 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and properties of long [n]cumulenes (n ≥ 5)
Johanna A.
Januszewski
and
Rik R.
Tykwinski
*
Department für Chemie und Pharmazie & Interdisciplinary Center for Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Henkestrasse 42, 91054 Erlangen, Germany. E-mail: rik.tykwinski@fau.de
First published on 27th March 2014
Abstract
Molecules composed of a contiguous sequence of double bonds, the [n]cumulenes, share structural similarities to both of their conjugated relatives, the polyenes and polyynes. The synthesis and properties of [n]cumulenes are, however, quite different from those of either polyenes or polyynes. At an infinite length, [n]cumulenes would provide one structural form of the hypothetical sp-hybridized carbon allotrope carbyne, while shorter derivatives offer model compounds to help to predict the properties of carbyne. Finally, derivatization of the π-electron framework of [n]cumulenes provides a number of different synthetic transformations, with cycloaddition reactions being the most common. In this review, both historical and recent synthetic achievements toward long [n]cumulenes (n ≥ 5) are discussed. This is followed by a description of our current understanding of the physical and electronic structure of [n]cumulenes based on UV/vis spectroscopy and X-ray crystallography. Finally, the reactivity of long [n]cumulenes is described.
 Johanna A. Januszewski | Johanna A. Januszewski was born in Bytów (Poland) in 1985. She received her diploma degree in 2010 from the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) under the supervision of Professor Rik R. Tykwinski. She is currently a PhD student in the Tykwinski group working in the field of cumulenes with a main focus on the synthesis and investigation of reactivity of these intriguing molecules. |
 Rik R. Tykwinski | Rik R. Tykwinski was born in 1965 in Marshall, MN. He completed his BS degree in 1987 at the University of Minnesota – Duluth and his PhD in 1994 at the University of Utah. He moved to ETH-Zürich for his post-doctoral research (1994–1997), and in 1997 he joined the faculty at the University of Alberta where he was promoted to Professor of Chemistry in 2005. In 2009, he began as Chair of Organic Chemistry at the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) in Germany. His interests focus on the development of synthetic methods for carbon-rich molecules and allotropes, characterization of their electronic properties, and applications of conjugated systems to molecular electronics, as well as mountain biking and entertaining his two sons. |
1. Introduction
For some years, we have concerned ourselves with the study of the one-dimensional carbon allotrope carbyne, which is constructed of only sp-hybridized carbon atoms (Fig. 1).1–4 The existence of carbyne has been a topic of much, and sometimes controversial,5,6 discussion over the years.7–14 Its natural existence has been proposed in, for example, meteorites,15,16 interstellar dust,17 and shock-compressed graphite,18 as well as terrestrial plant, fungal, and marine sources (in the case of polyynes).19–21 Furthermore, carbyne has been reportedly formed in the laboratory by a variety of processes, e.g., by solution-phase synthesis,12,22,23 laser irradiation of graphite,24 or gas-phase deposition methods.25 Whether naturally occurring or produced in a laboratory, carbyne remains rather poorly characterized as a material, and the specific properties of carbyne are thus often difficult to define (at least in the opinion of the authors of this review). There are, nevertheless, quite a number of fascinating properties predicted for carbyne structures (or shorter polyyne/cumulene oligomers) from both theory and experiment, based on the unusual electrical and optical nature of sp-hybridized carbon.26–32 This includes such applications as nanoelectronic or spintronic devices,33 nonlinear optical materials,34–37 molecular wires,38–47 and others.48–51 Recent theoretical calculations have also suggested that under tension, carbyne could be twice as stiff as the stiffest known materials, such as carbon nanotubes, graphene, and diamond.33,52 Furthermore, even though carbyne might be extremely stiff against strain, it might also allow for deformation through bending without influencing the properties.53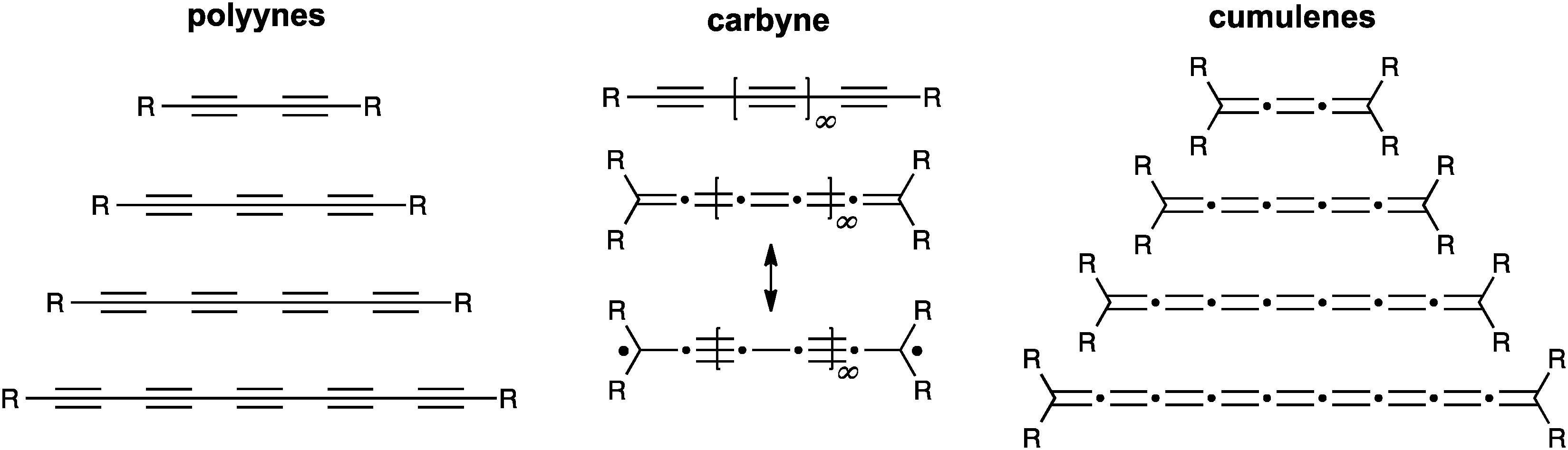 | ||
| Fig. 1 Schematic depiction of carbyne and homologous series of polyynes and cumulenes as model compounds for carbyne. | ||
In principle, two forms of carbyne might exist in a closed shell form (Fig. 1). One of these is the polyyne form which contains alternating triple and single bonds, giving a semiconductor at the carbyne limit, while the second possibility, the cumulene form, is composed of successive double bonds and should possess metallic behavior.54,55 It has also been suggested that the cumulenic form might exist with diradical character that, with bond length alternation restored, could structurally resemble a polyyne.56,57 Interestingly, Yakobson and coworkers predict a transition from cumulenic to acetylenic geometry as carbyne is stretched.33 The reverse process has also been predicted, and studies suggest a transition from a polyyne structure to a cumulene form, for example, under UV photoexcitation58 or charge transfer.54 Theoretical calculations typically predict a higher stability for the polyyne form of carbyne.33,59,60
As should be noted from the brief literature survey presented above, the structure of carbyne is on the one hand fundamental to its properties, and on the other hand not well understood. Thus, investigation of carbyne might be best approached through the rational synthesis and study of molecules with defined structure (Fig. 1). For this reason, model compounds have been synthesized and studied as a function of length to predict properties of carbyne. To date, most synthetic work has been directed toward polyynes,4,61–67 and the longest isolable polyyne so far contains 44 contiguous carbon atoms in a chain of 22 acetylene units.4 Even at this length, however, polyynes have not yet achieved a carbyne-like “status”. That is to say that UV/vis data show that saturation of the optical band gap has not yet been achieved, and extrapolation from this data predicts saturation at the point of ca. 48 acetylene units in order to form a carbyne-like compound, i.e., where elongation through additional acetylene units has no effect. The results from UV/vis analysis are also supported by other studies, such as crystallographic analysis of bond length alternation.68
In comparison to polyynes, the investigation of [n]cumulenes (where n is the number of cumulated double bonds in a chain constructed of n + 1 carbon atoms) as model compounds for carbyne has barely been discussed in the literature.69 To date, the longest cumulenes to be synthesized and studied are [9]cumulenes, i.e., molecules with nine consecutive double bonds in a chain of 10 carbons.70–73 On the basis of structure, a [9]cumulene is only approximately equal to the length of a rather short polyyne (a tetrayne). Unlike polyynes of this length, however, [9]cumulenes show dramatic instability under ambient conditions. The instability of [n]cumulenes has no doubt slowed progress on their synthesis and study. In this review, we discuss the history and evolution of cumulene synthesis, including current successes and limitations. The properties of long [n]cumulenes will then be discussed, in particular studies that address structural and electronic properties as they serve as model compounds for carbyne. As the structural motifs found throughout this review are rather large and appear numerous times, Fig. 2 offers a legend that introduces many of these structures and the associated nomenclature.
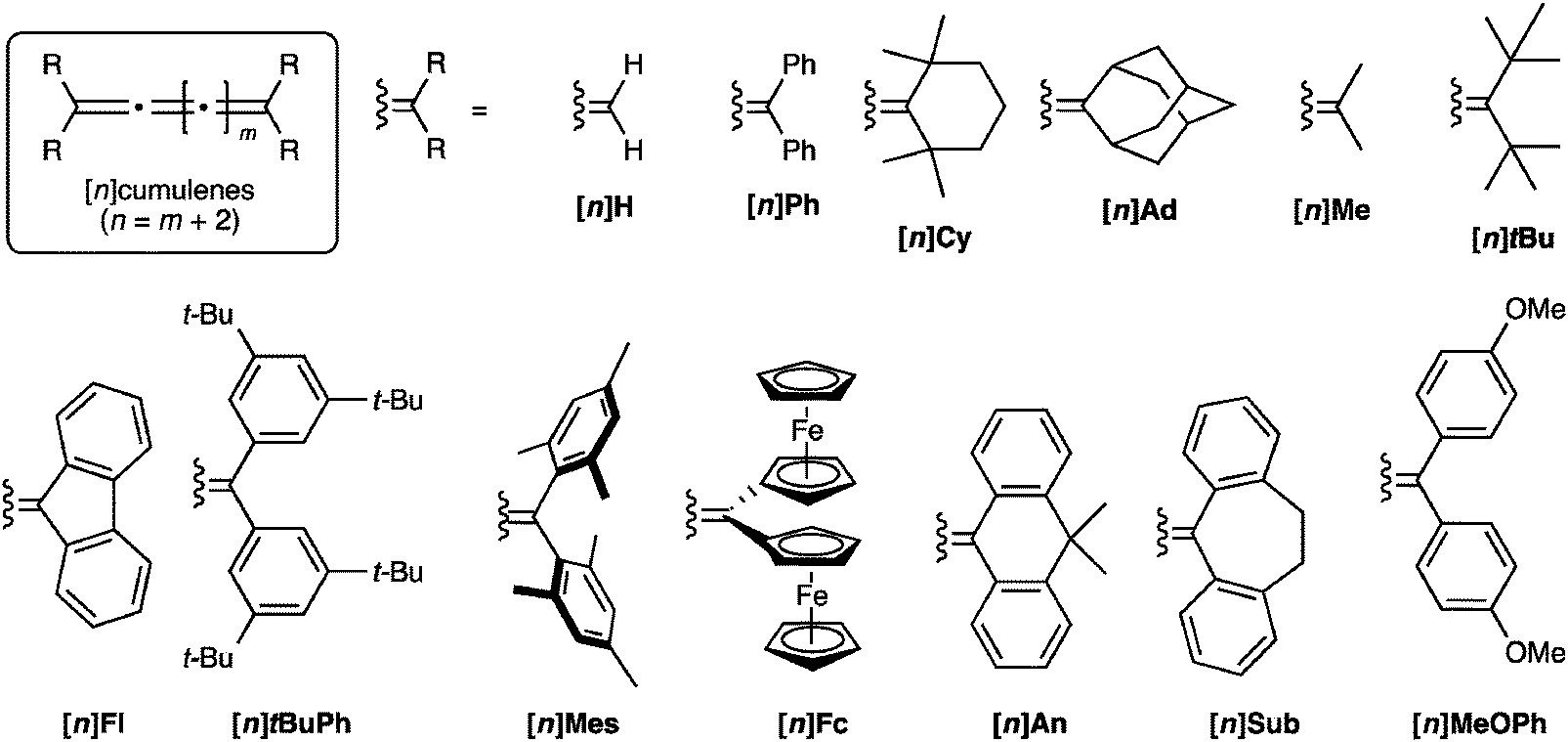 | ||
| Fig. 2 Schematic depiction of major structural classes of [n]cumulenes discussed in this review, where n is the number of cumulated double bonds in a chain constructed of n + 1 carbon atoms. | ||
2. Synthesis of [n]cumulenes
Since Kuhn's early work on cumulenes in the 1930s,74 quite a number of synthetic approaches have been developed to provide [n]cumulenes of various lengths. For the shorter analogues with n = 3 to 5, a number of common methods exist, mainly because of the greater stability of these cumulenes when compared to longer [n]cumulenes (n > 5). Better stability of the cumulene product also fosters greater functional group tolerance, and many more shorter cumulenes have been synthesized with n < 5. The synthesis of longer cumulenes benefited greatly from the pioneering advances for assembly of acetylenic compounds made by the groups of Bohlmann,61,75 Walton,62,63,76 and Jones,64,77 as well as others.78–80 From this era, acetylene compounds, and more precisely oligoyne diols,75,81 emerged as most convenient synthetic precursors for [n]cumulenes.The following overview gives a summary of synthetic methods for cumulenes and is presented based on ascending molecular length, starting from [3]cumulenes and concluding with the longest [n]cumulenes known to date, the [9]cumulenes.82 The goal of this synthetic section is twofold. First, the most common, or standard, synthetic pathways are presented, since these are most likely to be encountered in the current and future studies of cumulenes. Second, a number of alternative synthetic approaches are also described. These methods are often less general, but they offer insight into problems encountered with standard methods and provide a glimpse of the synthetic innovation that is often required for the synthesis of this challenging class of compounds. It is quickly recognized that most syntheses provide symmetrical substituted [n]cumulenes (four identical endgroups) with only a few exceptions. It is also noteworthy that the synthesis of tetraalkyl substituted [n]cumulenes is typically more difficult when compared to tetraaryl[n]cumulene, primarily due to stability of the products. Finally, the assembly of even-numbered [n]cumulenes (n = 4 and 6) is more complicated than that of odd-numbered [n]cumulenes (n = 5, 7, 9) due to synthetic accessibility of the precursors. Metallacumulenes are not included in this synthetic summary,83–87 although cumulenes used as ligands for metal coordination will be discussed briefly in the reactivity section. For more comprehensive descriptions of the syntheses of shorter [n]cumulenes, the reader is directed to reviews, for example, by Chauvin,88 Cadiot,89 Bruneau,90 and Ogasawara.82
2.1 Synthesis of [3]cumulenes
The synthesis of [3]cumulenes has recently been reviewed,88 and only selected examples are discussed here. One of the most common routes to [3]cumulenes is the metal catalyzed dimerization of carbenes/carbenoids (Scheme 1), as summarized in a review by Stang.91 These can be accomplished using the Cu(I) catalyst for aryl or alkynyl substituents as described by Diederich,92,93 as well as Kunieda and Takizawa.94 On the other hand, Iyoda95,96 used a Ni(II) catalyst to form [3]cumulenes. Tetraalkyl[3]cumulenes can be made directly from a lithium carbenoid as reported independently by, for example, Köbrich,97 Komatsu,98 and Oda.99 Stang, on the other hand, successfully formed a variety of alkyl substituted [3]cumulene via the carbenoid route using ethynylvinyl triflates as precursors.100,101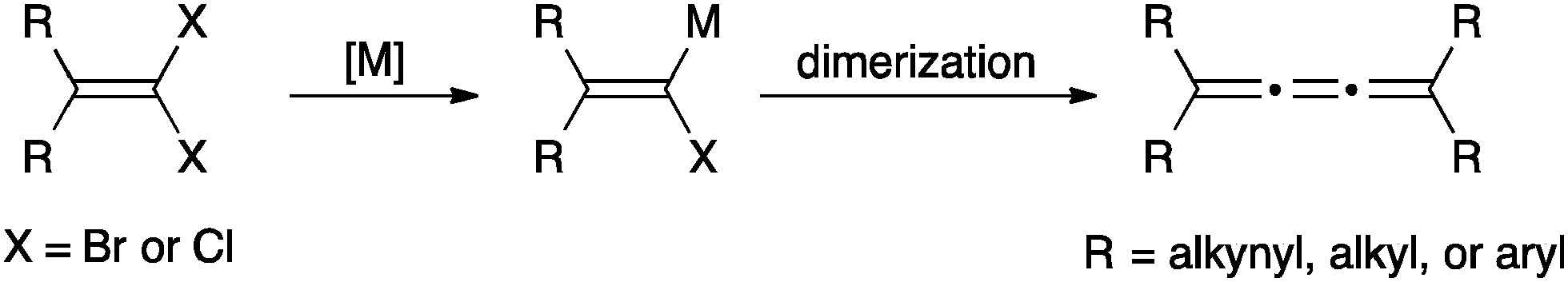 | ||
| Scheme 1 Synthesis of [3]cumulenes based on the carbene/carbenoid route (M = metal). | ||
An alternative method to generate carbenoids for the synthesis of [3]cumulenes is based on elimination of trihaloalkanes (Scheme 2). Direct elimination using a Cu/Cu(I) catalyst system gave [3]cumulene products in moderate yields (ca. 40–60%).94 Brand and coworkers,102,103 on the other hand, describe a route that proceeds through a dichloroalkene intermediate formed through reduction of the trihaloalkane, and this overall protocol gives higher yields (>85%) than the direct elimination although it requires three distinct synthetic steps.
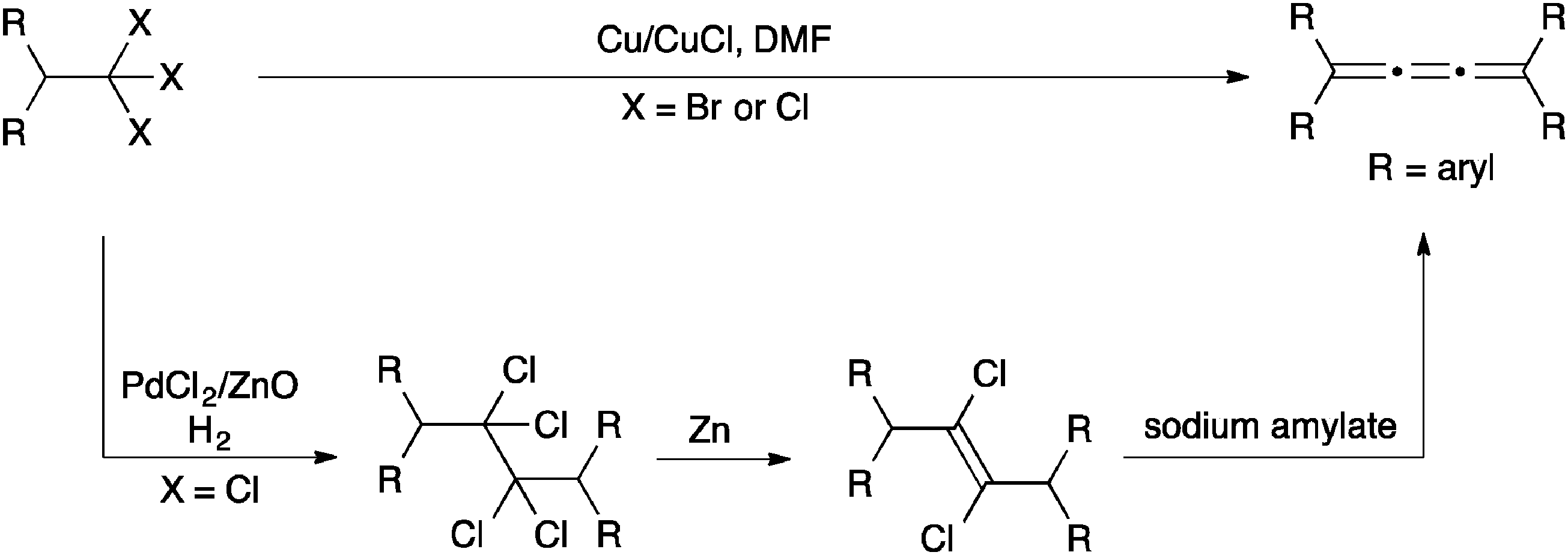 | ||
| Scheme 2 Synthesis of [3]cumulenes based on reductive elimination of trihaloalkanes. | ||
The most common synthetic route to [3]cumulenes is based on the reduction of an acetylenic diol or diether derivative (Scheme 3). The precursor is usually a Li- or Mg-acetylide,57,70,71,74,104–106 which is used in an addition reaction with a ketone that defines the endgroups. In cases where the free alcohols may not be compatible with subsequent synthetic steps, it can be blocked by the formation of, for example, methyl ethers through trapping with MeI.70 From either the diol or diether, aryl substituted [3]cumulenes can be formed directly via reduction with SnCl2,70,106 although P2I474 or HI and I2105 have also been used. Tetraalkyl[3]cumulenes are usually formed via halogenation, followed by reductive elimination (Scheme 3).71,107,108
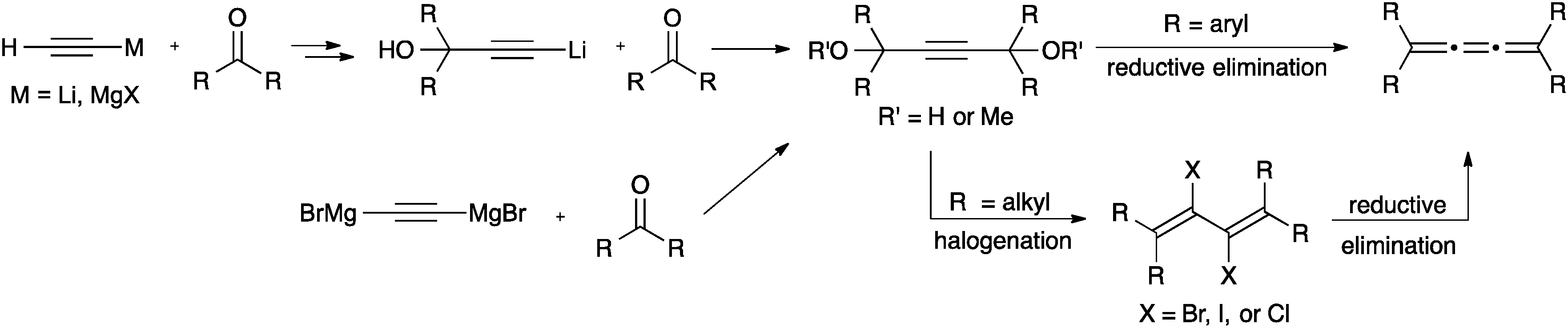 | ||
| Scheme 3 Synthesis of [3]cumulenes based on acetylenic diol derivatives. | ||
2.2 Synthesis of [4]cumulenes
Bildstein and coworkers have reported the formation of [4]Fc based on an adaptation of the diol approach described above for [3]cumulenes (Scheme 4).106,109 A homopropargylic ether is formed, lithiated, and then added to diferrocenylketone to complete the carbon skeleton. Treatment with acid (HBF4) results in the stabilized (and isolable) [3]cumulene intermediate, which can then be converted to [4]Fc through base induced elimination. Bildstein's approach is conceptually similar to that reported earlier by Nakagawa and coworkers,110 in which two different endgroups have been introduced in order to explore optical activity and racemization of [4]cumulenes.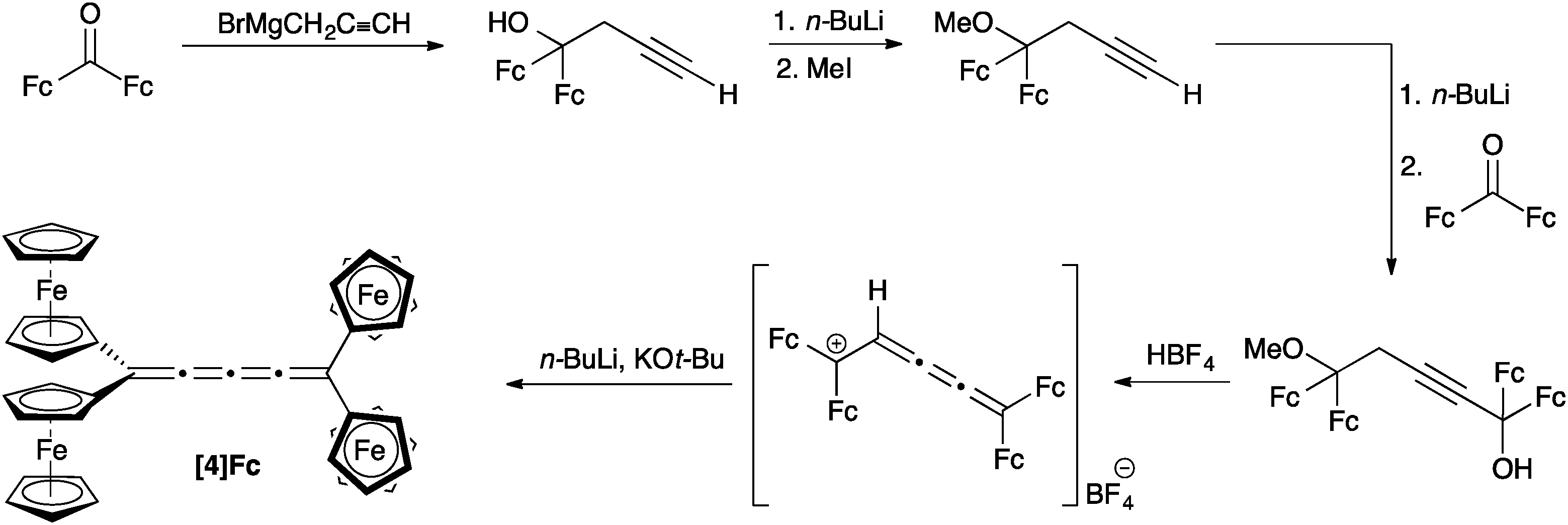 | ||
| Scheme 4 Synthesis of [4]Fc based on a diol derivative. | ||
Another possibility of the formation of tetraaryl[4]cumulenes has first been described by Kuhn111 and then Karich and Jochims112 and relies on the intermediate formation of dibromo-1,4-pentadienes from the appropriate unsubstituted dienes (Scheme 5). The diene is then converted to the [4]cumulene in good yield through base-induced elimination.
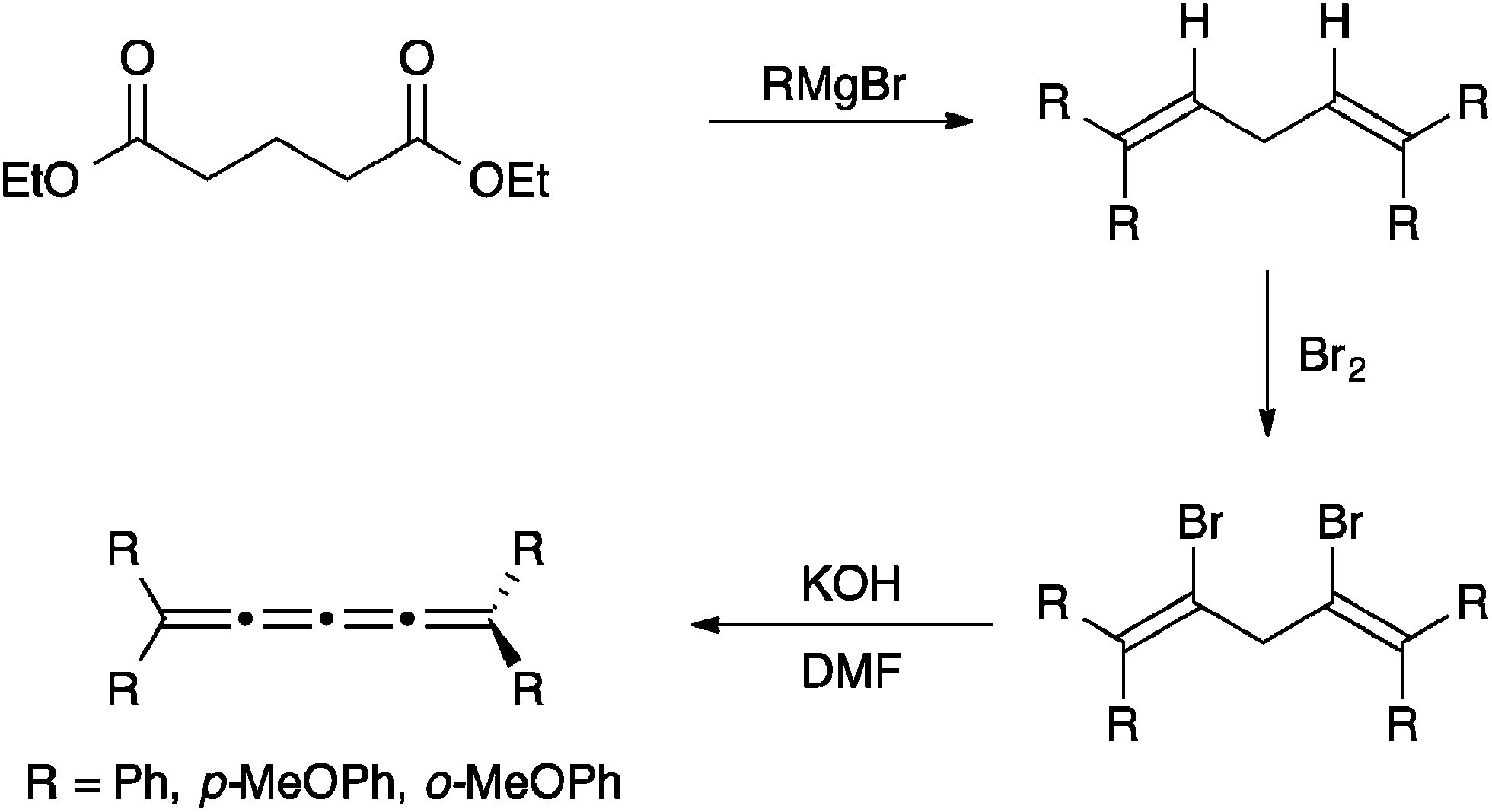 | ||
| Scheme 5 Synthesis of [4]cumulenes from diesters. | ||
[3]Cumulenes can be converted to [4]cumulenes via addition of dichlorocarbene to a [3]cumulene, followed by rearrangement or reductive elimination, depending on the structure of the precursor (Scheme 6). Jochims and coworkers highlight the potential effectiveness of this route through formation of [4]Ad, where MeLi gives a quantitative yield in the reduction step. Their route also provides rare examples of mixed dialkyl/diaryl endcapped [4]cumulenes.112 Irngartinger and Götzmann have developed a slightly modified version of this general protocol to synthesize [4]Cy, using Zn in the final reductive elimination step (Scheme 6).113
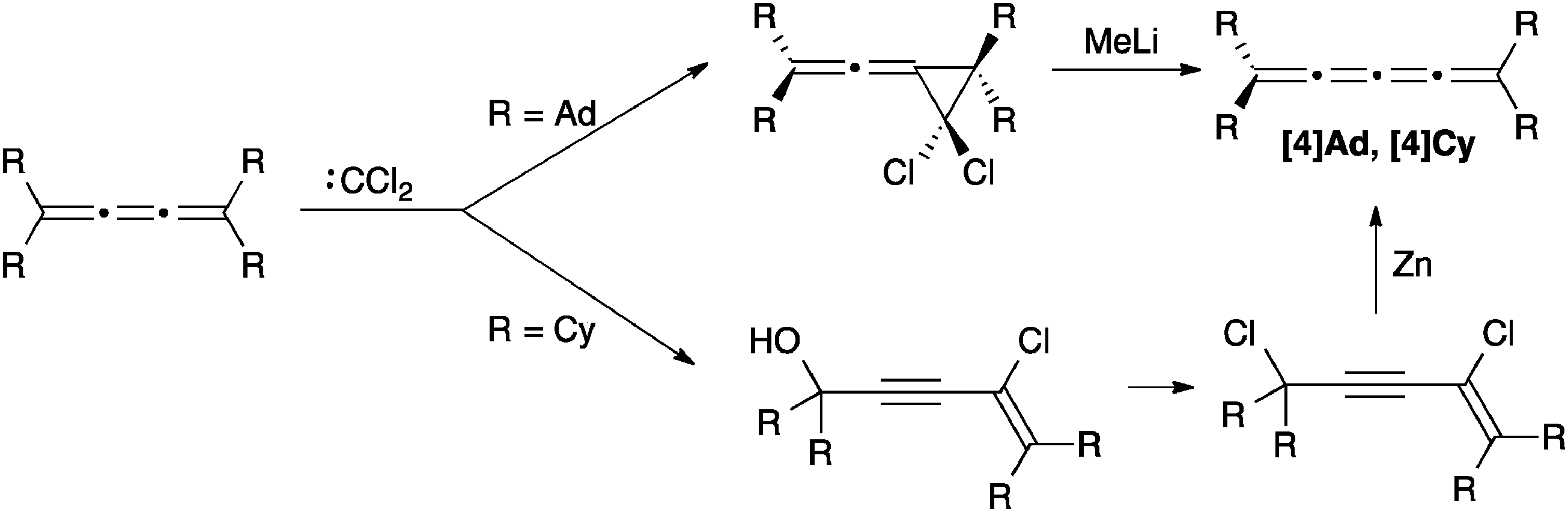 | ||
| Scheme 6 Synthesis of [4]cumulenes from [3]cumulenes. | ||
[4]Cumulenes can be obtained via carbene trapping as demonstrated by le Noble and coworkers (Scheme 7).114,115 The dialkylpentatetraenylidene intermediate is formed and reacts in situ with tetramethylethylene to give the [4]cumulene, which quickly converts to a radialene through dimerization (in the case of R = Me). The analogous reaction with 2-adamantylpentatetraenylidene, on the other hand, forms the stable [4]cumulene.
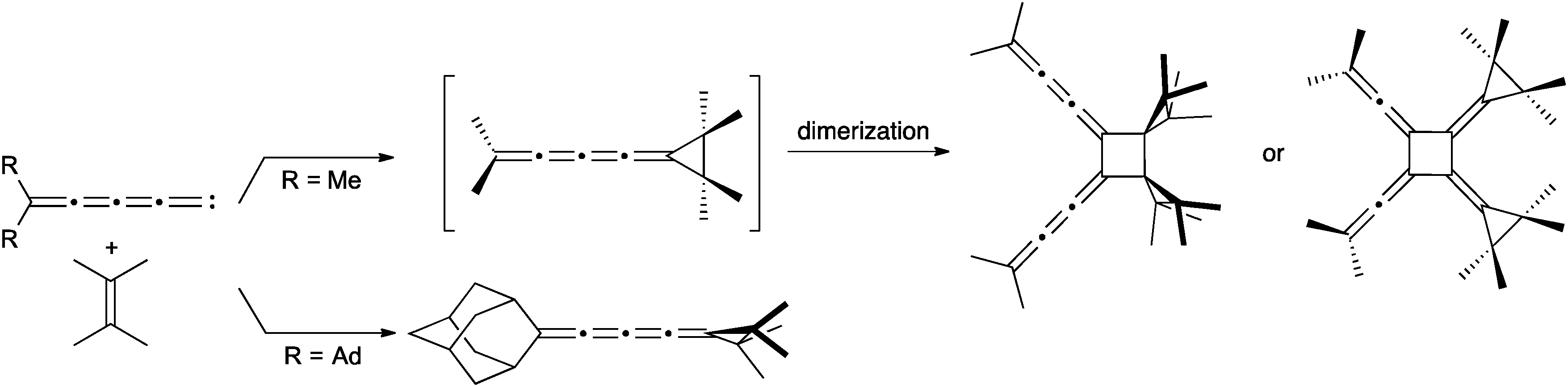 | ||
| Scheme 7 Synthesis of a [4]cumulene via carbene trapping with an olefin. | ||
2.3 Synthesis of [5]cumulenes
Of the higher [n]cumulenes (n ≥ 5), [5]cumulenes are by far the most studied and there are thus a number of efficient routes that have been developed for their synthesis.82 In analogy to the syntheses described for [3]cumulenes, dimerization reactions of carbenes have been used to give [5]cumulenes (Scheme 8).57,106,116–120 Starting with a terminal acetylene and a leaving group in the propargylic position, reaction with base produces a carbene intermediate, which leads to the [5]cumulene via dimerization. An alternative carbenoid route to [5]cumulenes has been described by Kollmar and Fischer,121 in which the vinylidene carbenoid is generated directly from a haloallene (Scheme 8). It is interesting to note that the influence of endgroups in this reaction is likely enhanced versus the analogous reaction to give [3]cumulenes, considering the mesomeric stabilization of this intermediate carbene (Scheme 8).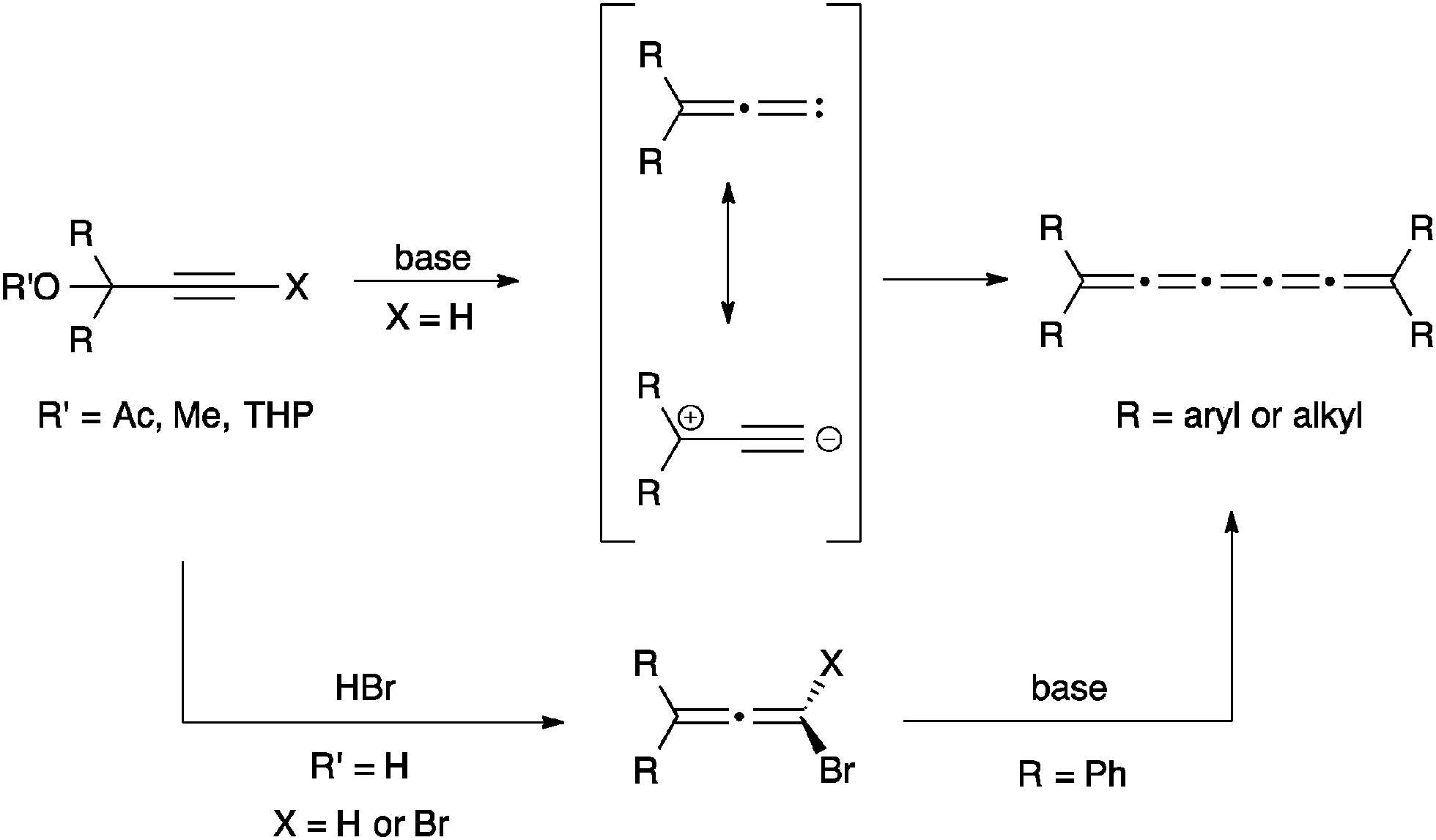 | ||
| Scheme 8 Synthesis of [5]cumulenes based on carbenes/carbenoids. | ||
Unsymmetrical [5]cumulenes can be synthesized through trapping of vinylidene carbenes, as reported by Stang and coworkers (Scheme 9).101,122,123 More specifically, elimination of a diyne vinyl triflate forms the intermediate carbene, which can be trapped by either addition to electron rich alkenes (i.e., tetramethylethylene and cyclohexene) or M–H bond insertion using R3MH (M = Si, Ge). In some cases, the resulting [5]cumulene is not stable and isomerizes or polymerizes during the reaction.
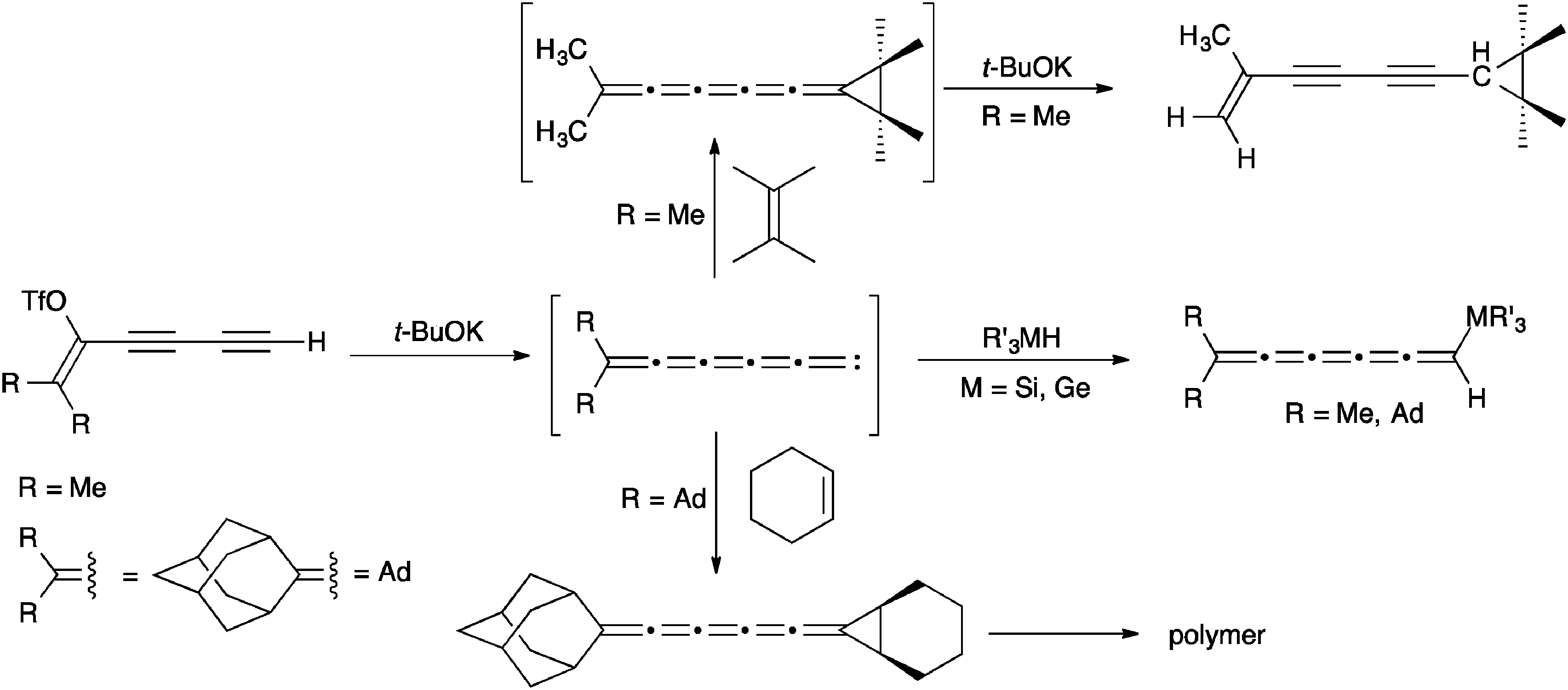 | ||
| Scheme 9 Synthesis of unsymmetrical substituted [5]cumulenes based on carbenoids. | ||
[5]Cumulenes can be formed based on the reaction of a [3]cumulene with dibromocarbene as described by Skattebol (Scheme 10).124 The addition of dibromocarbene to tetramethyl[3]cumulene gave the bicyclopropylidene product, and the subsequent rearrangement reaction, induced with MeLi, gives the unstable [5]Me product.
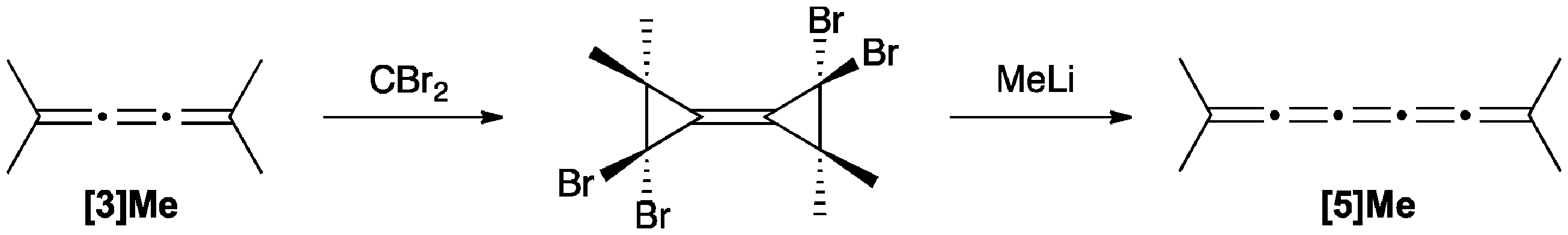 | ||
| Scheme 10 Synthesis of [5]Me from [3]Me. | ||
Assembly of [5]cumulenes based on acetylenic diol precursors is quite common (Scheme 11), in part because the necessary precursor, a diyne diol, can be efficiently formed via a number of routes. Often mimicking strategies described for [3]cumulenes, acetylenic diols are thus readily assembled through, for example, the addition of a Li- or Mg-acetylide to a ketone. Alternatively, oxidative homocoupling of propargyl alcohol derivatives is usually quite efficient.
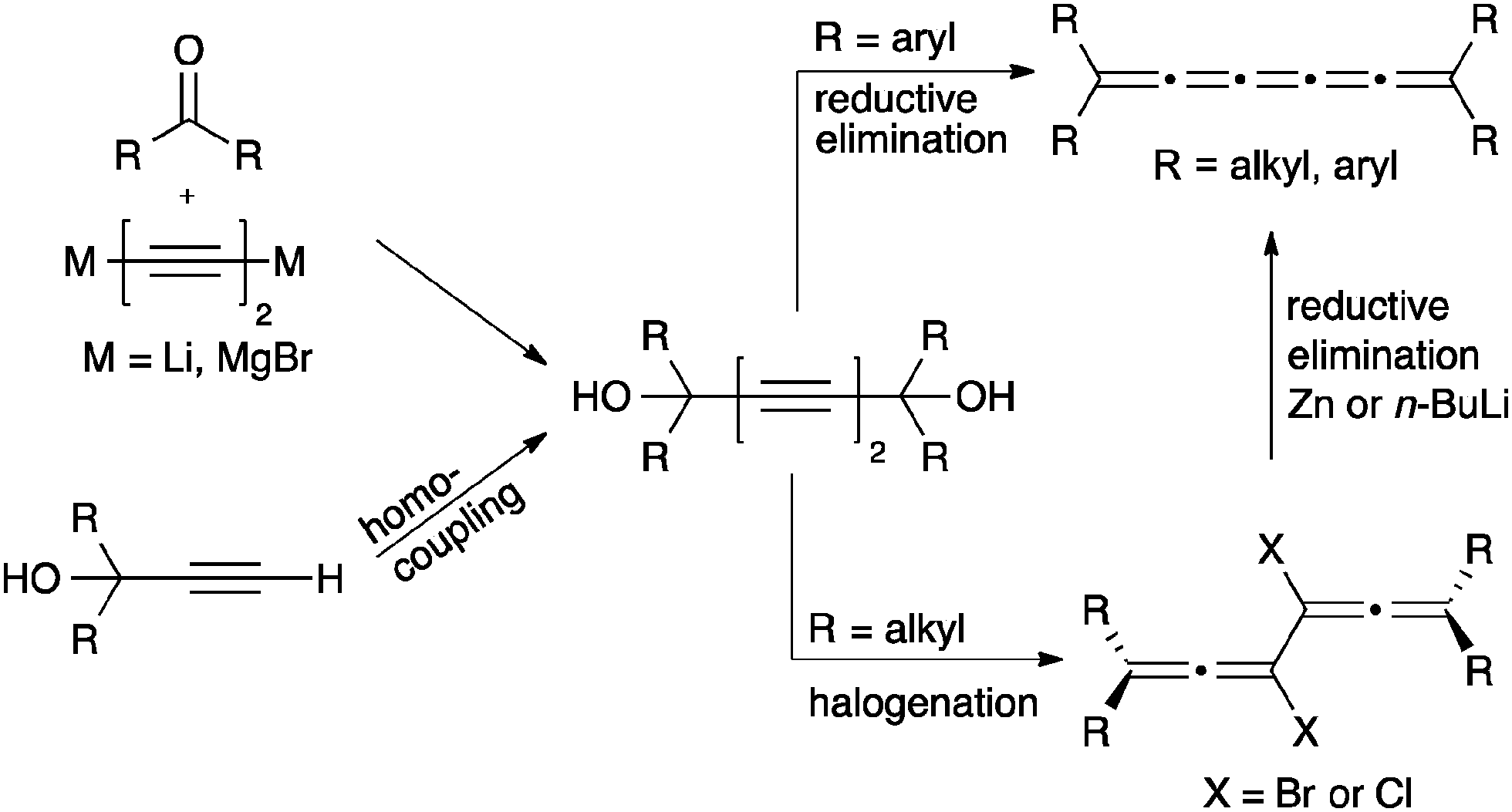 | ||
| Scheme 11 Synthesis of [5]cumulenes based on acetylenic diol derivatives. | ||
With the diyne diol in hand, conversion directly to an aryl substituted [5]cumulene is accomplished via reduction with P2I4,74 CrCl2,125 or SnCl2.70,126,127 In the majority of the recent studies, SnCl2 is the reductant of choice and usually gives good yields. In the case of alkyl substitution, conversion of the diyne diol to the corresponding dihalide with PI3,71 PBr3,71,127 HBr,127 or HCl127 is required, which is then followed by reductive elimination using Zn or n-BuLi.
2.4 Synthesis of [6]cumulenes
To date, only one synthesis has been reported for a [6]cumulene, namely [6]Fc (Scheme 12). Bildstein and coworkers128 have shown that a diyne diether can be readily assembled via an acetylenic cross-coupling reaction, and the [6]cumulene is then realized through an elimination process similar to the synthesis of the [4]Fc. It is worth mentioning that after the first elimination step, an unusual, air-stable cumulenium salt is obtained, and the stability is explained by the presence of four ferrocene donor groups and the unsaturated cumulene chain. Unfortunately, the resulting air-sensitive [6]Fc product could not be isolated, but UV/vis spectroscopy confirms the formation of the cumulene framework.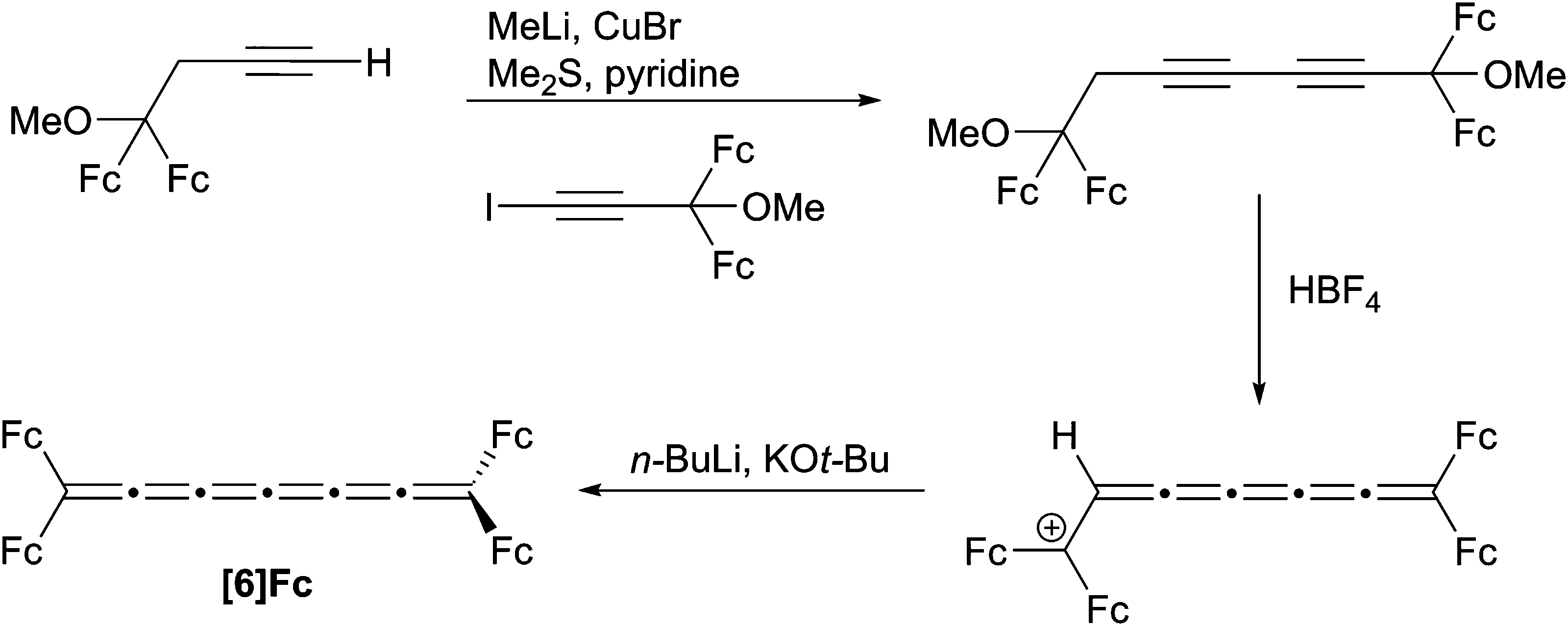 | ||
| Scheme 12 Synthesis of [6]Fc. | ||
2.5 Synthesis of [7]cumulenes
To our knowledge, only seven tetraaryl[7]cumulenes and one tetraalkyl[7]cumulene have been reported to date.70,71,129–131 All [7]cumulenes have been assembled from the corresponding triyne diol precursor (Scheme 13), which is typically formed as described above for [3]- and [5]cumulene syntheses. An alternative approach to the requisite diol has recently been reported, using a carbenoid Fritsch–Buttenberg–Wiechell (FBW) rearrangement1–3 in combination with Colvin's reagent132–134 to form the triyne framework.70 Following the trends established by [3]- and [5]cumulene syntheses, formation of the tetraalkyl[7]cumulene requires conversion of the diol to the dibromide, followed by reductive elimination with Zn. In contrast, the tetraaryl[7]cumulenes can be formed directly by reduction with either P2I4 or SnCl2. In most cases, the [7]cumulenes tend to decompose quickly, either in solution or in the solid state. In the most recent report on cumulene synthesis, however, Januszewski et al. show that [7]tBuPh and [7]Mes are sufficiently stabilized using sterically demanding aryl endcapping groups to provide crystalline products.70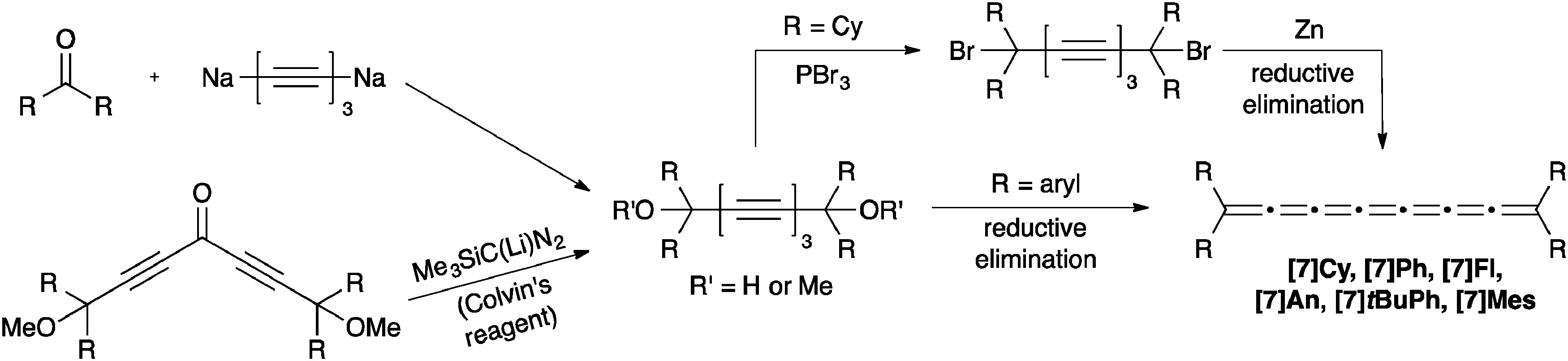 | ||
| Scheme 13 Synthesis of [7]cumulenes based on acetylenic diol derivatives. | ||
Perhaps the most structurally interesting [7]cumulene reported to date, [7]pPh, was synthesized by Cadiot and coworkers through a variation of the diol approach as outlined in Scheme 14.131 In this case, the acetylenic diol precursor was assembled using a Cu-catalyzed heterocoupling between a terminal diacetylene and a bromoacetylene derivative. While the final product could not be isolated, UV/vis spectroscopy confirmed formation, with a lowest energy absorption at 700 nm that is red-shifted versus that of all other [7]cumulenes (vide infra).
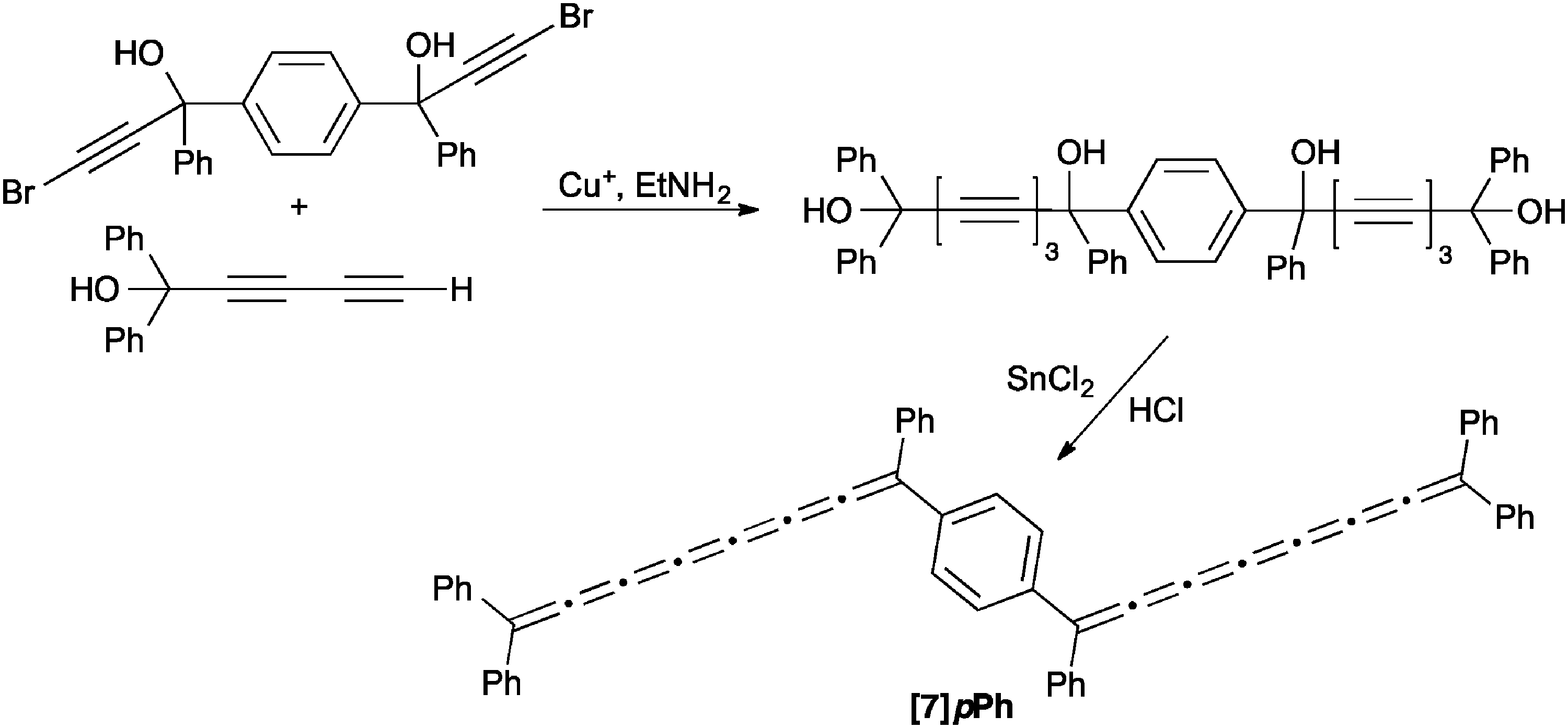 | ||
| Scheme 14 Synthesis of the bis[7]cumulene [7]pPh. | ||
2.6 Synthesis of [9]cumulenes
As a result of instability of the final product, very few [9]cumulenes have been successfully assembled, and there are only five examples to be found in the literature, including four tetraaryl- ([9]Ph, [9]Sub, [9]tBuPh, [9]Mes)70,72,73 and one tetraalkyl[9]cumulene ([9]Cy).71 The synthetic pathway to [9]cumulenes is, predictably, analogous to that of [5]cumulenes, except that diynes provide the precursors for the dimerization, rather than terminal alkynes (Scheme 15). As per usual, tetraaryl derivatives are formed directly from the diol via reduction using P2I4 or SnCl2, while the tetraalkyl[9]cumulene [9]Cy must be synthesized from the dibromide, via reduction with Zn. Isolated yields have not been reported for any of the [9]cumulenes due to the inability to isolate the unstable product.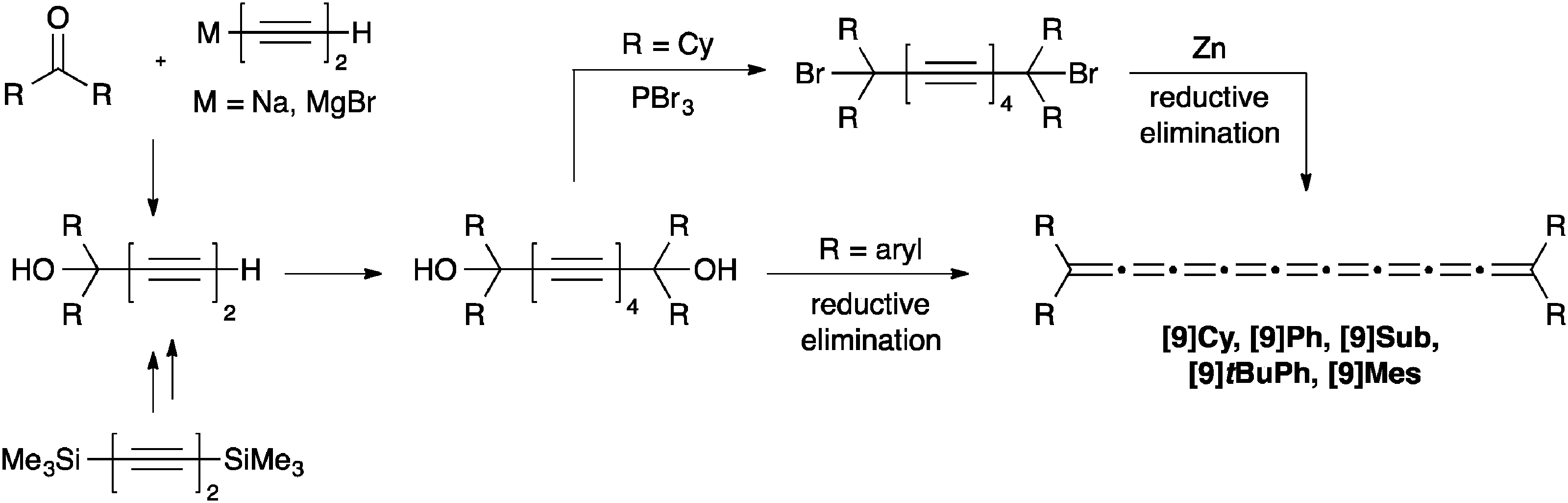 | ||
| Scheme 15 Synthesis of [9]cumulenes based on acetylenic diol derivatives. | ||
3. UV/vis spectroscopy
UV/vis spectroscopy has been the most common characterization method for long [n]cumulenes and in early studies it was the essential tool to confirm formation of [7]- and [9]-cumulenes.135 Using homologous series of [n]cumulenes, UV/vis spectroscopy allows analysis of electronic trends, i.e., optical band gap, as a function of cumulene length and substitution. Similar to that demonstrated for polyynes,136 UV/vis spectroscopic analysis versus molecular length might also allow extrapolation to an infinite chain length, which would offer a prediction of the band gap of the cumulenic version of carbyne. To date, however, this has not been possible due to the limited number of model compounds that are currently available.In principle, three major factors govern trends typically observed in the UV/vis spectra of [n]cumulenes, including (1) structure of the [n]cumulene (odd- versus even-numbered [n]cumulenes), (2) molecular length, and (3) the nature of the terminal substitution, i.e., endgroup effects (aryl versus alkyl giving mesomeric versus inductive effects, respectively).
3.1 Comparison of odd- versus even-numbered [n]cumulenes
The influence of odd- versus even-numbered [n]cumulenes is delineated schematically in Fig. 3, and this also demonstrates the potential mesomeric or inductive contribution from the endgroups. For even-numbered cumulenes, there are two π-systems that are degenerate and spatially orthogonal (Fig. 3a). In the case of aryl endcapping groups, each orthogonal π-system can conjugate with substituents at one end of the cumulenic framework, but not both. The situation is distinctly different for odd-numbered cumulenes (Fig. 3b), where the two π-systems of the sp-carbon framework are no longer degenerate. In this case, one π-system spans the length of the cumulene skeleton and can conjugate with both sets of endgroups (Fig. 3b, in red). The other π-system (in blue) does not communicate directly with the endgroups (i.e., via resonance) and is thus considerably shortened, although hyperconjugation with terminal groups is easily envisioned. It is worth noting that the influence of odd- versus even-numbered [n]cumulene structure should also be observed in the bond length alternation (BLA) of cumulenes. As shown by the mesomeric structures in Fig. 3b, increased BLA is expected for odd [n]cumulenes and should be further enhanced by groups able to conjugate to the cumulene core.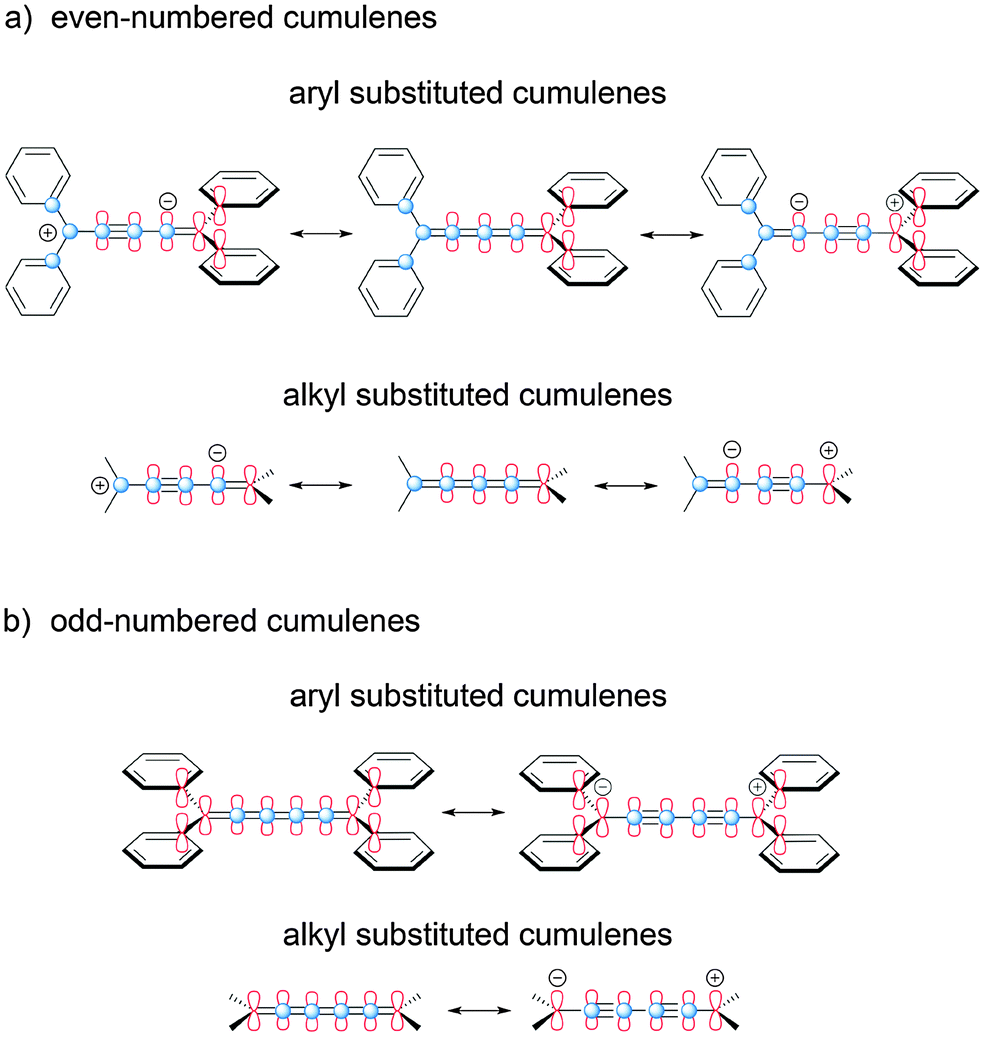 | ||
| Fig. 3 Electronic effects based on odd- and even-numbered [n]cumulenes, as demonstrated schematically with canonical structures for [4]- and [5]cumulenes. | ||
3.2 Comparison of [n]cumulenes as a function of length n
The UV/vis spectroscopic data of a series of tetraaryl- and tetraalkyl[n]cumulenes ([n]tBuPh and [n]Cy, respectively) are given in Fig. 4, and these two examples include the most complete analyses reported to date with n = 3, 5, 7, and 9 for both sets of molecules. A distinct signal pattern is immediately observed, showing two regions of absorption bands for both tetraaryl- and tetraalkyl[n]cumulenes, with the most intense absorptions mostly found at higher energy in the UV region (<400 nm). The observation that the fine structure of the absorption in the UV region often increases as a function of molecular length is noteworthy. The second set of absorptions is found in the visible region for tetraaryl[n]cumulenes from 420–670 nm, and at much higher energy for the tetraalkyl[n]cumulenes due to the absence of conjugation with the alkyl endgroups and the cumulene core (see Fig. 3).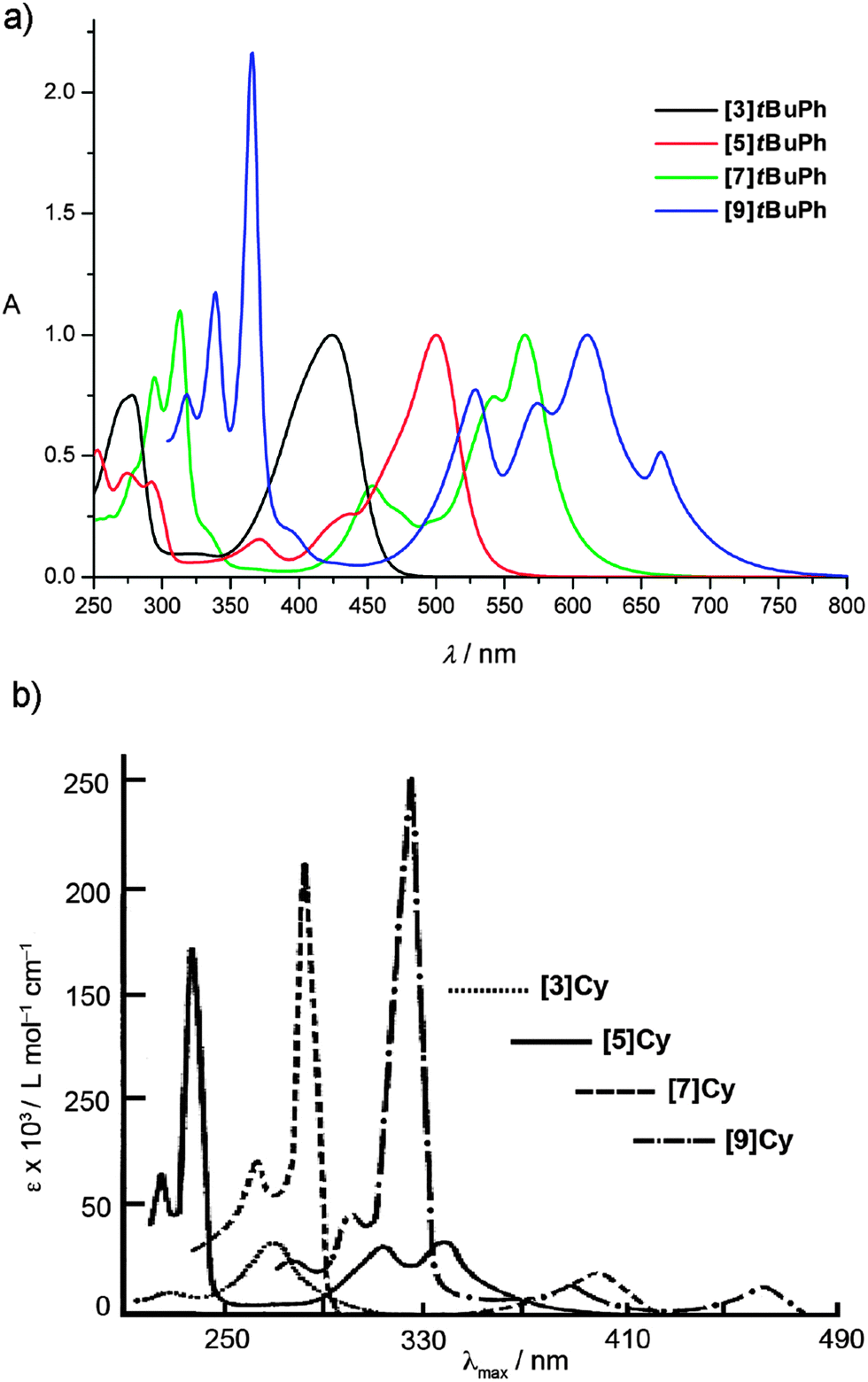 | ||
| Fig. 4 UV/vis spectra of [n]cumulenes. (a) [n]tBuPh measured in Et2O; spectra normalized to the most intense low energy absorption and (b) [n]Cy measured in Et2O (adapted with permission from ref. 135. Copyright 1964 John Wiley & Sons). | ||
The most obvious consequence of π-electron conjugation is observed in λmax values as a function of cumulene length chain, as demonstrated by values of the four [n]cumulene series listed in Table 1. As expected, λmax values of tetraarylcumulenes are found at much lower energy than those of the tetraalkylcumulenes, e.g., 663 nm versus 465 nm, respectively, for [9]Ph and [9]Cy. A monotonic red-shift in λmax is clearly visible as the cumulene length is increased within each series, indicating a decreasing HOMO → LUMO energy gap. It would be expected that at some cumulene length this effect reaches saturation, and λmax would reach a minimum and constant value. This limiting value would then represent an estimate of energy gap (Eg) of the material “cumulenic” carbyne. Several methods have been used for polyynes to describe the relationship between Eg, λmax, and n, such as an empirical power-law137,138 (Eg = 1/λmax ∼ n−x) or the exponential function proposed by Meier and coworkers.139 Unfortunately, attempts to apply these protocols to the cumulenes reported in Table 1 provide inconclusive estimates for Eg, and longer cumulenes (n > 9) are needed to complete this analysis. Thus, no experimental estimate for the cumulenic form of carbyne is currently available.
The nature of the four endgroups appended to the cumulene framework is expected to play an import role in λmax values (i.e., alkyl versus aryl). Within a series of cumulenes, however, experimental data show that the influence of the endgroups decreases rapidly as a function of cumulene length. For example, λmax values of the tetraaryl[5]cumulenes [5]Ph, [5]tBuPh, and [5]Mes (489, 500, and 460 nm, respectively), vary rather significantly (Table 1). The λmax values of the longer analogues [7]Ph/[7]tBuPh/[7]Mes and [9]Ph/[9]tBuPh/[9]Mes are, however, nearly identical, with values of about 560 nm and 665 nm, respectively.
3.3 Comparison of [5]cumulenes as a function of endcapping group
In Table 2, λmax values are given for a selection of [5]cumulenes, and the comparison of these values offers insight into the specific influence of the endgroup. The biggest shift to lower energy is observed for cumulenes in which the pendent aryl rings are forced to be coplanar to the cumulene framework, namely [5]An (λmax = 555 nm) and [5]Fl (λmax = 542 nm). Thus planarity of aryl endgroups appears to be a dominant factor. This supposition is also supported by λmax value measured for [5]Mes (465 nm), for which steric hindrance from the 2- and 6-methyl groups inhibits coplanarity of the aryl groups with the cumulene core, a fact supported by X-ray crystallographic analysis (vide infra).| [5]An | [5]Fl | [5]MeOPh | [5]tBuPh | [5]Ph | [5]Mes | [5]Cy | [5]tBu | [5]Me | |
|---|---|---|---|---|---|---|---|---|---|
| a CHCl3. b Benzene. c Pyridine. d Et2O. e THF. f Isooctane. g Ref. 57 reports the lowest energy absorption value as 417 nm, but with negligibly low ε value of 870 L mol−1 cm−1, thus the next higher energy value of 336 nm was taken here for comparison. h EtOH. | |||||||||
| 555a | 542a | 517a | 510a | 488a | 465a | 339d | 337d | 320h | |
| 540a | 500d | 489b | 460d | 336f,g | |||||
| 543b | 485e | ||||||||
| 547c | 493c | ||||||||
| Ref. | 130 | 74, 140, 120 | 120 | 70 | 119, 120, 127, 140 | 70 | 71 | 57, 141 | 142 |
Substituted [5]cumulenes [5]MeOPh and [5]tBuPh show red-shifted λmax values relative to [5]Ph with λmax = 517 and 510 nm, respectively. These features are readily explained by mesomeric and inductive effects of the MeO- and tBu endgroups, respectively, which donate electron density into the electron deficient sp-carbon framework.
Finally, the greatest blue shift in λmax is found for tetraalkyl cumulene [5]tBu (λmax = 337 nm in Et2O), which is at ca. 150 nm higher energy versus[5]Ph. There is, obviously, no mesomeric contribution from the alkyl endgroups to the conjugated structure of [5]tBu, but hyperconjugation with the four t-butyl groups appears to be present based on the comparison to [5]Me (λmax = 320 nm).
3.4 Comparison of [n]cumulenes to polyenes and polyynes
It is interesting to compare the UV/vis spectroscopic behavior of [n]cumulenes versus other conjugated oligomers of analogous size and substitution patterns, such as polyenes and polyynes (Table 3 and Fig. 5). The main goal of this analysis is to offer an empirical evaluation of electronic delocalization for the three compound classes; thus, comparisons of λmax values are made between molecules that share the same number of π-electrons in the longest conjugated segment. It is noted that for the phenyl endcapped series, however, a “fair” comparison is not possible due to the different valency requirements of the terminal atoms of each chain, i.e., termination with sp- versus sp2-carbon. It is also important to emphasize again the different π-electron system(s) present in each molecule, as described earlier in Fig. 3. Namely polyenes have only a single π-system, while polyynes possess two degenerate, orthogonal π-systems (Fig. 5). Finally, [n]cumulenes have two non-degenerate π-systems, i.e., one that extends the length of the molecule and conjugates to the endgroups (in red, Fig. 5) and a shorter π-system that is orthogonal and cannot conjugated to the endgroups (in blue, Fig. 5). The electron count for each molecule shown in Table 3 includes only electrons found in the longest conjugated π-system in each case.|
|
||||||
|---|---|---|---|---|---|---|
| π-Electrons | [n]Ph |
Ph(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )[n] )[n]
|
Ph(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) )[n] )[n]
|
[n]Cy |
tBu()[n]
|
tBu()[n]
|
| a Benzene. b Et2O. c n-Pentane. d Hexanes. e A [3]cumulene, diene, and diyne. f No measureable UV-absorption in solution (i.e., sample absorption lies under solvent absorption). g A [5]cumulene, triene, and triyne. h A [7]cumulene, tetraene, and tetrayne. i A [9]cumulene, pentaene, and pentayne. | ||||||
| 4e | 420 | 352 | 331 | 272 | 237 | —f |
| 6g | 489 | 377 | 363 | 339 | 276 | 213 |
| 8h | 557 | 404 | 402 | 401 | 311 | 240 |
| 10i | 663a | 424 | 437 | 465 | 343 | 266 |
| Ref. | 74, 103, 129, 72, 140 | 143, 144 | 144 | 71 | 145 | 68 |
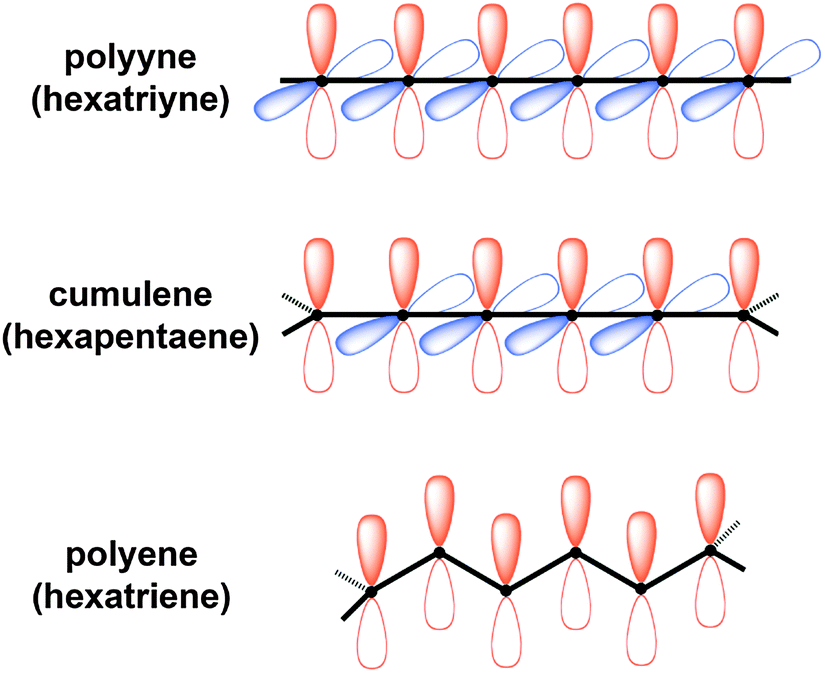 | ||
| Fig. 5 Schematic depiction of π-orbitals of structures with six π-electrons in the longest conjugated segment shown in red: a polyyne, cumulene, and polyene. The orthogonal and degenerate six π-electron segment of the hexatriyne and the orthogonal four π-electron segment of the [5]cumulene are shown in blue. | ||
The most interesting point to be gleaned from these comparisons is that for all series of molecules with equivalent π-electron counts, cumulenes show the lowest energy absorption, followed by polyenes and then polyynes (except for the pentayne Ph()[5]). The nature of the endcapping group also does play a role. In the presence of a conjugating endgroup, λmax for polyynes and polyynes (Ph()[n] and Ph()[n]) are nearly the same (Fig. 6a), while this is not the case with alkyl endcapping groups (Fig. 6b).
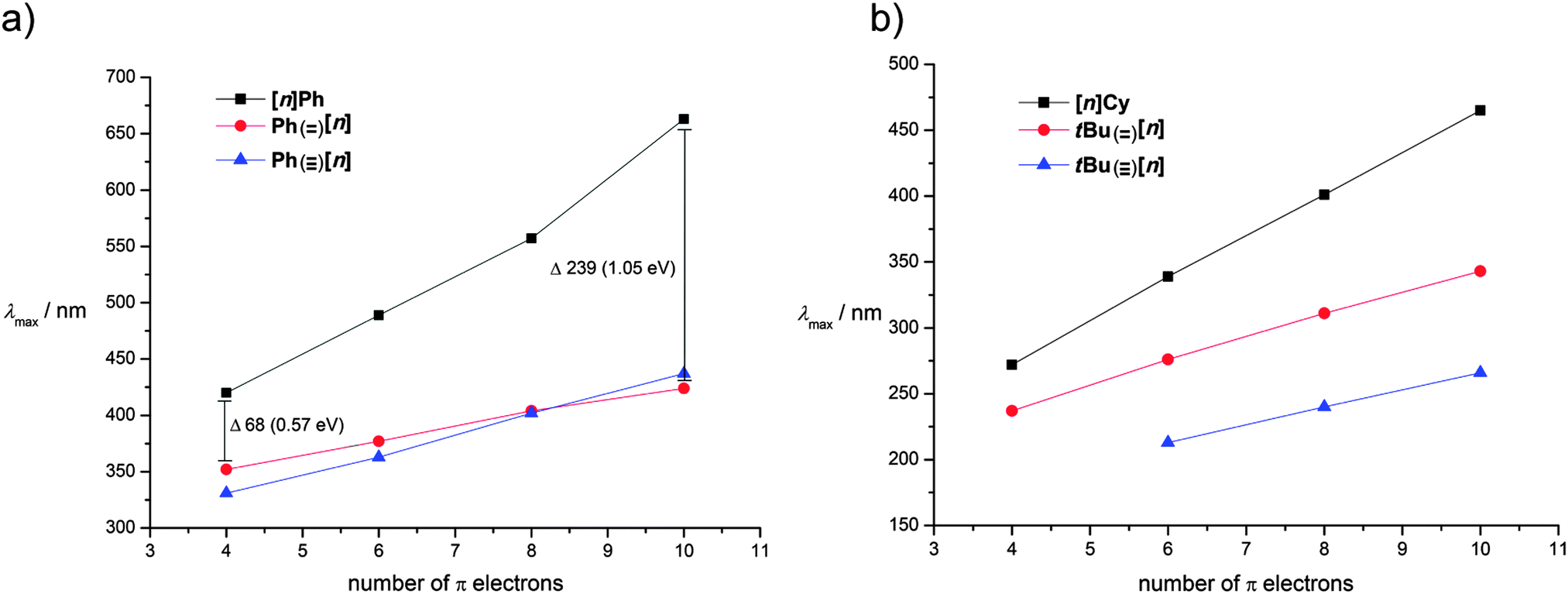 | ||
| Fig. 6 Plot of lowest energy absorption λmaxversus number of π-electrons for (a) phenyl- and (b) alkyl substituted cumulenes, polyenes, and polyynes. Values taken from Table 3. | ||
Interestingly, λmax values for phenyl substituted [n]cumulenes move to a lower energy in a much more dramatic fashion than those of either polyenes or polyynes (Fig. 6a). This demonstrates a significantly stronger increase of the conjugation with increase of chain length in the case of cumulenes than in polyenes or polyynes, a trend that is also supported by bond length alternation (BLA) data presented in the next section. For alkyl substituted [n]cumulenes, the same general trend is also observed but to a lesser extent than in phenyl substituted cumulenes.
4. Structural analysis by X-ray crystallography
X-ray crystallographic analysis is relatively uncommon for [n]cumulenes (n ≥ 5) due to instability under ambient conditions and limited synthetic accessibility. This method, however, offers profound insight into both the physical and electronic structure of cumulenes, especially via the analysis of bond length alternation (BLA) as a function of molecular length (BLA, defined as the bond length difference between the two central-most double bonds of the cumulene chain). As discussed above, UV/vis spectroscopy shows nicely that there is a relationship between the optical HOMO–LUMO gap and the length of the cumulene chain. In principle, this change in the HOMO–LUMO gap should coincide with structural changes of the cumulene framework. More specifically, BLA should diminish for longer cumulenes and eventually reach a constant value. Several theoretical calculations for cumulenes have suggested that BLA cumulenes should approach a value of nearly zero,55,146,147 although at the time of these reports experimental validation of these predictions was not possible. In this section, selected X-ray crystallographic data are discussed in terms of structural trends, followed by a summary of BLA analysis based on an overview of the experimental results available to date for cumulenes.In Fig. 7, bond lengths for [n]tBuPh (n = 3, 5, 7)70 and [n]Cy (n = 3, 4, 5)113,148 are summarized. First and foremost, it is clear that the cumulated double bonds are not equal in length in these molecules. Rather, there is an alternating sequence of longer and shorter bonds that is reminiscent of a polyyne structure, especially in [3]cumulenes. This trend is more pronounced in the tetraarylcumulenes,70 due to conjugation of the phenyl rings to the cumulene chain. The terminal cumulenic double bond (α) is consistently the longest and the observed values are only slightly longer than that found for a standard double bond. Conversely, the next double bond (β) is usually the shortest of the cumulene chain (except for [7]tBuPh and [7]Mes), with values slightly longer than a typical triple bond. Thus, bond length alternation is observed in all cumulenes studied to date.
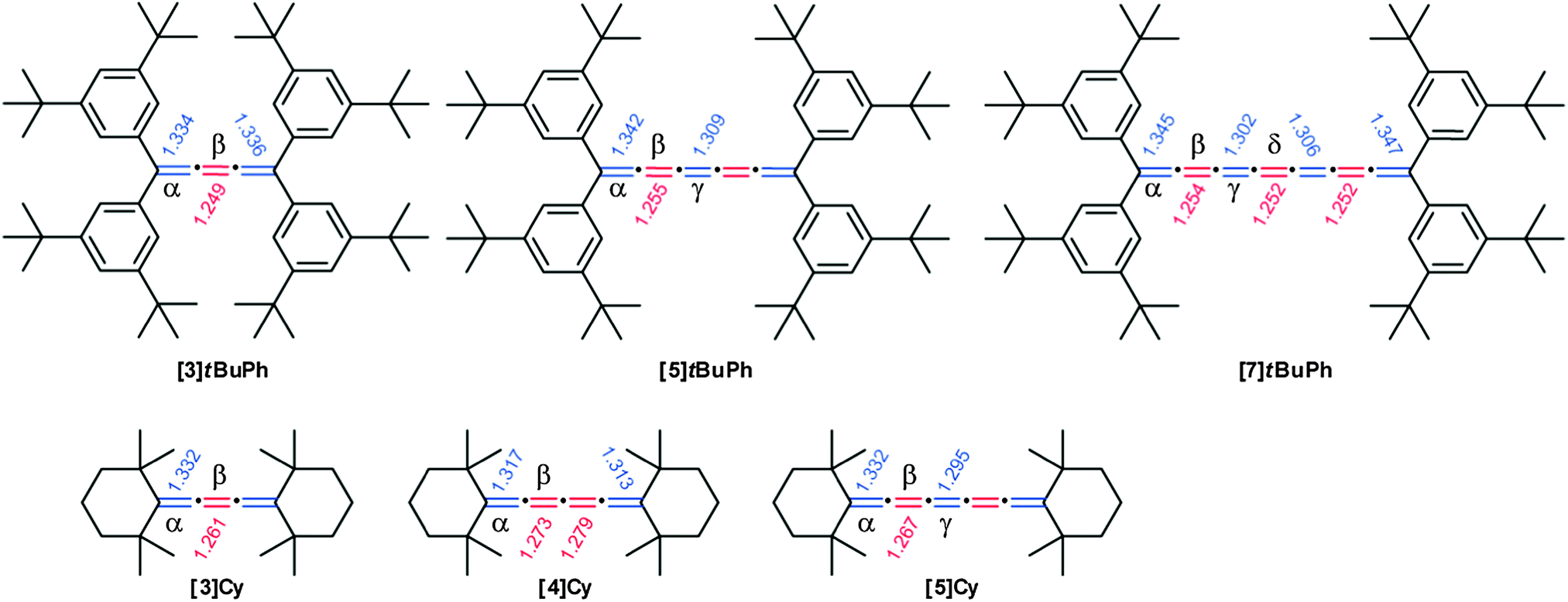 | ||
| Fig. 7 Chemical structures of [n]tBuPh and [n]Cy cumulenes including bond lengths in Å as determined by X-ray crystallography. | ||
In general, strongest bond length alternation can be expected from odd-numbered cumulenes in which a resonance contribution from the terminal groups is present, as discussed above for Fig. 3b. Conversely, reduced BLA is predicted for even-numbered tetraalkylcumulenes, as a result of the two orthogonal π-systems and restricted conjugation to the endgroups. A third situation also exists, however, in which restricted bond rotation would limit π-conjugation of terminal aryl rings to the cumulene core. In this case, the magnitude of the twist angles of the aryl endgroups should play a role in the observed BLA values. This is exactly the case for the [n]Mes series shown in Fig. 8,70 in which all four aryl groups are twisted significantly out of the plane of the cumulene skeleton, in the range of 45–52°.149 In comparison, only one aryl endgroup at each terminus of the [n]tBuPh cumulenes is appreciably twisted (44–55°), while the remaining two aryl groups are more coplanar (twist angles from 14–21°). Looking at bond lengths for the two series, the above hypothesis is confirmed, and the increased conjugation in the [n]tBuPh cumulenes results in reduced BLA for [n]Mes (see also Fig. 9).
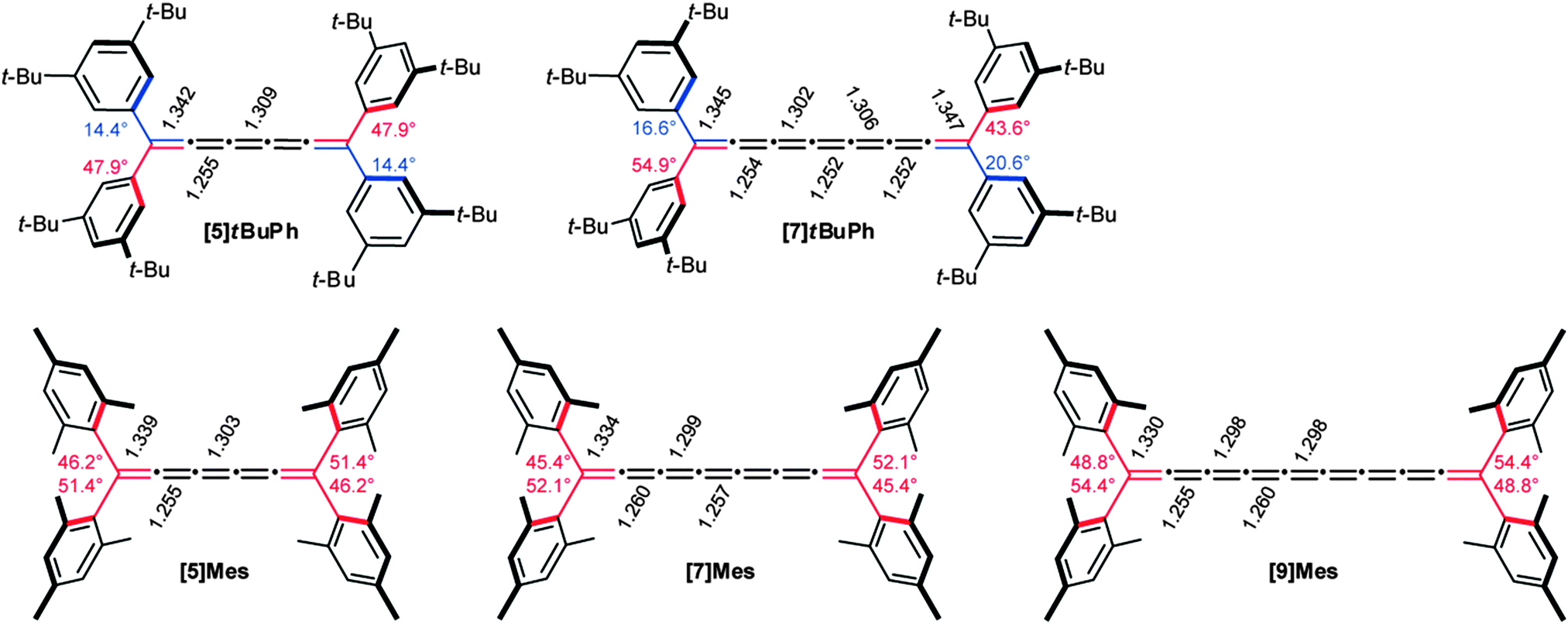 | ||
| Fig. 8 Twist angles of the aromatic ring relative to cumulenic framework of [5]- and [7]tBuPh (top) and [5]-, [7]-, and [9]Mes (bottom) as determined by X-ray crystallography. | ||
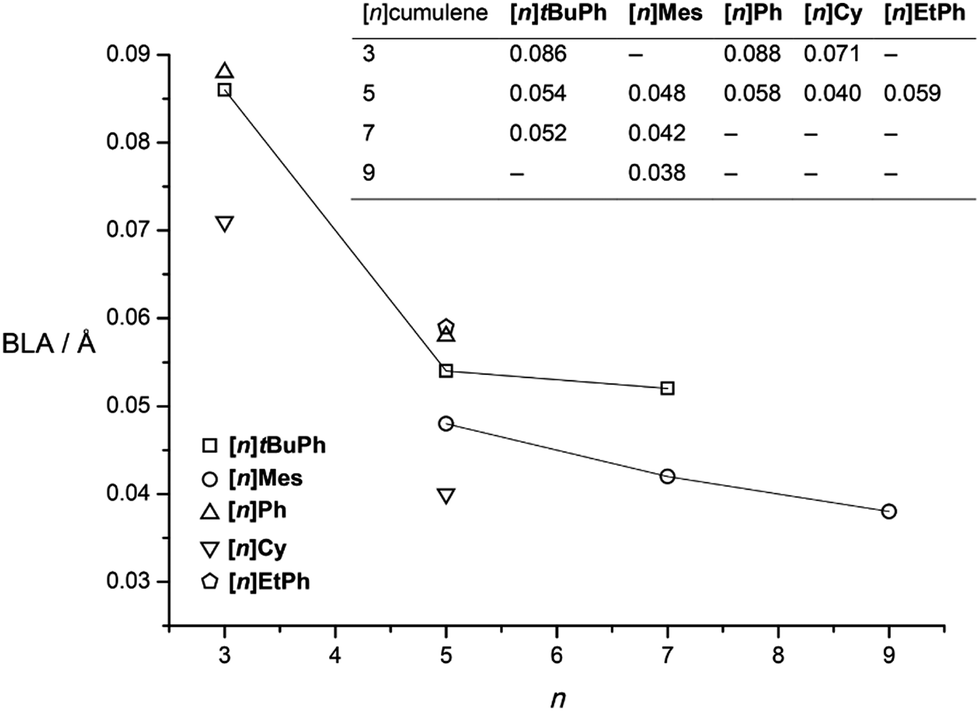 | ||
| Fig. 9 BLA values (insert) versus [n]cumulenic chain length, n (lines are only a guide for the eye). | ||
The analysis of data for [n]tBuPh cumulenes shows a steady decrease in BLA with increasing molecular length, ranging from 0.086 Å ([3]tBuPh) to 0.052 Å ([7]tBuPh, Fig. 9).70 The mesityl end-capped cumulenes show an analogous reduction in BLA versus length, from 0.048 Å ([5]Mes) to 0.038 Å ([9]Mes).70 In comparison to [n]tBuPh, however, BLA values for the [n]Mes derivatives are lower as discussed above. The X-ray data of other aryl substituted cumulenes are also included, and BLA values for [3]Ph,150[5]Ph,151 and [5]EtPh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 152 are also consistent with trends for the series [n]Mes and [n]tBuPh. In contrast to aryl substituted cumulenes, data for alkyl substituted cumulenes, [3]Cy and [5]Cy show the lowest BLA values known so far for cumulenes.113,148
152 are also consistent with trends for the series [n]Mes and [n]tBuPh. In contrast to aryl substituted cumulenes, data for alkyl substituted cumulenes, [3]Cy and [5]Cy show the lowest BLA values known so far for cumulenes.113,148
A plot of BLA values for [n]Mes and [n]tBuPh against the number of cumulated double bonds n suggests an asymptotic limit of 0.03–0.05 Å (Fig. 9). This prediction is somewhat higher than that from computational studies for the “parent” series of [n]cumulenes ([n]H). While computational results can differ depending on the method of analysis, the computational trend is clear that BLA ≤ 0.01 Å by the length of [9]H.146,147 The difference between experiment and theory likely arises from endgroup effects, but this hypothesis awaits confirmation.
5. Reactivity of longer [n]cumulenes (n ≥ 5)
The reactivity of [n]cumulenes with n = 2 and 3 has been reviewed by Diederich,153 Chauvin,88 and Ma.154 Due to the increasing instability in longer [n]cumulenes (n ≥ 5), the reactions of these molecules have been rare and are limited almost exclusively to reactions of [5]cumulenes.5.1 Miscellaneous reactions
Simple reactions of [5]cumulenes have been reported such as hydrogenations118,142 and partial hydrogenations155,156 of the cumulene core using H2/Rh/alumina and Al/Hg or the Lindlar catalyst, for example. The oxidation of a [5]cumulene via epoxidation has been described by Crandall and coworkers (Scheme 16).157 For example, the reaction of [5]tBu with m-CPBA gives a cyclopropanone intermediate, which goes on to give an allenic ester as the product. Epoxidation with dimethyldioxirane gives the cyclopropanone as a stable product, which can then be used to form [4]tBu through either thermal or photochemical loss of carbon monoxide. The [4]cumulene [4]tBu has also been subjected to oxidation with m-CPBA, and this reaction also gives the cyclopropanone product.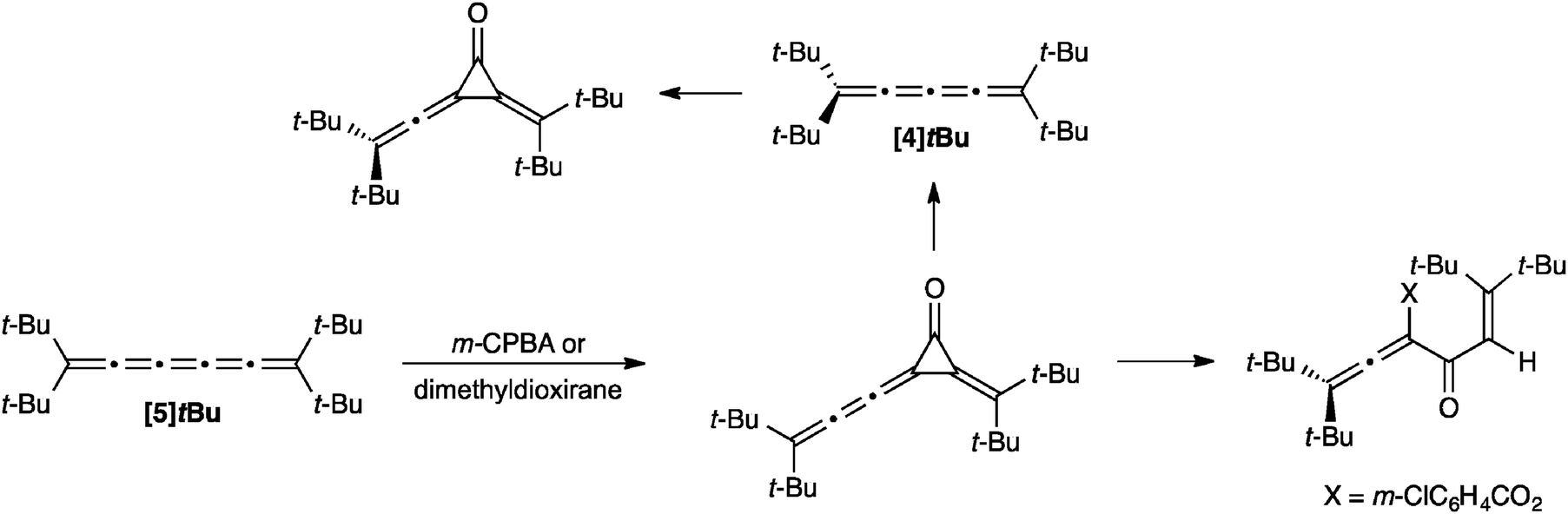 | ||
| Scheme 16 Oxidation products of [5]tBu. | ||
Theoretical predictions regarding the reactivity of cumulenes have been recently reported, particularly concerning oxygen sensitivity with respect to the carbon allotrope carbyne.158,159 Using density functional theory calculations, Moseler and coworkers report that reaction of O2 with the cumulene chain can cause cleavage, followed by repeated shortening of the chain through additional oxidation and loss of CO2.160
A variety of metal complexes can be formed through the reaction of an electrophilic metal with the π-rich skeleton of a [5]cumulene (Fig. 10).161 Complexes of [5]Ph with rhodium prefer bonding to the β-bond when triphenylphoshine is used as a ligand.162 The analogous system with i-Pr3P ligands shows rhodium bonded to the γ-bond at low temperature (the kinetic product), while complete conversion to thermodynamic product with Rh-complexation at the β-bond is achieved upon warming.163,164 Complexation of (Ph3P)2Pt to [5]Ph reveals similar behaviour, namely an equilibrium between the kinetic complex at the γ-bond and the thermodynamic complex at the β-bond.162 The reaction of [5]tBu with Fe2(CO)9 or Fe3(CO)12 gives a mixture of the mono- and dinuclear iron complexes,165 while Iyoda and coworkers have shown that under the appropriate conditions using non-sterically demanding endgroups, the reaction of either [5]H or [5]tBu with Fe3(CO)12 can be forced all the way to the tetranuclear iron complex.166,167 Finally, Suzuki and coworkers have shown that [5]cumulenes can be trapped with the low-valent zirconocene–bisphosphine complex [Cp2Zr(PMe3)2] to give the very strained zirconacyclopent-3-yne products.168–170
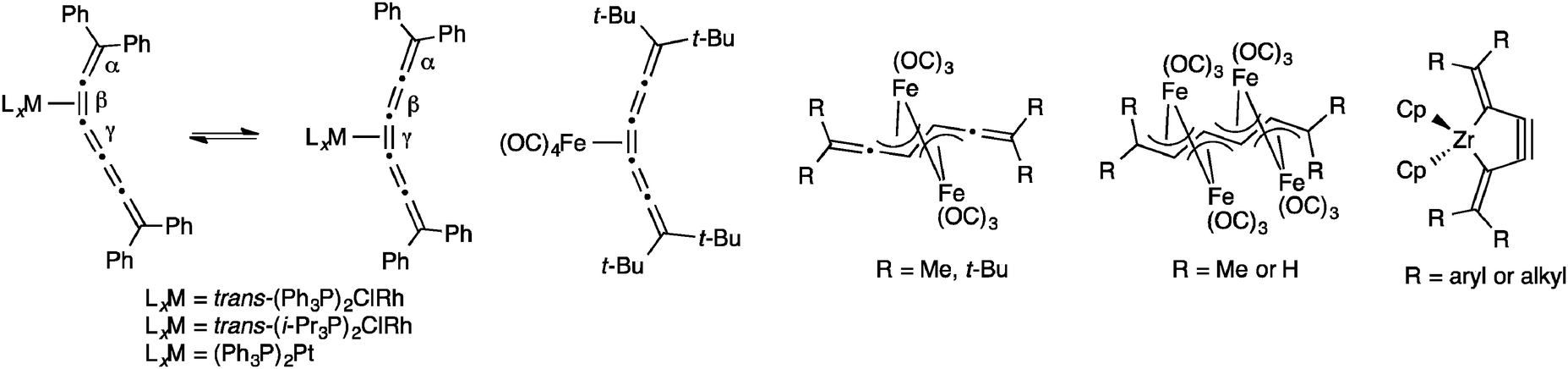 | ||
| Fig. 10 Selected metal complexes of [5]cumulenes. | ||
Finally, one example of the reactivity of a [6]cumulene has been reported.128 In this study, Bildstein and coworkers describe the hydrolysis of [6]Fc to give a heptatetraenone.
5.2 Cycloadditions with alkenes and alkynes
The most commonly investigated reactions for [5]cumulenes are cycloadditions, including cumulene oligomerization and either the addition of alkenes or alkynes. The presence of several double bonds in a cumulene framework offers several reaction sites, and products thus vary in symmetry and conjugation depending on the regiochemistry of the addition. For example, Scheme 17 describes the addition of alkenes to [5]cumulenes. On the one hand, it has been reported that tetrafluoroethylene (TFE) attacks the central γ-bond of [5]tBu to give a symmetric cyclobutane derivative.57,117 On the other hand, Bildstein and coworkers have shown that cycloaddition reaction of [5]Fc with either tetracyanoethylene (TCNE) or C60 (at a 6,6-ring junction) occurs at the β-bond, which affords the unsymmetrical cyclobutane derivatives.116 It is noteworthy that all three known examples in Scheme 17 utilize electron deficient alkenes, but nevertheless two different reactivity patterns are clearly operative, i.e., at the β- or γ-bond. Bildstein suggests that addition to the β-bond is the thermodynamic reaction pathway, while the alternative, addition to the γ-bond to give the symmetrical adduct, is described as the kinetic reaction pathway,171 similar to the situation described for metal complexes in Section 5.1.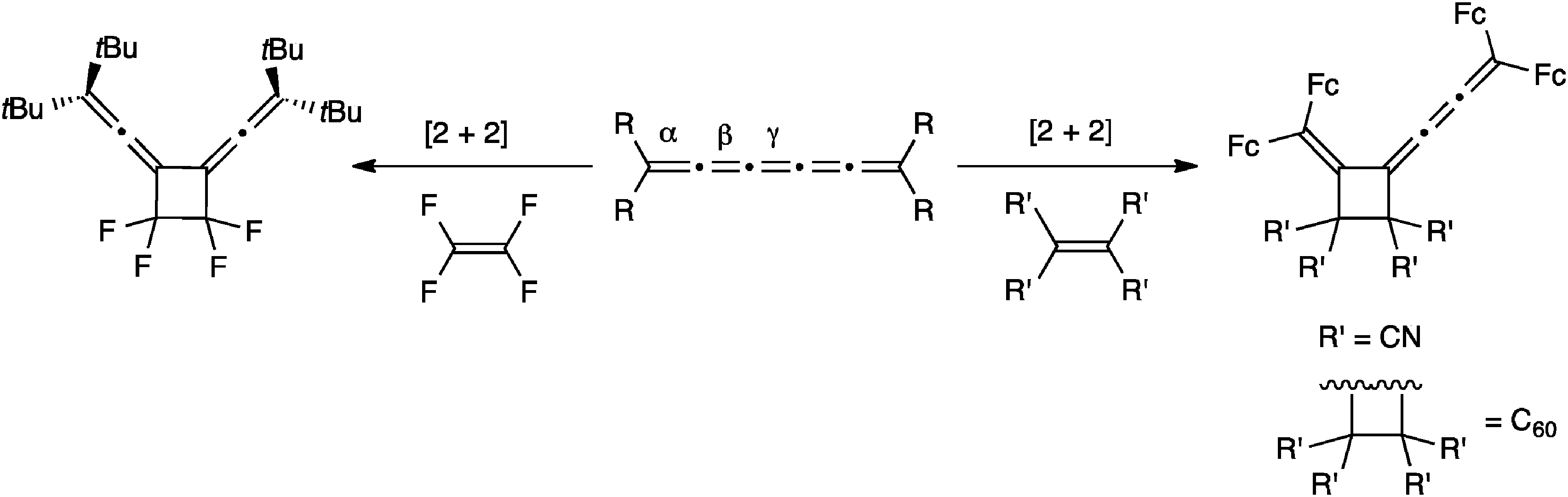 | ||
| Scheme 17 Cycloaddition of alkenes to a [5]cumulene (C60 = buckminsterfullerene). | ||
Hartzler, on the other hand, suggests that the [2+2] addition probably occurs by way of a thermally accessible diradical of the cumulene, and thus cycloaddition reactions at the central double bond might be expected, especially if the terminal carbon atoms are sterically hindered by substituents such as t-butyl (Fig. 11). This is consistent with the experimental results, which showed that addition of the highly reactive reagent tetrafluoroethylene occurs at the γ-bond.
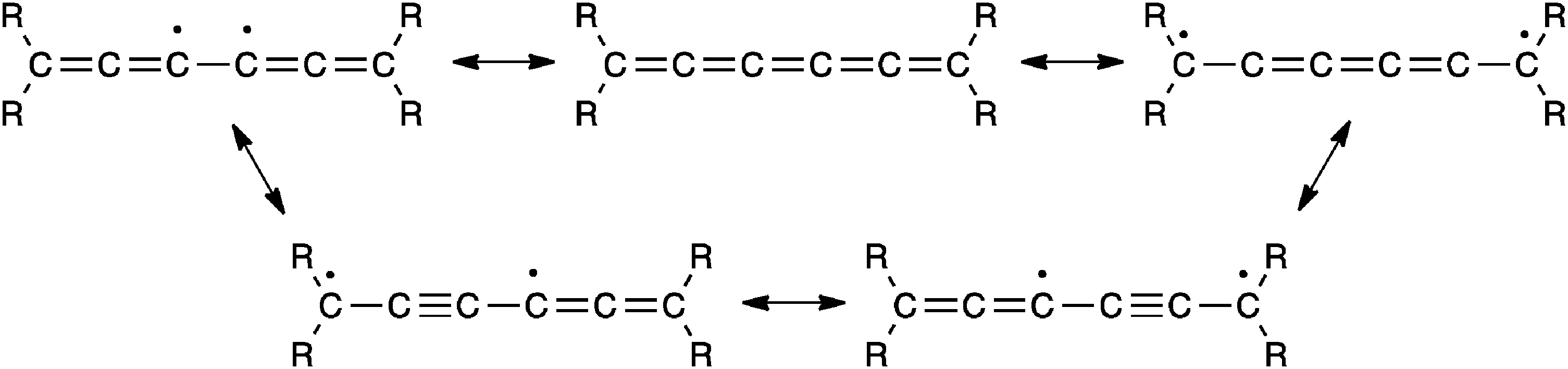 | ||
| Fig. 11 Diradical mesomeric structures as suggested by Hartzler for reactions of a [5]cumulene.57 | ||
Two examples of alkyne addition to [5]cumulenes have been reported, and both reactions use highly activated acetylenes to give a symmetrical [2+2] cyclobutene adduct via reaction at the central γ-bond (Scheme 18).57,116 The nature of the cumulene varied significantly in these two reactions, from R = t-Bu to R = Fc, but both authors suggest that the addition to the γ-bond is observed because of steric hindrance resulting from endgroups.
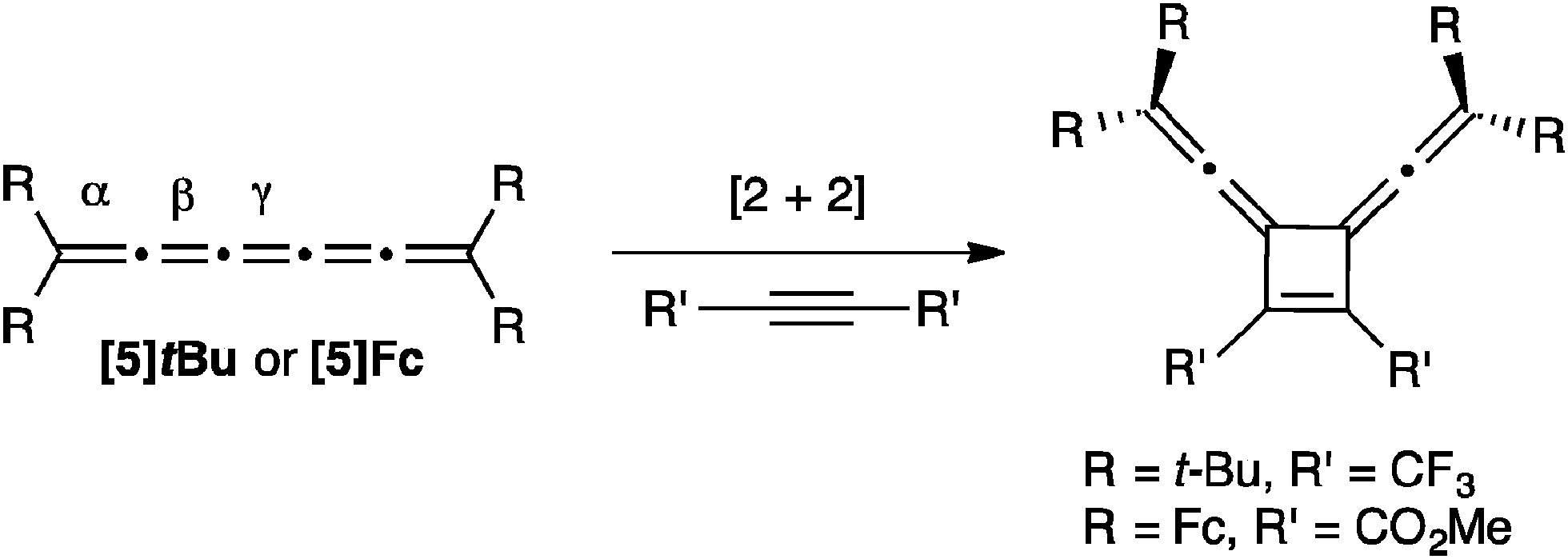 | ||
| Scheme 18 Addition of alkynes to [5]cumulenes. | ||
5.3 Di- and trimerizations
The reaction of [5]cumulenes often results in a formal cycloaddition between two cumulene molecules. Most commonly, such reactions proceed either thermally or via a Ni-catalyzed reaction (Scheme 19). Thermal dimerization is often observed for cumulenes with bulky alkyl substituents, giving a symmetrical vinylidene substituted [4]radialene 1, i.e., a cyclobutane ring that possesses four equally substituted exocyclic allene units.172,173 This was first demonstrated by Hartzler and coworkers for [5]tBu, i.e., when [5]tBu melts, the resulting liquid resolidifies with the formation of the radialene product.57 In a later report by Iyoda, it was suggested that the dimerization reaction occurs at the central, γ-double bond, due to the crisscross nature of the structure (transition state) that would be required for a thermal [2+2] reaction.127 It is interesting to ponder why the thermal reactions of tetraaryl[5]cumulenes do not seem to follow a similar pathway in the solid state, i.e., dimerization to give a radialene. It might be due to molecular structure and stronger BLA (vide supra), or perhaps steric factors based on the endgroups. Alternatively, it is also conceivable that favorable intermolecular stacking interactions of the aryl endgroups might prevent a crisscross orientation that would be necessary for a thermal [2+2] reaction.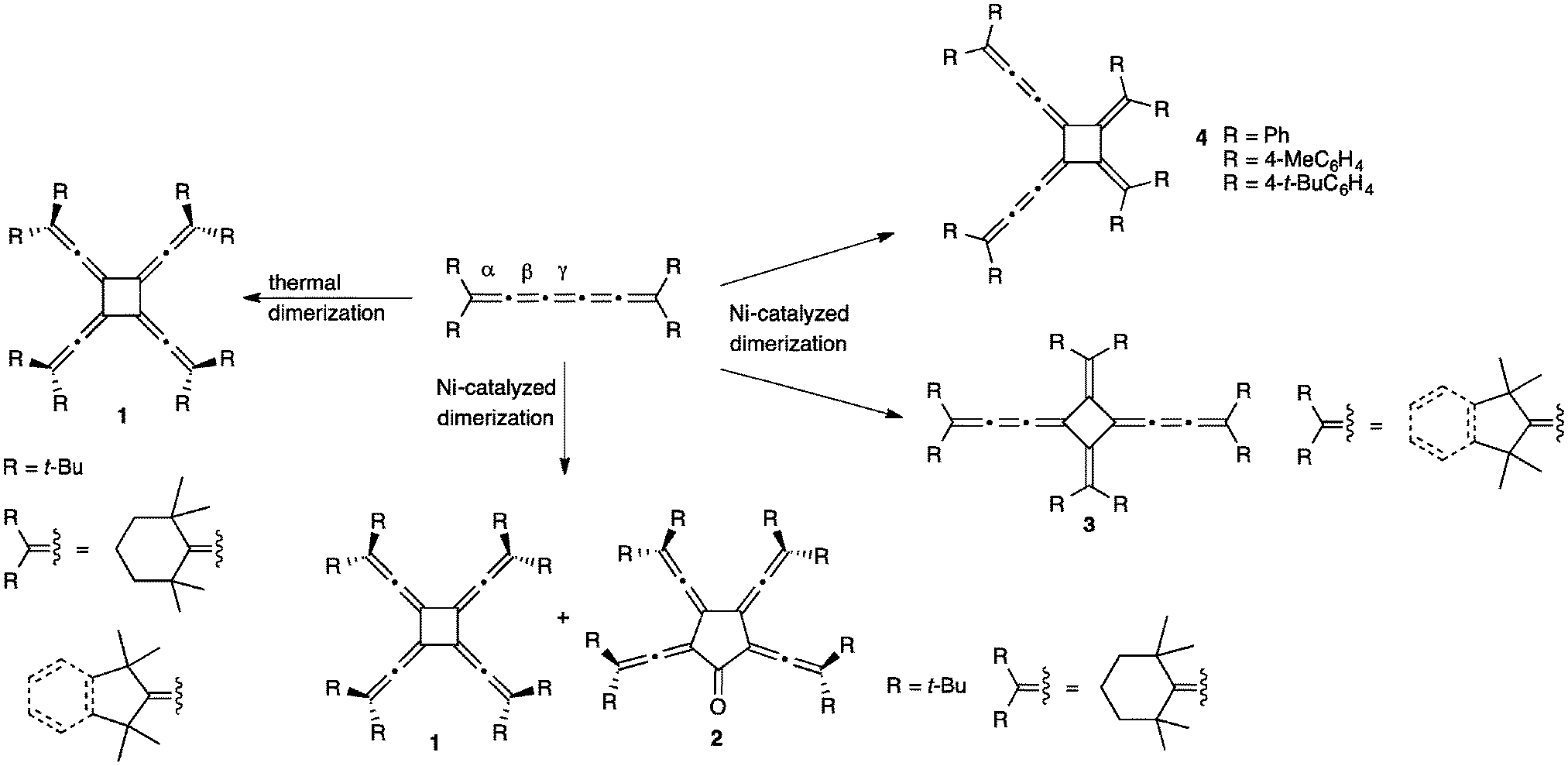 | ||
| Scheme 19 Dimerization reactions of [5]cumulenes. | ||
Aside from solid state reactions, radialenes 1 are also formed from alkyl substituted cumulenes in solution under Ni-catalysis, and these reactions also give [5]radialenones 2.127 With slightly less bulky alkyl endgroups, Ni-catalyzed head-to-tail dimerization at the β-bond results in the formation of [4]radialenes 3, while tetraaryl[5]cumulenes afford the deep blue head-to-head dimers 4. The authors suggest that the bulkiness of the terminal substituents in the [5]cumulenes controls the course of metal coordination, and leads to the selectivity observed in the oligomerization reactions.127
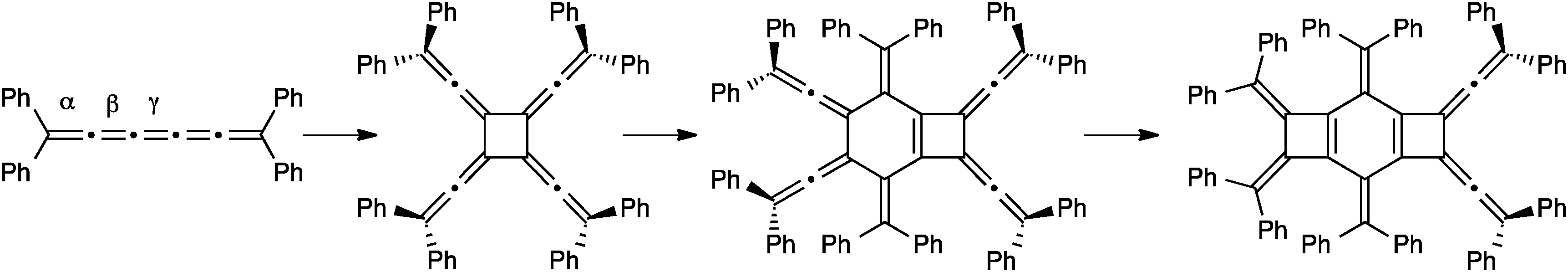
The final mode of dimerization reaction for [5]cumulenes has been documented independently by the studies of Stang174 and Scott,118 and later by Hopf and coworkers with the unsubstituted [5]H.175 In these three cases, the lack of sterically encumbered endgroups permits reaction at the α-bonds, with concomitant rearrangement of the cumulene framework to give a butadiyne moiety (Fig. 12). Scott has reported that the Cu(I) catalyzed cyclodimerization of [5]Me leads to the symmetrical 12-membered ring 5. Stang and coworkers have shown that no metal catalysis is necessary, and macrocycle 6 is isolated in a 31% yield as the only viable product. In the case of 6, a radical has been is suggested, which avoids the necessity of a symmetry forbidden [6+6] thermal cycloaddition. Finally, Hopf and coworkers report the formation of macrocycle 7, conceivably through dimerization of the parent system, [5]H, although the intermediate presence of [5]H has not been established.
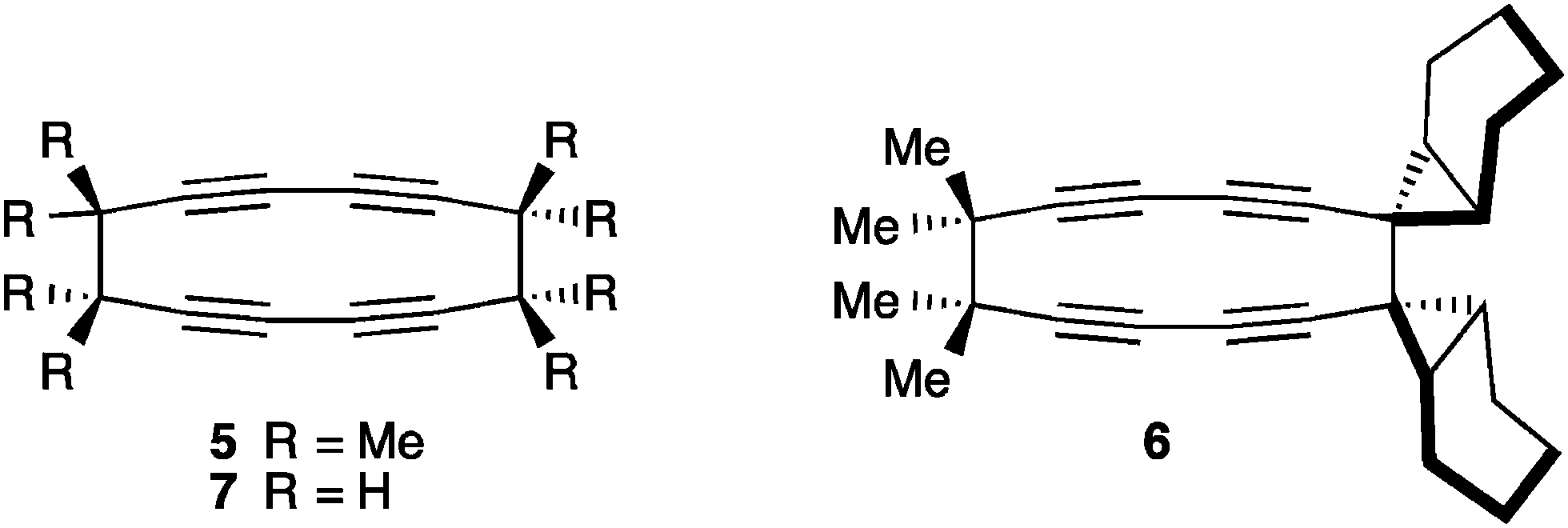 | ||
| Fig. 12 Dimerization reactions of [5]cumulenes. | ||
Similar to dimerization, [5]cumulenes have also been shown to formally undergo trimerization reactions, although in each of the two reported cases, dimerization precedes the formation of the trimer. The first example from Kawase et al. is outlined in Scheme 20 and forms a novel tricyclobutabenzene derivative from a precursor in which a [5]cumulene links two quinone rings.176 The authors suggest a mechanism in which the [5]cumulene is converted to a radical intermediate via oxidation. This intermediate then dimerizes to a cyclobutene-intermediate and subsequent addition of the third [5]cumulene unit gives a dicyclobutene intermediate. Finally, further oxidation and cyclization gives the tricyclobutabenzene product.
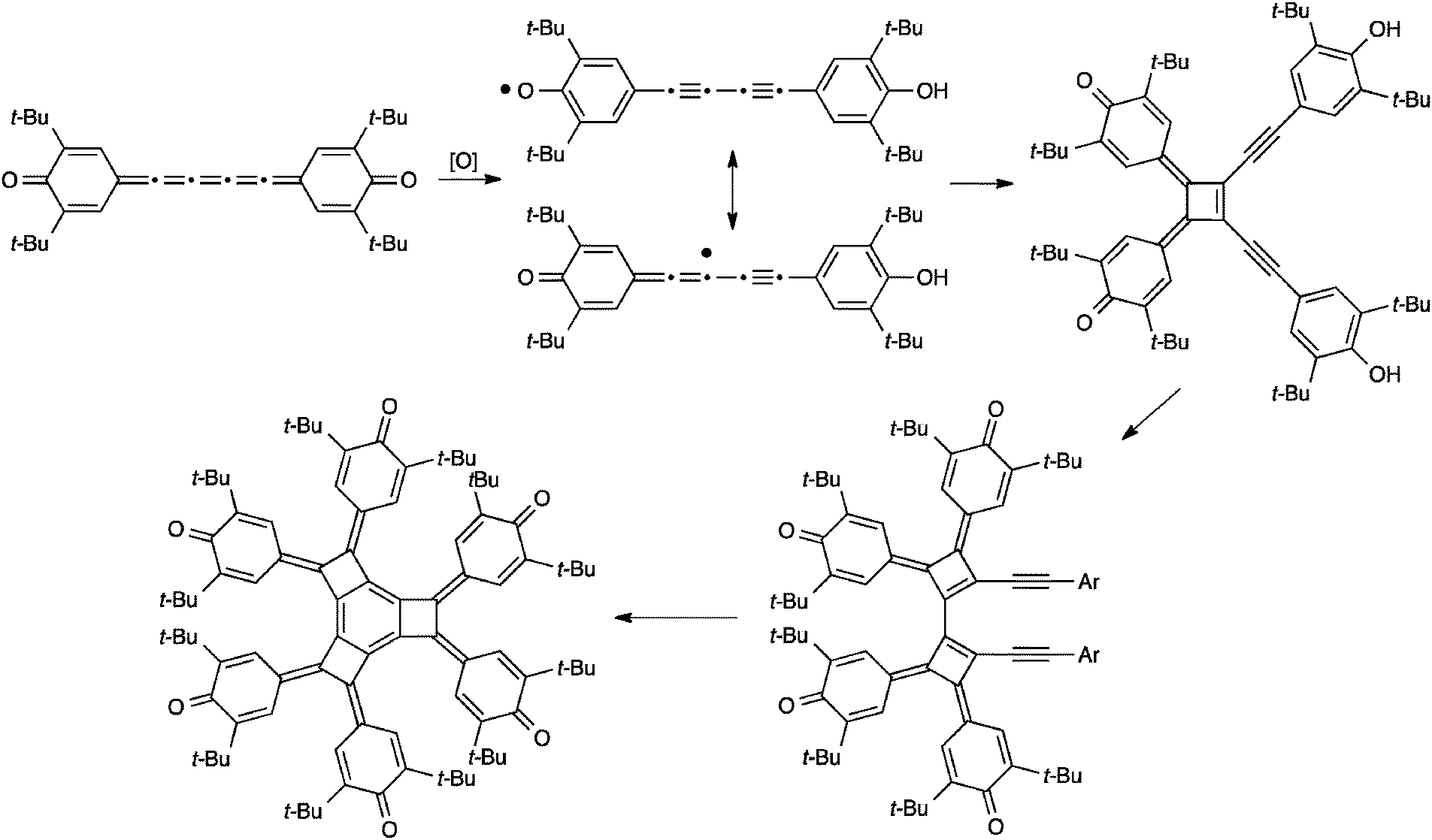 | ||
| Scheme 20 Trimerization reaction of a [5]cumulene. | ||
The second trimerization example describes the cyclotrimerization of [5]Ph to a tricyclodecadiene derivative as reported by Kawamura and coworkers (Scheme 21).177 The authors suggest that the reaction initiates with a solution-state dimerization of [5]Ph to give the symmetrical [4]radialene. A third equivalent of [5]Ph adds to this intermediate and gives the Diels–Alder adduct, which is ultimately converted to the product via an electrocyclization reaction. The reaction yield is quite reasonable (up to ca. 70%), as long as the concentration of [5]Ph is >13 mmol L−1. The product is rather stable, and subsequent Diels–Alder or electrocyclic reactions are not observed. This overall reaction is particularly unusual since it relies on the thermal dimerization of a tetraaryl[5]cumulene at the γ-bond, which is a reactivity pattern typically reserved for [5]cumulenes with sterically bulky alkyl groups.
 | ||
| Scheme 21 Trimerization reaction of [5]Ph. | ||
6. Conclusions and perspectives
The synthesis and study of long [n]cumulenes (n ≥ 5) is historically a rich area of organic chemistry that spans over 70 years. Much of what has been learned about these molecules has been reported in the early studies, while periodic advances have been recently described for both the synthesis and physical characteristics of long [n]cumulenes. There is a growing fundamental interest in long [n]cumulenes as model compounds for sp-carbon allotrope carbyne, and insight from the study of [n]cumulenes of defined length should allow researchers to compare and contrast the properties of the cumulenic and polyyne versions of carbyne. For example, the effect of structure and molecular length on the basic property of bond length alternation has recently been reported, and offers a platform for conducting further studies of physical structure and comparisons to theory. On the other hand, the unique π-electronic structure of cumulenes provides distinctive electronic and optical properties that suggest fascinating opportunities in molecular electronics and materials science. Key to exploiting this potential is the development of more stable cumulene structures, and synthetic methods to realize these targets.The interest in long [n]cumulenes is not, however, restricted to material applications. There is, without a doubt, a wealth of possible synthetic transformations that might exploit the reactive π-system offered by a cumulene, particularly in the realm of cycloaddition reactions. To date, however, there is insufficient data to outline a predictive scheme of what products might be expected, based on, for example, reactions that might be of either kinetic or thermodynamic control. In the studies of cumulene dimerization reactions, Professor Peter Stang offers perhaps the best assessment of the reactivity of [5]cumulenes, “What is not yet clear is what exactly controls the mode of cyclodimerizations and why seemingly similar cumulenes… result in such different cyclodimers.”174 The answer to this and many other questions await the future efforts of organic chemists.
Acknowledgements
The authors are grateful for support of this work from the Deutsche Forschungsgemeinschaft (DFG – SFB 953, “Synthetic Carbon Allotropes”), the DFG Cluster of Excellence “Engineering of Advanced Materials” at FAU, and “Solar Technologies go Hybrid” (an initiative of the Bavarian State Ministry for Science, Research and Art).References
- S. Eisler and R. R. Tykwinski, in Acetylene Chemistry: Chemistry, Biology, and Material Science, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, Germany, 2005, ch. 7 Search PubMed.
- W. A. Chalifoux and R. R. Tykwinski, C. R. Chim., 2009, 12, 341–358 CrossRef CAS.
- W. A. Chalifoux and R. R. Tykwinski, Chem. Rec., 2006, 6, 169–182 CrossRef CAS PubMed.
- W. A. Chalifoux and R. R. Tykwinski, Nat. Chem., 2010, 2, 967–971 CrossRef CAS PubMed.
- P. P. K. Smith and P. R. Buseck, Science, 1982, 216, 984–986 CAS.
- For an entertaining opinion on the subject of carbyne, see the comments by Sir Harold Kroto: http://www.rsc.org/chemistryworld/Issues/2010/November/CarbyneOtherMythsAboutCarbon.asp.
- H. O. Pierson, Handbook of Carbon, Graphite, Diamond and Fullerenes – Properties, Processing and Applications, William Andrew Publishing/Noyes, New Jersey, 1993 Search PubMed.
- E. H. L. Falcao and F. Wudl, J. Chem. Technol. Biotechnol., 2007, 82, 524–531 CrossRef CAS.
- S. Kim, Angew. Chem., Int. Ed., 2009, 48, 7740–7743 ( Angew. Chem. , 2009 , 121 , 7876–7879 ) CrossRef CAS PubMed.
- F. Cataldo, Polym. Int., 1997, 44, 191–200 CrossRef CAS.
- Y. P. Kudryavtsev, S. Evsyukoc, M. Gusevca, C. Babaev and C. Khvistov, in Chemistry and Physics of Carbon, ed. P. A. Thrower, Marcel Dekker, New York, 1997, pp. 1–99, vol. 25 Search PubMed.
- R. J. Lagow, J. J. Kampa, H.-C. Wei, S. L. Battle, J. W. Genge, D. A. Laude, C. J. Harper, R. Bau, R. C. Stevens, J. F. Haw and E. Munson, Science, 1995, 267, 362–367 CrossRef CAS PubMed.
- Y. Tobe and T. Wakabayashi, in Acetylene Chemistry: Chemistry, Biology, and Material Science, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, Germany, 2005, ch. 9 Search PubMed.
- Polyynes: Synthesis, Properties, and Applications, ed. F. Cataldo, Taylor & Francis, Boca Raton, FL, 2006 Search PubMed.
- A. G. Whittaker, E. J. Watts, R. S. Lewis and E. Anders, Science, 1980, 209, 1512–1514 CAS.
- R. Hayatsu, R. G. Scott, M. H. Studier, R. S. Lewis and E. Anders, Science, 1980, 209, 1515–1518 CAS.
- A. Webster, Mon. Not. R. Astron. Soc., 1980, 192, 7–9 CrossRef.
- A. El Goresy and G. Donnay, Science, 1968, 161, 363–364 CAS.
- A. L. K. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 ( Angew. Chem. , 2006 , 118 , 1050–1073 ) CrossRef PubMed.
- Y. Pan, T. L. Lowary and R. R. Tykwinski, Can. J. Chem., 2009, 87, 1565–1582 CrossRef CAS.
- B. W. Gung, C. R. Chim., 2009, 12, 489–505 CrossRef CAS.
- M. J. Wesolowski, S. Kuzmin, B. Wales, R. Karimi, A. A. Zaidi, Z. Leonenko, J. H. Sanderson and W. W. Duley, Carbon, 2011, 49, 625–630 CrossRef CAS.
- Y. Sato, T. Kodama, H. Shiromaru, J. H. Sanderson, T. Fujino, Y. Wada, T. Wakabayashi and Y. Achiba, Carbon, 2010, 48, 1673–1676 CrossRef CAS.
- A. Hu, M. Rybachuk, Q.-B. Lu and W. W. Duley, Appl. Phys. Lett., 2007, 91, 131906 CrossRef.
- L. Ravagnan, P. Piseri, M. Bruzzi, S. Miglio, G. Bongiorno, A. Baserga, C. S. Casari, A. Li Bassi, C. Lenardi, Y. Yamaguchi, T. Wakabayashi, C. E. Bottani and P. Milani, Phys. Rev. Lett., 2007, 98, 216103 CrossRef CAS PubMed.
- Carbyne and Carbynoid Structures, ed. R. B. Heimann, S. E. Evsyukov and L. Kavan, Kluwar Academic Press, London, 1999 Search PubMed.
- C. H. Hendon, D. Tiana, A. T. Murray, D. R. Carbery and A. Walsh, Chem. Sci., 2013, 4, 4278–4284 RSC.
- C. Motta, M. Cazzaniga, A. Bordoni and K. Gaál-Nagy, arXiv:0902.2573 [cond-mat.mtrl-sci].
- D. Usanmaz and G. P. Srivastava, J. Appl. Phys., 2013, 113, 193704 CrossRef.
- U. Mölder, P. Burk and I. A. Koppel, THEOCHEM, 2004, 712, 81–89 CrossRef.
- O. Yu. Podkopaeva and Yu. V. Chizhov, J. Struct. Chem., 2006, 47, 420–426 CrossRef CAS.
- I. Alkorta and J. Elguero, Struct. Chem., 2005, 16, 77–79 CrossRef CAS.
- M. Liu, V. I. Artyukhov, H. Lee, F. Xu and B. I. Yakobson, ACS Nano, 2013, 7, 10075–10082 CrossRef CAS PubMed.
- S. Eisler, A. D. Slepkov, E. Elliott, T. Luu, R. McDonald, F. A. Hegmann and R. R. Tykwinski, J. Am. Chem. Soc., 2005, 127, 2666–2676 CrossRef CAS PubMed.
- A. Lucotti, M. Tommasini, D. Fazzi, M. Del Zoppo, W. A. Chalifoux, R. R. Tykwinski and G. Zerbi, J. Raman Spectrosc., 2012, 43, 1293–1298 CrossRef CAS.
- I. Kminek, J. Klimovic and P. N. Prasad, Chem. Mater., 1993, 5, 357–360 CrossRef CAS.
- S. Ermer, S. Lovejoy, D. Leung, J. Altman, K. Aron, R. Spitzer and G. Hansen, Proc. SPIE, 1990, 1337, 89–98 CrossRef CAS.
- S. Ballmann, W. Hieringer, D. Secker, Q. L. Zheng, J. A. Gladysz, A. Görling and H. B. Weber, ChemPhysChem, 2010, 11, 2256–2260 CrossRef CAS PubMed.
- P. Moreno-Garcia, M. Gulcur, D. Zsolt Manrique, T. Pope, W. Hong, V. Kaliginedi, C. Huang, A. S. Batsanov, M. R. Bryce, C. Lambert and T. Wandlowski, J. Am. Chem. Soc., 2013, 135, 12228–12240 CrossRef CAS PubMed.
- N. D. Lang and Ph. Avouris, Phys. Rev. Lett., 1998, 81, 3515–3518 CrossRef CAS.
- F. Börrnert, C. Börrnert, S. Gorantla, X. Liu, A. Bachmatiuk, J.-O. Joswig, F. R. Wagner, F. Schäffel, J. H. Warner, R. Schönfelder, B. Rellinghaus, T. Gemming, J. Thomas, M. Knupfer, B. Büchner and M. H. Rümmeli, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085439 CrossRef.
- A. K. Nair, S. W. Cranford and M. J. Buehler, EPL, 2011, 95, 16002 CrossRef.
- O. Cretu, A. R. Botello-Mendez, I. Janowska, C. Pham-Huu, J.-C. Charlier and F. Banhart, Nano Lett., 2013, 13, 3487–3493 CrossRef CAS PubMed.
- B. Akdim and R. Pachter, ACS Nano, 2011, 5, 1769–1774 CrossRef CAS PubMed.
- J. Prasongkit, A. Grigoriev and R. Ahuja, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 155434 CrossRef.
- C. Ataca and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 235417 CrossRef.
- L. Ravagnan, N. Manini, E. Cinquanta, G. Onida, D. Sangalli, C. Motta, M. Devetta, A. Bordoni, P. Piseri and P. Milani, Phys. Rev. Lett., 2009, 102, 245502 CrossRef PubMed.
- S. Sitha, K. Bhanuprakash and B. M. Choudary, Synth. Met., 2005, 148, 227–235 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 10478–10486 CrossRef CAS PubMed.
- Y. H. Hu, J. Phys. Chem. C, 2011, 115, 1843–1850 CAS.
- S. Cahangirov, M. Topsakal and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195444 CrossRef.
- Y. Zhang, Y. Su, L. Wang, E. S.-W. Kong, X. Chen and Y. Zhang, Nanoscale Res. Lett., 2011, 6, 577 CrossRef PubMed.
- I. E. Castelli, P. Salvestrini and N. Manini, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 214110 CrossRef.
- A. Milani, A. Lucotti, V. Russo, M. Tommasini, F. Cataldo, A. Li Bassi and C. S. Casari, J. Phys. Chem. C, 2011, 115, 12836–12843 CAS.
- M. Weimer, W. Hieringer, F. Della Sala and A. Görling, Chem. Phys., 2005, 309, 77–87 CrossRef CAS.
- A. Auffrant, B. Jaun, P. D. Jarowski, K. N. Houk and F. Diederich, Chem.–Eur. J., 2004, 10, 2906–2911 CrossRef CAS PubMed.
- H. D. Hartzler, J. Am. Chem. Soc., 1971, 93, 4527–4531 CrossRef CAS.
- M. M. Yildizhan, D. Fazzi, A. Milani, L. Brambilla, M. Del Zoppo, W. A. Chalifoux, R. R. Tykwinski and G. Zerbi, J. Chem. Phys., 2011, 134, 124512 CrossRef PubMed.
- S. Yang and M. Kertesz, J. Phys. Chem. A, 2008, 112, 146–151 CrossRef CAS PubMed.
- M. Kertesz, J. Koller and A. Ažman, J. Chem. Phys., 1978, 68, 2779–2782 CrossRef CAS.
- F. Bohlmann, Angew. Chem., 1953, 65, 385–389 CrossRef CAS.
- T. R. Johnson and D. R. M. Walton, Tetrahedron, 1972, 28, 5221–5236 CrossRef CAS.
- R. Eastmond, T. R. Johnson and D. R. M. Walton, Tetrahedron, 1972, 28, 4601–4616 CrossRef CAS.
- E. R. H. Jones, Proc. Chem. Soc., 1960, 199–210 CAS.
- Q. Zheng, J. C. Bohling, T. B. Peters, A. C. Frisch, F. Hampel and J. A. Gladysz, Chem.–Eur. J., 2006, 12, 6486–6505 CrossRef CAS PubMed.
- M. I. Bruce, B. K. Nocholson and N. N. Zaitseva, C. R. Chim., 2009, 12, 1280–1286 CrossRef CAS.
- T. Gibtner, F. Hampel, J.-P. Gisselbrecht and A. Hirsch, Chem.–Eur. J., 2002, 8, 408–432 CrossRef CAS.
- W. A. Chalifoux, R. McDonald, M. J. Ferguson and R. R. Tykwinski, Angew. Chem., Int. Ed., 2009, 48, 7915–7919 ( Angew. Chem. , 2009 , 121 , 8056–8060 ) CrossRef CAS PubMed.
- X. Gu, R. I. Kaiser and A. M. Mebel, ChemPhysChem, 2008, 9, 350–369 CrossRef CAS PubMed.
- J. A. Januszewski, D. Wendinger, C. D. Methfessel, F. Hampel and R. R. Tykwinski, Angew. Chem., Int. Ed., 2013, 52, 1817–1821 ( Angew. Chem. , 2013 , 125 , 1862–1867 ) CrossRef CAS PubMed.
- F. Bohlmann and K. Kieslich, Chem. Ber., 1954, 87, 1363–1372 CrossRef CAS.
- F. Bohlmann and K. Kieslich, Abh. Braunschw. Wiss. Ges., 1957, 9, 147–166 Search PubMed.
- W. Ried, W. Schlegelmilch and S. Piesch, Chem. Ber., 1963, 96, 1221–1228 CrossRef CAS.
- R. Kuhn and K. Wallenfels, Chem. Ber., 1938, 71, 783–790 CrossRef.
- F. Bohlmann, Chem. Ber., 1951, 84, 785–794 CrossRef CAS.
- R. Eastmond, T. R. Johnson and D. R. M. Walton, J. Organomet. Chem., 1973, 50, 87–92 CrossRef CAS.
- E. R. H. Jones, M. C. Whiting, J. B. Armitage, C. L. Cook and N. Entwistle, Nature, 1951, 168, 900–903 CrossRef CAS.
- W. Hunsmann, Chem. Ber., 1950, 83, 213–217 CrossRef CAS.
- H. H. Schlubach and V. Franzen, Justus Liebigs Ann. Chem., 1951, 573, 105–109 CrossRef CAS.
- H. H. Schlubach and V. Wolf, Justus Liebigs Ann. Chem., 1950, 568, 141–159 CrossRef CAS.
- C. L. Cook, E. R. H. Jones and M. C. Whiting, J. Chem. Soc., 1952, 2883–2891 RSC.
- M. Ogasawara, in Science of synthesis. Cumulenes and allenes, ed. D. Bellus, Georg Thieme Verlag, 2008, vol. 44.1, pp. 9–70 Search PubMed.
- P. Aguirre-Etcheverry and D. O'Hare, Chem. Rev., 2010, 110, 4839–4864 CrossRef CAS PubMed.
- V. Cadierno and J. Gimeno, Chem. Rev., 2009, 109, 3512–3560 CrossRef CAS PubMed.
- C. M. Che, C. M. Ho and J. S. Huang, Coord. Chem. Rev., 2007, 251, 2145–2166 CrossRef CAS.
- C. Coletti, A. Marrone and N. Re, Acc. Chem. Res., 2012, 45, 139–149 CrossRef CAS PubMed.
- D. Touchard and P. H. Dixneuf, Coord. Chem. Rev., 1998, 178, 409–429 CrossRef.
- L. Leroyer, V. Maraval and R. Chauvin, Chem. Rev., 2012, 112, 1310–1343 CrossRef CAS PubMed.
- P. Cadiot, W. Chodkiewicz and J. Rauss-Godineau, Bull. Soc. Chim. Fr., 1961, 2176–2193 CAS.
- C. Bruneau and J.-L. Renaud, in Compr. Org. Funct. Group Transform. II, ed. A. R. Katritzky and R. J. K. Taylor, Elsevier, Oxford, 2005, vol. 1.20, pp. 1019–1081 Search PubMed.
- P. J. Stang, Chem. Rev., 1978, 78, 383–405 CrossRef CAS.
- J.-D. van Loon, P. Seiler and F. Diederich, Angew. Chem., Int. Ed. Engl., 1993, 32, 1187–1189 ( Angew. Chem. , 1993 , 105 , 1235–1238 ) CrossRef.
- A. Auffrant, F. Diederich, C. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 2004, 87, 3085–3105 CrossRef CAS.
- T. Kunieda and T. Takizawa, Chem. Pharm. Bull., 1977, 25, 1809–1810 CrossRef CAS.
- M. Iyoda, H. Otani and M. Oda, J. Am. Chem. Soc., 1986, 108, 5371–5372 CrossRef CAS.
- M. Iyoda, N. Nakamura, M. Todaka, S. Ohtsu, K. Hara, Y. Kuwatani, M. Yoshida, H. Matsuyama, M. Sugita, H. Tachibana and H. Inoue, Tetrahedron Lett., 2000, 41, 7059–7064 CrossRef CAS.
- G. Köbrich, H. Heinemann and W. Zündorf, Tetrahedron, 1967, 23, 565–584 CrossRef.
- K. Komatsu, H. Kamo, R. Tsuji and K. Takeuchi, J. Org. Chem., 1993, 58, 3219–3221 CrossRef CAS.
- H. Kurata, S. Muro, T. Enomoto, T. Kawase and M. Oda, Bull. Chem. Soc. Jpn., 2007, 80, 349–357 CrossRef CAS.
- P. J. Stang and T. E. Fisk, J. Am. Chem. Soc., 1980, 102, 6813–6816 CrossRef CAS.
- P. J. Stang, Acc. Chem. Res., 1982, 15, 348–354 CrossRef CAS.
- K. Brand and D. Krücke-Amelung, Chem. Ber., 1939, 72, 1036–1047 CrossRef.
- K. Brand and A. Busse-Sundermann, Chem. Ber., 1950, 83, 119–128 CrossRef CAS.
- G. Dupont, Ann. Chim. Phys., 1913, 8, 485–587 Search PubMed.
- E. Bergmann, H. Hoffmann and D. Winter, Chem. Ber., 1933, 66, 46–54 CrossRef.
- B. Bildstein, Coord. Chem. Rev., 2000, 206–207, 369–394 CrossRef CAS.
- M. Iyoda, K. Nishioka, M. Nose, S. Tanaka and M. Oda, Chem. Lett., 1984, 131–134 CrossRef CAS.
- J. Salkind and A. Kruglow, Chem. Ber., 1928, 61, 2306–2312 CrossRef.
- B. Bildstein, M. Schweiger, H. Kopacka, K.-H. Ongania and K. Wurst, Organometallics, 1998, 17, 2414–2424 CrossRef CAS.
- M. Nakagawa, K. Shingu and K. Naemura, Tetrahedron Lett., 1961, 22, 802–806 CrossRef.
- R. Kuhn, H. Fischer and H. Fischer, Chem. Ber., 1964, 97, 1760–1766 CrossRef CAS.
- G. Karich and J. C. Jochims, Chem. Ber., 1977, 110, 2680–2694 CrossRef CAS.
- H. Irngartinger and W. Götzmann, Angew. Chem., Int. Ed. Engl., 1986, 25, 340–342 ( Angew. Chem. , 1986 , 98 , 359–361 ) CrossRef.
- W. J. le Noble, S. Basak and S. Srivastava, J. Am. Chem. Soc., 1981, 103, 4638–4639 CrossRef CAS.
- S. Basak, S. Srivastava and W. J. le Noble, J. Org. Chem., 1987, 52, 5095–5099 CrossRef CAS.
- B. Bildstein, M. Schweiger, H. Angleitner, H. Kopacka, K. Wurst, K.-H. Ongania, M. Fontani and P. Zanello, Organometallics, 1999, 18, 4286–4295 CrossRef CAS.
- H. D. Hartzler, J. Am. Chem. Soc., 1966, 88, 3155–3156 CrossRef CAS.
- L. T. Scott and G. J. DeCicco, J. Org. Chem., 1980, 45, 4055–4056 CrossRef CAS.
- P. Cadiot and A. Willemart, Bull. Soc. Chim. Fr., 1951, 100–104 CAS.
- P. Cadiot, Ann. Chim., 1956, 13, 214–272 Search PubMed.
- H. Kollmar and H. Fischer, Tetrahedron Lett., 1968, 40, 4291–4294 CrossRef.
- P. J. Stang and M. Ladika, J. Am. Chem. Soc., 1980, 102, 5407–5409 CrossRef.
- P. J. Stang and M. Ladika, J. Am. Chem. Soc., 1981, 103, 6437–6443 CrossRef CAS.
- L. Skattebol, Tetrahedron Lett., 1965, 26, 2175–2179 CrossRef.
- R. Kuhn and K. Wallenfels, Chem. Ber., 1938, 71, 1510–1512 CrossRef.
- R. Kuhn and H. Krauch, Chem. Ber., 1955, 88, 309–315 CrossRef CAS.
- Y. Kuwatani, G. Yamamoto, M. Oda and M. Iyoda, Bull. Chem. Soc. Jpn., 2005, 78, 2188–2208 CrossRef CAS.
- B. Bildstein, W. Skibar, M. Schweiger, H. Kopacka and K. Wurst, J. Organomet. Chem., 2001, 622, 135–142 CrossRef CAS.
- R. Kuhn and H. Zahn, Chem. Ber., 1951, 84, 566–570 CrossRef CAS.
- H. Fischer and W. D. Hell, Angew. Chem., Int. Ed. Engl., 1967, 6, 954–955 ( Angew. Chem. , 1967 , 79 , 931–932 ) CrossRef CAS.
- J. Rauss-Godineau, W. Chodkiewicz and P. Cadiot, Bull. Soc. Chim. Fr., 1966, 9, 2877–2884 Search PubMed.
- E. W. Colvin and B. J. Hamill, J. Chem. Soc., Perkin Trans. 1, 1977, 865–869 Search PubMed.
- J. Kendall, R. McDonald, M. J. Ferguson and R. R. Tykwinski, Org. Lett., 2008, 10, 2163–2166 CrossRef CAS PubMed.
- P. Bichler, W. A. Chalifoux, S. Eisler, A. L. K. Shi Shun, E. T. Chernick and R. R. Tykwinski, Org. Lett., 2009, 11, 519–522 CrossRef CAS PubMed.
- H. Fischer, in The Chemistry of Alkenes, ed. S. Patai, John Wiley & Sons, New York, 1964, pp. 1025–1159 Search PubMed.
- R. R. Tykwinski, W. Chalifoux, S. Eisler, A. Lucotti, M. Tommasini, D. Fazzi, M. Del Zoppo and G. Zerbi, Pure Appl. Chem., 2010, 82, 891–904 CrossRef CAS.
- C. Bubeck, in Electronic Materials: The Oligomer Approach, ed. K. Müllen and G. Wegner, Wiley-VCH, Weinheim, New-York, 1998, ch. 8 Search PubMed.
- G. N. Lewis and M. Calvin, Chem. Rev., 1939, 25, 273–328 CrossRef CAS.
- H. Meier, U. Stalmach and H. Kolshorn, Acta Polym., 1997, 48, 379–384 CrossRef CAS.
- R. Kuhn and G. Platzer, Ber. Dtsch. Chem. Ges. B, 1940, 73, 1410–1417 Search PubMed.
- T. Negi, T. Kaneda, H. Mizuno, T. Toyoda, Y. Sakata and S. Misumi, Bull. Chem. Soc. Jpn., 1974, 47, 2398–2405 CrossRef CAS.
- L. T. Scott and G. J. DeCicco, Tetrahedron Lett., 1976, 31, 2663–2666 CrossRef.
- K. W. Hausser, R. Kuhn and A. Smakula, Z. Phys. Chem., Abt. B, 1935, 29, 384–390 Search PubMed.
- H. H. Schlubach and V. Franzen, Liebigs Ann., 1951, 573, 110–115 CrossRef CAS.
- K. Knoll and R. R. Schrock, J. Am. Chem. Soc., 1989, 111, 7989–8004 CrossRef CAS.
- F. Innocenti, A. Milani and C. Castiglioni, J. Raman Spectrosc., 2010, 41, 226–236 CAS.
- D. Nori-Shargh, F. Deyhimi, J. E. Boggs, S. Jameh-Bozorghi and R. Shakibazadeh, J. Phys. Org. Chem., 2007, 20, 355–364 CrossRef CAS.
- H. Irngartinger and H.-U. Jäger, Angew. Chem., Int. Ed. Engl., 1976, 15, 562–563 ( Angew. Chem. , 1976 , 88 , 615–616 ) CrossRef.
- Twist angles were calculated as the difference between planes generated from (a) the six carbons of the aryl ring and (b) the carbons of the cumulene skeleton, along with the four ipso-carbons of the aryl rings.
- Z. Berkovitch-Yellin and L. Leiserowitz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 3657–3669 CrossRef.
- The structure of [5]Ph was reported without values for bond length and angles, see: M. M. Woolfson, Acta Crystallogr., 1953, 6, 838–841 CrossRef CAS . Bond lengths from Januszewski, unpublished work.
- N. Suzuki, D. Hashizume and T. Chihara, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3436 CAS.
- P. Rivera-Fuentes and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 ( Angew. Chem. , 2012 , 124 , 2872–2882 ) CrossRef CAS PubMed.
- S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 ( Angew. Chem. , 2012 , 124 , 3128–3167 ) CrossRef CAS PubMed.
- R. Kuhn and H. Fischer, Chem. Ber., 1961, 94, 3060–3071 CrossRef CAS.
- R. Kuhn and H. Fischer, Chem. Ber., 1960, 93, 2285–2289 CrossRef CAS.
- J. K. Crandall, D. M. Coppert, T. Schuster and F. Lin, J. Am. Chem. Soc., 1992, 114, 5998–6002 CrossRef CAS.
- F. Cataldo, Polym. Degrad. Stab., 2006, 91, 317–323 CrossRef CAS.
- C. S. Casari, A. Li Bassi, L. Ravagnan, F. Siviero, C. Lenardi, P. Piseri, G. Bongiorno, C. E. Bottani and P. Milani, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075422 CrossRef.
- G. Moras, L. Pastewka, M. Walter, J. Schnagl, P. Gumbsch and M. Moseler, J. Phys. Chem. C, 2011, 115, 24653–24661 CAS.
- This topic has recently been reviewed, see: N. Suzuki and D. Hashizume, Coord. Chem. Rev., 2010, 254, 1307–1326 CrossRef CAS.
- L. Song, A. M. Arif and P. J. Stang, J. Organomet. Chem., 1990, 395, 219–226 CrossRef.
- H. Werner, R. Wiedemann, N. Mahr, P. Steinert and J. Wolf, Chem.–Eur. J., 1996, 2, 561–569 CrossRef CAS.
- I. Kovacik, M. Laubender and H. Werner, Organometallics, 1997, 16, 5607–5609 CrossRef CAS.
- R. B. King and C. A. Harmon, J. Organomet. Chem., 1975, 88, 93–100 CrossRef CAS.
- A. Nakamura, Bull. Chem. Soc. Jpn., 1965, 58, 1868–1873 CrossRef.
- M. Iyoda, Y. Kuwatani, M. Oda, K. Tatsumi and A. Nakamura, Angew. Chem., Int. Ed. Engl., 1991, 30, 1670–1672 ( Angew. Chem. , 1991 , 103 , 1697–1699 ) CrossRef.
- N. Suzuki, D. Hashizume, H. Yoshida, M. Tezuka, K. Ida, S. Nagashima and T. Chihara, J. Am. Chem. Soc., 2009, 131, 2050–2051 CrossRef CAS PubMed.
- N. Suzuki, N. Ohara, K. Nishimura, Y. Sakaguchi, S. Nanbu, S. Fukui, H. Nagao and Y. Masuyama, Organometallics, 2011, 30, 3544–3548 CrossRef CAS.
- N. Suzuki, D. Hashizume, H. Koshino and T. Chihara, Angew. Chem., Int. Ed., 2008, 47, 5198–5202 ( Angew. Chem. , 2008 , 120 , 5276–5280 ) CrossRef CAS PubMed.
- B. Bildstein, O. Loza and Y. Chizhov, Organometallics, 2004, 23, 1825–1835 CrossRef CAS.
- H. Hopf and G. Maas, Angew. Chem., Int. Ed. Engl., 1992, 31, 931–954 ( Angew. Chem. , 1992 , 104 , 953–977 ) CrossRef.
- M. Gholami and R. R. Tykwinski, Chem. Rev., 2006, 106, 4997–5027 CrossRef CAS PubMed.
- M. Kaftory, I. Agmon, M. Ladika and P. J. Stang, J. Am. Chem. Soc., 1987, 109, 782–787 CrossRef CAS.
- C. Werner, H. Hopf, I. Dix, P. Bubenitschek and P. G. Jones, Chem.–Eur. J., 2007, 13, 9462–9477 CrossRef CAS PubMed.
- T. Kawase, Y. Minami, N. Nishigaki, S. Okano, H. Kurata and M. Oda, Angew. Chem., Int. Ed., 2005, 44, 316–319 ( Angew. Chem. , 2005 , 117 , 320–323 ) CrossRef CAS PubMed.
- N. Islam, T. Ooi, T. Iwasawa, M. Nishiuchi and Y. Kawamura, Chem. Commun., 2009, 574–576 RSC.
| This journal is © The Royal Society of Chemistry 2014 |