Quinone/hydroquinone-functionalized biointerfaces for biological applications from the macro- to nano-scale
Wei Ma
and
Yi-Tao Long
*
State Key Laboratory of Bioreactor Engineering & Department of Chemistry, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, P. R. China. E-mail: ytlong@ecust.edu.cn; Fax: +86 2164252339; Tel: +86 2164252339
First published on 27th August 2013
Abstract
Quinones/hydroquinones (Q/HQ) are a class of prototypical redox molecules that play a variety of vital roles in biology, particularly in electron transfer and energy conservation systems. Biointerfaces supported on solid substrates or nanomaterials are widely used as biomimetic platforms that connect biological materials with artificial interfaces. Q/HQ-functionalized biointerfaces provide a new means for understanding the complex behaviors of Q/HQ at the interfaces of biological systems, and for further development of novel biomaterials and biosensors. This tutorial review provides a brief introduction to the recent advances in this field. We begin with the current methods used to functionalize Q/HQ on biointerfaces from the macro- to nano-scale (both solid substrates and nanomaterials), and then discuss their wide biological applications. Particularly exciting applications arise when Q/HQ-functionalized biointerface systems are coupled with biomimetic strategies. These systems can be used as models of other functionalized biointerfaces for biological applications, providing new insights into the future development of this area.
 Wei Ma | Dr Wei Ma is currently a postdoctoral researcher at East China University of Science and Technology (ECUST). She received her PhD degree from the Department of Chemistry, ECUST, in 2012. Her research interests mainly focus on biomolecular synthesis, bioelectrochemistry, biomimetic interface and biosensing. |
 Yi-Tao Long | Yi-Tao Long received his PhD in bioelectroanalytical chemistry from Nanjing University in 1998. After completing his two-year postdoctoral study at Heidelberg University, he moved to Canada, and worked for five years as a research scientist at the University of Saskatchewan and University of Alberta. Dr Long was appointed as full Professor at ECUST in 2007 after he worked in the Department of Bioengineering at UC Berkeley. His main research expertise involves nanopore single-molecule analysis, nanospectroscopy, spectroelectrochemistry, integrated biosensors and biointerfaces/biointerphases. |
Key learning points(1) The potent roles of Q/HQ-functionalized biointerfaces in various biological applications.(2) Currently available methods for functionalizing biointerfaces with Q/HQ (both solid substrate and nanomaterial-supported interfaces). (3) The broad applicability of Q/HQ-functionalized biointerfaces as models for biological applications employing versatile response mechanisms, as well as biomimetic strategies. |
1 Introduction
Quinones/hydroquinones (Q/HQ) are ubiquitous in nature and constitute an important class of natural evolutionary redox molecules. It is well known that Q/HQ fulfill a universal and possibly unique function in electron transfer and energy conservation systems.1 Q are the central cofactors in the enzymatic reaction of the electron-transport system that couples the transfer of electrons to the generation of trans-membrane electrochemical proton gradients that drive the production of adenosine triphosphate in photosynthesis and respiration.2 Furthermore, Q/HQ are also involved in a variety of critical biological processes, including brain activity and neurotransmission (i.e., dopamine), blood clotting (i.e., vitamin K), protein post-translational modification (i.e., topaquinone), cellular signaling molecule metabolism (i.e., estrogens and catecholamines), and antioxidant metabolism (i.e., ubiquinone and tocopherol congeners).Q/HQ function occurs across membranes or interfaces, from energy transduction to cell–cell interactions. However, the complexity of natural membranes or interfaces and their interactions with intra- and extracellular networks has to some extent been a challenge in conducting direct investigations. There has thus been increasing interest in creating various biomimetic interfaces (biointerfaces) on the surface of solid substrates and nanomaterials in basic and applied science. Biointerfaces have been explored for use in bio-sensing, bio-patterning and controllable drug release and delivery, yielding improved performance compared with solely either the organic or the inorganic systems.3 Biointerfaces can be used in biomimetic strategies to mimic native biological interfaces without compromising their structural function or integrity. Therefore, biointerfaces are widely used as biomimetic platforms that merge biological environments and artificial interfaces, facilitating the investigation of physiological processes.
Inspired by Nature's cue from the macroscopic to microscopic level, the fabrication of biointerfaces supported on solid substrates or nanomaterials has recently been suggested as a facile and universal strategy for furthering biological research and applications.4 Moreover, the robust capabilities of biointerfaces permit the elucidation of essential biological processes by studying the interactions between the biomimetic interface and biological coatings. Thus, to understand the complex interfacial behavior of Q/HQ in biological systems, biological and chemical modifications have been combined to create multi-functional Q/HQ biointerfaces. From a biomimetic point of view, Q/HQ-functionalized biointerfaces play a crucial role in elucidating Q/HQ behaviors in biological processes. For instance, functionalized biointerfaces have been utilized to investigate the electron transfer mechanism of coenzyme Q by mimicking the initial stages of respiration in mitochondria.5–7 Several reviews have discussed the physiological importance,8 pharmaceutical activities,9 and the proton-coupled electron transfer process10 of Q/HQ. This broad interest confirms the critical role of Q/HQ in life science; however, a review of the utilization of the properties of Q/HQ in biointerfacial models for biological applications is lacking. Here, we review the current states of fabrication and manipulation of Q/HQ-functionalized biointerfaces. We also outline the unique opportunities offered by these systems for studying the behaviors of Q/HQ at biointerfaces in biological processes and for practical biological applications. It is impossible to illustrate with certainty all of the biophysicochemical interactions of Q/HQ at a biological interface, but current techniques involving biointerface mimics provide a unique platform to guide these studies.
2 Solid-substrate-supported biointerfaces
A typical biointerface is composed of a supported substrate and a biological coating. As important molecules have been implicated in biological processes, Q/HQ are often incorporated into supported substrates as active components. A biointerface comprises the dynamic physicochemical interactions between a supported material surface and the surfaces of Q/HQ. For this field to evolve, we focus on supported materials, including solid substrates and nanomaterials, and explore the functionalization methods of Q/HQ and their biological applications.2.1 Self-assembled Q/HQ-monolayers (Q/HQ-SAMs) on solid substrates
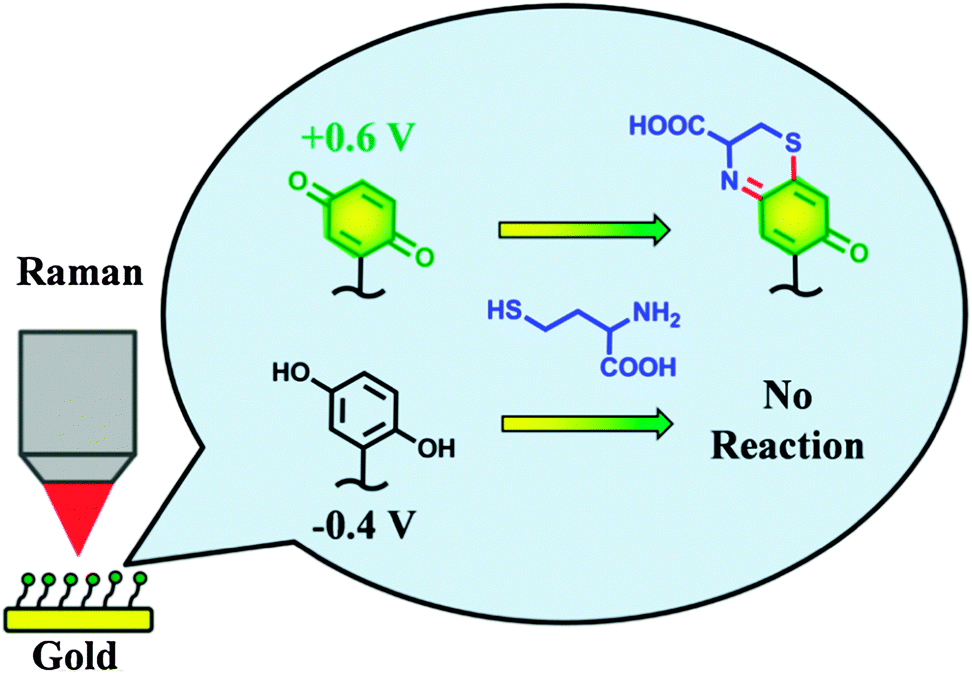 | ||
| Fig. 1 An electrochemically switched heterocyclization reaction of Q/HQ-SAMs on a gold electrode surface. Reproduced with permission from ref. 15. Copyright 2013 American Chemical Society. | ||
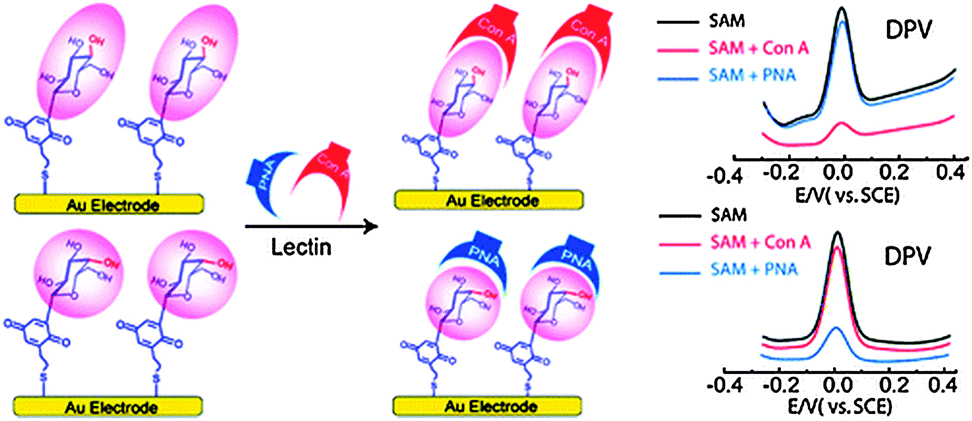 | ||
| Fig. 2 Fabrication of the epimeric monosaccharide–Q hybrids on a gold electrode surface using the SAM technique. Reproduced with permission from ref. 19. Copyright 2011 American Chemical Society. | ||
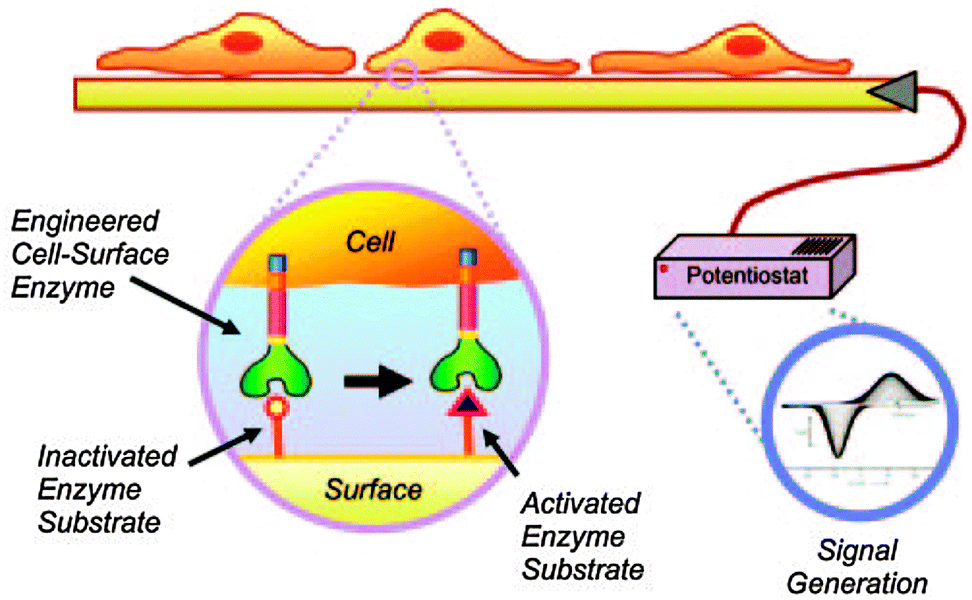 | ||
| Fig. 3 Schematic for transducing cellular activity to an electrical output through the enzymatic processing of a SAM electrode. The enzymatic activity of cutinase converts a functional group on the electrode surface from a redox-inactive hydroxyphenyl ester to a redox-active state HQ. Reproduced with permission from ref. 21. Copyright 2006 National Academy of Sciences, USA. | ||
Additionally, Yousaf's group has developed dynamic SAM substrates that “switch” the composition of cell-binding ligands on a substrate surface between an active and an inactive state.22–24 The switching could involve the isomerization of an immobilized ligand; alternatively, a substrate could be ‘turned on’ by ligand binding and could be ‘turned off’ by ligand release. For example, a redox-switchable SAM surface capable of altering the structure of small-molecules was designed using an electroactive oxime linkage and the Huisgen cycloaddition reaction (Fig. 4A).24 This approach was based on the conjugation of oxyamine-terminated ligands on HQ-terminated SAMs. HQ can be oxidized to the Q form via a mild oxidative potential and reacted with oxyamine to form a stable quinone–oxime linkage. For cell adhesion studies, the well known cell-adhesive peptide (RGD) was used as a dynamic ligand (Fig. 4B). By applying a non-invasive electrochemical potential, the affinity of the surface presenting cell-adhesive RGD peptides for cell integrin receptors was altered (from linear to cyclic RGD), modulating cell spreading and migration behavior. Therefore, achieving dynamic control of the redox-switchable SAM biointerface holds considerable promise in tissue engineering, medicine, cell biology and immunology.
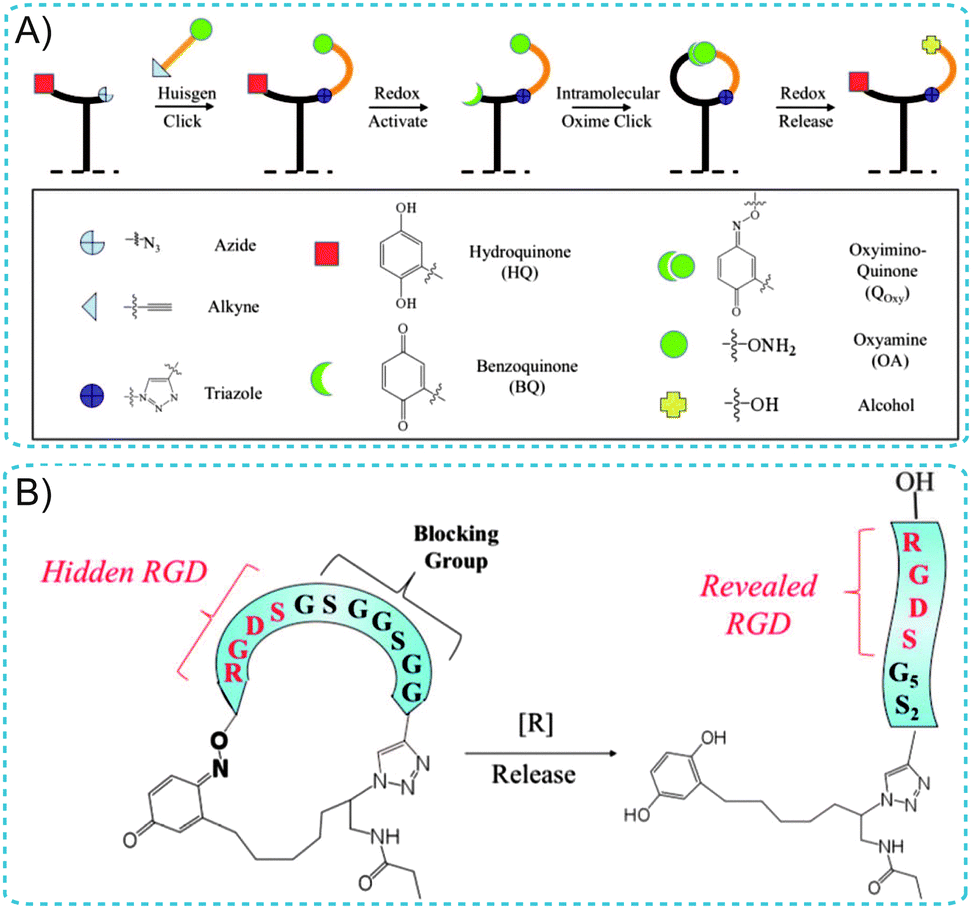 | ||
| Fig. 4 (A) Illustration of the immobilization and switchable control of ligand conformations on SAM surfaces. (B) A cell-adhesive RGD peptide was used as a dynamic ligand. A cyclized RGD peptide containing a blocking sequence does not permit cell adhesion (left). Reductive cleavage of the oxime bond reveals the adhesive RGD peptide for cell adhesion (right). Adapted with permission from ref. 24. Copyright 2011 American Chemical Society. | ||
2.2 Polyquinones/hydroquinones on solid substrates
Polyquinones/hydroquinones are particularly interesting redox polymers in a wide range of biological applications. For example, the electropolymerization of 6-vinyl coenzyme Q0 on electrodes gives rise to stable redox-active films, leading to electrocatalytic activity toward NADH oxidation.25 It should be noted that the fabricated polymeric materials based on polydopamine are of increasing interest in biological applications. In nature, many organisms exhibit reversible wet–dry adhesive properties. Among them, mussels and geckos are the most widely studied examples due to their high adhesive force. Messersmith and co-workers reported a hybrid biologically-inspired adhesive strategy employing arrays of gecko-mimetic nanoscale pillars coated with a thin mussel-mimetic polymer film (Fig. 5).26 The arrays of poly(dimethylsiloxane) (PDMS) pillars were successfully fabricated on a PMMA/Si master using electron-beam lithography. PDMS casting onto the master was followed by curing, and lift-off resulted in gecko-foot-mimetic nanopillar arrays. Finally, a mussel-adhesive-protein-mimetic polymer was coated onto the fabricated nanopillars. The topmost organic layer contained catechols, key components of wet adhesive proteins found in mussel holdfasts. This hybrid adhesive, which combined the salient design elements of both gecko and mussel adhesives, was useful for reversible attachment to a variety of surfaces in any environment. Furthermore, they also introduced a facile method for using dopamine self-polymerization to form multi-functional polydopamine films on a wide variety of inorganic and organic materials through the simple dip-coating of objects.27 Polydopamine coatings can serve as a versatile platform for secondary surface-mediated reactions, ultimately leading to metal, SAM, and grafted polymer coatings.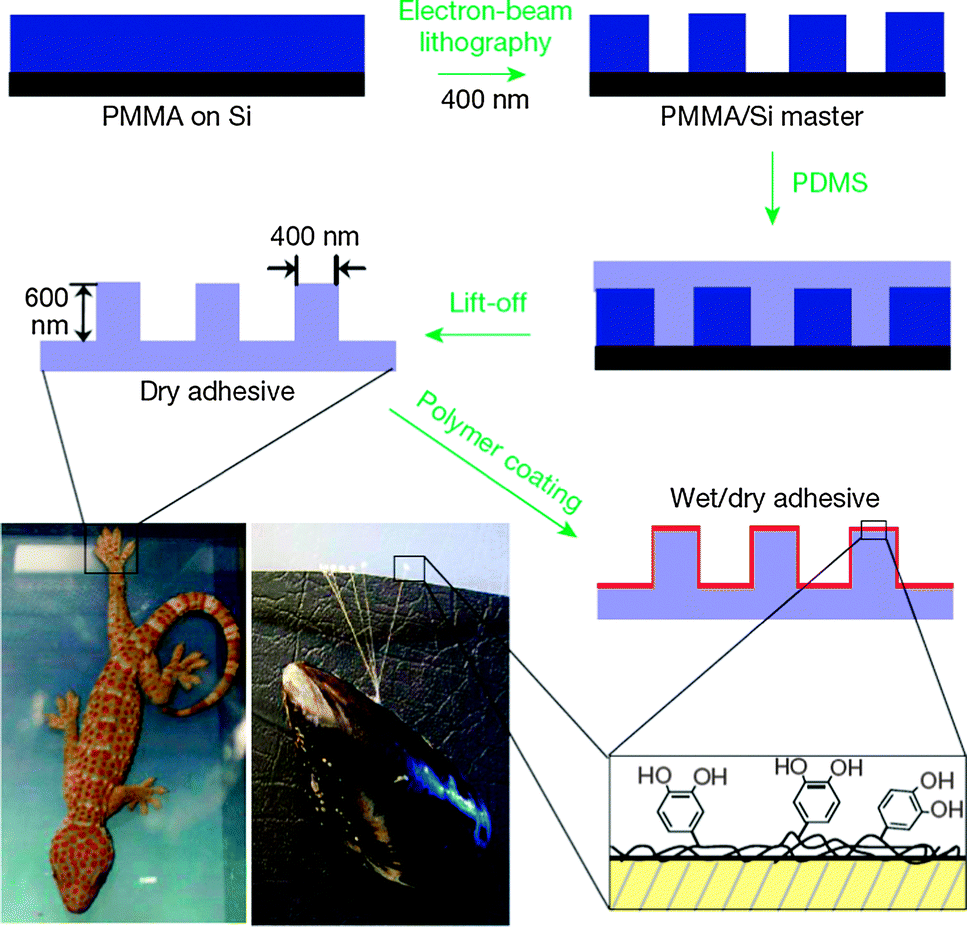 | ||
| Fig. 5 Fabrication of a wet–dry hybrid nanoadhesive inspired by mussels and geckos. Reproduced with permission from ref. 26. Copyright 2007 Nature Publishing Group. | ||
Similarly, polydopamine-assisted interfacial engineering has been synergistically integrated with block copolymer lithography for the surface nanopatterning of low-surface-energy substrate materials, such as Teflon, graphene, and gold.28 Both a hydrophobic cherry tomato and a synthetic Teflon film surface could be made dramatically hydrophilic through polydopamine treatment (Fig. 6A). Moreover, polydopamine-coated polyethylene separators generated using a simple dipping process overcame the poor wetting capabilities of conventional polyethylene separators (Fig. 6B).29 By harnessing the hydrophilic surface character of polydopamine, the modified polyethylene separators exhibited remarkably improved power performance without sacrificing the original advantages of polyethylene separators.
 | ||
| Fig. 6 (A) Mussel-inspired surface engineering of a cherry tomato and Teflon film using polydopamine treatment. (B) A wetting test of polyethylene separators before (left) and after (right) the polydopamine coating. Adapted with permission from ref. 28 and 29. Copyright 2011 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Further, living yeast cells were encapsulated with artificial polydopamine shells.30 Following encapsulation, the shells were functionalized with streptavidin by utilizing the chemical reactivity of polydopamine, and the functionalized cells were biospecifically immobilized onto biotin modified surfaces (Fig. 7). The viability of yeast cells was maintained within the polydopamine shells with a controllable thickness. In addition, the artificial shells increased the resistance of the cell to foreign aggression, such as lyticase. Thus, polydopamine encapsulation provides a valuable starting point for the fundamental investigation and applications of artificial spores because it endows living cells with durability against harsh environments, controllability in cell cycles, and reactivity for cell-surface modification.
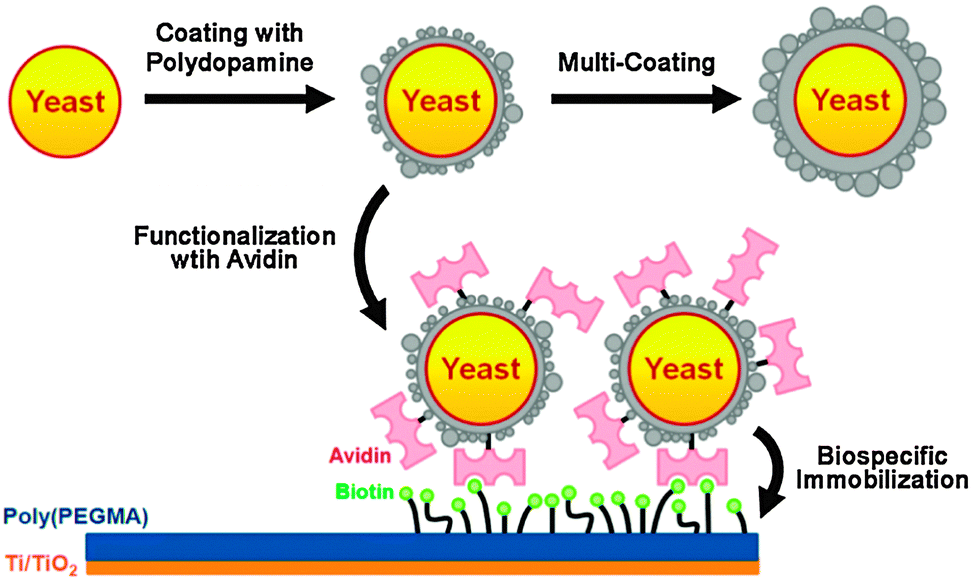 | ||
| Fig. 7 Polydopamine encapsulation of individual yeast cells and functionalization of the artificial polydopamine shells. Reproduced with permission from ref. 30. Copyright 2011 American Chemical Society. | ||
2.3 Q/HQ-supported artificial lipid bilayer membranes
As the vital components, biological membranes form the outer boundary or internal organelles of living cells. They consist largely of a lipid bilayer that imparts a two-dimensional fluid character; proteins embedded in the bilayer and carbohydrates located at its surface facilitate communication and transport across the membrane. The complexity of natural membranes makes direct investigations difficult. For this reason, artificial model membranes have provided a unique platform to unravel the crucial function of biological events in physiological processes.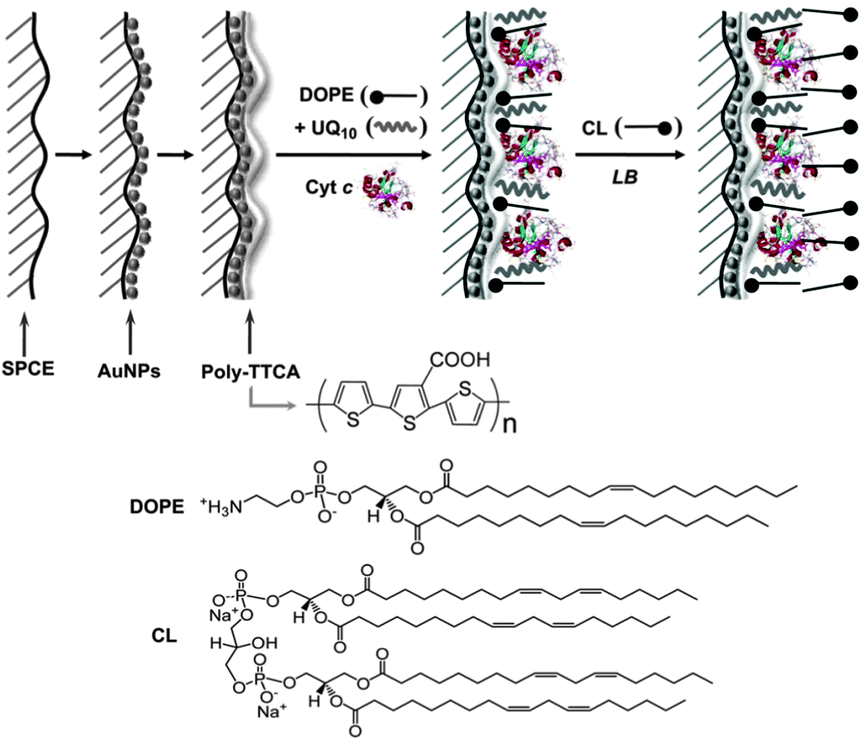
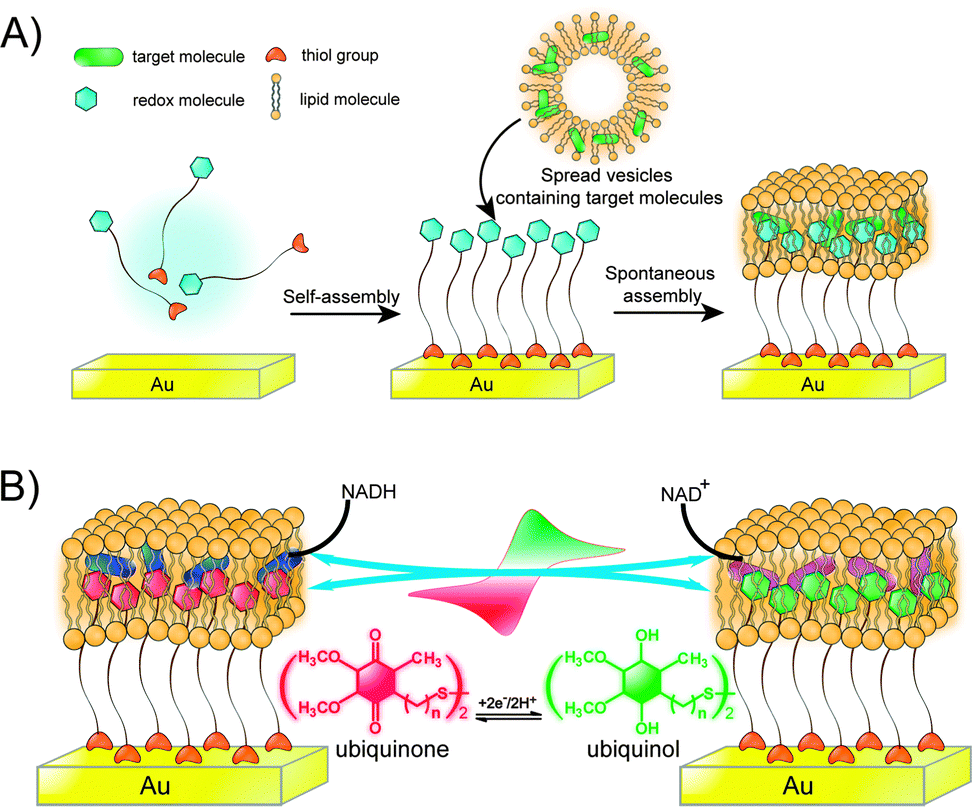 | ||
| Fig. 9 (A) Biomimetic membrane strategy of HBMs for electrochemical measurement. (B) Ubiquinone is embedded in a HBM system that contains NADH. Adapted with permission from ref. 34. Copyright 2013 Nature Publishing Group. | ||
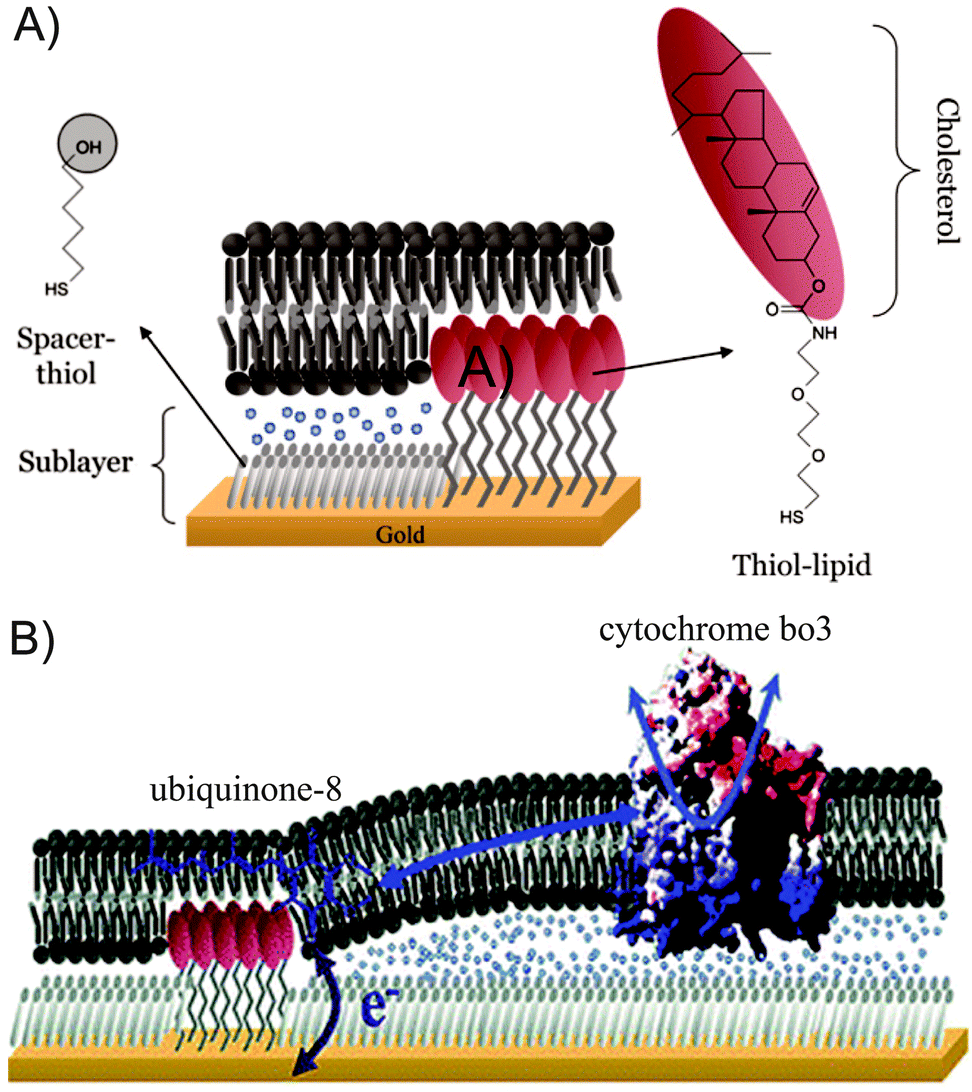 | ||
| Fig. 10 (A) Schematic representation of a tBLM system with thiol–lipids and spacers. (B) Co-immobilized ubiquinone-8 and cytochrome bo3 in tBLMs. Adapted with permission from ref. 35 and 36. Copyright 2007 Elsevier and 2006 American Chemical Society. | ||
3 Q/HQ-functionalized nanomaterial biointerfaces
Nanomaterials have generated intense interest due to their special physical, electronic, and chemical properties, which differ from bulk materials. Nanomaterial surfaces can function as an important interface to interact with the medium surrounding the nanomaterial. The most important characteristics of nanomaterial for a given medium depend on the biomolecules used in the surface functionalization, such as Q/HQ. Nanomaterials can present a multivalent array of Q/HQ and provide biointerfaces for Q/HQ display. In this section, we introduce Q/HQ-functionalized nanomaterials as scaffolds for the construction of biocompatible and target nano-tools for biological applications, particularly for the specific labeling and tracking of cells, tissues and organs.3.1 Q/HQ function in redox applications on nanomaterial biointerfaces
Among nanomaterials, quantum dots (QDs) are among the most extensively researched due to their unique optical properties, including a high emission quantum yield, sharp emission spectra, diameter-dependent emission wavelength tunability, and high chemical- and photostability. In addition, QDs are extremely sensitive to the presence of charges either on their surfaces or in the surrounding environment, leading to a variety of useful optical properties and electronic consequences. Reversible redox cycling between Q and HQ allows the Q/HQ molecule to function as a valuable charge mediator. The redox potential of capping Q/HQ molecules can be chosen to maximize the efficiency of charge transfer to promote transfer of external electrons and holes to QDs, resulting in QD's fluorescence changes. By exploiting this property, controlled charge transport across Q/HQ functionalized QDs can be employed in the development of biosensing applications.We have demonstrated that the enhancement or quenching of the fluorescence of QD bioconjugates can be switched by electrochemically modulating the redox state of surface-capping ubiquinone ligands (Fig. 11A).6 Interestingly, the emission of QD bioconjugates was enhanced when the surface-attached ubiquinone layer was reduced to ubiquinol in the presence of NADH and complex I in an effort to mimic the initial stages of mitochondrial respiration. In the cited study,6 emission of the reduced state ubiquinol-QDs was sensitive to the concentration of superoxide radical anions, and a decrease in the fluorescence intensity was observed. Importantly, the utility of this system was demonstrated by monitoring the change in fluorescence in response to trace reactive oxygen species (ROS) in living cells. 1,2,3-Triazole groups, which are similar to histidine, were incorporated into the ubiquinone ligands to enhance the compatibility of QD bioconjugates in biological systems. Fluorescence results indicated a time-dependent fluorescence enhancement similar to that of QDs assembled with ubiquinone, but a significant increase in the incubation time of complex I compared to the triazole-linked ubiquinone-QDs in the presence of NADH was observed (Fig. 11B). This behaviour arises because triazole groups behave similarly to histidine ligands and can be used to cap enzymes through proteins- or peptide-affinity coordination of triazole residues, leading to the triazole ubiquinone ligands improving binding affinity with complex I. This QD bioconjugate system could be used to probe in vitro and intracellular complex I levels by monitoring QD fluorescence changes. Epidemiological studies suggest that Parkinson's patients have reduced levels of complex I activity. Thus, this system could be employed as a powerful fluorescence biosensor for early stage Parkinson's disease diagnosis and progression monitoring by following complex I levels in SH-SY5Y cells.
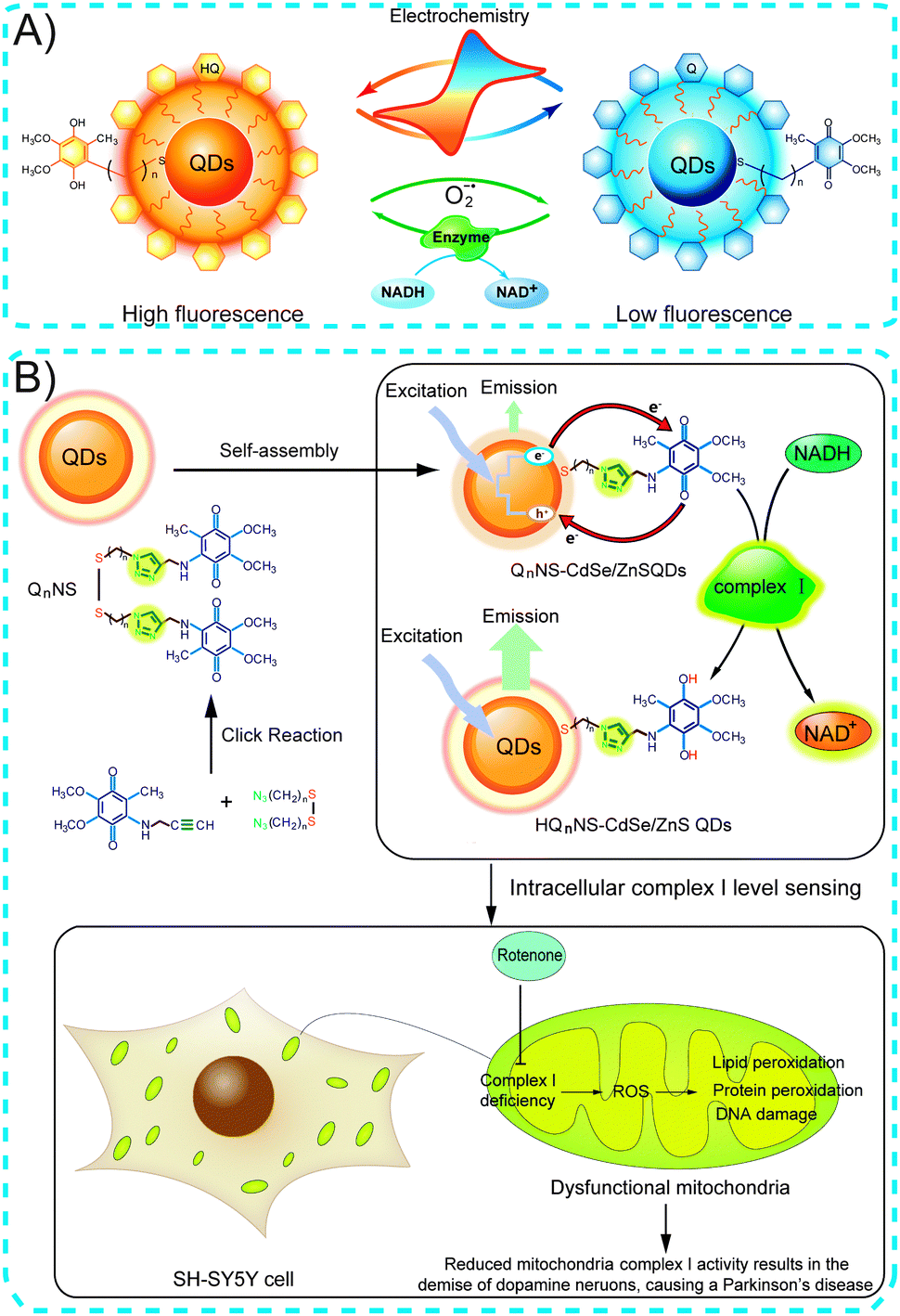 | ||
| Fig. 11 (A) Ubiquinone-functionalized QDs as redox fluorescence biosensors targeting intracellular reactive oxygen species. (B) Schematic of ubiquinone-functionalized QDs targeting Parkinson's disease diagnosis. Adapted with permission from ref. 6 and 7. Copyright 2011 Wiley-VCH Verlag GmbH & Co. KGaA and 2013 Nature Publishing Group. | ||
Another bioanalytical application involves the detection of enzymatic activity using the luminescence of QDs during biocatalytic processes. For example, tyrosinase activity and thrombin hydrolytic activity can be monitored by following the fluorescence changes using dopamine–QD conjugates. Tyrosinase can catalyze the oxidation of tyramine or tyrosine in the presence of O2 to their respective catechols (e.g., from tyramine to dopamine) and then to dopaquinone (Fig. 12A). Willner and co-workers demonstrated that dopaquinone conversion as an electron acceptor quenched the fluorescence of QDs.37 Decrease in QD fluorescence upon interaction with various concentrations of tyrosinase was used to measure tyrosinase activity (Fig. 12B).37,38 Following the cleavage of the dopaquinone unit of the peptide using thrombin, the dopaquinone molecules no longer provided efficient electron transfer to the QDs. As a result, the QD fluorescence recovery measured the activity of thrombin (Fig. 12C).37
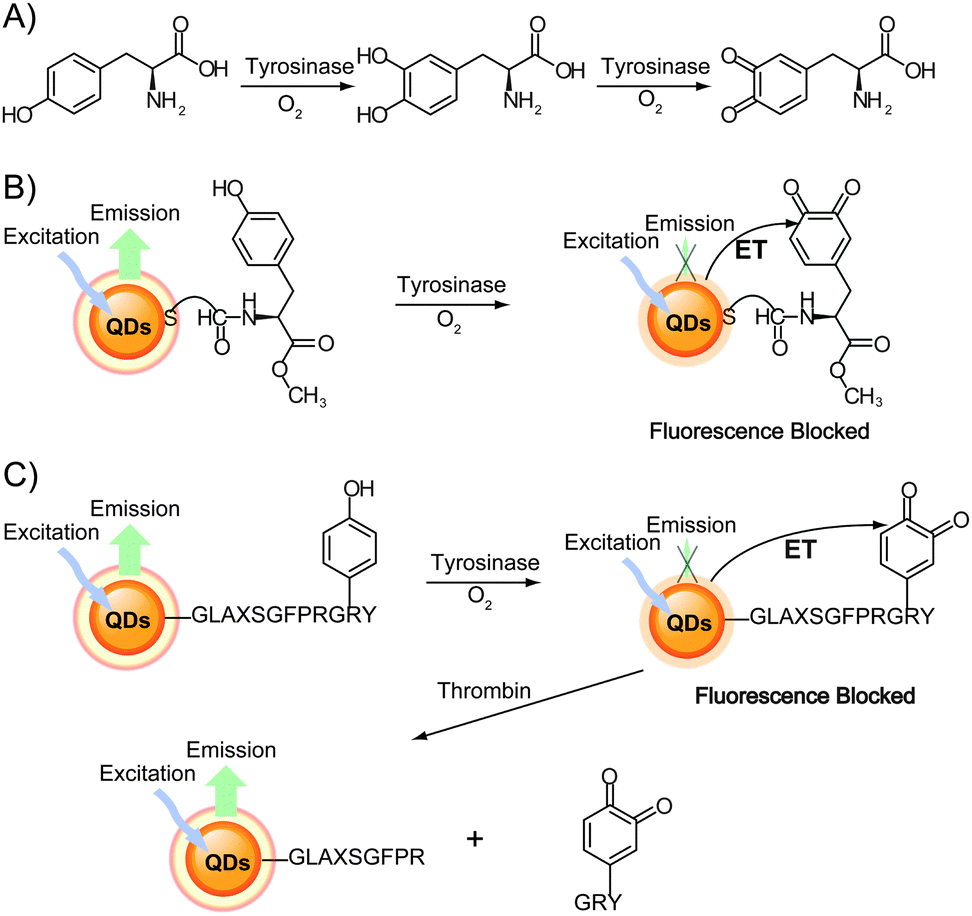 | ||
| Fig. 12 (A) Scheme of tyrosine oxidation by tyrosinase in the presence of O2. (B) Monitoring tyrosinase activity by the biocatalytic oxidation of tyrosine-functionalized QDs to dopaquinone, which results in electron transfer quenching of the QD fluorescence. (C) Sequential probing of tyrosinase activity and thrombin activity by the tyrosinase-induced oxidation of tyrosine, followed by the thrombin-induced cleavage of the dopaquinone portion of the peptide to recover the QD fluorescence. Adapted with permission from ref. 37. Copyright 2006 American Chemical Society. | ||
In addition, the transition between dopamine and dopaquinone can easily be achieved by varying the pH, thus enabling the dopamine-functionalized QD system to be used for pH sensing. Medintz and co-workers demonstrated the use of dopamine–QD conjugates as pH sensors to measure changes in cytoplasmic pH as cells undergo drug-induced alkalosis.39 At lower pH values, the dopamine primarily remained in a reduced state, leading to subtle fluorescence quenching. As the pH increased, the oxidized dopaquinone became more prevalent, and significant quenching was observed (Fig. 13A).39,40 The QD–dopamine bioconjugates were used to monitor progressive changes in cytosolic pH (from 6.6 to 12.8) by measuring the QD fluorescence (Fig. 13B). In addition, living cells were labeled with the QD–dopamine conjugates in a redox-sensitive pattern. When the cellular conditions were oxidized, QD fluorescence was observed throughout the cell, including the perinuclear region and the mitochondria. Under reducing conditions, only the cell periphery and the lysosomes exhibited fluorescence (Fig. 13C).41 This study provided a general framework for the creation of probes that label different areas of living cells according to their redox state.
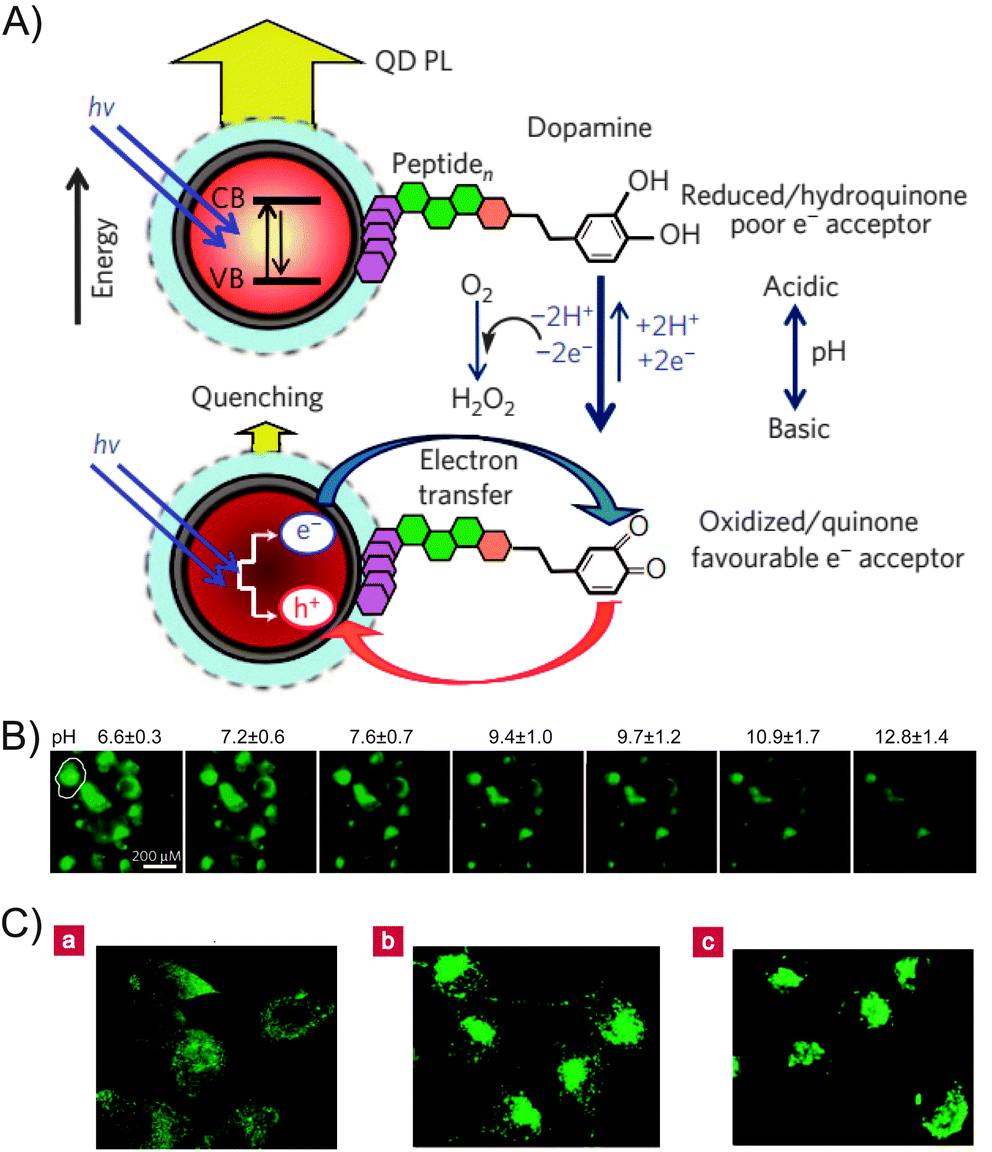 | ||
| Fig. 13 (A) Schematic representation of the use of QD–dopamine conjugates as a pH sensor. (B) Fluorescent image of intracellular pH sensing. (C) QD–dopamine labeled cells in a redox-sensitive pattern. (a) Cells under oxidizing conditions. (b) Untreated, actively proliferating cells. (c) Cells under reducing conditions. Adapted with permission from ref. 39 and 41. Copyright 2010 and 2006 Nature Publishing Group. | ||
A further competitive assay for monitoring changes in intracellular redox potential using surface-enhanced Raman scattering (SERS) nanosensors has been described.42 By modifying gold nanoparticles (NPs) with Q, whose SERS spectrum changes depend on its oxidation state, the proportions of oxidized and reduced species could be accurately measured, thus permitting the calculation of the intracellular redox potential. The localized temporal response to both reductive and oxidative stress was measured over a previously unattainable potential range, and correlated oxidative changes were measured during apoptosis by changes in caspase activity using the functionalized gold NPs in single fibroblast cells.
Moreover, dopamine can also be linked via its amino terminus to QDs using carbon disulfide (CS2), thereby quenching the QD fluorescence. Glutathione (GSH) can be used to cleave the disulfide bond and free the QDs, and then restore the fluorescence (Fig. 14).43 Here, an OFF–ON fluorescent probe was employed for GSH detection. However, this probe is unable to selectively detect GSH over cysteine or homocysteine.
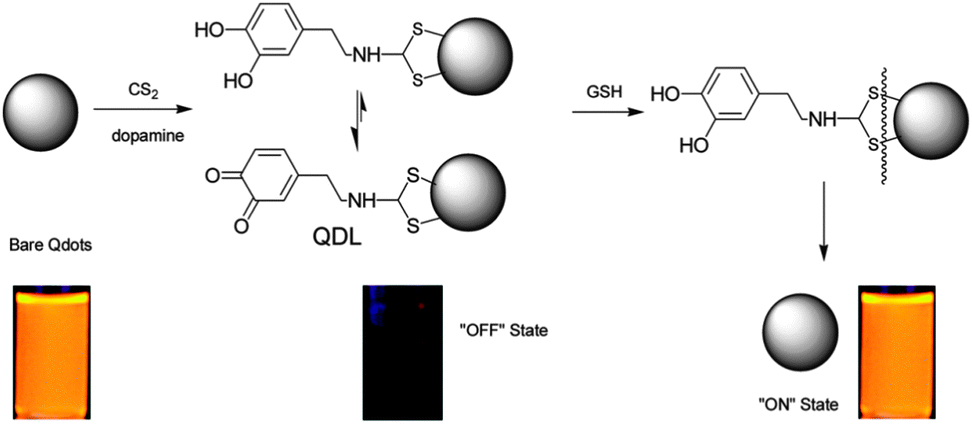 | ||
| Fig. 14 Scheme for the QD-based OFF–ON probe for the detection of glutathione. Reproduced with permission from ref. 43. Copyright 2009 American Chemical Society. | ||
The interaction between Q and NPs is frequently studied for their application based on mutual electron transfer. Recently we conducted an interesting study where thiol derivatives of ubiquinone induced a spontaneous fragmentation process of gold NPs in aqueous solution, due to the energetic electron injection from thiol-ubiquinone to the gold NPs.44 In addition, several studies have used QDs to determine charge separation and recombination rates via adsorbed Q. Weiss et al. quantitatively studied a Q–QD system that underwent both adsorbed and collisionally gated photo-induced charge transfer, resulting in the decay of the excited state of PbS QDs.45 The availability of a collisionally gated pathway improved the yield of electron transfer from PbS QDs to Q by an average factor of 2.5 compared with only the process of static electron transfer. A similar study of the electron transfer between type I core–shell QDs and adsorbed anthraquinone-2,3-dicarboxylic acid demonstrated that charge separation and recombination rates decrease dramatically due to the decrease in the electron and hole densities at the QD surface with increasing shell thickness.46 While using type II core–shell QDs, ultrafast electron separation and slow recombination with Q could be obtained by modulation on their shell thickness.47,48 These results reveal that QDs may be a promising material for solar energy conversion in photovoltaics and photocatalysis by enabling control over charge separation and recombination rates.
3.2 Q/HQ function in other applications on nanomaterial biointerfaces
Coenzyme Q10 acts as an effective inhibitor of oxidative damage by scavenging free radicals in both the mitochondria and lipid membranes. The potent antioxidant activity of coenzyme Q10 makes it a promising drug candidate for several diseases, but its poor aqueous solubility and low oral bioavailability have limited its development for oral administration. Various strategies are being developed to improve the bioavailability of coenzyme Q10. Jain et al. prepared stable coenzyme Q10-loaded polymeric NPs (poly lactic-co-glycolic acid) with sizes below 100 nm using a scalable emulsion–diffusion–evaporation method.49 The coenzyme Q10-NPs were found to improve oral bioavailability and ROS scavenging by 10- and 4.28-fold, respectively, compared with the free coenzyme Q10. Furthermore, the positively charged coenzyme Q10-NPs were localized in two major sources of ROS generation: the mitochondria and lysosomes. The remarkably greater hepatoprotective and anti-inflammatory activity of the coenzyme Q10-NPs compared with free coenzyme Q10 can be attributed to the mitigation of the deleterious effects associated with the generation of ROS by live noninvasive animal imaging. However, clinical trials must be conducted to further study coenzyme Q10-NP implementation for the treatment of ROS-generating diseases. Moreover, Maysinger et al. reported the design and construction of multi-functional nanocarriers for selective coenzyme Q10 delivery to the mitochondria using coenzyme Q10-loaded nanosized micelles.50 Confocal microscopy of the mitochondria-specific vital dye-labeled micelles demonstrated that the nanocarrier did indeed localize in the mitochondria. The high loading capacity and effectiveness of coenzyme Q10 allowed this mitochondria-targeting nanocarrier to be exploited for other novel site-directed nanodelivery systems employing various pharmacological agents.
The pharmaceutical activities of other Q/HQ compounds were also improved by exploiting nanomaterials as delivery vehicles. Choi et al. prepared biocompatible gold NPs containing the radiosensitizer β-lapachone (lap) and composed of a poly(ethylene glycol) (PEG) shell, per-6-thio-β-cyclodextrin (SH-CD) as a drug pocket, and anti-epidermal growth factor receptor (EGFR) antibody as a targeting ligand (Fig. 15).51 The preferential delivery of lap to tumor tissues was demonstrated using gold NP–lap conjugated anti-EGFR antibodies and cell lines expressing EGFR in varying levels. The intravenous injection of gold NP–lap conjugates resulted in highly enhanced radiotherapeutic efficacy and feasibility for preclinical evaluation for human cancer treatment through in vivo experiments. Another form of tumor-targeted delivery for doxorubicin (DOX) was designed using hyaluronate-tethered multi-walled carbon nanotube conjugates complexed with DOX via π–π stacking interactions.52 This nanodelivery system augmented the antitumor activity of DOX against cancer cells overexpressing hyaluronan receptors while reducing drug-associated cardiotoxicity. The gold NP–lap conjugates improved the tumor-growth inhibitory effect by 5 fold compared with free DOX at equivalent concentrations. The therapeutic conjugates were further labeled with the near-infrared fluorescent dye Alexa-Fluor-647 and the radiotracer Technetium-99m to track the intracellular trafficking and bio-distribution of the conjugates in real-time via optical imaging and scintigraphic techniques. Overall, the nano-carrier systems developed in the course of this research may expand the therapeutic possibilities for a broad spectrum of anticancer drugs.
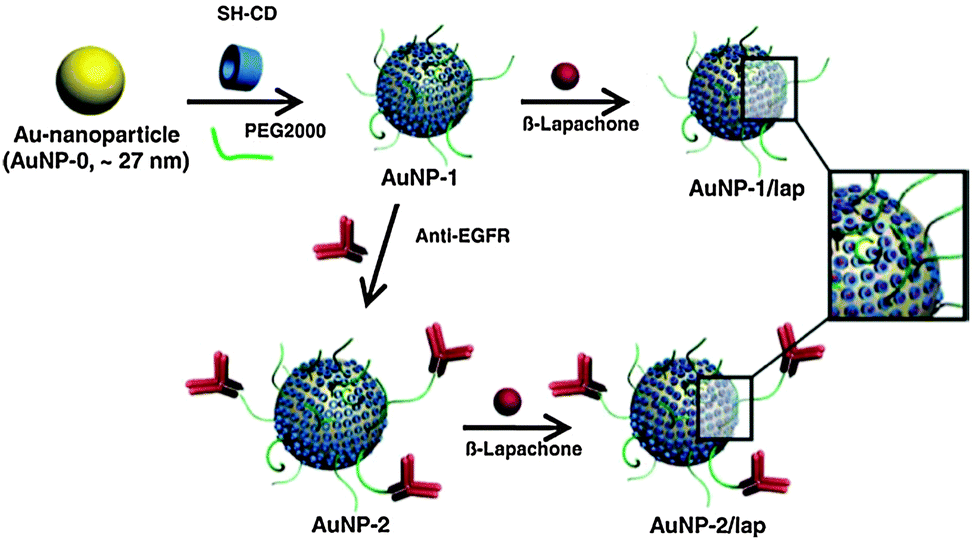 | ||
| Fig. 15 Schematic illustration of the preparation of gold NPs containing β-lapachone. Reproduced with permission from ref. 51. Copyright 2009 Elsevier. | ||
Additionally, the multiple bio-functionalization of quantum rods (QRs) for the specific bio-labeling and controlled drug release of dopamine has been reported.53 The QRs were made highly biocompatible through conjugation with PEG molecules and coating with a galactose shell with high affinity for the glucose transporter GLUT-1, which is over-expressed in many tumor cells. The controlled conjugation of dopamine to galactose through an ester bond also allowed facile hydrolysis by cellular esterases, as illustrated in Fig. 16. Thus, these dopamine glyco-QRs were able to selectively recognize specific cells and deliver active dopamine into them. This multi-functional system could be used as a platform to investigate both imaging and the therapeutic applications of drugs.
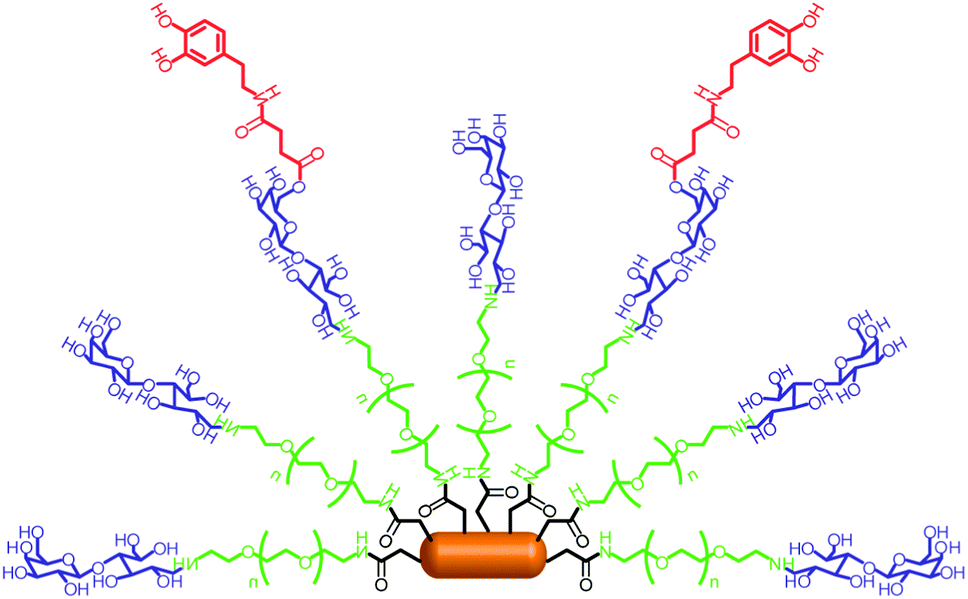 | ||
| Fig. 16 Schematic illustration of dopamine glyco-QRs. Adapted with permission from ref. 53. Copyright 2011 Royal Society of Chemistry. | ||
The polydopamine (PDA) on NPs of varying sizes can produce a PDA capsule.54,55 Based on this phenomenon, Caruso and co-workers prepared dopamine-mediated poly(L-glutamic acid) (PGA) conjugates (PGAPDA) as biodegradable capsules.56 The PGAPDA coating was assembled onto silica NPs, and stable PGAPDA capsules were obtained following the removal of the silica core. The capsules allowed the loading of cargo and its subsequent release upon capsule degradation. Pivotal to this work was the degradability of the capsule, which could be tuned by altering the percentage of dopamine within the PGAPDA, thus leading to potential medicinal applications. In addition, in vivo studies demonstrated that the use of cyclodextrins as “promoters” of the encapsulation of atovaquone in poly(anhydride) NPs created novel drug carriers for improving the oral delivery of this lipophilic drug.57 These studies indicate that Q/HQ-functionalized NP systems can improve bioavailability and represent promising oral delivery devices for treatment of numerous diseases.
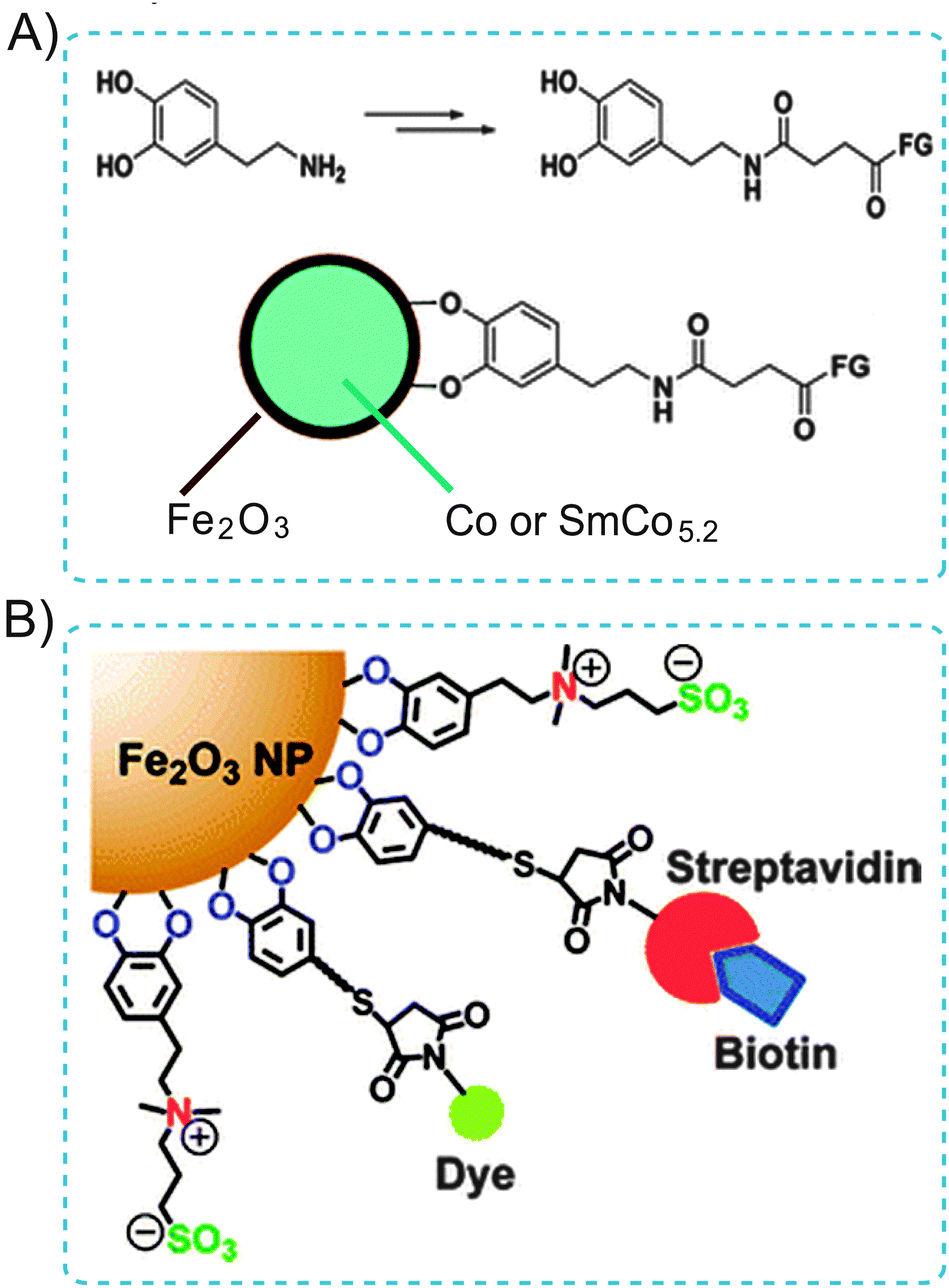 | ||
| Fig. 17 (A) Illustration of the use of dopamine as an anchor to link functional molecules to the Fe2O3 shell of magnetic NPs. (B) Schematic displaying the zwitterionic dopamine sulfonate (ZDS) ligand coated Fe2O3 NPs (ZDS-NPs). The streptavidin/dye functionalized ZDS-NPs specifically target biotin receptors. Adapted from ref. 58 and 59. Copyright 2004 and 2012 American Chemical Society. | ||
4 Conclusions and future prospects
A critical biointerface research is the development of coatings that alter the biocompatibility of interfaces or produce biomimetic platforms. For example, the self-assembled monolayers can be used to simulate cell membranes. Understanding the interactions between coatings and interfaces, and controlling the chemical and physical properties of biointerfaces, is critical for the development of biointerfaces for biological applications. In this tutorial review, we briefly discussed the recent progress in Q/HQ-functionalized biointerfaces made by our group and others. We focused on the support materials and functionalization methods typically employed in biointerface development. Particular attention was given to biointerfacial properties and their biological applications. Promising research directions for the biological application of Q/HQ-functionalized solid substrates and nanomaterials supported biointerfaces inspired by Nature are recommended. Q/HQ functionalized biointerfaces that have generated a fully artificial biomimetic interface are ideal for highly selective biosensing. The combination of biocompatible Q/HQ coatings and multi-functionalized biointerfaces provides valuable platforms that mimic the biological activity of Q/HQ and improve interfacial properties. Applications employing Q/HQ-functionalized biointerfaces have provided new insights into biological and molecular biological mechanisms. Based on these studies, it is possible to guide strategies into the development of novel high-performance biomaterials and biosensors and their applications. Although significant progress has been made in this field, the exploration of in vivo applications using Q/HQ-related biointerfaces is only at a starting point and deserves further study. The Q/HQ-based system can also be used as a model for functionalized biointerfaces, enabling numerous novel applications.Acknowledgements
This research was supported by the 973 Program (2013CB733700), the National Science Fund for Distinguished Young Scholars (21125522) and the Fundamental Research Funds for the Central Universities (222201313004).Notes and references
- H. Nohl, W. Jordan and R. J. Youngman, Adv. Free Radical Biol. Med., 1986, 2, 211–279 CrossRef CAS.
- L. A. Sazanov and P. Hinchliffe, Science, 2006, 311, 1430–1436 CrossRef CAS PubMed.
- A. Cattani-Scholz, D. Pedone, F. Blobner, G. Abstreiter, J. Schwartz, M. Tornow and L. Andruzzi, Biomacromolecules, 2009, 10, 489–496 CrossRef CAS PubMed.
- D. Chen, G. Wang and J. H. Li, J. Phys. Chem. C, 2007, 111, 2351–2367 CAS.
- W. Ma, D.-W. Li, T. C. Sutherland, Y. Li, Y.-T. Long and H.-Y. Chen, J. Am. Chem. Soc., 2011, 133, 12366–12369 CrossRef CAS PubMed.
- L.-X. Qin, W. Ma, D.-W. Li, Y. Li, X.-Y. Chen, H.-B. Kraatz, T. D. James and Y.-T. Long, Chem.–Eur. J., 2011, 19, 5262–5271 CrossRef PubMed.
- W. Ma, L.-X. Qin, F.-T. Liu, Z. Gu, J. Wang, Z. G. Pan, T. D. James and Y.-T. Long, Sci. Rep., 2013, 3, 1537–1544 CAS.
- W. Schultz, Trends Neurosci., 2007, 30, 203–210 CrossRef CAS PubMed.
- L. Roffe, K. Schmidt and E. Ernst, J. Clin. Oncol., 2004, 22, 4418–4424 CrossRef CAS PubMed.
- C. Costentin, Chem. Rev., 2008, 108, 2145–2179 CrossRef CAS PubMed.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- H. G. Hong and W. Park, Langmuir, 2001, 17, 2485–2492 CrossRef CAS.
- P. Petrangolini, A. Alessandrini, L. Berti and P. Facci, J. Am. Chem. Soc., 2010, 132, 7445–7453 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef PubMed.
- J. Li, C.-L. Sun, L. Tan, Y.-L. Xie and H.-L. Zhang, Langmuir, 2013, 29, 5199–5206 CrossRef CAS PubMed.
- S. A. Trammell, M. Moore, D. Lowy and N. Lebedev, J. Am. Chem. Soc., 2008, 130, 5579–5585 CrossRef CAS PubMed.
- Q. D. Zhang, G. March, V. Noel, B. Piro, S. Reisberg, L. D. Tran, L. V. Hai, E. Abadia, P. E. Nielsen, C. Sola and M. C. Phama, Biosens. Bioelectron., 2012, 32, 163–168 CrossRef CAS PubMed.
- R. S. Dey, S. Gupta, R. Paira and C. R. Raj, ACS Appl. Mater. Interfaces, 2010, 2, 1355–1360 CAS.
- X. P. He, X. W. Wang, X. P. Jin, H. Zhou, X. X. Shi, G. R. Chen and Y.-T. Long, J. Am. Chem. Soc., 2011, 133, 3649–3657 CrossRef CAS PubMed.
- J. Robertus, W. R. Browne and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 354–378 RSC.
- J. H. Collier and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2021–2025 CrossRef CAS PubMed.
- E. W. L. Chan, S. Park and M. N. Yousaf, Angew. Chem., Int. Ed., 2008, 47, 6267–6271 CrossRef CAS PubMed.
- W. Luo and M. N. Yousaf, J. Am. Chem. Soc., 2011, 133, 10780–10783 CrossRef CAS PubMed.
- B. M. Lamb and M. N. Yousaf, J. Am. Chem. Soc., 2011, 133, 8870–8873 CrossRef CAS PubMed.
- Y. Li, L. Shi, W. Ma, D.-W. Li, H.-B. Kraatz and Y.-T. Long, Bioelectrochemistry, 2011, 80, 128–131 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- B. H. Kim, D. H. Lee, J. Y. Kim, D. O. Shin, H. Y. Jeong, S. Hong, J. M. Yun, C. M. Koo, H. Lee and S. O. Kim, Adv. Mater., 2011, 23, 5618–5622 CrossRef CAS PubMed.
- M. H. Ryou, Y. M. Lee, J. K. Park and J. W. Choi, Adv. Mater., 2011, 23, 3066–3070 CrossRef CAS PubMed.
- S. H. Yang, S. M. Kang, K. B. Lee, T. D. Chung, H. Lee and I. S. Choi, J. Am. Chem. Soc., 2011, 133, 2795–2797 CrossRef CAS PubMed.
- K.-S. Lee, M.-S. Won, H.-B. Noh and Y.-B. Shim, Biomaterials, 2010, 31, 7827–7835 CrossRef CAS PubMed.
- T. W. McBee, L. Wang, C. Ge, B. M. Beam, A. L. Moore, D. Gust, T. A. Moore, N. R. Armstrong and S. S. Saavedra, J. Am. Chem. Soc., 2006, 128, 2184–2185 CrossRef CAS PubMed.
- I. Bogeski, R. Gulaboski, R. Kappl, V. Mirceski, M. Stefova, J. Petreska and M. Hoth, J. Am. Chem. Soc., 2011, 133, 9293–9303 CrossRef CAS PubMed.
- W. Ma, Y.-L. Ying, L.-X. Qin, Z. Gu, H. Zhou, D.-W. Li, T. C. Sutherland, H. Y. Chen and Y.-T. Long, Nat. Protocols, 2013, 8, 439–450 CAS.
- L. J. C. Jeuken, S. D. Connell, P. J. F. Henderson, R. B. Gennis, S. D. Evans and R. J. Bushby, J. Am. Chem. Soc., 2006, 128, 1711–1716 CrossRef CAS PubMed.
- L. J. C. Jeuken, R. J. Bushby and S. D. Evans, Electron. Commun., 2007, 9, 610–614 CrossRef CAS PubMed.
- R. Gill, R. Freeman, J.-P. Xu, I. Willner, S. Winograd, I. Shweky and U. Banin, J. Am. Chem. Soc., 2006, 128, 15376–15377 CrossRef CAS PubMed.
- X. Liu and H. X. Ju, Anal. Chem., 2008, 80, 5377–5382 CrossRef CAS PubMed.
- I. L. Medintz, M. H. Stewart, S. A. Trammell, K. Susumu, J. B. Delehanty, B. C. Mei, J. S. Melinger, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, Nat. Mater., 2010, 9, 676–684 CrossRef CAS PubMed.
- X. Ji, G. Palui, T. Avellini, H. B. Na, C. Y. Yi, K. L. Knappenberger, Jr. and H. Mattoussi, J. Am. Chem. Soc., 2012, 34, 6006–6017 CrossRef PubMed.
- S. J. Clarke, C. A. Hollmann, Z. J. Zhang, D. Suffern, S. E. Bradforth, N. M. Dimitrijevic, W. G. Minarik and J. L. Nadeau, Nat. Mater., 2006, 5, 409–417 CrossRef CAS PubMed.
- C. A. R. Auchinvole, P. Richardson, C. McGuinnes, V. Mallikarjun, K. Donaldson, H. McNab and C. J. Campbell, ACS Nano, 2012, 6, 888–896 CrossRef CAS PubMed.
- S. Banerjee, S. Kar, J. M. Perez and S. Santra, J. Phys. Chem. C, 2009, 113, 9659–9663 CAS.
- S. Riaz, W. Ma, C. Jing, M. H. Nawaz, D.-W. Li and Y.-T. Long, Chem. Commun., 2013, 49, 1738–1740 RSC.
- K. E. Knowles, M. Malicki and E. A. Weiss, J. Am. Chem. Soc., 2012, 134, 12470–12473 CrossRef CAS PubMed.
- H. Zhu, N. Song and T. Lian, J. Am. Chem. Soc., 2010, 132, 15038–15045 CrossRef CAS PubMed.
- S. Y. Jin, J. Zhang, R. D. Schaller, T. Rajh and G. P. Wiederrecht, J. Phys. Chem. Lett., 2012, 3, 2052–2058 CrossRef CAS.
- H. Zhu, N. Song and T. Lian, J. Am. Chem. Soc., 2011, 133, 8762–8771 CrossRef CAS PubMed.
- N. K. Swarnakar, A. K. Jain, R. P. Singh, C. Godugu, M. Das and S. Jain, Biomaterials, 2011, 32, 6860–6874 CrossRef CAS PubMed.
- A. Sharma, G. M. Soliman, N. Al-Hajaj, R. Sharma, D. Maysinger and A. Kakkar, Biomacromolecules, 2012, 13, 239–252 CrossRef CAS PubMed.
- S. Y. Jeong, S. J. Park, S. M. Yoon, J. Jung, H. N. Woo, S. L. Yi, S. Y. Song, H. J. Park, C. Kim, J. S. Lee, J. S. Lee and E. K. Choi, J. Controlled Release, 2009, 139, 239–245 CrossRef CAS PubMed.
- S. R. Datir, M. Das, R. Singh and S. Jain, Bioconjugate Chem., 2012, 23, 2201–2213 CrossRef CAS PubMed.
- M. A. Malvindi, R. Di Corato, A. Curcio, D. Melisi, M. G. Rimoli, C. Tortiglione, A. Tino, C. George, V. Brunetti, R. Cingolani, T. Pellegrino and A. Ragusa, Nanoscale, 2011, 3, 5110–5119 RSC.
- A. Postma, Y. Yan, Y. Wang, A. N. Zelikin, E. Tjipto and F. Caruso, Chem. Mater., 2009, 21, 3042–3044 CrossRef CAS.
- J. W. Cui, Y. J. Wang, A. Postma, J. C. Hao, L. Hosta-Rigau and F. Caruso, Adv. Funct. Mater., 2010, 20, 625–1631 Search PubMed.
- C. J. Ochs, T. Hong, G. K. Such, J. Cui, A. Postma and F. Caruso, Chem. Mater., 2011, 23, 3141–3143 CrossRef CAS.
- J. Calvo, J. L. Lavandera, M. Agüeros and J. M. Irache, Biomed. Microdevices, 2011, 13, 1015–1025 CrossRef CAS PubMed.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS PubMed.
- H. Wei, N. Insin, J. Lee, H. S. Han, J. M. Cordero, W. Liu and M. G. Bawendi, Nano Lett., 2012, 12, 22–25 CrossRef CAS PubMed.
- M. D. Shultz, J. U. Reveles, S. N. Khanna and E. E. Carpenter, J. Am. Chem. Soc., 2007, 129, 2482–2487 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |