 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pre-nucleation clusters as solute precursors in crystallisation
Denis
Gebauer
*a,
Matthias
Kellermeier
a,
Julian D.
Gale
b,
Lennart
Bergström
c and
Helmut
Cölfen
a
aDepartment of Chemistry, Physical Chemistry, University of Konstanz, Universitätsstrasse 10, Box 714, D-78464 Konstanz, Germany. E-mail: denis.gebauer@uni-konstanz.de; Fax: +49 7531 883139; Tel: +49 7531 882169
bDepartment of Chemistry, Nanochemistry Research Institute, Curtin University, PO Box U1987, Perth, WA 6845, Australia
cDepartment of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius väg 16C, S-106 91 Stockholm, Sweden
First published on 23rd January 2014
Abstract
Crystallisation is at the heart of various scientific disciplines, but still the understanding of the molecular mechanisms underlying phase separation and the formation of the first solid particles in aqueous solution is rather limited. In this review, classical nucleation theory, as well as established concepts of spinodal decomposition and liquid–liquid demixing, is introduced together with a description of the recently proposed pre-nucleation cluster pathway. The features of pre-nucleation clusters are presented and discussed in relation to recent modifications of the classical and established models for phase separation, together with a review of experimental work and computer simulations on the characteristics of pre-nucleation clusters of calcium phosphate, calcium carbonate, iron(oxy)(hydr)oxide, silica, and also amino acids as an example of small organic molecules. The role of pre-nucleation clusters as solute precursors in the emergence of a new phase is summarized, and the link between the chemical speciation of homogeneous solutions and the process of phase separation via pre-nucleation clusters is highlighted.
 Denis Gebauer | Denis Gebauer obtained his PhD in Physical Chemistry from the University of Potsdam (Germany), working at the Max-Planck-Institute of Colloids and Interfaces. After a postdoctoral stay at Stockholm University (Sweden), he started his Habilitation at the University of Konstanz in 2011. His research interests include concepts of nucleation and crystallization as well as biomineralization and materials chemistry in general. He is one of the awardees of the Heinz-Maier-Leibnitz Prizes 2012. Since the beginning of 2013, Denis Gebauer has been a Fellow of the Zukunftskolleg of the University of Konstanz. Besides science, he enjoys (wind-)surfing, cycling, and running. |
 Matthias Kellermeier | Matthias Kellermeier studied chemistry at the University of Regensburg (Germany) and received his PhD in the group of Werner Kunz, for which he was awarded the Starck Prize for Solid State Chemistry and Materials Research. In 2011, he moved to the University of Konstanz for a postdoctoral stay, before joining BASF SE (Ludwigshafen, Germany) in 2013 to work as a research scientist in the material physics department. His research interests range from self-assembled biomimetic materials and additive-controlled crystallization over general mechanisms of nucleation to the physical characterization of surfaces. Apart from science, he enjoys playing football and hiking. |
 Julian D. Gale | Julian Gale obtained his BA and DPhil from the University of Oxford, after which he was a postdoctoral research associate at the Royal Institution of Great Britain in collaboration with ICI Chemicals and Polymers. Following the award of a Royal Society University Research Fellowship, he moved to Imperial College London. In 2003 he moved to Curtin University in Western Australia to become a Premier's Research Fellow, and subsequently John Curtin Distinguished Professor in Computational Chemistry. His research interests include the development and application of computational techniques to problems in materials chemistry, geochemistry, and mineralogy. |
 Lennart Bergström | Lennart Bergström obtained his PhD in Physical Chemistry from the Royal Institute of Technology (Sweden), after which he held several positions at the Institute for Surface Chemistry, YKI, in Stockholm. In 2004, he became professor of Materials Chemistry at Stockholm University. Lennart Bergström is a Fellow of the Royal Society of Chemistry (2009) and a member of the Royal Swedish Academy of Engineering Sciences (2013), and received the Jacob Wallenberg Materials Award in 2007 and the Humboldt Research Award in 2011. His research interests include nanoparticle self-assembly, interparticle forces, colloidal processing, ceramics and porous materials, and nanocellulose hybrids. |
 Helmut Cölfen | Helmut Cölfen is full professor of Physical Chemistry at the University of Konstanz (Germany). His research interests are in the area of nucleation, classical and non-classical crystallization, biomineralization, synthesis of functional polymers, directed self-assembly of nanoparticles and fractionating methods of polymer and nanoparticle analysis—especially analytical ultracentrifugation. He has recently been listed in the Thomson Reuters ISI list of top 100 chemists worldwide for the years 2000–2010, and received the Academy Award of the Berlin-Brandenburg Academy of Sciences and Humanities in 2013. Besides science, he enjoys loud music. |
Introduction
The formation of solids by crystallisation from aqueous solution is a process that underlies natural phenomena, e.g. rock formation1 or biomineralisation,2 and also plays a pivotal role in industrial processes, such as the synthesis and purification of drugs.3 Crystallisation always involves nucleation, a first-order phase transition where solid particles are formed from their constituents—atoms, ions or (macro-)molecules. The importance of crystallisation in many fields of science and technology, including materials chemistry4 and structural biology,5 spurred a high level of research activity and led to the formulation of well-established theories for nucleation6–11 and crystal growth.12–16However, the arguably most consulted theoretical framework when it comes to nucleation from aqueous solution—classical nucleation theory (CNT)—has recently been challenged by observations primarily emanating from the biomineralisation and protein crystallisation research areas. Studies of e.g. calcium carbonates17 and calcium phosphates18,19 have shown that stable solute species, often called pre-nucleation clusters (PNCs), can exist in under- and supersaturated solutions, and participate in the process of phase separation.20 Essentially, the observation of PNCs is inconsistent with a fundamental consequence of the assumption underlying CNT that monomer association would lead to the generation of unstable species. Although a potential role for stable associates during nucleation was realised early on—and was accounted for in advanced treatments of nucleation in vapours,21 or in the kinetic theory of liquids,22 for example—recent research in this field has received much attention, with plenty of new publications constantly appearing.
In this review, we summarise both the early and recent experimental evidence, as well as results from computer simulations, on PNCs of calcium carbonate and phosphates, iron(oxy)(hydr)oxides, silica and amino acids. We provide fundamental definitions of PNCs, and discuss the basics of CNT, binodal demixing, spinodal decomposition, and liquid–liquid phase separation in relation to the characteristics of PNCs. This includes recent attempts to incorporate PNCs in the framework of classical models.
Our considerations show that PNCs are solute precursors to phases forming from homogeneous solution. The chemical notion of PNCs is an invaluable extension of the physics of phase separation, because it provides the molecular explanation for a nanoscopic phase separation. This event in turn triggers aggregation-based processes that subsequently lead to the emergence of amorphous, and/or crystalline, phases.
Classical nucleation theory
CNT6–11 stipulates that the formation of nuclei in supersaturated homogeneous solution is governed by the balance between the bulk and surface energy of the new phase. According to CNT, nuclei form in supersaturated solutions as a consequence of stochastic (microscopic) density fluctuations. These fluctuations occur by random collisions of the dissolved constituents and may be conceived of as monomer association in pseudo-equilibrium. Because the interfacial and the bulk energy of the nuclei scale with the square and the cube of the radius r (assuming nuclei to be spherical), respectively, the unfavourable interfacial contribution dominates at small sizes, and the favourable bulk term dominates at large sizes (Fig. 1). In summation, this leads to a positive excess free energy of small nuclei, ΔGex (blue curve, Fig. 1). The point at which the bulk contribution compensates for the energetic costs arising from the interfacial surface (maximum of the blue curve in Fig. 1) is called the critical size. Thermodynamically, the critical size reflects a metastable state (∂ΔGex/∂r = 0, ∂2ΔGex/∂r2 < 0), since any infinitesimal change towards either smaller or larger radii will render the system unstable and lead to nucleus dissolution or unlimited growth, respectively. By analogy with the notion of the activated complex in chemical kinetics,23 ΔGex can be regarded as the basis of the thermodynamic barrier for nucleation. Taking into account additional kinetic barriers (e.g. originating from dehydration of the constituent monomers or structural rearrangements within the nucleus), the rate of nucleation (J) can be expressed as;| J = A·exp(−EA/kBT)·exp(−ΔGex/kBT) | (1) |
The first exponent (−EA/kBT) is related to the kinetic barriers with an overall activation energy EA (kB: Boltzmann constant, T: absolute temperature), while the second exponent (−ΔGex/kBT) represents the thermodynamic barrier. The parameter A is a pre-exponential factor that depends on the properties of the investigated material.24
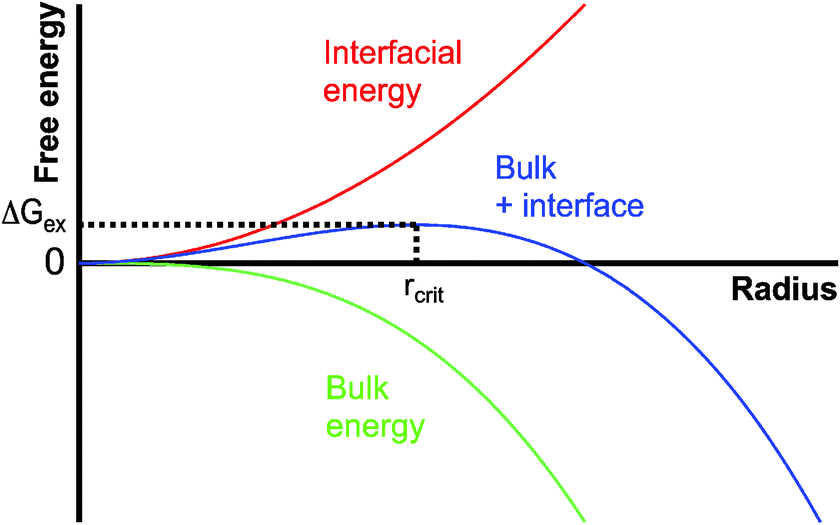 | ||
| Fig. 1 Schematic illustration of the size dependence of the energetics of nanoscopic nuclei within the framework of classical nucleation theory; for explanation see the text. | ||
The contributions to the kinetic barrier are difficult to quantify, and are therefore typically neglected. The thermodynamic term, on the other hand, can be determined based on the assumption that the nanoscopic nuclei behave like the macroscopic phase (crystal), i.e. have the same bulk structure and exhibit corresponding interfacial energetics. Indeed, this so-called capillary assumption is the foundation—and the crux—of CNT. The interfacial contribution to the excess free energy is usually taken to be the interfacial free energy of the (planar) boundary between the new phase and the solution. In more advanced treatments, the size dependence of the interfacial free energy may be accounted for.25–27 However, at typical critical nucleus sizes, the exact size dependence is unknown, or currently impossible to quantify.28,29
The bulk free energy of nuclei can be expressed by the so-called affinity (ϕ) term, which is defined as;
| ϕ = kBT·ln(IAP/Ksp) | (2) |
| ΔGex(S) ∼ αγ3ϕ−2 | (3) |
| rcrit ∼ γϕ−1 | (4) |
Based on the above considerations, it is possible to predict nucleation rates (eqn (1)) for any material at a given level of supersaturation. However, values calculated accordingly can differ by orders of magnitude from experimentally measured data,31,32 thus highlighting the limited ability of CNT to describe the behaviour of real systems in many cases.
Apart from the problematic assignment of the appropriate interfacial free energy of nuclei within the capillary assumption, the bulk term can also be difficult to quantify. The nucleated phase can have a structure (in terms of both aggregation state or polymorphism) that is distinctly different from the final product of the crystallisation process. This is often observed during precipitation of calcium carbonate, for instance, where amorphous calcium carbonate (ACC) is initially formed and subsequently transformed into more stable bulk crystalline polymorphs according to Ostwald's rule of stages.33–35 In this case, it is inherently difficult to assign a solubility product that is representative of the actually nucleated phase in the equations above (eqn (2)–(4)).30 This becomes even more complicated when considering that the thermodynamic stability, and, with it, the solubility of given polymorphs can change with particle size.36 While the Gibbs–Thomson effect may be taken into account in CNT,15,37,38 there is, for example, evidence for a crossover in thermodynamic stability of the different polymorphs of calcium carbonate at the nanoscale,39 suggesting that ACC could indeed become the stable modification at sizes below a few nm.40 This means that bulk values for Ksp might not be applicable to small nuclei at all, rendering the quantification of supersaturation and affinity according to eqn (2) difficult, if not impossible.
Finally, it should be noted that the above discussion is strictly concerned with homogeneous nucleation, which is the main focus of this review. The extension to heterogeneous nucleation of a solid at the interface between the solution and another phase—be it a solid (like dust particles) or vapour phase (such as air bubbles)—can be found elsewhere.24,28
Binodal demixing and spinodal decomposition
The thermodynamics underlying phase separation can be illustrated with phase diagrams as schematically shown for a generic two-phase system with a lower critical solution temperature (LCST) in Fig. 2. In the homogeneous region of the phase diagram (point A in Fig. 2), the solution is stable and there is no driving force for phase separation. This corresponds to the undersaturated regime in the language of CNT. The blue line is the so-called binodal curve, which marks the coexistence of, in this case, the solid and the liquid phase under a given set of conditions (corresponding to a saturated solution). As the binodal is crossed (point B in Fig. 2), e.g. by changing the composition at constant temperature (arrow in Fig. 2), the system becomes metastable and phase separation can take place. This region of the phase diagram, in which supersaturation increases continuously from point B to C, represents the situation described by CNT and indeed, binodal demixing is equivalent to classical nucleation for a nascent solid–liquid two-phase system.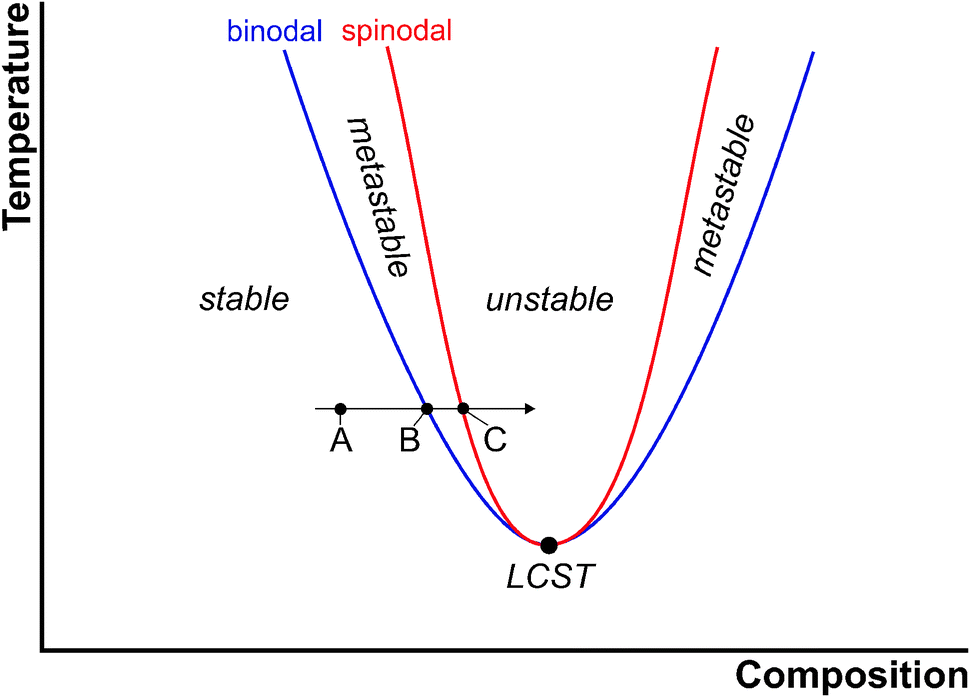 | ||
| Fig. 2 Schematic illustration of the phase diagram of a two-phase system with a lower critical solution temperature (LCST, large black circle). For explanation see the text. | ||
Point C in Fig. 2 marks the composition (or affinity, eqn (2)), at which the metastable system becomes unstable and phase separation must take place. The boundary between the metastable and unstable regime is referred to as the spinodal curve (red line). From a thermodynamic point of view, the binodal reflects regions of positive curvature in the free energy landscape, whereas the saddle point in free energy defines the spinodal. At the spinodal boundary, decomposition happens spontaneously, that is, the barrier for phase separation vanishes (as opposed to eqn (3)). This is, of course, only possible if the system can be brought to point C without nucleation occurring on the way.
In the case of electrolytes, the coexistence line (binodal) between cations Az+ and anions Bz− dissolved in the mother liquid (l1) and ions bound in the solid (s) salt is characterised by the phase equilibrium (for convenience, for a dissolved electrolyte consisting of cations and anions with equal valency) according to;
Az+(l1) + Bz−(l1) ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) AB(s) AB(s) | (5) |
In this case, the dissolution equilibrium, and with it, the liquid–solid coexistence line (blue line in Fig. 2), is characterised by the constant solubility Ksp = IAP(l1)/[AB(s)] = IAP(l1) of the stable modification, i.e. [AB(s)] = 1.41 This convention of basic thermodynamics essentially reflects the fact that the amount of the solid phase will not influence the solubility product. Hence, the equilibrium solubility is identical regardless of whether a tiny grain or a large crystal is present (although there may be size dependent effects on the solubility product, such as Gibbs–Thomson37 effects that may give rise to Ostwald ripening). It has to be emphasised, though, that Ksp does depend on composition and structure—i.e. (pseudo)-poly(a)morphism—of the solid. In global equilibrium, the system has to conform to Gibbs' phase rule (here, a single solid phase is allowed), albeit directly after phase separation, different (pseudo)-poly(a)morphs may be present in parallel, and distinct local equilibria across the phase interfaces co-exist. In this case, the most soluble (least stable) modification governs Ksp. Upon ripening toward global equilibrium, unstable modifications thus dissolve in favour of the stable one. The metastable regime of the phase diagram can hence further be subdivided into regions of accessibility for less stable (pseudo)-poly(a)morphs (not shown in Fig. 2 for the sake of clarity).
Considering potential crossovers of thermodynamic stability at the nanoscale (cf. section on CNT), the global nanoscopic phase equilibrium between the ion in the mother liquid (l1) and a nanoscopic liquid phase formed by the solutes (lnano) may also be written as:
| Az+(l1) + Bz−(l1) (AB)(lnano) | (6) |
Again, a constant solubility product applies according to Ksp(lnano) = IAP(l1)/[AB(lnano)] = IAP(l1), by analogy with eqn (5) and the considerations above. Note that the nanoscopic phase may also contain solvent molecules as in the case of crystalline pseudopolymorphism. However, for such a nanoscopic system, Fig. 2 has to be regarded as a phase diagram from a microscopic point of view; the binodal of the most stable modification is that of a nanoscopic liquid phase formed by the solute. When the nanoscopic liquid phase grows, it may change composition (and thereby solubility), and moreover, is expected to quickly become unstable with respect to solid modifications.40
Fundamentally, as originally introduced by Gibbs,43 and later developed by Cahn and Hilliard,44 phase separation in the binodal regime is based on statistical fluctuations that are large in degree (i.e. solute ions directly assemble to bulk-like structural units), but each one is small in extent. This means that nucleation leads to the stochastic formation of tiny nuclei, in accordance with the notions of CNT. Binodal fluctuations may overcome the barrier to nucleation (eqn (1)), where the probability for nucleation to take place increases with increasing supersaturation (eqn (3)). Spinodal fluctuations, on the other hand, are considered to be infinitesimal in degree but large in extent, corresponding to a diffusion-limited process of phase separation. Under these conditions, decomposition, rather than demixing, proceeds spontaneously and uniformly throughout the entire volume of a system that has become unstable. This can lead to the generation of characteristic channel-like bicontinuous patterns,45 which are distinctly different from the stochastic formation of separate nuclei in binodal demixing or homogeneous nucleation (Fig. 3).
 | ||
| Fig. 3 Spatial patterns produced upon spinodal decomposition (left) and binodal demixing/nucleation (right). Reproduced with permission from ref. 42. | ||
However, close to the spinodal, the notion of a nucleus itself is essentially lost. Inter alia, the composition in the centre of spinodal fluctuations approaches that of the surrounding solution, and their radius approaches infinity. Due to the molecular nature of the solvent and the solute, no part of the fluctuations can be regarded to be a homogeneous phase anymore. Hence, a nascent phase that directly results from spinodal decomposition is not supposed to have a solid-state structure resembling that of the macroscopic bulk.
Liquid–liquid phase separation
Spinodal decomposition in solution may be considered as a liquid–liquid-type phase separation, because of the spontaneous nature and diffusion-limited rate of demixing. However, the spinodal pathway is to be strictly differentiated from liquid–liquid separation in a metastable system, where a supersaturated solution divides into two liquid phases, one rich and one poor in solute. As opposed to the formation of one solid phase (particles) within one liquid phase (mother solution), we now consider the formation of two distinct liquid phases, that is, the formation of droplets of a second dense liquid phase in the mother solution, which consequently becomes lean in solute. Fig. 4 shows a corresponding binary phase diagram including a liquid–liquid miscibility gap.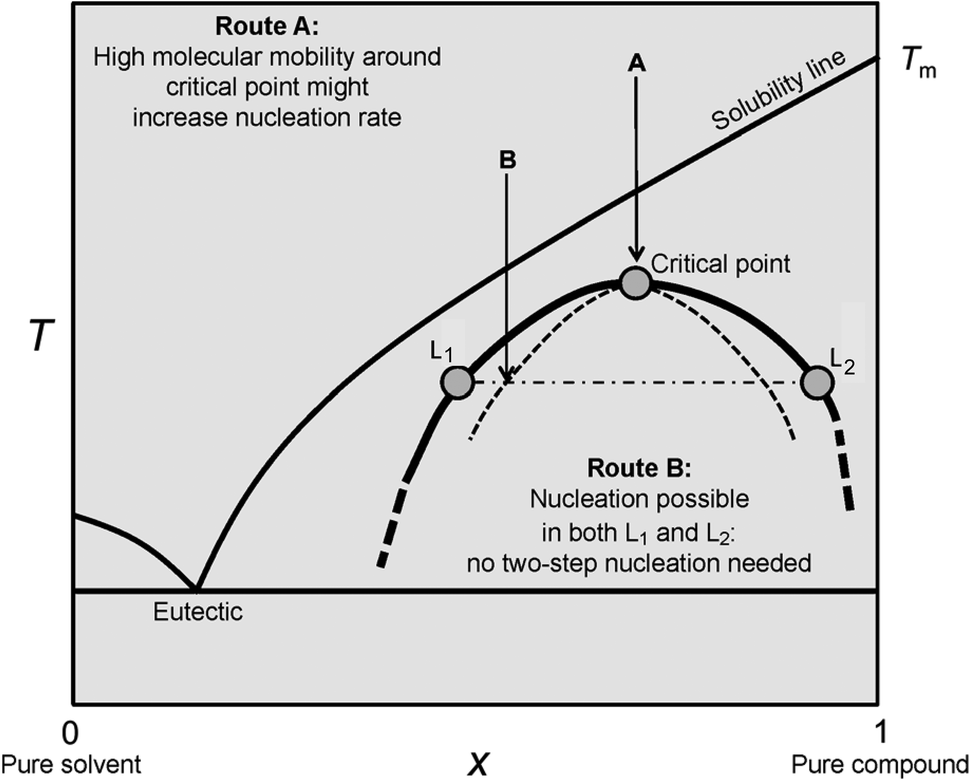 | ||
| Fig. 4 Schematic illustration of a binary phase diagram with a submerged liquid miscibility gap. When the solubility line of the solid solute is crossed upon cooling without nucleation in the mother liquid, the liquid–liquid coexistence line is approached. For explanation see the text. Reproduced with permission from ref. 46 Copyright 2013 John Wiley & Sons. | ||
While we do not address the special case of entering the miscibility gap via Route A here (see ref. 46 for explanation), in Route B, liquid–liquid separation may occur either via binodal demixing (upon crossing the bold line in Fig. 4) or through spinodal decomposition (crossing the dashed curve in Fig. 4). The liquid–liquid coexistence line (i.e. liquid–liquid binodal, bold line in Fig. 4) is characterised by the equilibrium between the two distinct liquid phases containing different amounts of the dissolved cations Az+ and anions Bz− in the case of electrolytes. In the following, we will denote the mother liquid as l1 and the dense liquid as l2. The phase equilibrium between l1 and l2 can be written as (again for an electrolyte consisting of cations and anions with equal valency);
| Az+(l1) + Bz−(l1) Az+(l2) + Bz−(l2) | (7) |
The corresponding equilibrium constant is K(l1,l2) = IAP(l2)/IAP(l1). In Fig. 4, the situation in liquid–liquid equilibrium—at a given temperature T—is exemplified by the compositions L1(T) and L2(T) of the solute-poor (l1) and the dense solute-rich phase (l2), respectively. From the point of view of CNT, the affinity for liquid–liquid separation ϕ(l1,l2) may thus be quantified according to;
| ϕ(l1,l2) = kBT·ln(IAP*(l1)/IAP(l1)) | (8) |
Importantly, there is no constant solubility product describing the liquid–liquid coexistence line, which in turn merely defines the equilibrium composition of the liquid phases l1 and l2 directly upon liquid–liquid phase separation at a given temperature (exemplified by L1(T) and L2(T), respectively, in Fig. 4). In other words, in liquid–liquid equilibrium, eqn (7) defines IAP*(l1) = IAP(l1) = IAP(l2)/K(l1,l2) ≠ const., and correspondingly ϕ = 0 for any given IAP(l1) (eqn (8)).
Comparing generic two-phase systems (Fig. 2, binodal curve according to eqn (5) or (6)) to liquid–liquid coexistence (Fig. 4, binodal curve according to eqn (7)), it should be emphasised that the liquid–liquid miscibility gap is always located in a metastable region of phase diagrams—in the stable region, the occurrence of additional bulk liquid phases violates Gibbs' phase rule, and must be regarded thermodynamically impossible.
The discovery of liquid precursor states during the precipitation of calcium carbonate47,48 suggests that liquid–liquid phase separation processes can be of pivotal importance for the crystallisation of inorganic minerals. This was first proposed by Gower,47,48 who observed liquid droplets rich in CaCO3 in the presence of certain poly(carboxylic acids),49 and therefore originally named these species “polymer-induced liquid precursors” (PILPs). Later, Wolf et al.50 and Bewernitz et al.51 showed that a dense liquid phase could indeed also form in the absence of any additives. Computer simulations52 suggest a liquid–liquid coexistence region in the phase diagram of aqueous CaCO3 solutions, in direct analogy to what has been described above. However, unlike Fig. 4, this liquid–liquid region is proposed to be bound by a lower critical solution temperature,52 as deduced from the temperature dependence of CaCO3 solubilities.53 The envisaged phase diagram is displayed in Fig. 5, and will be addressed in more depth below.
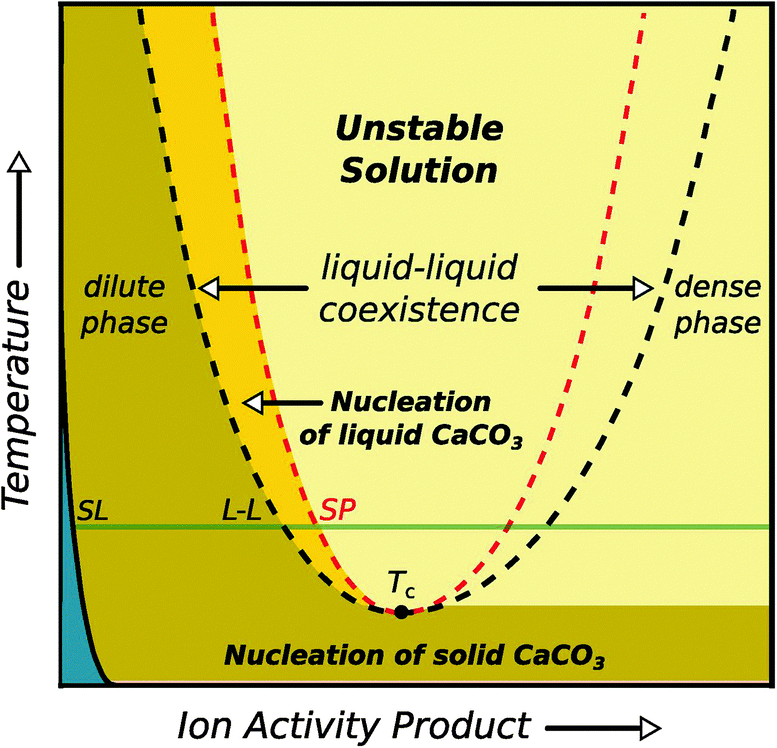 | ||
| Fig. 5 Schematic illustration of the binary phase diagram suggested for aqueous calcium carbonate solutions, including a liquid–liquid coexistence region. When entering this region, the liquid will split into a dense and dilute phase of dissolved calcium carbonate. The blue area corresponds to the undersaturated regime, SL and LL denote the solid–liquid and liquid–liquid binodal, respectively. Reproduced with permission from ref. 52 Copyright 2013 AAAS. | ||
The formation of liquid-like calcium carbonate has also been discussed in the framework of spinodal decomposition. For example, Faatz et al.54 argued that nanoparticles of ACC formed via liquid–liquid separation upon entering the spinodal regime depicted in Fig. 2, which may be accessible at very high levels of supersaturation. Similar conclusions were drawn by Rieger et al.,33 who observed liquid-like structures with patterns reminiscent of spinodal decomposition when quenching highly supersaturated CaCO3 solutions very rapidly after preparation. Subsequently, Wolf et al.55 demonstrated that liquid–liquid separation may also occur under conditions that involve the gradual increase of supersaturation, as opposed to the rapid mixing employed in prior studies, thereby pointing toward a binodal rather than a spinodal process.
While liquid–liquid separation, binodal demixing and spinodal decomposition can account for the occurrence and formation of liquid phases, they provide little insight into the molecular mechanisms underlying the respective processes of phase separation. While the occurrence of a liquid–liquid miscibility gap in metastable regions of phase diagrams may not be regarded a general phenomenon, all of these concepts fall short to accommodate PNCs—their existence in stable regions of the phase diagrams cannot be explained.20
Pre-nucleation clusters (PNCs)
In recent years, several experimental observations have been reported that cannot be thoroughly rationalised by the theories of phase separation processes outlined above. The basic point of conflict is the occurrence of (meta-)stable associates20,36 or (pseudo-)phases in the homogeneous region of the phase diagrams.32,56 The occurrence of stable solute species in homogeneous solutions has been evidenced for the most important biominerals, calcium phosphates and carbonates, iron(oxy)(hydr)oxides, silica, and also for organic compounds such as amino acids. The literature on these systems is reviewed in detail below. The occurrence of metastable (pseudo-)phases, on the other hand, has been predominantly observed in protein crystallisation,32,56,57 which shall not be reviewed here. Our tentative definition of PNCs shall be contemplated in the following sections, and comprises five major characteristics:(i) PNCs are composed of the constituent atoms, molecules, or ions of a forming solid, but can also contain additional chemical species.
(ii) PNCs are small, thermodynamically stable solutes, and there is thus formally no phase boundary between the clusters and the surrounding solution.
(iii) PNCs are molecular precursors to the phase nucleating from solution, and hence participate in the process of phase separation.
(iv) PNCs are highly dynamic entities, and change configuration on timescales typical for molecular rearrangements in solution (i.e., within hundreds of picoseconds).
(v) PNCs can have encoded structural motifs resembling, or relating to, one of the corresponding crystalline polymorphs.
Calcium carbonates and phosphates
In the case of calcium carbonate, the existence of PNCs was initially evidenced by means of a combination of potentiometric titrations and analytical ultracentrifugation (AUC).17,20 Measurements with a calcium ion-selective electrode (ISE) and pH-titration during the early stages of precipitation (pre- and post-nucleation) demonstrated that calcium and carbonate ions were bound at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in solution prior to the nucleation of solid CaCO3 (definition i), while AUC confirmed that species significantly larger than ion pairs had formed. A thermodynamic speciation model was introduced to account for cluster formation, which is compatible with experimentally observed ion binding profiles, and allows for solute cluster formation via multiple-binding.17 Recently, the simplifying assumption of ideal solutions, which was made in the initial investigation, has been shown to be acceptable by evaluation of the role of ionic activity.30 Moreover, claims58,59 that linear calcium binding profiles evidenced that association was limited to mononuclear calcium complexes have been challenged experimentally30 and theoretically.60 Indeed, the occurrence of larger associates beyond simple ion pairs was corroborated by means of cryogenic transmission electron microscopy (cryo-TEM),61 both in solutions saturated with respect to calcite, and also in the presence of solid CaCO3 particles.
:1 in solution prior to the nucleation of solid CaCO3 (definition i), while AUC confirmed that species significantly larger than ion pairs had formed. A thermodynamic speciation model was introduced to account for cluster formation, which is compatible with experimentally observed ion binding profiles, and allows for solute cluster formation via multiple-binding.17 Recently, the simplifying assumption of ideal solutions, which was made in the initial investigation, has been shown to be acceptable by evaluation of the role of ionic activity.30 Moreover, claims58,59 that linear calcium binding profiles evidenced that association was limited to mononuclear calcium complexes have been challenged experimentally30 and theoretically.60 Indeed, the occurrence of larger associates beyond simple ion pairs was corroborated by means of cryogenic transmission electron microscopy (cryo-TEM),61 both in solutions saturated with respect to calcite, and also in the presence of solid CaCO3 particles.
Evaluation of the experimentally determined ion association data utilising a multiple-binding speciation model shows that the clusters are thermodynamically stable (definition ii) within the solution phase (i.e. K ≫ 1 and ΔG = −RT·lnK < 0, where G is the free enthalpy, R the universal gas constant, and T the absolute temperature).17 A reassessment of the multiple-binding equilibrium later indicated that the coordination number of Ca2+ by CO32−, on average over the entire cluster, was approximately 2, independent of the pH level (cf. section on computer simulations below, where also the evidence for the cluster dynamics, definition iv, is discussed).60 The specific stability of PNCs, i.e. the free enthalpy for the formation of ion pairs within PNCs, was however experimentally found to depend on the pH of the solution. In fact, the clusters proved to be more stable at lower pH (K ∼ 1400 M−1 at pH 9) than at higher pH (K ∼ 1000 M−1 at pH 10). This pH-dependence of cluster stability correlates with the solubility of ACC nanoparticles initially precipitated at corresponding pH levels.17 That is, more stable PNCs yield less soluble (and hence more stable) ACC, and vice versa. Thereby, the more stable form of ACC turned out to be structurally related to calcite (the thermodynamically stable crystalline phase at ambient temperature and pressure), while the less stable ACC showed similarities to the structure of vaterite (a metastable crystalline polymorph) in terms of the very short-range order. This indicates that ACC, as well as PNCs, exhibit distinct proto-crystalline structures, a notion that was verified by a combination of NMR, EXAFS, and FT-IR measurements on solid ACC,62 leading to the establishment of the concept of amorphous polymorphism (i.e. polyamorphism) in the CaCO3 system.63
The obvious link between pre- and post-nucleation calcium carbonate speciation (definition v), and the notion of ACC polyamorphism, which was masterminded by Addadi and Weiner,65,66 suggests that nucleation can proceed through PNCs (while preserving structural features). Indeed, it appears that PNCs participate in one possible pathway to ACC in solution (definition iii). As PNCs do not have interfacial surfaces, cluster aggregation close to the point of nucleation—which was initially inferred from an increase of sedimentation coefficients measured by AUC17,61—requires the development of interfacial surfaces by phase separation (as a trigger to minimise the overall interfacial surface area). This context will be discussed in detail below.
The use of silica as a soluble additive at high pH levels was later able to lead to the collection of further and more direct evidence for this aggregation-based process. The rationale behind these experiments was that if phase separation occurs via aggregation-based processes, then it should be possible to inhibit this mechanism through colloidal stabilisation of the relevant species, e.g. by electrostatics via the introduction of charges. Indeed, the presence of silica was found to induce this effect. Stabilised solutions with a high concentration of calcium carbonate were obtained, which allowed straightforward characterisation by scattering techniques and imaging by cryo-TEM (Fig. 6).64 Interestingly, a rather broad size distribution of the species present under the respective conditions could be determined, whereas the average size seems to be only weakly affected by the high level of supersaturation, also when compared to PNCs imaged in undersaturated solution states.64 While silica oligomers (cf. below) may well affect the process of phase separation of calcium carbonate beyond the colloidal stabilisation of intermediates, no clear influence could be identified in a series of reference experiments.64 In any case, it appears that cryo-TEM imaging cannot discriminate between PNCs and phase-separated species—i.e. those that aggregate to form ACC. Since PNCs are regarded as solutes without an interface, there is no driving force for PNC aggregation, and it may hence be speculated that the majority of non-aggregated nanoscopic entities in Fig. 6 are solutes and do represent PNCs. However, the species that loosely pack to form diffuse aggregates in Fig. 6 have interfacial surfaces, which actually drive the observed aggregation process. Hence, they must be considered as nanophases, and not PNCs, and may in fact be nanodroplets that are stabilised colloidally against eventual coalescence by the presence of silica.
 | ||
| Fig. 6 Cryo-TEM images showing dynamic aggregation processes at high levels of supersaturation in strongly alkaline, silica-containing solutions (main image scale bar: 50 nm). Myriads of nanoclusters reversibly aggregate to form larger objects (typically 20–30 nm in size), which consist of assemblies of loosely packed clusters that appear to be PNCs (inset, scale bar: 10 nm). The assemblies later densify to yield compact spherical amorphous nanoparticles (not shown). Red circles (inset) highlight just some of the nanocluster constituents within the assemblies. Reproduced with permission from ref. 64 Copyright 2012, Wiley and Sons. | ||
For the calcium phosphate (CaP) system, it was found already in the mid-1970's that under various conditions, amorphous calcium phosphate (ACP) represents the first solid phase precipitated from solution.18,67 X-ray diffraction studies, specifically determining radial distribution functions, indicated the presence of small clusters as structural building units in the solid ACP precursor particles. Based on chemical analyses (essentially by measuring Ca/P ratios), the composition of these clusters was determined to be Ca9(PO4)6. These so-called “Posner's clusters” were suggested to be roughly spherical and closely packed, while randomly including water molecules in interstices to form the bulk of ACP. Thus, initial evidence of cluster-based mechanisms of ACP formation was entirely based on solid-state characterisation. Posner and Betts68 speculated that the cluster units would initially form in solution and nucleate ACP via aggregation. Later, Onuma and Ito19 were able to show that small clusters, 0.7 to 1.0 nm in size (commensurate with the theoretical size of Posner's clusters), existed in simulated body fluid, as well as in solutions that were undersaturated with respect to ACP and octacalcium phosphate (OCP, a metastable crystalline polymorph), but supersaturated with respect to hydroxyapatite (HAP, the thermodynamically stable phase under ambient conditions). By varying the composition of simulated body fluids, concurrent clustering of calcium phosphate (∼1 nm) and ACC (∼10–30 nm) could also be observed.69 We note that, in the context of aggregation, the discussed clusters have to have developed interfacial surfaces, and the observed solid-state structure almost certainly does not reflect the configuration of solute clusters in solution. Indeed, computer simulations (cf. also below) suggest that there is no reason to suppose that the dynamical configuration of PNCs of calcium carbonate should not also apply for the case of CaP.60
More recently, Dey et al.70 observed clusters with an average diameter of (0.9 ± 0.2) nm during the early stages of CaP nucleation under Langmuir monolayers using cryo-TEM. Even though the detected species were referred to as “pre-nucleation clusters”, it was concluded that the results of this work largely agreed with the original model of Posner et al.68 However, it was also emphasised that the sheer agreement in size between the proposed calcium phosphate PNCs and Posner's clusters did not prove their chemical or structural identity.70 Furthermore, no direct insight into the thermodynamic stability of the clusters could be gained beyond the generic observation of their existence in solution, corresponding cluster sizes, and of aggregation processes leading to nucleation of ACP, which subsequently appeared to crystallise via solid-state transformations. Again, it is crucial to note that the clusters observed to aggregate cannot qualify as PNCs, as here aggregation is evidence of the presence of interfacial surfaces (which is at odds with PNC definitions ii, iii and iv).
In another recent study on calcium phosphate precipitation in the presence of an excess of 2-amino-2-hydroxymethyl-propane-1,3-diol (TRIS), Habraken et al.59 have proposed that PNCs of calcium phosphate were in fact ion association complexes consisting of a single calcium ion and three coordinated hydrogen phosphate ions, i.e. [Ca(HPO4)3]4−. These highly charged species were suggested to undergo aggregation in solution, yielding ACP nanoparticles that later transformed into more stable (crystalline) phases by progressive inclusion of further calcium ions and concurrent deprotonation of the anions. This model should be regarded as an alternative to the PNC notion considered here, as it is at odds with all definitions above. Notably, the speciation developed by Habraken et al.59 relies on the assumption that CaP ion association constants documented in earlier literature did not include contributions of the newly postulated complexes, which in our opinion represents a critical aspect of their treatment. Moreover, the event triggering aggregation of the highly charged mononuclear complexes remains a conundrum from the point of view of colloid chemistry. Last, equilibrium constants describing the formation of [Ca(HPO4)3]4− were unfortunately not reported in this study,59 despite the presence of quantitative data, and thus, it is difficult to verify the proposed thermodynamic metastability of the proposed complexes (definition ii).
Finally, an interesting combination of the CaCO3 and CaP systems has been realised by Wang et al.,71 who studied the overgrowth of calcite with calcium phosphate by means of in situ atomic force microscopy (AFM). The results of this work suggest that small clusters are indeed also relevant for growth, a notion that will have to be confirmed for pure minerals in the future, and may possibly challenge the current understanding of crystal growth. Also whether or not these species qualify as PNCs within our definitions remains subject to further investigation.
Iron(oxy)(hydr)oxides
Iron (oxy)(hydr)oxides exhibit a more pronounced covalent character than CaCO3 and CaP. Aqueous solutions of iron(III), as well as the precipitation of iron(III) oxides from these solutions, have been studied extensively.72 In these systems, hydrolysis73–75 initially leads to the formation of several different molecular iron hydroxide species, rendering solutions of Fe3+/2+ highly acidic. At low pH, mono- or di-nuclear species are stabilised, but these tend to polymerise above ca. pH 6, leading to the occurrence of oxo-bridged poly-iron complexes, which are important in biology.76 Analytical ultracentrifugation measurements revealed that hydrolysed iron(III) solutions contain species with low sedimentation coefficients, equivalent to spheres of 2–4 nm diameter, which were also observed by electron microscopy.77–82 Upon ageing, the 2–4 nm polymeric spheres agglomerated to form rods. Later, the rods transformed into rafts that eventually flocculated. Hydrated amorphous iron(III) hydroxide phases can form in this manner, and may be converted into α-FeO(OH) (goethite) and α-Fe2O3 (hematite) through dehydration and crystallisation. This process is strongly influenced by additives and solution parameters such as ionic strength and pH.73 Thus, it is possible to synthesise well-defined nanoparticles of distinct iron oxides,83 including hematite,84 ferrihydrite,85 as well as their magnetic counterparts, maghemite86 and magnetite by fine-tuning the synthesis conditions.87 Very recently, Baumgartner et al.88 have shown that growth of magnetite occurs via rapid accretion of primary clusters, approximately 1 nm in size, along the rim of evolving nanoparticles (Fig. 7). | ||
| Fig. 7 Cryo-TEM image showing primary particles/clusters (arrows) that are attached to the surface of a larger magnetite particle (scale bar: 10 nm). Inset: fast Fourier transform (FFT) indicating crystallinity of the large particle. Reproduced with permission from ref. 88 Copyright 2013, Macmillan Publishers. | ||
Thus, magnetite formation apparently does not involve an extended amorphous phase as an intermediate, but rather proceeds directly via accretion of amorphous nanoclusters, which may or may not qualify as PNCs (cf. section on accommodation of PNCs within classical theories).
Silica
Silica is distinct from the other inorganic minerals discussed in this review in the sense that corresponding clusters (or oligomers) are based on covalent bonds with SiO4 tetrahedra as the basic structural motif, bridged by siloxane (Si–O–Si) linkages resulting from the condensation of free silanol (Si–OH) groups. When dissolved in water, silica forms silicic acid (Si(OH)4), which can become either protonated or deprotonated and condense to a variable extent, potentially yielding a broad range of dissolved species coexisting in equilibrium.89–91 These include simple monomers and dimers, but also polynuclear entities exhibiting linear, branched, ring- or cage-like structures, such as the cyclic tetramer (e.g. Si4O7(OH)53−), the prismatic hexamer (D3R, e.g. Si6O10(OH)5−) or the cubic octamer (D4R, e.g. Si8O13(OH)7−).92–96 The abundance of each of these species, as well as their degree of (de)protonation and hence charge, depends primarily on the pH and concentration of the system,97–100 but is also influenced by parameters like ionic strength,101–103 temperature,104–106 or the presence of multivalent cations,107–109 although the monomer represents the dominant population under most experimental conditions.110–112 Importantly, the formation of all of the above-mentioned oligomers occurs through a sequence of coupled condensation/hydrolysis and protonation/deprotonation reactions, each of which can be described by an equilibrium constant that defines the stability and amount of a particular species existing in solution at any given pH, temperature and ionic strength.97 Thus, the aqueous speciation of silica is characterised by an equilibrium distribution of solute oligomers that will respond to changes in conditions by shifting towards either smaller or larger average sizes (i.e. degrees of condensation), as well as higher or lower numbers of charge. These oligomers, or clusters, may indeed be conceived as PNCs; they consist of the atoms constituting solid silica (definition i), can be thermodynamically stable (i.e. can be associated with equilibrium formation constants larger than unity, while the particular position of the equilibrium is an intimate function of pH and ionic strength, definition ii),98,103 directly participate in the process of phase separation (see below, definition iii), and are certainly not nanoparticles at this stage. Moreover, silicate oligomers do also share a structural motif resembling that of the nascent solid material (bridged SiO4 tetrahedra, definition v).However, in regard to the dynamics of silica PNCs (definition iv), we have to differentiate them from other cases. This becomes clear when we consider another well-known example of cluster-based mineral formation. In the case of aqueous Al3+ solutions, hydrolysis also leads to the formation of polynuclear complexes,113 most notably the Keggin ion, Al13O4(OH)x(H2O)(31−x)+36−x, which is a structural motif that can aggregate to yield an amorphous gel, that upon ageing/dehydration results in crystalline aluminium hydroxides.114,115 While there is no immediate conflict with definitions (i)–(iii), the Keggin ion is a rather stable structure, and exhibits comparatively slow dynamics. From the point of view of dynamics (iv), the polynuclear Al3+ Keggin ion may hence not be regarded strictly a solute, thereby not qualifying as a PNC. In this sense, it may be rather regarded as a nanosolid that has formed from smaller oligomeric PNC precursors. Consistently, the Keggin ion will undergo aggregation as a result of the creation of energetically unfavourable interfacial surfaces during aluminium hydroxide precipitation.115
Returning to the case of silica, the covalent Si–O–Si bonds internally linking the silicic acid monomers within the oligomers are more stable than, for example, the interactions found in clusters of calcium carbonate or iron(oxy)(hydr)oxides, and also aluminium hydroxides. Hence, the exchange of the silica species with the surrounding solution is supposed to be relatively slow, and proceeds via hydrolysis and subsequent re-condensation reactions. However, silica chains as a whole may well display dynamic behaviour in terms of structural configuration and connectivity, even though certain favoured motifs (like the prismatic hexamer or the cubic octamer) are probably less dynamic than others (in analogy to the Keggin ion). Consequently, we argue that only silica species with a lower degree of condensation can be regarded as PNCs (predominantly those containing monomers linked to one or two neighbouring SiO4 tetrahedra, i.e. Q1 and Q2 units according to the notation introduced by Engelhardt et al.101). Structures with a higher degree of cross-linking (i.e. rich in Q3 and Q4 units) might indeed rather represent a second nanophase. In a narrow sense, they are no longer solutes with respect to dynamics anymore, and can consequently undergo agglomeration (see below).
In addition to dissolved oligomers, transparent solutions of silica (so-called sols) do almost always contain a certain amount of polymeric fragments and small colloids.36,64,116 These particles may display different degrees of hydration, have structures more or less similar to bulk silica, and exhibit surface charge distributions that again are determined predominantly by the pH and ionic strength of the solution.117 Depending on the level of saturation, they will either grow or dissolve over time, thus producing or consuming smaller oligomeric species. However, these processes are often very slow, so that the sol appears to be kinetically stable. The gradual segregation of a fluffy or gelatinous precipitate from solutions at moderate supersaturation, observed upon storing commercial stocks for several weeks, is commonly referred to as “ageing” of silica sols.118 On the other hand, when the supersaturation of the system is increased significantly (e.g. by changes in pH, concentration, or salt content), polycondensation of dissolved monomers and oligomers will be enhanced. This affords a larger number of colloidal particles (typically 1–3 nm in size), which can then either grow individually to give a more or less monodisperse suspension (sol),119 or become cross-linked and serve as building units for the formation of 3-D networks (gel),120 as illustrated in Fig. 8. In this context, the crucial parameter is the surface charge and mutual electrostatic repulsion of the particles, which can be fine-tuned by pH and the presence of charge-screening counter-ions, as described in detail elsewhere.89,121,122
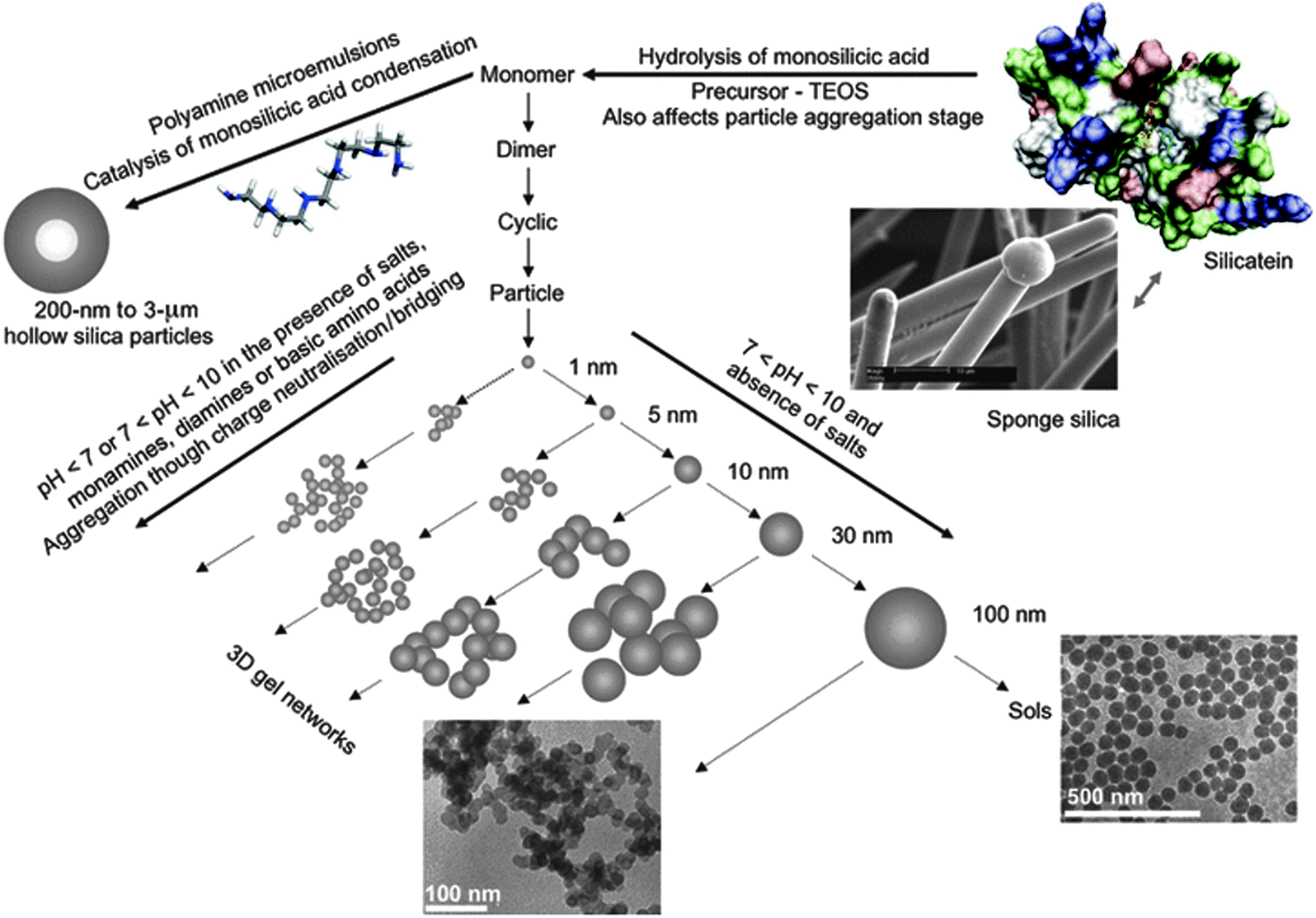 | ||
| Fig. 8 Illustration of the pathways to silica formation. For explanation see the text. Reproduced with permission from ref. 121 Copyright 2012 John Wiley & Sons. | ||
In a situation where a variety of dissolved oligomers and small nanoparticles coexist, it is inherently difficult to decide which of these species can actually still be considered as solutes and which ones would constitute a second phase, that is, to draw the borderline between pre- and post-nucleation stages. Under such circumstances, as already stated by Navrotsky,36 the notion of a critical nucleus giving rise to the emerging new phase loses its simple meaning.
The early stages of silica formation have been studied extensively due to its immanent relevance for important fields like biomineralisation,123 catalyst research,124,125 or construction industry,126 where the desired product can be an amorphous solid with well-defined shape, or a crystalline material with specific structure. However, to date, there is no clear-cut picture about the energetics of phase separation in any of these cases, i.e. whether, and if so, at which stage further growth is associated with an activation barrier that may be indicative of a nucleation event, at least in the “classical” sense. It has been suggested that coalescence of primary clusters, as well as dehydration and structural rearrangements of initially formed nanosized species, may be key steps in the early evolution of siliceous materials,36 but still much remains to be done to corroborate our understanding of silica precipitation from solution, and to allow for an unambiguous classification of corresponding solute precursors in the context of stable PNCs.
We note that the PNC concept allows the transition from pre- to post-nucleation states in silica solutions to be rationalised based on the dynamics of the nanoscale precursors as indicated above: species with fast dynamics are PNCs (pre-nucleation), while those showing less dynamic behaviour should be regarded as a separate (nano)phase (post-nucleation). Since the chemical speciation of solute precursors and nucleated (nano)phases is relatively similar in the case of silica, its precipitation from solution is somewhat more of a continuous process involving a gradual shift in the size distributions of both solute and solid species.127 Therefore, the nucleation of silica is typically not accompanied by an abrupt change in physical properties as usually observed for ionic minerals like calcium carbonate,17,20 although slow kinetics may also play a significant role in this context. The PNC pathway does not necessarily premise major barriers for phase separation, and thereby can inherently reconcile experimental observations of strong discontinuities in speciation (as seen e.g. for CaCO3) and seemingly continuous processes (silica) upon nucleation of the solid, or initially liquid, phase.
Before concluding this section, two more interesting aspects shall be pointed out. First, as already mentioned above, the formation of complex oxide–hydroxide clusters in aqueous solution is not a unique phenomenon of silica, alumina and iron oxides, but has also been observed during hydrolysis of a number of other multivalent cations such as titanium, chromium or uranium.128,129 Although the covalent character of these clusters is typically less pronounced than in the case of silica, they were found to contain structural characteristics reminiscent of the corresponding solid crystalline materials (definitions i, v) and play an active role during nucleation and growth of these phases (definition iii).36,129 Second, a common feature of many of these oxide minerals is their propensity to form hydrogels, i.e. networks of metal oxide–hydroxide-rich domains incorporating large amounts of water in their interstices.89,122 From a thermodynamic point of view, gels are rather ill-defined and may be regarded as pseudo single-phase systems, or alternatively, as phase-separated states comprising solid precipitate interspersed by liquid regions of saturated aqueous solution.130 In this sense, there is a certain conceptual analogy between gels and dense liquid nanodroplets proposed to occur during the onset of CaCO3 crystallisation:52 both represent intermediate, solvent-rich states on the way to the final anhydrous mineral, and share a similar basic chemical speciation. However, gels usually extend over much larger volumes, and may be considered a macroscopic manifestation of the initial stages of phase separation, which—in the case of silica—are kinetically accessible due to the relatively strongly pronounced covalent character of the underlying chemical interactions.
Amino acids
Apart from the inorganic compounds discussed above, there is also evidence for PNCs in aqueous solutions of organic molecules. In this section, we will focus on amino acids, but also point out other relevant organic systems. The most prominent—and also simple—example of an amino acid is glycine, where the existence and structure of the dimer, under different experimental conditions in solution, has been recurrently and heavily debated in the literature over the past century,131–134 due to potential implications for polymorph selection during glycine crystallisation.135,136 Nonetheless, in early studies it had already been proposed that the association of amino acids in solution may well proceed beyond simple dimerisation, even though larger species could at first only be detected in supersaturated systems close to the point of nucleation, primarily by means of diffusivity measurements and sedimentation analyses.137,138 Later on, the notion that non-covalent interactions between amino acid monomers lead to supramolecular assemblies, i.e. PNCs, was corroborated by a series of mass-spectrometric studies, where dilute solutions were analysed by means of electrospray ionisation (ESI) mass spectrometry (MS).139 Utilising this method, large oligomers could be observed for essentially any amino acid, regardless of its chemical functionality and widely independent of particular solution conditions like pH or solvent polarity,140–143 with entities comprising more than thirty monomeric units being detectable in some cases. Interestingly, it was reported that for selected amino acids, oligomers of a certain size were extraordinarily abundant in the gas phase and thus apparently showed enhanced stability compared to both smaller and larger species. In particular, this “magic-number” phenomenon was observed for the arginine tetramer,140 and the serine octamer.144 Furthermore, it has been suggested that amino acid clusters can exhibit strong homochiral preference and exclusively incorporate isomers of one given handedness, a process that might have deep implications for homochirogenesis during biochemical evolution.145Despite the fact that mass spectrometry has provided valuable insight into the nature of supramolecular amino acid assemblies, it still bears the major disadvantage of being a gas-phase technique that requires desolvation and ionisation of the solute analytes. In fact, results obtained in independent studies often differ considerably with respect to parameters like relative clustering propensities of distinct amino acids, average aggregate sizes, or the presence/absence of magic numbers and chiral selectivity, indicating that specific experimental instrument settings may have a profound influence on the data.142,146 This raises the fundamental question as to whether the associates seen by means of ESI-MS in the gas phase truly exist in the investigated solution, or if they rather formed during ionisation, or changed depending on the particular ionisation settings of the MS instrument utilised.
In order to confirm the clustering of amino acids in aqueous environments, complementary information must be collected directly in situ, as has been achieved with analytical ultracentrifugation analyses in a recent study.146 It was demonstrated that species with sedimentation coefficients typical for PNCs occur in solutions of all 20 natural amino acids far below saturation, while no consistent trends could be distinguished with respect to structure, charge and hydrophilicity of the monomeric molecules—in line with observations based on ESI-MS. However, detailed analyses of the data led to the conclusion that the clusters detected by AUC in solution are significantly larger on average (several tens of monomers) than the oligomers traced by ESI-MS in the gas phase (usually <10 monomeric units), most likely because the probability of successful ionisation in the ESI process decreases with the size of the associates.146 Evaluations of the concentrations of the different species, that is, of monomers and associates, further showed that the number of clusters co-existing with amino acid monomers is very low (typically <0.2 wt% next to an excess of >99.8 wt% monomers). Nevertheless, considerations based on the law of mass action suggest that the clusters are indeed thermodynamically stable solutes (i.e. that their equilibrium association constant is significantly larger than 1),38 and thus meet the definition (ii) of PNCs proposed here. Beyond that, it is worth noting that due to the rather high solubility of most amino acids, the concentration of clusters increases to the millimolar range as critical levels of supersaturation are approached, so that these species cannot be regarded as rare and of negligible population when it comes to phase separation processes.38
Molecular dynamics simulations have provided further support for the formation of larger associates in aqueous solutions of amino acids. For example, Hamad et al.147 reported equilibrium size distributions ranging from the monomer up to the pentamer for the case of glycine, and argued that these clusters were highly dynamic species, which assemble and disintegrate continuously on timescales typical for molecular rearrangements in solution—thereby meeting another criterion of our PNC definition (iv), which is strongly reminiscent of the dynamic polymer-like structural form envisaged for PNCs of calcium carbonate,60 which will be discussed in more detail below. However, unlike CaCO3 and other ionic (or covalent) minerals, amino acid monomers are connected by hydrogen bonds in clusters, and indeed, this is thought to drive clustering in these systems.147 Similar arguments were put forward to explain the occurrence of supramolecular polymers of aspartic acid in another study based on computer simulations.148
Despite all this evidence for the presence of clusters in amino acid solutions, it is still not clear to what extent these species are relevant for phase separation, and what particular role they may possibly play in this process. AUC analyses of arginine solutions showed that the clusters traced in dilute systems grow in size as the concentration increases toward the saturation limit.146 Moreover, beyond some critical threshold, the data indicated the occurrence of a second population of larger species (several nm in size), which could no longer be detected in the supersaturated regime. This was interpreted as evidence that the amino acid PNCs actively participate in the process of phase separation—a scenario quite analogous to what has been described above for the nucleation of calcium carbonate.17,20,61,64 Crystallisation precursors with sizes similar to those of the supposedly larger cluster aggregates have also been observed by means of neutron scattering in supersaturated solutions of glycine.149 In turn, related experiments based on small-angle X-ray scattering (SAXS) suggested that glycine dimers, present as equilibrium species along with monomers in the pre-nucleation stage, assemble into liquid-like clusters, which upon structural reorganisation serve as nucleation environments150—or in an alternative interpretation, progressively dehydrate and transform into solid nanoparticles (cf. below). In any case, there is significant evidence that the crystallisation of amino acids can involve PNCs, at least under certain conditions. In this context, it is also interesting to note that intense laser beams have been found to influence glycine nucleation, which was attributed to the electric field-based interaction of light with clusters (possibly PNCs) and which could even be utilised for polymorph selection.151,152 While all of the above underpins the role of clusters during amino acid nucleation, further work is needed to shed more light on the (thermodynamic) characteristics of these species as well as on the process of phase separation itself.
Another quite interesting observation made in recent studies is that solutions of amino acids can also contain much larger aggregated structures, typically 100–300 nm in size, which form spontaneously and appear to represent some kind of rare equilibrium population in these systems.154–156 Such mesoscale entities were detected by scattering techniques and tracking analyses, both at high dilution154 and near saturation in contact with solid amino acid crystals.155 In the latter case, clusters of about 1 nm were moreover found to coexist with the large aggregates. Hagmeyer et al.154 proposed that the formation of these unexpected species was driven by entropy, rendering them thermodynamically stable next to the much more abundant monomers. In turn, the work by Jawor-Baczynska et al.155,156 suggested that the observed mesostructures were actually ‘nanodroplets’ of an amino acid-rich phase exhibiting liquid-like properties. It was argued that nucleation occurs frequently within these dense environments, but that crystals able to grow can only be formed once the primary droplets (ca. 250 nm) have coalesced into larger volumes (ca. 750 nm). However, given the still limited amount of data on this phenomenon, it remains difficult to assess the role and relevance of these large aggregates in processes like PNC formation, or phase separation. In any case, it should be noted that liquid precursors of amino acids (PILPs) can indeed be stabilised with poly(carboxylic acids)—by analogy to the calcium carbonate system—,157–160 a feature that can be utilised to obtain mesocrystalline films (Fig. 9).153
 | ||
| Fig. 9 (Polarised) light microscopy images illustrating the transformation of polymer-induced liquid precursors (PILPs) of DL-lysine yielding mesocrystalline thin films. Reproduced with permission from ref. 153 Copyright 2013 John Wiley & Sons. | ||
Finally, it is worth mentioning that solute clustering has also been reported for a series of other small organic molecules, including urea,161,162 citrate,163 aminosulfonic acids,164 and many more.165–167 In some of these cases, a rather large degree of association was postulated, with species containing up to 100 monomeric units.132 However, in general, little is known regarding the nature of the clusters formed by these substances, let alone their relevance for phase separation, thus leaving an open and fascinating field for future investigations on cluster-based mechanisms in organic crystallisation.46
Computer simulation of PNCs
While experimental studies have provided considerable evidence for the existence of PNCs, obtaining direct structural information regarding the nature of these initial species can be challenging. Therefore, the use of computer simulation has become a powerful complementary tool to assist in the interpretation of experimental data. Simulations at the atomistic level span a range of techniques, from ab initio quantum mechanics through to simple force field methods. Because of the complexity and dynamical nature of ion speciation in aqueous solution, the use of more rigorous quantum-mechanical approaches has been largely restricted to ion pairing or situations where there is a well-defined structural motif, such as the corner-sharing tetrahedral oligomers that arise during polycondensation of silicates, as described above. Simulating the formation of PNCs from solution with as little a priori bias as possible currently requires the use of force-field methods, so as to make the computational exploration of configurations tractable.Here we will primarily focus on the case of calcium carbonate as an example of a system where computer simulation has played an important role in shedding light on the nature of PNCs. Force-field modelling of calcium carbonate has an extensive history, though until a decade ago studies were mainly concerned with the understanding of crystalline phases and their surfaces. More recently, the scope has expanded to also encompass nanoparticles, their structure, polymorphism and stability, as summarised elsewhere.168 Following the emergence of the concept of PNCs for calcium carbonate,17 it was therefore a natural extension to simulate the initial speciation of CaCO3 solutions. Arguably, the first study to appear employed density functional theory to perform molecular dynamics simulations on a small sample of ions in ∼50 water molecules.169 While the title suggested that this work addressed the “the onset of calcium carbonate nucleation”, in reality only a single calcium ion with varying numbers of (bi)carbonate anions was examined. In addition, the computational expense restricted the exploration to a few picoseconds, which is less than the average residence time of water in the first solvation shell of the calcium ion. Therefore, it proved difficult to draw conclusions regarding PNCs, let alone nucleation, from this study.
Through using force-field methods, Tribello et al.170 were able to perform the first extensive simulations of CaCO3 association in aqueous solution with the aim of investigating crystal growth. Because of the extended accessible timescale, it was possible to observe the formation of amorphous clusters, which exhibited signs of local order with domains that resembled both vaterite and aragonite, consistent with the idea of polyamorphism. The apparent discrepancy between these proto-structures and those found in more recent experimental studies62,63,171 can be explained by the fact that many force-field models incorrectly predict aragonite to be more stable than calcite. Subsequently, it also came to light that Ca2+ ions were under-solvated in the model of Tribello et al.,170 such that the solubility of calcium carbonate was underestimated by many orders of magnitude. As a result, it appears likely that what was observed in this work actually corresponds to spinodal decomposition (i.e. diffusion-limited phase separation).
Having recognised the importance of thermodynamic calibration, new force-field models emerged for calcium carbonate that yield the correct polymorphism and accurate solubility products for the crystalline phases. Based on these, Demichelis et al.60 were able to study the formation of PNCs in this system. By simulating a range of concentrations and pH values in the supersaturated regime, it was demonstrated that an equilibrium distribution of stable ion associates exists in solution prior to nucleation. These clusters consisted of a dynamic supramolecular ionic polymer, labelled DOLLOP (dynamically ordered liquid-like oxyanion polymers), which denotes the structural form of the PNCs, and is not supposed to give the phenomenon a new name. In DOLLOPs, both ions (Ca2+ and CO32−) and CaCO3 ion pairs rapidly attach and detach to form a combination of linear chains, rings and occasionally branched structures (Fig. 10). Often, ion detachment occurs to a solvent-separated state, followed by reattachment to either the same or a different binding site. Water remains an integral part of these structurally disordered PNCs. Several key observations arise from these DOLLOP species, which support the interpretation of experimental data previously made:
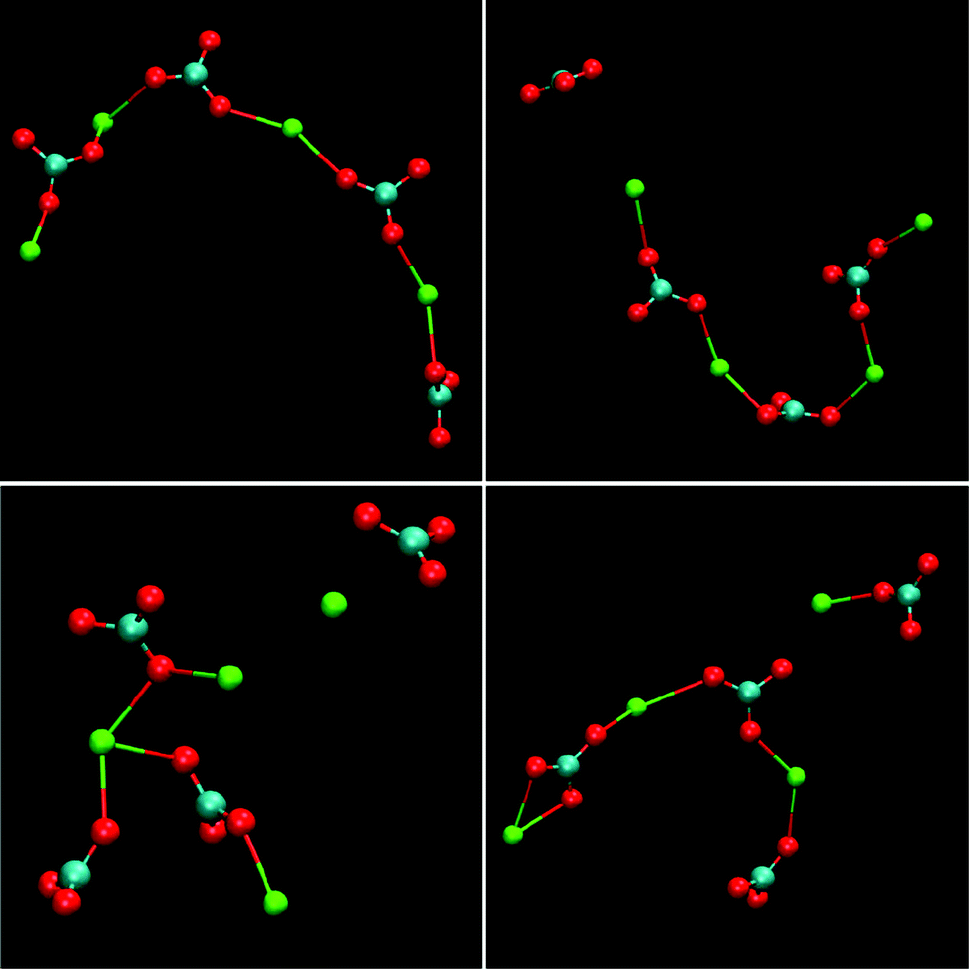 | ||
| Fig. 10 Examples of the structures of PNCs for calcium carbonate as obtained from molecular dynamics simulations. The structures represent the configuration of four separate, four formula unit PNCs after 1 ns of simulation under experimental conditions, [Ca2+] = 0.4 mM, [HCO3−] = 10 mM, pH = 10. Reproduced with permission from ref. 60 Copyright 2011, Macmillan Publishers. | ||
(a) The ions are in dynamic equilibrium between the PNCs and the free (unbound) state, demonstrating that there is no phase boundary.
(b) Fitting the observed distribution of cluster sizes to a speciation model gives equilibrium constants in good quantitative agreement with those obtained experimentally, confirming that K ≫ 1 and that the PNCs are thermodynamically stable.
(c) The average ratio of calcium to carbonate in the PNCs is approximately 1:1, while the coordination number of Ca2+ by CO32− is close to 2 under most conditions.
(d) The structures observed for these PNCs are distinct from solid amorphous calcium carbonate based on the radial density of calcium carbonate within the species.63
In the DOLLOP model for PNCs, the probability of a cluster containing on average a given number of formula units decays exponentially with increasing size. This is at odds with initial evidence from analytical ultracentrifugation (AUC) and cryo-TEM that, at first sight, indicate the presence of discrete preferential cluster sizes at the nanoscale.17,61 However, the duration of AUC experiments is in the range of hours, and given the very high cluster dynamics, AUC data can only reflect time average cluster sizes leading to the observed apparently monodisperse particle size distribution. This renders the determination of exact cluster size distributions impossible based on this technique.20,172 Cryo-TEM, on the other hand, is a “snapshot technique”,173 which should be capable of resolving actual size distributions, but the technique suffers from poor statistics, especially at very low cluster concentrations, and potential issues in terms of electron contrast of the PNCs in the background of the vitrified buffer.20 In fact, PNCs imaged utilising cryo-TEM at high levels of supersaturation in silica stabilised environments, thanks to better statistics, indeed evidence a broader, potentially exponentially decreasing PNC size distribution.64 Still, experimental determinations of the exact size distribution of PNCs do remain a challenge, and will be of high importance to provide evidence regarding the underlying molecular mechanisms.
In the work of Demichelis et al.,60 there was a change in the behaviour of the associated species noted beyond a certain carbonate concentration. Specifically, this corresponds to the breakdown of equilibrium between the solution and the PNCs, at least on the timescales accessible to unbiased molecular dynamics. Recently, this problem has been addressed in more detail by Wallace et al.,52 who used an accelerated sampling scheme to model the growth of clusters by ion pair addition. In doing so, they were able to overcome the challenges of restricted timescale and allow simulations to be performed at more realistic degrees of supersaturation. This study has demonstrated the smooth transition of the calcium coordination number by carbonate from 2 toward higher values, closer to those expected for solid phases, though water remains extensively present within the PNCs. Indeed, the diffusion coefficients of calcium ions in the clusters were found to be considerably higher than those expected for solid ACC, consistent with the pre-nucleation species becoming a dense liquid phase at some point. Based on this, Wallace et al.52 were able to show that for larger clusters, thermodynamics favours liquid–liquid separation leading to droplets of a dense liquid phase immersed in a solution of lower concentration. PNCs (i.e. DOLLOPs) can then, in principle, still be formed within the ion-depleted solution, but their concentration will be low owing to the law of mass action.
A further key finding of Wallace et al.52 is that the free energy of CaCO3 clusters in solution essentially decreases monotonically with increasing size, within the precision of the computations. This confirms the intrinsic lack of “magic numbers” in the cluster size distribution. Hence there can always potentially be a distribution of sizes that will vary as a function of the level of supersaturation. Despite this, use of an Ising lattice gas model provided evidence that liquid–liquid separation can give rise to both distinct small and large cluster sizes under appropriate conditions. It can therefore be conjectured that the clusters observed in cryo-TEM might be frozen droplets of the dense liquid phase, or even an amorphous solid, rather than the initially formed PNCs that exist before the binodal is reached. By progressively dehydrating the dense liquid phase, it was furthermore shown that agglomeration of these droplets/clusters gives pair-distribution functions that agree with experimental data for ACC once the appropriate composition is reached. While this is indirect evidence, since no mechanistic pathway has been simulated, it still strongly suggests that nucleation of ACC from the dense liquid droplets is possible.
Besides calcium carbonate, simulation methods have been applied to the (pre-)nucleation behaviour of a range of other materials, though the majority have been single-component systems. However, arguably even the simplest two-component model, a mixture of two types of Lennard-Jones particles, offers valuable insights. Here, Anwar and Boateng174 demonstrated that crystallisation may proceed through the formation of small amorphous clusters (possibly representing PNCs), which subsequently aggregate leading to liquid–liquid separation. Nucleation of the crystalline phase then occurs within the dense phase. Another popular example for the simulation of mineral precipitation from aqueous solution is sodium chloride. In this case, both unbiased and enhanced sampling molecular dynamics schemes have been employed. When NaCl is grown by gradual addition of ions to the simulation, then amorphous clusters initially appear that develop crystalline regions as time progresses.175 When enhanced sampling is used,176 more ordered clusters with elements of the rock salt structure are found, even at small sizes, though other work has suggested that wurtzite may be more stable in this size regime.177 Many of these clusters persist for reasonable lengths of time, suggesting that perhaps they may be stable PNCs, though a rigorous quantification of the equilibrium constants is required to be certain.60
As a final note in this section, computer simulation has identified PNCs for alkaline-earth minerals other than carbonates, such as barite (BaSO4). Here, relatively ordered species emerge that consist of only four formula units.178 However, this compact cluster is less symmetric than the eventual structure of barite, likely due to increased hydration.
Accommodating PNCs within classical theories
In this section, we review recent attempts to accommodate the occurrence of PNCs within the framework of CNT. The considerations are rather technical, and non-expert readers may directly proceed to the section on the caveats of classical theories.So as to accommodate stable, or metastable, clusters within the framework of CNT, Hu et al.28 reformulated the classical expression for the excess free energy (ΔGex(S)) in eqn (3) as;
| ΔGex,cluster(S) ∼ γ3(ϕ ± C)−2 | (9) |
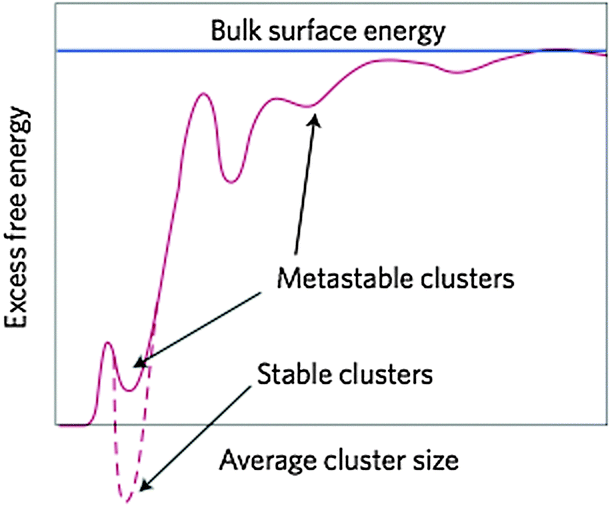 | ||
| Fig. 11 Schematic representation of the excess free energy of clusters ΔGex,cluster, which are conceived of as nuclei in the framework of an extended CNT (eqn (9) and (10)), as a function of size (d). The excess free energy is drawn relative to the free ions in solution, contains only surface contributions, and is normalised to the surface energy of the bulk (blue line). Stable clusters lie in a global minimum, whereas metastable clusters populate local minima. Note that the hypothetical variations of ΔGex,cluster in the graph are due to presumed (though as yet unknown) changes in the surface energy of the clusters with increasing size. In-between the two limiting cases (i.e. γcluster = 0 for d = 0 (dissolved state), and γcluster = γbulk for d → ∞), there may be certain favourable structural configurations that correspond to global or local minima. From ref. 179, reproduced with permission from Macmillan Publishers, Copyright 2013. | ||
Habraken et al.59 have used the concept of Hu et al.28 to explain the nucleation of amorphous calcium phosphate, which forms at levels of supersaturation too low to be rationalised by CNT. It was proposed that [Ca(HPO4)3]4− complexes possess an interfacial surface that renders them higher in free energy than their dissolved constituent ions, and thus metastable (case 1). This was inferred from the fractal nature of assemblies that formed from the clusters after nucleation, rather than from actual equilibrium constants (cf. above).
In the work of Baumgartner et al.,88 a similar treatment has been presented for the nucleation of magnetite from solution, assuming a given level of supersaturation (i.e. a given IAP, eqn (3)), from which, in principle, either a metastable amorphous (A) or stable crystalline phase (C) can be nucleated. These two distinct possibilities are shown as pathways (I) and (II), respectively, in Fig. 12a for the case of nucleation involving monomers as building units. The dashed arrow between A and C indicates that the amorphous phase can subsequently transform into the crystalline phase according to Ostwald's step rule.35 Depending on the relative height of the barriers associated with the nucleation of the two possible phases (ΔGex = ΔGA/C), either the crystalline (ΔGA > ΔGC) or the amorphous modification (ΔGA < ΔGC) will initially be formed. With the affinity for the two phases (ϕA and ϕC) and their respective surface energies (γA and γC), expressions similar to eqn (3) can be used to predict the turnover between the two scenarios, as indicated by the bold solid line in Fig. 12b for different relative values of the relevant parameters (γA/γC = (gA/gC)2/3, where gA and gC are the bulk free energies of the amorphous and crystalline phases, respectively).
 | ||
| Fig. 12 (a) Schematic representation of the four major potential pathways from atoms or molecules to bulk phases. (b) Parameter space for different ratios of surface and bulk energies, which determines whether an amorphous or crystalline phase is preferentially nucleated at a level of supersaturation according to a given IAP (eqn (3)). For monomer-based scenarios, the solid line gives the turnover point between the two possibilities. Dashed lines mark corresponding boundaries for cluster-based mechanism with different relative cluster stabilities fP(fP/gC = −0.25, 0.25, and 0.50, respectively). Bold and regular letters indicate the favoured phases for nucleation from clusters and monomers, respectively. From ref. 88, reproduced with permission from Macmillan Publishers, Copyright 2013. | ||
On the other hand, when nucleation occurs through primary particles or clusters, accretion of such species can also yield either an amorphous intermediate (pathway (III) in Fig. 12a) or directly the crystalline phase (pathway (IV) in Fig. 12a). Again, the choice of which polymorph is nucleated will be determined by the difference between the free energy barriers (Δ![[G with combining tilde]](https://www.rsc.org/images/entities/i_char_0047_0303.gif) A and ΔC for cluster-based nucleation of the amorphous and crystalline phase, respectively), which can be written as follows:
A and ΔC for cluster-based nucleation of the amorphous and crystalline phase, respectively), which can be written as follows:
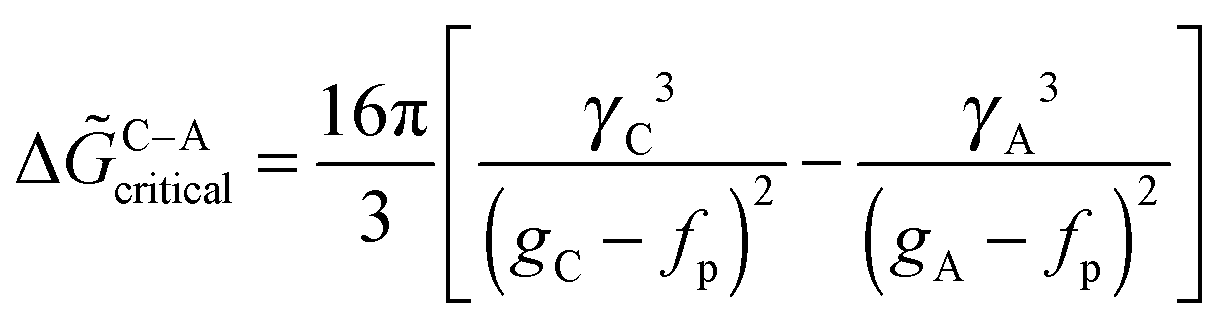 | (10) |
Here, fP represents the free energy of monodispersed clusters, where a positive value indicates that this species is stable relative to the free ions in solution. Thus, depending on the energetics of the clusters (given as fP/gC in Fig. 12b), the boundary for the transition between the two possible scenarios can be shifted in either direction (γA/γC = ((gA − fP)/(gC − fP))2/3). If the free energy difference defined by eqn (10) is negative, then the crystalline phase would nucleate in preference to the amorphous one. Furthermore, it is evident from Fig. 12b that the direct formation of the stable crystalline phase becomes more and more favourable as the stability of the clusters increases at a given ratio of surface energies γA/γC. In a way, this is consistent with Ostwald's rule of stages, which predicts the phase that is energetically closest to the initial state to be kinetically most readily accessible.
Physically, the balance between the activation energies for the formation of an amorphous or crystalline critical nucleus (eqn (10)) is controlled by the size of the critical nucleus, since larger nuclei will require a higher number of clusters to be consumed, in analogy to the model of Hu et al.28 Given that the interfacial free energy of a crystalline phase is considerably larger than that of an amorphous phase (i.e. γC ≫ γA), this requires the free energy of the primary particles/clusters to be a substantial fraction of the bulk free energy of the amorphous phase (i.e. gA − fP → 0) in order to lead to direct nucleation of the crystalline modification according to eqn (10). In turn, this implies either that the amorphous state must be much less stable than the crystalline one, or that the clusters have a near bulk-like free energy. The latter scenario is likely to be true for strongly bonded covalent molecular fragments with a well-defined structure, resembling the structure of the bulk material. However, such clusters are unlikely to be in fast equilibrium with the ions in solution, and therefore should not to be part of a single homogeneous phase anymore. If this is the case, then a phase separation (i.e. a nucleation event) has already taken place, and the considered primary particles/clusters are no longer PNCs, at least in the sense defined here.
Based on the above arguments, it appears that direct formation of the crystalline bulk material—bypassing the amorphous phase where one exists—may occur due to the presence of stable pre-organised clusters, though not PNCs. However, this is more likely to occur via pathway (IV) in Fig. 12a, which relies on accretion of nanoparticles, rather than dissolution and subsequent nucleation of the crystalline phase, as this avoids having to overcome the activation barrier to return to the state of separated dissolved ions in solution. In any case, the predictive and explanatory power of modified CNT models, especially when it comes to polymorph selection, needs to be evaluated in more depth in the future.
Caveats of classical models
The treatments presented in the previous section are classical in the sense that the balance between the surface and bulk energy of nanoscopic nuclei is considered to be the basis for nucleation kinetics. The analyses of Baumgartner et al.,88 Hu et al.28 and De Yoreo179 focus on the thermodynamic barrier of nucleation, which scales with particle/cluster size and the level of supersaturation. However, as indicated by Hu et al. and De Yoreo, the problem of this approach is that we cannot know exactly the size dependence of the interfacial free energy, which will certainly exhibit minima at sizes corresponding to, for instance, clusters with high coordination and beneficial arrangement of hydration layers (as indicated schematically in Fig. 11). On the other hand, considering nanoscopic nuclei to behave as if they were macroscopic (i.e. the capillary assumption made in CNT) and thus approximating their interfacial surface free energy by that of the bulk material is questionable and may lead to unrealistic predictions of e.g. nucleation rates in many systems. At very small volumes and high curvature, interfaces are ill defined, and for species dissolved in water, the notion of an interface actually merges with the concept of the hydration of atoms, ions, or molecules. Quantitatively, a combined nanoscopic picture of the free enthalpies of hydration and interfacial surface may be described by the differential quotient ∂G/∂x, where G is the free enthalpy and x is the distance from the centre of a solute species. The problems discussed above now boil down to the question as to how this expression may be integrated in the case of non-infinitesimal distances, that is, for distances that cover the dimensions of hydration layers or those of the interface. In classical nucleation theory, it is assumed that the formation of any species larger than the single monomers will be associated with energetic costs arising from the generation of an interfacial surface. Considering chemical reactions in aqueous solution, such as ion-pairing, complex formation, or polymerisation, all chemical species larger than monomers resulting from these reactions, though well known to be stable in many cases, would be inherently unstable according to the capillary assumption, owing to the supposed creation of interfacial surface. In this regard, the models of Hu et al.28 and Baumgartner et al.88 in a way transfer the problem of the emergence of an interface to another level, that is, from monomers to clusters.Although it is very difficult to theoretically estimate the kinetic barriers of nucleation within mechanistic models, such effects could play a decisive role in the process of phase separation. For example, in the case of calcium carbonate, partial dehydration of Ca2+ and CO32− ions upon PNC formation (as well as concurrent structural rearrangements) result in a state that is (relatively) close to the nucleated phase. Thus, the kinetic activation barrier for the transition from partially dehydrated PNCs to (even less hydrated, potentially liquid) ACC may well be significantly lower than nucleation from the more hydrated dissociated ions. Either way, in our opinion, this notion directly reflects the empirical Ostwald–Volmer rule, which states that, in the case of energetically similar modifications, the one with the lowest density will form first.
In the following sections, we are going to confront the concept of stable PNCs with the distinct “classical” models of nucleation outlined above, and discuss discrepancies and commonalities between the different approaches on the basis of experimental and theoretical observations reviewed above. In doing so, we refer to the different characteristic definitions of PNCs.
PNCs and CNT (binodal liquid–solid demixing)
First of all, PNCs differ from classical nuclei in terms of composition (definition i), as they are strongly hydrated species with a liquid-like character—in contrast to the capillary assumption of CNT. While the size distribution of classical nuclei and PNCs may both depend on the level of supersaturation, only PNCs exist to a significant extent in (under-)saturated solutions. Classically, nuclei are considered to be unstable and become metastable at a given critical size. PNCs are stable solutes in dynamic equilibrium with the free ions (ii). Hence, the average concentration of PNCs in solution is much higher than expected for classical nuclei, which are in fact supposed to be very rare species. This directly follows from the corresponding equilibrium constants, with K ≫ 1 (stable) in the case of PNCs, and 0 < K < 1 in the case of classical critical nuclei (that represent a metastable transitional state). Moreover, PNC-induced liquid–liquid phase transition and subsequent accretion (iii) is not accounted for in CNT, and can even be deemed thermodynamically impossible within the classical framework.180 Hence, in contrast to classical nuclei, PNCs should not be regarded as solid particles, but rather as polynuclear solute ion association complexes exhibiting a highly dynamic character (iv). Last, but not least, in principle, distinct structures of classical nuclei can be accounted for when corresponding stabilities, and with it, solubility products of the different modifications are known (eqn (2)). However, the link between pre- and post-nucleation speciation (v) can hardly be explained based upon classical considerations, whereas the explanatory power of the PNC concept is very appealing—especially when it comes to the occurrence of polyamorphism in minerals.63PNCs and binodal liquid–liquid demixing
Binodal liquid–liquid demixing (with a nanoscopic phase equilibrium according to eqn (6)) is different from binodal liquid–solid demixing in regard to the structure of nuclei (i). The structure of liquid-like (highly hydrated) minerals may be quite close to that of PNCs. In fact, PNCs have been suggested to exhibit liquid-like characteristics based upon computer simulations, where the radius of gyration of the PNC could vary by almost a factor of two at an energetic cost less than the thermal energy per degree of freedom—reminiscent of distorting the shape of a liquid droplet.60 Other than that, comparing the characteristics of binodal liquid droplets to the characteristics of PNCs, similar analogies and discrepancies as discussed for solid nuclei in the different contexts should apply: as opposed to PNCs, binodal fluctuations are unstable, and metastable at the critical size (ii). Binodal fluctuations do not lead to aggregation-based phase separations (iii), but may exhibit very similar dynamics as PNCs (iv). Also the link between pre- and post-nucleation speciation can hardly be explained based upon un- and meta-stable liquid binodal fluctuations (v).PNCs and spinodal decomposition
Spinodal fluctuations may have several commonalities with PNCs. First, both spinodal fluctuations and PNCs may exhibit a liquid-like character, and may be similar in terms of their structure (i), in analogy to the notion outlined in the previous sub-section. Size and size distributions, however, are very difficult to compare, since spinodal fluctuations are not associated with a barrier for phase separation, and can ultimately lead to the formation of bicontinuous patterns (Fig. 3). PNCs, on the other hand, are solutes associated with actual cluster size distributions in equilibrium (ii). Having said that, a generic commonality is that both PNCs and spinodal fluctuations spatially form throughout a given system, whereas the key difference is that PNC formation is a process yielding stable solute clusters (ii), while spinodal fluctuations are non-equilibrium states. Furthermore, it is important to realise that spinodal fluctuations do not occur in undersaturated solutions as well as close to the solid–liquid (eqn (5)) or liquid–liquid (eqn (8)) coexistence line (binodal). PNCs do exist under these conditions. PNCs are direct molecular precursors to nanodroplets and hence participate in the process of phase separation (iii). The nanodroplets aggregate and form larger species that may still exhibit a liquid-like character, which is to some extent commensurate with spinodal demixing. However, spinodal processes generally lead to the formation of two distinct homogeneous phases through inhomogeneous fluctuations. This appears to be in stark contrast to the fractal, sub-structured entities produced upon nanodroplet aggregation (Fig. 6), thus arguing against a spinodal mechanism. It has to be noted, however, that the structural features of droplet aggregates have so far only been observed in cryo-TEM micrographs, which represent quenched states and do hence not necessarily reflect the situation in solution. The dynamics (iv) of spinodal fluctuations and PNCs are in fact supposed to be similar. However, being a non-equilibrium process, spinodal fluctuations cannot explain the link between pre- and post-nucleation speciation observed experimentally (v).PNCs and liquid–liquid phase separation
At first glance, the commonalities and differences between PNCs and liquid–liquid binodal fluctuations, as well as spinodal fluctuations outlined in the previous sub-sections, also appear to apply for the corresponding processes in liquid–liquid phase separation. The basic difference with regard to the above types of phase separations, however, is that nucleated liquid droplets, regardless of whether they have formed via a binodal or spinodal mechanism, do not exhibit constant solubility (eqn (7)). In other words, an additional discrepancy arises from the specific characteristics of the liquid–liquid phase equilibrium: PNCs are solutes, whereas liquid droplets produced upon liquid–liquid separation represent a second phase—without associated solubility. The essence of this context can be illustrated by experimental data, where dilute calcium solution is continuously dosed into dilute carbonate buffer of constant pH as shown in Fig. 13. In fact, pre-nucleation ion binding in PNCs according to;| Az+(l1) + Bz−(l1) (AB)cluster(l1) | (11) |
 | ||
| Fig. 13 Free calcium detected with an ion selective electrode in 25 mL 10 mM carbonate buffer at pH 9.00, 9.25, 9.50, 9.75 and 10.0 as indicated, during addition of aqueous 10 mM calcium chloride solution at a constant rate of 10 μL min−1. Arrows indicate the time of nucleation of solid calcium carbonate. Before this point in time, there are no obvious indications of phase separation, as the detected calcium develops continuously. The difference between the added calcium (dashed line) and detected calcium prior to nucleation is ascribed to the binding in stable PNCs. Reproduced with permission from ref. 20. | ||
When the equilibrium constants of ion binding within PNCs (eqn (11)) and liquid–liquid coexistence (eqn (7)) are similar, and the liquid droplets are nucleated essentially without supersaturation (eqn (8)), the crossing of the liquid–liquid binodal may not affect the behaviour of pre-nucleation ion binding. Alternatively, the liquid–liquid equilibrium may also be established very slowly. In either case, this type of liquid–liquid binodal demixing might happen anywhere before the distinct change indicated by arrows in Fig. 13. As a further alternative, the corresponding PNC-to-nanodroplet transition may be reflected in the development after nucleation (arrows), where the ion binding behaviour establishes the post-nucleation solubility of the formed solid (eqn (5)), or liquid, phase (eqn (6)).
Hence, regardless at which point in the curves in Fig. 13 liquid–liquid separation occurs exactly, PNCs can be regarded as the direct precursors of liquid nanodroplets. This is probably most evident in the case of CaCO3 precipitation, where liquid precursor phases have frequently been observed.49,51,55 The notions introduced by Wallace et al.52 can thus provide the crucial information to explain how a nanoscopic liquid–liquid separation can be possible from a molecular point of view. In fact, the phase equilibrium in eqn (7) only considers the presence of two distinct liquid phases l1 and l2 of the solvent with its solute monomers. Without active association equilibria (eqn (11), albeit within both l1 and l2), PNC formation will not occur in either liquid phase. In other words, the formation of larger PNCs—which have been suggested to undergo a transition to less dynamic species with higher ionic coordination (cf. section on computer simulations)—within the dense liquid phase l2 is ultimately assisted by the formation of the dense liquid phase itself, that is, by a higher IAP(l2). This idea essentially reflects the clarification of Davey et al.46—i.e. that two distinct liquid phases do not have any bearing on the nucleation process other than to offer two distinct environments, where both classical and “non-classical” mechanisms may be active, or heterogeneous nucleation may be promoted owing to the presence of interfaces.
A central question is whether PNCs form in the nanoscopic dense liquid droplets and change speciation, or is the formation of nanoscopic dense liquid droplets induced or promoted by the PNCs? In the work of Wallace et al.,52 this issue is avoided by referring to the phenomenon as a “microscopic liquid–liquid separation”, that is, a purely nanoscopic phenomenon, where also the liquid–liquid phase interface becomes ill-defined, and inhomogeneous fluctuations may be envisaged to occur upon the establishment of a liquid–liquid equilibrium within the notions of Cahn and Hilliard.44 It is important to emphasise that liquid–liquid separation cannot explain the experimentally observed occurrence of PNCs in the stable region of the phase diagrams. Furthermore, as indicated above, so as to be consistent with experimental observations, the proposed liquid–liquid equilibrium (eqn (7)) needs to be associated with similar equilibrium constants as pre-nucleation ion association (eqn (11)), as well as occur essentially without supersaturation via a binodal mechanism. Indeed, this necessity highlights the molecular precursor character of the PNCs. Thus, we must conclude that either way, the phenomena cannot be explained based upon strictly “classical” considerations, or without the occurrence of PNCs. The change in speciation of PNCs may indeed be labelled as liquid–liquid separation, if the pre-requisites outlined above do apply, but this ultimately becomes a question of semantics. Phase separation via PNCs can proceed via liquid–liquid demixing, and the PNC notion explains how nanoscopic dense liquid droplets can be formed from a molecular point of view.
Towards an alternative model of phase separation
As discussed in detail above, the occurrence of PNCs cannot be explained thoroughly within any of the existing models of phase separation. It becomes obvious that PNC formation relies on chemical interactions in aqueous solution. It is difficult to give generic values for the change in free energy upon clustering; however, very strong interactions that are more exergonic than approximately −20 kBT appear improbable in aqueous solutions. For example, Busenberg and Plummer181 gave a summary of common ion pairing constants, which should be regarded as a good measure, where the largest association constant, found for the case of magnesium phosphate, corresponds to a change in free energy of ca. −16 kBT. The key to PNC formation is the balance between the free energies of hydration and interaction of the constituents, which should be valid for both distinctly covalent and ionic interactions, as well as hydrogen bonding. Aluminium hydroxides, iron(oxy)(hydr)oxides, as well as silica, undergo polycondensation reactions, forming chemical bonds with more pronounced, or even distinctly covalent character. In contrast, calcium carbonate and calcium phosphate exhibit interactions that are almost purely ionic in nature. Last, but not least, the example of amino acids shows that PNC formation can also rely on specific intermolecular interactions such as hydrogen bonding.Indeed, the balance between monomer–monomer interaction and monomer hydration should determine the actual degree of association. In cases where the interactions between the monomers are of moderate strength, as is the case in aqueous solution (see above), the outcome of the ion association will be a distribution of clusters or oligomers of different sizes, as described e.g. by the polycondensation theory of Flory.182,183 Since there is no interface that must be minimised, a population of oligomers with various sizes is indeed entropically favourable. At the same time, PNCs do not grow without limit to yield macroscopic particles as they are solutes, and there is no need to minimise interfacial surfaces—at least, in the initial stages.182
Computer simulations have shown that large PNCs become distinct from the smaller ones,52,60,63 as they develop a higher coordination number to species other than water. Moreover, there may be certain configurations that may be more stable than others (e.g. Keggin ion), which also slows down the dynamics, and these species do not qualify as PNCs anymore. At this point, the PNCs become nanoscopic droplets, which can aggregate and form larger entities that undergo progressive dehydration to give solid nanoparticles, as described above. For example, the crystallisation of calcium carbonate along the PNC pathway can be understood chemically and structurally as a progressive, step-wise loss of hydration water, according to the sequence Ca2+(aq)/HCO3−(aq)/CO32−(aq) → PNCs → dense liquid nanodroplets → liquid ACC → solid ACC → anhydrous crystalline polymorphs. In this scenario, different fundamental barriers may be envisaged that inhibit phase separation from proceeding spontaneously, as observed experimentally. During the actual step of phase separation, i.e. from PNCs to nanodroplets, the nanodroplets may aggregate into larger assemblies, which ultimately yield nanoparticles of ACC by coalescence and fusion of individual nanodroplets into a continuous phase. Depending on certain experimental parameters and/or the particular state at which the process is quenched, the initially obtained phase can still contain higher or lower amounts of water, and thus display a more or less liquid-like character. For example, there is evidence that at near-neutral pH (typically between 7 and 8), nucleation of CaCO3 preferentially results in liquid-like intermediates,50 possibly due to the influence of bicarbonate ions.51 These may be either incorporated into the nascent phase, owing to non-equilibrium conditions upon liquid–liquid phase separation, or stabilise the droplets intrinsically by binding to their interfacial surfaces.51 Likewise, additives such as polyelectrolytes were shown to be capable of stabilising liquid CaCO3 as well as amino acid precursors,49,153 probably because they can effectively inhibit the release of hydration water or stabilise the nanodroplets colloidally.64,184 Finally, it has been suggested that precipitation from high levels of supersaturation can result in liquid-like initial structures,33,54 albeit the pathway could change toward a spinodal one under these conditions.
Open questions and challenges
There are several open questions that need to be answered to further our understanding of the molecular processes that underlie phase separation. In the tentative list below, we have simply listed some of the questions and challenges without going into the details of the connections to the above discussions for the sake of brevity:• Is there a crossover in thermodynamic stability of polymorphs at the nanoscale?
• What is the dependence of the PNC size distribution on supersaturation?
• Where is the locus of the liquid–liquid binodal?
• What is the size distribution of PNCs, or nanodroplets, beyond the liquid–liquid binodal?
• What is the average lifetime of PNCs and nanodroplets?
• Is the pathway envisaged in CNT blocked owing to the formation of PNCs?
• Are PNC-like pathways relevant in heterogeneous nucleation?
• Do, or can, liquid phases form that exhibit solubility?
• Are there liquid–liquid transitions in initially formed phases?
• Are the phases formed via aggregation of nanodroplets homogeneous or sub-structured at the nanoscale?
• Does the formation of solid amorphous phases from liquid nanodroplets proceed via nucleation or simply dehydration (solidification)?
• Where is the locus of the solid–liquid spinodal?
• Does polyamorphism rely on the presence of PNCs?
• What is the molecular structural explanation for polyamorphism?
• What is the role of the hydration network in the PNC pathway?
• Are there amorphous–amorphous (solid–solid) transitions upon Ostwald ripening?
• Do, or can, crystals nucleate directly from PNCs, or are phase-separated nanodroplets a requirement for the nucleation of solids?
• Are nanodroplets, or PNCs, relevant for crystal growth?
Conclusions
In conclusion, there is vast experimental evidence for the occurrence of PNCs in various systems, including the most important biominerals, calcium carbonate and phosphate, iron(oxy)(hydr)oxides, and silica, as well as organic molecules such as amino acids. Computer simulations shed light on the structural features of PNCs, and the molecular mechanisms that underlie phase separation processes via PNCs.When it comes to current attempts to include PNCs in the framework of classical nucleation theory, the predictive and explanatory power of these models will have to be tested in the future. In this context, it should be noted that the (non-)88 existence of (amorphous) intermediate phases may be difficult to detect. Attempts to trace meta- or unstable precursor phases usually rely on quenching techniques, as performed amongst others for calcium sulfate185 or iron oxide88 crystallisation. Such methods are generally liable to artefacts, and transient, and/or highly dynamic, intermediates can simply be missed although they in fact exist. Historically, this has been one of the reasons why amorphous intermediates have for a long time been overlooked in the case of calcium carbonate, where their importance is generally accepted today.49
PNCs can be regarded as molecular solute precursors to binodal fluctuations in nanoscopic liquid–liquid separation.52 While this mechanism can rationalise many observations, it appears that theories of phase separation in general need to be expanded by chemical notions, because the physical notion of the generation of an interface alone cannot explain the experimentally observed behaviour, in many cases. Chemical interactions in solution can lead to association events, which are not linked to the problem of phase separation at all, and can proceed significantly beyond the dimer. However, these chemical solute species, PNCs, can change their structure and/or dynamics, and thereby lay the foundation to the process of phase separation at advanced stages. We thus propose that phase separation via PNCs encompasses the following three major steps:
(a) Solute association occurs in accordance with dynamic polycondensation reactions. The formed associates are stable and exist in equilibrium with their constituent monomers. Association can be based upon covalent, ionic or hydrogen-bonding interactions, while in the case of very weak interactions, significant association may only be achieved once sufficiently high concentrations of monomers are present.
(b) Large PNCs can form only upon reaching a certain critical composition (i.e. critical IAP or concentration depending on the species), which may define the locus of the liquid–liquid binodal (eqn (7)). At this point, the PNCs (can) change speciation and become less dynamic; they develop interfaces, and become nanodroplets. However, the direct creation of a nanoscopic solid phase from PNCs should not be excluded categorically.
(c) The nanoscopic intermediate phase undergoes concurrent accretion and dehydration,186 which can lead to either sub-structured (aggregation) or homogeneous (coalescence) intermediates. Eventually, solid amorphous nanoparticles are formed, which subsequently may crystallise. Depending on the specific kinetics, various intermediate forms may be accessible.
At this point, we take the opportunity to deliberate on our basic PNC definitions. The definition of the composition of PNCs (definition i) is intentionally wide. The stoichiometry (if applicable) of constituents in the PNCs may resemble that of the bulk phase with the addition of hydration water, as well as hydroxide or hydronium ions. These may be regarded as additional chemical species bound in PNCs, but may also directly participate in the early stages of crystallisation via acid–base equilibria with the primary PNC constituents. The coordination of spectator ions to PNCs should not be categorically excluded, but since they are not a part of the forming solid, they should not be regarded to be a part of PNCs either. It is inherently difficult to decide a priori if a given complex or cluster does play a role in phase separation or not, and experimental evidence is necessary. However, the vast majority of mononuclear coordination complexes187 almost certainly do not qualify as PNCs, as they clearly do not form corresponding solids, and may exhibit very slow dynamics (e.g. Cr(III) aqua complexes).188 The latter point highlights that the dynamics of PNCs is a very important definition (iv), which is central to pinpointing the event of phase separation. The highly dynamic change in configuration influences both the connectivity of the PNCs and the exchange rate of the constituent species with the solution. Essentially, the dynamic nature of PNCs underpins the notion that they do not have interfacial surfaces (definition ii and iii), which would typically bring about much slower exchange rates. So as to qualify as a PNC, either a dynamic connectivity, or a highly dynamic exchange of constituents with the solution (or both) has to be fulfilled. With this requirement, also rather small clusters may not qualify as PNCs—such as the Keggin ion. A crucial question in this context is at what size solutes with slow dynamics will become nanophases, and can be associated with an interfacial surface rather than a hydration layer? This will intimately depend on the structure of the solvent, i.e. water. Indeed, considerations of hydrogen bonding maintained around small solutes, but not around species larger than 0.5–1 nm, could be one criterion to discriminate between hydration layers and interfaces for ‘solutes’ with slow dynamics, as discussed in detail elsewhere.189,190
In realising that “classical” notions of nucleation have been modified—empirically, semi-empirically, and sometimes with profound theoretical foundation,11 so as to successfully describe experimental data, we suggest to use the unambiguous term “pre-nucleation cluster pathway” for the process of phase separation described here. Finally, we would like to point out that all of the above shows that minerals—the benchmark of hard materials—do behave like soft matter191 during the early stages of precipitation, which involves the discussion of liquid precursors to mesocrystalline intermediates and single crystals.192,193 Including the “in-between”194 of molecular and solid-state chemistry appears to be strictly required for the description of phase separation processes. These aspects are especially appealing to further our understanding of biomineralisation, where soft matter interactions may be the basis of the sophisticated and concerted control of mineralization in organisms.195
Acknowledgements
DG thanks the Zukunftskolleg of the University of Konstanz for a Fellowship as well as the Young Scholar Fund (University of Konstanz) and the Fonds der Chemischen Industrie for financial support. MK is grateful to BASF SE for funding of a postdoc position. JDG thanks the Australian Research Council for support through the Discovery Program. LB thanks the Swedish Research Council (VR) and the Humboldt Foundation for financial support.Notes and references
- H. Blatt, R. J. Tracy and B. E. Owens, Petrology: igneous, sedimentary, and metamorphic, W. H. Freeman, New York, 3rd edn, 2006 Search PubMed.
- H. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, New York, 1989 Search PubMed.
- B. Y. Shekunov and P. York, J. Cryst. Growth, 2000, 211, 122–136 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- W. Hendrickson, Science, 1991, 254, 51–58 CAS.
- J. W. Gibbs, Trans. Conn. Acad. Arts Sci., 1876, 3, 108–248 Search PubMed.
- M. Volmer and A. Weber, Z. Phys. Chem., 1925, 119, 277–301 Search PubMed.
- R. Becker and W. Döring, Ann. Phys., 1935, 24, 719–752 CrossRef CAS.
- J. Frenkel, J. Chem. Phys., 1939, 7, 538–547 CrossRef PubMed.
- J. B. Zeldovich, Acta Physicochim. URSS, 1943, 18, 1–22 Search PubMed.
- D. Kashchiev, Nucleation: Basic theory with applications, Butterworth-Heinemann, Oxford, 2000 Search PubMed.
- P. Hartman and W. G. Perdok, Acta Crystallogr., 1955, 8, 49–52 CrossRef.
- P. Hartman and W. G. Perdok, Acta Crystallogr., 1955, 8, 521–524 CrossRef CAS.
- P. Hartman and W. G. Perdok, Acta Crystallogr., 1955, 8, 525–529 CrossRef CAS.
- W. K. Burton, N. Cabrera and F. C. Frank, Philos. Trans. R. Soc. London, Ser. A, 1951, 243, 299–358 CrossRef.
- A. W. Vere, Crystal growth: principles and progress, Plenum Press, New York, 1987 Search PubMed.
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- F. Betts and A. Posner, Mater. Res. Bull., 1974, 9, 353–360 CrossRef CAS.
- K. Onuma and A. Ito, Chem. Mater., 1998, 10, 3346–3351 CrossRef CAS.
- D. Gebauer and H. Cölfen, Nano Today, 2011, 6, 564–584 CrossRef CAS PubMed.
- J. L. Katz, H. Saltsburg and H. Reiss, J. Colloid Interface Sci., 1966, 21, 560–568 CrossRef CAS.
- J. Frenkel, Kinetic theory of liquids, Dover Publications, New York, 1955 Search PubMed.
- H. Eyring, Chem. Rev., 1935, 17, 65–77 CrossRef CAS.
- J. J. De Yoreo and P. G. Vekilov, Rev. Mineral. Geochem., 2003, 54, 57–93 CrossRef CAS.
- A. Dillmann and G. E. A. Meier, Chem. Phys. Lett., 1989, 160, 71–74 CrossRef CAS.
- A. Dillmann and G. E. A. Meier, J. Chem. Phys., 1991, 94, 3872–3884 CrossRef CAS PubMed.
- I. J. Ford, A. Laaksonen and M. Kulmala, J. Chem. Phys., 1993, 99, 764–765 CrossRef CAS PubMed.
- Q. Hu, M. H. Nielsen, C. L. Freeman, L. M. Hamm, J. Tao, J. R. I. Lee, T. Y. J. Han, U. Becker, J. H. Harding, P. M. Dove and J. J. De Yoreo, Faraday Discuss., 2012, 159, 509–523 RSC.
- H. Z. Zhang and J. F. Banfield, J. Phys. Chem. Lett., 2012, 3, 2882–2886 CrossRef CAS.
- M. Kellermeier, A. Picker, A. Kempter, H. Cölfen and D. Gebauer, Adv. Mater., 2013 DOI:10.1002/adma.201303643.
- D. Kashchiev, J. Chem. Phys., 2008, 129, 164701 CrossRef PubMed.
- P. G. Vekilov, Nanoscale, 2010, 2, 2346–2357 RSC.
- J. Rieger, T. Frechen, G. Cox, W. Heckmann, C. Schmidt and J. Thieme, Faraday Discuss., 2007, 136, 265–277 RSC.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Nanoscale, 2011, 3, 265–271 RSC.
- W. Ostwald, Z. Phys. Chem., 1897, 22, 289–330 CAS.
- A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12096–12101 CrossRef CAS PubMed.
- H. H. Teng, P. M. Dove, C. A. Orme and J. J. De Yoreo, Science, 1998, 282, 724–727 CrossRef CAS.
- P. G. Vekilov, D. Frenkel, K. J. Roberts, J. J. De Yoreo, C. Virone, I. Rosbottom, R. P. Sear, D. Gebauer, H. Cölfen and J. D. Gale, Faraday Discuss., 2012, 159, 139–180 RSC.
- A. V. Radha, A. Fernandez-Martinez, Y. Hu, Y.-S. Jun, G. A. Waychunas and A. Navrotsky, Geochim. Cosmochim. Acta, 2012, 90, 83–95 CrossRef CAS PubMed.
- P. Raiteri and J. D. Gale, J. Am. Chem. Soc., 2010, 132, 17623–17634 CrossRef CAS PubMed.
- P. W. Atkins and J. De Paula, Physical chemistry, Oxford University Press, Oxford, New York, 7th edn, 2002 Search PubMed.
- J. García-Ojalvo, A. M. Lacasta, J. M. Sancho and R. Toral, Europhys. Lett., 1998, 42, 125–130 CrossRef.
- J. W. Gibbs, The collected works of J. Willard Gibbs, Yale University Press, New Haven, 1948, vol. 1 Search PubMed.
- J. W. Cahn and J. E. Hilliard, J. Chem. Phys., 1959, 31, 688 CrossRef CAS PubMed.
- J. W. Cahn, Acta Metall., 1961, 9, 795–801 CrossRef CAS.
- R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166–2179 CrossRef CAS PubMed.
- L. B. Gower and D. A. Tirrell, J. Cryst. Growth, 1998, 191, 153–160 CrossRef CAS.
- L. B. Gower and D. J. Odom, J. Cryst. Growth, 2000, 210, 719–734 CrossRef CAS.
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed.
- S. E. Wolf, L. Müller, R. Barrea, C. J. Kampf, J. Leiterer, U. Panne, T. Hoffmann, F. Emmerling and W. Tremel, Nanoscale, 2011, 3, 1158–1165 RSC.
- M. A. Bewernitz, D. Gebauer, J. R. Long, H. Cölfen and L. B. Gower, Faraday Discuss., 2012, 159, 291–312 RSC.
- A. F. Wallace, L. O. Hedges, A. Fernandez-Martinez, P. Raiteri, J. D. Gale, G. A. Waychunas, S. Whitelam, J. F. Banfield and J. J. De Yoreo, Science, 2013, 341, 885–889 CrossRef CAS PubMed.
- J. Kawano, N. Shimobayashi, A. Miyake and M. Kitamura, J. Phys.: Condens. Matter, 2009, 21, 425102 CrossRef PubMed.
- M. Faatz, F. Gröhn and G. Wegner, Adv. Mater., 2004, 16, 996–1000 CrossRef CAS.
- S. E. Wolf, J. Leiterer, M. Kappl, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2008, 130, 12342–12347 CrossRef CAS PubMed.
- O. Galkin, W. Pan, L. Filobelo, R. E. Hirsch, R. L. Nagel and P. G. Vekilov, Biophys. J., 2007, 93, 902–913 CrossRef CAS PubMed.
- P. G. Vekilov, Cryst. Growth Des., 2010, 10, 5007–5019 CAS.
- E. W. Moore and H. J. Verine, Am. J. Physiol.: Gastrointest. Liver Physiol., 1981, 241, G182–G190 CAS.
- W. J. E. M. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. H. Bomans, P. M. Frederik, J. Laven, P. van der Schoot, B. Aichmayer, G. de With, J. J. DeYoreo and N. A. J. M. Sommerdijk, Nat. Commun., 2013, 4, 1507 CrossRef PubMed.
- R. Demichelis, P. Raiteri, J. D. Gale, D. Quigley and D. Gebauer, Nat. Commun., 2011, 2, 590 CrossRef PubMed.
- E. M. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Science, 2009, 323, 1455–1458 CrossRef CAS PubMed.
- D. Gebauer, P. N. Gunawidjaja, J. Y. P. Ko, Z. Bacsik, B. Aziz, L. J. Liu, Y. F. Hu, L. Bergström, C. W. Tai, T. K. Sham, M. Edén and N. Hedin, Angew. Chem., Int. Ed., 2010, 49, 8889–8891 CrossRef CAS PubMed.
- J. H. E. Cartwright, A. G. Checa, J. D. Gale, D. Gebauer and C. I. Sainz-Díaz, Angew. Chem., Int. Ed., 2012, 51, 11960–11970 CrossRef CAS PubMed.
- M. Kellermeier, D. Gebauer, E. Melero-García, M. Drechsler, Y. Talmon, L. Kienle, H. Cölfen, J. M. García-Ruiz and W. Kunz, Adv. Funct. Mater., 2012, 22, 4301–4311 CrossRef CAS.
- S. Weiner, I. Sagi and L. Addadi, Science, 2005, 309, 1027–1028 CrossRef CAS PubMed.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS.
- F. Betts, N. Blumenthal, A. Posner, G. Becker and A. Lehninger, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2088–2090 CrossRef CAS.
- A. S. Posner and F. Betts, Acc. Chem. Res., 1975, 8, 273–281 CrossRef CAS.
- A. Oyane, K. Onuma, A. Ito, H.-M. Kim, T. Kokubo and T. Nakamura, J. Biomed. Mater. Res., Part A, 2003, 64, 339–348 Search PubMed.
- A. Dey, P. H. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1010–1014 CrossRef CAS PubMed.
- L. Wang, S. Li, E. Ruiz-Agudo, C. V. Putnis and A. Putnis, CrystEngComm, 2012, 14, 6252–6256 RSC.
- J. Jolivet, E. Tronc and C. Chaneac, C. R. Geosci., 2006, 338, 488–497 CrossRef CAS PubMed.
- R. M. Cornell, R. Giovanoli and W. Schneider, J. Chem. Technol. Biotechnol., 2007, 46, 115–134 CrossRef.
- C. M. Flynn, Chem. Rev., 1984, 84, 31–41 CrossRef CAS.
- K. S. Murray, Coord. Chem. Rev., 1974, 12, 1–35 CrossRef CAS.
- S. J. Lippard, Angew. Chem., Int. Ed. Engl., 1988, 27, 344–361 CrossRef.
- P. J. Murphy, A. M. Posner and J. P. Quirk, J. Colloid Interface Sci., 1976, 56, 270–283 CrossRef CAS.
- P. J. Murphy, A. M. Posner and J. P. Quirk, J. Colloid Interface Sci., 1976, 56, 284–297 CrossRef CAS.
- P. J. Murphy, A. M. Posner and J. P. Quirk, J. Colloid Interface Sci., 1976, 56, 298–311 CrossRef CAS.
- J. Dousma and P. L. De Bruyn, J. Colloid Interface Sci., 1978, 64, 154–170 CrossRef CAS.
- B. A. Sommer, D. W. Margerum, J. Renner, P. Saltman and T. G. Spiro, Bioinorg. Chem., 1973, 2, 295–309 CrossRef CAS.
- T. G. Spiro, S. E. Allerton, J. Renner, A. Terzis, R. Bils and P. Saltman, J. Am. Chem. Soc., 1966, 88, 2721–2726 CrossRef CAS.
- X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805–1813 CrossRef CAS.
- M. P. Morales, T. González-Carreño and C. J. Serna, J. Mater. Res., 2011, 7, 2538–2545 CrossRef.
- V. M. Yuwono, N. D. Burrows, J. A. Soltis and R. L. Penn, J. Am. Chem. Soc., 2010, 132, 2163–2165 CrossRef CAS PubMed.
- S. Lefebure, E. Dubois, V. Cabuil, S. Neveu and R. Massart, J. Mater. Res., 2011, 13, 2975–2981 CrossRef.
- L. Vayssières, C. Chaneac, E. Tronc and J. P. Jolivet, J. Colloid Interface Sci., 1998, 205, 205–212 CrossRef PubMed.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- R. K. Iler, The chemistry of silica: solubility, polymerization, colloid and surface properties and biochemistry, Wiley, New York, 1979 Search PubMed.
- P. M. Dove and J. D. Rimstidt, in Silica: physical behavior, geochemistry and materials applications, ed. P. J. Heaney, C. T. Prewitt and G. V. Gibbs, Mineralogical Society of America, Washington, DC, 1994, vol. 29, pp. 259–301 Search PubMed.
- W. Stumm and J. J. Morgan, Aquatic chemistry: chemical equilibria and rates in natural waters, Wiley, New York, 3rd edn, 1996 Search PubMed.
- C. W. Lentz, Inorg. Chem., 1964, 3, 574–579 CrossRef CAS.
- R. K. Harris, C. T. G. Knight and W. E. Hull, J. Am. Chem. Soc., 1981, 103, 1577–1578 CrossRef CAS.
- J. L. Bass and G. L. Turner, J. Phys. Chem. B, 1997, 101, 10638–10644 CrossRef CAS.
- H. Cho, A. R. Felmy, R. Craciun, J. P. Keenum, N. Shah and D. A. Dixon, J. Am. Chem. Soc., 2006, 128, 2324–2335 CrossRef CAS PubMed.
- P. Bussian, F. Sobott, B. Brutschy, W. Schrader and F. Schüth, Angew. Chem., Int. Ed., 2000, 39, 3901–3905 CrossRef CAS.
- S. Sjöberg, J. Non-Cryst. Solids, 1996, 196, 51–57 CrossRef.
- S. Sjöberg, L.-O. Öhman and N. Ingri, Acta Chem. Scand., Ser. A, 1985, 39, 93–107 CrossRef PubMed.
- M. Kellermeier, E. Melero-García, F. Glaab, R. Klein, M. Drechsler, R. Rachel, J. M. García-Ruiz and W. Kunz, J. Am. Chem. Soc., 2010, 132, 17859–17866 CrossRef CAS PubMed.
- I. L. Svensson, S. Sjöberg and L.-O. Öhman, J. Chem. Soc., Faraday Trans., 1986, 82, 3635–3646 RSC.
- L. G. Engelhardt, D. Zeigan, H. Jancke, W. Wieker and D. Hoebbel, Z. Anorg. Allg. Chem., 1975, 418, 17–28 CrossRef.
- G. A. Icopini, S. L. Brantley and P. J. Heaney, Geochim. Cosmochim. Acta, 2005, 69, 293–303 CrossRef CAS PubMed.
- S. Sjöberg, Y. Hägglund, A. Nordin and N. Ingri, Mar. Chem., 1983, 13, 35–44 CrossRef.
- G. B. Alexander, J. Am. Chem. Soc., 1954, 76, 2094–2096 CrossRef CAS.
- S. A. Greenberg and D. Sinclair, J. Phys. Chem., 1955, 59, 435–440 CrossRef CAS.
- D. J. Belton, O. Deschaume, S. V. Patwardhan and C. C. Perry, J. Phys. Chem. B, 2010, 114, 9947–9955 CrossRef CAS PubMed.
- W. L. Marshall and J. M. Warakomski, Geochim. Cosmochim. Acta, 1980, 44, 915–924 CrossRef CAS.
- M. Tanaka and K. Takahashi, J. Trace Microprobe Tech., 2001, 19, 581–589 CrossRef CAS PubMed.
- M. Tanaka, K. Takahashi and Y. V. Sahoo, Anal. Bioanal. Chem., 2004, 378, 789–797 CrossRef CAS PubMed.
- L. W. Gary, B. H. W. S. de Jong and E. Dibble Walter, Geochim. Cosmochim. Acta, 1982, 46, 1317–1320 CrossRef.
- A. R. Felmy, H. Cho, J. R. Rustad and M. J. Mason, J. Solution Chem., 2001, 30, 509–525 CrossRef CAS.
- X. Yang, P. Roonasi and A. Holmgren, J. Colloid Interface Sci., 2008, 328, 41–47 CrossRef CAS PubMed.
- J. W. Akitt, N. N. Greenwood, B. L. Khandelwal and G. D. Lester, J. Chem. Soc., Dalton Trans., 1972, 604–610 RSC.
- S. M. Bradley, R. A. Kydd and R. F. Howe, J. Colloid Interface Sci., 1993, 159, 405–412 CrossRef CAS.
- J. Y. Bottero, M. Axelos, D. Tchoubar, J. M. Cases, J. J. Fripiat and F. Fiessinger, J. Colloid Interface Sci., 1987, 117, 47–57 CrossRef CAS.
- The colloid chemistry of silica, ed. H. E. Bergna, American Chemical Society, Washington, DC, 1994 Search PubMed.
- P. J. Scales, F. Grieser, T. W. Healy, L. R. White and D. Y. C. Chan, Langmuir, 1992, 8, 965–974 CrossRef CAS.
- E. Melero-García, R. Santisteban-Bailón and J. M. García-Ruiz, Cryst. Growth Des., 2009, 9, 4730–4734 Search PubMed.
- G. Bogush, M. Tracy and C. Zukoski, J. Non-Cryst. Solids, 1988, 104, 95–106 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, J. Non-Cryst. Solids, 1985, 70, 301–322 CrossRef CAS.
- D. J. Belton, O. Deschaume and C. C. Perry, FEBS J., 2012, 279, 1710–1720 CrossRef CAS PubMed.
- C. J. Brinker and G. W. Scherer, Sol-gel science: the physics and chemistry of sol-gel processing, Academic Press, Boston, 1990 Search PubMed.
- C. C. Perry, Rev. Mineral. Geochem., 2003, 54, 291–327 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- S. Y. Yang and A. Navrotsky, Chem. Mater., 2002, 14, 2803–2811 CrossRef CAS.
- M.-C. Schlegel, A. Sarfraz, U. Müller, U. Panne and F. Emmerling, Angew. Chem., Int. Ed., 2012, 51, 4993–4996 CrossRef CAS PubMed.
- S. A. Pelster, R. Kalamajka, W. Schrader and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 2299–2302 CrossRef CAS PubMed.
- G. Furrer, B. L. Phillips, K.-U. Ulrich, R. Pöthig and W. H. Casey, Science, 2002, 297, 2245–2247 CrossRef CAS PubMed.
- W. H. Casey and T. W. Swaddle, Rev. Geophys., 2003, 41, 1–20 CrossRef.
- H.-D. Dörfler, Grenzflächen- und Kolloidchemie, VCH, Weinheim, 1994 Search PubMed.
- W. C. M. Lewis, Chem. Rev., 1931, 8, 81–165 CrossRef CAS.
- R. M. Ginde and A. S. Myerson, J. Cryst. Growth, 1992, 116, 41–47 CrossRef CAS.
- D. Erdemir, S. Chattopadhyay, L. Guo, J. Ilavsky, H. Amenitsch, C. Segre and A. S. Myerson, Phys. Rev. Lett., 2007, 99, 115702 CrossRef.
- J. Huang, T. C. Stringfellow and L. Yu, J. Am. Chem. Soc., 2008, 130, 13973–13980 CrossRef CAS PubMed.
- I. Weissbuch, V. Y. Torbeev, L. Leiserowitz and M. Lahav, Angew. Chem., Int. Ed., 2005, 44, 3226–3229 CrossRef CAS PubMed.
- C. S. Towler, R. J. Davey, R. W. Lancaster and C. J. Price, J. Am. Chem. Soc., 2004, 126, 13347–13353 CrossRef CAS PubMed.
- Y. C. Chang and A. S. Myerson, AIChE J., 1986, 32, 1567–1569 CrossRef CAS.
- A. S. Myerson and P. Y. Lo, J. Cryst. Growth, 1991, 110, 26–33 CrossRef CAS.
- C. K. Meng and J. B. Fenn, Org. Mass Spectrom., 1991, 26, 542–549 CrossRef CAS.
- D. Zhang, L. Wu, K. Koch and R. Cooks, Eur. J. Mass. Spectrom., 1999, 5, 353–361 CrossRef CAS.
- Z. Takats, S. C. Nanita, R. G. Cooks, G. Schlosser and K. Vékey, Anal. Chem., 2003, 75, 1514–1523 CrossRef CAS.
- P. Nemes, G. Schlosser and K. Vékey, J. Mass Spectrom., 2005, 40, 43–49 CrossRef CAS PubMed.
- N. Toyama, J. Kohno, F. Mafuné and T. Kondow, Chem. Phys. Lett., 2006, 419, 369–373 CrossRef CAS PubMed.
- P. X. Yang, R. F. Xu, S. C. Nanita and R. G. Cooks, J. Am. Chem. Soc., 2006, 128, 17074–17086 CrossRef CAS PubMed.
- K. J. Koch, F. C. Gozzo, S. C. Nanita, Z. Takats, M. N. Eberlin and R. G. Cooks, Angew. Chem., Int. Ed., 2002, 41, 1721–1724 CrossRef CAS.
- M. Kellermeier, R. Rosenberg, A. Moise, U. Anders, M. Przybylski and H. Cölfen, Faraday Discuss., 2012, 159, 23–45 RSC.
- S. Hamad, C. E. Hughes, C. R. A. Catlow and K. D. M. Harris, J. Phys. Chem. B, 2008, 112, 7280–7288 CrossRef CAS PubMed.
- P. Raiteri, R. Demichelis, J. D. Gale, M. Kellermeier, D. Gebauer, D. Quigley, L. B. Wright and T. R. Walsh, Faraday Discuss., 2012, 159, 61–85 RSC.
- C. E. Hughes, S. Hamad, K. D. M. Harris, C. R. A. Catlow and P. C. Griffiths, Faraday Discuss., 2007, 136, 71–89 RSC.
- S. Chattopadhyay, D. Erdemir, J. M. B. Evans, J. Ilavsky, H. Amenitsch, C. U. Segre and A. S. Myerson, Cryst. Growth Des., 2005, 5, 523–527 CAS.
- B. A. Garetz, J. Matic and A. S. Myerson, Phys. Rev. Lett., 2002, 89, 175501 CrossRef.
- T. Sugiyama, T. Adachi and H. Masuhara, Chem. Lett., 2007, 36, 1480–1481 CrossRef CAS.
- Y. Jiang, H. Gong, M. Grzywa, D. Volkmer, L. Gower and H. Cölfen, Adv. Funct. Mater., 2013, 23, 1547–1555 CrossRef CAS.
- D. Hagmeyer, J. Ruesing, T. Fenske, H.-W. Klein, C. Schmuck, W. Schrader, M. E. M. da Piedade and M. Epple, RSC Adv., 2012, 2, 4690–4696 RSC.
- A. Jawor-Baczynska, J. Sefcik and B. D. Moore, Cryst. Growth Des., 2013, 13, 470–478 CAS.
- A. Jawor-Baczynska, B. D. Moore, H. S. Lee, A. V. McCormick and J. Sefcik, Faraday Discuss., 2013, 167, 425–440 RSC.
- S. Wohlrab, H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 4087–4092 CrossRef CAS PubMed.
- Y. Jiang, L. Gower, D. Volkmer and H. Cölfen, Phys. Chem. Chem. Phys., 2012, 14, 914–919 RSC.
- Y. Jiang, H. Gong, D. Volkmer, L. B. Gower and H. Cölfen, Adv. Mater., 2011, 23, 3548–3552 CrossRef CAS PubMed.
- Y. Jiang, L. B. Gower, D. Volkmer and H. Cölfen, Cryst. Growth Des., 2011, 11, 3243–3249 CAS.
- L. S. Sorell and A. S. Myerson, AIChE J., 1982, 28, 772–779 CrossRef CAS.
- M. Sedlák, J. Phys. Chem. B, 2006, 110, 4329–4338 CrossRef PubMed.
- M. A. Larson and J. Garside, Chem. Eng. Sci., 1986, 41, 1285–1289 CrossRef CAS.
- K. Haupa, Z. Szewczuk and Z. Mielke, Rapid Commun. Mass Spectrom., 2013, 27, 1993–1998 CrossRef CAS PubMed.
- A. T. Allen, R. M. Wood and M. P. McDonald, Sugar Technol. Rev., 1974, 2, 165–180 CAS.
- S. Parveen, R. J. Davey, G. Dent and R. G. Pritchard, Chem. Commun., 2005, 1531–1533 RSC.
- R. A. Chiarella, A. L. Gillon, R. C. Burton, R. J. Davey, G. Sadiq, A. Auffret, M. Cioffi and C. A. Hunter, Faraday Discuss., 2007, 136, 179–193 RSC.
- D. Spagnoli and J. D. Gale, Nanoscale, 2012, 4, 1051–1067 RSC.
- D. Di Tommaso and N. H. de Leeuw, J. Phys. Chem. B, 2008, 112, 6965–6975 CrossRef CAS PubMed.
- G. A. Tribello, F. Bruneval, C. Liew and M. Parrinello, J. Phys. Chem. B, 2009, 113, 11680–11687 CrossRef CAS PubMed.
- A. Fernandez-Martinez, B. Kalkan, S. M. Clark and G. A. Waychunas, Angew. Chem., Int. Ed., 2013, 52, 8354–8357 CrossRef CAS PubMed.
- M. Kellermeier, H. Cölfen and D. Gebauer, Methods in Enzymology, Academic Press Inc, Burlington, 2013, vol. 532, pp. 45–69 Search PubMed.
- H. Friedrich, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2010, 49, 7850–7858 CrossRef CAS PubMed.
- J. Anwar and P. K. Boateng, J. Am. Chem. Soc., 1998, 120, 9600–9604 CrossRef CAS.
- S. A. Hassan, J. Chem. Phys., 2011, 134, 114508 CrossRef PubMed.
- D. Zahn, Phys. Rev. Lett., 2004, 92, 040801 CrossRef.
- F. Giberti, G. A. Tribello and M. Parrinello, J. Chem. Theory Comput., 2013, 9, 2526–2530 CrossRef CAS.
- F. Jones, S. Piana and J. D. Gale, Cryst. Growth Des., 2008, 8, 817–822 CAS.
- J. J. De Yoreo, Nat. Mater., 2013, 12, 284–285 CrossRef CAS PubMed.
- A. E. Nielsen, Kinetics of Precepitation, Pergamon Press, New York, 1964 Search PubMed.
- E. Busenberg and L. N. Plummer, Geochim. Cosmochim. Acta, 1989, 53, 1189–1208 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1936, 58, 1877–1885 CrossRef CAS.
- P. J. Flory, Chem. Rev., 1946, 39, 137–197 CrossRef CAS.
- D. Gebauer, H. Cölfen, A. Verch and M. Antonietti, Adv. Mater., 2009, 21, 435–439 CrossRef CAS.
- A. E. S. Van Driessche, L. G. Benning, J. D. Rodriguez-Blanco, M. Ossorio, P. Bots and J. M. García-Ruiz, Science, 2012, 336, 69–72 CrossRef CAS PubMed.
- A. V. Radha, T. Z. Forbes, C. E. Killian, P. U. P. A. Gilbert and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16438–16443 CrossRef CAS PubMed.
- E. Wiberg, N. Wiberg and A. F. Holleman, Inorganic chemistry, Academic Press, De Gruyter, San Diego, Berlin, New York, 1st English edn, 2001 Search PubMed.
- J. N. Israelachvili, Intermolecular and surface forces, Academic Press, Burlington, MA, 3rd edn, 2011 Search PubMed.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- F. H. Stillinger, J. Solution Chem., 1973, 2, 141–158 CrossRef CAS.
- S. Shaw and L. Cademartiri, Adv. Mater., 2013, 25, 4829–4844 CrossRef CAS PubMed.
- R.-Q. Song, H. Cölfen, A.-W. Xu, J. Hartmann and M. Antonietti, ACS Nano, 2009, 3, 1966–1978 CrossRef CAS PubMed.
- R.-Q. Song, A.-W. Xu, M. Antonietti and H. Cölfen, Angew. Chem., Int. Ed., 2009, 48, 395–399 CrossRef CAS PubMed.
- M. Antonietti and G. A. Ozin, Chem.–Eur. J., 2004, 10, 28–41 CrossRef CAS PubMed.
- J. S. Evans, CrystEngComm, 2013, 15, 8388–8394 RSC.
| This journal is © The Royal Society of Chemistry 2014 |