 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic triangulenes and helicenes: synthesis, chemical stability, optical properties and extended applications of these unusual dyes
Johann
Bosson
,
Jérôme
Gouin
and
Jérôme
Lacour
*
Department of Organic Chemistry, University of Geneva, quai Ernest Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: jerome.lacour@unige.ch; Fax: +41 22 379 32 15; Tel: +41 22 379 60 62
First published on 5th February 2014
Abstract
Cationic triangulenes and helicenes are highly stable carbocations with planar and helical conformations respectively. These moieties are effective dyes with original absorption and emission properties. Over the last decade, they have received greater attention and are considered as valuable tools for the development of innovative applications. In this review, the synthesis of these unique compounds is presented together with their core chemical and physical properties. Representative applications spanning from surface sciences to biology and chemistry are presented.
 Johann Bosson | Johann Bosson completed his BSc at ESCPE-Lyon and received his PhD from the University of Lyon under the guidance of Philippe Belmont. He then moved to the University of St Andrews for a post-doctoral stay in the group of Steven P. Nolan, investigating new transformations catalyzed by gold and ruthenium NHC complexes. In 2011, he joined the group of Jérôme Lacour at the University of Geneva where he is currently developing new stable carbocations and applications thereof. |
 Jérôme Gouin | Jérôme Gouin was born in Orsay, France, in 1985 and studied at the University of Orsay and received his MSc degree in 2009. He is a PhD student in the group of Prof. J. Lacour. His research interests consist of synthesizing new compounds in the family of cationic [4]helicene to study their properties and find applications. |
 Jérôme Lacour | Jérôme Lacour obtained in 1993 his PhD in Chemistry from the University of Texas at Austin. After post-doctoral studies at Harvard University, he joined the Organic Chemistry Department of the University of Geneva in 1995. He is the author or coauthor of more than 150 publications and 200 lectures. He is a member of the Editorial Advisory Board of Chemical Society Reviews. He is also a member of the International Scientific Committee of the International Symposium on Chirality and a member of the Organizing Committee of the EUCHEM “Bürgenstock” Conference on Stereochemistry. |
Introduction
Since the pioneering studies of Hofmann, Verguin and Lauth on the preparation of fuchsine and crystal violet, cationic triarylmethyl organic dyes have been topics of academic and industrial importance.1 The carbocations are isolable due to the presence of donor groups on the aromatic rings that stabilize the positive charge (i.e. the para amino groups on the specified derivatives, Fig. 1).2 However, steric repulsions enforce a twist of the aromatic rings and the triarylmethyl carbenium ions adopt therefore three-bladed propeller conformations. A reduction of the efficacy of the delocalization and the electronic stabilization results.Such a drawback was realized as early as 1964 by Martin and Smith who proposed to prepare planarized structures instead.3 Sesquixanthylium 1 was prepared and shown to be more stable than its propeller analogue 2 (Fig. 1). Despite the originality and the obvious benefits associated with the structure, compound 1 remained somewhat confidential until the 1990s when a series of studies on functionalized derivatives of 1 and on nitrogen-containing analogues, compounds 3 to 5 (Fig. 2), were reported.
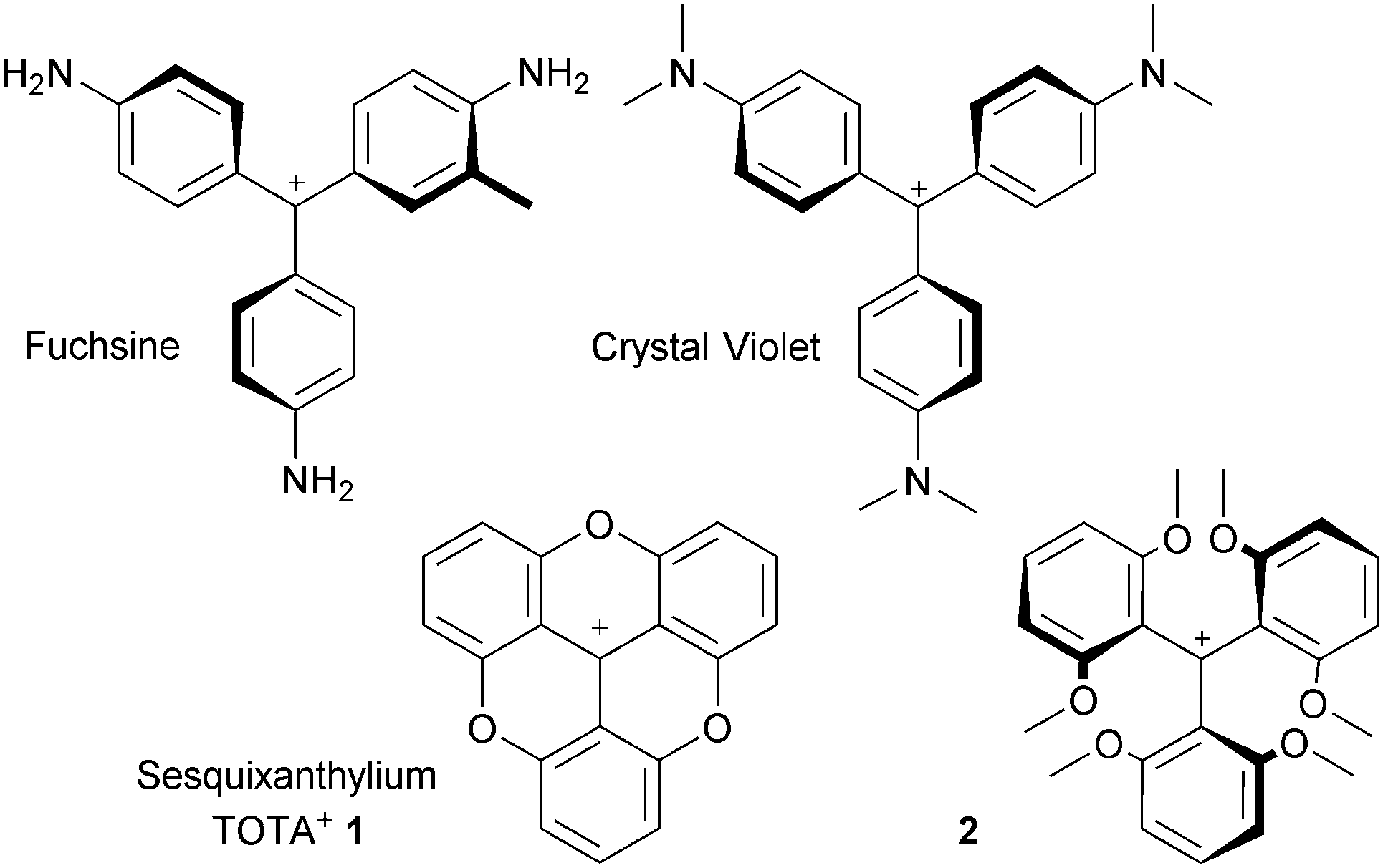 | ||
| Fig. 1 Fuchsine and crystal violet (CV), TOTA+1 and its precursor 2. | ||
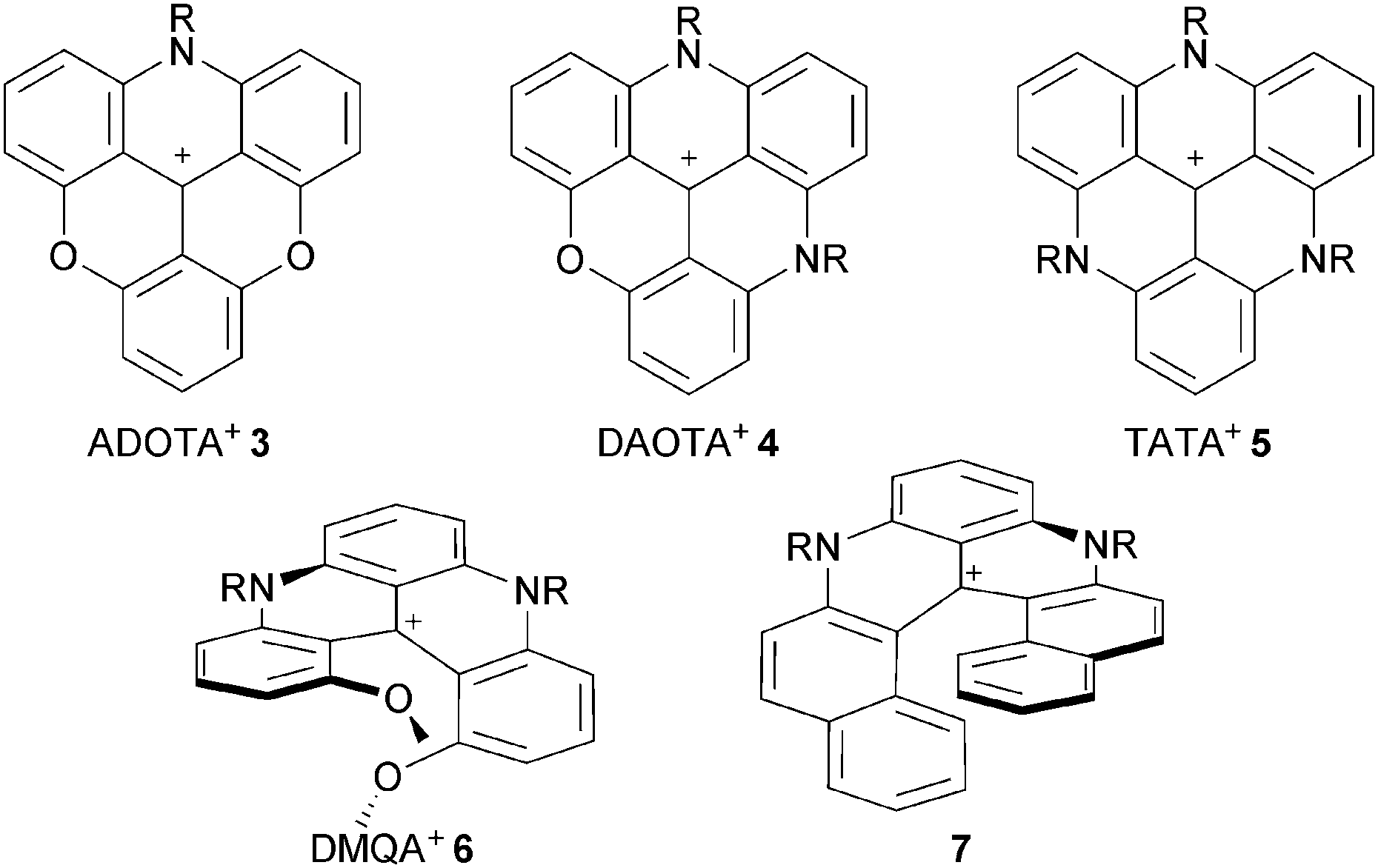 | ||
| Fig. 2 N-containing cationic triangulenes (3, 4, 5) and helicenes (6, 7). | ||
Compounds 1, 3, 4 and 5 are the subject of this review. These moieties are called triangulenes due to their structural triangular geometry and hence the names TOTA+, ADOTA+, DAOTA+ and TATA+ that are used to describe them.4 The chemistry of structurally (and synthetically) related cationic [4] and [6]helicenes 6 and 7 will also be detailed.5 Syntheses will be described. Chemical stability and optical properties will be discussed as these compounds are cationic dyes of tremendous stability under basic conditions. Applications of these core structures and derivatives in the fields of chirality, physical organic chemistry, catalysis, photophysics, optoelectronic and biology will be exemplified.
Synthesis
Preamble: on the importance of nucleophilic aromatic substitution reactions (SNAr)
In the seminal publication by Martin and Smith, trioxatriangulenium 1 was prepared in two steps only. First, diethylcarbonate and ortho-lithiated 1,3-dimethoxybenzene were reacted together using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 stoichiometry.3 Then, the resulting triaryl carbinol 8 was dissolved in molten pyridinium hydrochloride to generate the desired, fully ring-closed, planar structure 1. Importantly, these two steps outline the strategy employed in all the procedures used to make compounds of this type that is (i) the construction of an appropriate triarylmethyl carbenium precursor and then (ii) SNAr to perform ring closures (planarization) and functionalizations at the periphery of the core structures (Scheme 1).
:3 stoichiometry.3 Then, the resulting triaryl carbinol 8 was dissolved in molten pyridinium hydrochloride to generate the desired, fully ring-closed, planar structure 1. Importantly, these two steps outline the strategy employed in all the procedures used to make compounds of this type that is (i) the construction of an appropriate triarylmethyl carbenium precursor and then (ii) SNAr to perform ring closures (planarization) and functionalizations at the periphery of the core structures (Scheme 1).
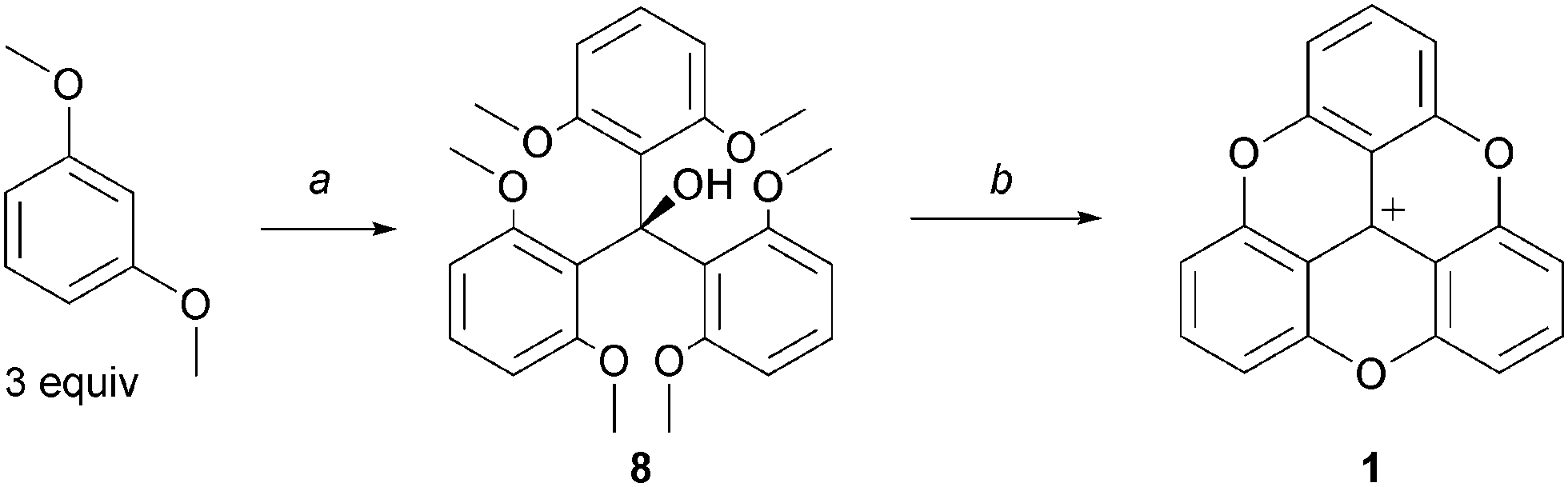 | ||
| Scheme 1 Historical preparation of 1via triaryl carbinol 8. a: PhLi (3.5 equiv.), (EtO)2CO, Et2O/benzene, reflux, 3 days; b: Pyr·HCl, 205 °C, 1 h. | ||
In fact, triaryl carbenium ions substituted by alkoxy or hydroxyl groups undergo facile substitution reactions of the OR or OH moieties in the presence of nucleophiles, as these substituents are effective nucleofuges in this context (Scheme 2, X = OMe typically). The first elemental step, the addition (a, 2 to 9, see Scheme 2), is usually rate-determining. It is rendered possible by the electron-withdrawing ability of the central positive charge. The second elemental step, the elimination of X (b, 9 to 10, see Scheme 2), transforms the transient sp3-hybridized site of attack back to sp2 reestablishing the aromaticity of the ring. Importantly for this chemistry, alkoxy substituents X are both sufficiently electron-donating to stabilize the original cationic structures and yet good enough leaving groups. The nucleophiles must be able to add at sterically hindered (ortho) positions and be also excellent donating groups to drive the reactions to completion by a better stabilization of the positive charge after the substitution (e.g., 10 must be more stable than 2).
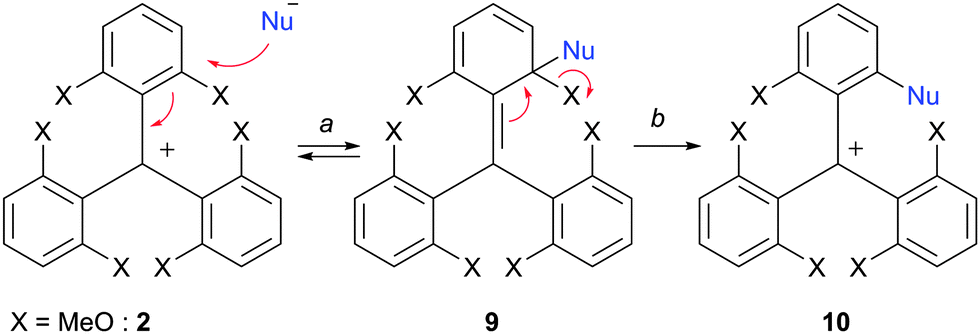 | ||
| Scheme 2 SNAr as transformations of choice. | ||
This method of ortho substitution can then lead to ring closures if the introduced nucleophile reacts consecutively with a second (neighboring) X substituent in another sequence of addition/elimination. It can also be applied to substrates possessing alkoxy groups at para positions. Many examples will be shown in the following paragraphs.
Peripheral functionalization of central trioxo cores
After the initial report on 1,3 the first novel derivatives to be prepared based on a TOTA+ skeleton were derivatives with amino donor groups at positions para to the formal positive charge, and tris-dialkylamino A3-TOTA+11 in particular.6 Adducts A1-TOTA+12 and A2-TOTA+13 with one and two NR2 groups respectively were reported only later (Fig. 3).7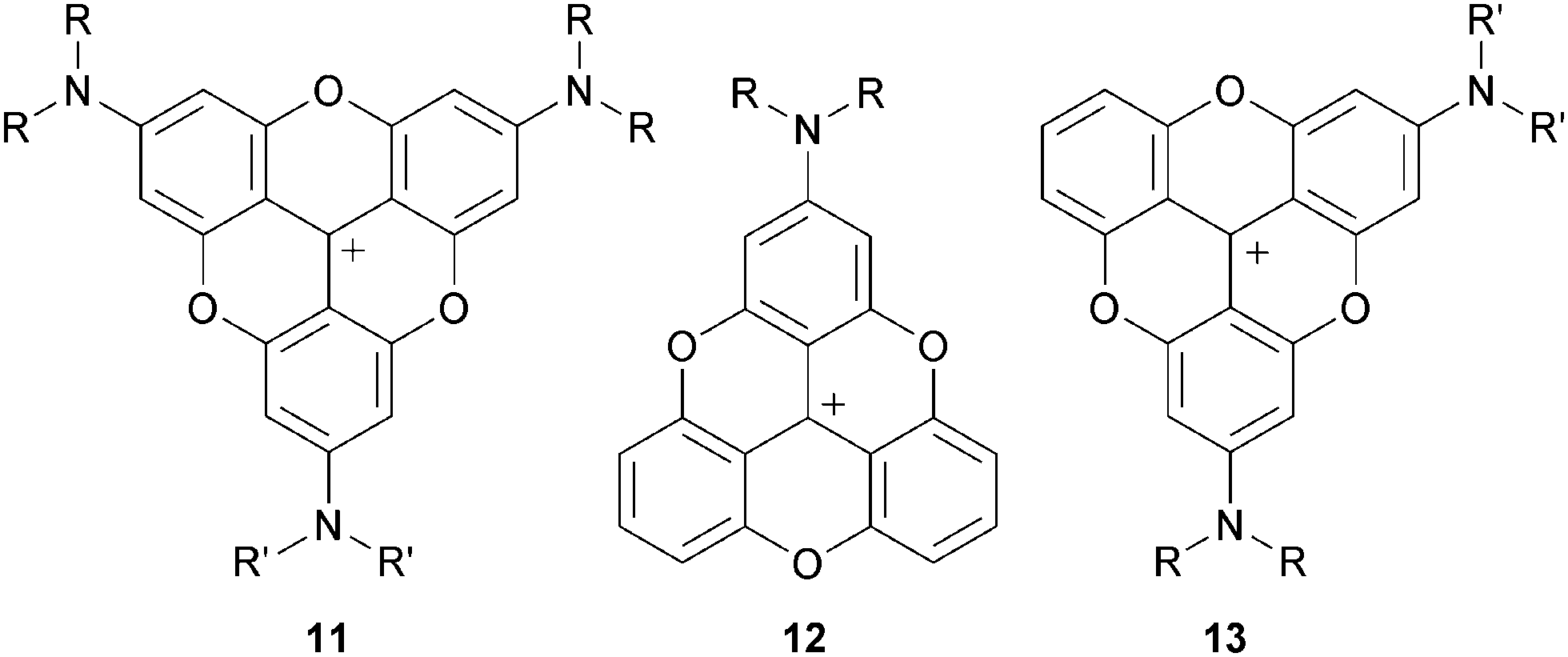 | ||
| Fig. 3 Structures of An-TOTA+ (n = 3) 11, (n = 1) 12 and (n = 2) 13. | ||
A3-TOTA+11 was synthesized from 2,4,6,2′,4′,6′,2′′,4′′,6′′-nonamethoxytriphenylcarbenium ion 14 (Scheme 3). Compound 14 was prepared by addition of an excess of ortho-lithiated 1,3,5-trimethoxybenzene to diethyl carbonate, the resulting carbinol being converted to 14 in 70% overall yield by acid treatment.8 Upon treatment with secondary amines, sequential SNAr of the three methoxy groups para to the central carbon atom was then achieved to provide mono-, bis-, and tris-NR2 substituted triaryl carbenium ions, 15, 16 and 17 respectively, in good yields (77–93%). To isolate 15 and 16 selectively, a careful choice of solvent and a control of the reaction time were necessary. With 17 in hand, conversion to yellow-orange A3-TOTA+11 was achieved by treatment with LiI at 170 °C. A3-TOTA+ ions are highly stable carbocations; the precise measurement of their chemical stability will be discussed later in the article.
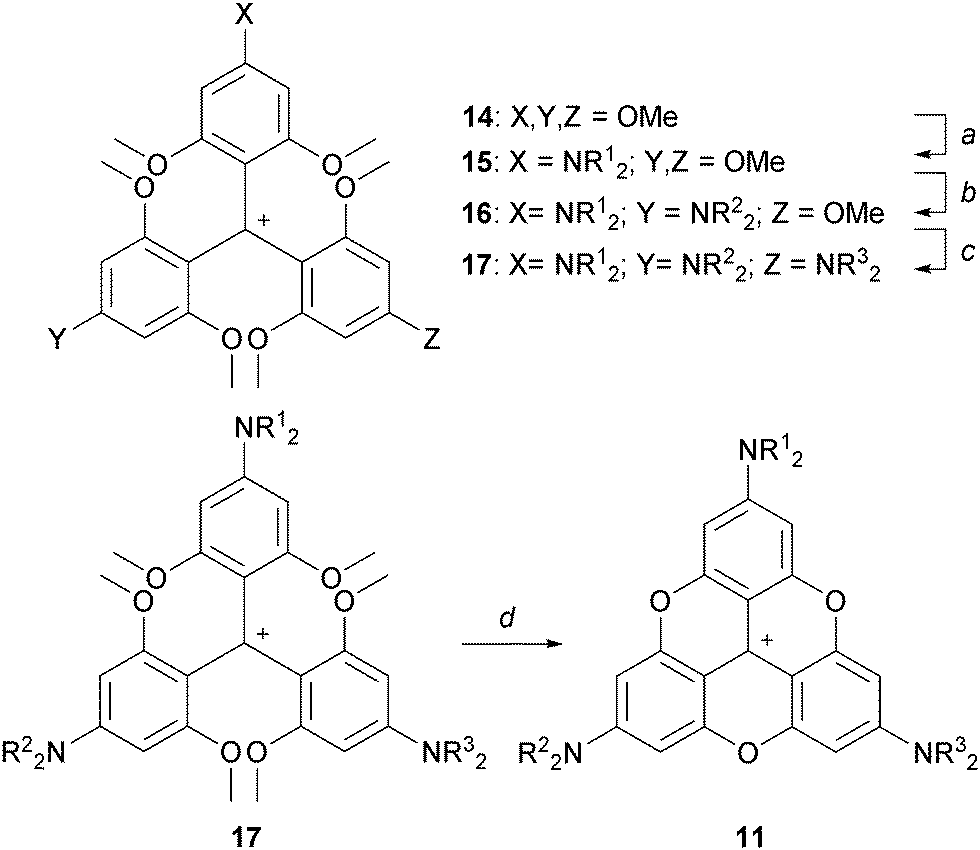 | ||
| Scheme 3 Synthesis of A3-TOTA+. a: R12NH, CH3CN, 5 min; b. R22NH, CH3CN, 20 h; c: R32NH, CH3CN or DMF or NMP, up to 9 days; d: LiI, NMP, 170 °C, 4 h. | ||
Mono- and bis-dialkylamino TOTA+12 and 13 required the synthesis of specific triaryl carbenium precursors 18 and 19 which were, this time, prepared using stepwise protocols.3,7 For instance, to make precursor 18, ortho-lithiated 1,3,5-trimethoxybenzene was submitted to a single carbonation reaction yielding ethyl 2,4,6-trimethoxybenzoate 20 in moderate yield; this compound being subjected to an excess of ortho-lithiated 1,5-dimethoxy benzene to form 18 (Scheme 4). For 19, a similar approach was used; the p-OMe groups being introduced this time on the nucleophilic fragments rather than on the electrophilic ester building block 21.
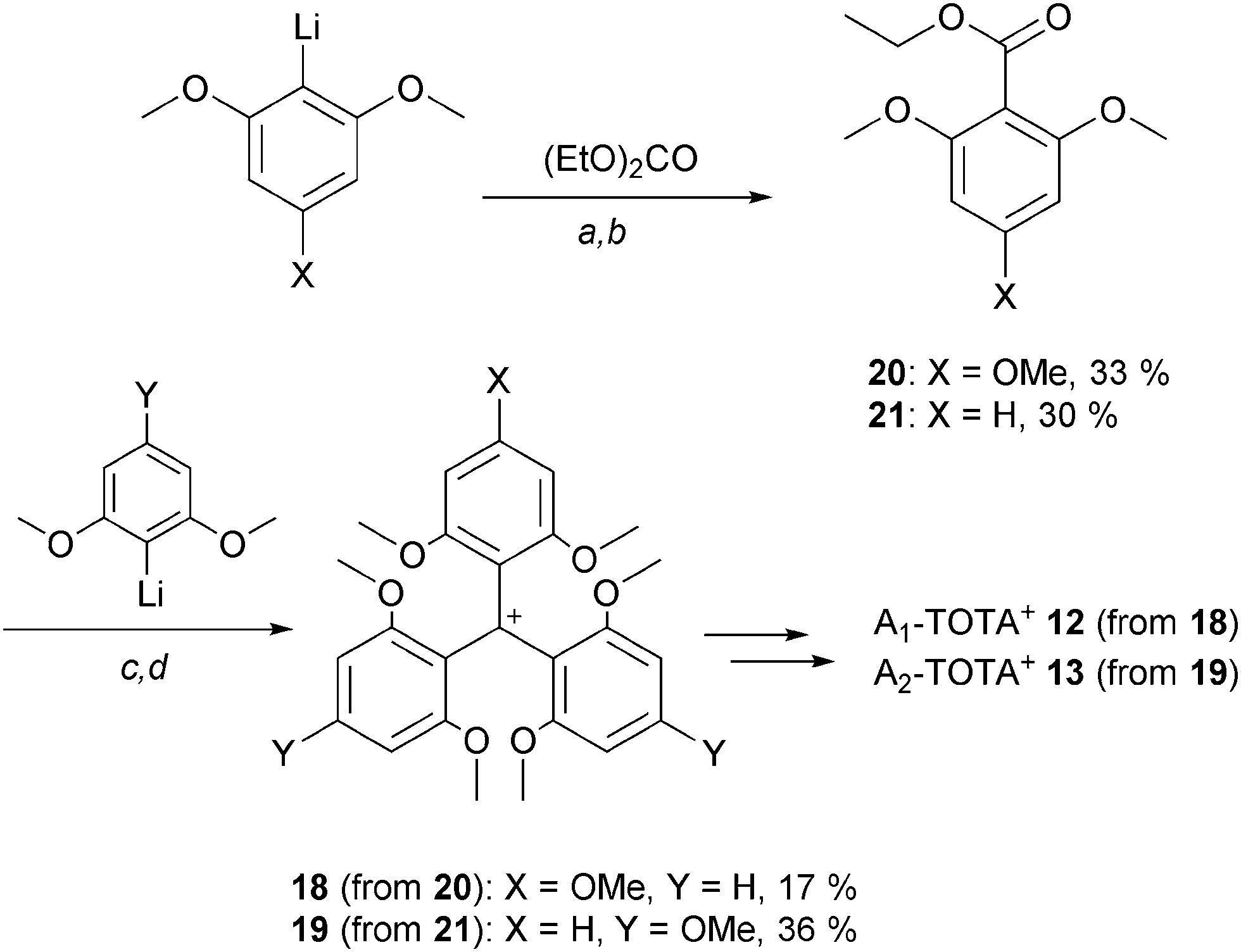 | ||
| Scheme 4 Hepta- and octamethoxy triaryl carbenium ions. a: 1,3,5- (for 20) or 1,3-dimethoxy benzene (for 21), n-BuLi, Et2O/benzene (1:1), 20 °C, 3 h; b: diethyl carbonate, Et2O, 20 °C, 16 h. c: 1,3- (for 18) or 1,3,5-dimethoxy benzene (for 19), n-BuLi, Et2O, 20 °C, 48 h; d: 20 (for 18) or 21 (for 19), Et2O, 20 °C, 16 h. | ||
With cations 18 and 19 in hand, substitution of the p-OMe group(s) by a secondary amine and subsequent LiI-mediated cyclizations proceeded to afford A1-TOTA+ and A2-TOTA+ ions respectively, albeit in moderate yields (23% and 35%, with R = decyl for 12 and R = R′ = ethyl for 13). Unlike in the preparation of 11, a need for collidine as an additive in the LiI-mediated reaction was noted.7
In a recent study, precursor 14 was converted into the tris hydroxylated analog H3-TOTA+22. In this case, both cyclization of the internal core and hydrolysis of the peripheral MeO groups occurred upon treatment with molten pyridine hydrochloride.9,10 In DMSO (6% v/v H2O), 22 exists in either cationic, neutral, anionic or bis-anionic forms as a function of pH (Scheme 5).11
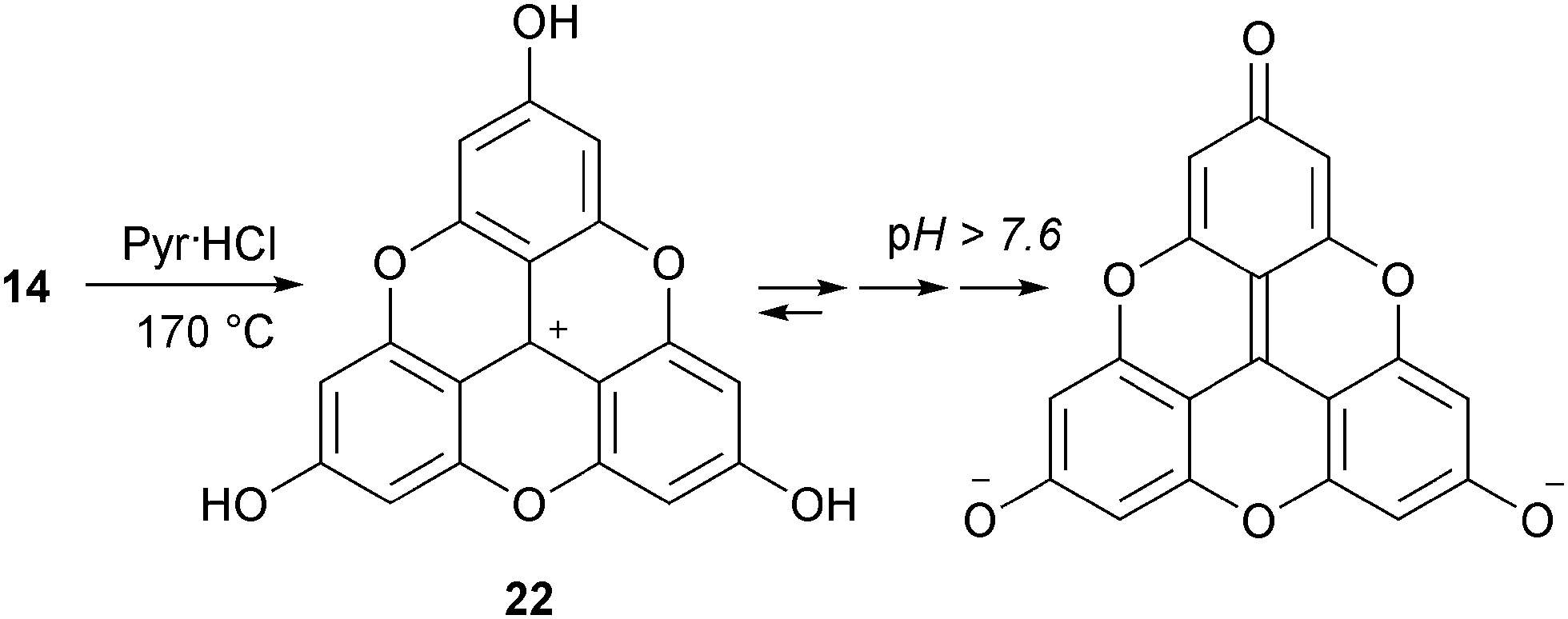 | ||
| Scheme 5 Formation of H3-TOTA+22. | ||
Construction of nitrogen-containing triangulene cores
An important breakthrough was disclosed in 2000 by Laursen and Krebs with the synthesis of cationic triazatriangulene 5 (TATA+) and related derivatives.12 It was shown that the treatment of 2 with primary alkylamines formed sequentially acridinium ions 23, dimethoxyquinacridinium ions (DMQA+) 6 and TATA+ adducts 5 (Scheme 6). In terms of mechanism, these compounds clearly result from SNAr of the MeO groups by the amines as depicted in Scheme 2. Under appropriate reaction conditions, the addition of one, two or three amino groups can be achieved. The first amino bridge is obtained at room temperature. A moderate (80–110 °C) or a strong heating (>140 °C) are however necessary for the making of 6 and 5 respectively as substitution reactions of MeO groups become less and less facile with each N-atom introduction. Large excesses of amines RNH2 are also usually necessary. At elevated temperatures and in the case of low boiling point amines, it was mentioned that the addition of additives such as benzoic acid is useful.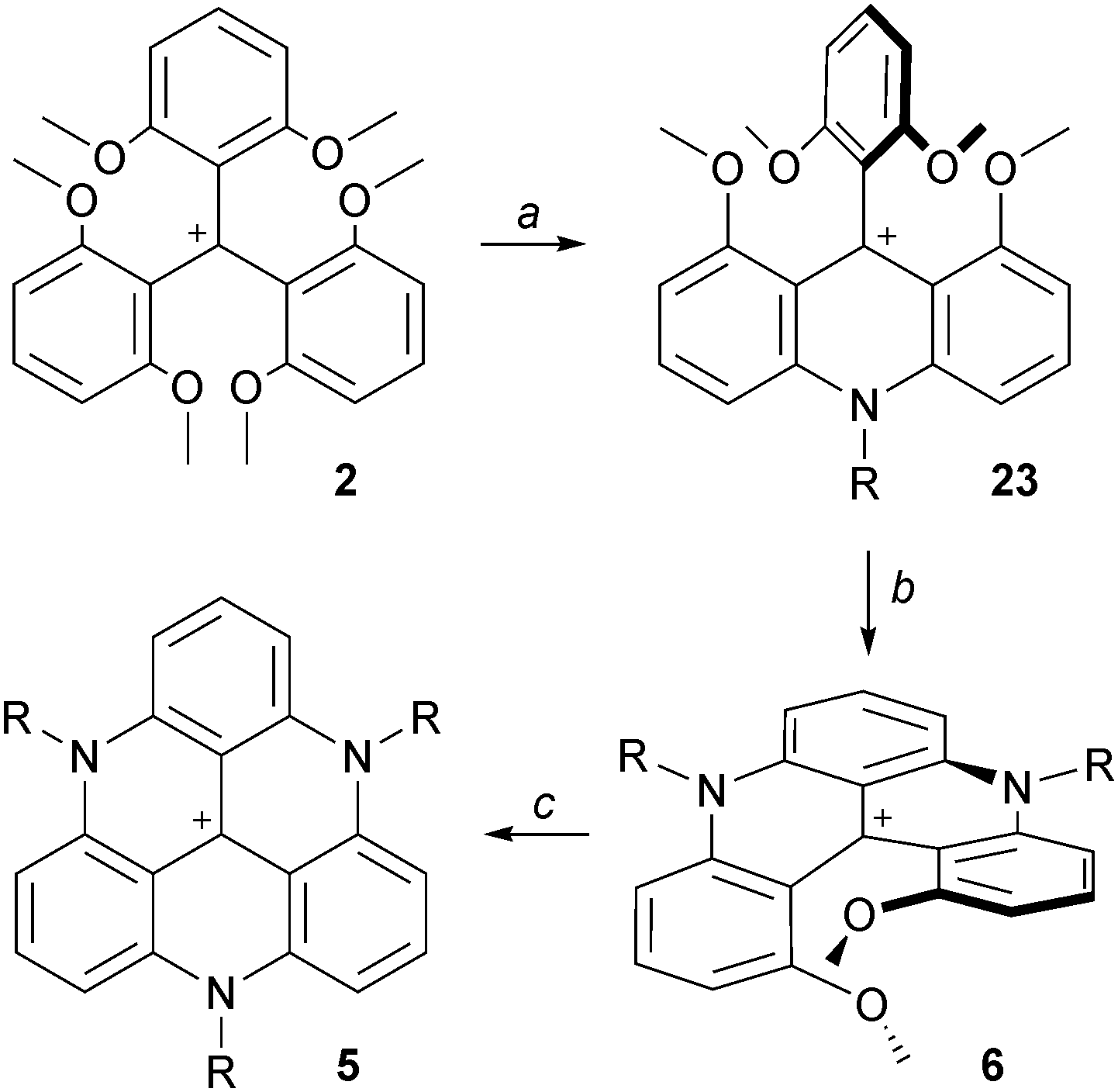 | ||
| Scheme 6 Synthesis of TATA+ core 5. a: RNH2 (5 equiv.), NMP, 25 °C, 15 h; b: RNH2 (25 equiv.), NMP, 100 °C, 45 min; c: RNH2 (25 equiv.), NMP, reflux, 10–24 h. | ||
The possibility to isolate acridinium 23 and quinacridinium 6 ions in good yields could then be used for the preparation of ADOTA+3, DAOTA+4 and unsymmetrical TATA+ ions (Scheme 7).13 In fact, treatments of 23 and 6 under conditions used for the preparation of TOTA+ (typically molten Pyr·HCl or LiI/DMF) yielded the fully ring-closed azadioxa 3 (80% yield) and diazaoxa 4 (85% yield) analogs that have interesting properties of their own (vide infra). Using with 23 an amine different from the one first introduced afforded C2-symmetrical TATA+ derivatives after ring closures.13,14
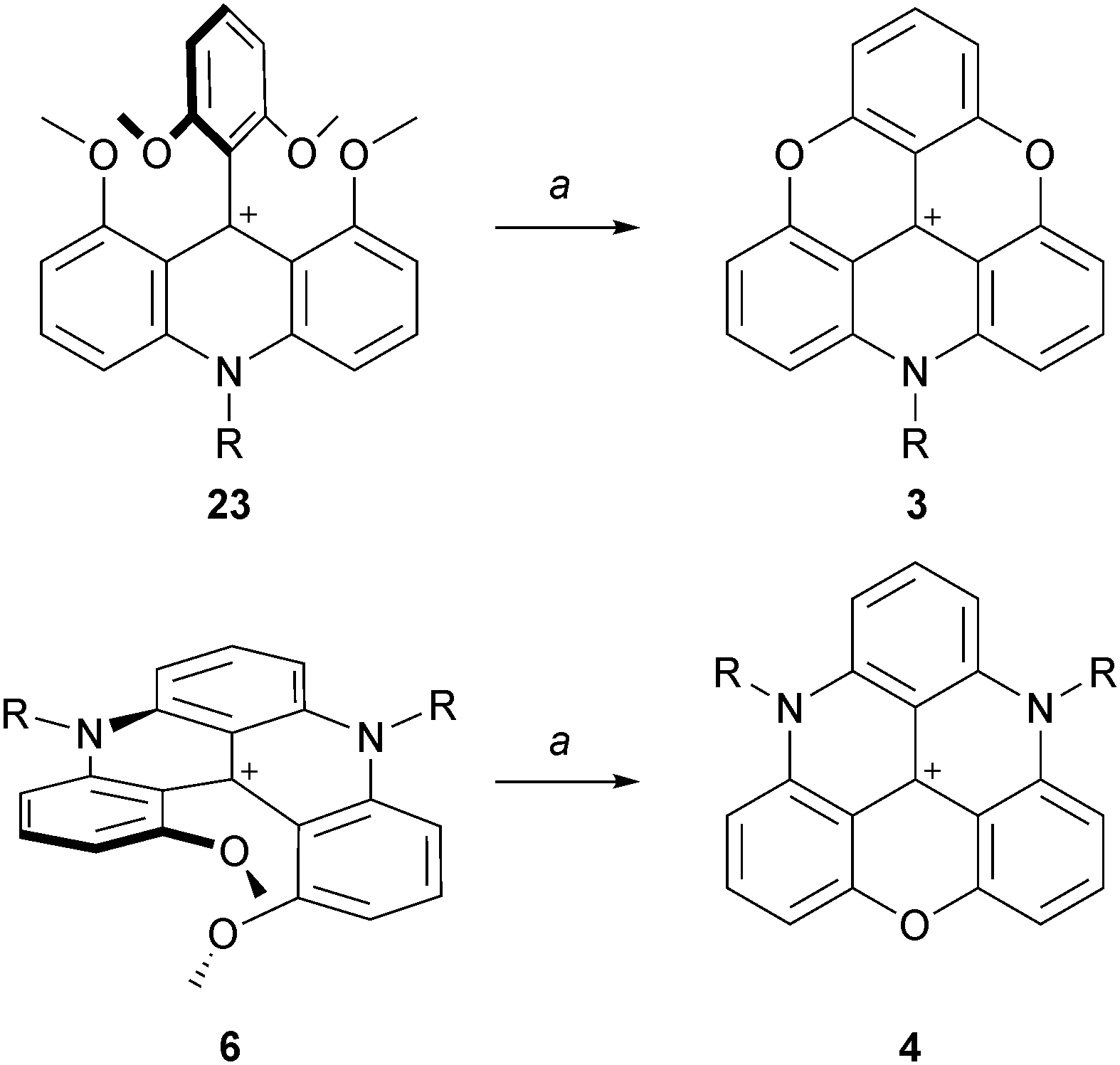 | ||
| Scheme 7 Preparation of ADOTA+3 and DAOTA+4. a: Pyr·HCl, 200 °C, 1 h. | ||
Also, it was shown that an acridinium cation of type 24 can be made from precursor 17 of A3-TOTA+ moieties.1517 reacted (only) once with alkyl amines to generate 24 which could then be converted to A3-ADOTA+25 (Scheme 8, 80% yield). Product 25 is again a particularly stable carbocationic species; this property will be discussed in detail later in this review.
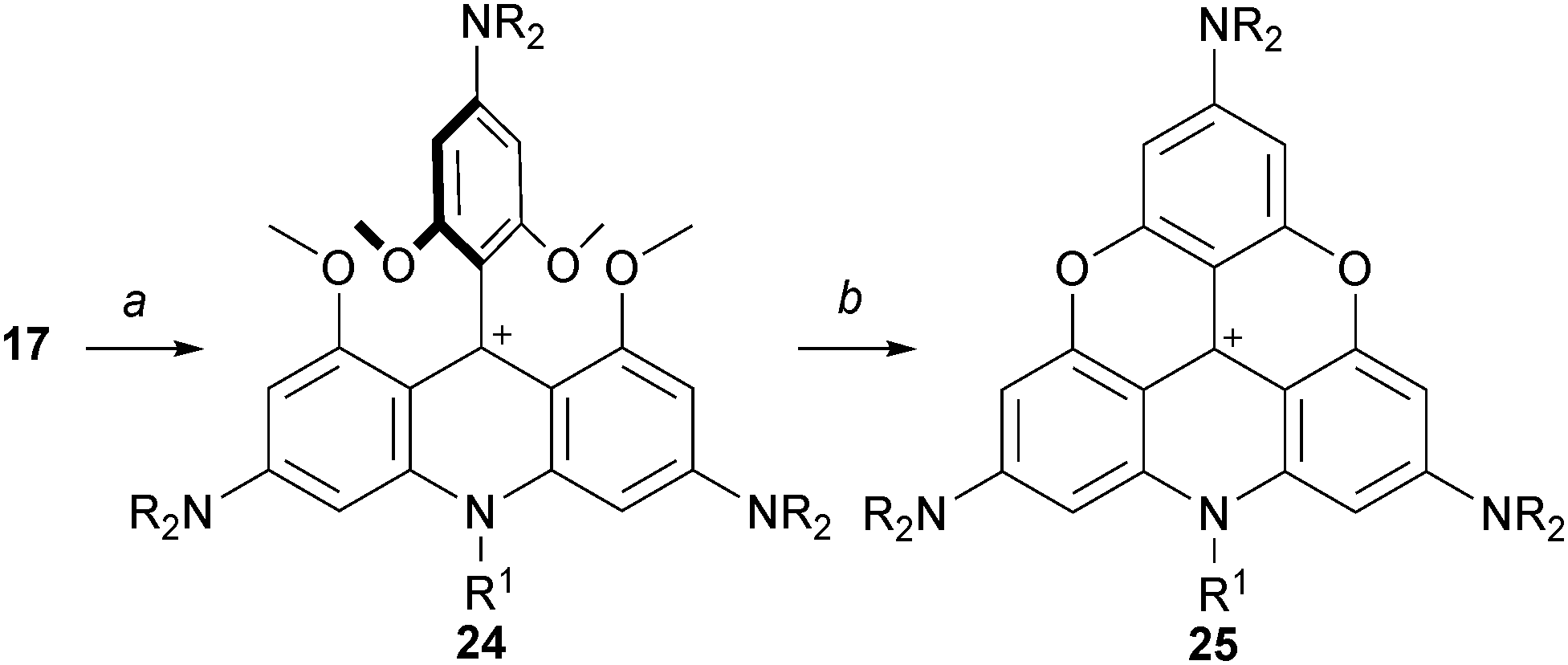 | ||
| Scheme 8 Preparation of A3-ADOTA+25. a: R1-NH2, 20 °C, 4 days; b: LiI (10 equiv.), collidine, 180 °C, 2 h. | ||
With anilines that are less nucleophilic than alkyl amines, more forcing conditions are necessary for the preparation of the corresponding derivatives. For instance, Krebs used boiling 2,6-lutidine as solvent for the reaction of anilines with 2 to afford the acridinium ions 26, which were then converted into their ADOTA+ ions 27 (Scheme 9).16 Of note, compound 27 derived from 4-bromoaniline could be engaged in palladium-catalyzed Suzuki cross-coupling reactions for further investigation. It was later shown that temperatures as low as 50 °C over 12 hours could be used for the transformation of 2 into 26.17
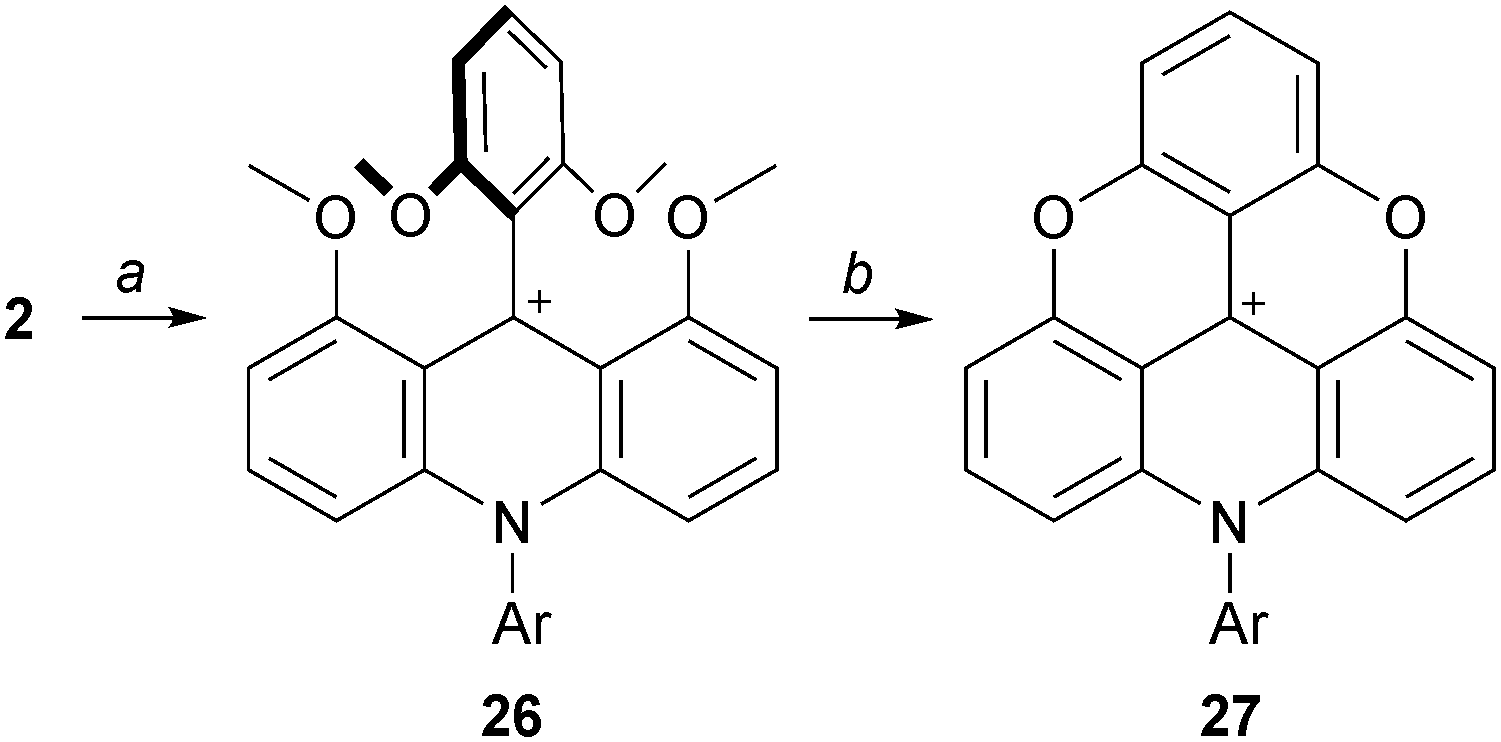 | ||
| Scheme 9 Preparation of ADOTA+27 using anilines as nucleophiles. a: ArNH2, 2,6-lutidine, reflux, 30 min; b: Pyr·HCl, 180 °C, 1 h. | ||
More recently, Laursen and co-workers managed to prepare TATA+ ions with three aryl substituents on the N-atoms by treatment at 200 °C (20 min) of mixtures of cation 2, NaH (3.1 equiv.) and the corresponding anilines (Scheme 10).18 This procedure seems to be limited to arylamines with melting points lower than 200 °C since they are used as solvents. As in the previous case, further functionalization via palladium-catalyzed cross coupling reactions was possible.
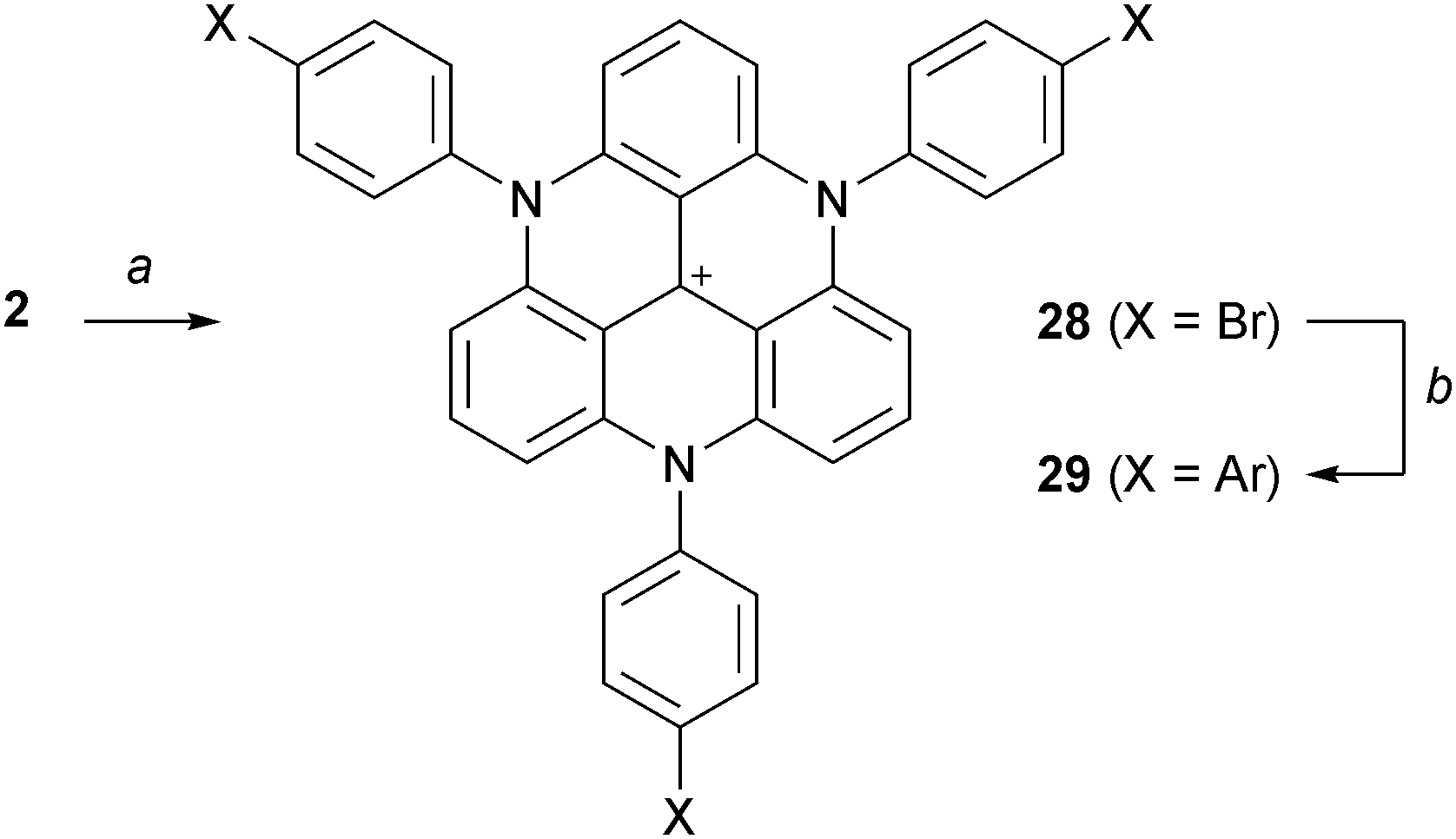 | ||
| Scheme 10 Preparation of triaryl TATA+28 and cross-coupling reactions. a: aniline, NaH, 190–210 °C, 20 min; b: ArB(OH)2, PdCl2(PPh3)2, K2CO3, toluene/H2O, reflux, 12 h. | ||
Finally, the formation of sulfur-bridged analogues was reported (Scheme 11).19 It was necessary to include the sulfur atom early in the synthesis and tetramethoxyphenylthioxanthenium ion 30 was prepared from 2-methoxybenzoic acid in six steps. 30 was reacted with aniline (neat, 110 °C, 30%) to yield the dimethoxythiachromenoacridinium cation 31. Treatment of 30 at 200 °C in pyridinium hydrochloride yielded cationic O,O,S-triangulene 32 (73% yield). For the analog 33 of ADOTA+, in sharp contrast with the situation detailed above, only with anilines (neat, 190 °C, 22 h) could the corresponding cationic N,O,S-triangulene 33 be afforded (30%).20
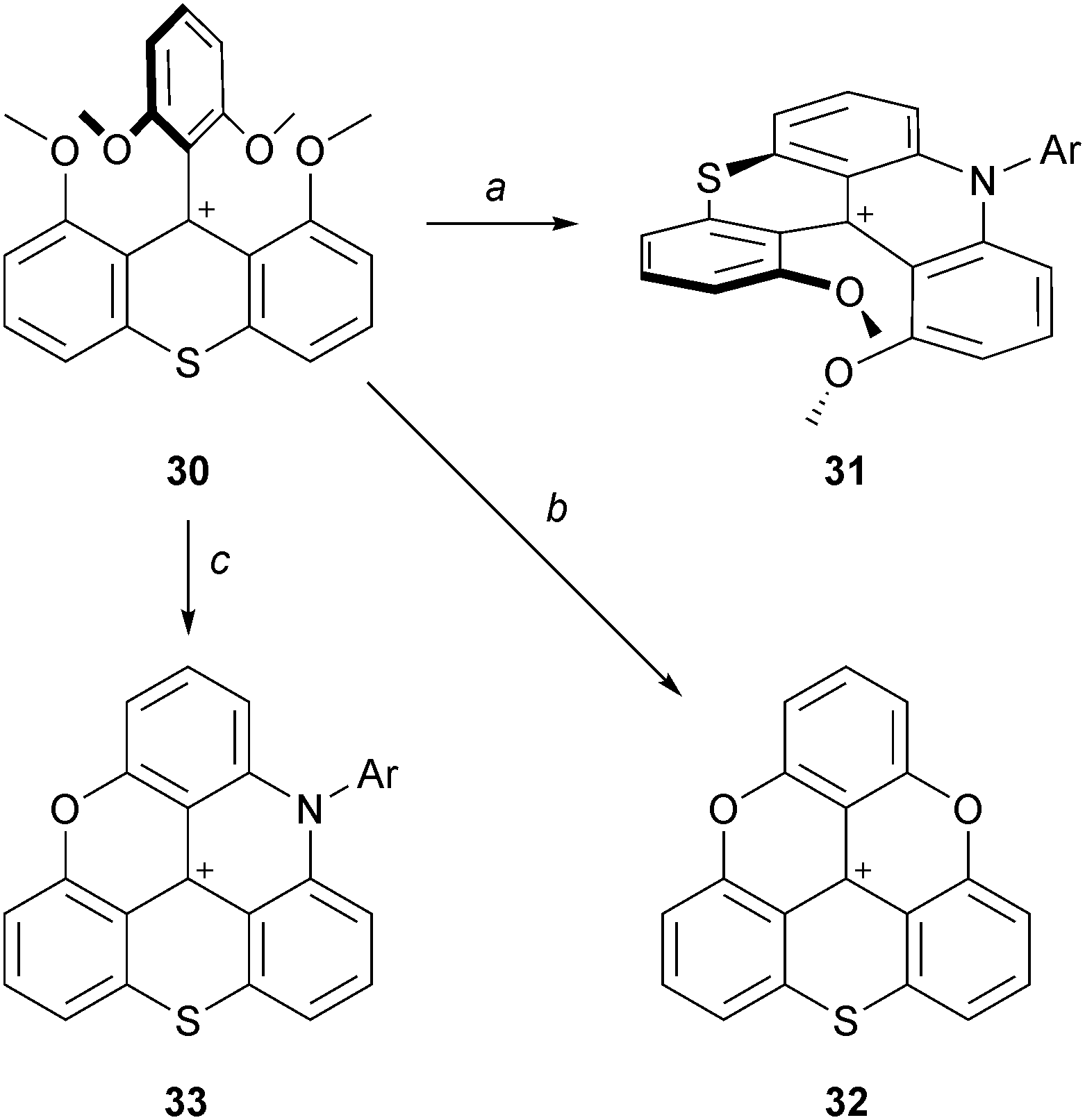 | ||
| Scheme 11 Sulfur-bridged analogues. a: aniline (30 equiv.), PhCO2H (3 equiv.), 110 °C, 22 h; b: Pyr:HCl, 200 °C, 4 h; c: anilines (20 equiv.), 190 °C, 22 h. | ||
Cationic [4] and [6]helicenes
As just presented, using slightly modified protocols, it is possible to isolate quinacridinium ion 6 or its thioaza analog 31 in moderate to good yields (vide supra). Unlike triangulenes, these compounds adopt a helical conformation due to the steric repulsion between the remaining methoxy groups. As such, they can be considered as cationic [4]helicenes. The characterization of their chiral nature and properties will be the subject of later chapters.Lacour and co-workers reported the synthesis of 1,13-dimethoxychromenoxanthenium ion 34 (DMCX), the dioxo analog of 6 (Scheme 12).21 A stepwise synthesis was necessary as it was found that 34 converted rapidly into TOTA+ under rather mild conditions. In fact, 34 had never been isolated in previously reported synthesis of 1 despite being an obvious intermediate. Practically, precursor 2 was treated with BBr3 (excess) to provide a mixture of mono- and biphenols 35. Upon ion exchange and heating at 100 °C, 34 was obtained as a mixture with TOTA+. To achieve the separation of 34 and 1, a reduction of the crude with NaBH4, a chromatography of the neutral adducts and a final oxidation with I2 were necessary. Starting from 2, compound 34 was obtained in 15% yield.
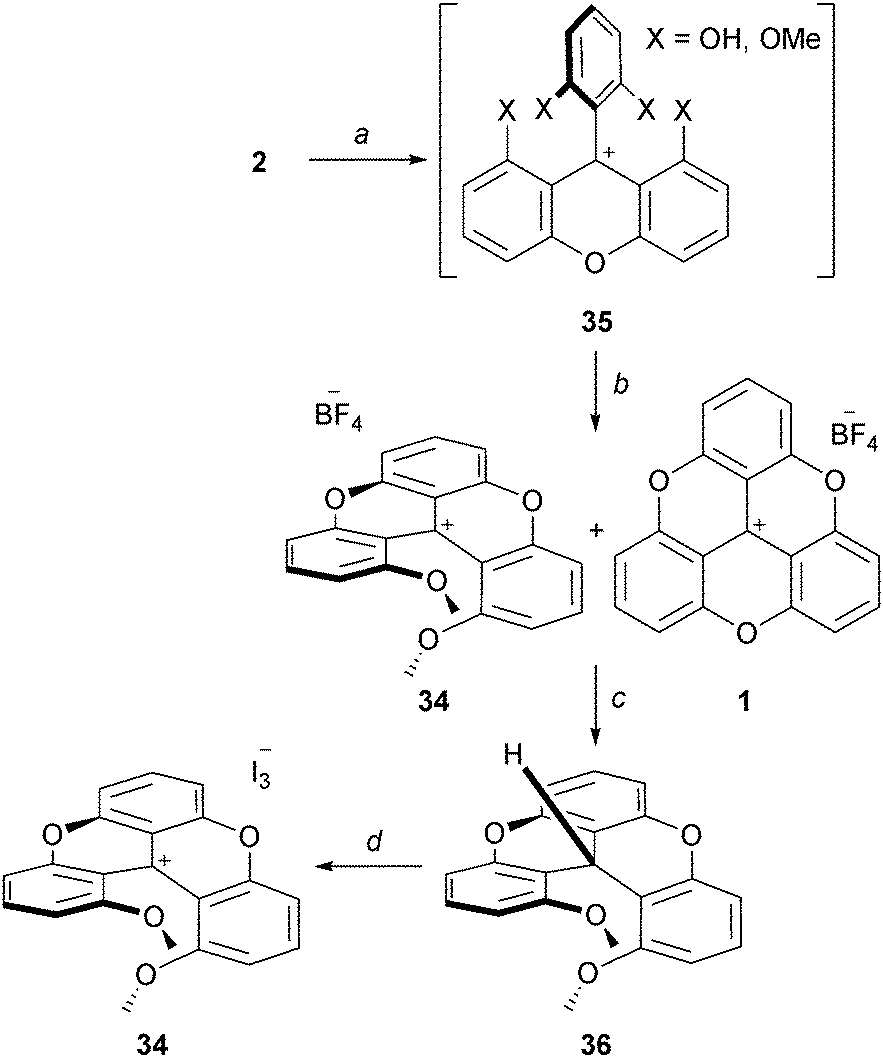 | ||
| Scheme 12 Preparation of the DMCX ion 34. a: BBr3, CH2Cl2, 0 to 20 °C; b: 1. HBF4 aq, 2. neat, 100 °C; c: NaBH4, EtOH, 20 °C, then separation by chromatography on SiO2; d: I2, Et2O, 20 °C. | ||
Recently, dioxo-, azaoxa- and diaza[6]helicenes, compounds 37, 38 and 7 respectively, were prepared from a common identical advanced intermediate 39 (Scheme 13).22 This derivative was obtained in five steps (59% overall yield, 10 g scale) from commercially available materials. Rapid treatment of 39 in molten pyridinium hydrochloride afforded 37 in 95% yield. Mixed azaoxa 38 was obtained in a two-step sequence (40% overall yield). First, 39 was reacted with a primary amine at 50 °C followed, after evaporation of the excess of amine, by a thermal treatment at 200 °C. Finally, treatment of 39 with a large excess of RNH2 (25 equiv., NMP, 170 °C, 5 min, μW) afforded the diaza[6]helicenes 7 (40–50% yield).
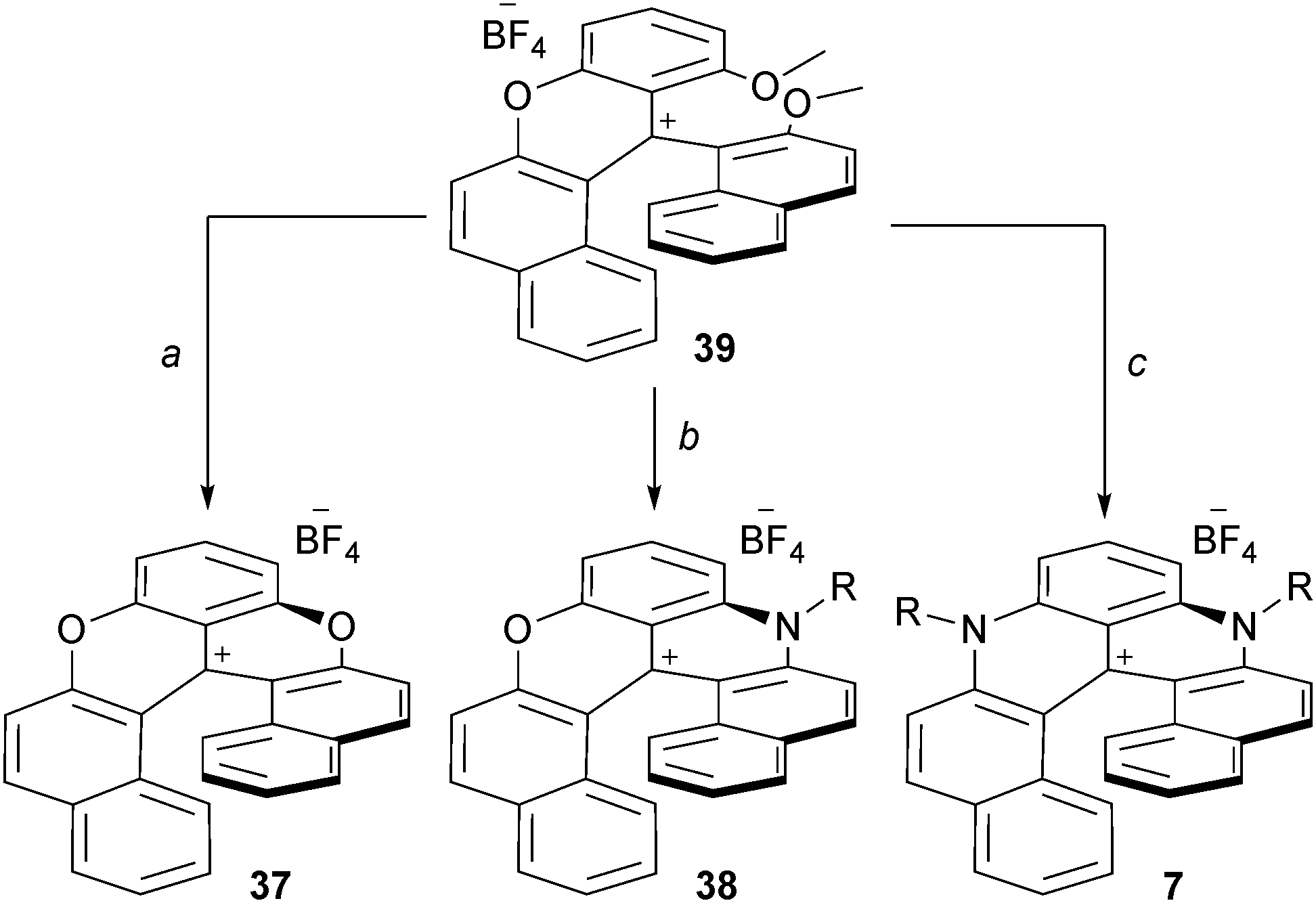 | ||
| Scheme 13 Preparation of [6]helicenes 37, 38 and 7 from 39. a: Pyr·HCl, 224 °C, 2 min; b: 1. RNH2, 50 °C, 30 min, 2. neat, 200 °C, 5 min; c: RNH2, NMP, 170 °C (μW), 5 min. | ||
Interestingly, and quite unusually in both cationic triangulene and helicene chemistry, regioselective functionalization of compounds of type 7 was possible. Diaza[6]helicenes substituted at positions 5 and 13, or 8 and 10 were readily obtained (Fig. 4). In fact, thanks to the contribution and proximity of the two nitrogen atoms, the more electron-rich benzo group can react with electrophilic reagents while the naphthyl subunits undergo reactions with nucleophilic moieties. Further transformations were possible through cross-coupling chemistry using halogen-containing derivatives (made by reaction of 7 with NBS for instance). All these compounds possess quite unique properties that will be detailed in the following paragraphs.
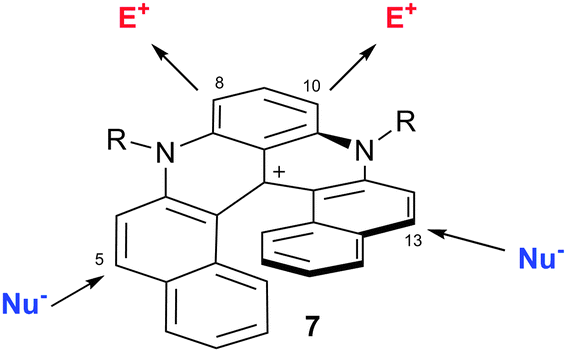 | ||
| Fig. 4 Post-functionalization of cationic diaza[6]helicene 7. | ||
Core properties
Exceptional stability in basic media
One of the most interesting and intriguing characteristics of triangulenium ions and their related derivatives is their extremely high stability as carbenium species in basic solutions. Many of these moieties are actually so unreactive that their corresponding carbinols cannot be obtained in straight basic aqueous solutions (pH ≤ 14). This “resistance” to the nucleophilic attacks of water and the hydroxide anion to the central carbon is best expressed by the very large and positive pKR+ values that these compounds possess. As a reminder, the pKR+ value is defined by the equilibrium between the cationic species and their corresponding carbinols as described in eqn (1).| R+ + H2O ⇄ ROH + H+ |
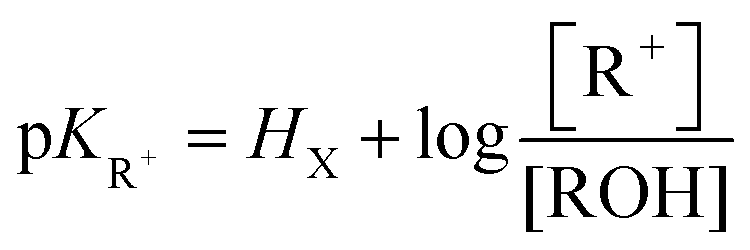 | (1) |
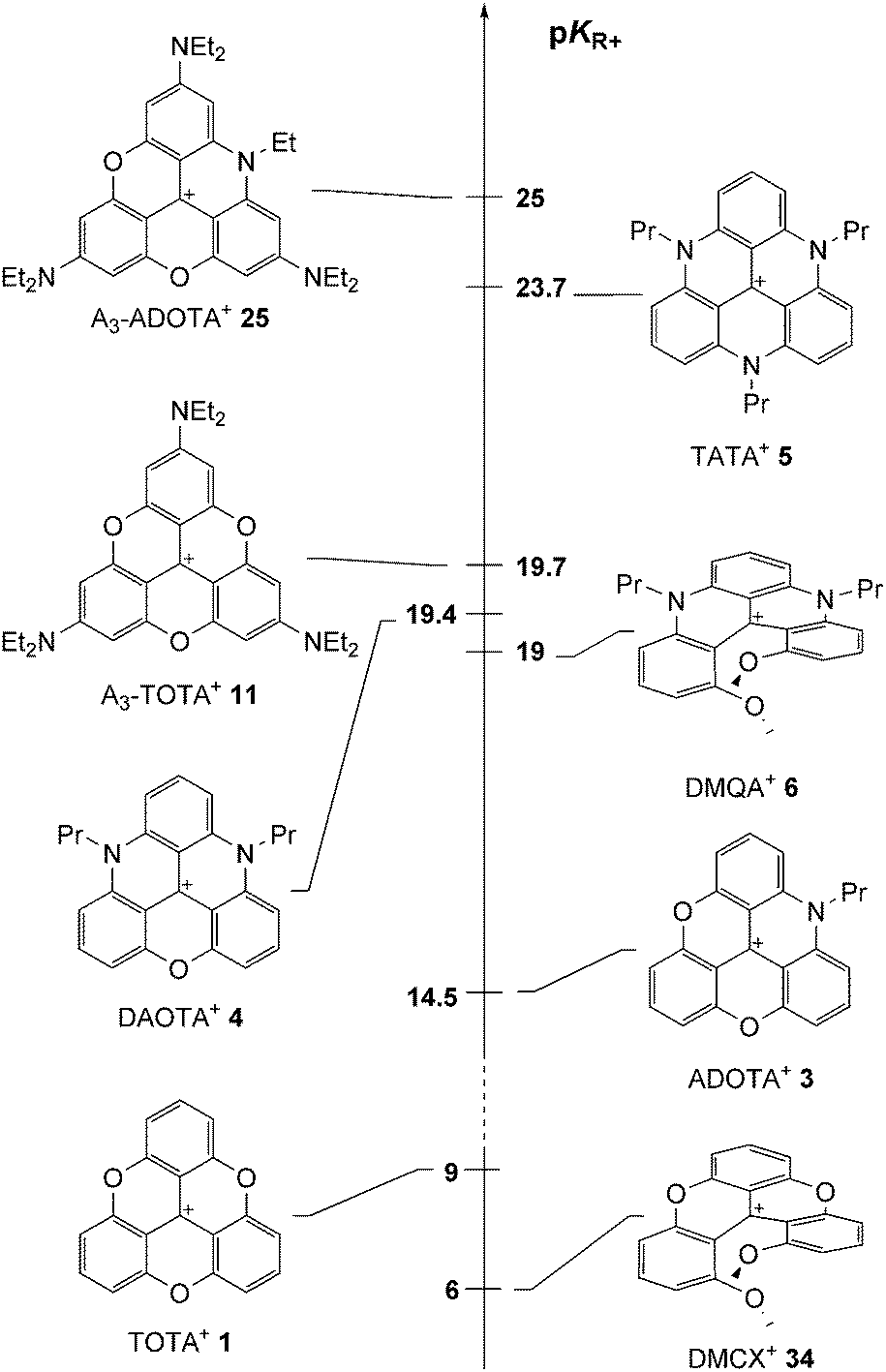 | ||
| Fig. 5 pKR+ values of cationic helicenes and triangulenes. | ||
Absorption and fluorescence
Cationic triangulenes and related helicenes are effective dyes. Their optical properties are summarized in Table 1. They usually absorb light in the visible region at wavelengths between 450 and 650 nm. In a general fashion, as observed in the spectra of many triaryl carbenium ions, the lowest energy absorption band presents a shoulder. Different explanations have been proposed to rationalize this behavior. For instance, in the D3h symmetric structures (i.e. TOTA+1, A3-TOTA+11 or TATA+5), the excited state could be doubly degenerated and the shoulder would then result from two different electronic π → π* transitions from one single ground state to two close-lying excited states.26 Alternatively, the lowest excited state could possess a degenerated C2v symmetry and undergo a stabilizing Jahn–Teller-type distortion. This would influence the HOMO ground state and, in fine, it would be responsible for the lift of its degeneracy.27 Furthermore, the nature of the medium surrounding the dyes has a strong influence on the shape of the band. Depending on the solvent, the dye may exist as an independent solvated ion, as a tight ion pair or even as an ion quadruple. It is then immediately reflected in the absorption spectra (e.g. in the case of A3-TOTA+11, Fig. 6).28| Entry | Compound | Solvent | λ max (nm) | ε λ max (M−1 cm−1) | λ flu (nm) | ϕ (%) | τ (ns) |
|---|---|---|---|---|---|---|---|
| 17 | TOTA+1 | CH2Cl2 | 475 | 8200 | 520 | 11 | 11.7 |
| 27 | A1-TOTA+12 | CH2Cl2 | 507 | 41700 |
529 | 5.1 | 4.2 |
| 37 | A2-TOTA+13 | CH2Cl2 | 513 | 59700 |
544 | 100 | 4.16 |
| 47 | A2-TOTA+13 | CH3CN | 512 | 46000 |
541 | 44 | 1.34 |
| 57 | A3-TOTA+11 | CH2Cl2 | 471 | 132900 |
494 | 67 | 3.6 |
| 69 | H3-TOTA+22 | DMSO | 416 | 57000 |
491 | <1 | 3.2 |
| 79 | H3-TOTA+22 | H2O | 413 | 38000 |
477 | 30 | 2.9 |
| 89 | H3-TOTA022 | DMSO | 477 | 35000 |
493 | 62 | 4.3 |
| 99 | H3-TOTA022 | H2O | 429 | 23000 |
474 | 5 | 3.8 |
| 109 | H3-TOTA−22 | DMSO | 485 | 55000 |
505 | 87 | 4.2 |
| 119 | H3-TOTA2−22 | DMSO | 451 | 76000 |
471 | 65 | 3.3 |
| 129 | H3-TOTA2−22 | H2O | 425 | 63000 |
477 | 30 | 2.9 |
| 1330 | ADOTA+3 | CH3CN | 540 | 9800 | 555 | 42 | 26.8 |
| 1430 | DAOTA+4 | CH3CN | 557 | 14800 |
590 | 46 | 24.8 |
| 1530 | TATA+5 | CH3CN | 525 | 18200 |
557 | 21 | 9.4 |
| 1633 | DMQA+6 | CH2Cl2 | 617 | 656 | 20.5 | 12 | |
| 1733 | DMQA+6 | CH3CN | 617 | 13800 |
668 | 7.6 | 5.5 |
| 1834 | DMCX+34 | CH2Cl2 | 580 | 3100 | 613 | 1.5 | 2.3 |
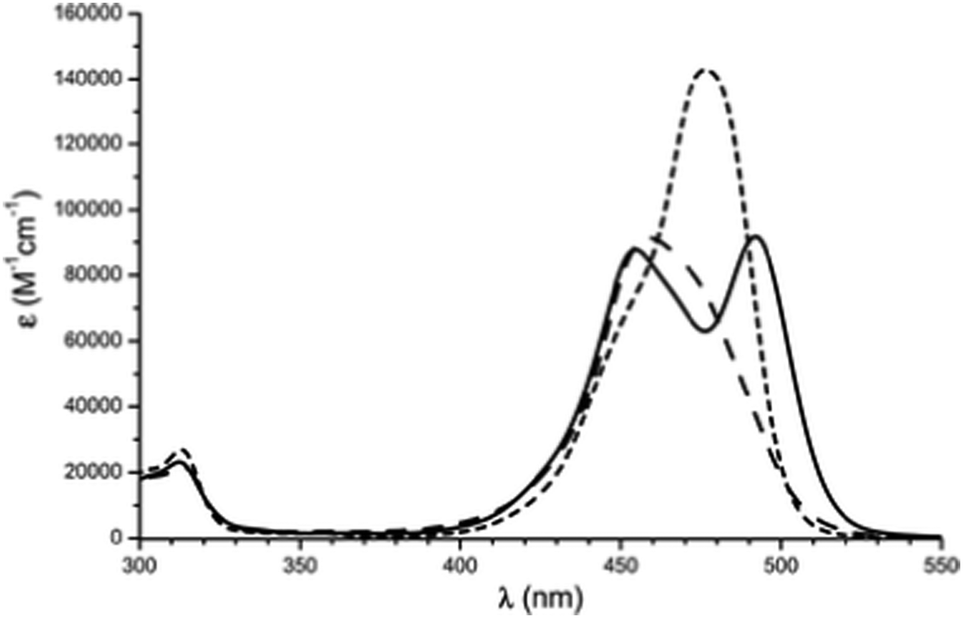 | ||
| Fig. 6 Absorption spectra of A3-TOTA+ (tris-didecylamino-11) in CH2Cl2 (dot line), benzene (plain line) and heptanes (dot dashed line). Reproduced from ref. 28 with permission from the European Society for Photobiology, the European Photochemistry Association, and The Royal Society of Chemistry. | ||
Symmetry is definitely essential. D3h-symmetric compounds (see above) absorb at higher energies than C2v-symmetric A1-TOTA+12, A2-TOTA+13, ADOTA+3 and DAOTA+4. Similarly, cationic helicenes (of lower symmetry) absorb at higher wavelengths than the corresponding triangulenes. In terms of electronic influence, introduction of electron donating groups in the para position related to the central carbon atom induces an enhancement of the absorption coefficient in the A-TOTA+ series.7 Also, replacing oxygen by nitrogen atoms in the cationic cores induces a gradual red shift in the absorption spectra. This is particularly well observed in the triangulene series from TOTA+ to DAOTA+ (Fig. 7) or in the helicenes from DMCX+34 to DMQA+6. However, as the influence of the symmetry is predominant, D3h-symmetric TATA+ absorbs at higher energy than C2v-symmetric DAOTA+. It is also worth mentioning that the replacement of a N-atom by a sulfur induces a red shift in the absorption properties as seen for compounds 32 and 33.19
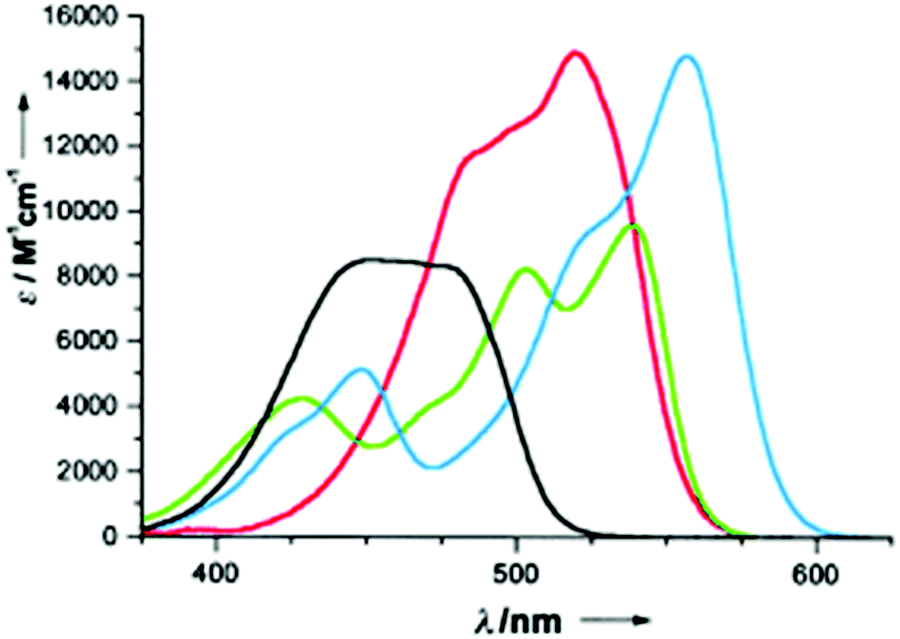 | ||
| Fig. 7 Absorption spectra in acetonitrile of TOTA+1 (black), ADOTA+3 (green), DAOTA+4 (blue) and TATA+5 (red). Reproduced from ref. 13. | ||
Laursen and co-workers went further into details in their analysis of the relation between symmetry and optical properties.29C2v-symmetric ADOTA+ and DAOTA+ ions present S0 to S1 electronic transitions at wavelengths superior to 500 nm and S0 to S2 transitions in the 400–450 nm region. The S0 to S1 transition involves an electron movement from the nitrogen atoms toward the central carbon atom whereas the S0 to S2 transition is due to electron movement from the oxygen atoms with a lower absorption coefficient due to a lower electron donating ability. Fluorescence anisotropy measurement further showed that the emissions of those two transitions are perpendicular. TOTA+ and TATA+, which belong to the D3h group point, exhibit on the other hand degenerate transitions (vide supra). Absorption and emission spectra are not mirror images, indicating that the transitions are actually overlapping. Fluorescence anisotropy measurement showed that, most probably, the excited state is C2v-symmetric.
Cationic triangulenes and related helicenes are moderately to highly fluorescent. In the A-TOTA+ series, introduction of electron donating groups at the periphery of the cationic core in the para position to the formal cationic charge induced an enhancement of the fluorescence quantum yields.7 ADOTA+ and DAOTA+ are particularly effective fluorophores with quantum yields higher than 40% in CH3CN and fluorescence lifetimes higher than 20 ns.30 As a consequence, applications in bioimaging have been developed and will be detailed later (vide infra).
Fluorescence enhancement and fluorescence quenching
Fluorescence can be enhanced on metal surfaces. This has been investigated by Sørensen, Gryczynski and co-workers in the case of ADOTA+ adsorbed on silver nanoparticles on a gold film (Ag-SACs).31 The metal enhanced fluorescence is 54 times stronger than on glass. On the other hand, Dileesh and Gopidas have shown that the fluorescence of the electron acceptor TOTA+ ion is effectively quenched using a variety of electron donating quenchers through a photoinduced electron transfer (PET) mechanism.32 In this case, a reductive quenching occurs as the quencher transfers one electron to the cation. The same authors further investigated the fluorescence quenching of the complete TOTA+, ADOTA+, DAOTA+ to TATA+ series.30 In a general fashion, from “electron poor” TOTA+ to “electron rich” TATA+, the reductive quenching becomes less and less efficient while the oxidative fluorescence quenching is more prompt to occur.13Electrochemistry
A rather rich electro reduction and oxidation chemistry has also been reported. The carbenium ions are in fact readily studied by cyclic voltammetry or other related methods. It is possible to observe up to two one-electron oxidations and two one-electron reductions. Strong differences are observed among the different cations. However, and quite unfortunately, values of the measured potentials diverge quite strongly from one study to the next as different sets of conditions and methods have been used over the past 30 years. As such, only the most recent values measured by cyclic voltammetry vs. SCE (Saturated Calomel Electrode) are reported. In general, the first one-electron oxidation and reduction waves appear chemically reversible. The derived potentials are summarized in Fig. 8 for triangulenium 1,353,304,305,30116 and DMQA+6.36 Clearly, the more the nitrogen atoms present in the triangulene core or as substituents, the easier it is to oxidize the corresponding carbenium ions. Similarly to the pKR+ values, this can be explained by the better donor ability of N-atoms vs. O-atoms. Alternatively, with more oxygen substituents, compounds are reduced readily. For instance, TOTA+ and TATA+ are reduced at −0.385 and −1.40 V respectively. | ||
| Fig. 8 Oxidation and reduction potentials of cationic helicenes and triangulenes vs. SCE. | ||
Counterion influence in solution and solid states
All the compounds under study are cationic and, not too surprisingly, negative counterions may have a noticeable influence in solution on the photophysical properties of the salts. The equilibrium between tight, loose, dissociated ion pairs and aggregates depends not only on the polarity of the solvent but also on the gegenion.37 Solubility of the salts in either organic solvents and aqueous solutions or their elution behavior on chromatographic phases can also be addressed through the nature of the anion. If the cation is chiral and racemic, then enantiopure counterions can help differentiate the enantiomers in NMR spectroscopy and assist possibly in the resolution via, for instance, a selective precipitation of one of the diastereomeric salts (vide infra).Negative counterions may also have an influence on condensed state structures. For instance, in monocrystals of TOTA+ salts 1, monodentate and “small” counterions are localized close to the central carbon atom whereas they are more likely to be positioned at the periphery of TATA+ or A3-TOTA+ derivatives 5 and 11, near the electron-rich N-atoms. For TOTA+ specifically,38 strong differences were observed in the X-ray diffraction analyses of salts with monovalent (I−, BF4−, AsF6−, PF6−, HNO3−, NO3−, CF3SO3−) and divalent (Mo6Cl142−, S2O62−) anions. For the first class, a zigzag pattern between cations and anions was noticed while divalent counterions tended to be “sandwiched” between two cations.
Recently, in association with TATA+ cations, interesting results were observed with a particular class of counterions, that of planarizable anionic complexes. The complexes are comprised of a small anionic atom (typically chloride) onto which is wrapped an acidic organic ligand (e.g., a BF2 complex of dipyrrol-1,3-dienone). Those anionic entities can be prepared by the in situ reaction of the neutral ligand with a [TATA][Cl] salt for instance. The result is a tight association of a planar cation and a planarized “supramolecular anion” which leads to the formation of charge-by-charge assemblies.39 Both ions stack on top of each other (Fig. 9). In some instances, via electrostatic and π–π interactions, a well-ordered gel is produced which acts as an energy trap. Several applications (i.e. formation of mesophases, preparation of soft materials, charge carrier properties) of these charge-by-charge or charge-segregated assemblies have been reported.40
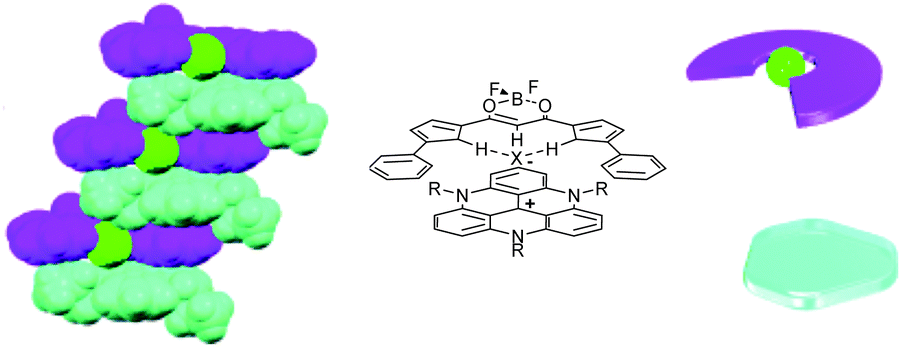 | ||
| Fig. 9 Solid state structure of a charge-by-charge assembly from the BF2 complex of dipyrrol-1,3-dienone (purple) planarized with a Cl− anion (green) and tris-propyl-TATA+ (turquoise-blue). Adapted from ref. 39. | ||
Triangulenes: 2D and 3D structures
Yet, in many other instances, the anions seem to have little effect on the modes of assembly adopted by the carbenium ions, and that of TATA+, A3-TOTA+ and ADOTA+, 5, 11 and 3 in particular. For instance, X-ray structure analyses showed that salts of 5 adopt smectic-like double layers in the solid state.13,41 The distance between the two layers was controlled by the length of the alkyl side chains that are positioned essentially perpendicular to the aromatic cores. The layers were interdigitated due to the positive dispersive interactions between the alkyl chains leading, as a consequence, to a parallel stacking of two TATA+ together (see Fig. 10). Globally, hexagonally ordered bilayers were seen. When dichloromethane solutions of 5 (tris n-propyl and n-octyl derivatives) were spin-casted, highly-ordered thin films were obtained. A macroscopic homogeneity was obtained over centimeters and the films were composed of highly anisotropic polycrystalline materials. Optoelectronic applications were evidenced thanks to the high fluorescence and efficient exciton transport properties.41,42 In the case of tri-aryl-TATA+28 and unlike 5, the aryl groups are perpendicular to the triangulene core and this conformation forbids the “classical” multi-lamellar stacking.18 In the case of A3-TOTA+11 and similarly to TOTA+, the cations stack in columnar structures, the anions being segregated around the amino groups.6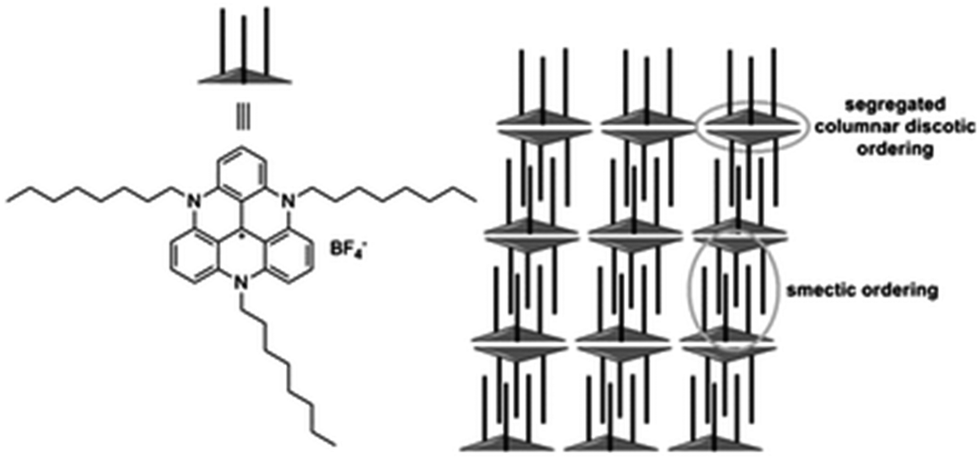 | ||
| Fig. 10 Parallel stacking of smectic-like double layers of tris-n-octyl-TATA+5. Adapted from ref. 41. | ||
Amphiphilic A3-TOTA+ possessing both short and long alkyl chains were further prepared and studied in 2D structures. PF6 salts of 39, 40 and 41 formed Langmuir monolayers at the water–air surface (Fig. 11).4339 presented a columnar π stack and a clear π–π stacking distance of 3.46 Å. Very similar films were obtained with 40, but the π stacking was revealed to be less pronounced. For 41, well-defined 2D structures could not be observed. Langmuir–Blodgett (LB) films were prepared from monolayer Langmuir films of PF6 salts of 39 and 40.44 No difference in terms of stacking distance was noticed compared to Langmuir films. The UV-vis absorption in LB films was blue shifted compared to the dyes in solution whereas, in fluorescence, a red-shift up to 100 nm was observed. It is consistent with a H-type aggregation. Polarized fluorescence indicated that the π–π interactions induced a macroscopic alignment and that the counterions were located between the A3-TOTA+ layers. Water soluble amphiphilic A3-TOTA+ were also prepared by integrating sulfonate groups. When mixed (up to 10%) with 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and vortexed at 40 °C, the formation of unilamellar vesicles (ULVs) occurred.45 Due to the optical properties of A3-TOTA+ ions (vide supra), the highly stable and unimodal size distribution ULVs obtained were fluorescent, making this strategy a new tool for the study of vesicles.
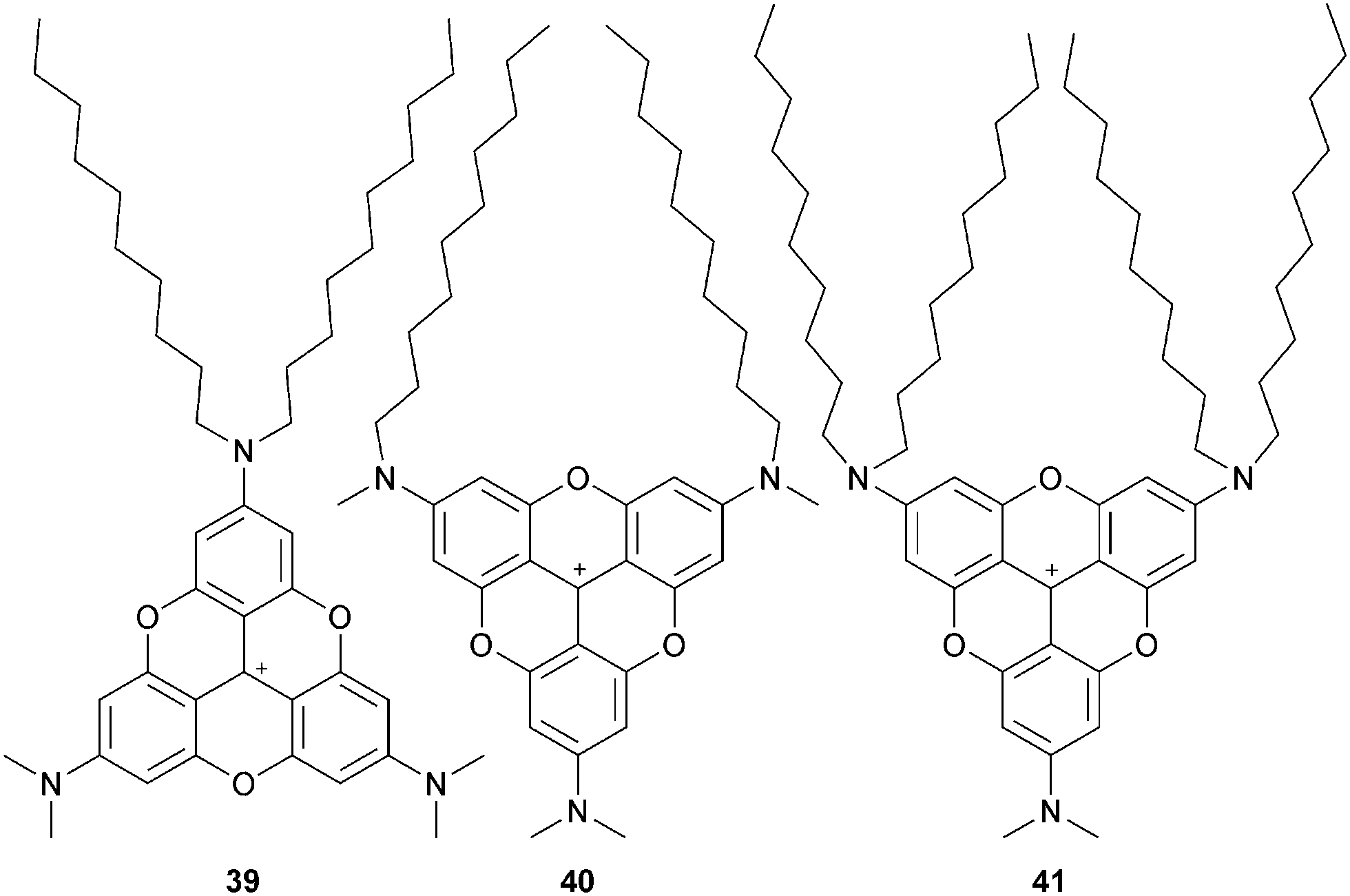 | ||
| Fig. 11 Amphiphilic A3-TOTA+39, 40 and 41. | ||
Finally, another type of amphiphiles were prepared based on ADOTA+ cores (called A-domain) linked to poly-2′,5′-dioctyl-4,4′′-terphenylenecyanovinylene tails (called J-domain). Those JA-entities 42 also led to self-assembled Langmuir films at the water–air interface (Fig. 12).46 The J-domain could serve as a light-harvesting antenna (absorbing at 350 nm) able to transfer the energy to the fluorescent A-domain (emitting at 570 nm). In Langmuir–Blodgett (LB) films deposited on glass surfaces, a current–voltage characteristic of a rectifying behaviour was observed. Electron transport applications were further evaluated.47
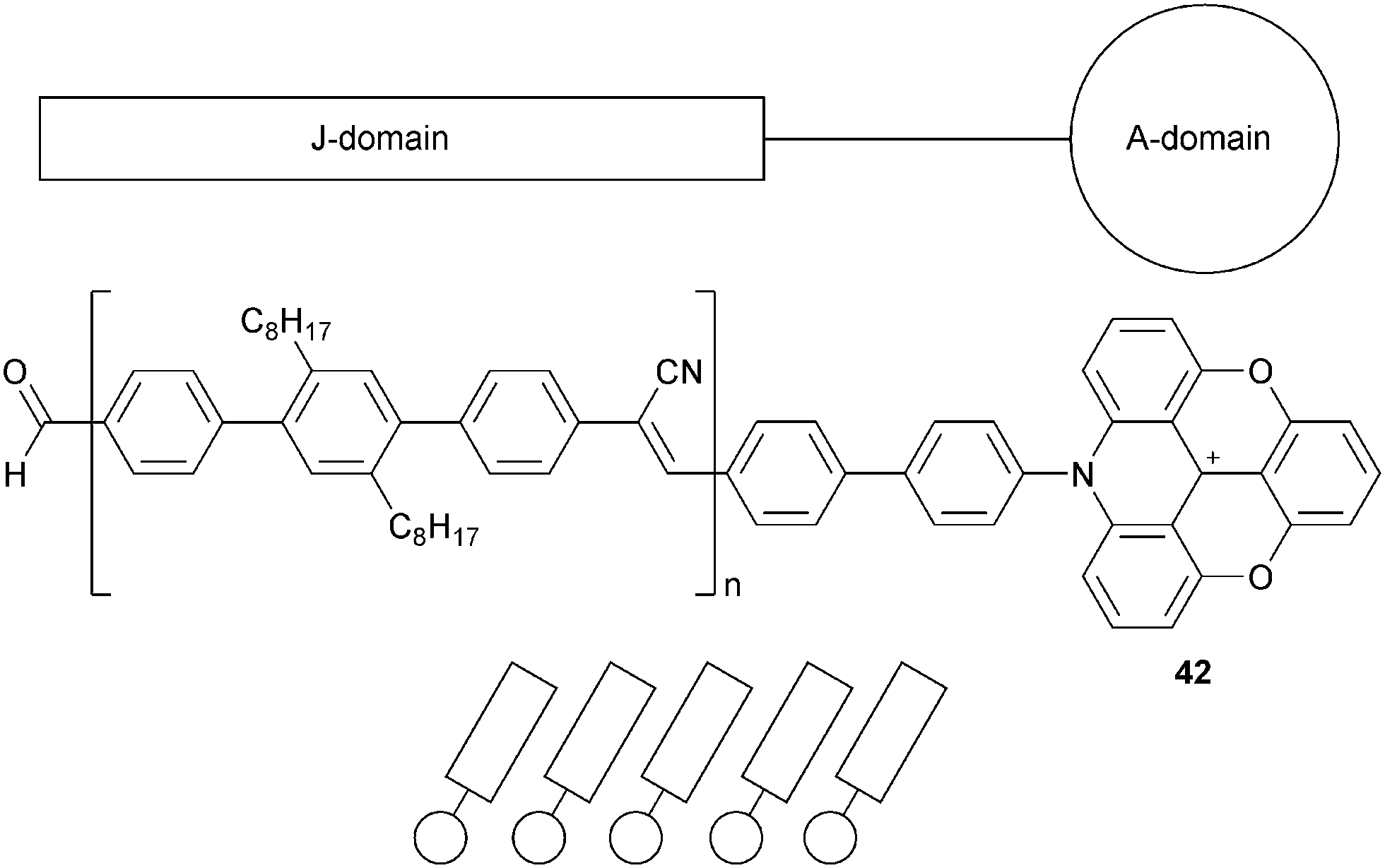 | ||
| Fig. 12 JA-assemblies based on ADOTA+42. | ||
Helicenes and chirality
As discussed previously, due to strong repulsions occurring between the methoxy or benzo extremities, compounds 6, 31, 34, 7, 37 and 38 adopt chiral helical conformations. As such, they can be considered as cationic [4] and [6]helicenes. The left- and right-handed enantiomers are characterized by their M and P configurations respectively (Fig. 13 with 6 as an example). In this series, DMQA+ salts of type 6 were the first to be resolved using an asymmetric ion pairing strategy.48 Associating racemic 6 with enantiopure BINPHAT anions (Fig. 14)49 resulted in a selective precipitation of a single diastereomeric salt (d.r. > 49:1).50 Ion exchange metathesis with HPF6 then afforded enantiopure (+)- and (−)-[6][PF6] salts for which P and M configurations were assigned by vibrational circular dichroism (VCD). Recently, similar resolution and absolute configuration assignment protocols were used for the enantiomeric separation of [6]helicene 7.22
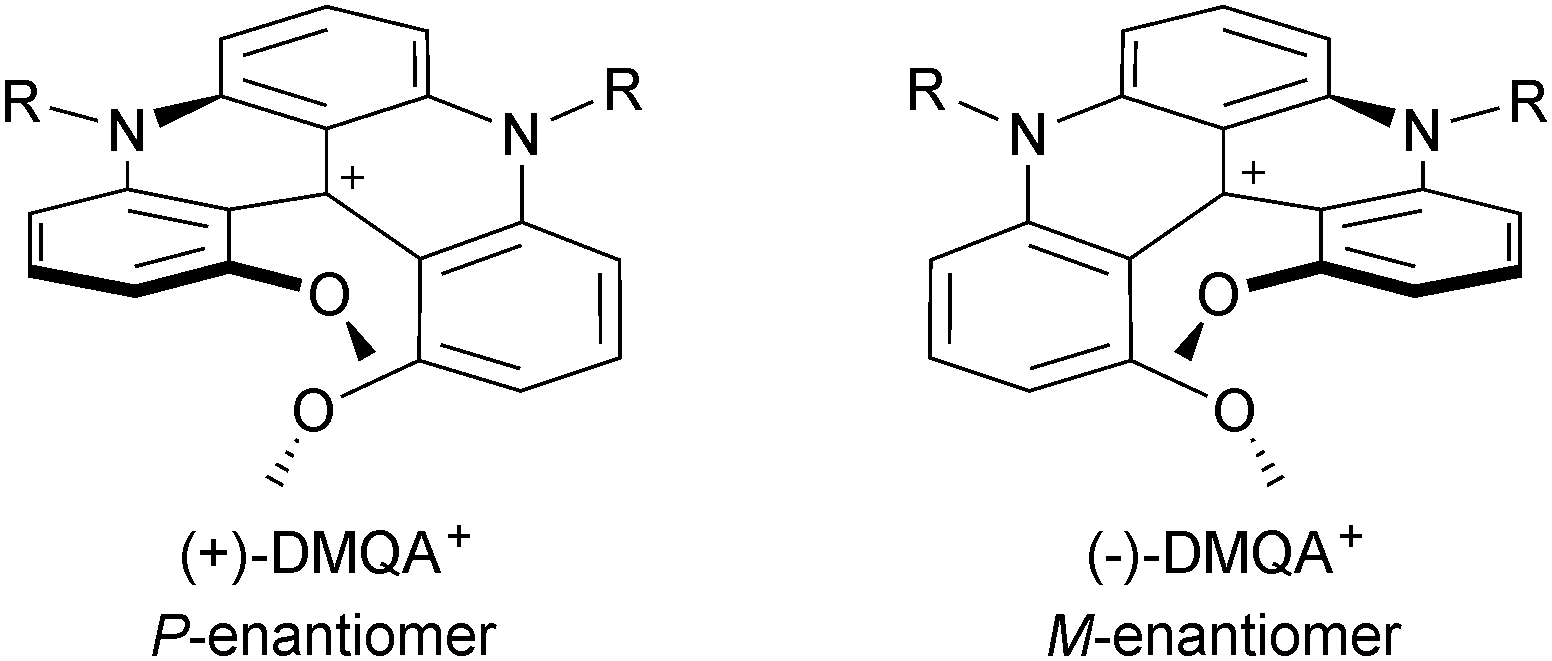 | ||
| Fig. 13 (+)-(P) and (−)-(M) enantiomers of DMQA+6. | ||
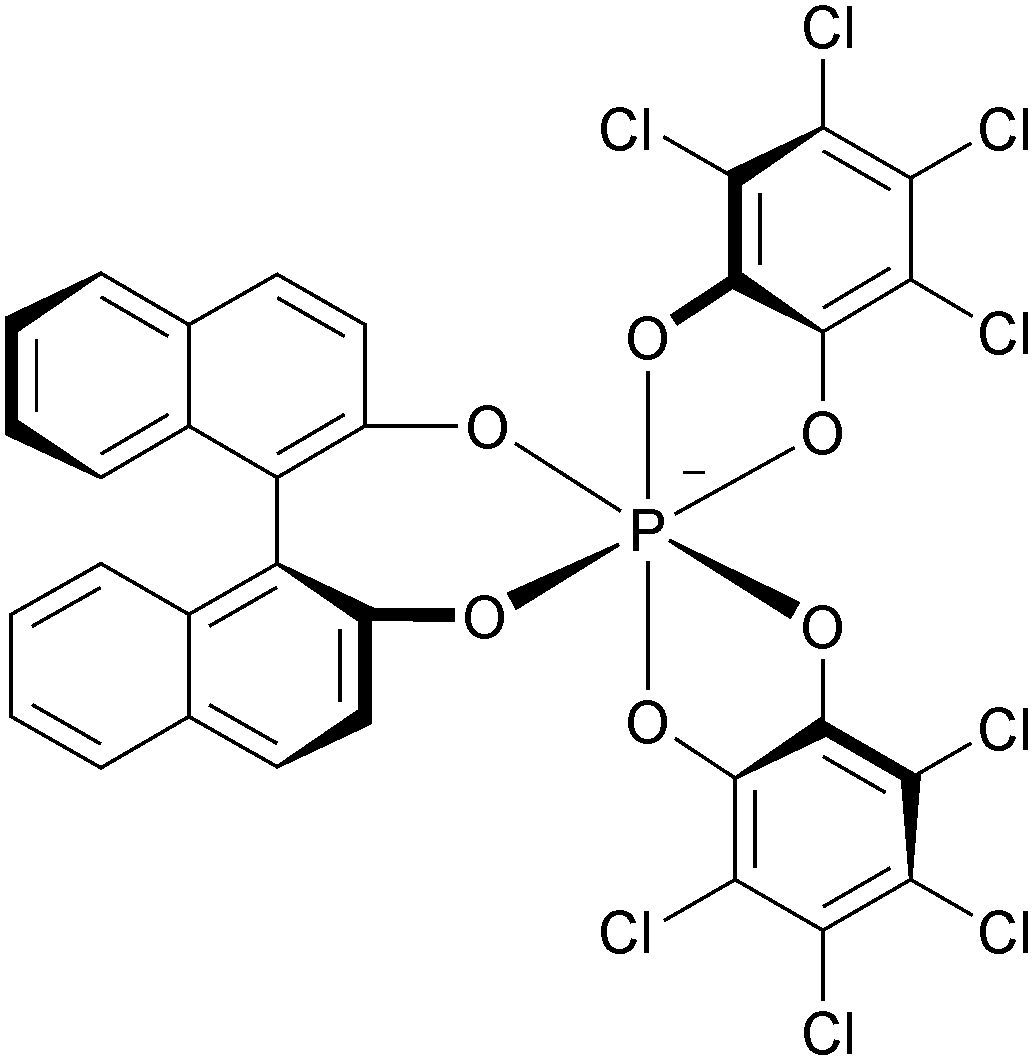 | ||
| Fig. 14 Structure of the (Δ)-BINPHAT anion. | ||
Two other resolution strategies were developed. Chiral stationary phase (CSP) chromatography was employed to separate the enantiomers. For instance, with 6, an efficient separation was obtained on an analytical scale using reverse CSP conditions.51 However, on a preparative scale, it was not practical. Salts 6 were thus transformed into neutral adducts by reduction or alkylation reactions prior to the separation (with hydride or strongly nucleophilic reagents, see section “Neutral Products of Addition”). Then, a larger panel of stationary phases could be used and the separations proceeded well under normal phase conditions. This three-step strategy (reduction, separation, reoxydation) was utilized for the isolation of the enantiomers of dioxo[4]helicene 34.21 Alternatively, in an approach which is classical to helicene chemistry, a chiral auxiliary can be introduced to promote the direct separation. In this context, racemic 6 was reacted with the carbanion of enantiopure (R)-methyl p-tolyl sulfoxide. The resulting diastereomeric products of addition to the central carbon (R,M)- and (R,P)-43 were easily separated by column chromatography (SiO2, Et2O, ΔRf = 0.3).52 Removal of the chiral sulfoxide moiety through a Pummerer fragmentation afforded then the enantiopure cationic helicenes (see section “Neutral adducts of [4] and [6]helicenes”) (Scheme 14).
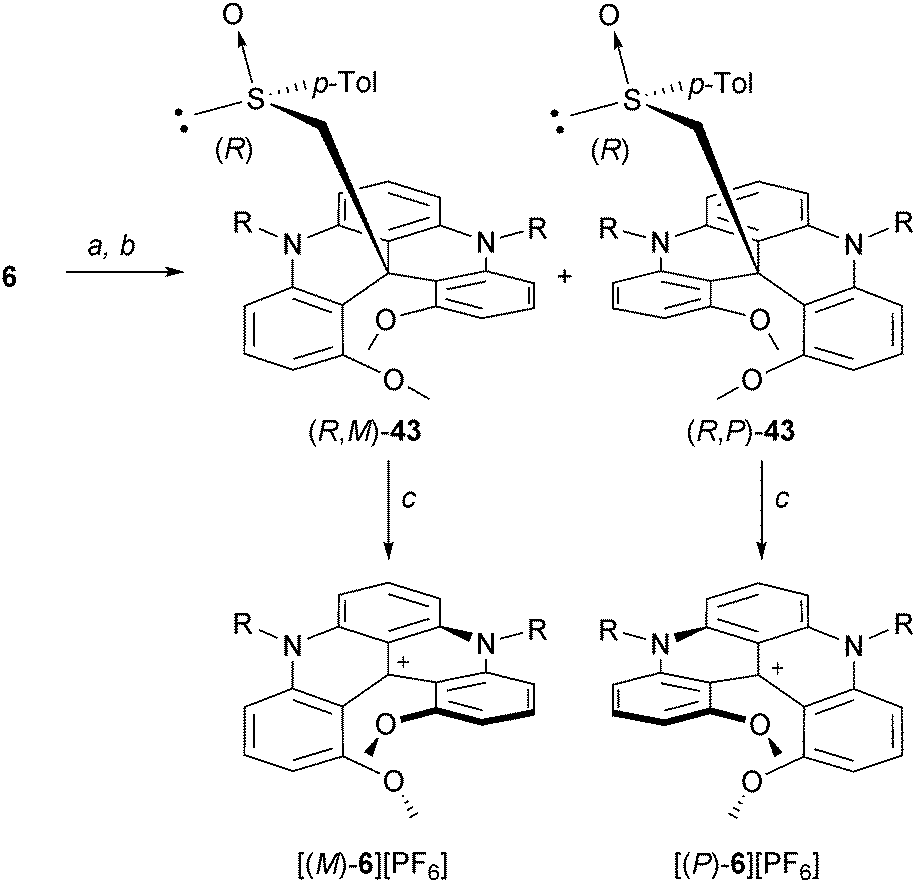 | ||
| Scheme 14 Resolution of 6via a chiral sulfoxide auxiliary approach. a: (+)-(R)-methyl p-tolyl sulfoxide, LDA, THF, 0 °C; b: chromatography (SiO2); c: aq HPF6, acetone. | ||
With enantiopure dioxo 34 and diaza[4]helicenes 6 in hand, a barrier of racemization was measured, 34 and 6 racemizing in the 90–120 and 200–230 °C range of temperatures respectively. For 34, at 120 °C, kinetic data afforded half-life (t1/2 = 3.6 min) and ΔG‡ (115.9 kJ mol−1) values while for 6 at 200 °C, t1/2 = 183 h and ΔG‡ = 172.8 kJ mol−1.53 For diaza[6]helicene 7, it was not possible to determine these parameters as the compound started to decompose (>190 °C, dibutyl sulfoxide) before the enantiomerization. This indicates that these compounds are highly configurationally stable, in particular in direct comparison with, for instance, classical [6]helicene (t1/2 = 13.4 min, ΔG‡ = 154.3 kJ mol−1 at 196 °C).
Recently, these experimental values were compared with theoretical ones as the racemization mechanism for 34, 6 and putative azaoxa[4]helicene 44 was determined in silico (B3LYP, 6-31+G*).54 In Fig. 15 are reported the calculated ground state and transition state structures. For the N,N′-dimethyldiaza[4]helicenium ion of type 6, the barrier calculated (166.4 kJ mol−1) was in very good agreement with the experimental value. Generally speaking, the enantiomeric stability of the [4]helicene ions depends on the nature of the heteroatoms within the helical framework – the stability increasing with the van der Waals radii and donor ability of the bridging atoms. As a consequence, the dioxo 34 ion is less stable than (putative) dicarbo and diaza[4]helicene ions. If the disulfo[4]helicenium ion could be made, it was predicted that it would be the most stable of the series. In the particular case of nitrogen, the resonance effects contribute strongly to the configurational stability.
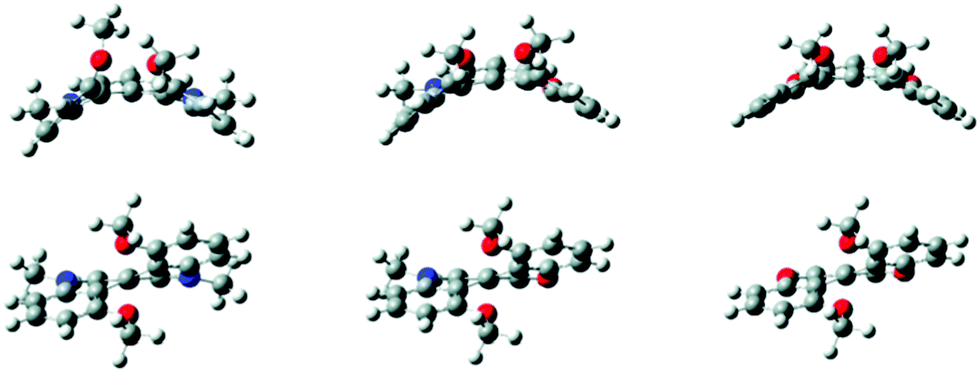 | ||
| Fig. 15 Calculated transition state (top) and equilibrium (bottom) structures of cationic diaza-, azaoxa- and dioxo[4]helicenes 6, 44, 34. Reproduced from ref. 54. | ||
Some of the chiroptical properties of 6, 34 and 44, such as the electronic circular dichroism (ECD) and VCD spectra, were furthermore measured. It was shown that VCD is particularly favorable for the determination of the absolute configuration of the enantiomers as the rigidity of the skeletons induces few conformations and hence relatively straightforward calculations of the theoretical spectra. A thorough study on the ECD spectra of 6, 34 and 44 was also performed detailing the influence of the heteroatoms on the parameters that control the rotational strength of the absorption peaks.54,55 In a related study, a series of compounds was screened in silico to determine which derivatives would present stronger circular polarized luminescence.56
Applications of cationic triangulenes and helicenes
With these moieties possessing rather unusual chemical and physical properties in hand, quite a few applications were conceived spanning several domains of chemistry, from synthesis to biological chemistry through supramolecular, coordination, chirality, etc.Synthetic applications: catalysis and photo-oxidation
For instance, TATA+ ions 5 (pKR+ > 20, vide supra), which are among the most stable carbenium salts of the series, can be used in phase-transfer catalysis (PTC). PTC is generally popular because it is a reaction method that typically requires simple procedures, mild conditions, inexpensive reagents and solvents, and has the ability to scale up the reaction. Unreactive cationic species are required. The fact that TATA+ ions do not react in strongly basic and nucleophilic reaction mixtures was thus seen as an advantage for a PTC application. Lipophilic salts of [5][BF4] (Fig. 1, R = Pr, Hex, Dec) performed equally well or better than usual PTC catalysts such as tetrabutylammonium bromide (TBAB) and 18-crown-6.57 Alkylation, epoxidation, cyclopropanation and aziridination reactions were all successfully achieved (Scheme 15).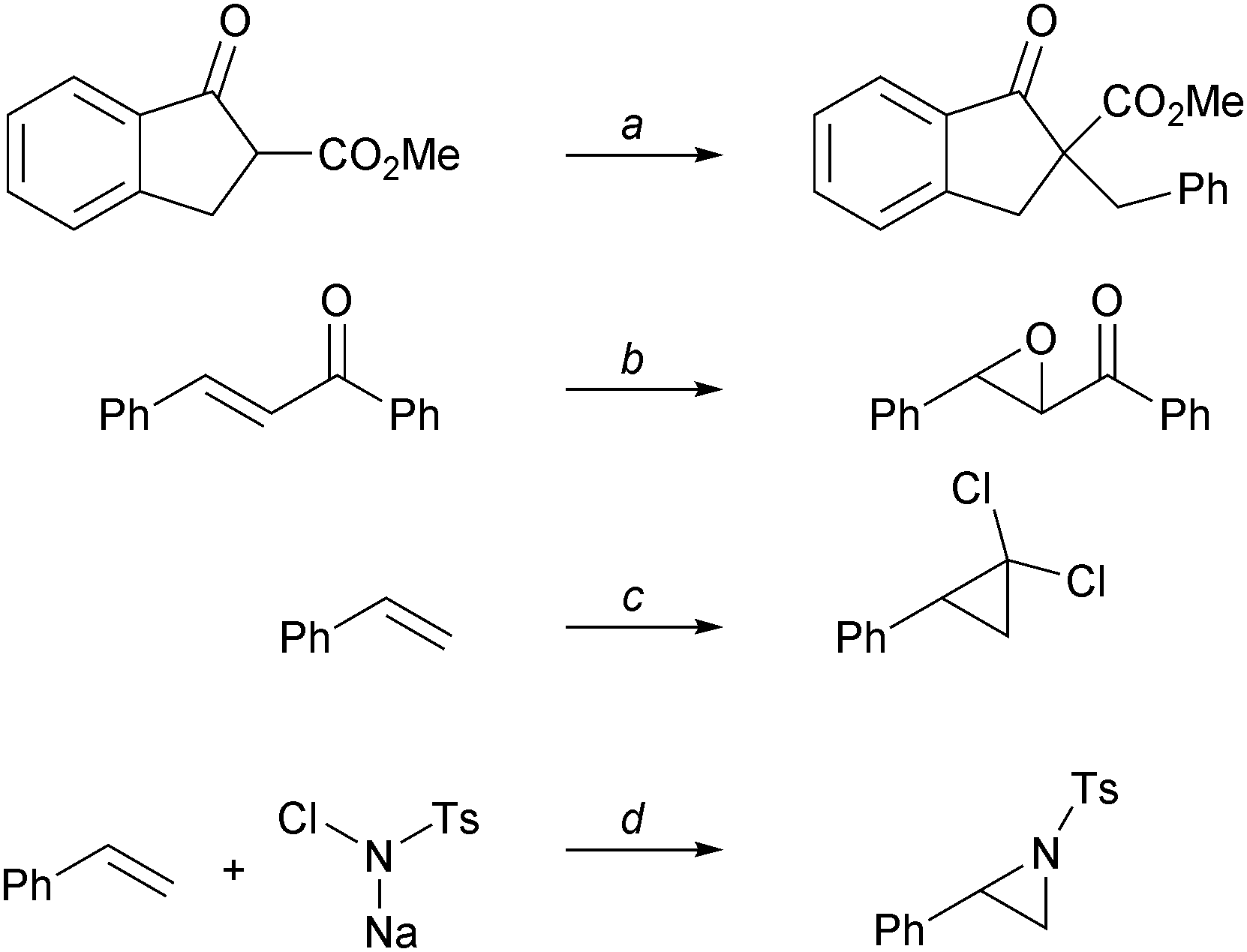 | ||
| Scheme 15 Phase transfer catalysis. a: [5][BF4] (1–10 mol%), PhCH2Br, 50% KOH aq, 20 °C; b: [5][BF4] (10 mol%), 30% H2O2, 50% KOH aq, triton X-100 (1 mol%), iPr2O, 20 °C; c: KOH powder, CHCl3, [5][BF4] (2 mol%), CH2Cl2, 40 °C; d: [5][BF4] (10 mol%), I2, CH2Cl2/H2O, 20 °C. | ||
In these previous reactions, it was not necessary to pay attention to the presence or the absence of light in the laboratory. This was not the case however for the air oxidation reaction of benzylic primary amines to imines that occurred in the presence of catalytic amounts of acridinium intermediates 23 or DMQA+6.58 A slow but effective photo-oxidation of the C–NH2 bond was observed when a source of light was present (summer sunlight in Geneva was sufficient for instance).
Biological applications: nucleic acid interactions and bioimaging
A series of studies on the interactions of the carbenium ions with nucleic acids were reported, and that of TOTA+1 and [4]helicene 6 in particular. First, independent reports demonstrated, through principally fluorescence quenching experiments,32 that TOTA+ ions interact strongly with G-residues as single nucleotides or within DNA sequences. Under irradiation, the interaction resulted in an electron-transfer from the base to the cation and hence in a photo-induced oxidation reaction.59 Precise mechanistic information was afforded through induced circular dichroism (CD) and X-ray diffraction analyses demonstrating that an intercalation of TOTA+ in DNA has occurred (Fig. 16). A binding constant of 41000 M−1 was determined and, not surprisingly, the preferred binding sites in duplex DNA were C–G base pairs. It was suggested that the base radical cation formed after photo-induced oxidation by TOTA+ migrates to the 5′-GG-3′ sequence via a hopping mechanism.60 Extension of this chemistry to telomeric sequences, in particular the G-quadruplex, was afforded.61 However, the interaction between TOTA+ and quadruplex DNA was weaker than between duplex DNA and TOTA+. Surprisingly, TOTA+ mediated photo-oxidation of a guanine rich sequence was more prompt to occur in duplex DNA than in quadruplex DNA, which is against the general observation of the protective capability of the telomers.
 | ||
| Fig. 16 [d(CGATCG)]2–TOTA+2 complex. Reproduced from ref. 60. | ||
Double-stranded DNA is of course chiral and its interaction with cationic helicenes as single enantiomers in place of achiral triangulene ions was thus deemed interesting.62 Three different ions 6 were utilized (Fig. 1, R = Me, Pr, CH2CH2OH).48 In all instances, fluorescence quantum yields and lifetimes of 6 increased significantly in the presence of right-handed B-DNA and the binding constants were found to be larger for the M- over the P-enantiomers of 6 by a factor of 1.2 to 2.3. Intercalation is only likely with the less-sterically hindered moiety (R = Me) whereas aggregates and more hindered helicenes interact with the major groove. It was further shown that the racemic 6 aggregated quite effectively in water in comparison with the enantiopure materials leading to the lowest binding values of all.33
Near infra-red fluorescent dyes (700–1100 nm, NIR) have also gained a large interest in bioimaging over the past decade as safe, noninvasive imaging/contrasting probes. Unfortunately, when such fluorophores possess high molar extinction coefficients, they tend to be characterized by moderate quantum yields and short fluorescence lifetimes. It was thus interesting to study ADOTA+3 and DAOTA+4 (red absorption, NIR emission, high ε values, quantum yields ca. 60%, fluorescence lifetime >20 ns)30 in the context of in vitro and in vivo imaging.63 Derivatives with butyric acid side chains 45 and 46 were prepared and functionalized by trans-amidation reactions with hexadecyl amine or arginine (Fig. 17, top). Direct labeling of proteins such as avidin, streptavidin and Immunoglobulin G (IgG) could be achieved. Amphiphilic 45 and 46 (R = hexadecyl) were used to detect small lipid vesicles while arginine-modified cations were utilized for intra-cellular localization. Helped by the long fluorescence lifetime (>20 ns), it was possible to perform in vivo fluorescence lifetime imaging (FLIM) under time-gated detection (10 ns after excitation). Clearer imaging resulted thanks to a silencing of the background emission (Fig. 17, bottom).
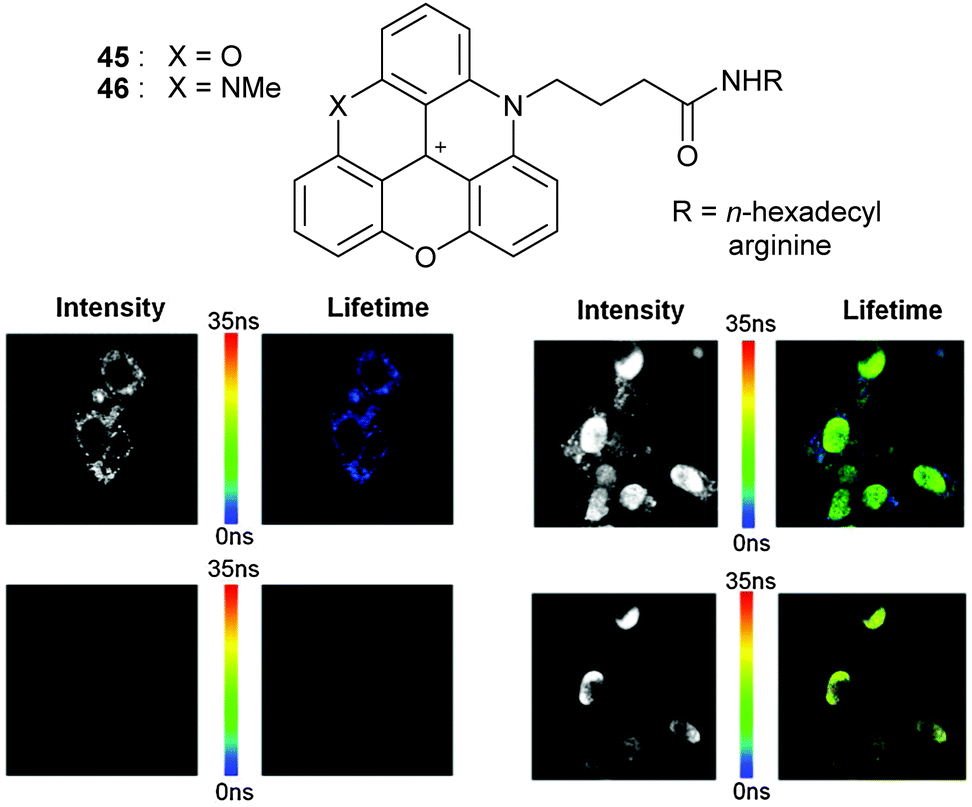 | ||
| Fig. 17 FLIM of the 4T1 endothelial cell line without (left) and with (right) DAOTA-arginine with immediate detection (top row) or 10 ns gated detection (bottom row). In short, background fluorescence is only observed on the top images. Only DAOTA-arg is detected on the two bottom right pictures. Adapted from ref. 63. | ||
In another study, 45 was conjugated to an anti-rabbit Immuglogulin G (antiIgG). As mentioned (vide supra), ADOTA+ ions possess marked fluorescence anisotropy properties and the formation of a complex between 45 (antiIgG) and its target protein rabbit IgG could be monitored by steady-state anisotropy and time-resolved methods.64 Fluorescence anisotropy being a ratiometric measurement, this method allowed an accurate detection of the binding values.
Supramolecular assemblies
Tripodal ligands for metallic cations play an important role in biology and chemistry alike. Researchers in this field are often interested in developing multimodal probes that can, for instance, combine optical (fluorescence) and MRI (paramagnetic metal ion) capabilities. In this context, TATA+5 was appropriately modified for the complexation of lanthanide cations through the incorporation of pyridyldicarbonyl (PDC) chains (Fig. 18).65 Complexation of 47 with a Eu(III) cation was evidenced in solution and in the solid-state. In terms of photophysical properties, it was shown that 47 and [Eu47]4+ fluoresced alike. However, the europium-centred emission was weak due to the concurrent energy transfer from pyridyldicarbonyl moieties to the extended aromatic platform and, unfortunately, a sensitization of Eu(III) did not happen upon excitation of the triangulene core.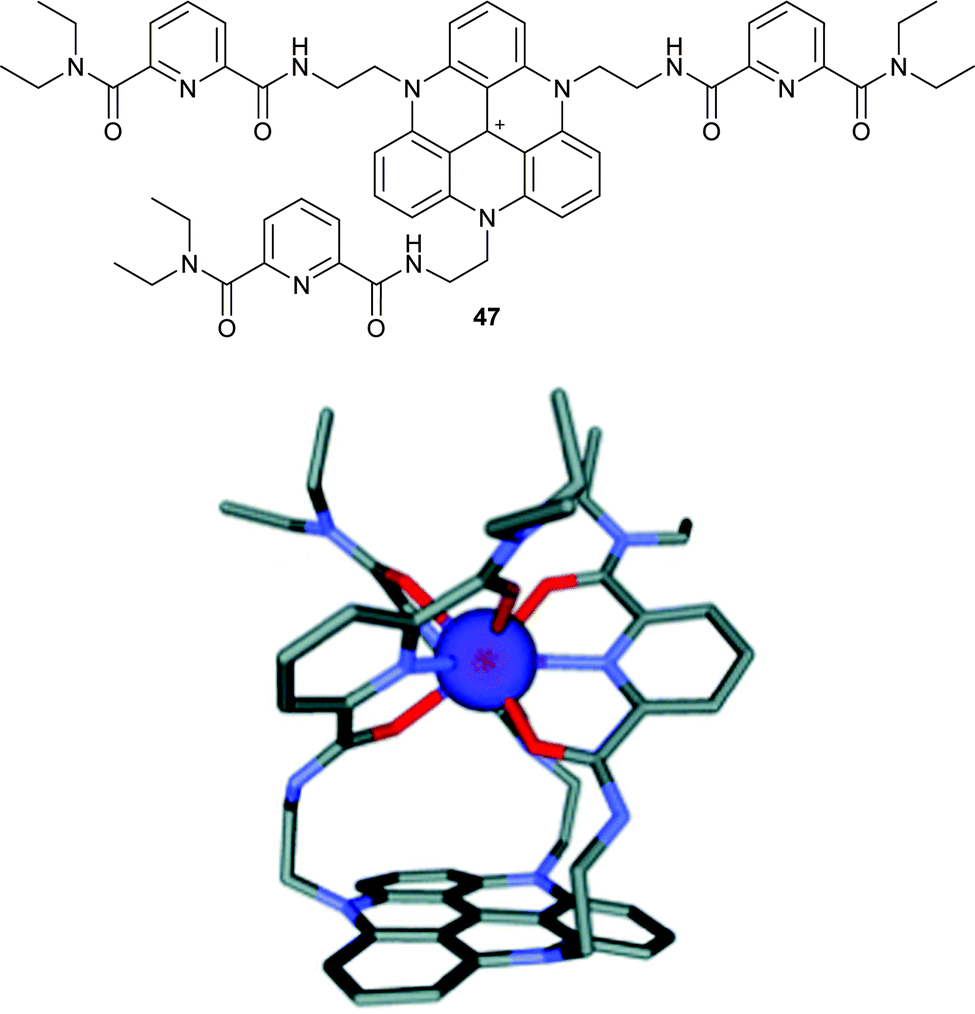 | ||
| Fig. 18 Structure of 47 (top) and the model of the [Eu47]4+ complex (bottom). Reproduced from ref. 65. | ||
Neutral products of nucleophilic addition: synthesis and applications
Preamble
In previous parts of this review, care was taken to emphasize the stability of the carbenium ions in (highly) basic media. It was stated that ions 3, 4, 5, 6, 11, or 25 undergo only with difficulty addition reactions at the central carbon atom. However, it was briefly mentioned that hydrides21 or sulfoxide carbanions48 can react with some of the carbocations to generate neutral adducts. It is now our purpose to show that this transformation leads to interesting structures of their own; the products being useful for a variety of applications.First, it is necessary to realize that the additions of nucleophiles to the central carbon of the carbenium derivatives always proceed with a highly noticeable signature which is a loss of color. In fact, transformation of the sp2-hybridized centers into sp3-carbon atoms interrupts effectively the extended conjugation. The adducts become colorless and are hence denominated leuco for that reason.66 The change of hybridization also modifies profoundly the geometries. For instance, planar triangulene precursors are transformed into cup-like molecules. Several applications making use of the resulting geometries or of the added appendages will now be detailed.
Cup-like molecules and applications
The first mention of the creation of bowl shaped molecules upon addition of alkyl groups to cationic triangulenes was reported by Siegel and coworkers.67 It was shown that TOTA+1 reacts with RLi or RMgX reagents to provide the corresponding adducts 48 named trioxatricornan (Scheme 16, R = Me, vinyl, allyl). Interestingly and importantly, the new electron-rich derivatives underwent readily electrophilic substitution reactions such as brominations or nitrations. Regioselectivity was not controlled at that stage as 1:1 mixtures of products were obtained. However, treatment of the mixture of brominated isomers 49 with sodium amide or (in situ generated) lithium dimethylamide led to an interesting and elegant triple cine substitution and the isolation of the tris para-amino 50. An alternative route for the making of derivatives of type 50 was described later taking advantage of the facile synthesis A3-TOTA+ compounds 11 and their reactivity with MeLi.6
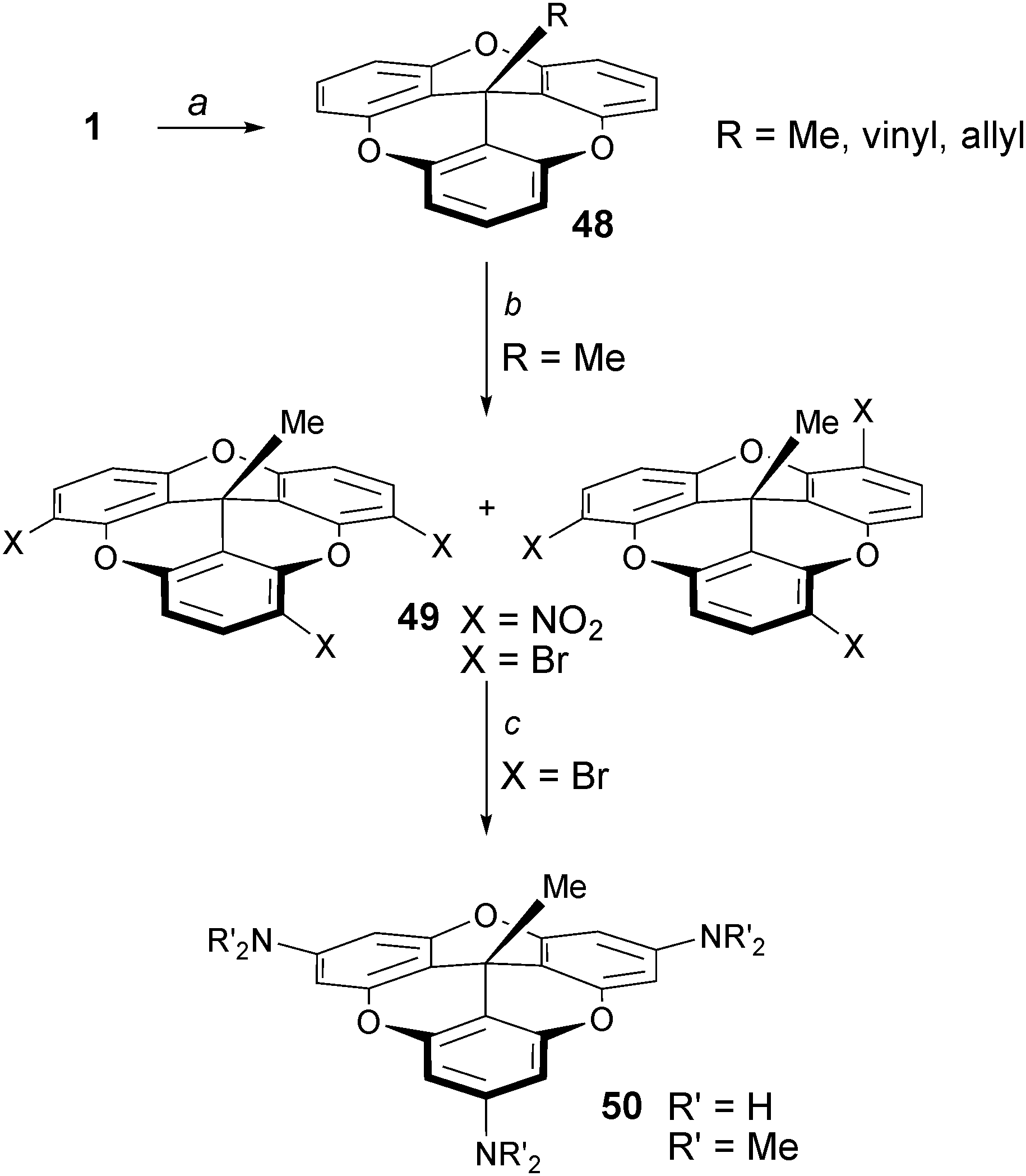 | ||
| Scheme 16 Synthesis of trioxatricornan 50. a: MeLi-LiBr, Et2O, THF, 20 °C, 1 h or CH2CHMgBr, THF, 20 °C, 7 h or CH2CHCH2MgBr, THF/Et2O, 0 to 20 °C, 16 h; b: HNO3 or Br2, CH3COOH, 80 °C, 30 min; c: NaNH2, liq NH3, THF, −78 to 20 °C, sealed tube, 19 h or HNMe2, n-BuLi, THF, −78 to 20 °C, sealed tube, 48 h. | ||
Compound 50 (R′ = H) was then used for the preparation of macrocyclophane 51 using a three-step protocol involving a protection of the amino groups, alkylations of the N atoms with flexible terminal alkynyl chains and a final cyclotrimerization reaction installing a central benzo group (Fig. 19).68 Solution and solid-state structures were investigated and compared with the calculated geometry. The newly formed aryl ring maintained a certain distance with the tricornan unit and a cavity was formed. Important changes were observed upon modifying the linkers between the benzo ring and the tricornan core. For instance, introduction of heteroatoms induced a collapse of the cavity.69 In another study, precursor 50 was used for the formation of the tris-catecholamide ligand 52 which underwent facile complexation with ferric ions (Fe3+) to form metallo-macrocyclophane complexes.70
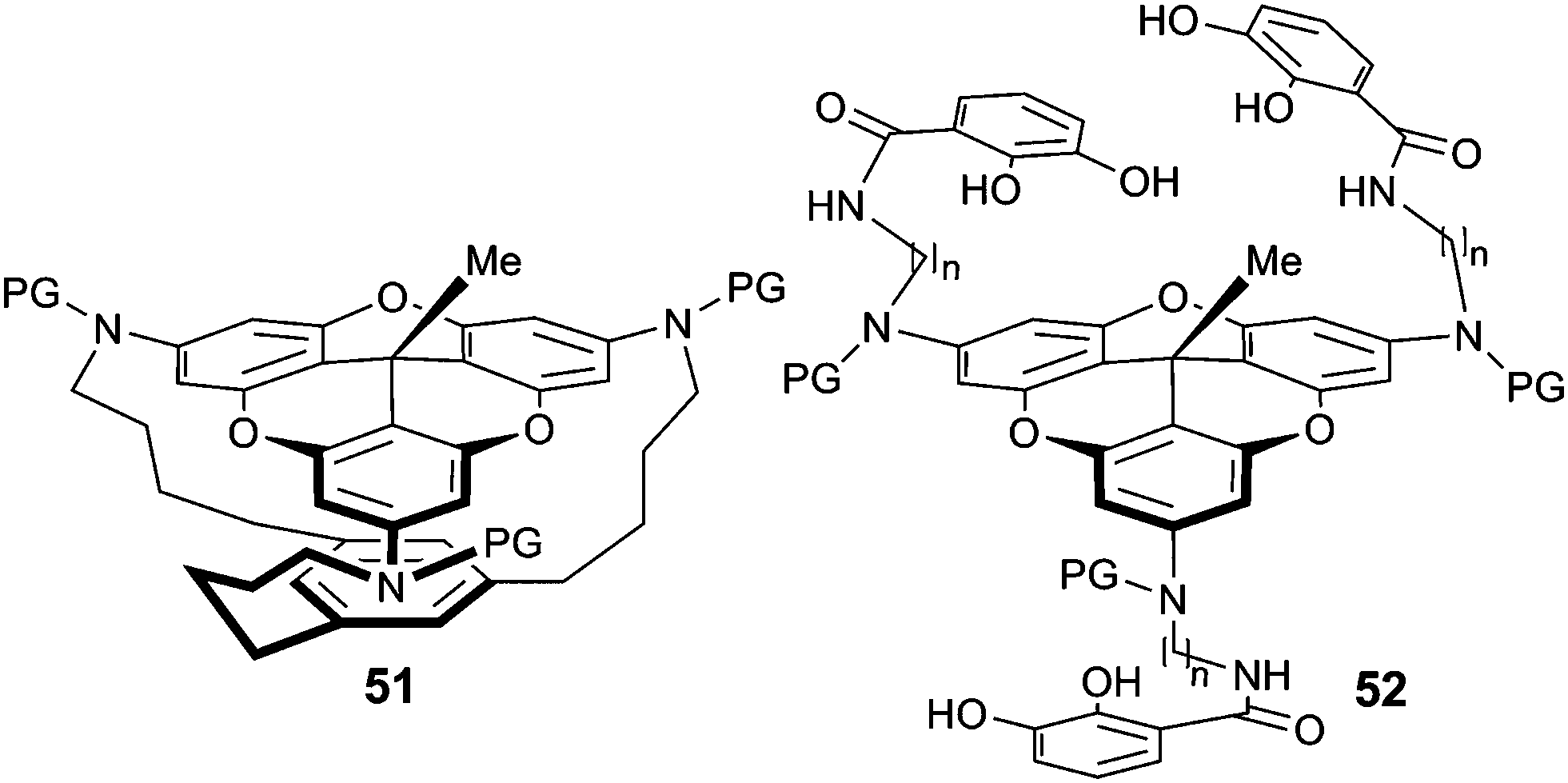 | ||
| Fig. 19 Macrocyclophanes 51 and 52. | ||
The regioselective formation of substituted trioxatricornan adducts can also be achieved using an ortho lithiation/halogenation route.71 Treatment of a compound of type 48 (R = CH2CH2OMOM) with t-BuLi/TMEDA followed by an addition of diiodoethane afforded exclusively chiral (racemic) C3-symmetric triiodo 53 (Scheme 17). This compound was further transformed into macromolecules 54 containing folded secondary structures. This is promoted by intramolecular hydrogen bonding interactions between the oxygen atoms of the tricornan core and the hydrogens of the external amide groups.
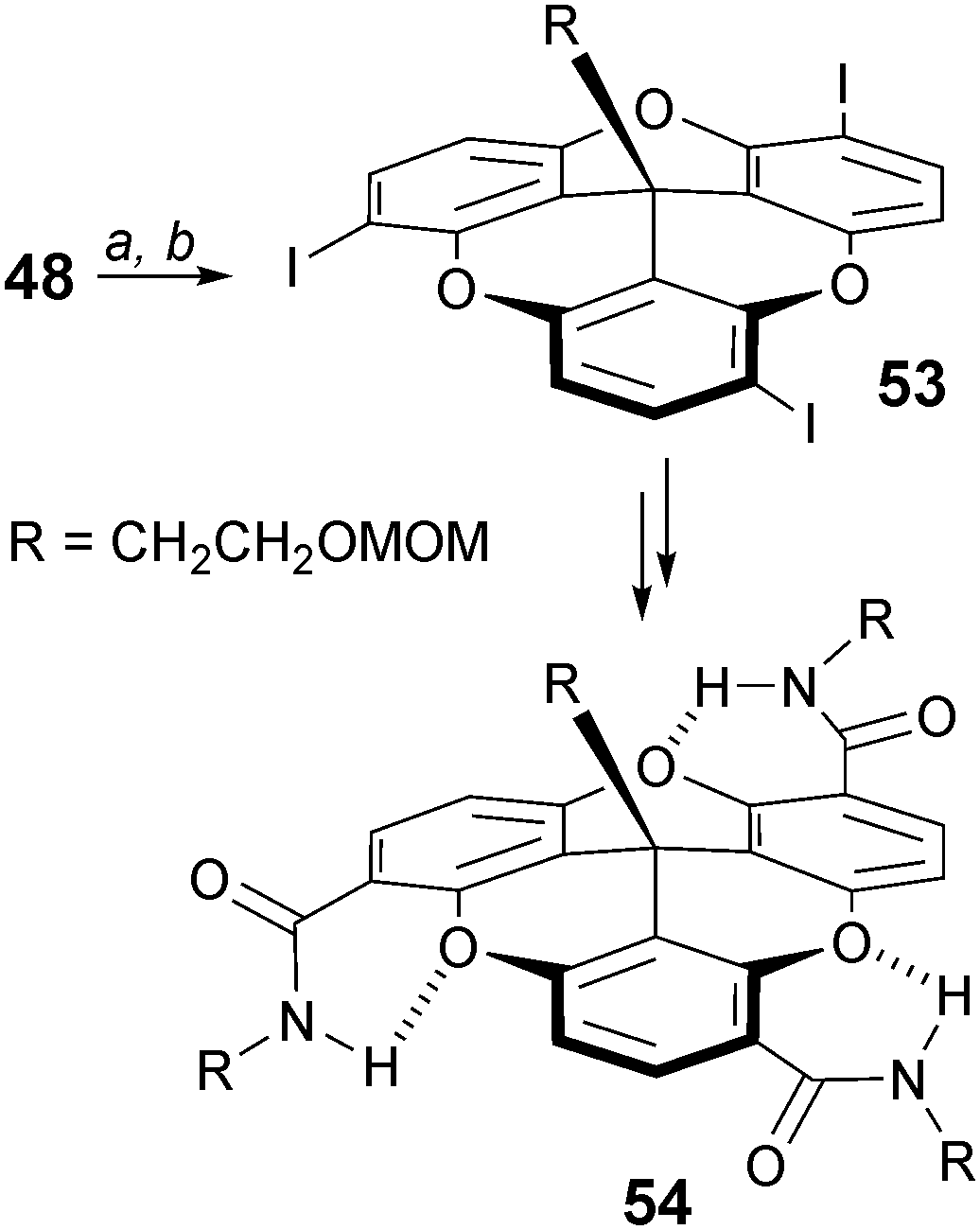 | ||
| Scheme 17 C 3-symmetric folded 54. a: TMEDA, t-BuLi, Et2O, −78 to 0 °C, 12 h; b: ICH2CH2I, 0 °C, 12 h. | ||
Another type of chiral cup-like molecule can be prepared using unsymmetrical DAOTA+55. This precursor was prepared in three steps from 2 by sequential additions of aniline, propyl amine and a final oxo ring closure (combined yield 28%).72 Addition of MeLi proceeded well to form chiral 56 (Scheme 18). The enantiomers were separated by CSP-HPLC and the absolute configuration was determined by VCD.
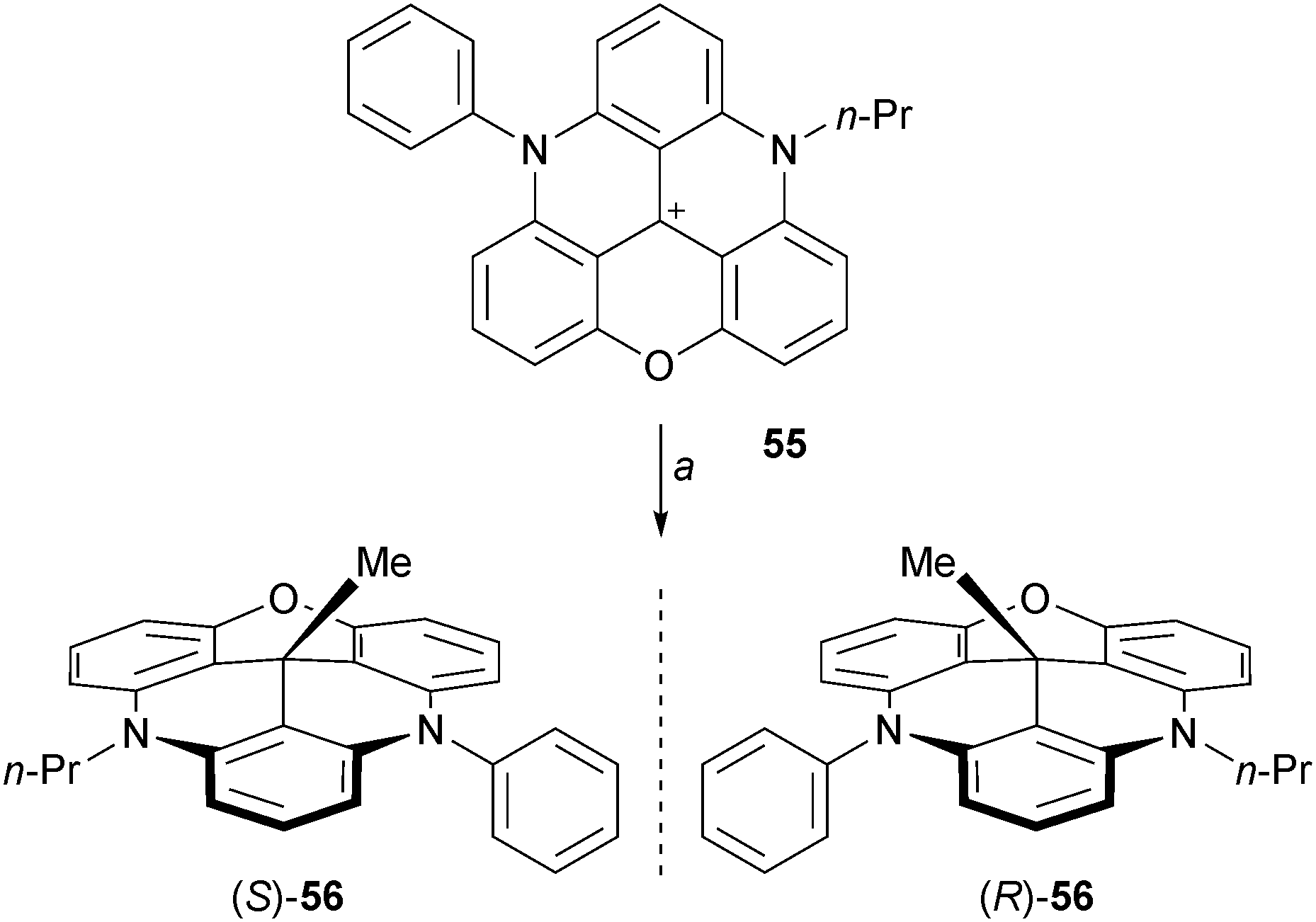 | ||
| Scheme 18 Chiral diazaoxatricornan 56. a: MeLi (1.3 equiv.), THF, 20 °C, 16 h, 97%. | ||
TATA+ cations also reacted with alkynyl and aryllithium reagents to form products of additions at the central carbon. Interestingly, these adducts 57, and their TATA+ precursors, formed hexagonally ordered adlayers on Au(111) surfaces (Fig. 20).73 The triangulene cores adsorbed in a planar fashion. The length of the flexible side chain controlled the space between the central units and, as a consequence, the coverage density. The groups introduced during the nucleophilic attacks are orthogonal to the surface. The adlayers were stable, suggesting a strong adsorption. Further studies were reported with, for instance, photo-switchable appendages and evidence for chiral arrangements was noted with tris-octyl-TATA+ derivatives.74
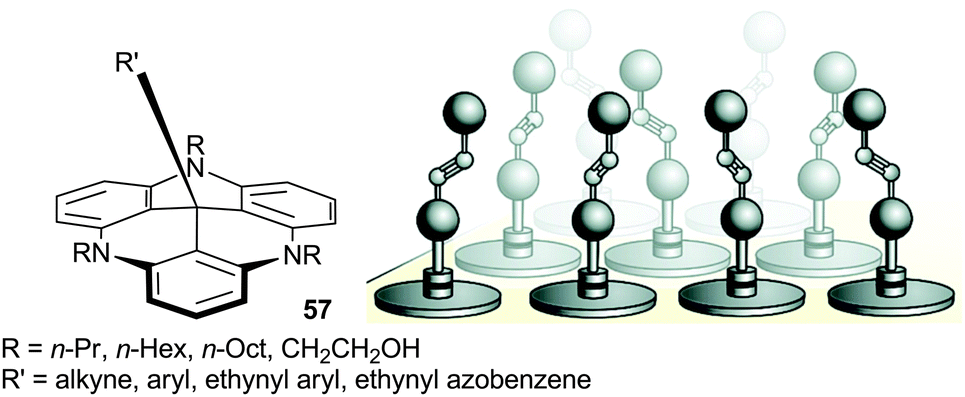 | ||
| Fig. 20 Representation of the adsorption of functionalized 57 on the Au(111) surface. Adapted from ref. 73. | ||
Finally, as previously mentioned in the discussion concerning the chirality of cationic [4] and [6]helicenes, the carbanion from methyl p-tolyl sulfoxide is an excellent nucleophile able to react with the carbenium ions of study. In the context of this section, this anion was reacted with ADOTA+ and TATA+ cations 3 and 5. Adducts 58 and 59 were isolated and submitted to conditions that favor typically Pummerer rearrangements (TFAA, CH2Cl2). Interestingly, these two compounds behaved differently. While 59 reacted through a fragmentation pathway only, ADOTA+ derivative 58 presented a split reactivity; half-rearrangement (compound 60) and half-fragmentation towards 3 (Scheme 19).75 This (and other experiments) indicated that the driving force towards a Pummerer fragmentation pathway is a strong electrofugal character of the cation; a pKR+ value higher or equal to 14 being necessary.
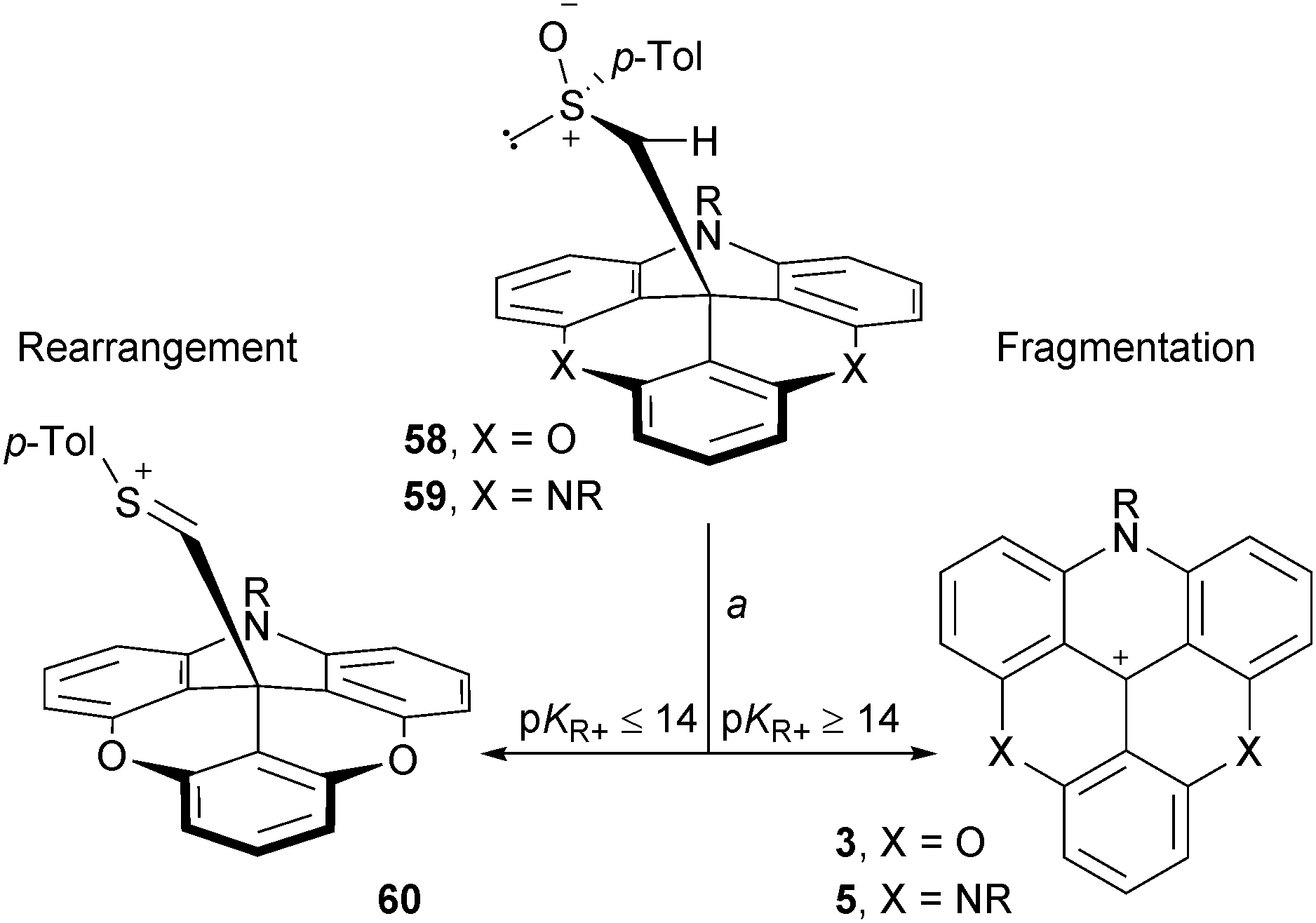 | ||
| Scheme 19 Classical Pummerer rearrangement vs. unusual Pummerer fragmentation reactions. a: (CF3C(O))2O, CH2Cl2, 20 °C, 20 min. | ||
Neutral adducts of [4] and [6]helicenes
Rotaxanes and pseudo-rotaxanes are captivating objects composed minimally of a thread-like molecule surrounded by a macrocycle. Usually, (pseudo)rotaxanes are achiral when either the thread or macrocycle (ring) are non-oriented. In contrast, if a dissymmetry is present in both structural elements, the resulting rotaxane becomes inherently chiral. In this context, an ammonium thread-like molecule 61 was prepared by (i) addition of the carbanion of acetonitrile onto DMQA+6, (ii) reduction of the nitrile moiety, (iii) formation of a secondary amine by reductive amination and finally (iv) by acidification with HPF6 (Scheme 20). The resulting chiral thread 61 was mixed with both non-oriented and oriented crown ether rings.76 Formation of pseudo rotaxane adducts was clearly evidenced by NMR spectroscopy. While a stereocontrol could not be evidenced in the presence of the oriented ring with the PF6 salt of 61, a change to a more lipophilic counterion (TRISPHAT77) induced a low but definite diastereoselectivity (d.r. 54:46).
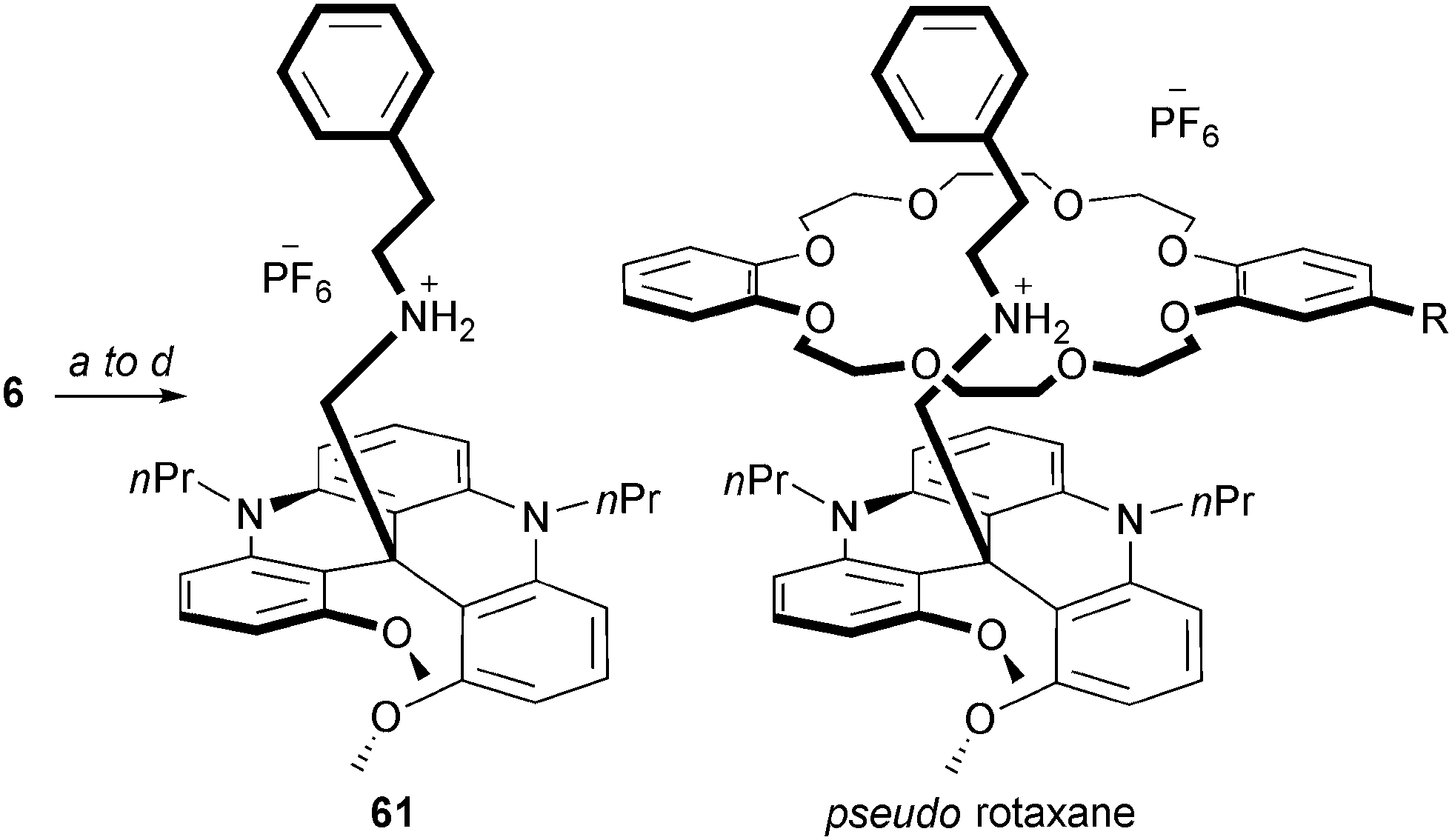 | ||
| Scheme 20 Preparation of pseudo-rotaxanes. Achiral ring: R = H, chiral ring: R = (3,5-bistrifluoromethyl)phenyl. a: NaH, CH3CN, 20 °C; b: LiAlH4, Et2O, 20 °C; c: PhCHO, MgSO4, NaBH4, MeOH, 20 °C; d: HPF6, acetone, 20 °C. | ||
Using DMQA+ cations as electrophilic substrates, it was also possible to tackle another question of “fundamental” stereochemistry. It concerned the possibility to influence the facial selectivity of the addition reaction onto the trigonal center through the helicity of the skeleton only.17 Using unsymmetrical derivatives of type 6 (two different N–R side-chains), it was demonstrated that the helical framework can be indeed effective in the stereocontrol of the reaction (Fig. 21). Hydride and organolithium nucleophiles afforded adducts with diastereoisomeric ratios up to and higher than 49:1. This geometrical distinction of the two diastereotopic faces is not due to a deformation of the helical framework but is linked to a difference in the substituents on two nitrogen atoms. The N-atom linked to the (bulky) aromatic substituent remains sp2-hybridized from the starting cation to the preferred diastereoisomeric product whereas the more flexible N-atom attached to an alkyl group bends to accommodate the strain induced by the developing nucleophilic attack.
 | ||
| Fig. 21 Nucleophilic attack on the re or si face of the M-enantiomer of 6. | ||
Finally, using a two-step reduction/metalation procedure, it was possible to transform chiral carbenium ions 6 into reactive carbanion intermediates.78 First, a reduction with NaBH4 afforded the leuco methine products which were reacted with an excess of n-BuLi. This umpolung procedure afforded highly nucleophilic carbanions 62 that reacted with “soft” electrophilic reagents such as isothiocyanates and carboxylic acid anhydrides (Scheme 21). Unprecedented thioamide and ketone products of type 63 were obtained in good yields and a complete retention of configuration was observed starting with enantiopure (M)- or (P)-6.
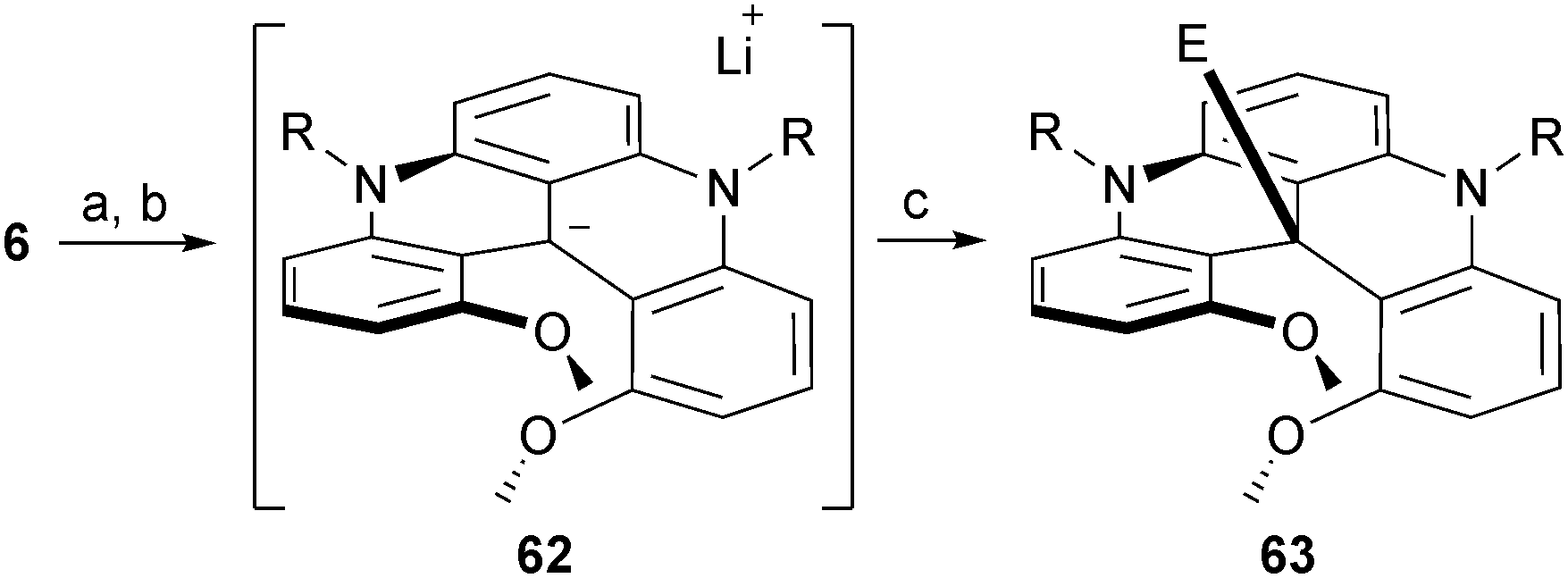 | ||
| Scheme 21 Formation of carbanion 62 and trapping with electrophiles. a: NaBH4, EtOH, 25 °C, 1 h; b: n-BuLi, Et2O, −78 to 0 °C, 30 min; electrophile (10 equiv.), 0 °C, 20 min. | ||
Conclusion
Cationic triangulenes and helicenes are highly stable carbenium ions. They can be stored as carbocations under air and most of them are inert in strongly alkaline aqueous solutions. Triangulenium ions are planar fully conjugated structures whereas the helicene moieties adopt a helical conformation and are chiral as a consequence. Although the first stable triangulene ion (TOTA+) was reported already in the 1960's, the field gained momentum only in the past decade with the preparation of nitrogen containing analogues; the introduction of the nitrogen atom(s) either within the cationic skeleton or at the periphery increasing the intrinsic stability of the cations even further. Besides, these moieties are colorful dyes and their electronic absorption can be modulated as a function of the heteroatom patterns. Moreover, they are fluorescent molecules with moderate to excellent quantum yields; fluorescence lifetime superior to 20 ns being observed for some of them.Combinations of the planar or helical conformations together with the (very) high chemical stability and excellent optical properties have led to multiple applications in the recent years. For instance, the planar triangulenes were used as keystones for the preparation of metallo-macrocyclophanes or for the functionalization of surfaces. Their cationic character has been exploited for charge transport or for phase transfer catalysis. Applications as fluorophores for bioimaging and interactions with DNA were investigated. The chirality of the cationic helicenes was advantageously availed for the enantioselective recognition of B-DNA or for the preparation of chiral supramolecules. As further improvements in triangulene and helicene ion synthesis have been recently reported, the preparation of even more tailored compounds can be expected that will certainly find numerous other (new) applications in the coming years.
We thank the University of Geneva, the Swiss National Science Foundation, the NCCR Chemical Biology (JB, JL) for financial support.
Notes and references
- D. F. Duxbury, Chem. Rev., 1993, 93, 381–433 CrossRef CAS.
- In these examples and all along this review, the anionic counterions that are of course present to balance the positive charge are omitted for clarity reasons, unless otherwise stated.
- J. C. Martin and R. G. Smith, J. Am. Chem. Soc., 1964, 86, 2252–2256 CrossRef CAS.
- The terms TOTA+, ADOTA+, DAOTA+ and TATA+ correspond to trioxa-, azadioxa-, diazaoxa- and triazatriangulenium ions, DMQA+ quotes for dimethoxyquinacridinium ion.
- Helicenes are polycyclic aromatic compounds with nonplanar screw-shaped skeletons formed by ortho-fused benzene or other aromatic rings. For a general review on the topic, see: Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B. Christensen and N. Harrit, J. Am. Chem. Soc., 1998, 120, 12255–12263 CrossRef CAS.
- T. J. Sørensen and B. W. Laursen, J. Org. Chem., 2010, 75, 6182–6190 CrossRef PubMed.
- Alternatively, the carbinol can be obtained from 1,3,5-trimethoxybenzene using n-BuLi in toluene for the ortho-lithiation and diphenyl carbonate as the electrophile, see: M. Wada, H. Konishi, K. Kirishima, H. Takeuchi, S. Natsume and T. Erabi, Bull. Chem. Soc. Jpn., 1997, 70, 2737–2741 CrossRef CAS.
- F. Westerlund, C. B. Hildebrandt, T. J. Sørensen and B. W. Laursen, Chem.–Eur. J., 2010, 16, 2992–2996 CrossRef CAS PubMed.
- It had been earlier noted that MeO groups at the para positions of (formal) carbocationic atoms can be converted into hydroxyl groups by a simple treatment under acidic conditions, see R. Levine and J. R. Sommers, J. Org. Chem., 1974, 39, 3559–3564 CrossRef CAS.
- In water, H-TOTA− is not observed. For H-TOTA+/H-TOTA0, pKa = 3.7; for H-TOTA0/H-TOTA2−, pKa = 7.6.
- B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS.
- B. W. Laursen and F. C. Krebs, Chem.–Eur. J., 2001, 7, 1773–1783 CrossRef CAS.
- Interestingly, it is possible to convert DAOTA+ derivatives into TATA+. In other words, oxo-bridges can be transformed directly into aza-bridges.
- B. W. Laursen and T. J. Sørensen, J. Org. Chem., 2009, 74, 3183–3185 CrossRef CAS PubMed.
- F. C. Krebs, Tetrahedron Lett., 2003, 44, 17–21 CrossRef CAS.
- J. Guin, C. Besnard, P. Pattison and J. Lacour, Chem. Sci., 2011, 2, 425–428 RSC.
- P. Hammershoj, T. J. Sørensen, B. H. Han and B. W. Laursen, J. Org. Chem., 2012, 77, 5606–5612 CrossRef PubMed.
- C. Nicolas, G. Bernardinelli and J. Lacour, J. Phys. Org. Chem., 2010, 23, 1049–1056 CrossRef CAS.
- Tetramethoxyphenylthioxanthenium ion 30 is quite more electrophilic than other precursors and aliphatic amines react with it in irreversible nucleophilic additions to the central carbon atoms.
- J. Guin, C. Besnard and J. Lacour, Org. Lett., 2010, 12, 1748–1751 CrossRef CAS PubMed.
- F. Torricelli, J. Bosson, C. Besnard, M. Chekini, T. Bürgi and J. Lacour, Angew. Chem., Int. Ed., 2013, 52, 1796–1800 CrossRef CAS PubMed.
- R. Breslow and H. Won Chang, J. Am. Chem. Soc., 1961, 83, 2367–2375 CrossRef CAS.
- R. J. Goldacre and J. N. Phillips, J. Chem. Soc., 1949, 1724–1732 RSC.
- For the precise experimental determination of the pKR+ values for the titled compounds and the correspondence between the DMSO:water ratio and the HX values, see: B. W. Laursen and F. C. Krebs, Chem.–Eur. J., 2001, 7, 1773–1783 CrossRef CAS.
- J. Reynisson, G. Balakrishnan, R. Wilbrandt and N. Harrit, J. Mol. Struct., 2000, 520, 63–73 CrossRef CAS.
- J. Reynisson, R. Wilbrandt, V. Brinck, B. W. Laursen, K. Nørgaard, N. Harrit and A. M. Brouwer, Photochem. Photobiol. Sci., 2002, 1, 763–773 CAS.
- B. W. Laursen, J. Reynisson, K. V. Mikkelsen, K. Bechgaard and N. Harrit, Photochem. Photobiol. Sci., 2005, 4, 568–576 CAS.
- E. Thyrhaug, T. J. Sørensen, I. Gryczynski, Z. Gryczynski and B. W. Laursen, J. Phys. Chem. A, 2013, 117, 2160–2168 CrossRef CAS PubMed.
- S. Dileesh and K. R. Gopidas, J. Photochem. Photobiol., A, 2004, 162, 115–120 CrossRef CAS.
- T. J. Sørensen, B. W. Laursen, R. Luchowski, T. Shtoyko, I. Akopova, Z. Gryczynski and I. Gryczynski, Chem. Phys. Lett., 2009, 476, 46–50 CrossRef PubMed.
- S. Dileesh and K. R. Gopidas, Chem. Phys. Lett., 2000, 330, 397–402 CrossRef CAS.
- O. Kel, P. Sherin, N. Mehanna, B. Laleu, J. Lacour and E. Vauthey, Photochem. Photobiol. Sci., 2012, 11, 623–631 CAS.
- T. J. Sørensen, A. Ø. Madsen and B. W. Laursen, Tetrahedron Lett., 2013, 54, 587–590 CrossRef.
- I. Němcová and I. Němec, J. Electroanal. Chem. Interfacial Electrochem., 1971, 30, 506–510 CrossRef.
- B. W. Laursen, PhD thesis, University of Copenhagen (Risø), 2001.
- F. Westerlund, J. Elm, J. Lykkebo, N. Carlsson, E. Thyrhaug, B. Akerman, T. J. Sorensen, K. V. Mikkelsen and B. W. Laursen, Photochem. Photobiol. Sci., 2011, 10, 1963–1973 CAS.
- F. C. Krebs, B. W. Laursen, I. Johannsen, A. Faldt, K. Bechgaard, C. S. Jacobsen, N. Thorup and K. Boubekeur, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 410–423 CrossRef.
- Y. Haketa, S. Sasaki, N. Ohta, H. Masunaga, H. Ogawa, N. Mizuno, F. Araoka, H. Takezoe and H. Maeda, Angew. Chem., Int. Ed., 2010, 49, 10079–10083 CrossRef CAS PubMed.
- B. Dong and H. Maeda, Chem. Commun., 2013, 49, 4085–4099 RSC.
- T. J. Sørensen, C. B. Hildebrandt, J. Elm, J. W. Andreasen, A. O. Madsen, F. Westerlund and B. W. Laursen, J. Mater. Chem., 2012, 22, 4797–4805 RSC.
- T. J. Sørensen, C. B. Hildebrandt, M. Glyvradal and B. W. Laursen, Dyes Pigm., 2013, 98, 297–303 CrossRef.
- J. B. Simonsen, K. Kjær, P. Howes, K. Nørgaard, T. Bjørnholm, N. Harrit and B. W. Laursen, Langmuir, 2009, 25, 3584–3592 CrossRef CAS PubMed.
- J. B. Simonsen, F. Westerlund, D. W. Breiby, N. Harrit and B. W. Laursen, Langmuir, 2011, 27, 792–799 CrossRef CAS PubMed.
- D. Shi, G. Sfintes, B. W. Laursen and J. B. Simonsen, Langmuir, 2012, 28, 8608–8615 CrossRef CAS PubMed.
- F. C. Krebs, H. Spanggaard, N. Rozlosnik, N. B. Larsen and M. Jørgensen, Langmuir, 2003, 19, 7873–7880 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2003, 80, 257–264 CrossRef CAS.
- C. Herse, D. Bas, F. C. Krebs, T. Bürgi, J. Weber, T. Wesolowski, B. W. Laursen and J. Lacour, Angew. Chem., Int. Ed., 2003, 42, 3162–3166 CrossRef CAS PubMed.
- J. Lacour, A. Londez, C. Goujon-Ginglinger, V. Buss and G. Bernardinelli, Org. Lett., 2000, 2, 4185–4188 CrossRef CAS PubMed.
- With (Λ,R)- and (Δ,S)-BINPHAT anions, diastereomerically pure [(M)-6][(Λ,R)-BINPHAT] and [(P)-6][(Δ,S)-BINPHAT] salts can be obtained by precipitation in mixtures of benzene and THF. In the mother liquor, selectivity of the chiral recognition is however only moderate (d.r. 2.4:1).
- C. Villani, B. Laleu, P. Mobian and J. Lacour, Chirality, 2007, 19, 601–606 CrossRef CAS PubMed.
- B. Laleu, P. Mobian, C. Herse, B. W. Laursen, G. Hopfgartner, G. Bernardinelli and J. Lacour, Angew. Chem., Int. Ed., 2005, 44, 1879–1883 CrossRef CAS PubMed.
- Racemization studies were performed by direct VT-ECD monitoring at a single wavelength when possible (e.g.34) or, more traditionally, by taking aliquots at given period of times and analyzing the enantiomeric proportion by CSP-HPLC (e.g.6).
- J. Elm, J. Lykkebo, T. J. Sørensen, B. W. Laursen and K. V. Mikkelsen, J. Phys. Chem. A, 2011, 115, 12025–12033 CrossRef CAS PubMed.
- The rotational strength (R) depends on the electric |μ| and magnetic |m| transition dipole moments and the angle between them (θ) according to the formula: R ∝ |μ|·|m|.cos(θ).
- J. Elm, J. Lykkebo, T. J. Sørensen, B. W. Laursen and K. V. Mikkelsen, J. Phys. Chem. A, 2012, 116, 8744–8752 CrossRef CAS PubMed.
- C. Nicolas and J. Lacour, Org. Lett., 2006, 8, 4343–4346 CrossRef CAS PubMed.
- C. Nicolas, C. Herse and J. Lacour, Tetrahedron Lett., 2005, 46, 4605–4608 CrossRef CAS.
- A. Pothukuchy, S. Ellapan, K. R. Gopidas and M. Salazar, Bioorg. Med. Chem. Lett., 2003, 13, 1491–1494 CrossRef CAS PubMed.
- J. Reynisson, G. B. Schuster, S. B. Howerton, L. D. Williams, R. N. Barnett, C. L. Cleveland, U. Landman, N. Harrit and J. B. Chaires, J. Am. Chem. Soc., 2003, 125, 2072–2083 CrossRef CAS PubMed.
- A. Pothukuchy, C. L. Mazzitelli, M. L. Rodriguez, B. Tuesuwan, M. Salazar, J. S. Brodbelt and S. M. Kerwin, Biochemistry, 2005, 44, 2163–2172 CrossRef CAS PubMed.
- O. Kel, A. Fürstenberg, N. Mehanna, C. Nicolas, B. Laleu, M. Hammarson, B. Albinsson, J. Lacour and E. Vauthey, Chem.–Eur. J., 2013, 19, 7173–7180 CrossRef CAS PubMed.
- B. P. Maliwal, R. Fudala, S. Raut, R. Kokate, T. J. Sørensen, B. W. Laursen, Z. Gryczynski and I. Gryczynski, PLoS One, 2013, 8, e63043 CAS.
- T. J. Sørensen, E. Thyrhaug, M. Szabelski, R. Luchowski, I. Gryczynski, Z. Gryczynski and B. W. Laursen, Methods Appl. Fluoresc., 2013, 1, 025001 CrossRef PubMed.
- J. Hamacek, C. Besnard, N. Mehanna and J. Lacour, Dalton Trans., 2012, 41, 6777–6782 RSC.
- This loss of colour can be advantageously used for the measurement of the pKR+ values using a UV-Vis absorption monitoring method.
- M. Lofthagen, R. VernonClark, K. K. Baldridge and J. S. Siegel, J. Org. Chem., 1992, 57, 61–69 CrossRef CAS.
- M. Lofthagen, R. Chadha and J. S. Siegel, J. Am. Chem. Soc., 1991, 113, 8785–8790 CrossRef CAS.
- M. Lofthagen and J. S. Siegel, J. Org. Chem., 1995, 60, 2885–2890 CrossRef CAS.
- M. Lofthagen, J. S. Siegel and M. Hackett, Tetrahedron, 1995, 51, 6195–6208 CrossRef CAS.
- S. K. Narasimhan, D. J. Kerwood, L. Wu, J. Li, R. Lombardi, T. B. Freedman and Y.-Y. Luk, J. Org. Chem., 2009, 74, 7023–7033 CrossRef CAS PubMed; S. K. Narasimhan, D. J. Kerwood, L. Wu, J. Li, Y. Han, S. Zhu, A. Shah, R. Lombardi, T. B. Freedman and Y.-Y. Luk, J. Org. Chem., 2010, 75, 2138 CrossRef.
- P. Mobian, C. Nicolas, E. Francotte, T. Bürgi and J. Lacour, J. Am. Chem. Soc., 2008, 130, 6507–6514 CrossRef CAS PubMed.
- B. Baisch, D. Raffa, U. Jung, O. M. Magnussen, C. Nicolas, J. Lacour, J. Kubitschke and R. Herges, J. Am. Chem. Soc., 2009, 131, 442–443 CrossRef CAS PubMed.
- N. Hauptmann, K. Scheil, T. G. Gopakumar, F. L. Otte, C. Schütt, R. Herges and R. Berndt, J. Am. Chem. Soc., 2013, 135, 8814–8817 CrossRef CAS PubMed and references therein.
- B. Laleu, M. S. Machado and J. Lacour, Chem. Commun., 2006, 2786–2788 RSC.
- P. Mobian, N. Banerji, G. Bernardinelli and J. Lacour, Org. Biomol. Chem., 2006, 4, 224–231 CAS.
- J. Lacour, C. Ginglinger, C. Grivet and G. Bernardinelli, Angew. Chem., Int. Ed., 1997, 36, 608–610 CrossRef CAS.
- D. Conreaux, N. Mehanna, C. Herse and J. Lacour, J. Org. Chem., 2011, 76, 2716–2722 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |