Synthesis, structure and reactivity of rare-earth metal complexes containing anionic phosphorus ligands
Tianshu Li
,
Sabrina Kaercher
and
Peter W. Roesky
*
Institut für Anorganische Chemie, Karlsruhe Institute of Technology (KIT), Engesserstrasse 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 15th August 2013
Abstract
A comprehensive review of structurally characterized rare-earth metal complexes containing anionic phosphorus ligands is presented. Since rare-earth elements form hard ions and phosphorus is considered as a soft ligand, the rare-earth metal phosphorus coordination is regarded as a less favorite combination. Three classes of phosphorus ligands, (1) the monoanionic organophosphide ligands (PR2−) bearing one negative charge on the phosphorus atom; (2) the dianionic phosphinidene (PR2−) and P3− ligands; and (3) the pure inorganic polyphosphide ligands (Pnx−), are included here. Particular attention has been paid to the synthesis, structure, and reactivity of the rare-earth metal phosphides.
 Tianshu Li | Tianshu Li received her doctoral degree in organometallic chemistry and catalysis with Prof. Robert H. Morris from the University of Toronto (Toronto, ON, Canada) in 2005. From 2006 to 2009, she was a postdoctoral researcher with Dr Greg Kubas and Prof. Tom Baker at Los Alamos National Laboratory (Los Alamos, NM, USA), where she worked on catalyst design for the biomimetic hydrogen production project. Since 2009 she has been working as a researcher with Prof. Peter Roesky at Karlsruhe Institute of Technology (Karlsruhe, Germany). Her current research interests include characterization and properties of rare-earth metal phosphide complexes. |
 Sabrina Kaercher | Sabrina Kaercher was born as Sculfort in Strasbourg in 1980 and graduated from the University of Strasbourg. She obtained her doctoral degree from the University of Strasbourg (Dr Pierre Braunstein) in 2009. She was as a postdoc first with Prof. Dr Doris Kunz at University of Tübbingen (2010) and then with Prof. Dr Peter W. Roesky (2011–2013) at the Karlsruhe Institute of Technology. |
 Peter W. Roesky | Peter W. Roesky obtained his diploma in 1992 from the University of Würzburg and his doctoral degree from the Technical University of Munich (with Prof. W. A. Herrmann) in 1994. He was a postdoc with Prof. T. J. Marks at Northwestern University (1995–1996). In 1999 he completed his Habilitation at the University of Karlsruhe. As a full professor he joined the faculty of the Freie Universität Berlin in 2001. Since 2008 he has held the chair of inorganic functional materials at the University of Karlsruhe. He received in 1996 a Liebig-Stipendium, in 1999 a Heisenberg-Stipendium, and in 2000 a Karl-Winnacker-Stipendium. |
List of the key learning points(1) Going beyond the classical Pearsons HSAB concept(2) Recent developments in rare-earth metal chemistry (3) Recent developments in phosphide chemistry (4) Polyphosphides of the rare-earth metals (5) Adapting the Zintl concept from solid-state chemistry to molecular organometallic and inorganic chemistry |
1. Introduction
Phosphorus is one of the most useful and well-established donor atoms in coordination chemistry, and can form strong bonds with soft metals. Many phosphorus transition metal complexes are used as catalysts in various industrial processes.1 In contrast, the phosphorus rare-earth metal complexes are far less common. The rare-earth metals (Ln) have recently gained public interest because of the export restriction of these elements by the Chinese government since 2005. In the last three decades, many inorganic and organometallic rare-earth metal complexes have been synthesized and applied in homogeneous catalysis.2 According to Pearsons HSAB concept, rare-earth elements form hard ions and phosphorus is considered as a soft ligand.3 Thus, the rare-earth metal phosphorus coordination is regarded as a less favorite combination. Marks and coworkers established a series of absolute metal ligand bond disruption enthalpies (BDE) in Cp*2Sm–X (Cp* = η5-C5Me5) compounds, which demonstrate the bonding preferences of Sm–X (kcal mol−1): Cl (97.1) > C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh (93.2) > Br (83.6) > OtBu (82.4) > S(nPr) (73.4) > I (69.4) > H (54.2) > NMe2 (48.2) > PEt2 (32.6).4 Ln–P bonded complexes, shown to be the thermodynamically weakest link, are considerably challenging but exciting to access. In spite of the difficulty in the synthesis and the characterization of rare-earth metal phosphorus complexes, the chemistry of rare-earth metal complexes with phosphorus donor ligands has seen significant advances during the last two decades; however, only a limited number of reviews have been distributed.5–10 Early reports on the neutral phosphine adduct of inorganic and organometallic rare-earth metal complexes have been reviewed by Fryzuk and coworkers in 1990.9 In 1998 and 2001, Nief reviewed the rare-earth metal complexes containing neutral and anionic phosphorus ligands8 and the heterocyclopentadienyl phospholyl ligands,7 respectively. In 2006, Le Floch reviewed phospholyl ligands used in the coordination chemistry.5
CPh (93.2) > Br (83.6) > OtBu (82.4) > S(nPr) (73.4) > I (69.4) > H (54.2) > NMe2 (48.2) > PEt2 (32.6).4 Ln–P bonded complexes, shown to be the thermodynamically weakest link, are considerably challenging but exciting to access. In spite of the difficulty in the synthesis and the characterization of rare-earth metal phosphorus complexes, the chemistry of rare-earth metal complexes with phosphorus donor ligands has seen significant advances during the last two decades; however, only a limited number of reviews have been distributed.5–10 Early reports on the neutral phosphine adduct of inorganic and organometallic rare-earth metal complexes have been reviewed by Fryzuk and coworkers in 1990.9 In 1998 and 2001, Nief reviewed the rare-earth metal complexes containing neutral and anionic phosphorus ligands8 and the heterocyclopentadienyl phospholyl ligands,7 respectively. In 2006, Le Floch reviewed phospholyl ligands used in the coordination chemistry.5
Currently, the goal of this contribution is to present a comprehensive coverage of structurally characterized rare-earth metal complexes with anionic phosphorus ligands, which are less common than complexes with neutral phosphine ligands. Three classes of ligands are reviewed here according to the formal charge on the phosphorus atom: (1) the monoanionic organophosphide ligands (PR2−); (2) the dianionic phosphinidene (PR2−) and P3− ligands; and (3) the pure inorganic polyphosphide ligands (Pnx−). Since the rare-earth metal phosphide complexes were reviewed by Nief in 1998,8 the complexes before 1998 will be mentioned briefly. Specifically excluded are phospholyl derivatives.
2. Rare-earth metal complexes containing monoanionic organophosphide ligands (PR2−)
With few exceptions, rare-earth metal phosphide complexes can be prepared by two general methods: (1) salt metathesis (salt elimination) reaction between a rare-earth metal halide precursor and an alkali phosphide compound (eqn (1)); and (2) intermolecular deprotonation (alkane or amine elimination) reaction between a rare-earth metal complex containing a basic ligand (alkyl or amido) and an acidic protonated phosphide compound (eqn (2)).| [R′LnX] + KPR2 → [R′Ln(PR2)] + KX | (1) |
| [R′LnR′′] + HPR2 → [R′Ln(PR2)] + HR′′ | (2) |
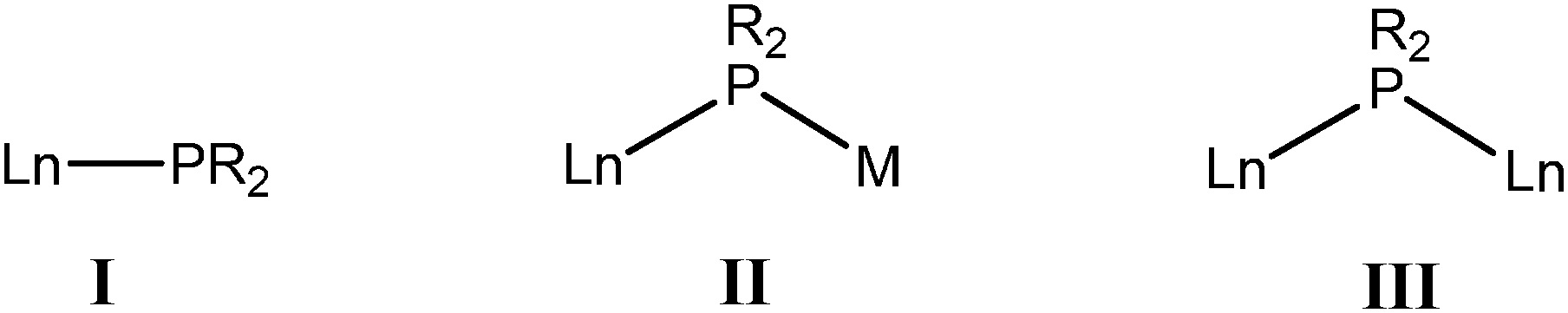
Rare-earth metals as hard cations are commonly σ-bonded to anionic phosphide ligands with substantial ionic character. The structure of the complex adopted in each case is strongly dependent upon the steric and electrostatic properties of the ligands and the metals. The three most common classes of phosphide coordination modes in the rare-earth metal complexes are: terminally coordinated (I); bridged between a rare-earth metal and an alkali metal (II); and bridged between two rare-earth metals (III).
2.1 Monodentate phosphide complexes
Monodentate phosphide ligands reviewed in this section consist of PR2− (R = alkyl, aryl and silyl), HPR− (R = aryl and silyl) and H2P−. With one exception, HPR− (R = aryl and silyl) or H2P− ligand coordinates to a rare-earth metal almost exclusively in mode II or III as a bridging phosphide ligand.Since 1976, many chemists have made efforts to synthesize rare-earth metal phosphide complexes. [Cp2Ln(PtBu2)] (Cp = η5-C5H5), [Cp2Ln(PPhtBu)], [(THF)xLn(PtBu2)3], [Cp2Yb(P(C6H11)2)], [(H3CC5H4)2Sm(PPh2)] and [Ln{N(SiMe3)2}2(PPh2)] are among the first rare-earth metal phosphide compounds.8 However, due to their air-sensitivity or the disordered crystals obtained, these compounds were only characterized by analytical and spectroscopic methods. The first structurally characterized rare-earth metal complex containing an anionic phosphide ligand was reported in 1986 by Schumann and coworkers.11 [Cp2Lu{μ-PPh2}2Li(tmeda)] (1) (tmeda = N,N,N′,N′-tetramethylethylenediamine) was obtained from the protonation reaction of [Cp2Lu{μ-CH3}2Li(tmeda)] with 2 equiv. of HPPh2. The dinuclear lutetium–lithium complex is bridged by two diphenyl phosphide ligands in mode II (Scheme 1 and Fig. 1).
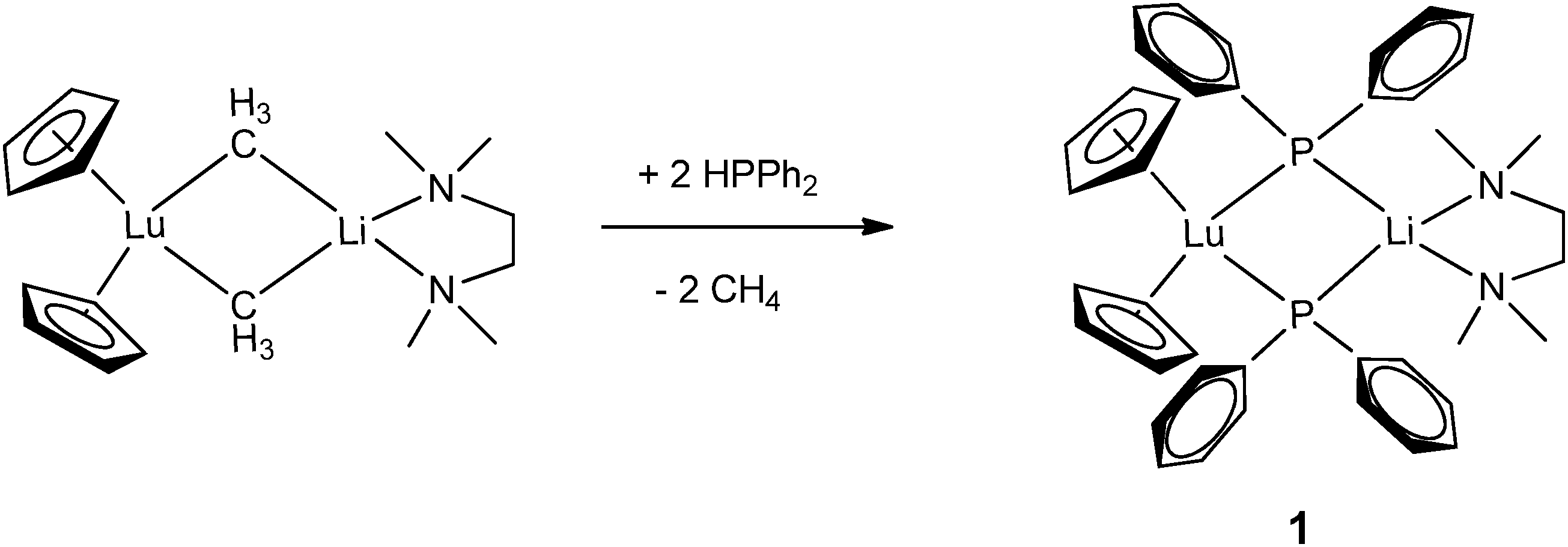 | ||
| Scheme 1 | ||
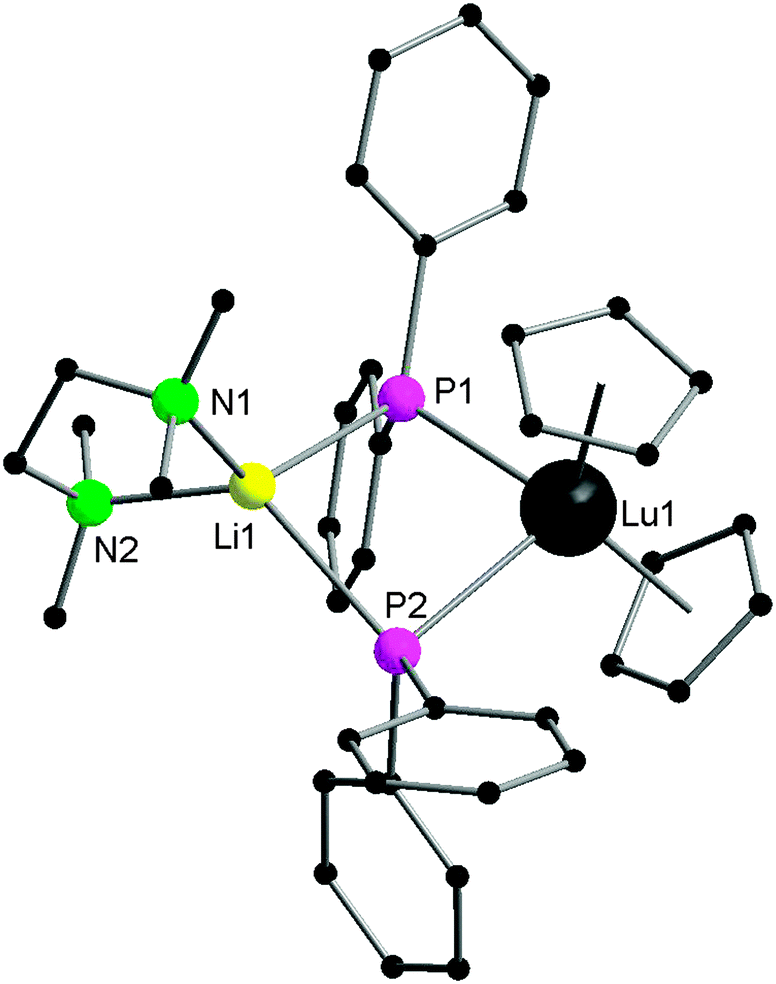 | ||
| Fig. 1 Solid-state structure of 1. Hydrogen atoms are omitted for clarity. | ||
As shown in Fig. 1, the lutetium atom is surrounded by a distorted tetrahedral array of the centroids of the two cyclopentadienyl rings and the two bridging phosphorus atoms. The first reported lengths of the lutetium–phosphide bond distances are 2.782(1) and 2.813(2) Å. An angle of 148.7° is formed between the P1–Lu–P2 and P1–Li–P2 planes.11
In the 1990s, other “ate” complexes [(tBu2P)2La{(μ-PtBu2)2Li(THF)}] (2)12 and [Ln{(μ-PtBu2)2Li(THF)}2]13,14 (Ln = Yb (3), Eu (4), Sm (5)) with the lanthanide metal in the formal oxidation states +3 and +2, respectively, were reported by Rabe and coworkers from the salt metathesis of the corresponding metal triflate complexes with excess LitBu2P, and reduction of the trivalent lanthanides as well in the case of formation of 3–5. Compounds 2–5 are the first homoleptic rare-earth metal complexes surrounded by phosphide ligands (Scheme 2).
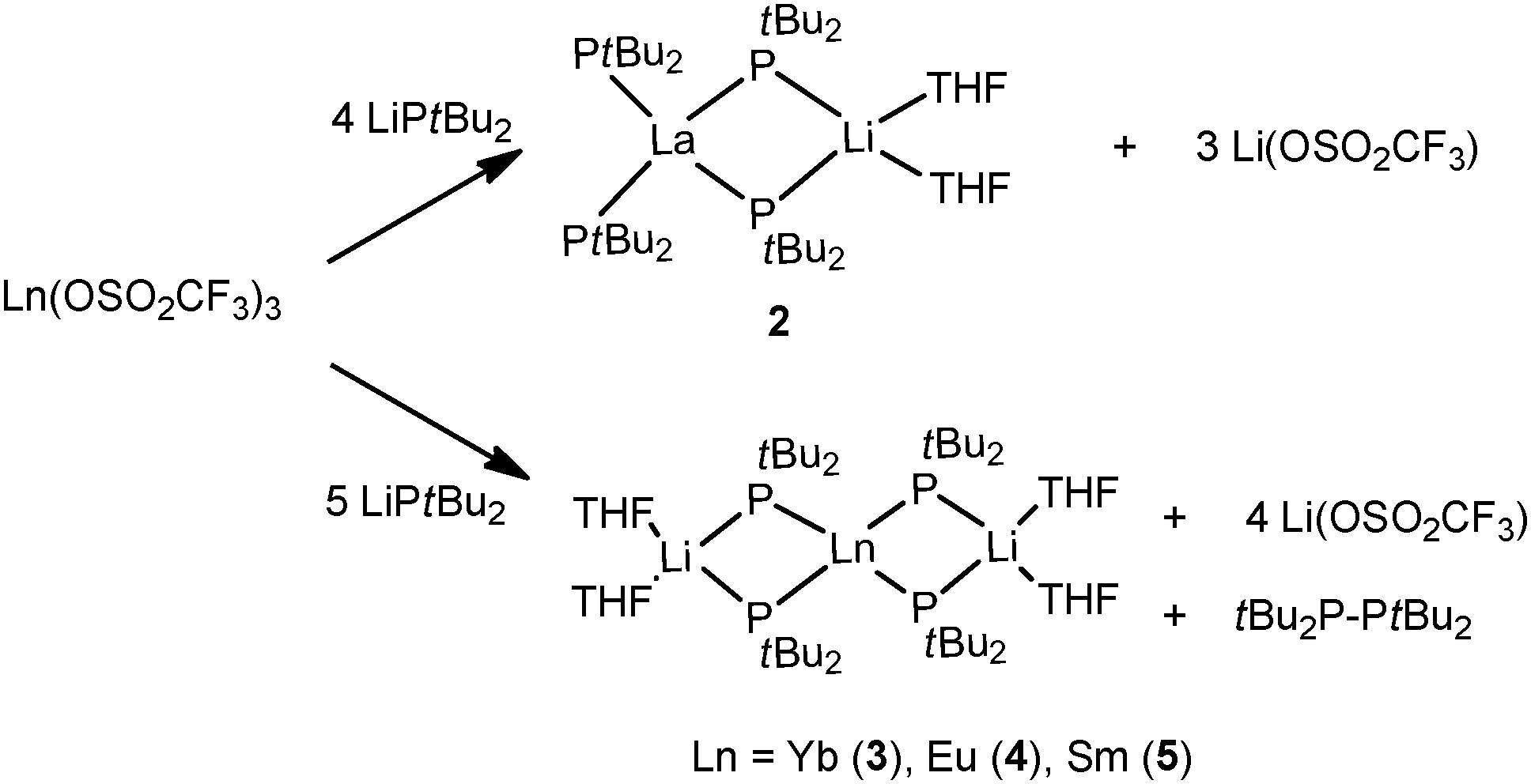 | ||
| Scheme 2 | ||
Westerhausen and coworkers focused on the synthesis of yttrium phosphide complexes. In 2002, they prepared a homoleptic mode III complex [Y{P(SiMe3)2}3]2 (6) by protonation of [Y{CH(SiMe3)2}3] with HP(SiMe3)2. The yttrium atoms are in a distorted tetrahedral environment with Y–P bond lengths of 2.677 and 2.848 Å for the terminal and bridging ligands, respectively.15 Nevertheless, the homoleptic phosphide complexes are still rare.
Three mixed ligated mode II cyclopentadienyl yttriate complexes [Cp′′2Y(μ-PHSiiPr3)2M(sol)] (1,3-(Me3Si)2C5H3 = Cp′′; M = Li, sol = THF (7); M = K, sol = C6H6 (8)) and [Cp′′2Y(μ-PH2)2Li2(μ-Cl)(TMEDA)2] (9) were prepared and structurally characterized by Westerhausen and coworkers (Scheme 3). 7 and 8 were obtained from salt metathesis of [Cp′′2Y(μ-Cl)2Li(THF)2] with 2 and 3 equiv. of KPHSiiPr3, respectively. Complex 9 isolated from the reaction of [Cp′′2Y(μ-Cl)2Li(THF)2] with TMEDA and (DME)LiPH2 (DME = 1,2-dimethoxyethane) is extremely sensitive toward moisture and air and decomposes at room temperature even under an argon atmosphere (Scheme 3).16
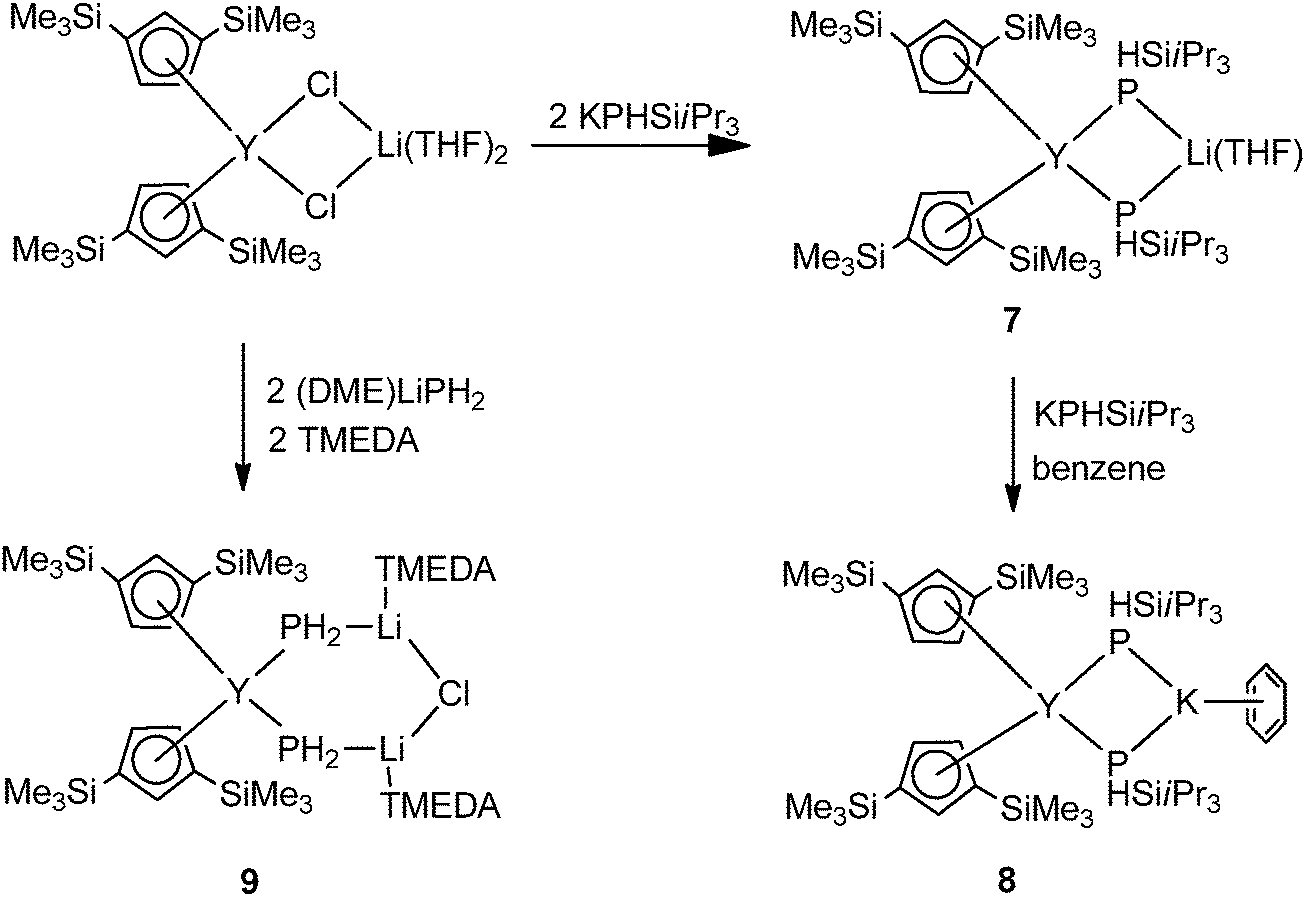 | ||
| Scheme 3 | ||
In contrast to other “ate” complexes, Hou and coworkers showed an example of a divalent mono-phosphide bridged polymeric mode II complex. [Cp*Sm(THF)(μ-PHAr)KCp*(THF)]∞ (10) was prepared by reacting [Cp*2Sm(THF)2] with 1 equiv. of KPHAr (Ar = C6H2tBu3-2,4,6) (Scheme 4).6,17 The phosphide ligand is bridged between K and Sm possibly due to the strong electron donating ability of the phosphide ligand. Due to the formation of an “ate” complex, the Sm–P bond in 10 (3.234(2) Å) is significantly longer than those in previously reported samarium(II) bis(phosphide) complexes such as [Sm(PAr2)2(THF)4] (3.139(3) Å when Ar = Ph18 and 3.034(2) Å when Ar = Mes19). The K–P bond distance of 3.350(3) Å is comparable with those in the polymeric potassium phosphide starting compound [KPHAr]∞ (3.181(2)–3.357(2) Å).20
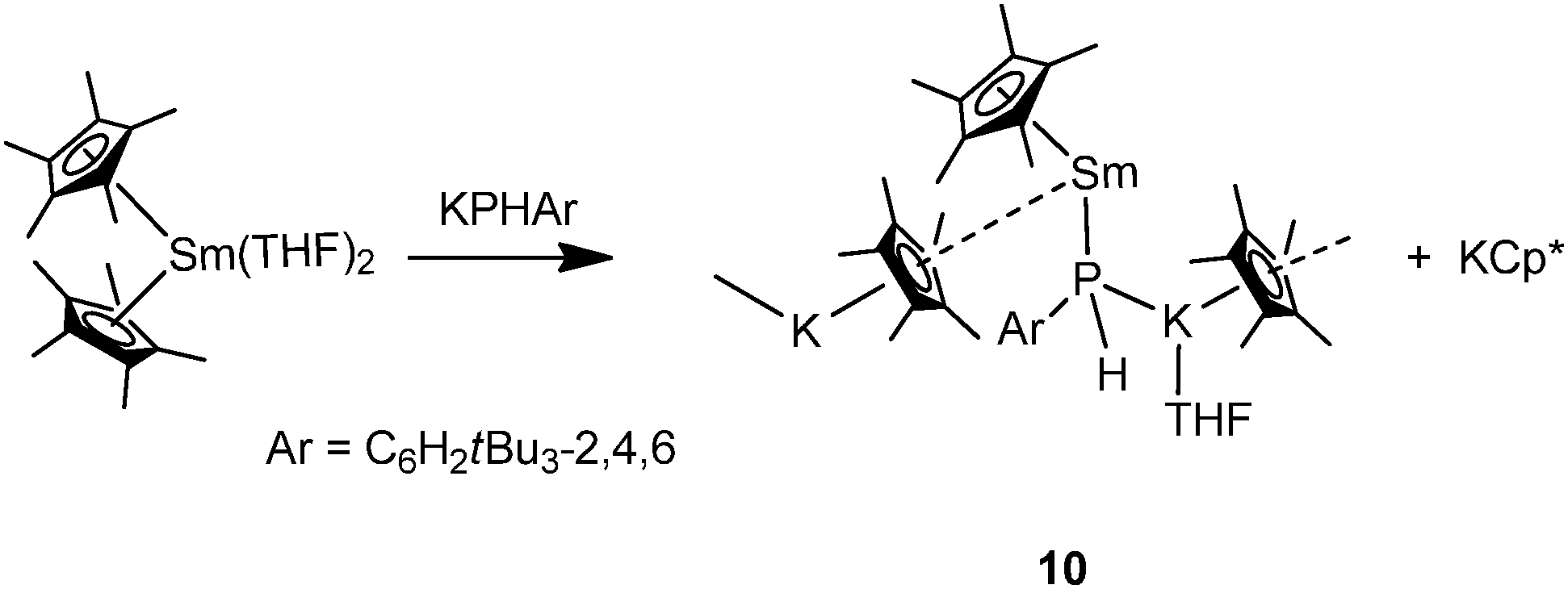 | ||
| Scheme 4 | ||
The first structurally characterized terminally bound mode I phosphide ligand rare-earth metal complex, [La{N(SiMe3)2}2(PPh2)(Ph3PO)2] (11), was synthesized in 1992 by Aspinall and coworkers from the amine elimination reaction of [La{N(SiMe3)2}3(Ph3PO)] with HPPh2 followed by Ph3PO.21
In the 1990s, many other mode I complexes were prepared by salt metathesis reaction. Rabe and coworkers have synthesized and structurally characterized a series of terminally bound trivalent complexes, [Ln{P(SiMe3)2}3(THF)2] (Ln = Tm, Nd), and divalent complexes [Ln{PPh2}2(L)4] (Ln = Sm, Yb, Eu; L = THF or N-methylimidazole) and [Ln{PHMes}2(THF)4] (Ln = Yb, Eu; Mes = mesityl, C6H2-2,4,6-Me3). Nief and coworkers also reported two divalent complexes [Ln{PMes2}2(THF)4] (Ln = Yb, Sm). The complexes above have been included in the early Nief's review.8
In 2002, Westerhausen and coworkers reported the only terminally bound HPR− complex [Cp′′2Y{P(H)SitBu3}] (12) by the salt metathesis reaction of [Cp′′2YCl] with KHPSitBu3. The yttrium atoms are in a distorted tetrahedral environment with a Y–P bond length of 2.770 Å (Fig. 2). The NMR spectroscopy of 12 shows the coupling constants of 1J(Y,P) and 1J(P,H) with values of 144.0 Hz and 201.0 Hz, respectively.15
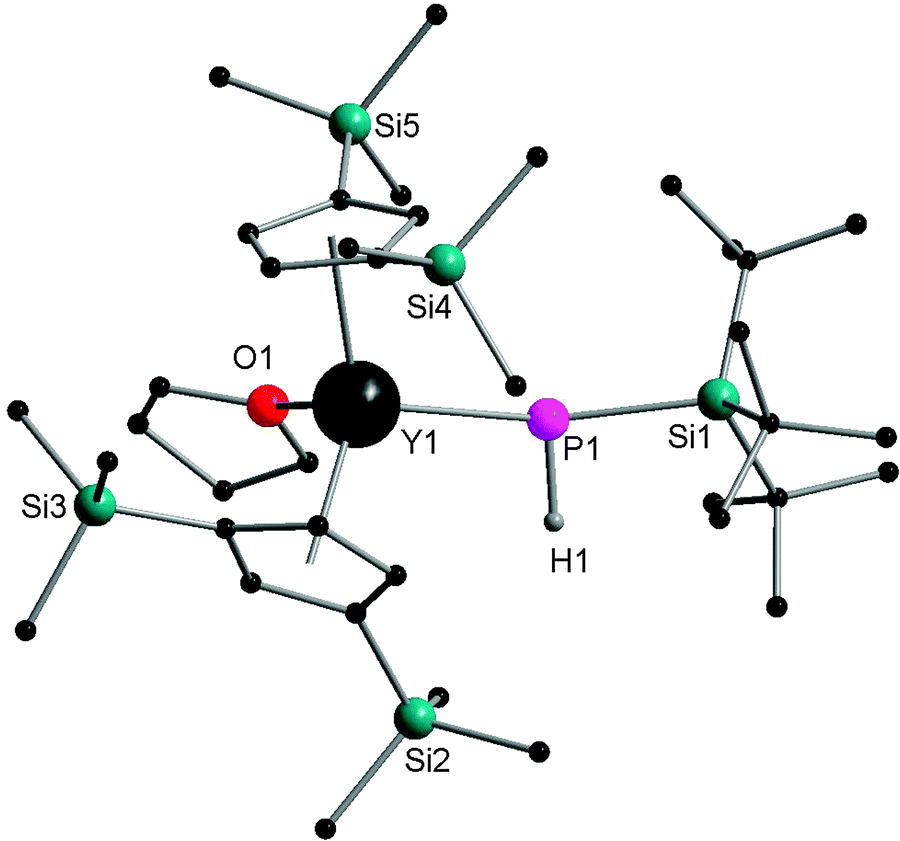 | ||
| Fig. 2 Solid-state structure of 12. Hydrogen atoms are omitted for clarity. | ||
Recently, an equilibrium between mode I and mode III complexes was revealed by Hou and coworkers. The reaction of [{Me2Si(C5Me4)(NMes)}La(CH2C6H4NMe2-o)(THF)] with 1 equiv. of HPPh2 shown in Scheme 5 yielded the corresponding mode I monomeric phosphide complex [{Me2Si(C5Me4)(NMes)}La(PPh2)(THF)2] (13) which recrystallized from benzene as the dimeric analogue mode III half-sandwich complex [{Me2Si(C5Me4)(NMes)}La(PPh2)]2 (14). Treatment of 14 with THF again resulted in 13.22
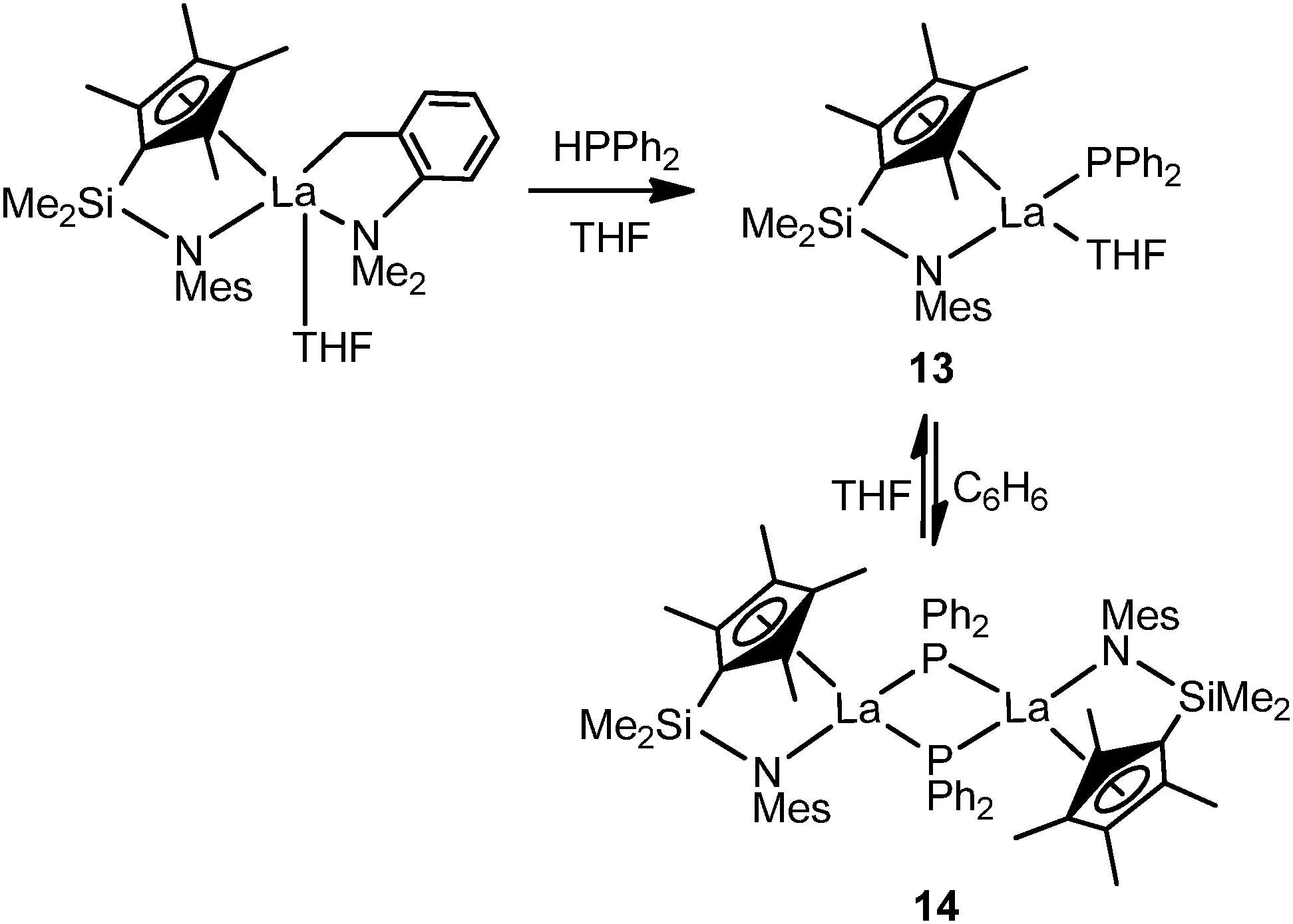 | ||
| Scheme 5 | ||
Two terminally bound yttrium phosphide complexes were reported by the Zhou and the Chen groups. As shown in Scheme 6, by increasing the steric bulkiness of the co-ligands, the PPh2− anion coordinates only as a terminal ligand in mode I. [(TpMe2)(Cp)Y(PPh2)(THF)] (15) (TpMe2 = tris(3,5-dimethylpyrazolyl)borate), was formed in the reaction of [(TpMe2)(Cp)YPh(THF)] with 1 equiv. of HPPh2 in THF at ambient temperature. The reactivity of the Ln–P bond in 15 was studied and is reviewed at the end of Section 2.1.23
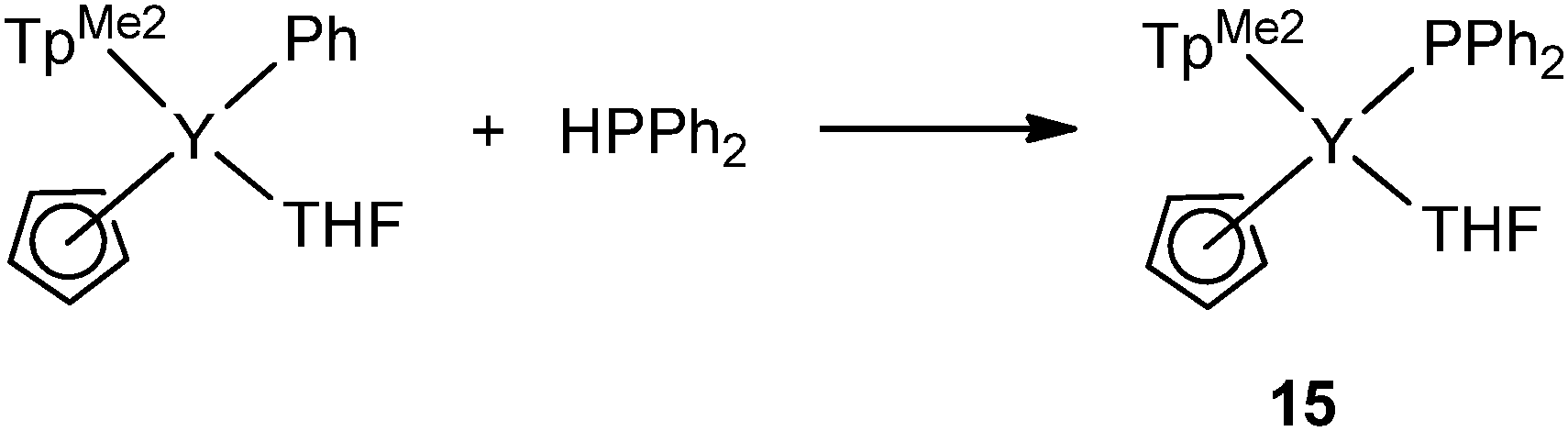 | ||
| Scheme 6 | ||
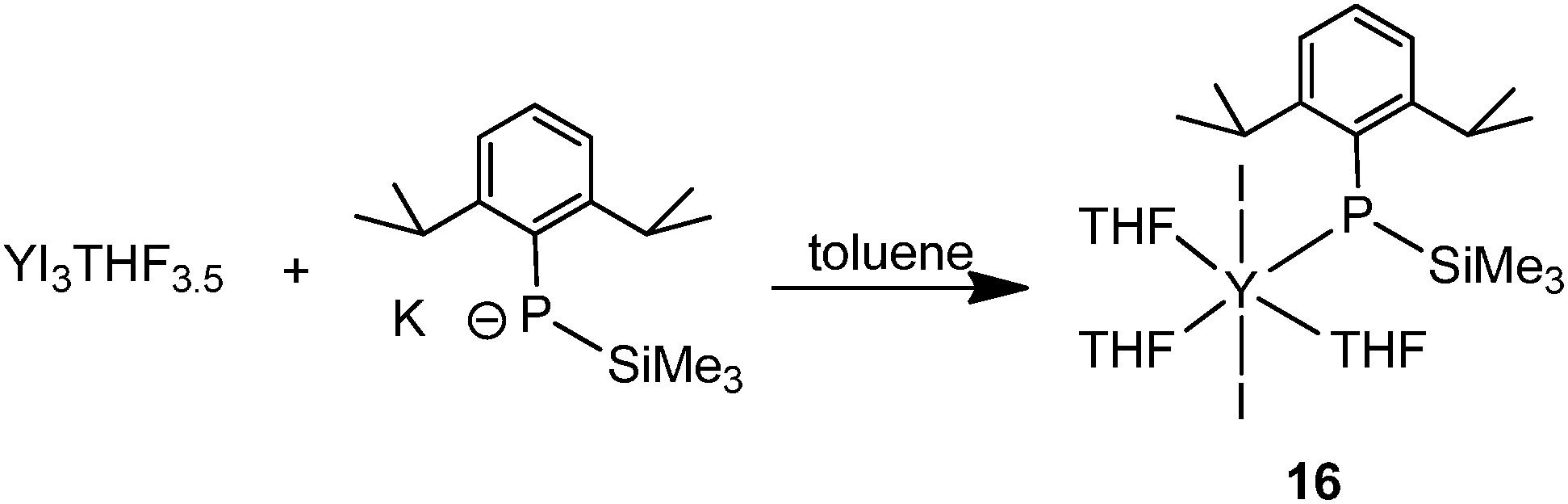
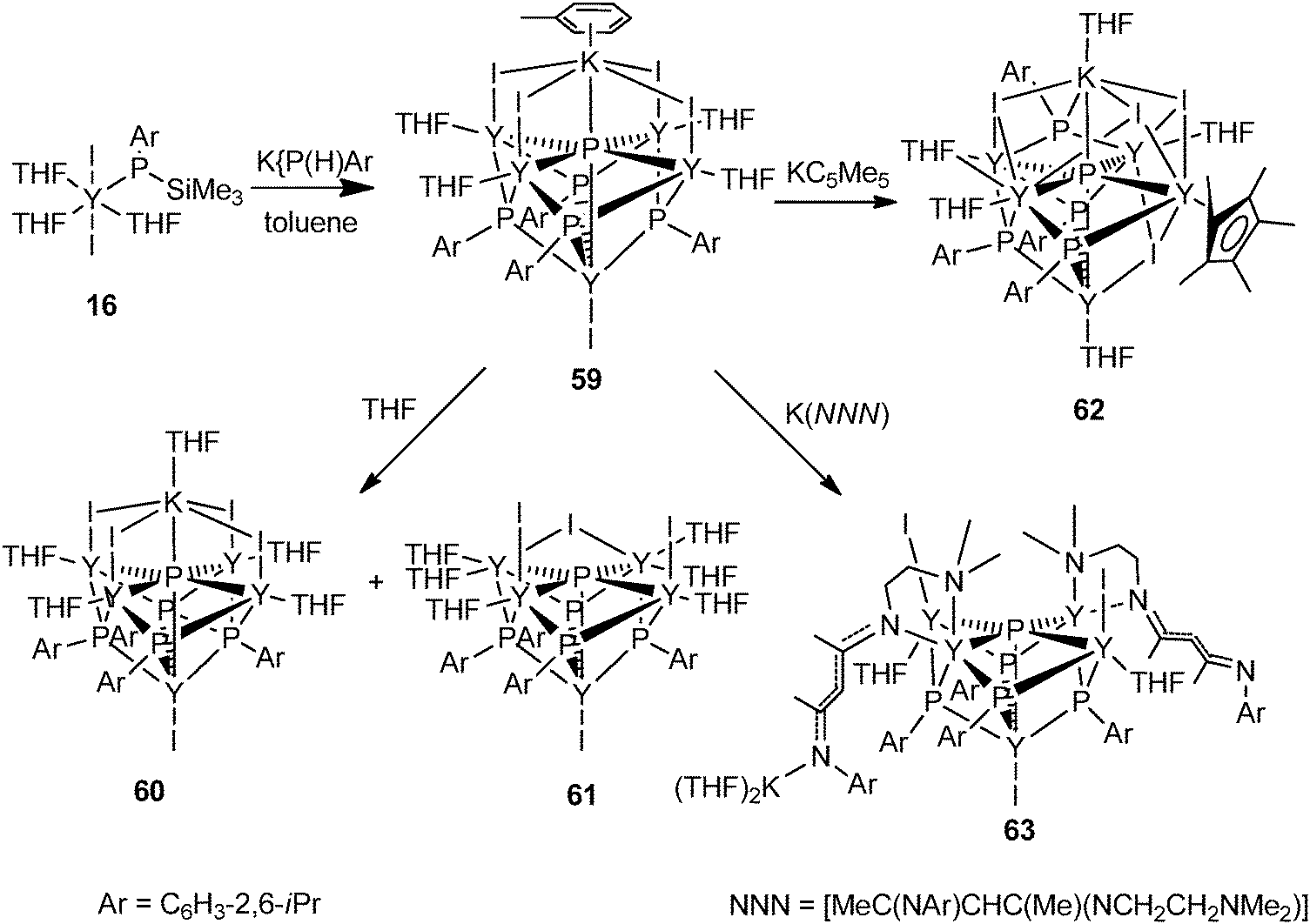
The halide compound [Y{P(SiMe3)(Ar)}I2(THF)3] (16) (Ar = 2,6-iPr2C6H3) without a co-ligand was isolated from the salt metathesis reaction of YI3(THF)3.5 with 1 equiv. of K{P(SiMe3)(Ar)} in toluene (Scheme 7).24 This complex has been used as a precursor to prepare yttrium phosphinidene phosphide and the first soluble rare-earth metal P3−-containing compounds, which are reviewed in Section 3.
 | ||
| Scheme 7 | ||
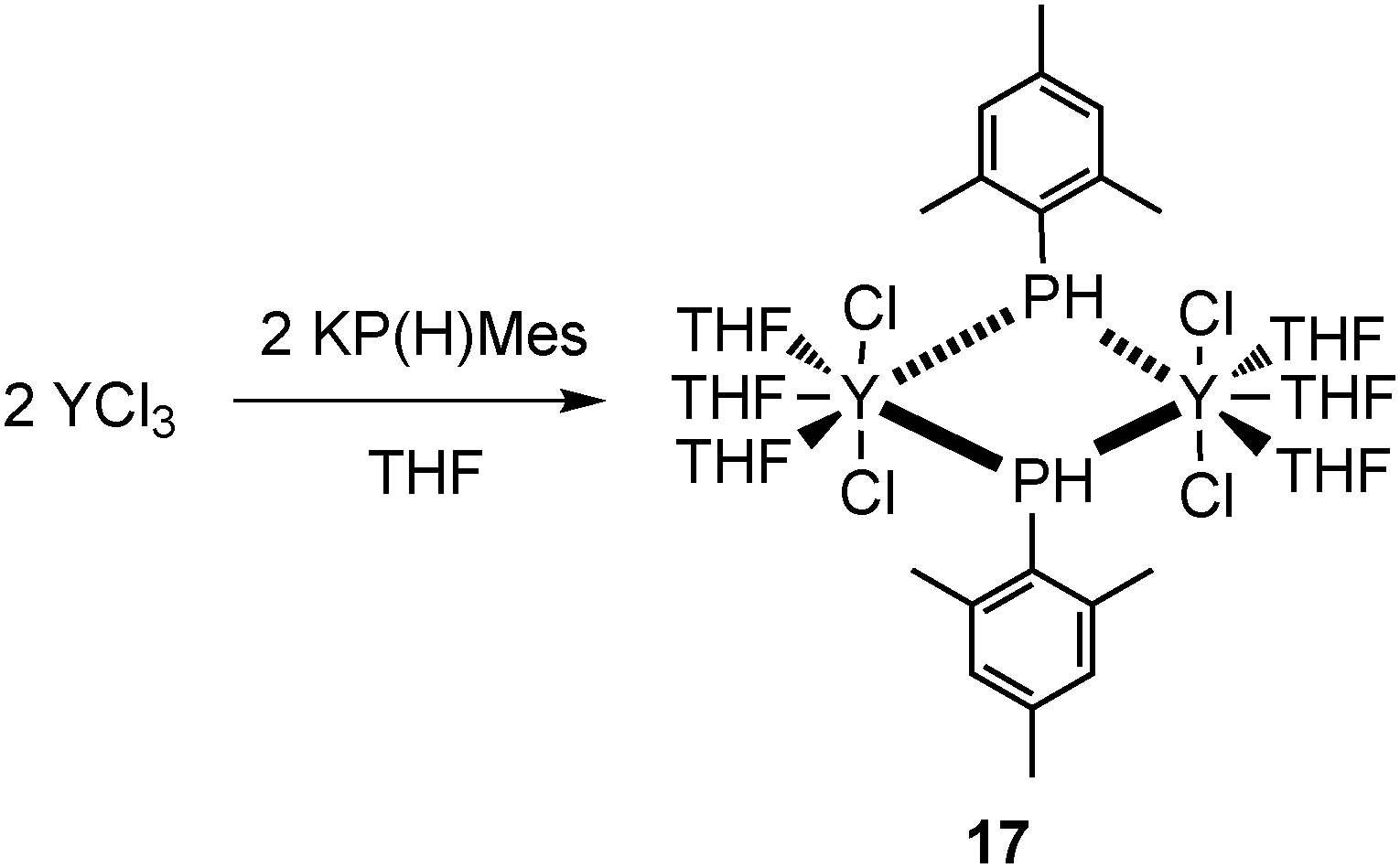
Another complex without a co-ligand is a seven-coordinate dinuclear mode III yttrium complex [(THF)3YCl2{μ-P(H)Mes}]2 (17), which was prepared by salt metathesis of KPHMes with YCl3 in THF (Scheme 8). The yttrium atoms exhibit pentagonal bipyramidal coordination spheres with bridging mesitylphosphanide substituents.25 Similarly to terminally bound phosphide complex 16 and the phosphinidene complex 55, compound 17 contains neither sterically demanding nor strong π-electronic donating ancillary ligands which are commonly believed to be responsible for the stabilization of the lanthanide phosphide complexes.26
 | ||
| Scheme 8 | ||
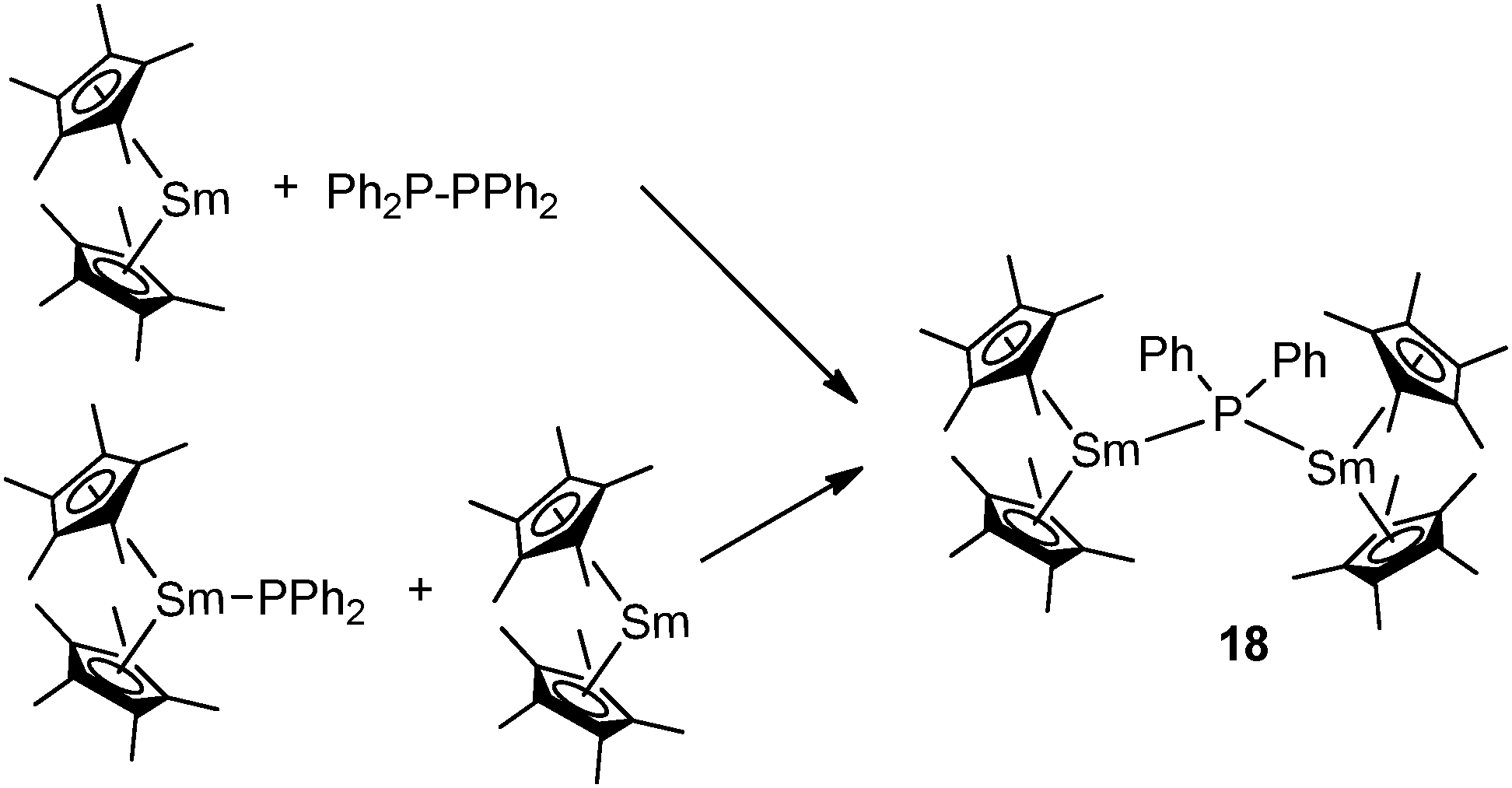
The Evans group prepared the first mixed-valent rare-earth metal phosphide complex. The dinuclear mode III complex [Cp*2Sm(μ-PPh2)SmCp*2] (18), which contains Sm(II) and Sm(III) atoms bridged by a diphenylphosphide ligand, can be prepared by the redox reaction of Ph2P–PPh2 and [Cp*2Sm] or by the reaction of [Cp*2Sm] with [Cp*2Sm(PPh2)] (Scheme 9).27
 | ||
| Scheme 9 | ||
Another unusual dinuclear divalent samarium complex, [{(Me3Si)2P}Sm{μ-P(SiMe3)2}3Sm(THF)3] (19), was obtained from salt metathesis of [SmI2(THF)2] and 2 equiv. of KP(SiMe3)2 (Scheme 10). The two samarium centers in 19 bridged by three PPh2 ligands are both divalent but in different coordination environments, which is unique in rare-earth metal chemistry.28
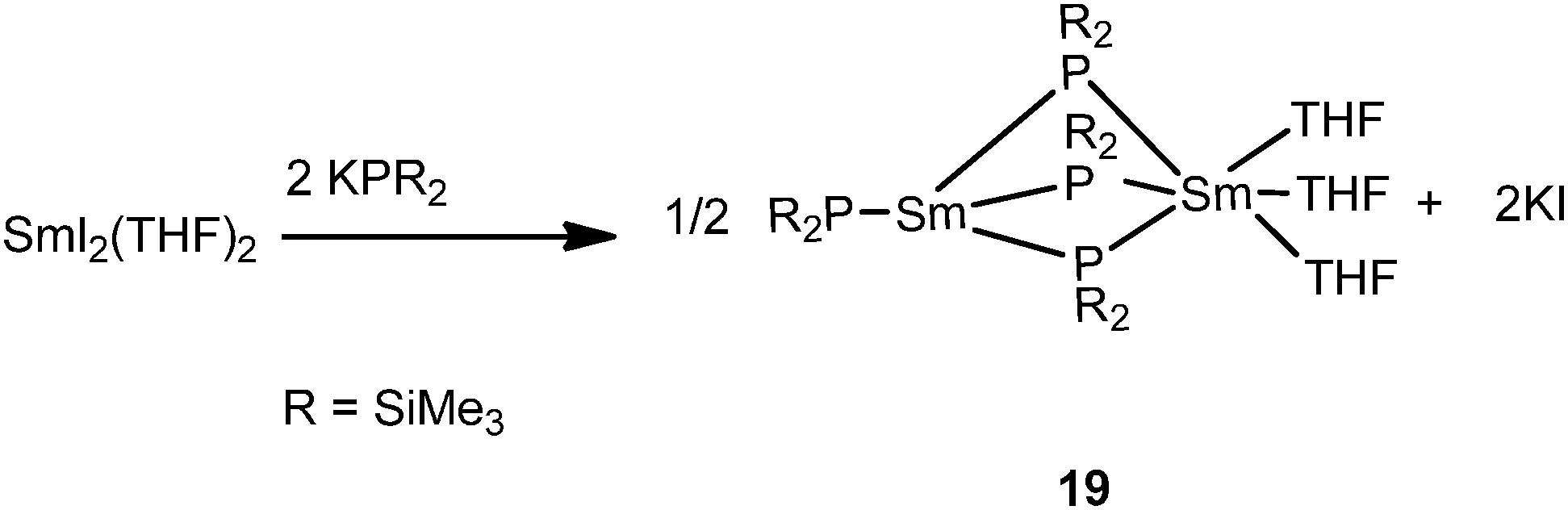 | ||
| Scheme 10 | ||
Several rare-earth phosphides were studied for catalytic transformations. In 2001, Marks and coworkers reported a novel mode III lanthanocene phosphide complex likely playing a role as the catalyst resting state in intramolecular hydrophosphination/cyclization of phosphinoalkenes and phosphinoalkynes (Scheme 11). Dimeric [Cp*2YP(H)Ph]2 (20) was isolated and crystallographically characterized from the protonation reaction of the hydride precatalyst [Cp*2YH]2 with excess PhPH2 (Scheme 12).29
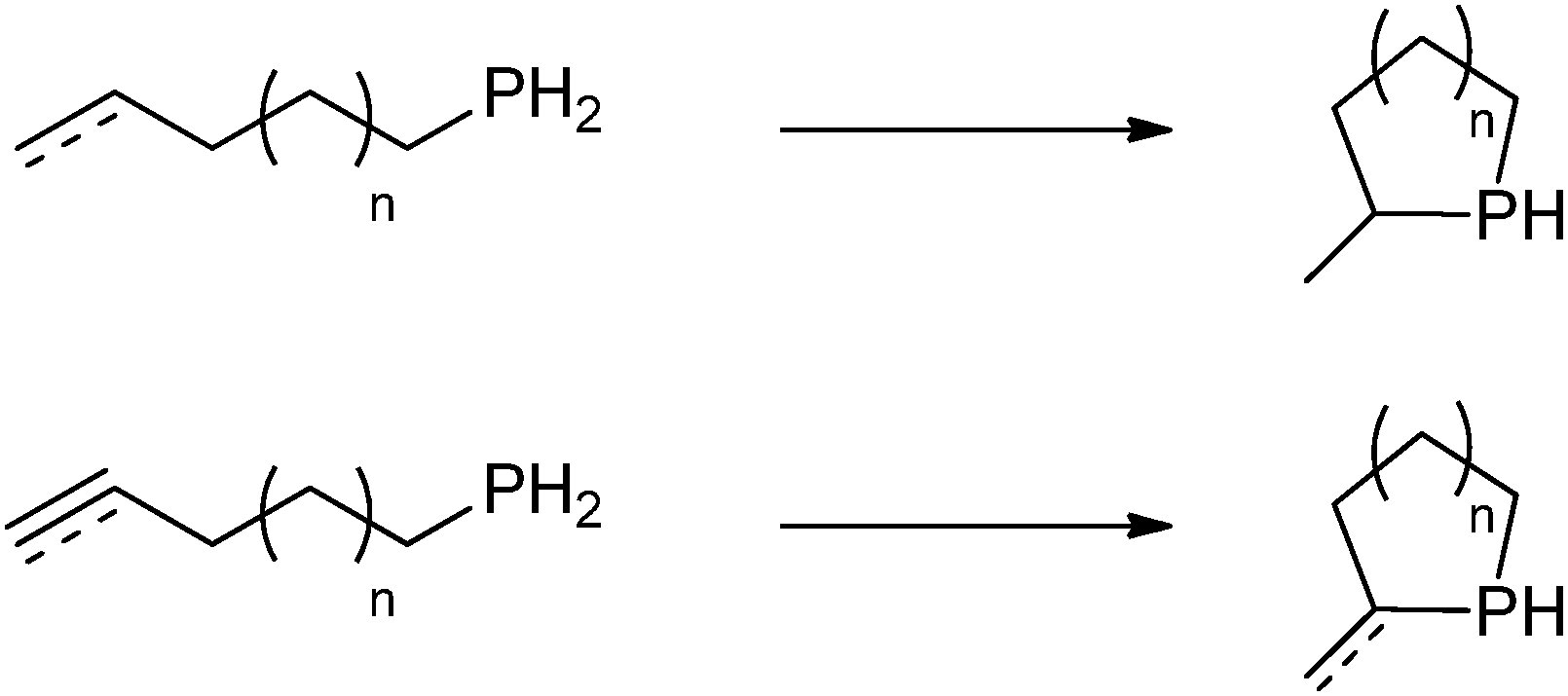 | ||
| Scheme 11 | ||
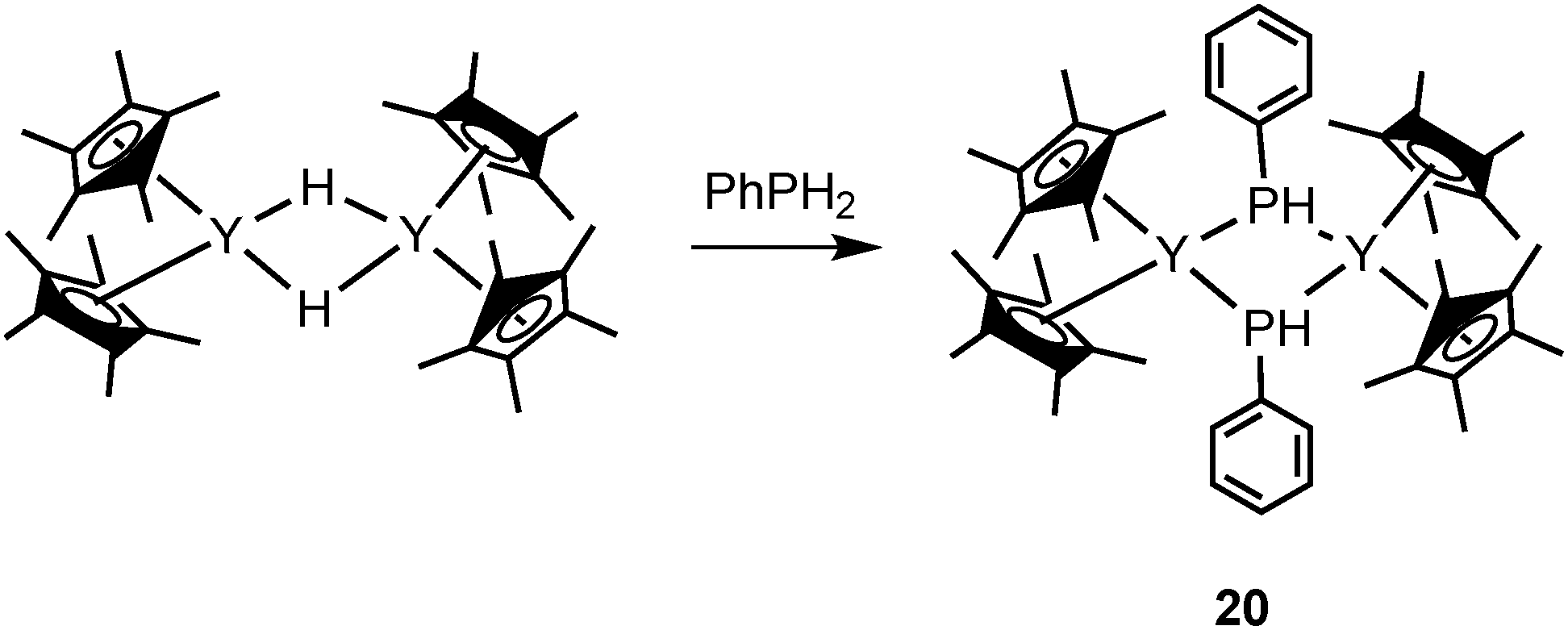 | ||
| Scheme 12 | ||
The lanthanum terminally bound phosphide complex 13 was reported to be the catalyst for the addition of a phosphine P–H bond to a carbodiimide compound to form a series of phosphaguanidine derivatives (Scheme 13).22
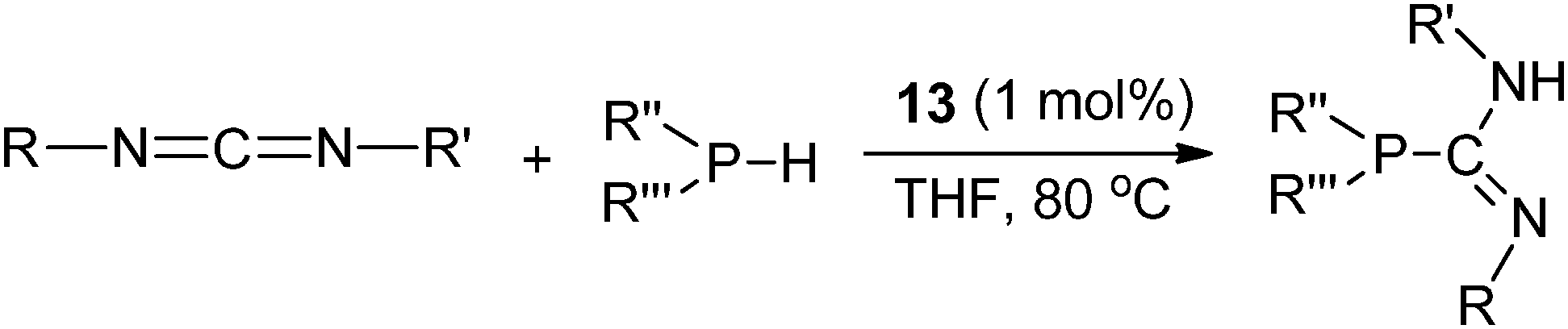 | ||
| Scheme 13 | ||
As shown in Scheme 14, the first example of isocyanate and isothiocyanate insertions into the Ln–P σ-bond was studied by Zhou and coworkers. The yttrium terminally bound phosphide complex 15 can catalyze the cyclotrimerization of PhNCO under mild conditions through stepwise insertions of isocyanates into the Y–P or N bond.23
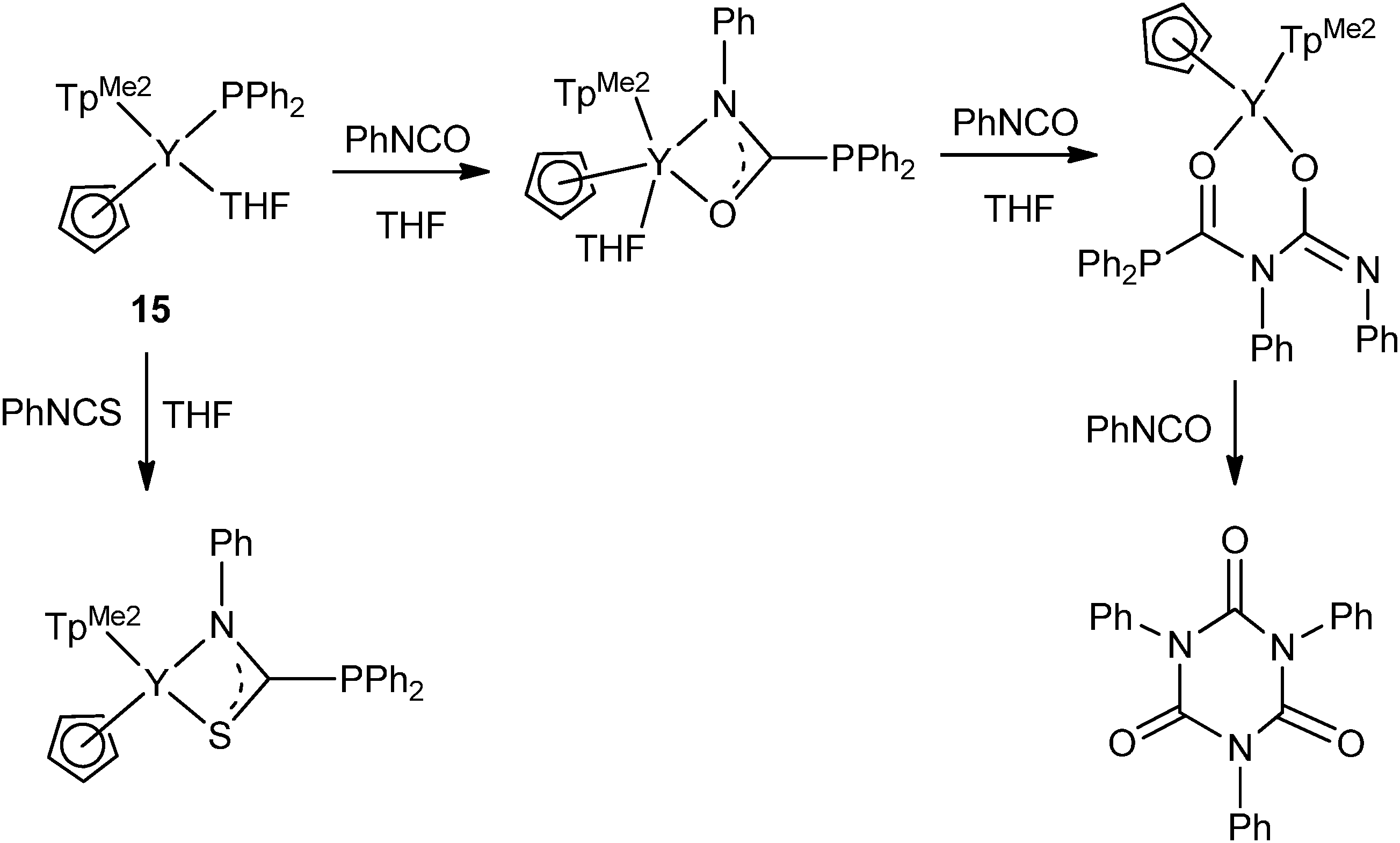 | ||
| Scheme 14 | ||
2.2 Chelating complexes
Two main categories of chelating phosphide ligands are included in this section: (1) amino (N-) or alkoxo (O-) functionalized phenylphosphide ligands; and (2) silylene-linked cyclopentadienyl phosphide Cp–P ligands. In general, the complexes with chelating phosphide have been much more intensively investigated in terms of their reactivity than the compounds described in the previous section.In 1993, Aspinall and coworkers reported the first chelating phosphide complex [{(CHMe2)2N}2La{μ-P(C6H4OMe-o)2}2Li(THF)] (21) prepared by protonation of Li[La{N(CHMe2)2}4] with 2 equiv. of HP(C6H4OMe-o)2 which contains both hard (O) and soft (P) donor sites (Scheme 15). Due to the increased size and chelating ability of the phosphide ligand, 21 is significantly more stable than the mono-dentate lanthanide phosphide complex.21,30
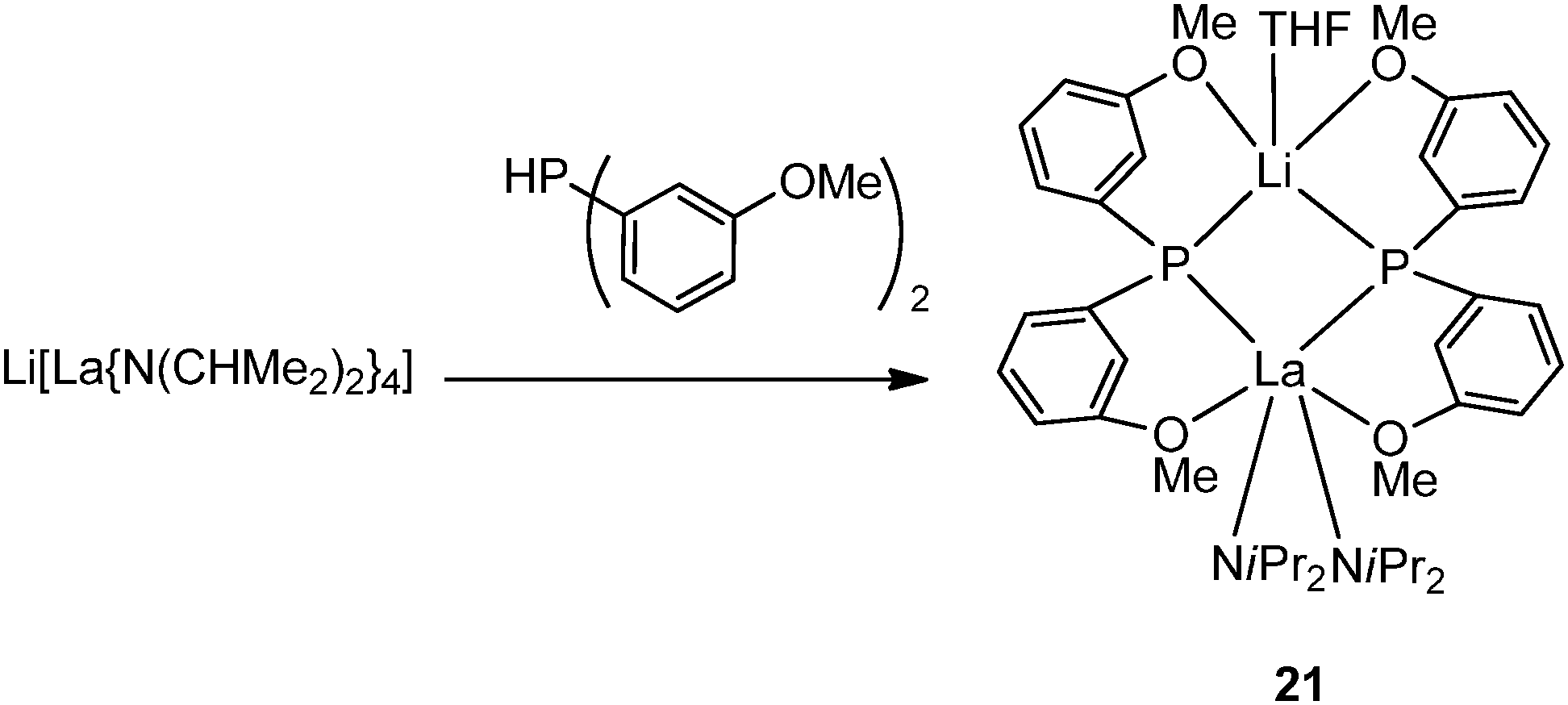 | ||
| Scheme 15 | ||
Izod and coworkers focused on the divalent rare-earth metal complexes containing O- or N-functionalized chelating phosphide ligands. Samarium(II) and ytterbium(II) complexes are readily accessible by metathesis reactions of LnI2 and potassium phosphides.
Salt metathesis of the potassium salt of alkoxophosphide K[P{(Me3Si)2CH}(C6H3-2-OMe-3-Me)] and YbI2 affords [Yb{P{(Me3Si)2CH}(C6H3-2-OMe-3-Me)}2(THF)2] (22) containing two P–O bidentate ligands (Scheme 16). In contrast, a similar reaction between the closely related but less steric hindered potassium phosphide K[P{(Me3Si)2CH}(C6H4-2-OMe)] and YbI2 in THF–ether yields an unprecedented alkoxophosphide complex of ytterbium(II) [Yb{P{(Me3Si)2CH}(C6H4-2-OMe)}(THF)]4·4Et2O (23) due to an unusual cleavage reaction between the potassium phosphide and YbI2, which contains a cuboidal core in the solid state structure (Fig. 3). As shown in Fig. 4, the Yb atoms sit at alternative corners of the Yb4O4 “cube”. Each metal atom coordinates to three μ3-alkoxide groups, two μ2-phosphide groups, and a molecule of THF.31
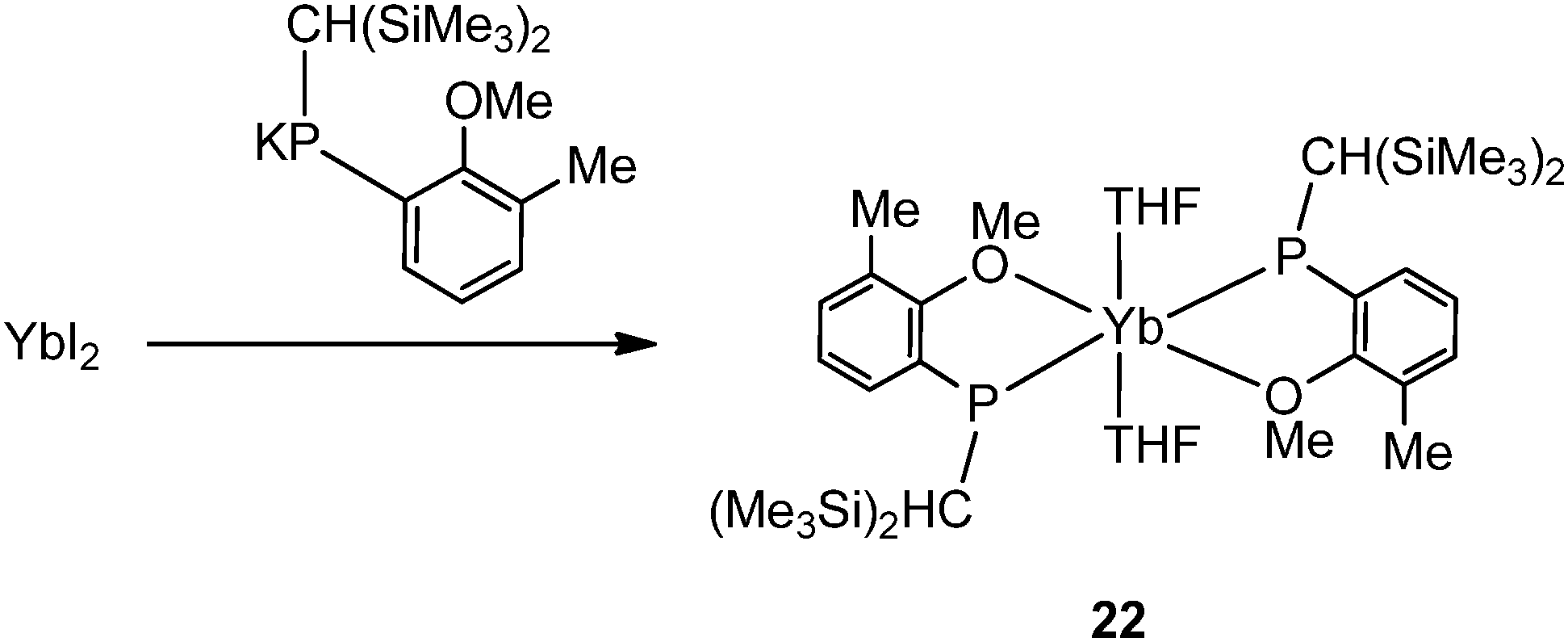 | ||
| Scheme 16 | ||
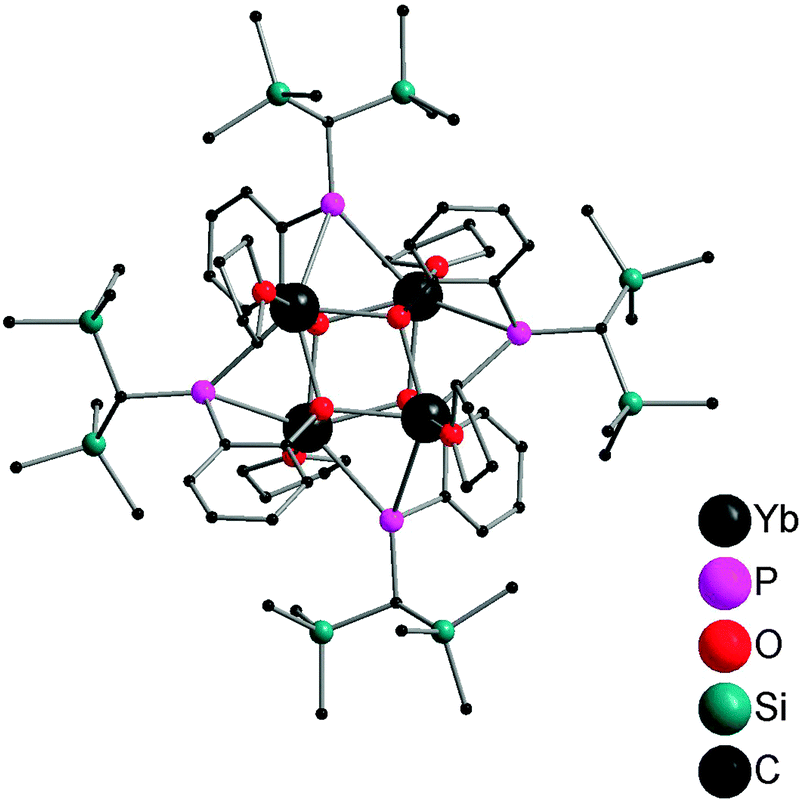 | ||
| Fig. 3 Solid-state structure of 23. Hydrogen atoms are omitted for clarity. | ||
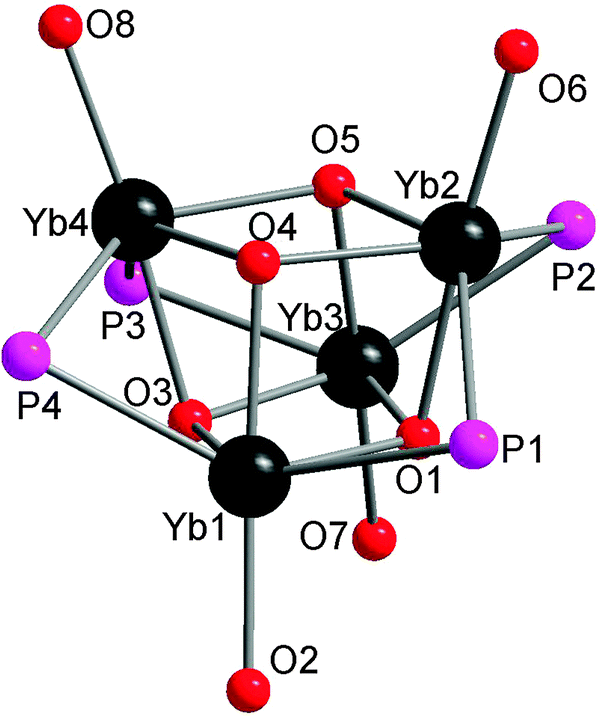 | ||
| Fig. 4 Structure of the cuboidal core of 23. | ||
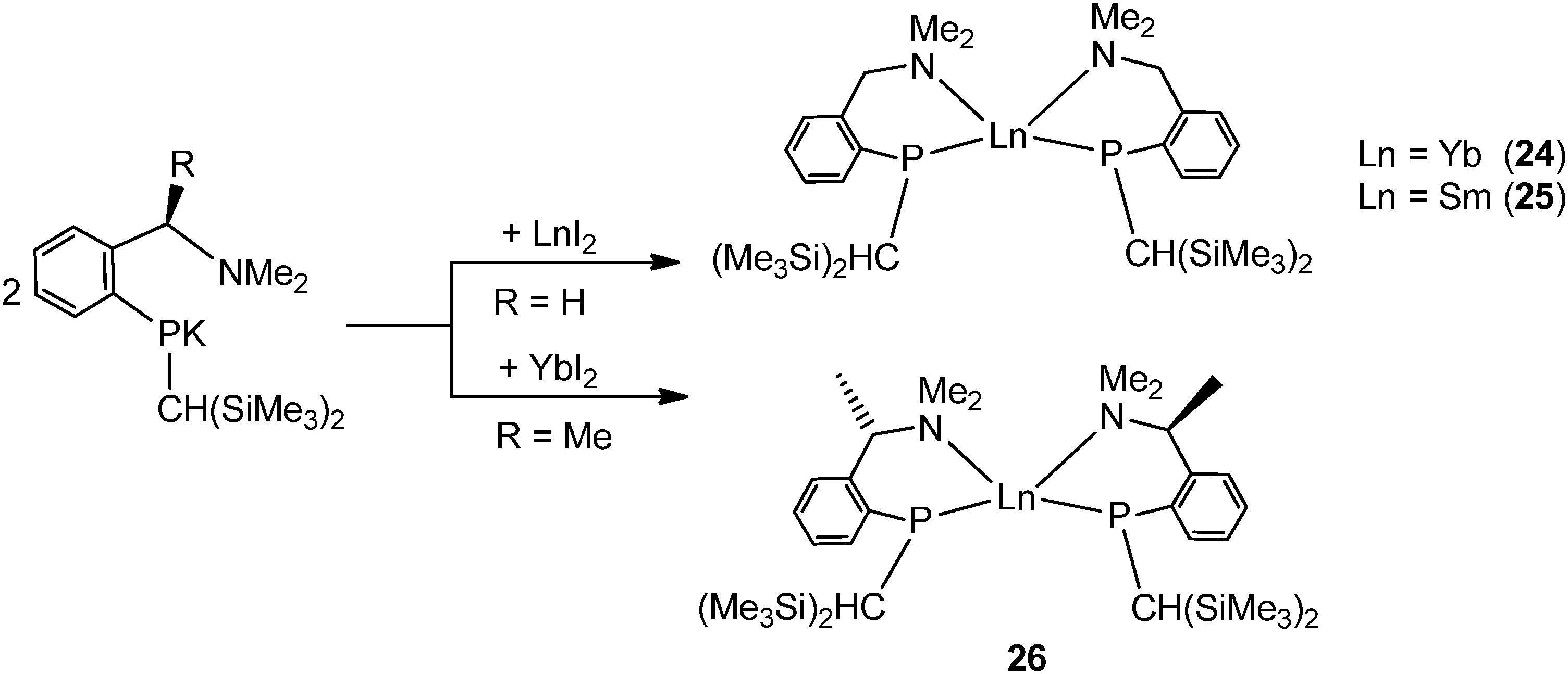
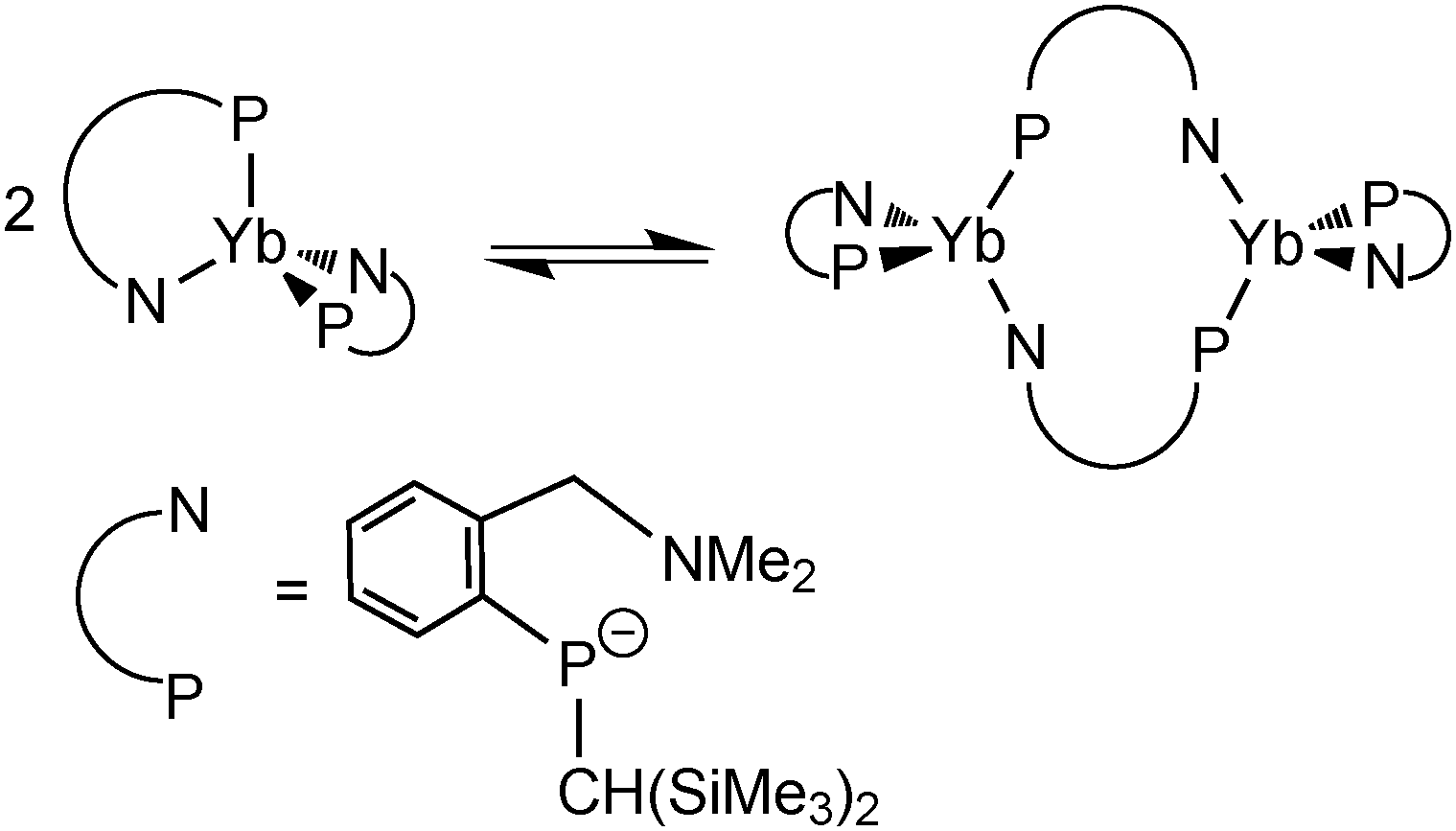
By using the amino-functionalized P–N ligands {(Me3Si)2CH}(C6H4-2-CHRNMe2)P (R = H or R/S-Me), the first examples of homoleptic, solvent- and alkali-metal-free divalent rare-earth metal phosphide complexes, [{{(Me3Si)2CH}(C6H4-2-CH2NMe2)P}2Ln] (Ln = Yb32 (24); Sm33 (25)) and the chiral modified [{(Me3Si)2CH}{C6H4-2-(R/S)-CH(Me)NMe2}P]2Yb34 (26) were prepared by salt metathesis of corresponding potassium phosphide and LnI2 followed by heating in the case of the samarium compound (Scheme 17). X-ray crystallography shows complex 25 to be monomeric, with a four-coordinate, distorted tetrahedral Sm(II) center (Fig. 5).33 The monomer–dimer equilibrium observed for the Yb complex in solution suggests that the P–N chelating ligand provides just enough steric hindrance which allows the complex to lie on the edge of steric saturation and aggregation (Scheme 18).32
 | ||
| Scheme 17 | ||
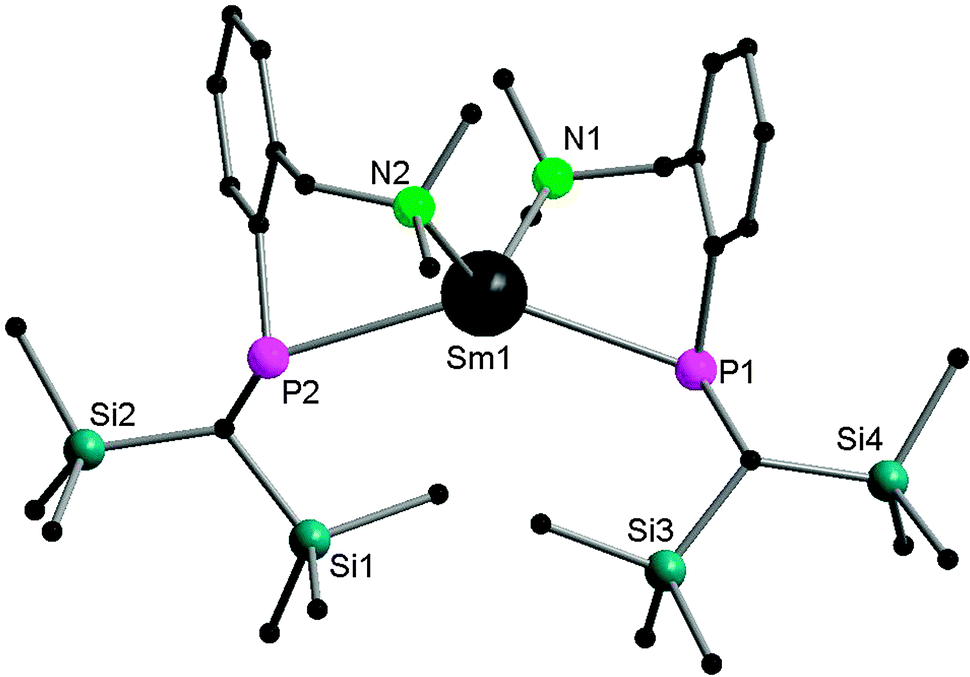 | ||
| Fig. 5 Solid-state structure of 25. Hydrogen atoms are omitted for clarity. | ||
 | ||
| Scheme 18 | ||
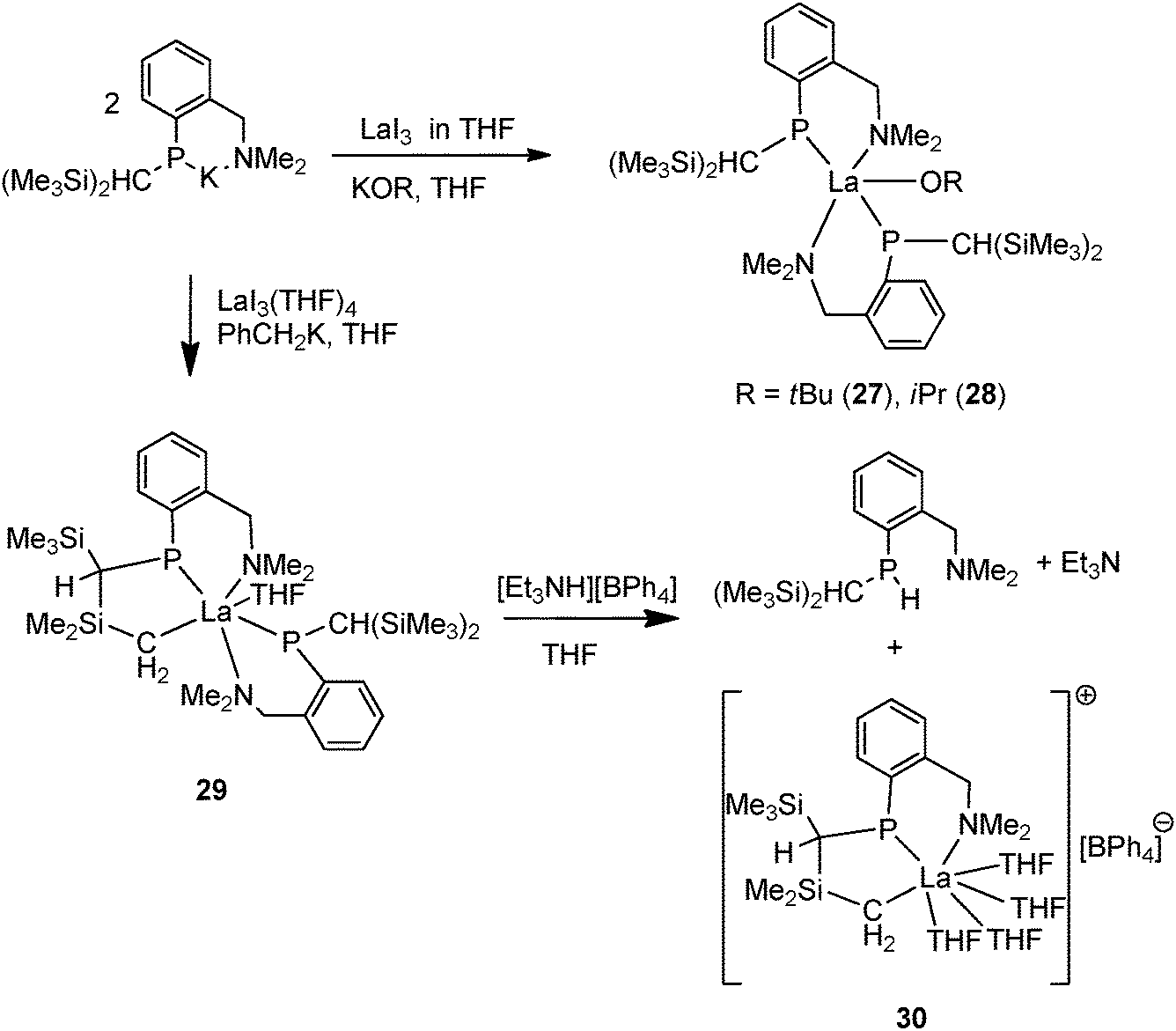
In addition to homoleptic 24–26, Izod and coworkers synthesized several heteroleptic lanthanum complexes with the same P–N ligand [{(Me3Si)2CH}(C6H4-2-CH2NMe2)P]. The salt metathesis of LaI3 with potassium phosphide followed by KOR led to [{{(Me3Si)2CH}(C6H4-2-CH2NMe2)P}2La(OR)] (R = tBu (27), iPr (28)). However, the reaction of the ligand and LaI3 followed by CH2PhK forms a cyclometalated lanthanum phosphide alkyl complex 29 as a result of intramolecular C–H activation (Scheme 19).35 Complex 29 undergoes protonation by [Et3NH][BPh4] at the P–La rather than the C–La bond, which affords an unusual N–P–C tridentate phosphide supported lanthanum alkyl cationic complex 30 along with a phosphine byproduct of the ligand {(Me3Si)2CH}(C6H4-2-CH2NMe2)PH (Scheme 19).36
 | ||
| Scheme 19 | ||
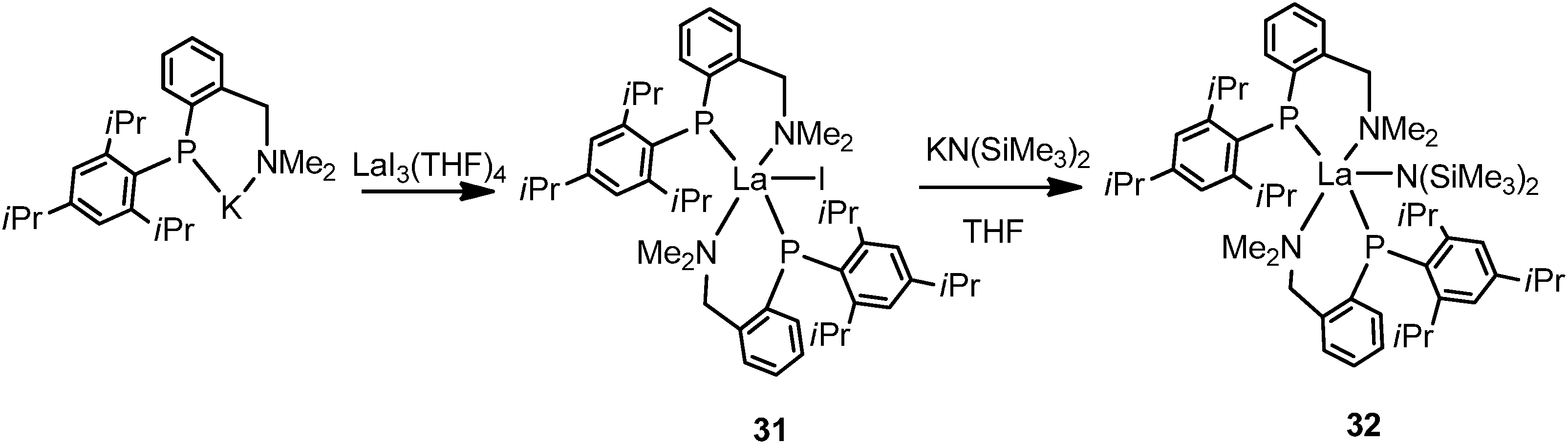
By alternating the substituent to C6H2-2,4,6-iPr3 (Tripp) on the phosphide, another lanthanum phosphide complex [{(Tripp)(C6H4-2-CH2NMe2)P}2LaI] (31) was isolated by salt metathesis of LaI3(THF)4 with potassium salt of the phosphide in THF. Treatment of 31 with one equivalent of KN(SiMe3)2 in THF yields the amido complex [{(Tripp)(C6H4-2-CH2NMe2)P}2LaN(SiMe3)2] (32) (Scheme 20).37
 | ||
| Scheme 20 | ||
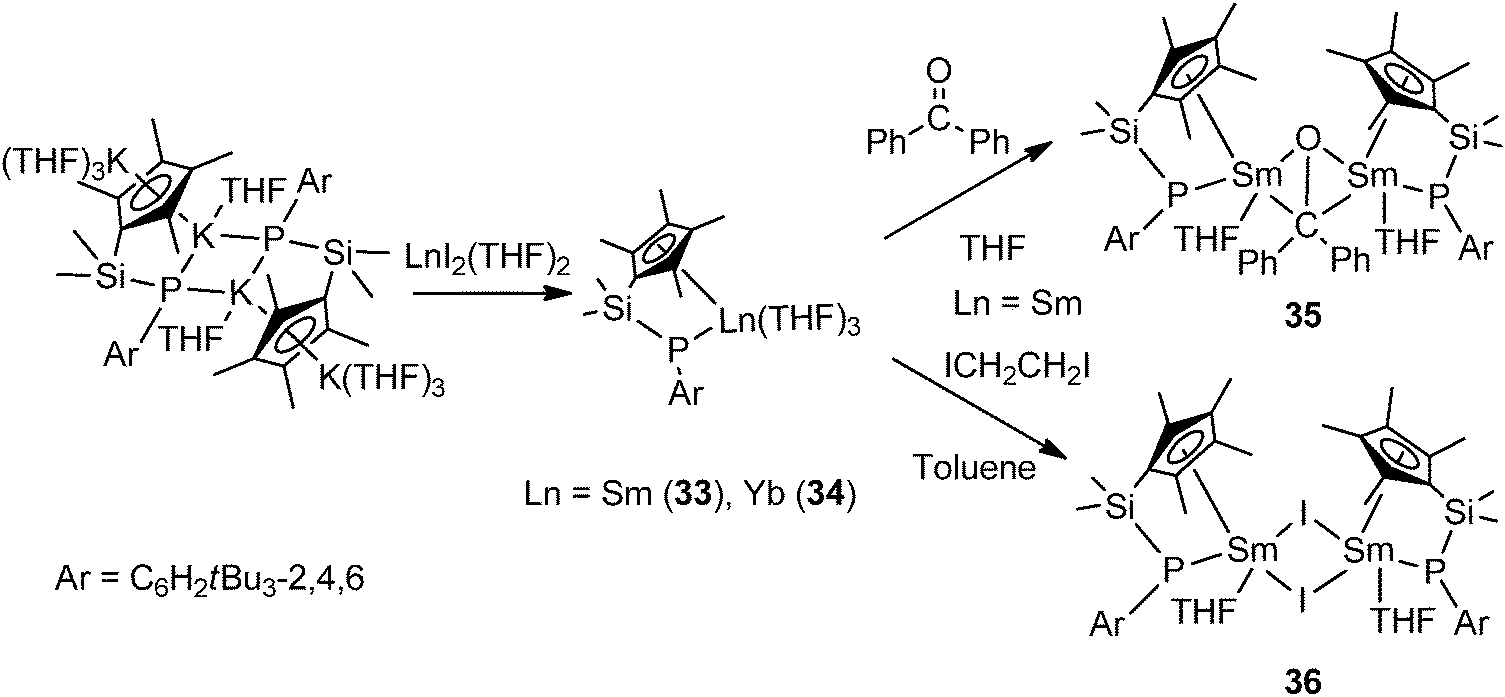
Hou and coworkers reported the first chelating silylene-linked cyclopentadienyl–phosphido (Cp–P) rare-earth complexes. Salt metathesis of dipotassium Cp–P complex [Me2Si(C5Me4)(PAr)K2(THF)4]2 (Ar = C6H2tBu3-2,4,6) with 2 equiv. of [LnI2(THF)2] led to the formation of Cp–P divalent lanthanide complexes [Me2Si(C5Me4)(PAr)Ln(THF)3] (Ln = Sm (33), Yb (34)). The electron transfer reaction of the Sm complex 33 with benzophenone in THF afforded the first structurally characterized disamarium(III) ketone-dianionic complex 35. Treatment of 33 with ICH2CH2I in toluene easily yielded the corresponding samarium(III) iodide complex 36 (Scheme 21).6,38
 | ||
| Scheme 21 | ||
The protonation reaction of [Ln(CH2SiMe3)3(THF)2] with Me2Si(C5Me4H)PHR (R = C6H2tBu3-2,4,6) in 2.5 h afforded [{Me2Si(C5Me4)PHR}Ln(CH2SiMe3)2THF] (Ln = Y (37), Lu (38)). The NMR and crystallographic studies of 37 and 38 reveal that the metal center is only bonded to one Cp unit and the proton is untouched in the PHR unit. The C–H activation product, CpP–C tridentate complex [{Me2Si(C5Me4)P(C6H2tBu2CMe2H2-2,4,6)}Y(THF)2] (39), was resulted from the same reaction or from the solution of 37 after 2 days. By using a similar but less sterically hindered CpP ligand, dinuclear complexes [{Me2Si(C5Me4)PR}2Ln(CH2SiMe3)(THF)n]2 (Ln = Y, R = Ph, n = 1 (40); Ln = Yb, R = Ph, n = 0 (41); Ln = Lu, R = Ph, n = 0 (42); Ln = Y, R = Cy (Cy = cyclohexyl), n = 0 (43); Ln = Yb, R = Cy, n = 0 (44); Ln = Lu, R = Cy, n = 0 (45)) were prepared by protonation of [Ln(CH2SiMe3)3(THF)2] with Me2Si(C5Me4)PHR in hexane. The molecular structure of 45 is shown in Fig. 6. Complexes 40–45 adopt a similar dimeric structure, in which the two metal centers are bridged by the two phosphides and each of them bears a terminal alkyl ligand. The phosphide bridges are asymmetric. The “intermolecular” Ln–P bonds are shorter than the corresponding “intramolecular” chelating Ln–P bonds (2.796(5) vs. 2.836(6) Å in 41, 2.786(1) vs. 2.826(1) Å in 42). The reactions of the alkyl complexes 40–42 with 2 equiv. of PhSiH3 in THF led to the corresponding isostructural hydride complexes 46–48 containing one phosphide and two hydride bridges, while the other phosphide ligand is bonded to only one metal center. The benzene solution of 43 treated with PhSiH3 afforded a THF free tetranuclear hydride complex 49 which is almost insoluble in all the common organic solvents (Scheme 22). Two other bridging hydride phosphide complexes [Me2Si(C5Me4)(PR)LnH(THF)]2 (R = C6H2tBu3-2,4,6; Ln = Y (50), Lu (51)) were prepared by a similar reaction of 37 and 38 with PhSiH3 in benzene (Scheme 22).39
 | ||
| Fig. 6 Solid-state structure of 45. Hydrogen atoms are omitted for clarity. | ||
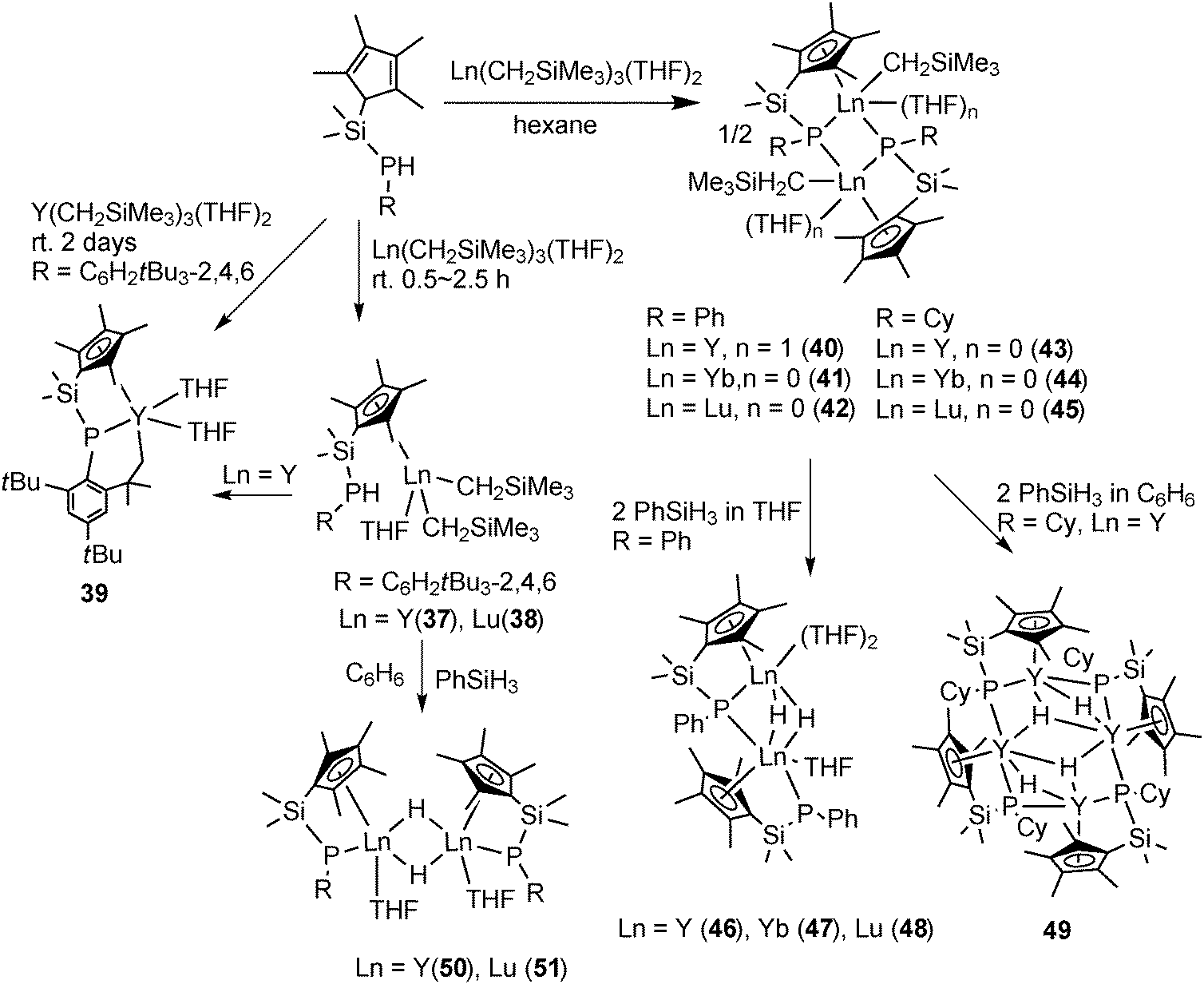 | ||
| Scheme 22 | ||
All the CpP alkyl and hydride complexes mentioned above are active catalysts for hydrosilylation of olefin or the polymerization of ethylene.39
Complexes 43 and 45 have shown extremely high regio- and stereoselectivity in the catalytic polymerization of isoprene to isotactic 3,4-polyisoprene (Scheme 23). This has been achieved for the first time by use of a combination of a binuclear rare earth metal dialkyl complex and an equimolar amount of [Ph3C][B(C6F5)4] as a catalyst system. A DFT calculation suggested that a binuclear monocationic monoalkyl species [Me2Si(C5Me4)(μ-PCy)Y(μ-CH2SiMe3)Y(μ-PCy)(C5Me4)SiMe2]+ obtained by the reaction of 40 with [Ph3C][B(C6F5)4] could be the true catalyst species (Scheme 24).40
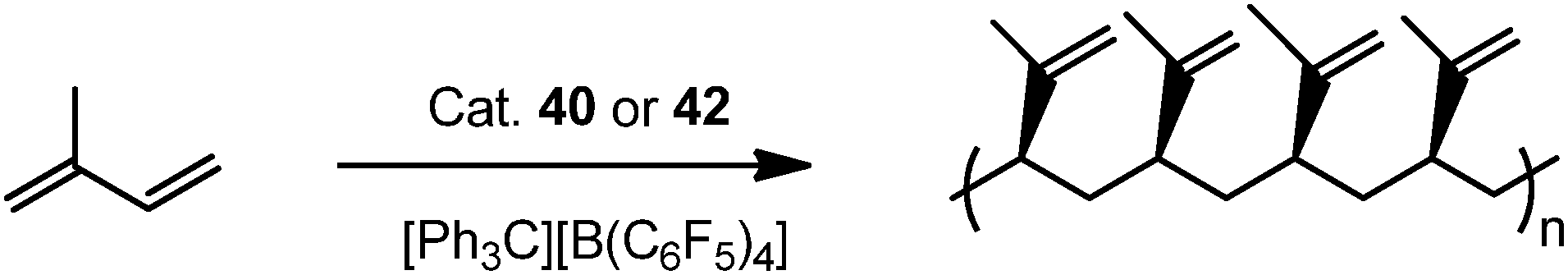 | ||
| Scheme 23 | ||
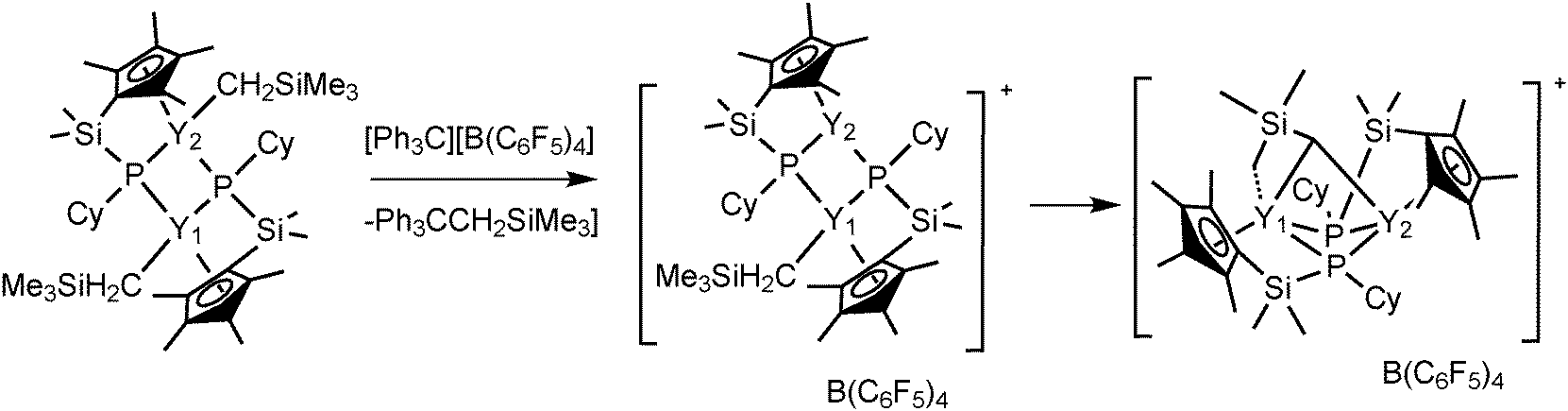 | ||
| Scheme 24 | ||
3. Rare-earth metal complexes containing dianionic phosphide (phosphinidene) (PR2−) or P3−
Phosphinidenes are the phosphorus analogs of carbenes and nitrenes, which bear two electrons on the phosphorus atom. Similarly to the rare-earth metal mono-anionic phosphide complexes, phosphinidene complexes can also be prepared by salt metathesis or intermolecular protonation reaction. Based on the limited number of complexes, only bridging phosphinidene PR2− complexes as modes II and III are reported, in which the R group on the ligand is restricted to an aryl group. No mode I compounds with a formal Ln–P double bond are known.The chemistry of rare-earth metal phosphinidene remained unknown until 2008 Kiplinger and coworkers reported the first lanthanide phosphinidene complex [(PNPiPr)Lu]2(μ-PMes)2 (52) (PNPiPr = {2-(iPr2P)-4-Me-C6H3}2N). The α-hydrogen abstraction reaction of [(PNPiPr)Lu(CH2SiMe3)2] with MesPH2 in toluene afforded the lutetium phosphinidene complex 52 (Scheme 25).
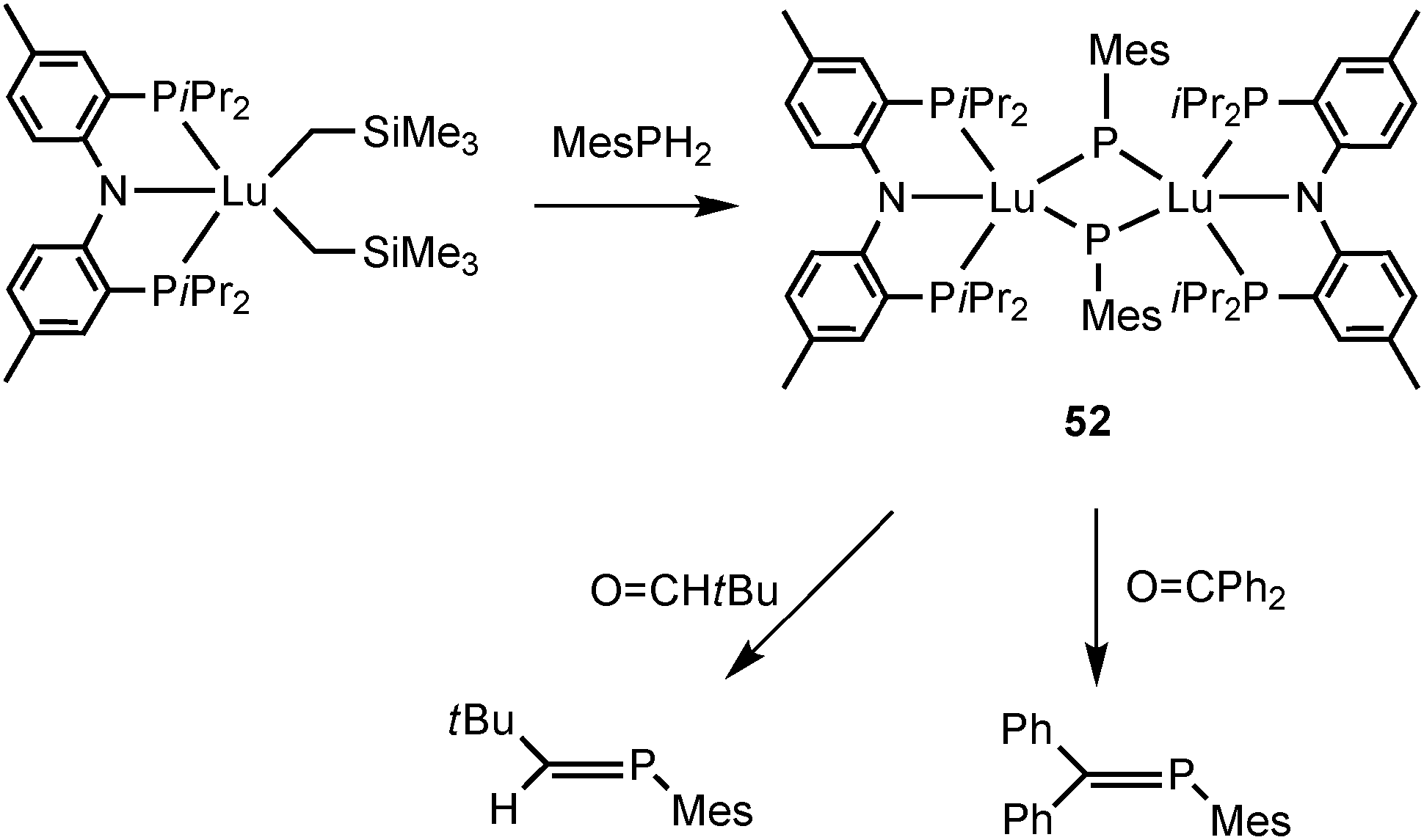 | ||
| Scheme 25 | ||
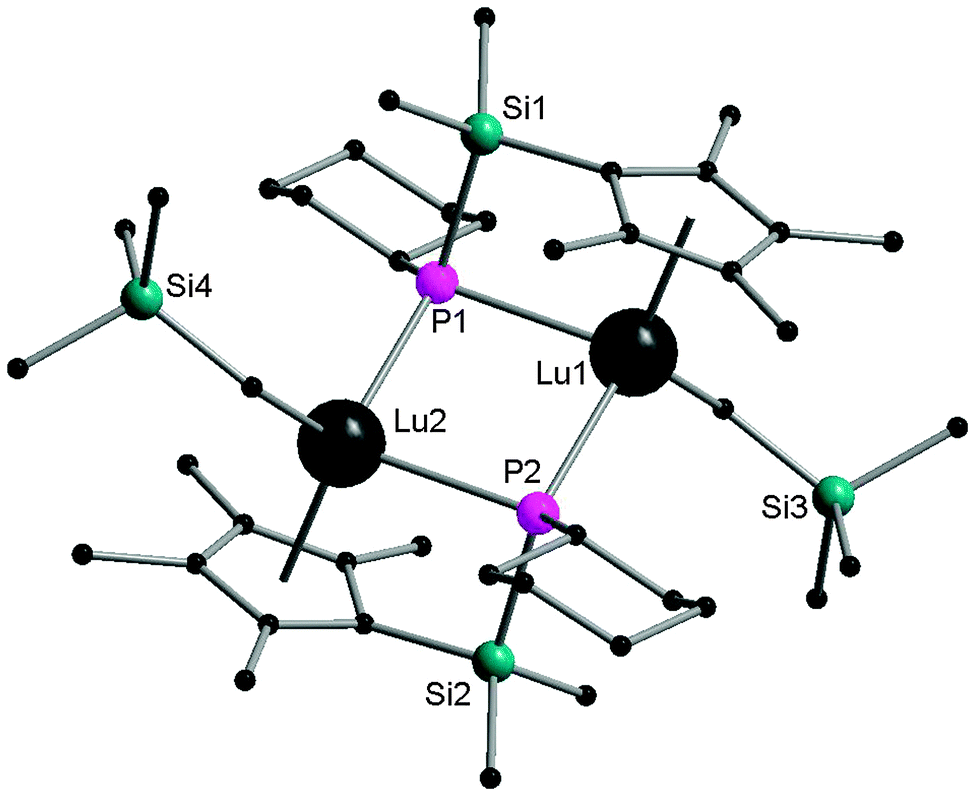
Interestingly, the Lu2P2 core is asymmetric (Fig. 7). The two shorter Lu–P bonds of the core (2.6031(16) and 2.5973(15) Å) are on average ∼0.06 Å shorter than the other two Lu–P bonds (2.6724(14) and 2.6527(16) Å) as well as shorter than the Lu–P bond distances reported for the lutetium phosphide complexes (e.g., 1, Lu–P = 2.782(1), 2.813(2) Å; 42, Lu–P = 2.826(1), 2.786(1) Å; 45, Lu–P = 2.817(1), 2.789(1) Å; 48, Lu–P = 2.788(2), 2.861(2) Å). The sum of the angles around two phosphorus atoms in phosphinidene ligands are 358.9° and 356.5°. Complex 52 is a nucleophilic phosphinidene transfer reagent, which reacts as a phospha-Wittig reagent with aldehydes and ketones to give the corresponding phosphaalkenes (Scheme 25).41
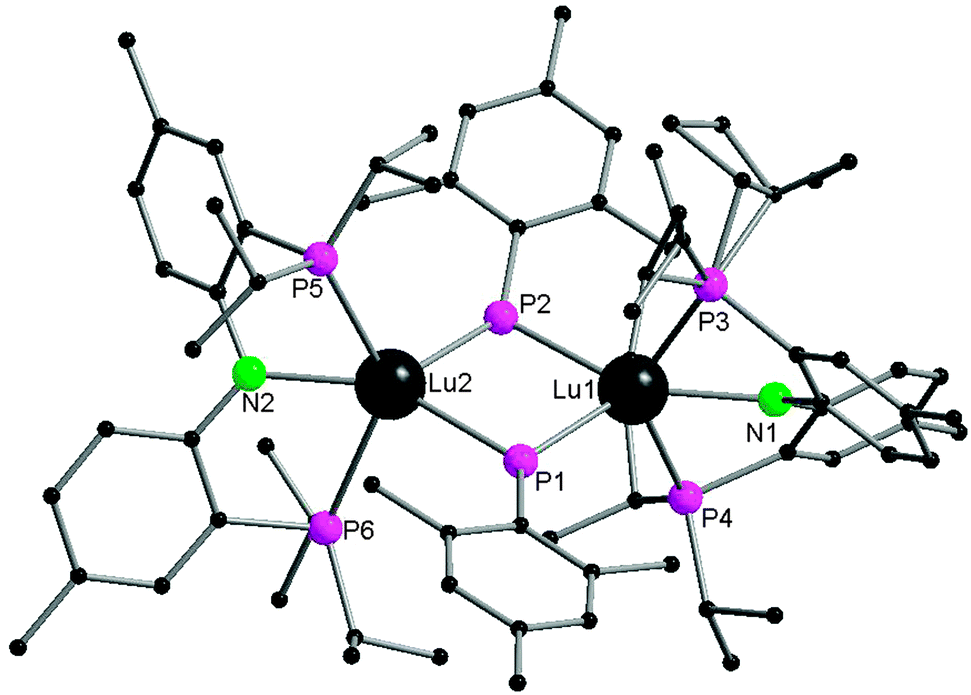 | ||
| Fig. 7 Solid-state structure of 52. Hydrogen atoms are omitted for clarity. | ||
Mindiola and coworkers have presented the first examples of phosphinidene complexes of scandium in both neutral and ionic forms. The same ancillary ligand PNPiPr as in 52 was utilized in the scandium examples. The combined salt metathesis and protonation reaction of [(PNPiPr)Sc(CH3)Br] and LiPH(Tripp) led to formation of a dinuclear scandium phosphinidene complex [(PNPiPr)Sc(μ-P(Tripp))]2 (53). Treating [(PNPiPr)Sc(CH3)Br] with a bulkier phosphide LiP(H)(DMP) (DMP = 2,6-Mes2C6H3) afforded a mononuclear, phosphinidene “ate” complex, [(PNPiPr)Sc(μ-P(DMP))(μ-Br)Li] (54) wherein a LiBr residue has interacted with the shortest known Sc–P bond (2.338(2) Å). The Li cation in 54 can be partially encapsulated with DME to provide a phosphinidene salt derivative, [(PNPiPr)Sc(μ-P(DMP))(μ-Br)Li(DME)] (55) (Scheme 26). Complex 54 shows clean phospha-Wittig chemistry toward ketones and phosphorus dichlorides.42
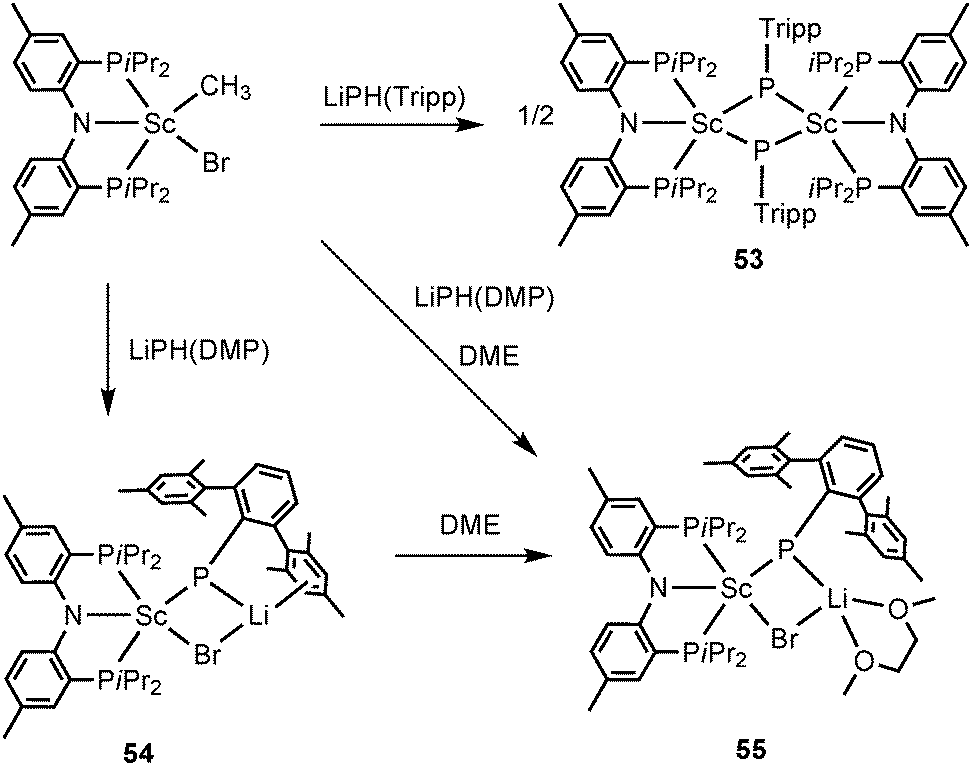 | ||
| Scheme 26 | ||
Chen and coworkers reported the synthesis and structural characterization of first early lanthanide phosphinidene species [(THF)3(I)Nd(μ-PAr)]2 (56) (Ar = 2,6-iPr2C6H3) by the reaction of NdI3(THF)3.5 with 2 equiv. of K[ArPSiMe3] via salt metathesis and a silyl exchange in THF (Scheme 27). The X-ray crystal structure of complex 56 shows a centrosymmetric dimer bridging by two phosphinidene units. An iodide and three THF molecules complete the pseudo-octahedral coordination sphere of each Nd(III). Complex 56 demonstrates that neither sterically demanding nor strong π-electronic donating ancillary ligands are necessary for the stabilization of the lanthanide phosphinidene species.43 A similar situation was observed for the phosphide complexes 16 and 17. Due to the presence of the Nd–I bond, 56 has been used as a precursor for the synthesis of Nd phosphinidene complexes supported by Cp* and TpPh (hydrotris(pyrazolyl)borate) ligands, [(μ-PAr)(Cp*)Nd(THF)]2 (57) (Ar = 2,6-iPr2C6H3) and [(μ-PAr)(TpPh)Nd(THF)]2 (58) via a salt elimination (Scheme 27). Both complexes possess the bridging phosphinidene structure, and in 58 one pyrazolyl of the TpPh ligand undergoes a “1,2-borotropic” shift to minimize the steric hinders around the metal center.26
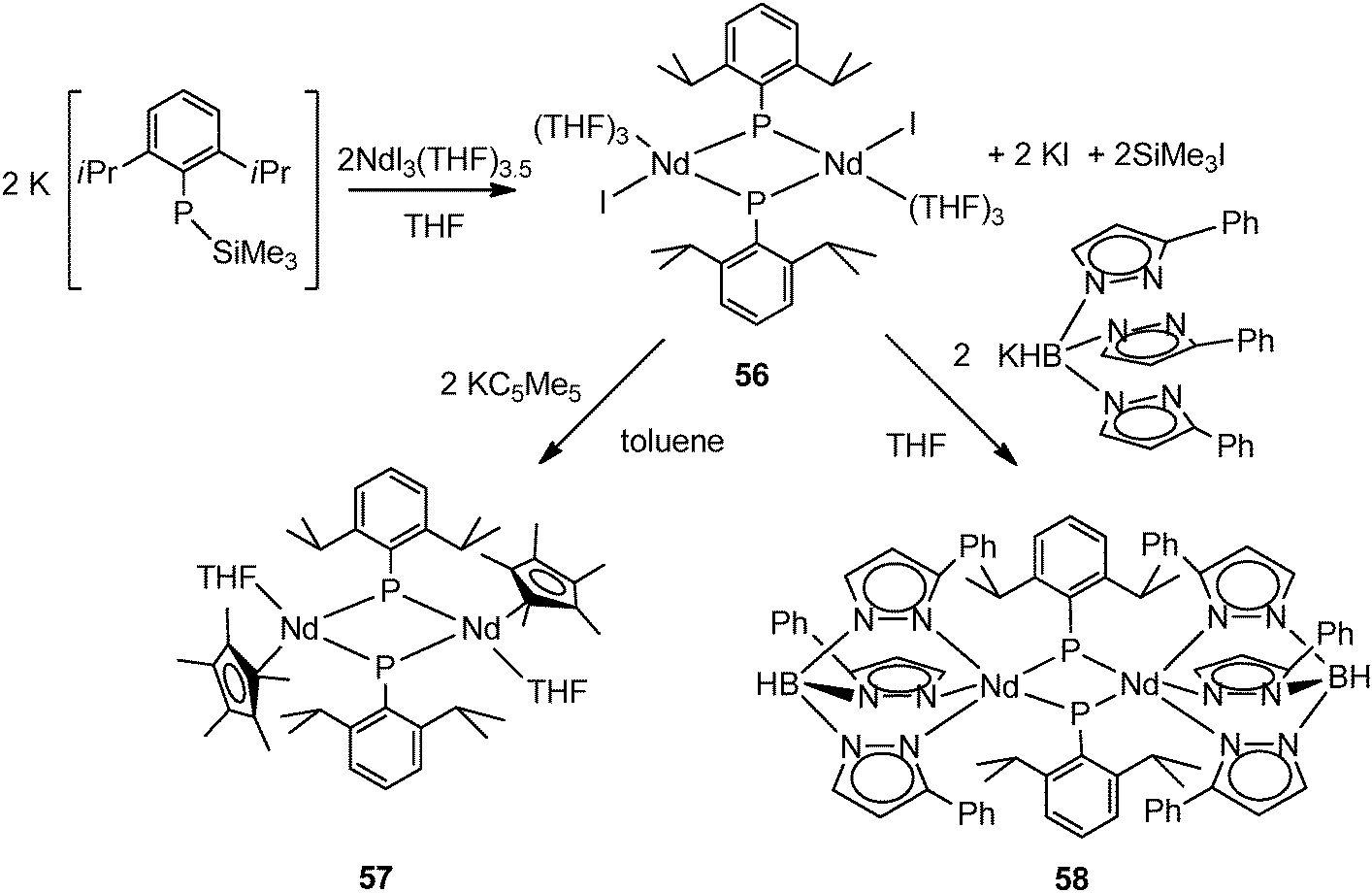 | ||
| Scheme 27 | ||
The first well-defined P3−-containing rare-earth metal compounds were synthesized by Chen and coworkers in 2011. A P3−-containing polymetallic yttrium iodide complex 59 was obtained from reacting 16 with 1 equiv. of K[P(H)Ar] (Ar = 2,6-iPr2C6H3) in toluene via P–Si (or H) and P–C bond cleavage. As shown in Fig. 8, compound 59 adopts a pseudo-C4v point group symmetry. Five Y3+ centers occupy the vertices of a square pyramid while the four μ3-[PR]2− ligands cap the side faces of this pyramid. Each of the basal Y3+ centers coordinates to the unique μ6-P3− ligand (2.7770(4) Å), two μ3-[PR]2− ligands, one μ-I− ligand, and a THF ligand, while the apical Y3+ center bounds to the μ6-P3− ligand (2.8850(19) Å), four μ3-[PR]2− ligands, and a terminal I− ligand. A K+ center in a pseudo-octahedron is bridged by four μ-I− ligands with four basal Y3+ atoms and forms an interaction with the μ6-P3− ligand. As shown in Scheme 28, this compound can be transferred into other P3−-containing yttrium coordination compounds by metathesis reactions. Treating 59 with THF afforded two other polymetallic yttrium phosphinidene phosphides 60 and 61. The structure of 60 resembles that of 59, with toluene coordinated to K replaced by a THF molecule. 61 possesses a pseudo-Cs point group symmetry as a result of the loss of KI. One of the I− ligands bridges two Y3+ centers in μ-mode. In 61, the P3− ligand adopts a rectangular pyramid coordination mode. Yttrium phosphinidene phosphide 62 or 63 was obtained from the reaction of compound 59 with KCp* or a potassium salt of β-diketiminato based tridentate ligand K[MeC(NAr)CHC(Me)(NCH2CH2NMe2)] (Ar = 2,6-iPr2C6H3), respectively. The P3− ligand in the soluble coordination compounds displays two types of coordination modes: octahedral in complexes 59, 60 and 62 and rectangular pyramidal in complexes 61 and 63.24
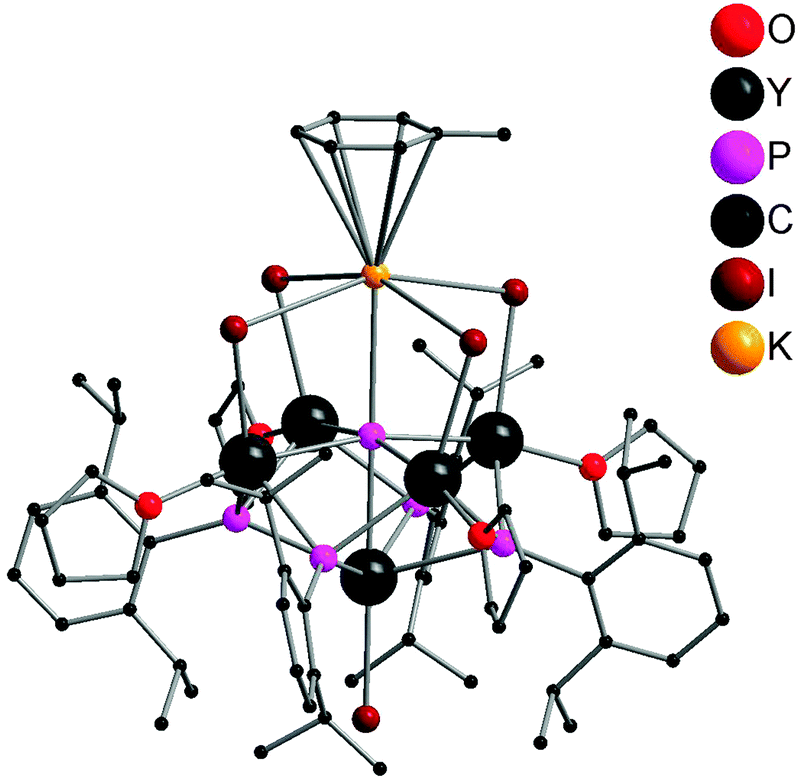 | ||
| Fig. 8 Solid-state structure of 59. Hydrogen atoms are omitted for clarity. | ||
 | ||
| Scheme 28 | ||
4. Rare-earth metal complexes containing polyphosphide pure inorganic ligands (Pnx−)
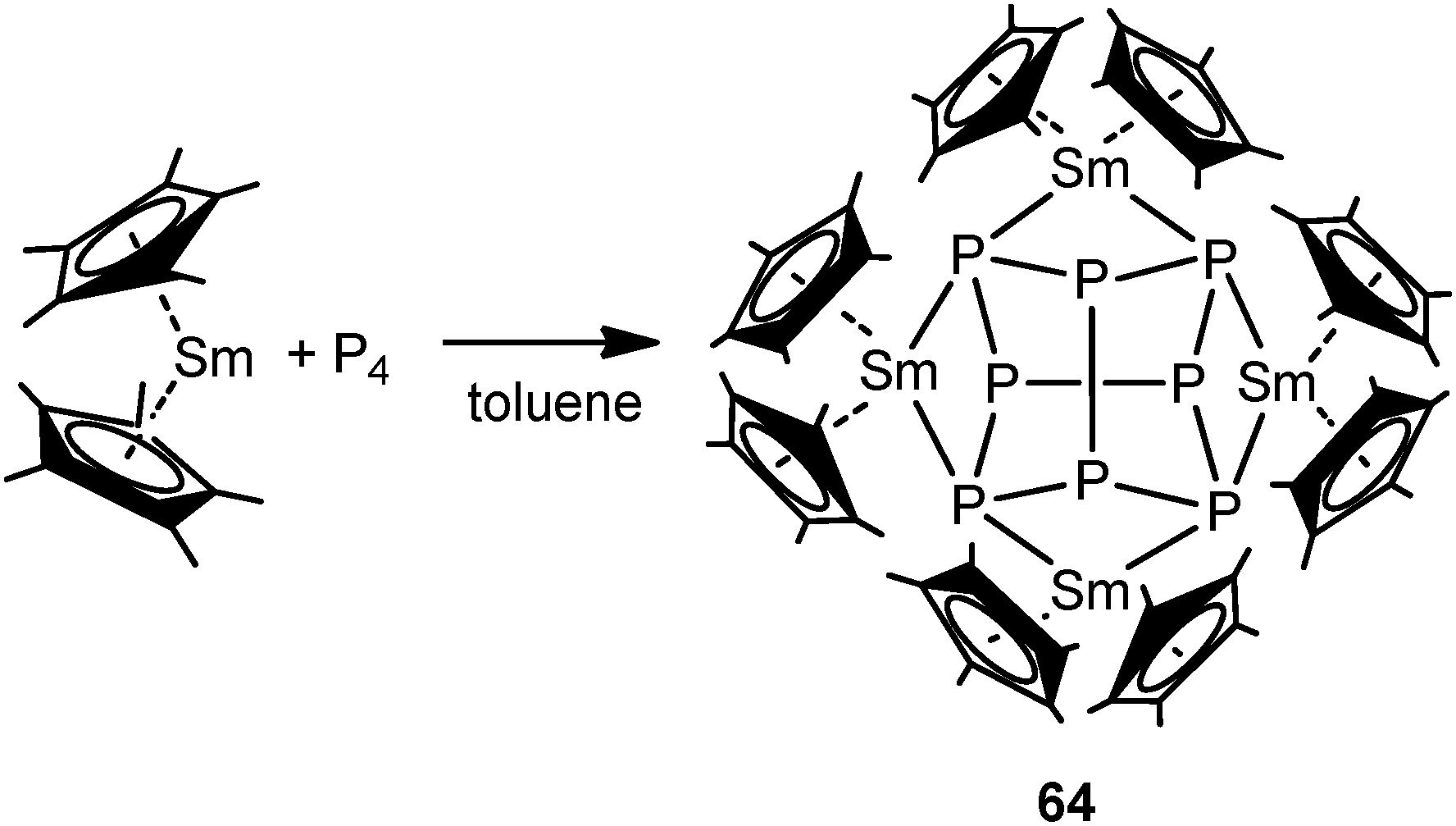
Recently, there has been great interest in the rare-earth metal coordination chemistry of monocyclic and polycyclic “naked” polyphosphide ligands. Low valent rare-earth metals are well known for their reducing potential. Reported rare-earth metal polyphosphide complexes were prepared by redox reactions either with P4 or with transition metal polyphosphides.In 2009, the first molecular rare-earth metal polyphosphide complex, [{Cp*2Sm}4P8] (64), was reported by Roesky and coworkers. Diffusion of P4 vapor into a toluene solution of solvate-free samarocene led to the complex 64 (Scheme 29). The formation of compound 64 is induced by one electron transfer from each divalent samarium atom to a phosphorus atom. As shown in Fig. 9, the structure of the complex is very rare and can be described as a realgar-type P84− ligand trapped in a cage of four samarocenes. The P84− ligand is isostructural and isoelectronic with the P4S4 molecule and can be considered as the larger analogue of the well-established Zintl anion P73−. The Sm–P distances within 64 range from 2.997(2) Å to 3.100(2) Å. Each {Cp*2Sm} unit bridges the central P8 cage giving P–Sm–P angles of P2–Sm1–P4 63.55(5)° and P2–Sm2–P4′ 64.27(5)°. In the center of the P8 cage, a crystallographic C2 axis through the P1–P1′ and P3–P3′ bonds is observed.44
 | ||
| Scheme 29 | ||
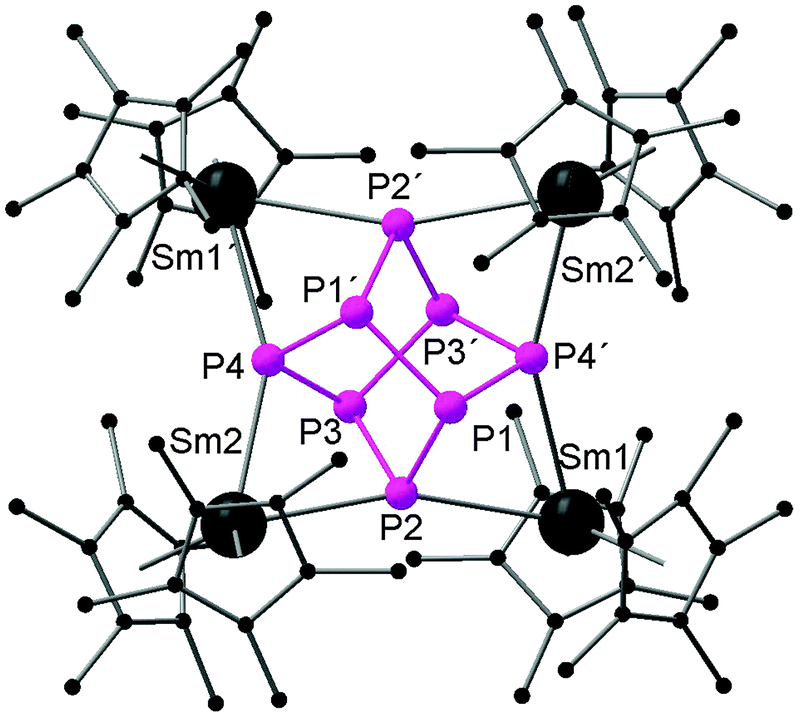 | ||
| Fig. 9 Solid-state structure of 64. Hydrogen atoms are omitted for clarity. | ||
In 2012, another P84− rare-earth metal complex was prepared by Diaconescu and coworkers. [{(NNfc)Sc}4P8] (65) (NNfc = 1,1-fc(NSitBuMe2)2, fc = ferrocenylene) was obtained by the reaction of P4 and 1 equiv. of the scandium naphthalene complex [{(NNfc)Sc}2(μ-C10H8)] which is an excellent two-electron reducing agent. The core P84− ligand in 65 resembles the realgar type structure in 64. The P73− compounds [{(NNfc)Ln(THF)n}3P7] (Ln = Sc, n = 0 (66); Ln = Y, n = 1 (67)) were obtained with excess P4 in the reaction with [{(NNfc)Sc}2(μ-C10H8)] and [{(NNfc)Y(THF)}2(μ-C10H8)], respectively (Scheme 30). The molecular structure of 66 is shown in Fig. 10. The P73− unit in 66 and 67 is similar to the Li3P7 solid state structure45 and they are the first examples of a rare-earth metal Zintl P73− compound directly obtained from P4 activation. DFT calculations on the polyphosphide compounds 64–67 revealed that there is mainly ionic interaction between the rare-earth metals and the Zintl P84− or P73− unit.44,46
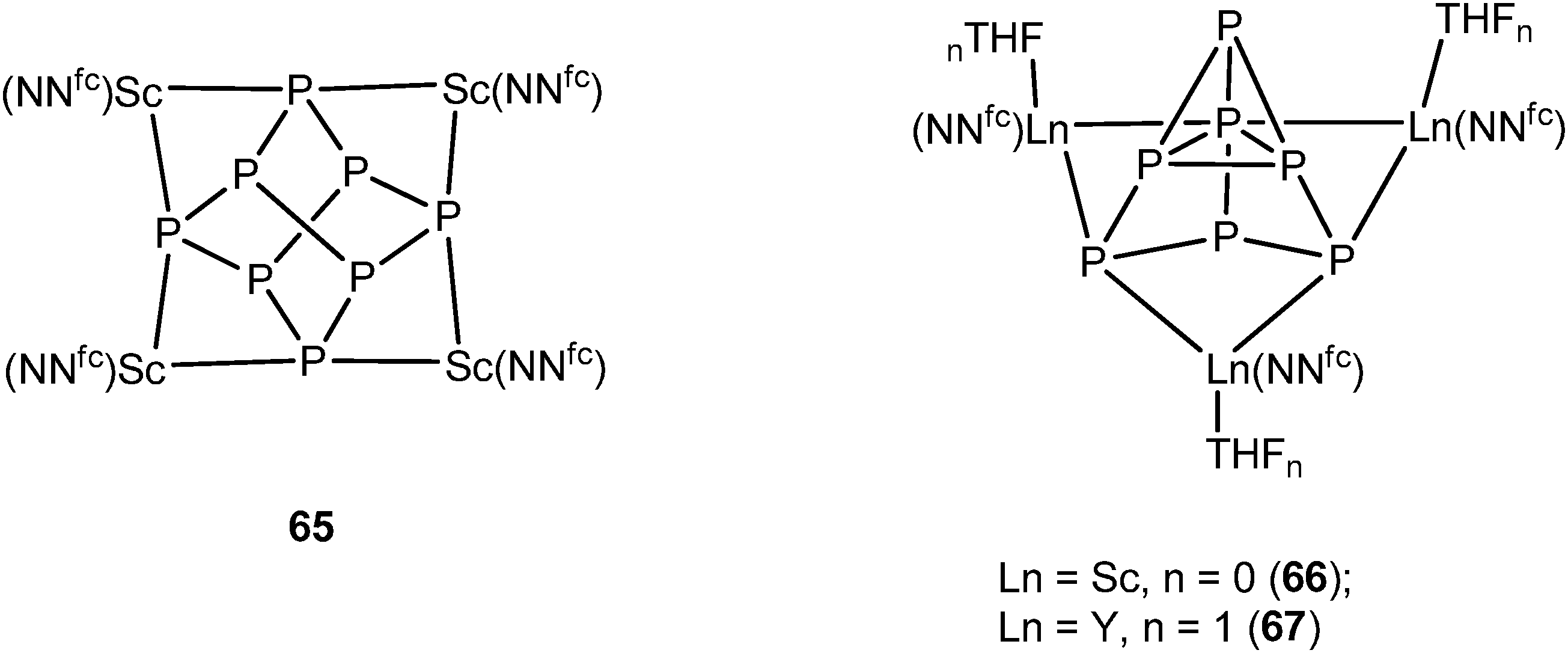 | ||
| Scheme 30 | ||
 | ||
| Fig. 10 Solid-state structure of 66. Hydrogen atoms are omitted for clarity. | ||
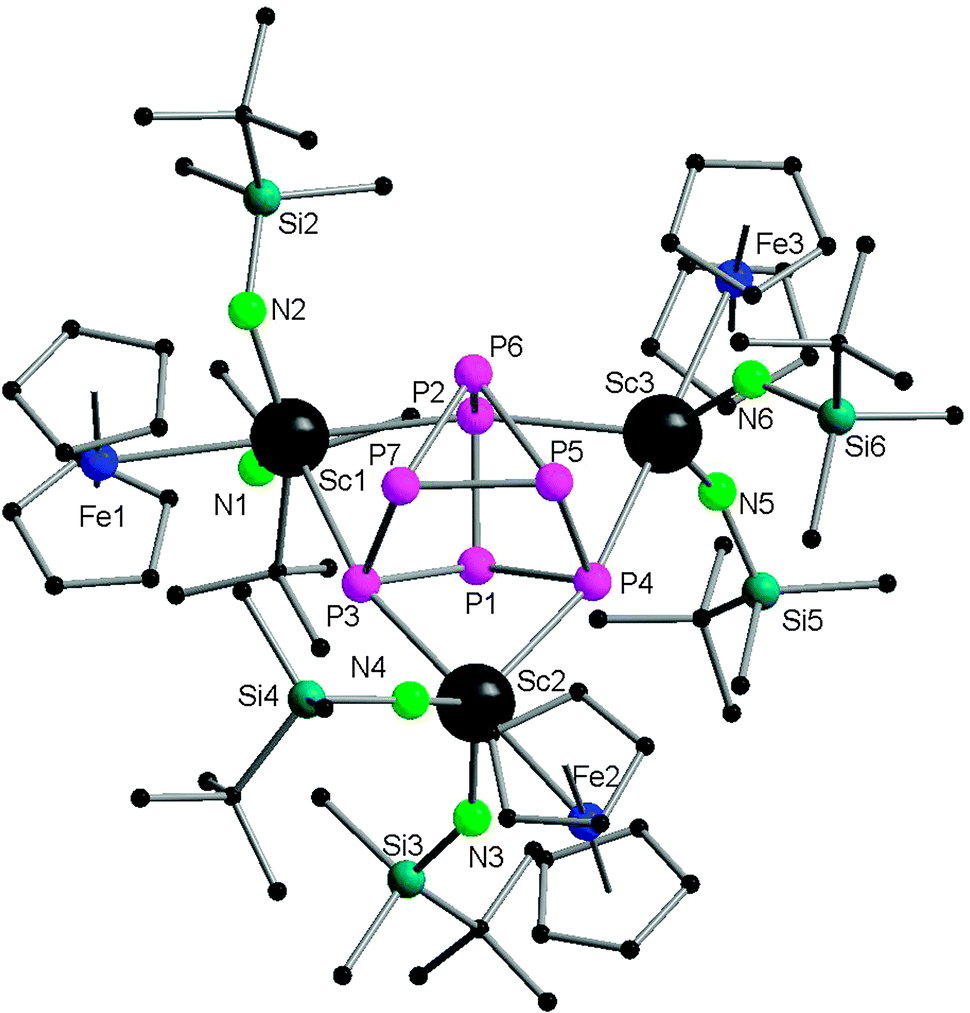
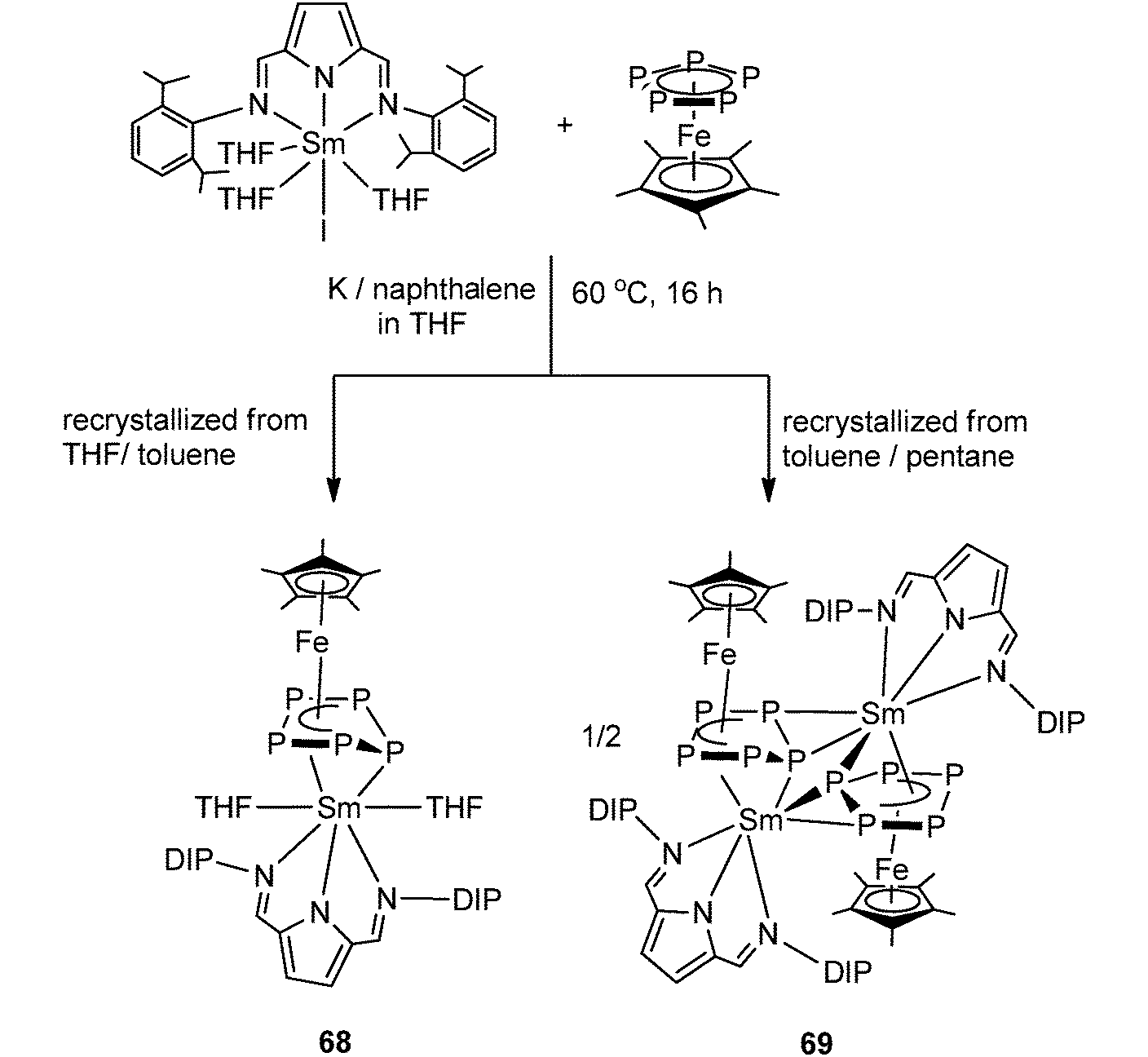
Very recently, Roesky and coworkers have prepared four new 3d–4f polyphosphide complexes by one or two electron reducing pentaphosphaferrocene [Cp*Fe(η5-P5)], with a divalent samarium complex. The first triple-decker polyphosphide bridging complexes of rare earth elements and 3d transition metals were formed by reacting [(DIP2pyr)SmI(thf)3] (DIP2pyr = 2,5-bis{N-(2,6-diisopropylphenyl)iminomethyl}pyrrolyl) and 1 equiv. of [Cp*Fe(η5-P5)] in THF in the presence of potassium–naphthalene. While the new monomeric cyclo-P5 bridging complex [Cp*FeP5Sm(DIP2pyr)(THF)2] (68) was recrystallized from THF and toluene, the dimeric complex 69 was obtained from toluene and pentane (Scheme 31). In the reaction, potassium–naphthalene acts as a reducing agent and an iodine abstracting reagent. As a result of the two electrons reduction by potassium and samarium, the cyclo-P5 is transformed from planar to an open envelope conformation, which links to the iron atom in an η4-coordination mode and to the samarium atom in 68 in an η3-coordination mode (Fig. 11). In 69 an additional η2-coordination of the second cyclo-P5 unit is observed as a result of the dimerization. These two coordination sites are occupied by THF molecules in the monomeric compound 68. In both compounds, reduction of the cyclo-P5 unit leads to the {Cp*FeP5}2− subunit, in which the phosphorous ligand may be formally considered as a cyclo-P53− polyphosphide anion. As shown in Fig. 11, P1 is bent out from the P4 plane at an angle of 127.6°. The P3–P4 bond (2.164(2) Å) in complex 68 is slightly shorter than the other P–P bonds (2.175(2) Å–2.228(2) Å). Three Sm–P distances in 68 are ranged from 2.847(2) Å to 3.0703(15) Å.47 Scheer and coworkers have conducted a redox chemistry study on [Cp*Fe(η5-P5)] and they successfully prepared dianionic species K2{Cp*FeP5}, in which the cyclo-P5 unit exhibits the same open-envelope geometry with comparable P–P bond distances to those in complexes 68 and 69.48
 | ||
| Scheme 31 | ||
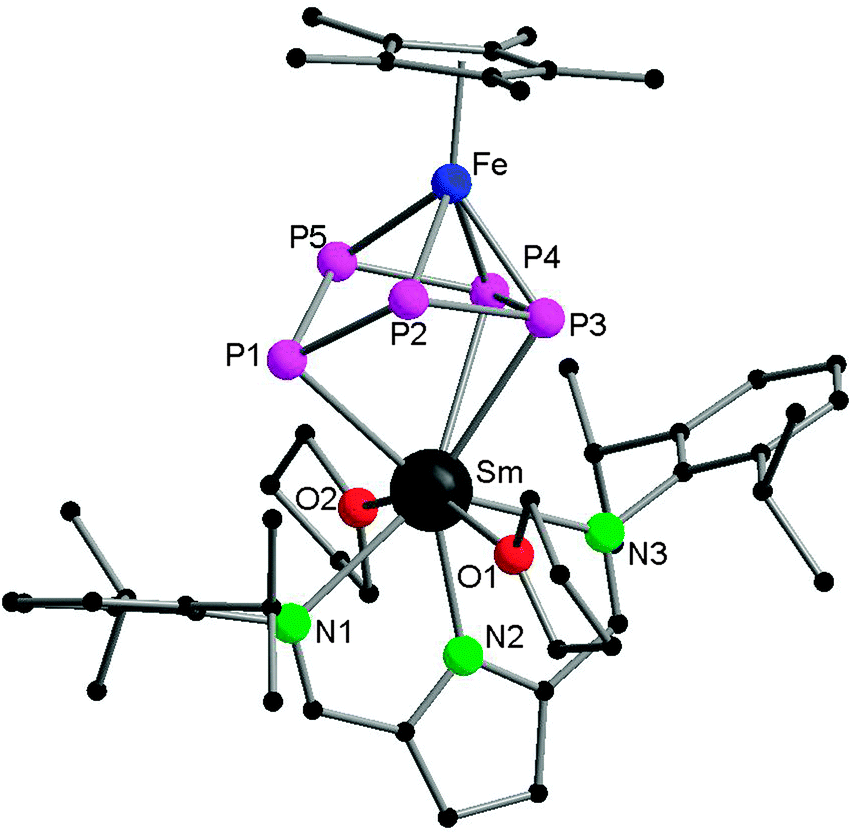 | ||
| Fig. 11 Solid-state structure of 68. Hydrogen atoms are omitted for clarity. | ||
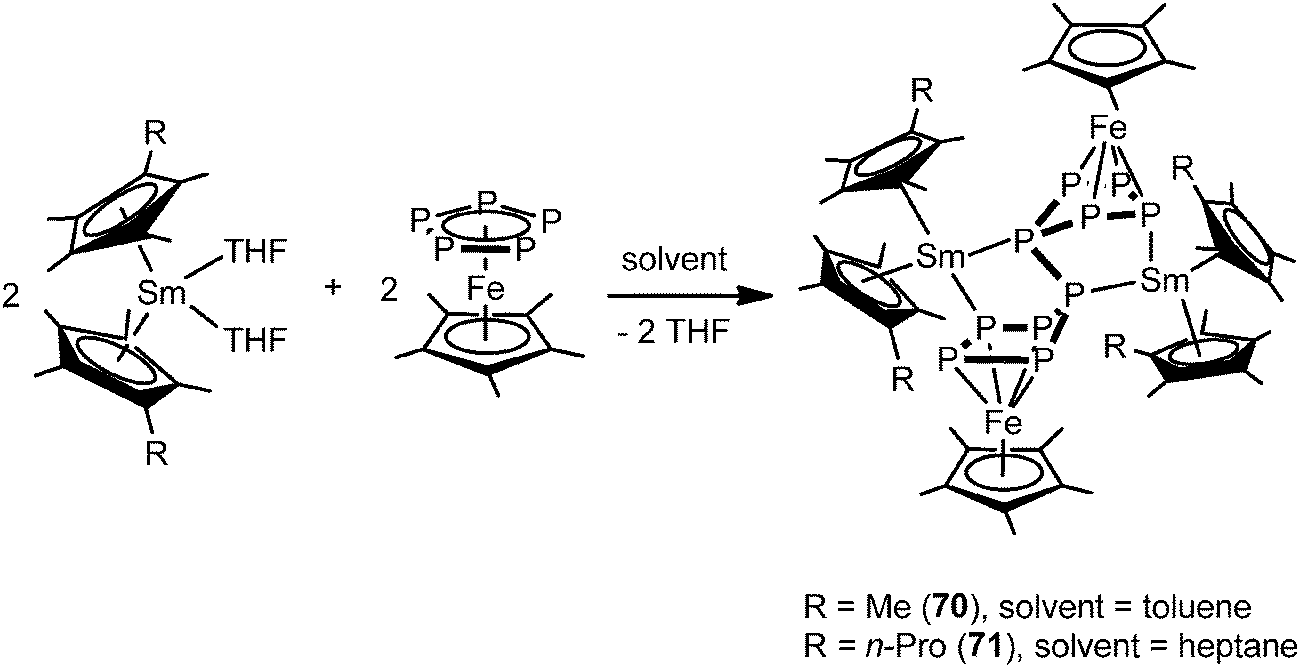
In contrast to the two electron reduction of [Cp*Fe(η5-P5)], the one electron reduction by samarocenes led to [(Cp*Fe)2P10{Sm(η5-C5Me4R)2}2] (R = Me (70), nPr (71)) containing a P104− unit (Scheme 32). They are the first P10 bridging 3d–4f metal complexes. They are also the first examples of reductive coupling of polyphosphide complexes by divalent lanthanides. Each cyclo-P5 unit has been reduced once to force one phosphorus atom to bend away from the iron center and changed the confirmation from planar to open envelope shaped. The newly formed P–P bonds between two bent atoms with distances of P5–P6 2.2089(13) Å (70) in agreement with a single bond are created between the two off-plane phosphorus atoms (Fig. 12). The P–P bond distances around cyclo-P5 rings in 70 are in the range of 2.137(2) to 2.1866(14) Å and on average 0.03 Å shorter than those in 68. The core polyphosphide unit is formally considered as a P104− anion. The P10 systems in 70 and that in [K2(dme)3][(Cp*Fe)2(P10)]48 are comparable. The Sm coordinated compounds show slightly longer P–P bonds than the potassium salt of the dianion.49
 | ||
| Scheme 32 | ||
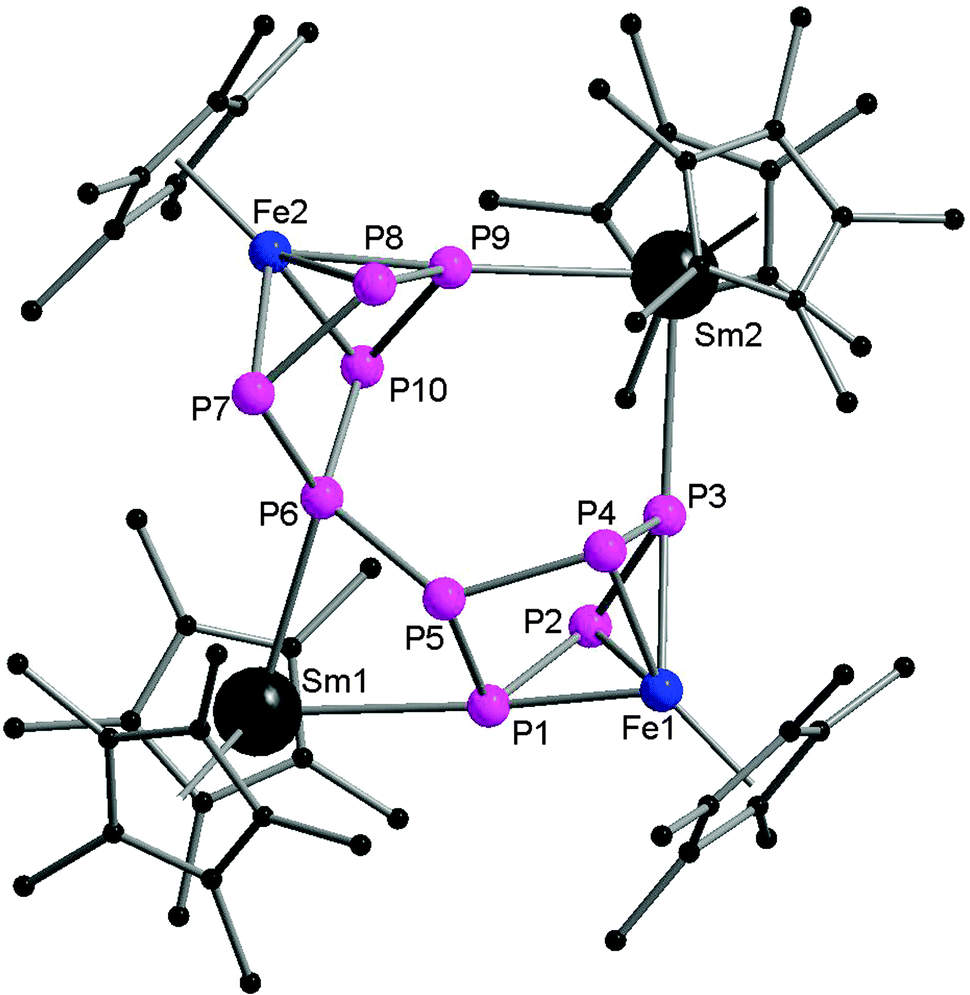 | ||
| Fig. 12 Solid-state structure of 70. Hydrogen atoms are omitted for clarity. | ||
5. Conclusion
The hard rare-earth metals are almost exclusively ligated by hard donor atoms from the second row of the periodic table (oxygen, nitrogen, carbon) and the halides. Preparation of rare-earth metal complexes containing phosphorus ligands was considered as a synthetic challenge. The rare-earth metal complexes with anionic phosphorus ligands are even less common than complexes with neutral phosphine ligands. Since Schumann and coworkers reported the first structurally characterized rare-earth metal phosphide in 1986, many new rare-earth metal phosphides have been developed. The discoveries of the first phosphinidene complex in 2008 and of the first polyphosphide complex in 2009 have opened new perspectives for the rare-earth metal complexes containing anionic phosphides. Moreover, the catalytic applications and the reactivity of certain rare-earth metal phosphides have also seen progress. Based on this fast development, we expect a rich synthetic chemistry of Ln–P bond species as well as new applications of these compounds in catalysis and material science in the near future.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie. Ms Petra Smie is acknowledged for preparing the figures.References
- Phosphorus Compounds-Advanced Tools in Catalysis and Material Sciences, ed. M. Peruzzini and L. Gonsalvi, Springer, Netherlands, 2011, vol. 37 Search PubMed.
- Molecular Catalysis of Rare-Earth Elements, Structure and Bonding, ed. P. W. Roesky, Springer, Berlin, Heidelberg, 2010 Search PubMed.
- R. G. Pearson, Inorg. Chim. Acta, 1995, 240, 93–98 CrossRef CAS.
- S. P. Nolan, D. Stern and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 7844–7853 CrossRef CAS.
- P. L. Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef PubMed.
- Z. Hou and Y. Wakatsuki, J. Organomet. Chem., 2002, 647, 61–70 CrossRef CAS.
- F. Nief, Eur. J. Inorg. Chem., 2001, 891–904 CrossRef CAS.
- F. Nief, Coord. Chem. Rev., 1998, 178–180, 13–81 CrossRef CAS.
- M. D. Fryzuk, T. S. Haddad and D. J. Berg, Coord. Chem. Rev., 1990, 99, 137–212 CrossRef CAS.
- H. Schumann, I. Albrecht, M. Gallagher, E. Hahn, C. Janiak, C. Kolax, J. Loebel, S. Nickel and E. Palamidis, Polyhedron, 1988, 7, 2307–2315 CrossRef CAS.
- H. Schumann, E. Palamidis, G. Schmid and R. Boese, Angew. Chem., Int. Ed. Engl., 1986, 25, 718–719 CrossRef.
- G. W. Rabe, J. Riede and A. Schier, Inorg. Chem., 1996, 35, 40–45 CrossRef CAS PubMed.
- G. W. Rabe, G. P. A. Yap and A. L. Rheingold, Inorg. Chem., 1997, 36, 3212–3215 CrossRef CAS PubMed.
- G. W. Rabe, J. Riede and A. Schier, Inorg. Chem., 1996, 35, 2680–2681 CrossRef CAS.
- M. Westerhausen, S. Schneiderbauer, M. Hartmann, M. Warchhold and H. Noeth, Z. Anorg. Allg. Chem., 2002, 628, 330–332 CrossRef CAS.
- M. Westerhausen, S. Schneiderbauer, N. Makropoulos, M. Warchhold, H. Noeth, H. Piotrowski and K. Karaghiosoff, Organometallics, 2002, 21, 4335–4341 CrossRef CAS.
- Z. Hou, Y. Zhang, H. Tezuka, P. Xie, O. Tardif, T.-a. Koizumi, H. Yamazaki and Y. Wakatsuki, J. Am. Chem. Soc., 2000, 122, 10533–10543 CrossRef CAS.
- G. W. Rabe, G. P. A. Yap and A. L. Rheingold, Inorg. Chem., 1995, 34, 4521–4522 CrossRef CAS.
- F. Nief and L. Ricard, J. Organomet. Chem., 1997, 529, 357–360 CrossRef CAS.
- G. W. Rabe, G. P. A. Yap and A. L. Rheingold, Inorg. Chem., 1997, 36, 1990–1991 CrossRef CAS.
- H. C. Aspinall, S. R. Moore and A. K. Smith, J. Chem. Soc., Dalton Trans., 1992, 153–156 RSC.
- W.-X. Zhang, M. Nishiura, T. Mashiko and Z. Hou, Chem.–Eur. J., 2008, 14, 2167–2179 CrossRef CAS PubMed.
- W. Yi, J. Zhang, L. Hong, Z. Chen and X. Zhou, Organometallics, 2011, 30, 5809–5814 CrossRef CAS.
- Y. Lv, X. Xu, Y. Chen, X. Leng and M. V. Borzov, Angew. Chem., Int. Ed., 2011, 50, 11227–11229 CrossRef CAS PubMed.
- S. Krieck, H. Goerls and M. Westerhausen, Inorg. Chem. Commun., 2009, 12, 409–411 CrossRef CAS PubMed.
- P. Cui, Y. Chen and M. V. Borzov, Dalton Trans., 2010, 39, 6886–6890 RSC.
- W. J. Evans, J. T. Leman, J. W. Ziller and S. I. Khan, Inorg. Chem., 1996, 35, 4283–4291 CrossRef CAS PubMed.
- G. W. Rabe, J. Riede and A. Schier, Organometallics, 1996, 15, 439–441 CrossRef CAS.
- M. R. Douglass, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 2001, 123, 10221–10238 CrossRef CAS PubMed.
- H. C. Aspinall, S. R. Moore and A. K. Smith, J. Chem. Soc., Dalton Trans., 1993, 993–996 RSC.
- W. Clegg, K. Izod, S. T. Liddle, P. O'Shaughnessy and J. M. Sheffield, Organometallics, 2000, 19, 2090–2096 CrossRef CAS.
- K. Izod, P. O'Shaughnessy, J. M. Sheffield, W. Clegg and S. T. Liddle, Inorg. Chem., 2000, 39, 4741–4748 CrossRef CAS.
- W. Clegg, K. Izod and S. T. Liddle, J. Organomet. Chem., 2000, 613, 128–131 CrossRef CAS.
- S. Blair, K. Izod and W. Clegg, J. Organomet. Chem., 2003, 688, 92–99 CrossRef CAS PubMed.
- K. Izod, S. T. Liddle, W. McFarlane and W. Clegg, Organometallics, 2004, 23, 2734–2743 CrossRef CAS.
- K. Izod, S. T. Liddle and W. Clegg, Chem. Commun., 2004, 1748–1749 RSC.
- K. Izod, S. T. Liddle, W. Clegg and R. W. Harrington, Dalton Trans., 2006, 3431–3437 RSC.
- O. Tardif, Z. Hou, M. Nishiura, T.-a. Koizumi and Y. Wakatsuki, Organometallics, 2001, 20, 4565–4573 CrossRef CAS.
- O. Tardif, M. Nishiura and Z. Hou, Tetrahedron, 2003, 59, 10525–10539 CrossRef CAS PubMed.
- L. Zhang, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2005, 127, 14562–14563 CrossRef CAS PubMed.
- J. D. Masuda, K. C. Jantunen, O. V. Ozerov, K. J. T. Noonan, D. P. Gates, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 2408–2409 CrossRef CAS PubMed.
- B. F. Wicker, J. Scott, J. G. Andino, X. Gao, H. Park, M. Pink and D. J. Mindiola, J. Am. Chem. Soc., 2010, 132, 3691–3693 CrossRef CAS PubMed.
- P. Cui, Y. Chen, X. Xu and J. Sun, Chem. Commun., 2008, 5547–5549 RSC.
- S. N. Konchenko, N. A. Pushkarevsky, M. T. Gamer, R. Koeppe, H. Schnoeckel and P. W. Roesky, J. Am. Chem. Soc., 2009, 131, 5740–5741 CrossRef CAS PubMed.
- W. Hönle, H. G. von Schnering, A. Schmidpeter and G. Burget, Angew. Chem., Int. Ed. Engl., 1984, 23, 817–818 CrossRef.
- W. Huang and P. L. Diaconescu, Chem. Commun., 2012, 48, 2216–2218 RSC.
- T. Li, J. Wiecko, N. A. Pushkarevsky, M. T. Gamer, R. Koeppe, S. N. Konchenko, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2011, 50, 9491–9495 CrossRef CAS PubMed.
- M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 2972–2976 CrossRef CAS PubMed.
- T. Li, M. T. Gamer, M. Scheer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2013, 49, 2183–2185 RSC.
| This journal is © The Royal Society of Chemistry 2014 |