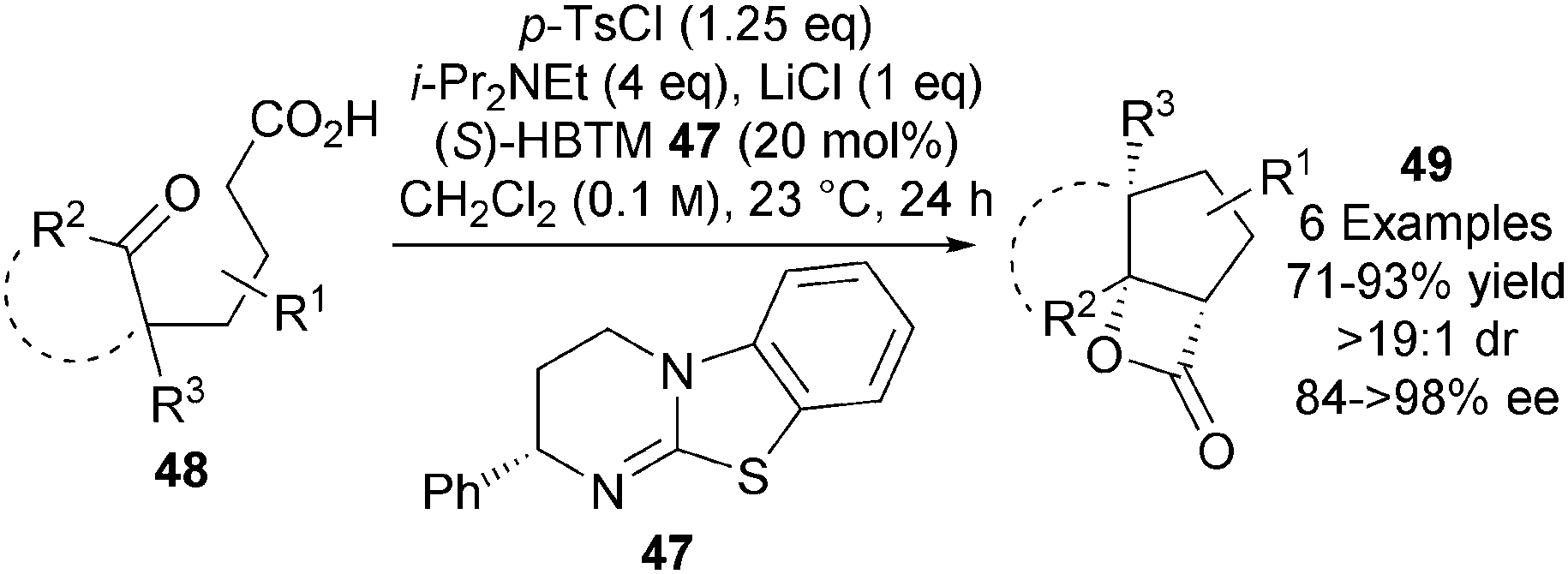 | ||
| Scheme 14 Catalytic asymmetric synthesis of bi- and tricyclic β-lactones. | ||
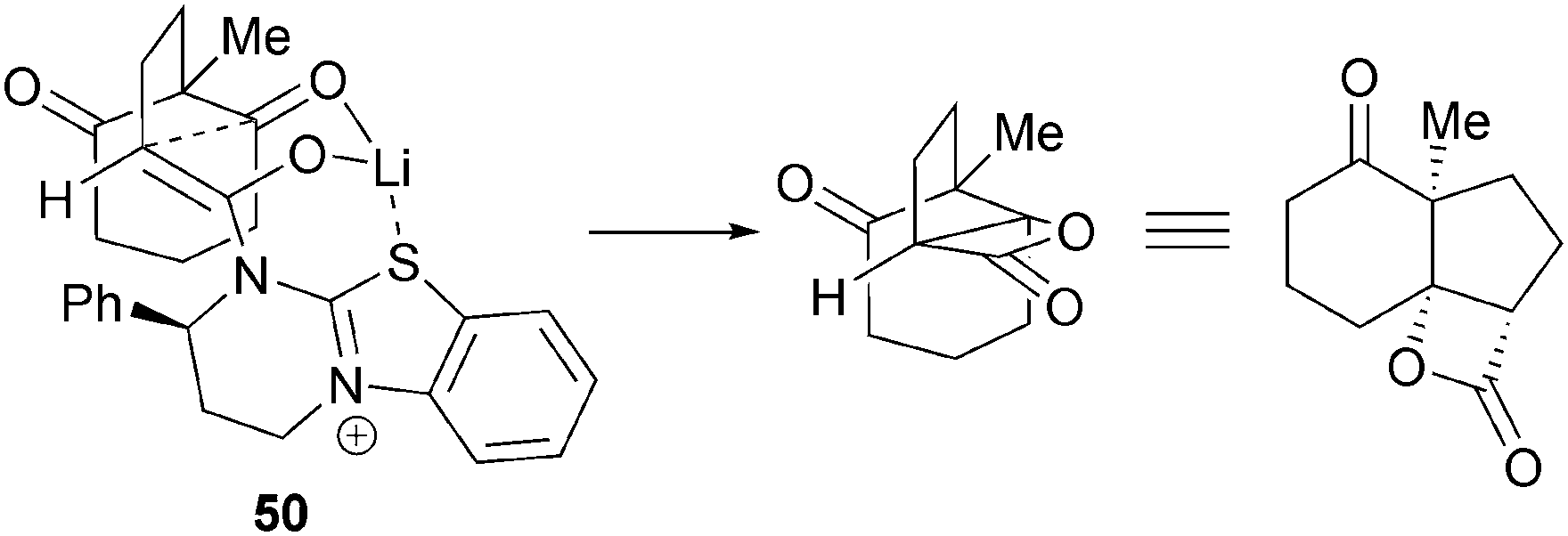
A model is provided that explains the observed relative and absolute configuration (Fig. 6). Formation of the (Z)-ammonium enolate, with Li–S chelation results in a bicyclic chair-like transition state 50 that includes ketone activation by Li, rationalising the observed increase in yield.
 | ||
| Fig. 6 Stereochemical rationale for the asymmetric NCAL reaction. | ||
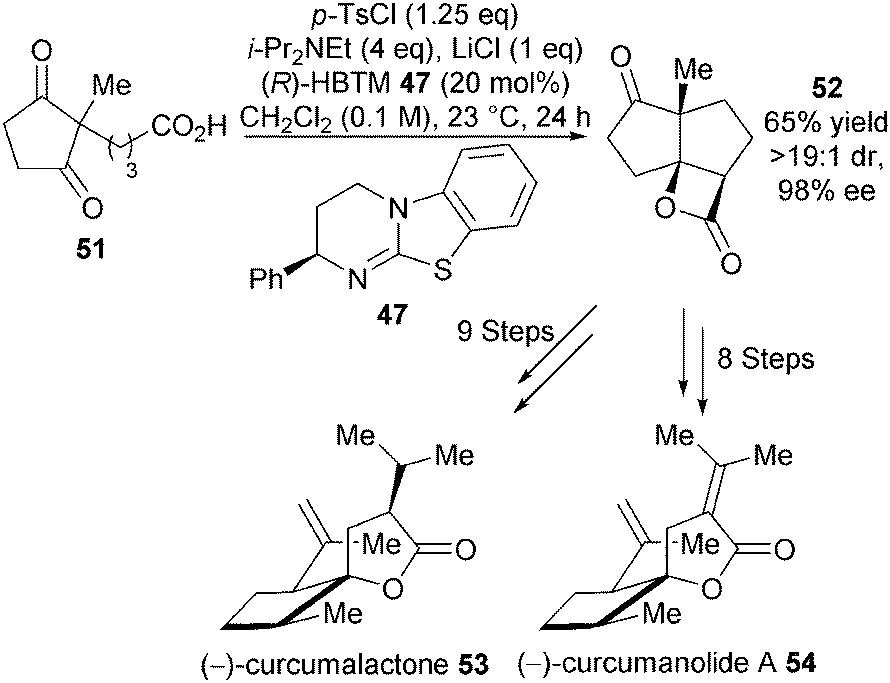
In a powerful demonstration of utility, this catalytic asymmetric NCAL reaction was later used by the Romo group as a key step towards their asymmetric synthesis of (−)-curcumanolide A and (−)-curcumalactone.27 Desymmetrisation of dione 51 using (R)-HBTM 47 (20 mol%), p-TsCl as activating agent and LiCl as Lewis acid additive gave the desired enantiomer of tricyclic β-lactone 52 in 65% yield, >19:1 dr and 98% ee on gram scale (Scheme 15). This key building block was further elaborated into (−)-curcumalactone 53 and (−)-curcumanolide A 54 in 9 and 8 steps, respectively.
 | ||
| Scheme 15 Application of the catalytic asymmetric NCAL reaction. | ||
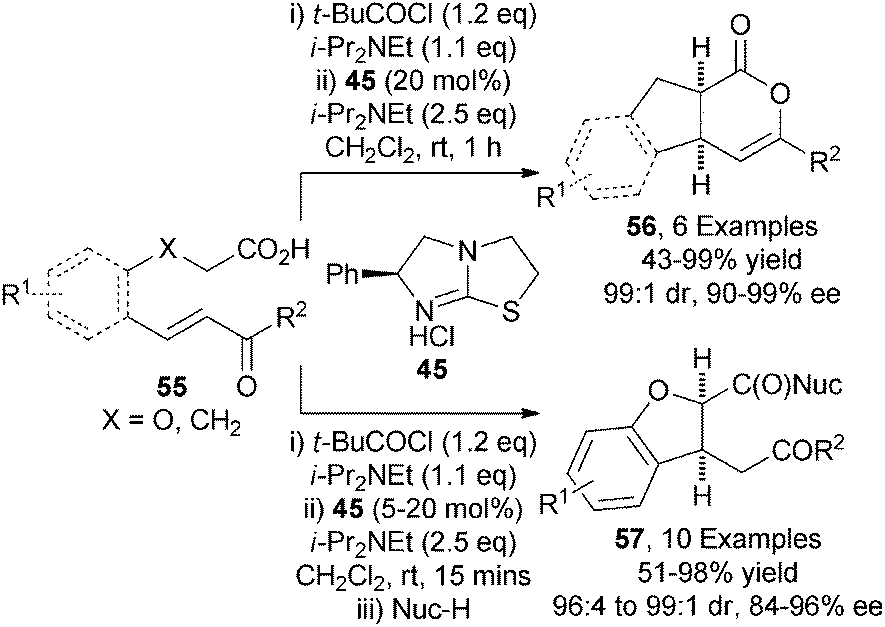
Building upon these precedents, the first application of carboxylic acid derived ammonium enolates in Michael addition processes was demonstrated by the Smith group.28 Using commercially available pivaloyl chloride as activating agent and (S)-tetramisole hydrochloride 45 as nucleophilic precatalyst, a range of enone-acids 55 undergo intramolecular Michael addition-lactonisation to give functionalised indenes 56 or dihydrofuran carboxylates 57 (after in situ ring-opening) with excellent diastereo- and enantiocontrol (up to 99:1 dr, up to 99% ee) (Scheme 16).
 | ||
| Scheme 16 Intramolecular Michael addition-lactonisation. | ||
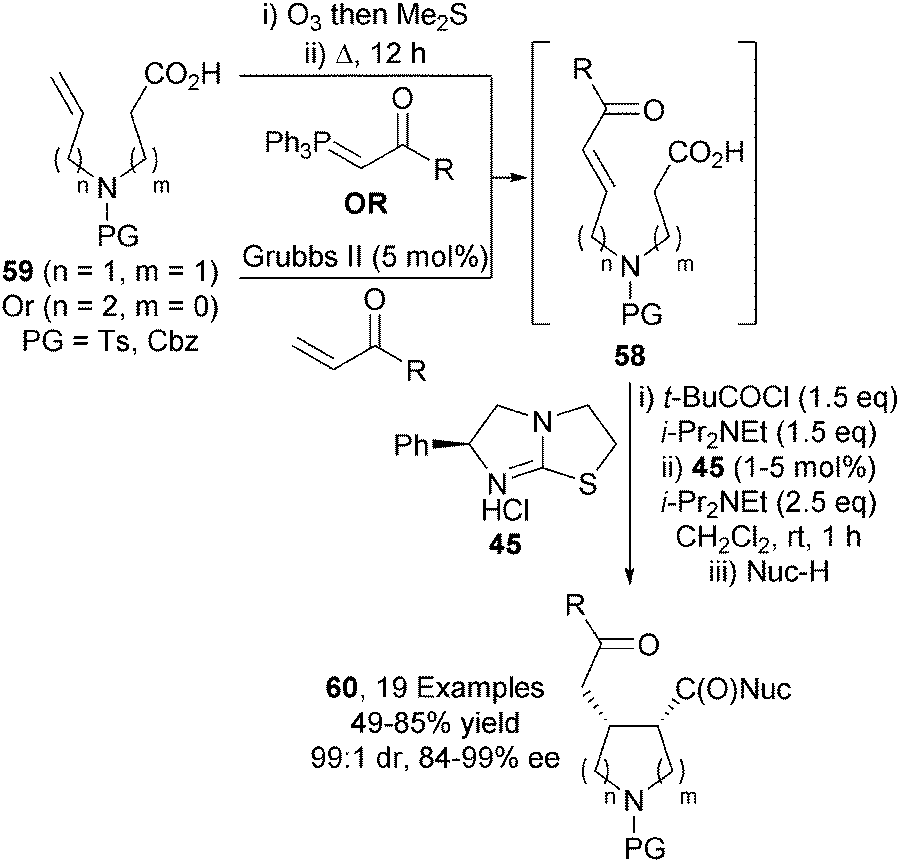
This approach was later applied to the synthesis of disubstituted pyrrolidines.29 Enone-acids 58, made in situ via either ozonolysis/Wittig olefination or cross metathesis from 59, were transformed into 2,3- or 3,4-syn-disubstituted pyrrolidines 60 under similar catalytic conditions in excellent diastereo- and enantiocontrol (up to 99:1 dr, up to 99% ee) (Scheme 17).
 | ||
| Scheme 17 Telescoped synthesis of stereodefined pyrrolidines. | ||
Notably, the diastereoselectivity of this transformation can be reversed through judicious choice of nucleophilic catalyst. Using modified Mukaiyama's reagent 61 as activating agent and OTMS-quinidine 62 as catalyst, enone-acid 63 gives 3,4-anti-disubstituted pyrrolidine 64 preferentially in modest diastereoselectivity (67:33 dr) with the major diastereoisomer formed in excellent enantioselectivity (99% ee) (Scheme 18).
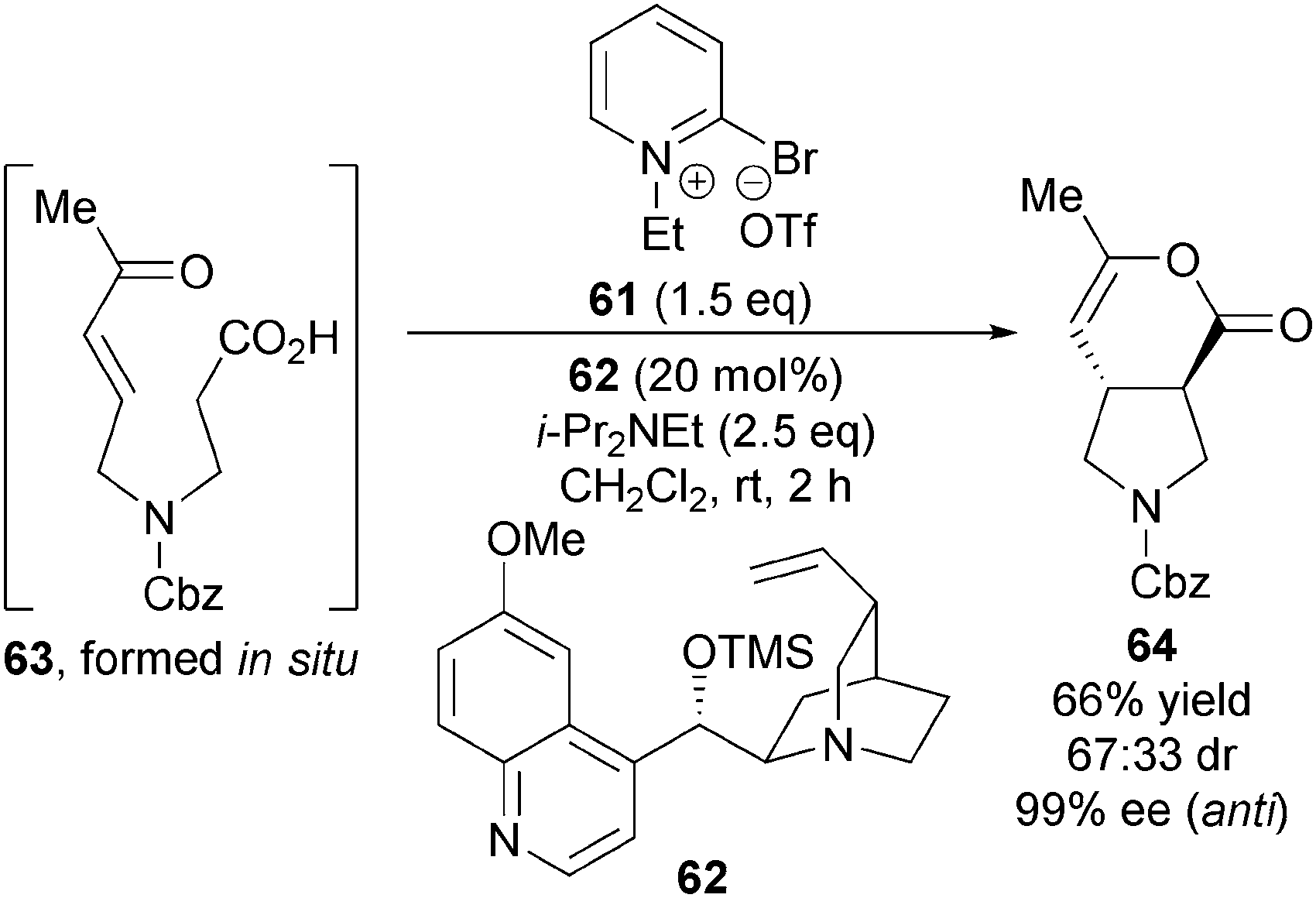 | ||
| Scheme 18 Accessing the 3,4-anti diastereoisomer. | ||
In the tetramisole promoted cyclisations, the observed stereoselectivity can be explained via pre-transition state assembly 65 in which the enolate oxygen is orientated syn to the sulfur atom of the catalyst, allowing for a stabilising no to σ*C–S interaction (or electrostatic stabilisation). Michael addition then proceeds anti to the stereodirecting phenyl substituent via the enolate Si-face generating the syn-product (Fig. 7). For the OTMS-quinidine catalysed reaction, cyclisation proceeds preferentially via the enolate Re-face (pre-transition state assembly 66), which minimises steric clashes with the ethylene bridge within the quinidine skeleton, giving the anti-diastereoisomer as the major product in high ee.
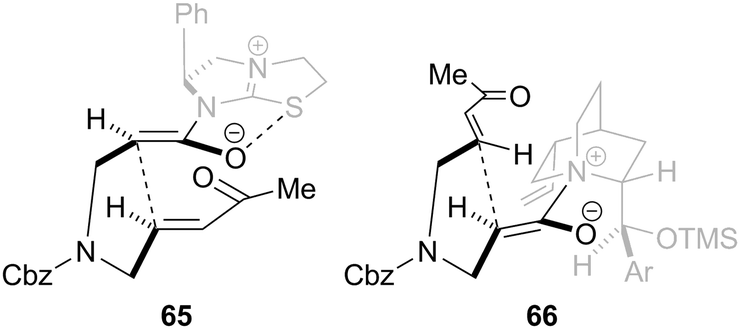 | ||
| Fig. 7 Stereochemical rationale for Lewis base promoted intramolecular Michael addition-lactonisations. | ||
2.2. Intermolecular processes
The intramolecular NCAL and related Michael addition-lactonisation methodologies showcase the power of this carboxylic acid functionalisation strategy. Reviewing Romo's intramolecular NCAL strategy employing aldehyde-acid starting materials and cinchona alkaloid Lewis bases, Gaunt and Johansson remarked “The moderate reactivity of the zwitterionic ammonium enolates is bypassed in the intramolecular reaction because of the proximity of the aldehyde, but it cannot be avoided in the intermolecular process”.6 To extend this protocol to intermolecular processes remained elusive until 2011 when the Smith group extended their methodology towards the first application of carboxylic acid derived ammonium enolates in intermolecular processes.28 HBTM-2.1 67 effectively catalyses the intermolecular Michael addition-lactonisation of arylacetic acids 68 and α-keto-β,γ-unsaturated esters 69 in the presence pivaloyl chloride, giving anti-dihydropyranones 70 with excellent diastereo- and enantiocontrol (up to 98:2 dr, up to 99% ee) (Scheme 19). The isothiourea derived ammonium enolate formed by employing HBTM-2.1 67 is seemingly sufficiently nucleophilic to promote the challenging intermolecular bond forming process.
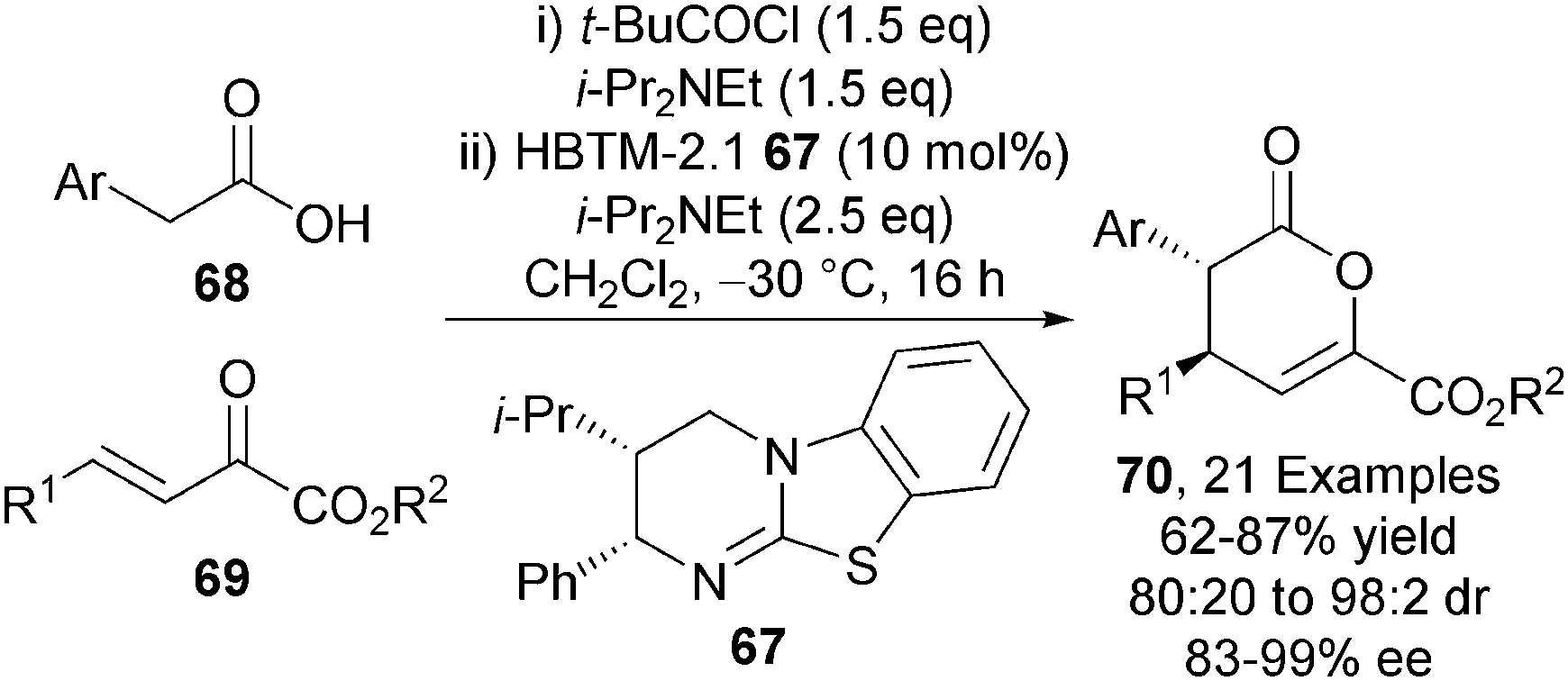 | ||
| Scheme 19 Isothiourea-catalysed intermolecular Michael addition-lactonisation. | ||
This intermolecular protocol was extended towards alternative electron deficient Michael acceptors. Employing trifluoromethyl enones 71 under closely related reaction conditions generates trifluoromethyl bearing anti-dihydropyranones 72 with high stereocontrol (up to 95:5 dr, up to >99% ee) (Scheme 20).30 Notably, these heterocyclic products could be readily derivatised into those containing CF3-stereogenicity via highly diastereoselective methods including hydrogenation of 73 to form saturated lactone 74 or reduction with LiAlH4 to give lactol 75 with no erosion in enantiopurity.
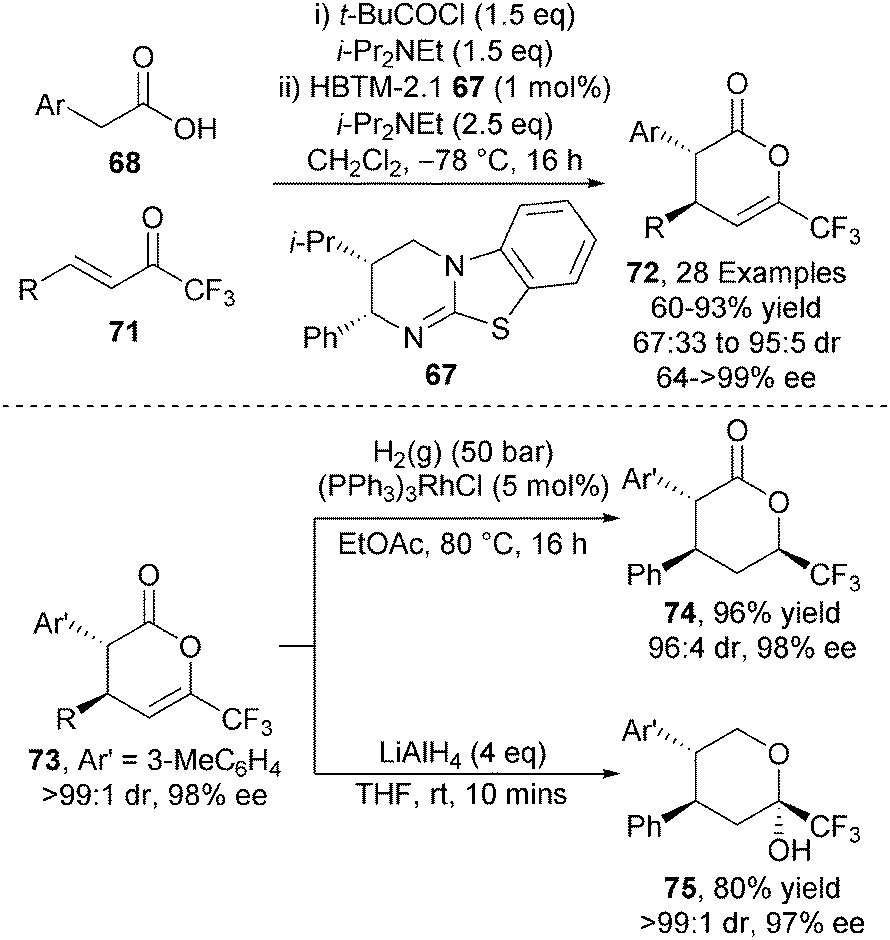 | ||
| Scheme 20 CF3-containing heterocycle formation. | ||
Kinetic investigations indicated this process to be first order in mixed anhydride 76 and HBTM-2.1 67, whilst being zero order in trifluoromethyl enone 77 (Scheme 21). Increasing the stoichiometry of i-Pr2NEt (up to 8 equivalents) had a negligible effect upon reaction rate, consistent with the rate-determining transition structure being constructed from the catalyst and the mixed anhydride. Importantly, a primary kinetic isotope effect kobsH/kobsD = 3.8 is observed when the reaction is performed using α,α-di-deuterio 4-fluorophenylacetic acid, consistent with deprotonation being rate determining.
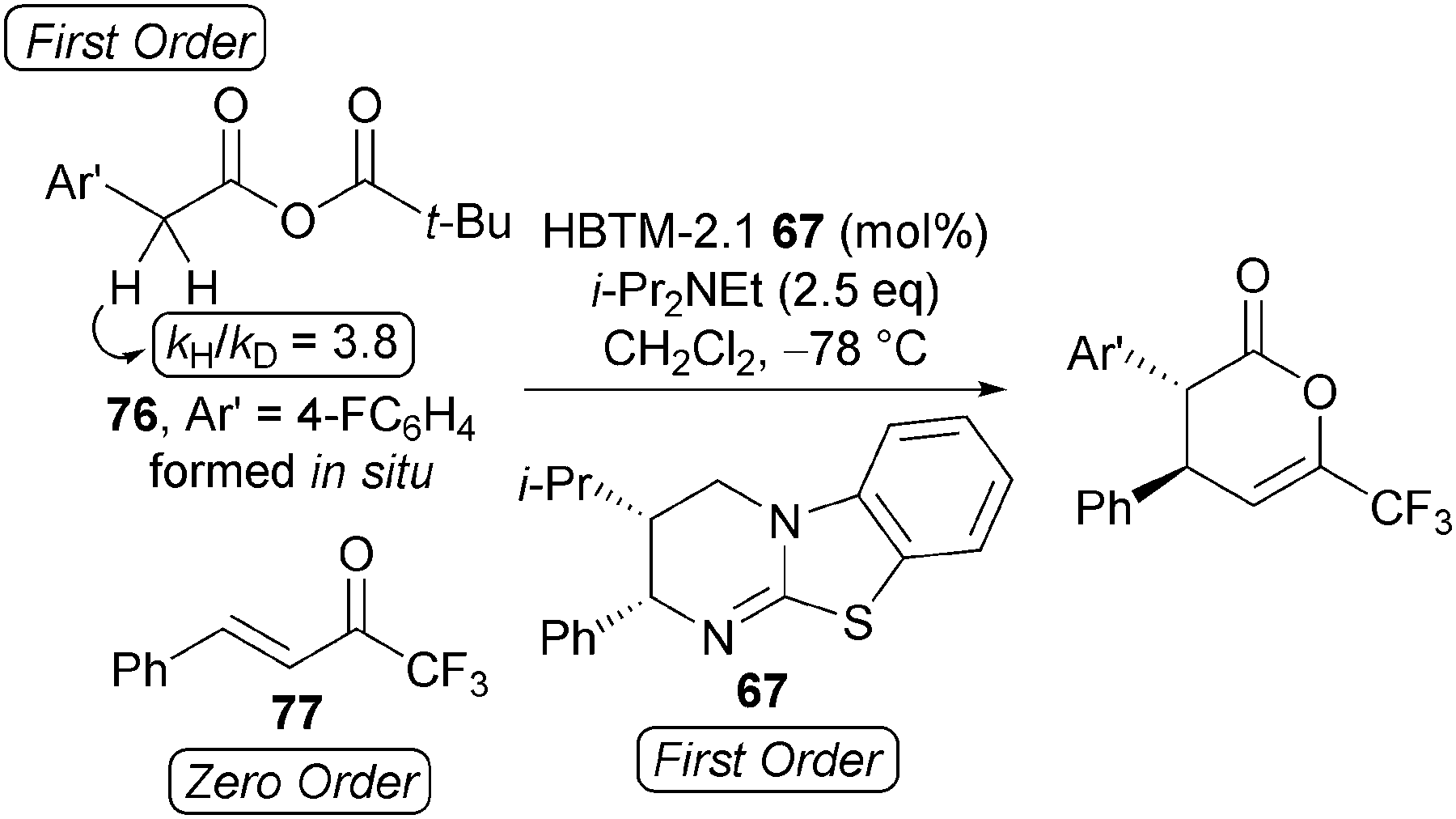 | ||
| Scheme 21 Kinetic investigations. | ||
Based upon these kinetic observations, a catalytic cycle for the reaction has been proposed that proceeds via initial in situ formation of mixed anhydride 78, which is intercepted by HBTM-2.1 67 to form the corresponding acyl ammonium ion 79 (Scheme 22). Rate-determining deprotonation by pivalate generates the (Z)-ammonium enolate 80 which undergoes stereoselective Michael addition with trifluoromethyl enone 71, followed by intramolecular cyclisation to afford anti-dihydropyranone 72 with regeneration of the catalyst.
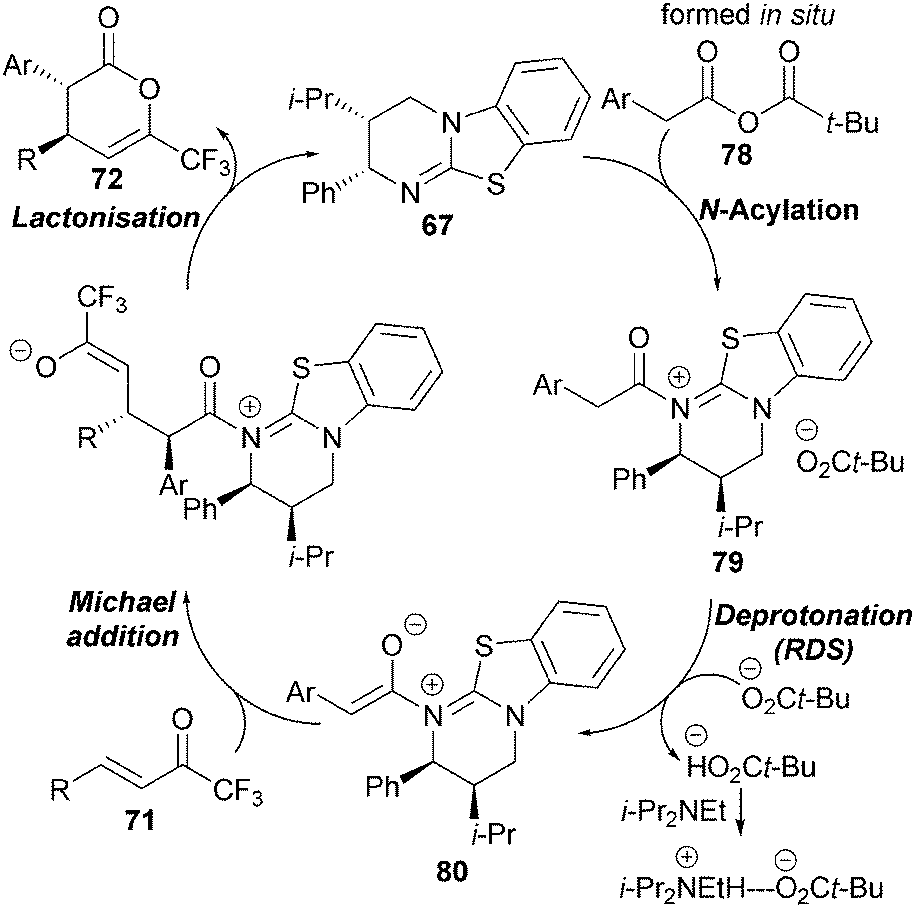 | ||
| Scheme 22 Proposed catalytic cycle. | ||
Evidence for the presence of an acyl isothiouronium ion such as 79 in the catalytic cycle was provided through its use as a precatalyst. 81 was prepared and isolated by reaction of HBTM 2.1 with the corresponding acid chloride, and used as precatalyst, giving anti-dihydropyranone 82 in identical diastereo- and enantiocontrol (90:10 dr and 99% ee) to that employing HBTM-2.1 67 directly in this protocol (Scheme 23).
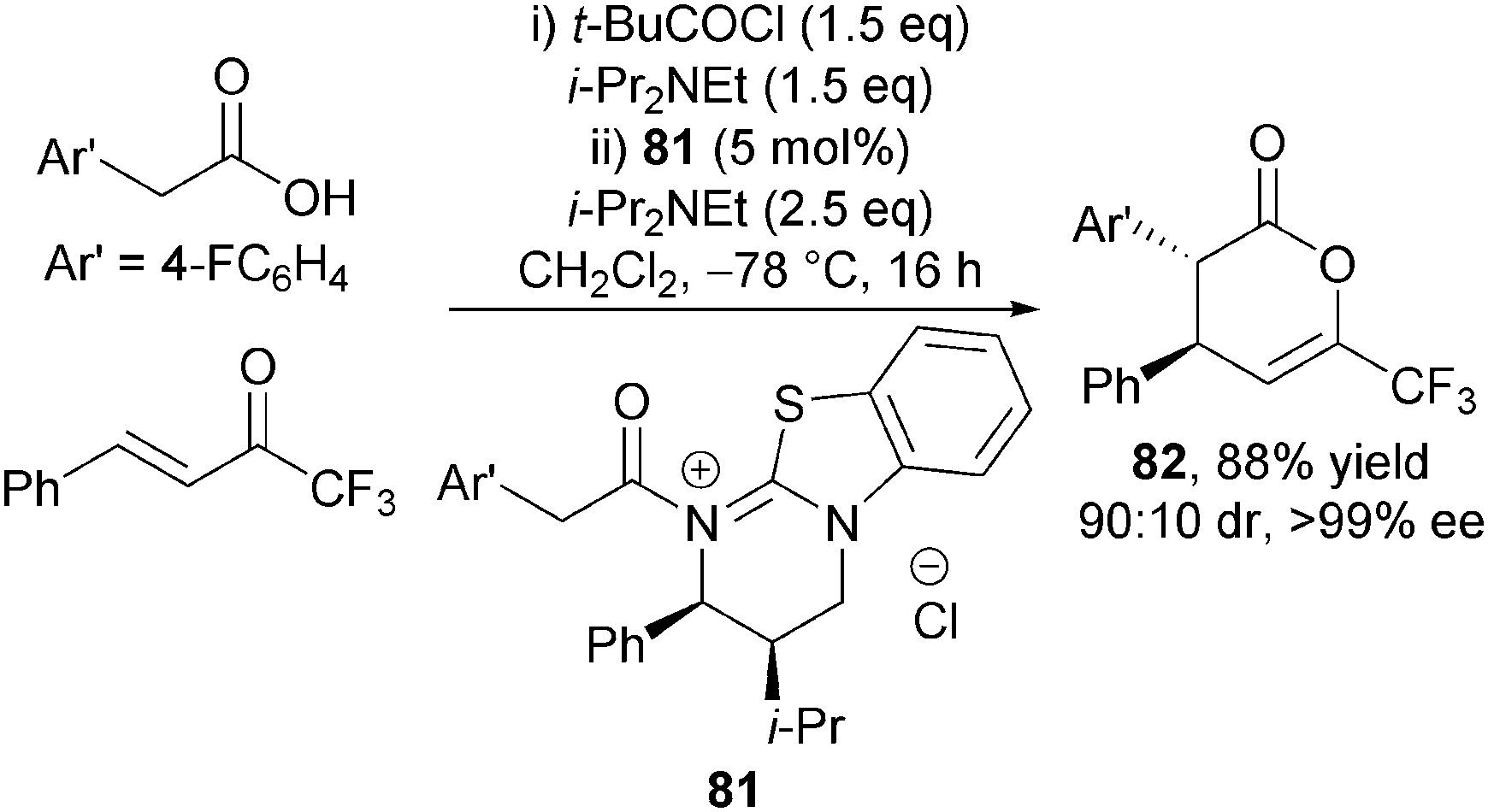 | ||
| Scheme 23 Validation of isothiouronium ion as a plausible intermediate. | ||
Furthermore, the organocatalytic reaction was shown to be stereospecific, with the (Z)-enone 83 affording syn-dihydropyranone 84 in high diastereoselectivity (85:15 dr) with the major diastereoisomer formed in excellent enantioselectivity (99% ee) (Scheme 24). This useful experiment revealed that the either diastereoisomer of the product can be obtained via judicious choice of starting material configuration.
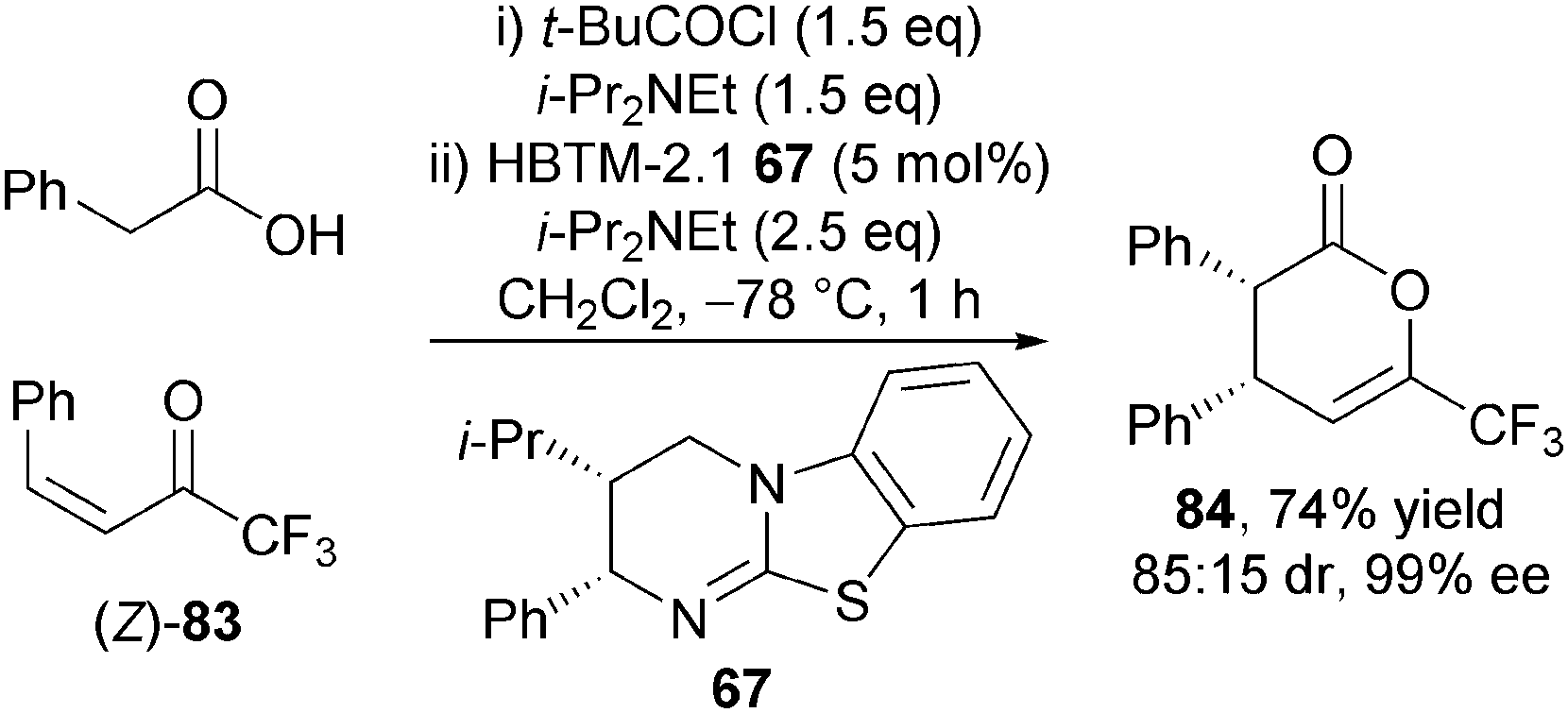 | ||
| Scheme 24 Reaction stereospecificity. | ||
Models explaining the observed stereoselectivity involve the isothiouronium heterocycle adopting a half-chair type conformation with the C(2)-phenyl substituent pseudoaxial to minimise 1,2 steric interactions and the C(3)-i-Pr unit pseudoequatorial (Fig. 8).31 Within the (Z)-ammonium enolate, the oxygen atom preferentially lies syn to the sulfur atom within the isothiouronium ion, allowing a stabilising no to σ*C–S interaction. The reaction proceeds though the diastereomeric transition states shown, giving the observed anti-diastereoisomer (from (E)-enone) or syn-diastereoisomer (from (Z)-enone) in high enantiomeric excess.
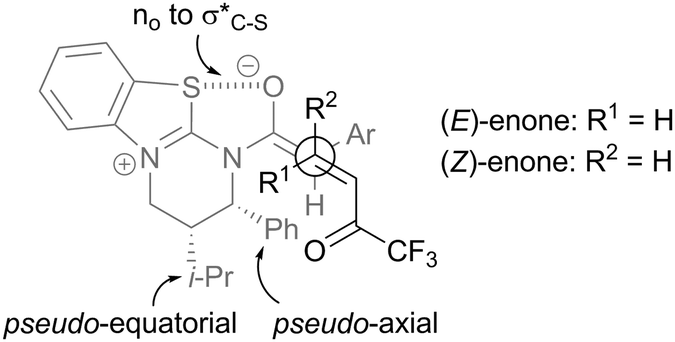 | ||
| Fig. 8 Stereochemical rationale. | ||
In addition to carbon-carbon bond forming reactions, the catalytic asymmetric α-amination of carboxylic acids using isothioureas has been demonstrated.32 HBTM-2.1 67 efficiently catalyses the α-functionalisation of a range of carboxylic acids with N-aryl-N-aroyl diazenes 85 at low catalyst loadings (as low as 0.25 mol%), giving either 1,3,4-oxadiazin-6-ones 86 or N-protected α-amino acid derivatives 87 upon ring-opening with excellent enantiocontrol (up to >99% ee) (Scheme 25). Importantly, non-arylacetic acids could be incorporated in this protocol through the use of a highly electron deficient Michael acceptor and PS-BEMP as base. The N–N bond within the hydrazide products 88 could be readily cleaved using SmI2 to afford bespoke N-aryl-α-aryl glycine building blocks 89 in excellent enantioselectivity.
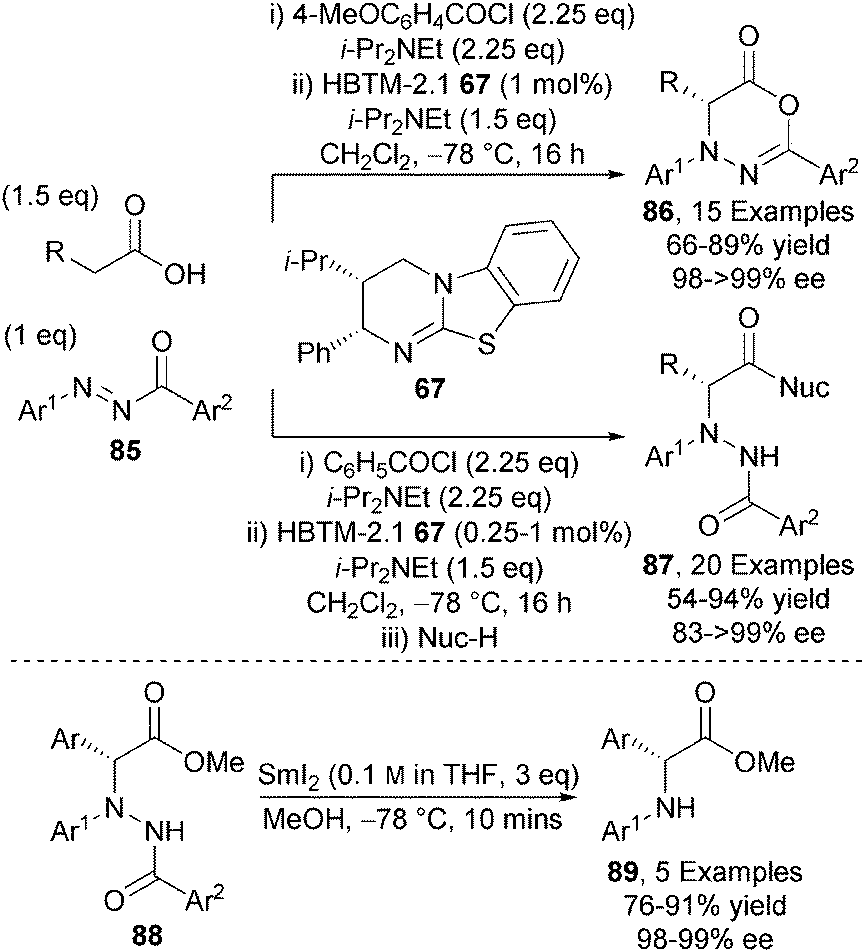 | ||
| Scheme 25 Isothiourea-catalysed α-amination of carboxylic acids. | ||
Carboxylic acid derived ammonium enolates have also been used in Michael addition-lactamisation processes.33 BTM 90 efficiently catalysed the asymmetric formal [4+2] cycloaddition between arylacetic acids 68 and ketimines 91, giving a range of anti-dihydropyridones 92 with good diastereocontrol (typically 85:15 dr) and excellent enantiocontrol (up to 99% ee) (Scheme 26).
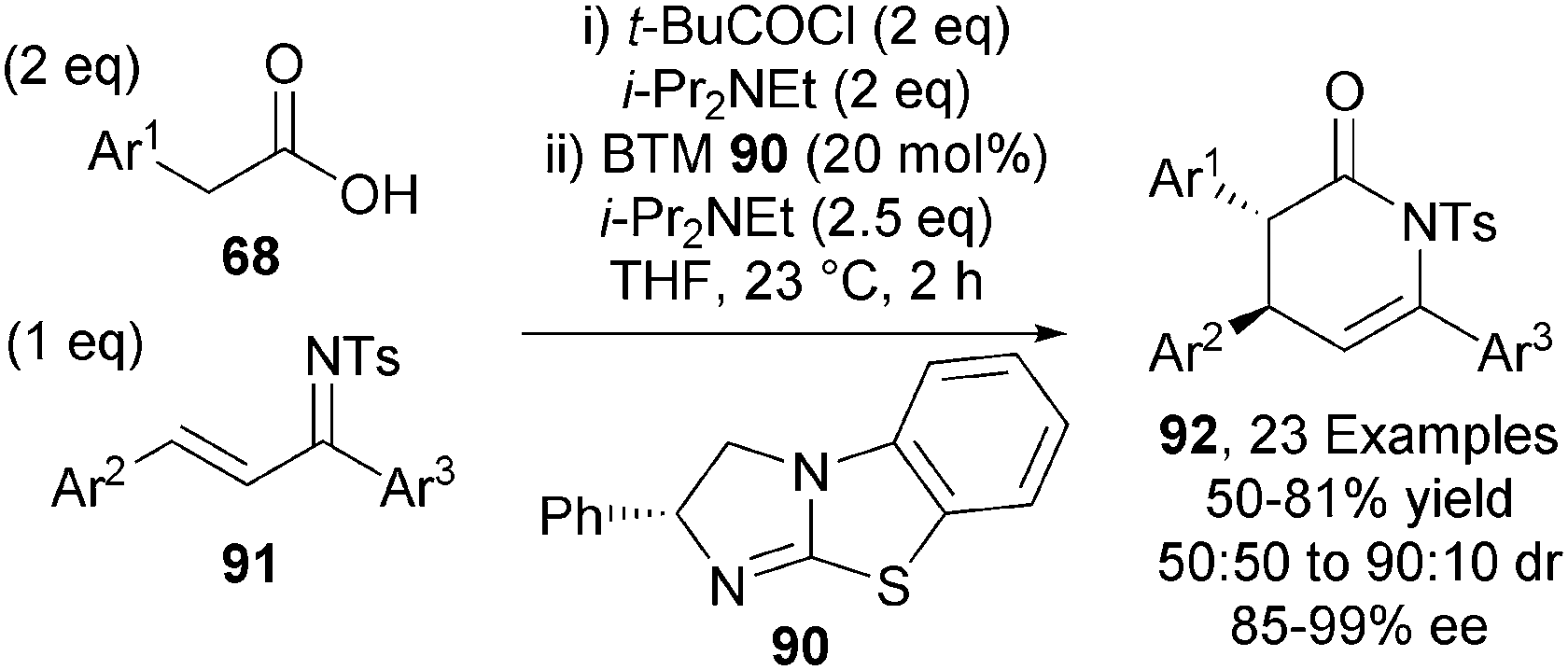 | ||
| Scheme 26 Intermolecular Michael addition-lactamisation. | ||
Building upon this work, this Michael addition-lactamisation protocol was applied to the synthesis of functionalised pyridines.34 Achiral isothiourea DHPB 93 efficiently catalyses the Michael addition-lactamisation process between (phenylthio)acetic acid 94 and ketimines 95, initially giving dihydropyridones 96 (Scheme 27). These products undergo rapid elimination of thiophenol to give pyridones 97, followed by intramolecular N- to O-sulfonyl transfer upon heating, affording a range of 2,4,6-substituted pyridines 98 in acceptable yields (40–69%) over 3 steps in one-pot. A closely related one-pot protocol was later developed to access a range of trifluoromethyl substituted 2-pyrones.35
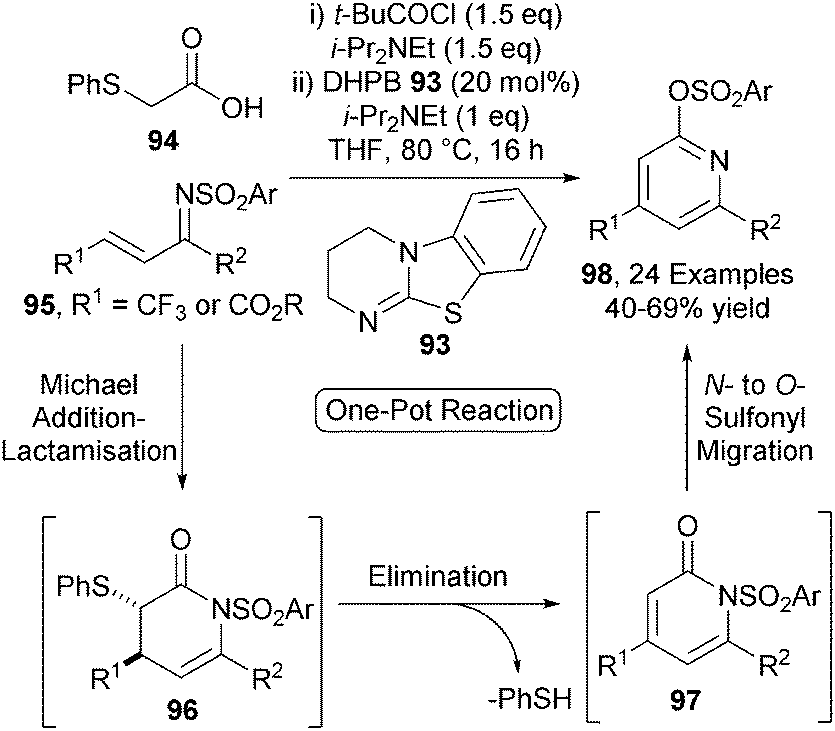 | ||
| Scheme 27 One-pot organocatalytic pyridine synthesis. | ||
Of particular note in this chemistry is that the activating sulfonyl group on the ketimine is transformed into the synthetically useful functional handle (the 2-sulfonate group) in the resulting pyridines. A number of derivatisations showcasing the versatility of this group were demonstrated, including a host of cross-coupling methodologies. This methodology was also applied to the synthesis of 99 – a pyridine with known biological activity as a COX-2 inhibitor (Scheme 28). Pyridine 100 was accessed from ketimine 101 in 53% yield using the optimised reaction conditions and subsequent SNAr with cyclohexylamine afforded 99 in 92% yield.
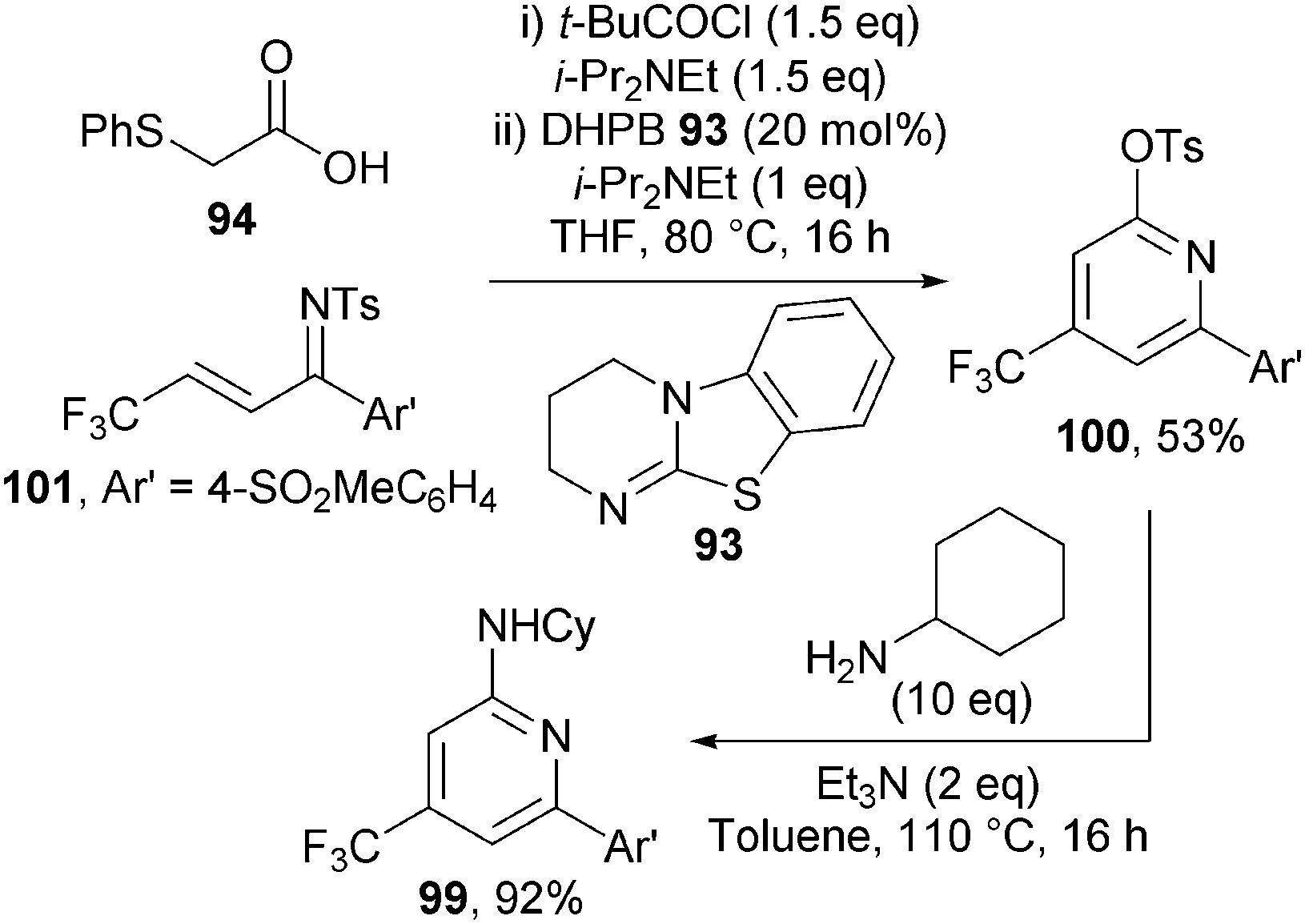 | ||
| Scheme 28 Rapid assembly of biologically active pyridine. | ||
In 2014, the first intermolecular formal [2+2] cycloaddition involving a carboxylic acid derived ammonium enolate was demonstrated.36 BTM 90 effectively catalyses the annulation of arylacetic acids 68 and N-sulfonyl imines 102, in the presence of tosyl chloride, giving β-lactams 103 with excellent diastereocontrol and high enantiocontrol (up to >95:5 dr, up to 88% ee) (Scheme 29). Alternatively, using HBTM-2.1 67 as catalyst, a range of β-amino esters 105 could be accessed after in situ ring-opening with n-BuLi/MeOH in excellent diastereo- and enantiocontrol (up to >95:5 dr, up to 99% ee).
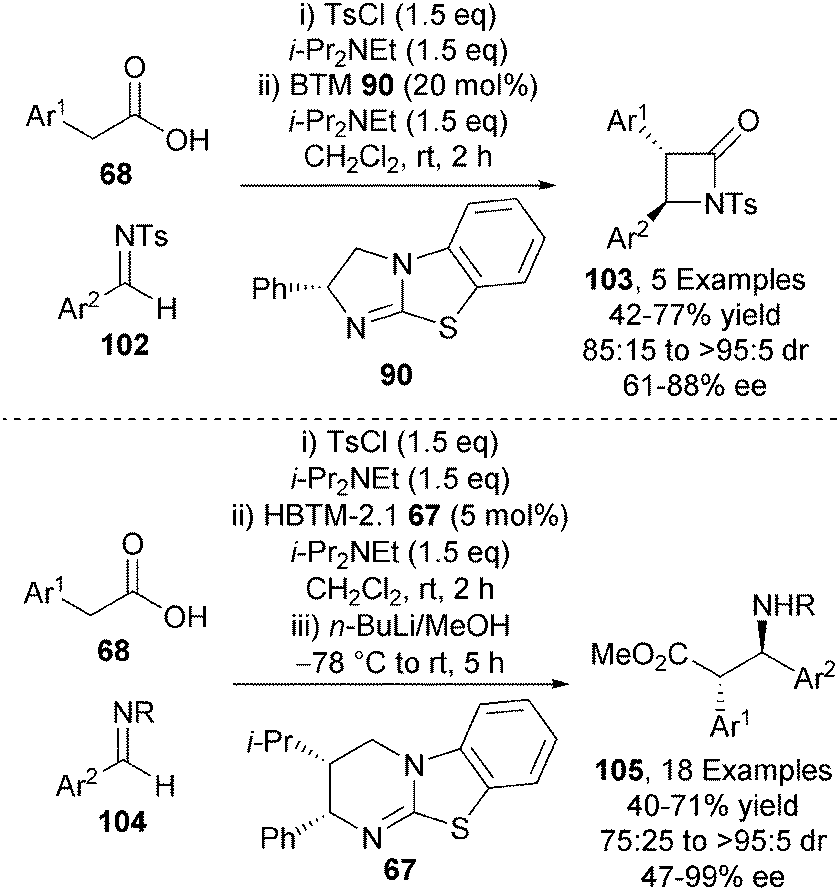 | ||
| Scheme 29 Formal [2+2] cycloadditions using N-sulfonyl imines. | ||
The isothiourea-mediated functionalisation of 3-alkenoic acids proceeding via an ammonium dienolate has also been disclosed by Smith and co-workers.37 α-Functionalisation of the intermediate dienolate derived from 3-alkenoic acids 106 and HBTM-2.1 67 with N-tosyl aldimines 102 gives a range of β-lactams 107 with modest diastereocontrol (up to 82:18 dr), although each diastereoisomer is formed with excellent enantioselectivity (typically >95% ee) (Scheme 30).
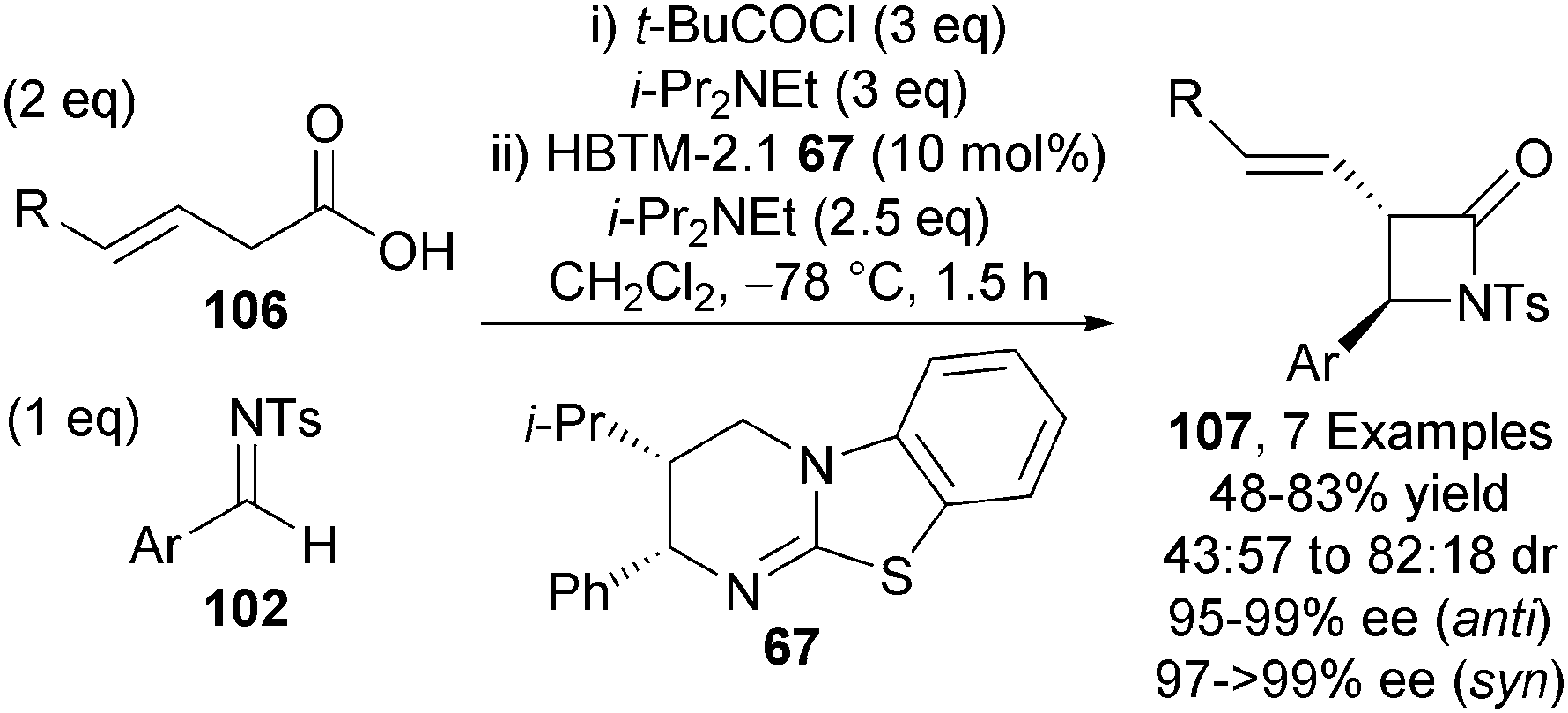 | ||
| Scheme 30 α-Functionalisation of 3-alkenoic acids. | ||
3. Anhydrides as C1-ammonium enolate precursors
One notable disadvantage of the in situ acid activation strategy is the formation of by-products such as pyridones (from Mukaiyama-type reagents) and pivalic anhydride (from pivaloyl chloride) which can in some cases be difficult to separate from the desired products. Anhydrides are already pre-disposed towards nucleophilic attack by a Lewis base and so do not need to be pre-activated in the same manner as carboxylic acids. Upon addition of a Lewis base, the only by-product is one equivalent of parent carboxylic acid, which is easily removed from the product via basic work-up. With this advantage in mind, the Smith group demonstrated that HBTM-2.1 67 effectively catalyses the intermolecular Michael addition-lactonisation of 2-arylacetic anhydrides 108 and a range of Michael acceptors, namely α-keto-β,γ-unsaturated esters 69, trifluoromethyl enones 71 and N-aryl-N-aroyl diazenes 85, giving diverse synthetic building blocks with high diastereo- and enantiocontrol (up to 98:2 dr, up to >99% ee) (Scheme 31).38 C1-ammonium enolates derived from 2-arylacetic anhydrides were also applied towards formal [2+2] cycloadditions with N-sulfonyl imines, giving β-lactams with high diastereo- and enantiocontrol (up to >95:5 dr, up to 99% ee).36
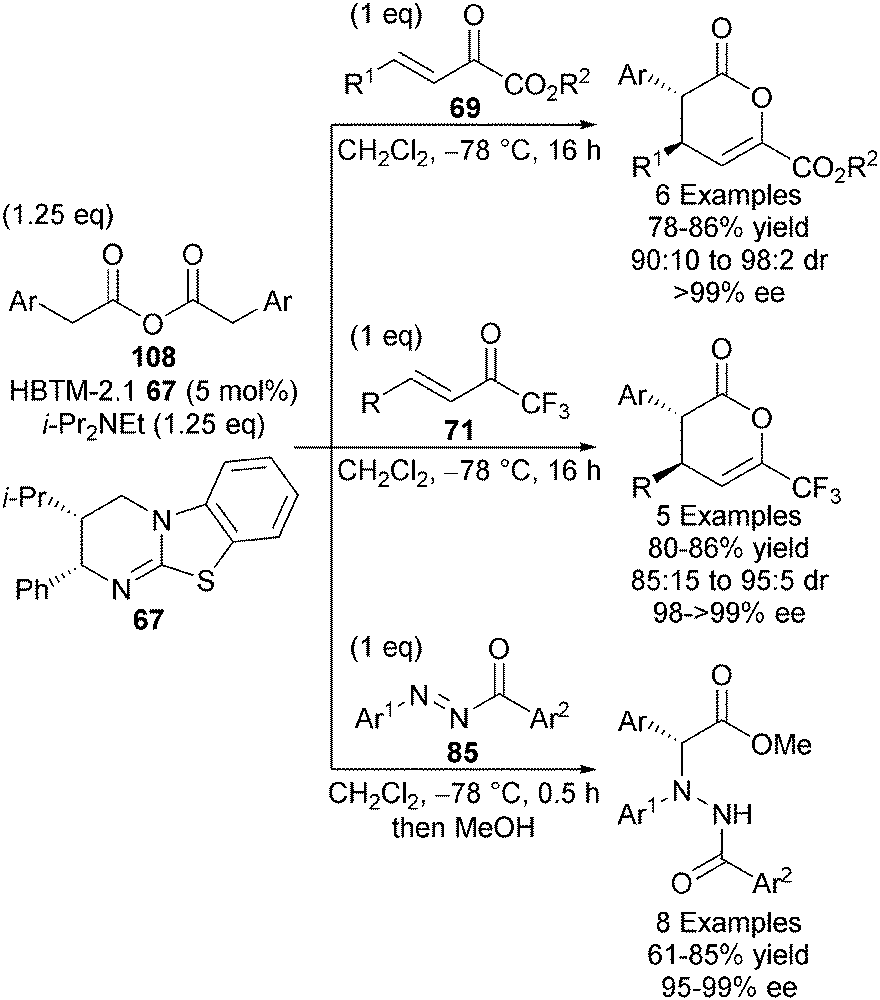 | ||
| Scheme 31 2-Arylacetic anhydrides as C1-ammonium enolate precursors. | ||
4. Carboxylic esters as C1-ammonium and C1-azolium enolate precursors
Activated esters are susceptible towards nucleophilic attack by a Lewis base tertiary amine or NHC and so have found utility as C1-ammonium and azolium enolate precursors.4.1. C1-azolium enolates from carboxylic esters
This section will focus on the generation and utility of C1-azolium enolates from carboxylic ester starting materials using NHC catalysts. C1-azolium enolates are commonly accessed from aldehyde, enal and ketene starting materials. Despite the potential practical advantages of accessing these enolates from bench stable esters or alternative carboxylic ester derivatives, this area has received little attention.39 In 2012, Chi reported the first NHC mediated activation of stable carboxylic esters to generate C1-azolium enolates.40 After initial optimisation, triazolium-based chiral precatalyst 109 (30 mol%) promoted the asymmetric formal [4+2] cycloaddition of activated arylacetic esters 110 and ketimines 91 giving a range of anti-dihydropyridones 92 in high diastereoselectivity (10:1 to >20:1 dr), with the major diastereoisomer formed in moderate to excellent enantioselectivity (60 to >98% ee) (Scheme 32). Notably, the use of both Me4NCl (1 eq.) and excess i-Pr2NEt (5 eq.) were necessary for achieving high stereoselectivity. The use of high catalyst loadings (30 mol%) and the formation of toxic p-nitrophenol by-product are notable drawbacks of this entry to C1-azolium enolates.
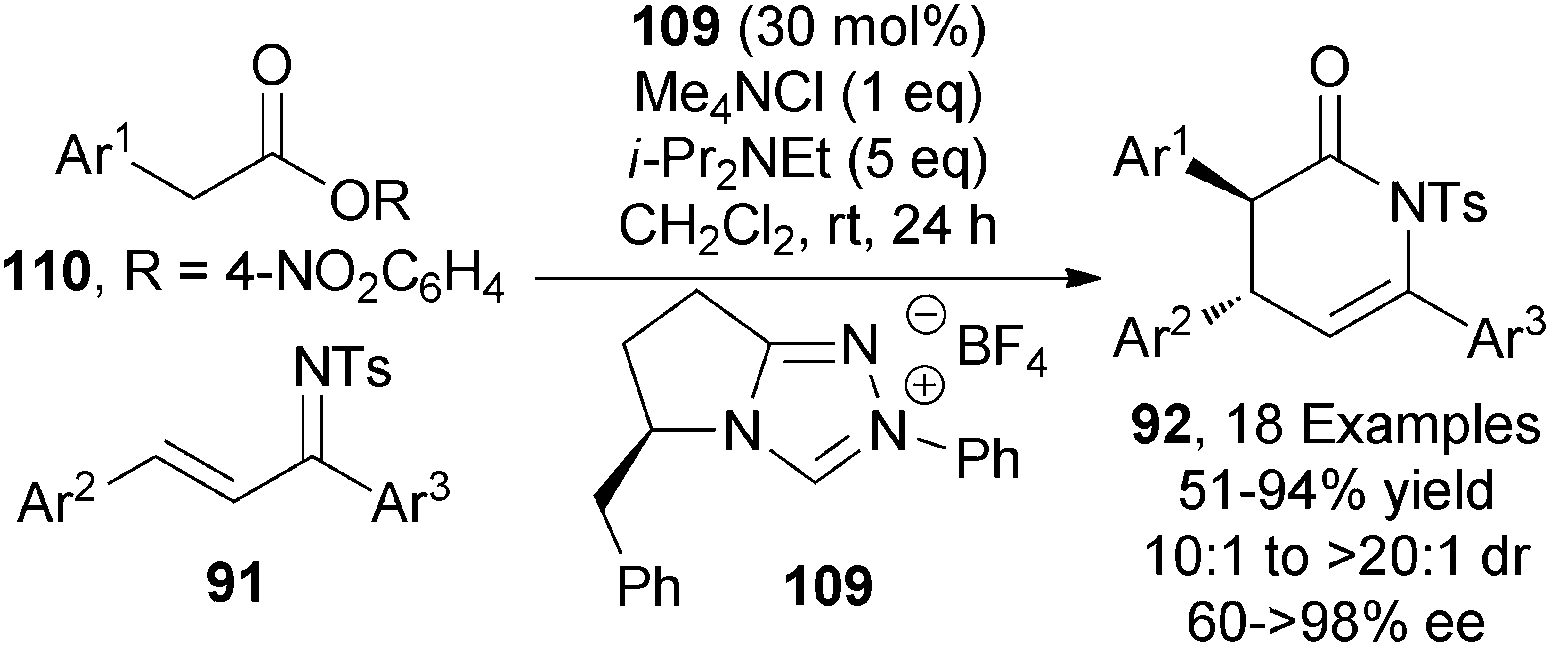 | ||
| Scheme 32 NHC-catalysed functionalisation of activated arylacetic esters. | ||
The Chi group later expanded the scope of this reaction to include simple activated alkylacetic esters 111 through the use triazolium precatalyst 112 (30 mol%). In the presence of DBU, anti-dihydropyridines 113 are obtained in excellent yields and stereoselectivities (up to 18:1 dr, up to >99% ee) (Scheme 33).41 Further work by the same group highlighted the ability to organocatalytically functionalise the simplest activated acetic ester (R1 = H in Scheme 33) using similar reaction conditions.42
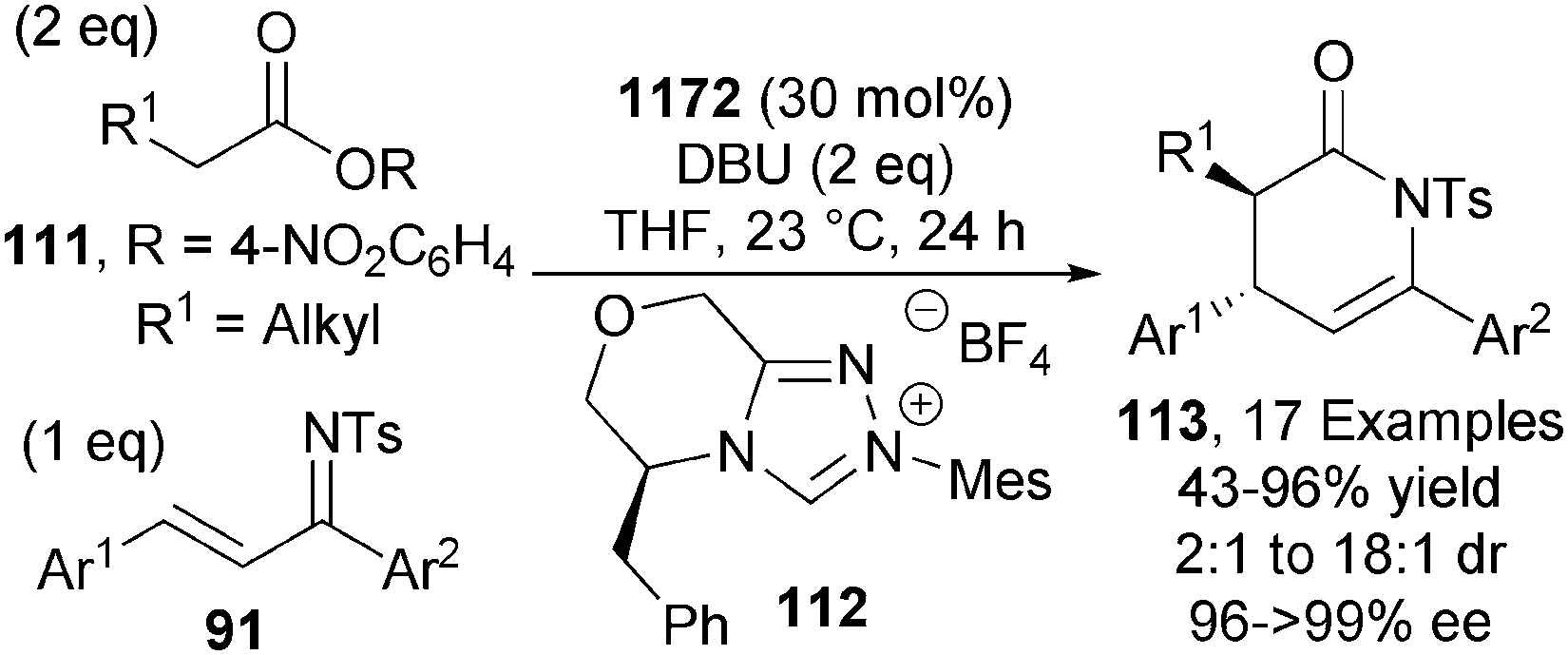 | ||
| Scheme 33 Activated alkylacetic esters as C1-azolium enolate precursors. | ||
Chi and co-workers also applied ester-derived C1-azolium enolates towards the synthesis of spyrocyclic oxindoles.43 Achiral NHC precatalyst 114 (30 mol%), in the presence of base, promotes the reaction between activated arylacetic esters 110 and isatin-derived azadienes 115 giving a range of spyrocyclic oxindoles 116 in modest diastereoselectivities (63:37 to 72:28 dr) (Scheme 34). Attempts to render the process asymmetric were thwarted by the low reactivity of the azadienes. The best result was obtained using L-phenylalanine-derived triazolium salt 117, affording δ-lactam 118 in 45% yield with modest diastereo- and enantiocontrol (70:30 dr and 62% ee).
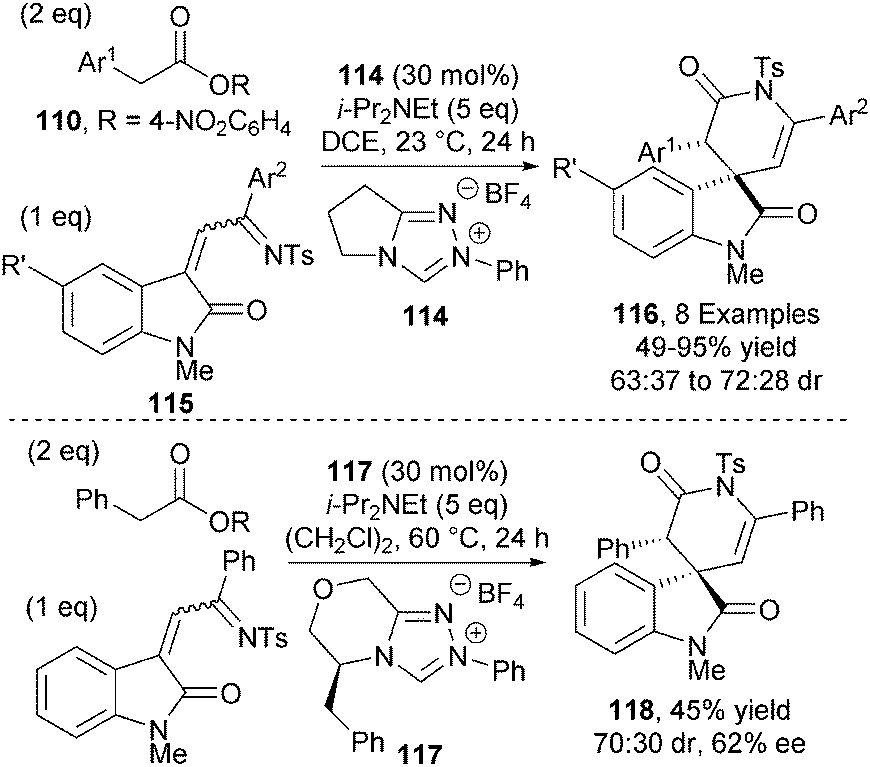 | ||
| Scheme 34 NHC-catalysed spirocyclic oxindole formation. | ||
In 2013, Chi reported the first NHC-catalysed asymmetric γ-functionalisation of α,β-unsaturated esters via an azolium dienolate intermediate.44 Triazolium NHC precatalyst 119, in the presence of K2CO3, effectively promotes the γ-activation of α,β-unsaturated esters 120 that undergo addition to hydrazones 121 to give δ-lactam products 122 in high yield and with excellent levels of enantioselectivity (up to >98% ee) (Scheme 35). Using slightly modified reaction conditions, γ-substituted and β,β′-dialkyl substituted α,β-unsaturated esters could also be used in this protocol, giving the corresponding δ-lactams in moderate to good yields and enantioselectivity.
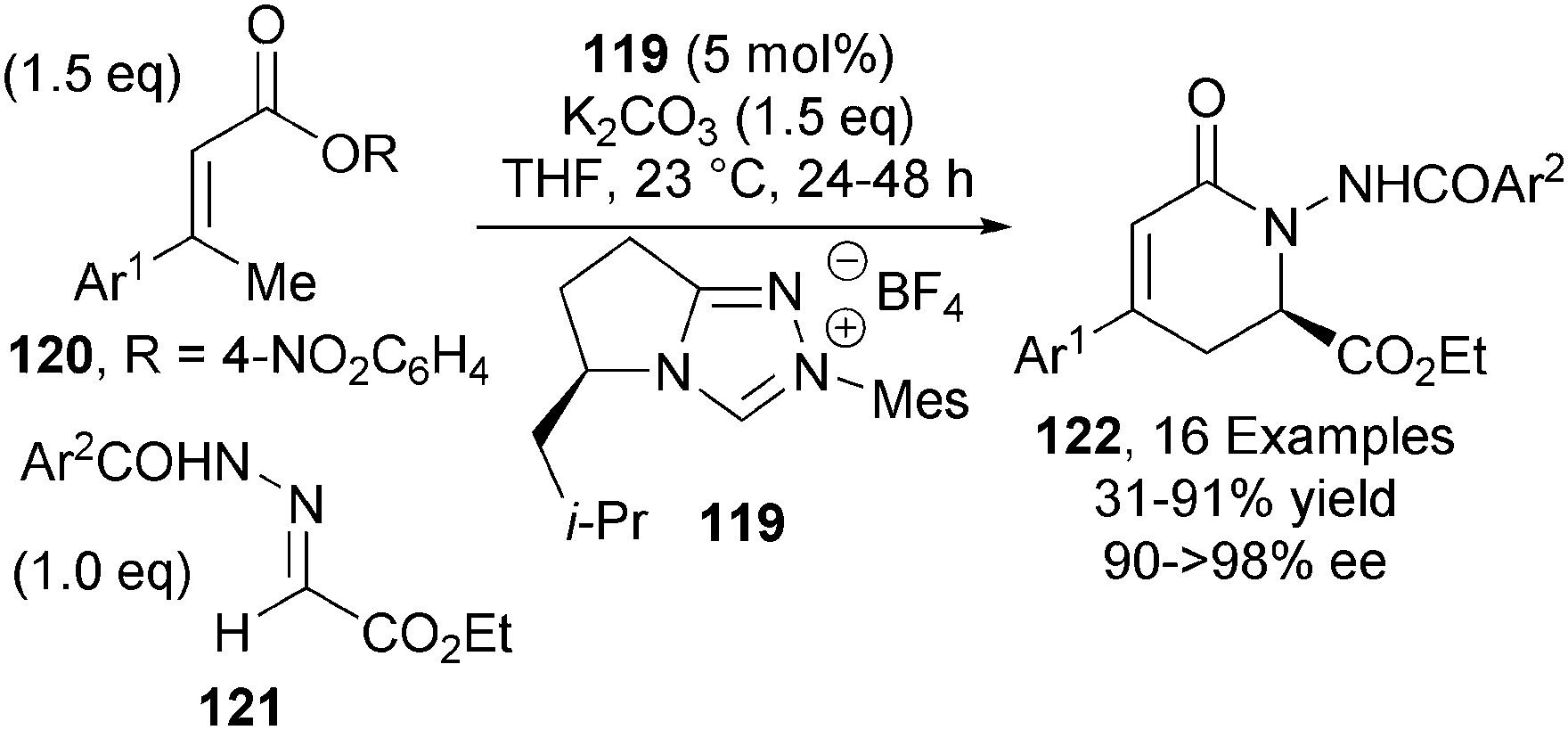 | ||
| Scheme 35 NHC-catalysed γ-activation of activated esters. | ||
A proposed catalytic cycle for this process involves initial formation of acyl azolium 123, which undergoes γ-deprotonation to form the key C1-azolium dienolate 124 (Scheme 36). Subsequent stereoselective nucleophilic attack towards hydrazone 121 gives NHC-bound adduct 125 that rapidly collapses to give δ-lactam 122 with regeneration of the catalyst.
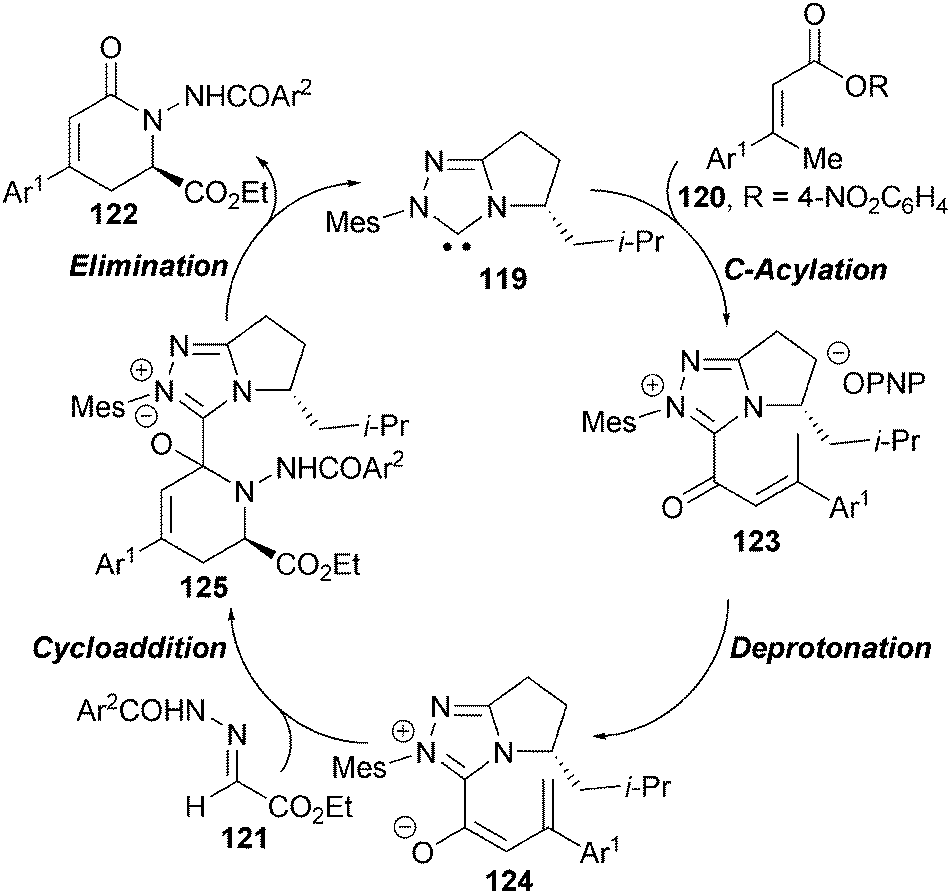 | ||
| Scheme 36 Proposed catalytic cycle. | ||
4.2. C1-ammonium enolates from carboxylic esters
C(1)-ammonium enolates can also be generated from a tertiary amine and a 4-nitrophenol ester. In a closely related process to that outlined previously,34 the Chi group demonstrated that DMAP 40 catalyses the one-pot formation of pyridines 126 from activated α-chloro acetic esters 127 and chalcone-derived imines 128 in moderate to excellent yields (32–99%) (Scheme 37).45 The use of α-chloro acetic esters avoids the formation of thiophenol (in Smith's system) but this advantage is offset by the formation of the toxic p-nitrophenol by-product and the need to prepare the ester from the acid in a separate synthetic step.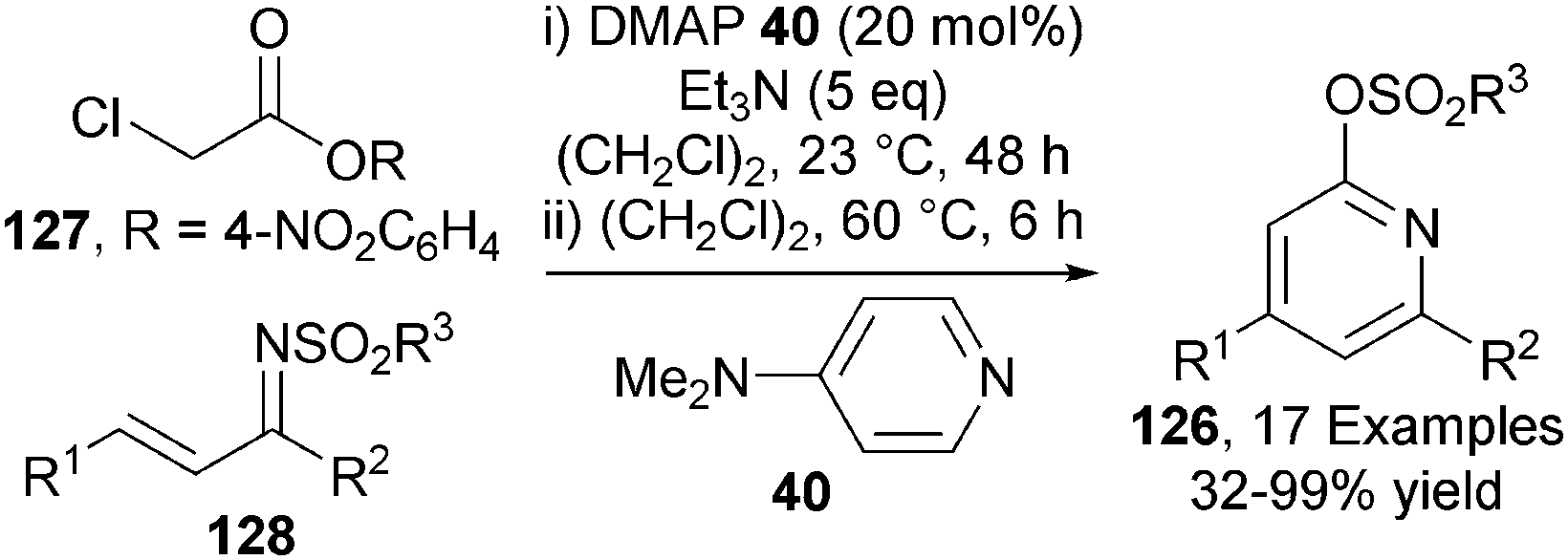 | ||
| Scheme 37 Pyridines from activated α-chloro acetic esters. | ||
The only asymmetric use of C(1)-ammonium enolates from activated esters has been through the demonstration of an isothiourea-catalysed asymmetric [2,3]-rearrangement of allylic ammonium ylides.46 A range of allylic quaternary ammonium salts 129 (either isolated or made in situ), undergo BTM 90-catalysed [2,3]-rearrangement in the presence of i-Pr2NH as base and HOBt additive to afford a range of syn-α-amino acid derivatives 130 in excellent diastereo- and enantiocontrol (up to >95:5 dr, up to >99% ee) (Scheme 38).
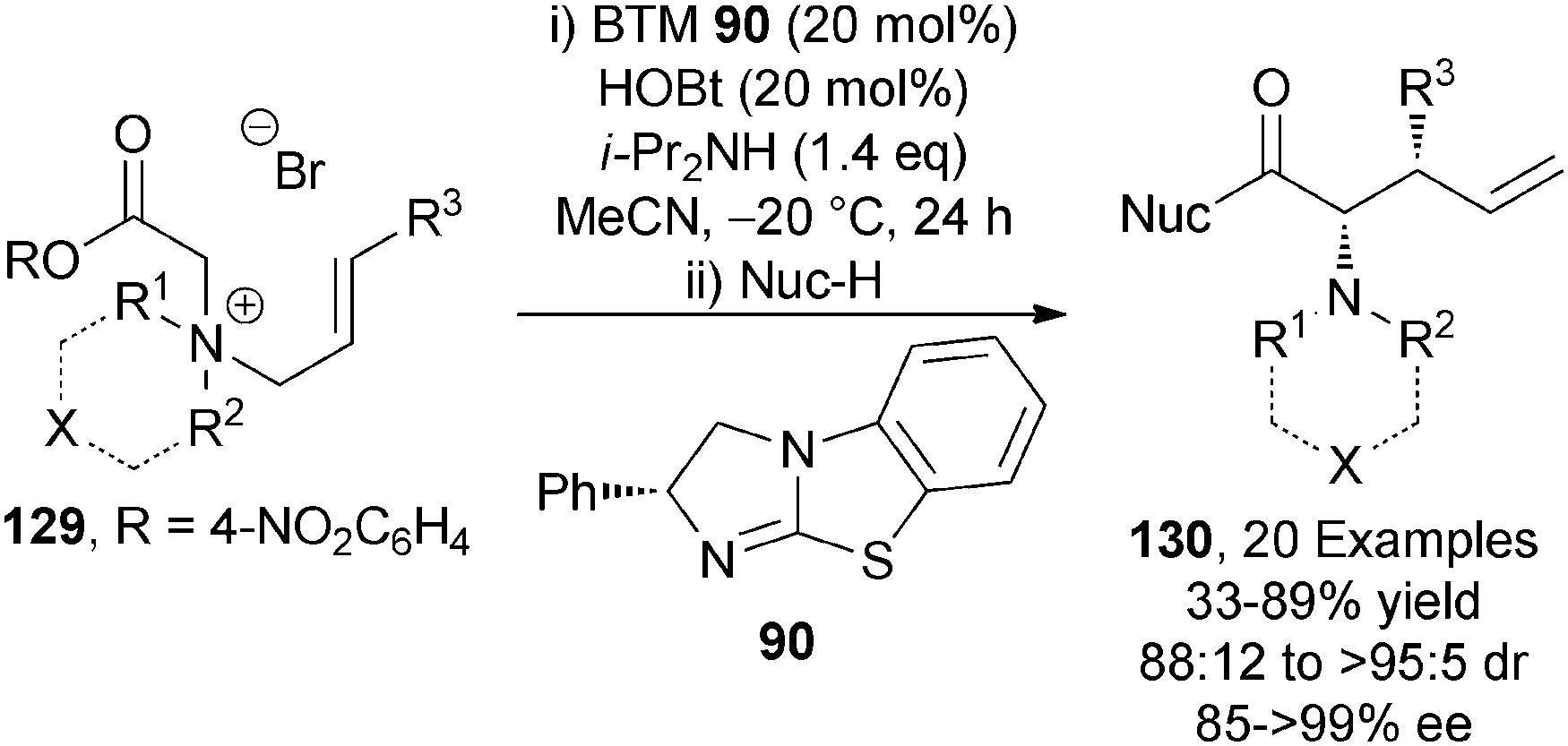 | ||
| Scheme 38 [2,3]-Rearrangement of allylic ammonium ylides. | ||
5. Future challenges
This tutorial review has summarised a number of Lewis base strategies employing tertiary amine and NHC catalysts and carboxylic acid, anhydride or ester starting materials. Despite the range of innovative strategies that have been developed in this area, challenges and opportunities remain. The drive to maximise reaction effectiveness and catalyst efficiency will demand catalytic procedures that proceed routinely at low catalyst loadings and that are amenable to large-scale product formation. The application of these strategies to natural product targets and other relevant biologically active materials will continue to inspire new strategies and disconnections. Other key fundamental questions also remain within this research area. In the relentless drive for efficiency and atom economy can a catalytic activating agent be used to promote the functionalisation of carboxylic acids rather than a stoichiometric reagent? In the use of carboxylic esters can other less toxic starting materials be used as alternatives to electron deficient phenols? Can we use renewable feedstocks as convenient starting materials for this chemistry and prepare value-added products? Can the repertoire of catalysts that promote these reaction processes be extended to other Lewis base families? In this context, the generation of C1-ammonium and azolium enolates from carboxylic acids, anhydrides or esters offers many opportunities for future reaction development as well as the invention of new modes of chemical reactivity and discovery.6. Conclusions
The use of carboxylic acids, esters and anhydrides as bench stable C1-ammonium/azolium enolate precursors is a rapidly expanding area of research. The potential advantages of using these precursors over traditional starting materials such as ketenes strengthens the potential appeal of this chemistry towards industry. Due to the diverse nature of reactions discovered from these nucleophilic intermediates, it is clear that many new applications remain to be discovered in the coming years.Notes and references
- D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed and references therein.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed and references therein.
- S. Mukherkee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef PubMed and references therein.
- A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef PubMed and references therein.
- For a leading reference see T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582 CrossRef CAS PubMed.
- M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596 CrossRef CAS PubMed and references therein.
- J. Douglas, G. Churchill and A. D. Smith, Synthesis, 2012, 2295 CAS and references therein.
- D. H. Paull, A. Weatherwax and T. Lectka, Tetrahedron, 2009, 65, 6771 CrossRef CAS PubMed and references therein.
- X. Zhao, K. E. Ruhl and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 12330 CrossRef CAS PubMed.
- H. Wynberg and E. G. J. Staring, J. Am. Chem. Soc., 1982, 104, 116 CrossRef.
- G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945 CrossRef CAS.
- T. Mukaiyama, M. Usui and K. Saigo, Chem. Lett., 1976, 5, 49 CrossRef.
- G. S. Cortez, S. H. Oh and D. Romo, Synthesis, 2001, 1731 CrossRef CAS PubMed.
- S. H. Oh, G. S. Cortez and D. Romo, J. Org. Chem., 2005, 70, 2835 CrossRef CAS PubMed.
- H. Nguyen, S. H. Oh, H. Henry-Riyad, D. Sepulveda and D. Romo, Org. Synth., 2011, 88, 121 CrossRef CAS.
- D. Sikriwal and D. K. Dikshit, Tetrahedron, 2011, 67, 210 CrossRef CAS PubMed.
- K. A. Morris, K. M. Arendt, S. H. Oh and D. Romo, Org. Lett., 2010, 12, 3764 CrossRef CAS PubMed.
- H. Henry-Riyad, C. Lee, V. C. Purohit and D. Romo, Org. Lett., 2006, 8, 4363 CrossRef CAS PubMed.
- G. Ma, H. Nguyen and D. Romo, Org. Lett., 2007, 9, 2143 CrossRef CAS PubMed.
- H. Nguyen, G. Ma and D. Romo, Chem. Commun., 2010, 46, 4803 RSC.
- H. Nguyen, G. Ma and D. Romo, J. Org. Chem., 2011, 76, 2 CrossRef CAS PubMed.
- G. Liu and D. Romo, Angew. Chem., Int. Ed., 2011, 50, 7537 CrossRef CAS PubMed.
- G. Liu, M. E. Shirley and D. Romo, J. Org. Chem., 2012, 77, 2496 CrossRef CAS PubMed.
- V. C. Purohit, A. S. Malta and D. Romo, J. Am. Chem. Soc., 2008, 130, 10478 CrossRef CAS PubMed.
- C. A. Leverett, V. C. Purohit and D. Romo, Angew. Chem., Int. Ed., 2010, 49, 9479 CrossRef CAS PubMed.
- X. Yang, G. Lu and V. Birman, Org. Lett., 2010, 12, 892 CrossRef CAS PubMed.
- C. A. Leverett, V. C. Purohit, A. G. Johnson, R. L. Davis, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2012, 134, 13348 CrossRef CAS PubMed.
- D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714 CrossRef CAS PubMed.
- D. Belmessieri, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2013, 15, 3472 CrossRef CAS PubMed.
- L. C. Morrill, J. Douglas, T. Lebl, A. M. Z. Slawin, D. J. Fox and A. D. Smith, Chem. Sci., 2013, 4, 4146 RSC.
- C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914 CrossRef CAS PubMed.
- L. C. Morrill, T. Lebl, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2012, 3, 2088 RSC.
- C. Simal, T. Lebl, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2012, 51, 3653 CrossRef CAS PubMed.
- D. G. Stark, L. C. Morrill, P.-P. Yeh, A. M. Z. Slawin, T. J. C. O'Riordan and A. D. Smith, Angew. Chem., Int. Ed., 2013, 52, 11642 CrossRef CAS PubMed.
- P.-P. Yeh, D. S. B. Daniels, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2014, 16, 964 CrossRef CAS PubMed.
- S. R. Smith, J. Douglas, H. Prevet, P. Shapland, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1626 CrossRef CAS PubMed.
- L. C. Morrill, S. M. Smith, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1640 CrossRef CAS PubMed.
- L. C. Morrill, L. A. Ledingham, J.-P. Couturier, J. Bickel, A. D. Harper, C. Fallan and A. D. Smith, Org. Biomol. Chem., 2014, 12, 624 CAS.
- During the preparation of this manuscript, a review detailing the NHC catalysed activation of esters was disclosed: P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2014, 53, 1485 CrossRef CAS PubMed.
- L. Hao, Y. Du, X. Chen, H. Jiang, Y. Shao and Y. R. Chi, Org. Lett., 2012, 14, 2154 CrossRef CAS PubMed.
- L. Hao, S. Chen, J. Xu, B. Tiwari, Z. Fu, T. Li, J. Lim and Y. R. Chi, Org. Lett., 2013, 15, 4956 CrossRef CAS PubMed.
- S. Chen, L. Hao, Y. Zhang, B. Tiwari and Y. R. Chi, Org. Lett., 2013, 15, 5822 CrossRef CAS PubMed.
- L. Hao, C. W. Chuen, R. Ganguly and Y. R. Chi, Synlett, 2013, 1197 CAS.
- J. Xu, Z. Jin and Y. R. Chi, Org. Lett., 2013, 15, 5028 CrossRef CAS PubMed.
- L. Hao, X. Chen, S. Chen, K. Jiang, J. Torres and Y. R. Chi, Org. Chem. Front., 2014, 1, 148 RSC.
- T. H. West, D. S. B. Daniels, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476–4479 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |