
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of the maximum entropy principle to determine ensembles of intrinsically disordered proteins from residual dipolar couplings†
M.
Sanchez-Martinez
and
R.
Crehuet
 *
*
Institute of Advanced Chemistry of Catalunya (IQAC), CSIC, Spain. E-mail: ramon.crehuet@iqac.csic.es
First published on 13th October 2014
Abstract
We present a method based on the maximum entropy principle that can re-weight an ensemble of protein structures based on data from residual dipolar couplings (RDCs). The RDCs of intrinsically disordered proteins (IDPs) provide information on the secondary structure elements present in an ensemble; however even two sets of RDCs are not enough to fully determine the distribution of conformations, and the force field used to generate the structures has a pervasive influence on the refined ensemble. Two physics-based coarse-grained force fields, Profasi and Campari, are able to predict the secondary structure elements present in an IDP, but even after including the RDC data, the re-weighted ensembles differ between both force fields. Thus the spread of IDP ensembles highlights the need for better force fields. We distribute our algorithm in an open-source Python code.
Introduction
Intrinsically disordered proteins (IDPs) are an emerging family of proteins characterized by their ability to adopt a vast number of configurations in solution. Their role in cell signalling, transcription and aggregation turns them into key proteins in cancer and neurodegenerative diseases.1,2 One would expect many of them to be drug targets; however very few studies have addressed the interaction of IDPs with small molecules.1,3 One reason for this is the difficulty in both generating and characterizing the ensemble of configurations that turn an IDP functional.4 A common mechanism of IDPs is a folding transition upon binding partner proteins.5 The amount of secondary structure elements in the unbound IDPs governs the kinetics of this binding process,6 thus the need to understand IDP secondary structure elements in solution. These regions are also called MoRFs7,8 and many studies aim at their identification.A very suitable technique to characterize the secondary structure at a residue level is the NMR residual dipolar couplings (RDCs),9 a technique that has been thoroughly developed by Blackledge10–13 and Forman-Kay14–17 groups, among others. In an isotropic medium, such as liquid water, dipolar couplings average out to zero. But if the medium has some preferential directions, then there is a partial alignment of the molecules and a residual coupling can be measured.
Contrary to what is the case for folded proteins, in IDPs the alignment tensor is essentially determined by the local (secondary) structure.16 When the main mechanism of alignment is steric, repulsion between the protein and the alignment medium tends to align secondary structure elements parallel to the medium. For this reason N–H couplings convey important information on the secondary structure. When the alignment medium is parallel to the field they are positive in α-helices – as all N–H are parallel to the helix – negative in β-sheets – as N–H are perpendicular to the sheet – and are very low for regions without any secondary structure, where residue orientations are random. A qualitative interpretation of RDCs can be based on these principles, but a quantitative explanation can be achieved if one is able to generate an ensemble of configurations that reproduce the measured RDCs.11,12,15,16
The generation of the ensemble that fit the RDCs is the crux of several approximations used in this field.18 A common approach is to sample random coil regions of the Ramachandran plot with codes such as Flexible Meccano,13,19 TraDES,16,20 or BEGR21 and then introduce secondary structure regions and weight them with a statistical analysis11,17 or a genetic algorithm.13 This is because the physics behind these force fields is very simple and cannot predict secondary or tertiary structure. These methods have proved extremely successful in interpreting several IDP studies, but lack predictive value in terms of secondary structure elements.
The problem of optimizing an ensemble is a case of inferential structure determination,22 albeit with a much broader probability distribution. If this distribution comes from a simulation, we would like to modify it so that it agrees with the experimental data. Ideally, the inclusion of the experimental data should create ensembles that agree among themselves, even if coming from different simulation methods. Here we explore to which extent this is true.
We present a method based on the maximum entropy principle (MaxEnt) to fit RDC data to simulated ensembles. Maximum entropy is a logically consistent way to fit a distribution to previously known values introducing the minimum possible modifications.23,24 It has been advocated very recently as a powerful technique to solve structural problems25 and it has already been applied to SAXS ensemble determination.26
We generated our ensembles from two coarse-grained force fields, which have more accurate physical terms than TRaDES or Flexible Meccano while remaining computationally affordable. Coarse-grained methods allow sampling of the large conformational space essential to describe IDPs and converge RDC data. However the simulation force-field does not influence the validity of the presented selection procedure, which can be applied to all types of ensembles.
Our aim of this work is three-fold. First, we develop a fitting algorithm to adjust experimental RDCs to an ensemble of conformations. We implement our method in a publicly available code so that it can be compared to others, and can be used by any research group.27 Second, we explore the information content of RDC data and the influence of our force field; in other words, how much do the RDCs constrain the initial ensemble. Considerable efforts have been made to determine how much different experimental data determine the properties of the ensembles.17 Here we want to highlight the relevance of the underlying model, which is often overlooked. And third, we test whether some coarse grained methods can produce more accurate ensembles than random-coil-based force fields and thus increase the prediction of RDCs.
Methods
The maximum entropy (MaxEnt) principle derives from minimizing the information included in an ensemble to fit certain observables. It was first introduced by Jaynes23 and was recently applied as a way to constrain molecular dynamics on-the-fly.28,29 Roux and co-workers showed that under certain circumstances, their results were equivalent to the more traditional constrains with harmonic potentials, used also in molecular dynamics,30 while Vendruscolo and co-workers showed that the restraint strength can be related to the experimental error.31 Here we present the application of the MaxEnt to the a posteriori re-weighting of an ensemble that has already been calculated. We also add some modifications needed to treat RDC data.We decided to implement an a posteriori re-weighting so that our method could be applied to ensembles generated with any software or force field. A second reason is that when applying the constraints on-the-fly, one usually averages by the number of replicas running in parallel32,33 but the number of replicas needed to converge the RDC values for IDPs is of the order of thousands (see Results section), which means that constraint molecular dynamics could only be run in supercomputers.
In our a posteriori re-weighting we assume we have a set of N structures {Xj=1,N} that we have previously calculated with a Monte Carlo or molecular dynamics simulation. As such, they have already been generated with a probability proportional to their Boltzmann factor, which depends on each specific force field. For a set of M observables q = {qi=1,M}, Pitera and Chodera showed that the application of the MaxEnt principle resulted in a reweighting of the probability of each structure j by a term:28
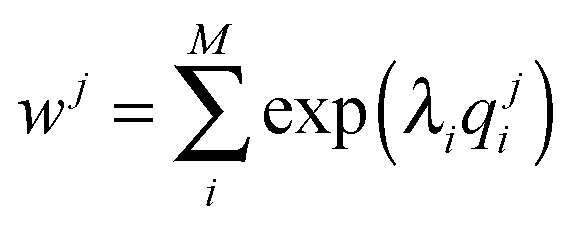 | (1) |
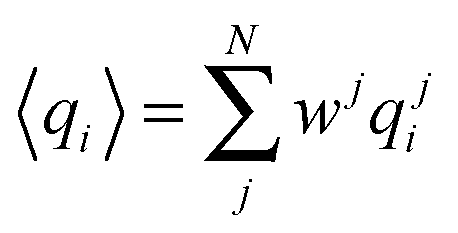 | (2) |
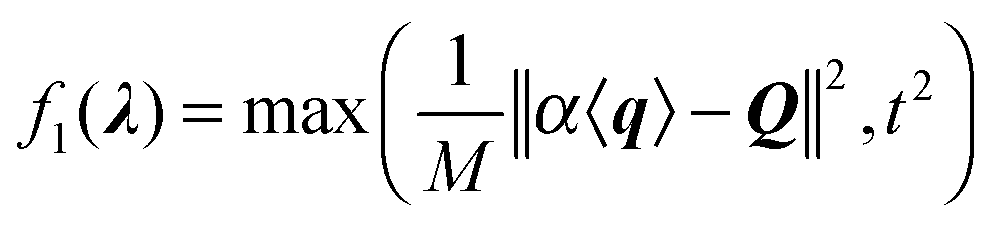 | (3) |
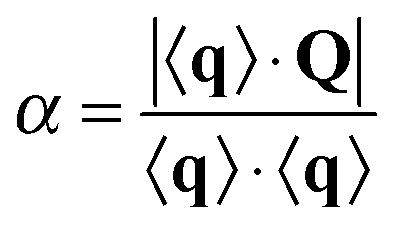 | (4) |
Because the scaling adds one degree of freedom, the set of λ = {λi} that minimize f1 lies on a 1-dimensional curve. Based on the MaxEnt principle, we seek λ that minimally modifies the ensemble. By eqn (1) these are the λ as close as possible to 0. Therefore we add a penalty term:
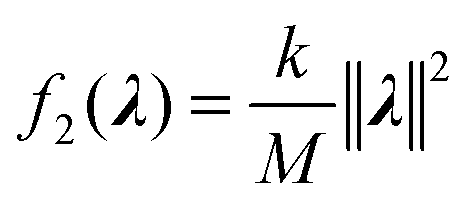 | (5) |
Our implementation converges in less than 10 seconds for the ensembles used in this work in a 1 processor Xeon machine. This is to be compared with the Bayesian method developed by Stultz,36,37 which being their most efficient method takes about 30 minutes in an 8 processor Xeon machine with an ensemble of 299 structures. At the time of writing this paper, Das et al.38 published an interesting paper with a full Bayesian approach (called FitEnsemble) based on Monte Carlo sampling and implemented in pyMC.39 In the results section we compare our method with theirs and we show that the full Bayesian approach does not convey any essentially new information. At present their method cannot deal with scale-invariant quantities such as RDCs, but we do not see any fundamental reason why it could not be extended to treat them and we plan to explore this possibility. That would allow a cleaner way to introduce the uncertainty of RDCs' prediction and the experimental error, which are cumbersome to include in a maximum entropy formalism40 in an ad hoc manner. As the comparison with FitEnsemble38 will show, both of these terms are small for RDCs and the MaxEnt principle results in a fast algorithm. The extension of generative probabilistic models40,41 or maximum likelihood approaches42 to IDPs is also an attractive alternative, but it is beyond the scope of this work to evaluate them. The MaxEnt principle gives results in agreement with the Sparse Ensemble Selection algorithm,43 but the latter is computationally more expensive and needs some further development to be applicable to IDPs.43
Data
As N–H RDCs are the most discussed RDCs for IDPs we focus on these data, but we also explore the additional information carried by Cα–Hα RDCs. We use two kinds of data. First, we test our method with synthetic data, as that allows comparisons with the exact result. Then we apply the method to experimental RDCs to see how it performs. In both cases we use a 53 residue sequence from the nucleocapsid-binding domain of Sendai virus phosphoprotein. This protein has a crucial role in the replication and transcription of the negative strand RNA genome.11,44 The N-terminal domain of this protein is unstructured but contains some partial secondary structure. The sequence of the simulated fragment is FVTLHGAERLEEETNDEDVSDIERRIAMRLAERRQEDSATHGDEGRNNGVDHE (the charges at the end of the sequence were removed as it is part of a larger protein). This fragment corresponds to the residue numbering 458 to 510 in ref. 11. We have analysed only this region as it contains secondary structure elements11,44 that cannot be predicted with a simple force field such as Flexible Meccano.Synthetic data
We run a parallel tempering simulation using the Profasi force field45,46 in the Profasi code47 with 16 replicas, from 270 to 330 K.We take T1 = 325.6 K as our reference or “experimental” ensemble. We calculated the RDCs for 8000 uncorrelated structures with PALES48 using steric alignment, because of the NMR setup used (see ESI† for the PALES options used). Then, we have used the ensembles of structures at T0 = 317.0 K to fit the RDC data at T1.
Because we have simulated both ensembles, we know that the weight of a given structure j with energy Ej from the T1-ensemble at temperature T0 is given by the Boltzmann factor, namely
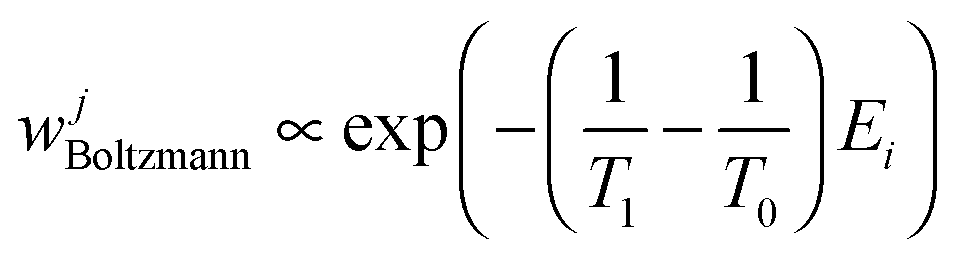 | (6) |
Experimental data
The experimental data for this study were obtained from the work of Blackledge and co-workers.11 In their study they measured N–H and Cα–Hα RDCs and made a statistical analysis to evaluate which regions of the α-helix needed to be added to explain the observed results. When comparing with experimental data, our residue number 1 corresponds to residue number 458 in ref. 11. In this region, 31 N–H RDCs and 25 Cα–Hα RDCs were measured. RDCs for the 11 terminal residues are not calculated nor taken into account for the fit side to eliminate the boundary effects in the RDCs.49,50The most interesting part corresponds to residues 18 to 34, because of their tendency to form partial α-helices, also known as MoRFs.7,8
These data have been simulated with two different coarse-grained force fields: Profasi45–47 and Campari.51 Profasi was chosen for its focus on reproducing the folding behaviour of proteins based on physical terms. We think that using a physics-based force field is important to work with IDPs as knowledge-based force fields are biased towards folded proteins. Profasi has also been applied to IDPs.52,53 The choice of Campari is justified because it was specifically designed to work with IDPs and has been applied in several studies.51,54 The Campari system contains 9 sodium ions to neutralize the charge.
The RDCs were calculated from the PDBs with the PALES software.48 As the alignment media, poly(ethylene glycol), is dominated by steric interactions, we used the steric alignment in PALES (see the ESI† for further details).
Data and code availability
The Profasi and Campari ensembles re-weighted to fit the experimental data have been deposited in the Protein Ensemble Database (pE-DB)55 with the code 4AAB. Because the pE-DB does not support weighted ensembles, the deposited structures are those structures with weights larger than 0.75 (see below).Cross-validation
We have performed two types of cross-validation. First, we use experimental N–H RDCs as a training set and leave the experimental Cα–Hα as the test set. Second, we use a set of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structures as a test set and use a variable number of structures in the training set. We tried the following sizes for the training set: {100, 250, 500, 750, 1000, 2500, 5000, 7500, 10000}. When using smaller sets, MaxEnt could not converge to the requested accuracy in the training set. Note that the training set is not a subset of the test set, and in the final case, we have a total of 20000 structures. We compare the error in the fit in the test set with the λ = {λi} and the scale factor coming from the training set with respect to the error in that training set. This procedure can tell us the adequate size of the training set and an estimation of the error.
000 structures as a test set and use a variable number of structures in the training set. We tried the following sizes for the training set: {100, 250, 500, 750, 1000, 2500, 5000, 7500, 10000}. When using smaller sets, MaxEnt could not converge to the requested accuracy in the training set. Note that the training set is not a subset of the test set, and in the final case, we have a total of 20000 structures. We compare the error in the fit in the test set with the λ = {λi} and the scale factor coming from the training set with respect to the error in that training set. This procedure can tell us the adequate size of the training set and an estimation of the error.
Results and discussion
Size of the ensemble and error estimation
The number of molecules in an NMR experiment is orders of magnitude larger than what can be simulated. How many structures should an ensemble contain? We seek the minimum number of structures needed so that when we add more structures to the ensemble (sampling from the probability distribution given by our force field) the results do not change appreciably.56 This depends on both the property we measure and the shape of the probability distribution of the ensemble. For example, for several folded proteins, a single structure can reproduce a SAXS curve or a diffraction pattern.
Fig. 1 shows the error in the test set when using different number of structures for the training set to fit N–H RDCs with the Campari ensemble. We can see that for training sets smaller than several thousands, the errors in the test set remain very large, and increase as we improve the fit in the training set. In other words, the optimized {λi} are not transferable. This shows us that we need training sets at least of 7000 structures to determine parameters that do not overfit the experimental results until an RMS error of approximately 1 Hz. Because this number is close to the experimental error, we consider ensemble sizes of 7000–10000 as adequate.
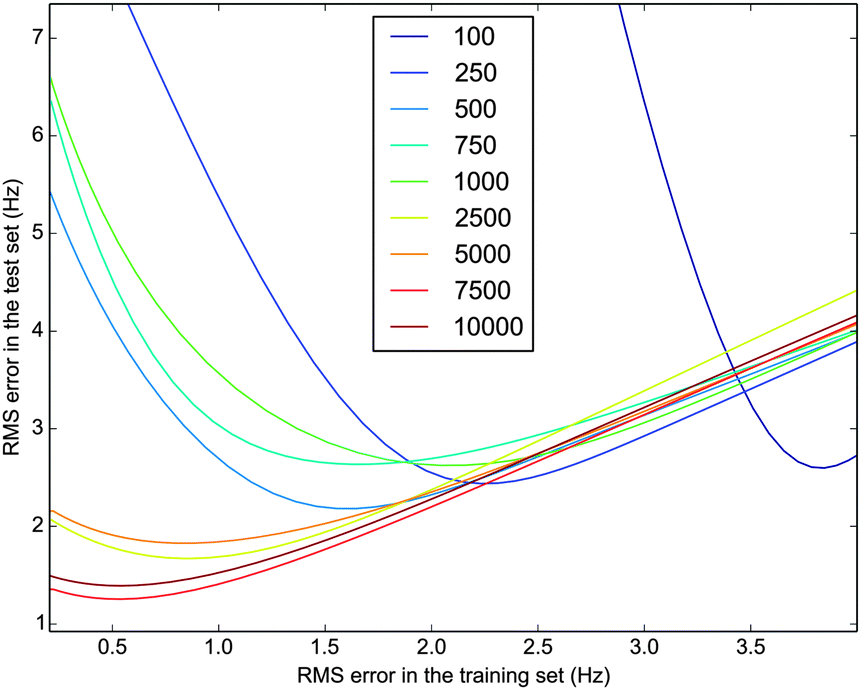 | ||
| Fig. 1 Plot of the root mean square (RMS) error allowed when fitting the training set with respect to the error in the test set. The test set is always of 10000 structures whereas the training set increases from 100 to 10000 structures. Results seem converged above 7500 structures and trying to fit below 1 Hz results in overfitting even for the largest ensembles. | ||
Alternatively, we can estimate the error when calculating the mean value for an RDC: the standard error of the mean. There is certain ambiguity in this value as RDCs can be scaled, but we take here a fixed scale factor obtained from the fit of the 10000 structures (α = 2.08). Fig. S2 (ESI†) agrees with our conclusion that several thousands of structures are needed to get a mean RDC value of the same order of the experimental error. This result is independent of the residue we are measuring: the convergence of all RDCs is the same. Other studies have also found that the underlying ensembles are more heterogeneous than what the measured mean value may suggest.56–58
Several previous studies used a smaller ensemble size31,32 to successfully simulate IDPs. The size of the ensemble in these MD restrained simulations depends not only on the dispersion of the measured property but also on the other parameters used for the restrain, namely its force constant.30,31 These studies run simulations in parallel and were limited by computational resources, but formally their results are exact only when the number of replicas tends to infinity. Other computational methods are expensive, thus limiting the size of the ensembles.4,14,15,17,37 Our method is efficient for thousands of structures so that we prefer to use the full simulated ensemble.
A second important reason to limit the size of the ensembles is to reduce the overfitting. This is an issue when the weights of the structures are the parameters to be optimized, because new structures introduce new parameters, with the obvious risk of overfitting. With the MaxEnt algorithm, the number of parameters is fixed by the number of experimental data and not by the number of structures in the ensemble, which again does not prevent the use of large ensembles.
Synthetic data. What are the RDCs re-weighting?
In this section we analyse to which extent the MaxEnt can recover an unknown ensemble, using some experimental data from that ensemble.To analyse the secondary structure (SS) content of the ensemble, we use SS-map.59 SS-map is a software that plots the SS fraction of a given residue on the y axis and the length of the SS element on the x axis, thus providing a picture of the SS distribution of an ensemble with the information of the cooperativity of different SS of individual residues. By plotting both the fraction of SS and its length, it allows to distinguish, for example, a fully formed helix of 10 residues present 50% of the time from 2 fragments of 5 residues spanning the same range.
The ensemble at T1 represents what in a real situation would be the unknown ensemble, from which we only know the measured RDCs. T0 is a calculated ensemble that presumably will be similar, but does not have to reproduce the data exactly. MaxEnt should be able to reweight the T0-ensemble so that it fits the “measured” RDCs. Will the T0 re-weighted ensemble be more similar to the T1 ensemble?
Fig. 2 shows the SS-map of the synthetic ensembles at temperatures T0 and T1 and the re-weighted T0-ensemble to fit T1 N–H RDCs. Because T0 is a lower temperature, this ensemble presents longer helices. Fig. 3 shows that the application of the MaxEnt principle returns a set of weights that can reproduce the final RDCs.
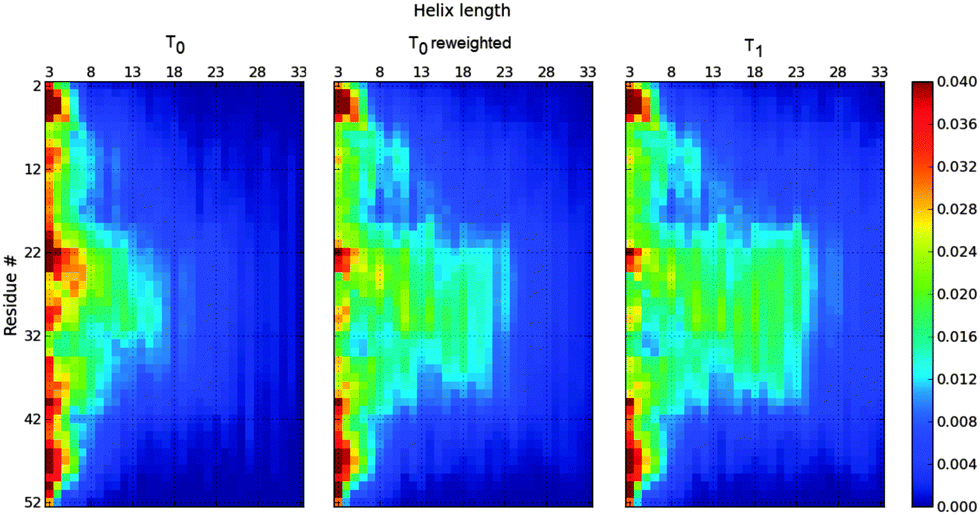 | ||
| Fig. 2 SS-map of the Profasi ensemble at T0 = 317.0 K (right) and T1 = 325.6 K (left) and the T0 MaxEnt re-weighted ensemble to fit T1 N–H RDCs (middle). The latter ensemble has fewer long helices than the T0 ensemble, but it still contains more long helices than the T1 ensemble despite reproducing the RDCs at T1. | ||
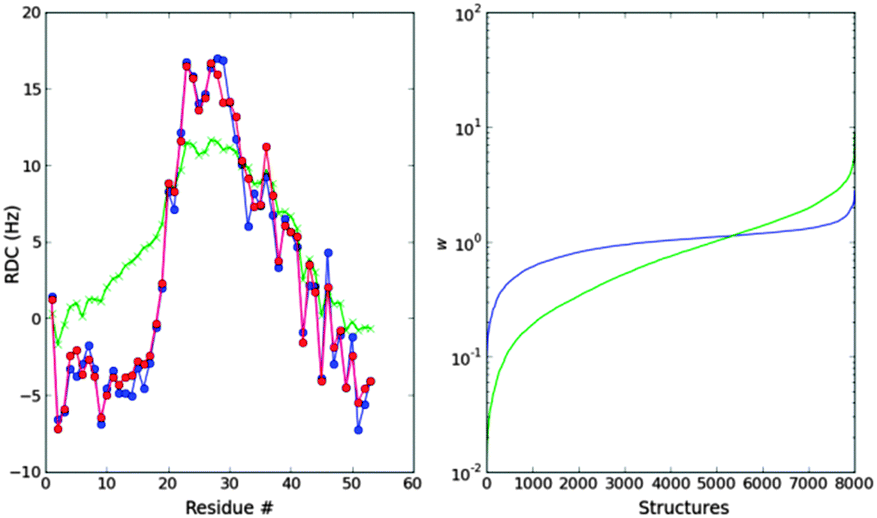 | ||
| Fig. 3 Left: MaxEnt fit of the Profasi T0 = 317.0 K ensemble to the Profasi T1 = 325.6 K average N–H RDCs (blue). The unweighted ensemble (green) has too many long alpha-helices compared to the optimized ensemble (red). Right: distribution of the weights after the ME optimization (blue) compared to the exact Boltzmann weights. | ||
The re-weighting needed to fit the data gives a set of weights that are closer to 1 than the exact Boltzmann reweighting (see Fig. 3). In other words, although the exact Boltzmann weights can reproduce the RDCs of the objective T1-ensemble (see Fig. S3, ESI†), the MaxEnt principle tells us that, based on the data, we do not need to change the weights that much, and that a lower modification of the ensemble is enough and consistent with the data.
As Fig. 3 suggests, the energy distribution of the reweighted T0-ensemble is still closer to that of the T0-ensemble than to that of the objective T1. On average the energy increases but remains lower than the T1-energy distribution (see Fig. S4, ESI†). Fig. S5 (ESI†) shows that most of the structures do not get re-weighted, and only a few do. For those that get re-weighted there is a certain correlation between the Boltzmann re-weighting and the re-weighting given by the N–H RDCs. Of course, if more data are used, for example Cα–Hα RDCs, the reweighting will increase, but even when doubling or tripling the number of experimental data, the degrees of freedom of the ensemble are much higher. We explore this in the following section.
The N–H RDCs do not give information on the energy but on the SS content of the structures; thus we expect the re-weighting to change the SS distribution. Fig. 2 and Fig. S6 (ESI†) reveal that the re-weighting of the data produced goes in the expected directions: the T0 ensemble gets depleted from the long helices that give too large RDCs. But these figures also show that the SS-map of the resulting ensemble remains different from that of the objective T1-ensemble. There are still regions of long helices much less populated in the T1-ensemble. In the following section we will give a reason why the reweighting is not complete and only affects some of the structures.
The results from this section suggest that the RDCs give some information on the SS content of an ensemble, but this information is limited and cannot fully determine the helical propensity nor the helical lengths of an ensemble.
Application to experimental RDCs
We now focus on the reproduction of the experimental RDCs. First we use N–H RDCs and then we include Cα–Hα RDC either as a form of cross-validation or as a source of further structural information. Here, we treat the temperature of the simulation as a parameter, so that we first select the ensemble that best fits the N–H RDCs. For Profasi, this temperature is 325.6 K, and for Campari, the temperature is closer to the experimental one: 300.5 K. As these are the only ensembles we will use from now on, we will refer to them as Profasi and Campari ensembles. Previous studies showed that some force fields need higher-than-experimental temperatures to agree with the data;57 however this adds a parameter that limits the predictive power of Profasi.The Profasi ensemble fits the N–H RDCs reasonably well, but shows a region, around residue 35, of too much alpha helices. The MaxEnt algorithm produces a small reweighting of this ensemble, with most of the structures retaining a weight close to one. Therefore the SS-map of the ensemble is visually indistinguishable from the one shown in Fig. 2.
We can use the Cα–Hα RDCs to cross-validate this refined ensemble. The Cα–Hα RDCs are very similar to the original ones, showing that we did not incur overfitting, but differ significantly from the experimental RDCs (Fig. S7, ESI†). This shows that Cα–Hα and N–H RDCs are not correlated, and depend on different structural properties of the ensemble. The lack of agreement with Cα–Hα indicates that the Profasi ensemble does not correctly represent the real structural ensemble.
As fitting one set of RDCs does not affect the other, we can use MaxEnt to also fit Cα–Hα RDCs The resulting ensemble is reweighted to a stronger extent and correctly fits the 56 RDCs (Fig. S8, ESI†). However Fig. 5 shows that despite the use of the additional 25 Cα–Hα RDCs, the fitted Profasi ensemble has only changed its composition slightly (compare with Fig. 2). This change went in the expected direction, increasing the long helices in the region of residues 20–27 and depleting the ensemble from helices in the region 31–39 (Fig. S9, ESI†). However this change was minor compared to the overall composition of the ensemble. Thus, even the use of 56 RDC data does not qualitatively change the Profasi ensemble and hints that it is still far from the real ensemble. We believe that this information can be used by developers to improve the quality of this force field. The spread of IDPs' energy landscape makes them a good target to find the balance between secondary structure populations and lengths versus random coils.
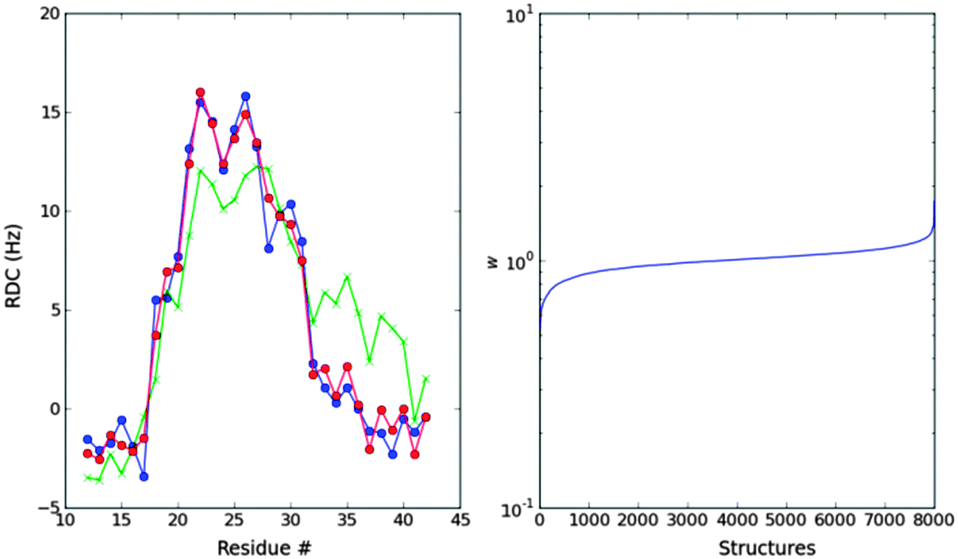 | ||
| Fig. 4 Left: MaxEnt fit of the Profasi T1 = 325.6 K ensemble to the experimental N–H RDCs (blue). The unweighted ensemble (green) has a region of too much alpha-helices compared to the optimized ensemble (red) between residues 32 and 40. Right: distribution of the weights after the optimization. | ||
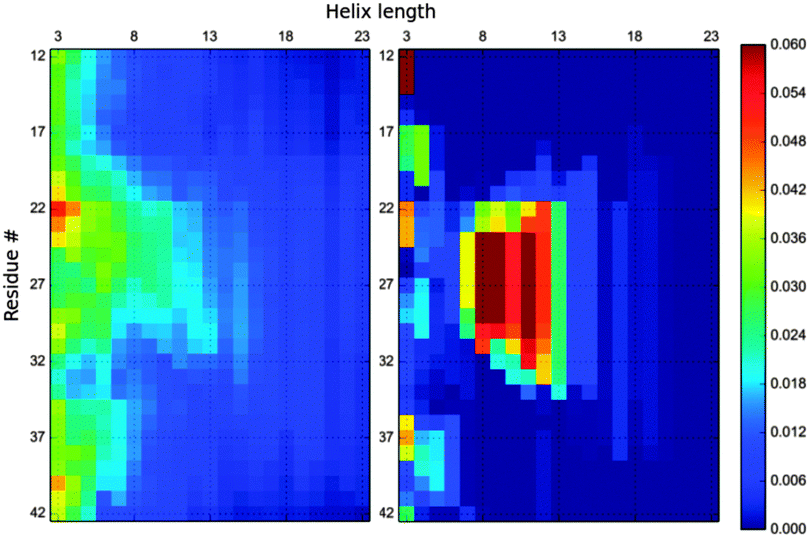 | ||
| Fig. 5 SS-map of the MaxEnt re-weighted Profasi (left) and Campari (right) ensembles using 31 N–H RDCs and 25 Cα–Hα RDCs. Both ensembles fit the experimental RDCs to the same accuracy. | ||
The Profasi ensemble differs from the ensemble deduced by Blackledge and co-workers,11,44 which was mainly composed of random coil regions and three long helices. Their helices add up to 75% of the ensemble, and the longest helix has a population of 11% and ranges from residue 20 to 35. The robustness of their choice was checked by statistically significant improvement compared to other helical combinations. Despite Profasi being able to reproduce the folding of peptides and small proteins ab initio,45,60 it does not predict the long helical elements suggested by Blackledge and co-workers.
The introduction of the experimental data does not reweight all the structures equally, because the weight of a structure depends on its RDC values.
The set of RDCs forms a 31-component vector that is difficult to compare to weight of the structures. We can compress the information of this vector in its root-mean-square (RMS) value. If we plot the optimized weights vs. the RMS of the RDC vector for each structure, a clear trend appears (Fig. 6): the higher the RMS(RDC) the more reweighted the structure is. This makes sense, as reweighting a structure with small RDCs does not improve the fit. In other words, MaxEnt (or any other fitting procedure) is blind to structures that have low RDCs. Because RDCs can be scaled, “low” or “high” RDC refers to the value with respect to the other structures. As is well known, large RDCs correspond to long helices, and these structures are the ones MaxEnt finally re-weights to a larger extent.
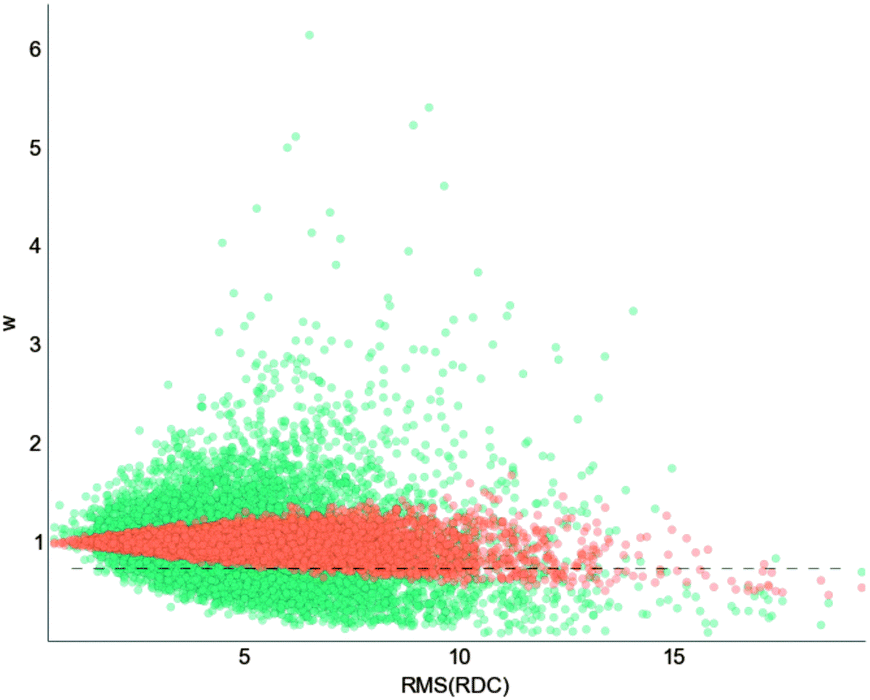 | ||
| Fig. 6 Optimized weights for the Profasi ensemble to fit the experimental RDCs. The x-axis represents the root-mean-square of the RDCs for each of the 8000 structures, showing that the structures that get significantly reweighted are the ones that have large RDCs. When using only N–H RDCs (orange) the reweighting is smaller than when also using Cα–Hα RDCs. The dotted lines are set at w = 0.75, and define a fraction of structures that, if removed, improve significantly the fit. See the text for more details. | ||
Only 208 structures out of 8000 have a weight lower than 0.75 (see Fig. 7) when fitting N–H RDCs. Just by removing these structures from the ensembles and letting the others unchanged, the fit is almost as good as the optimized one in Fig. 4 (RMSD = 1.96 Hz compared to the optimized 1.00 Hz, Fig. S10, ESI†). The SS-map of these structures (Fig. S11, ESI†) reveals that these 208 structures are mainly long helices in the region of residues 32–40, just where the original Profasi ensemble gives RDCs that are too large. Thus the MaxEnt re-weighting agrees with our biophysical intuition.
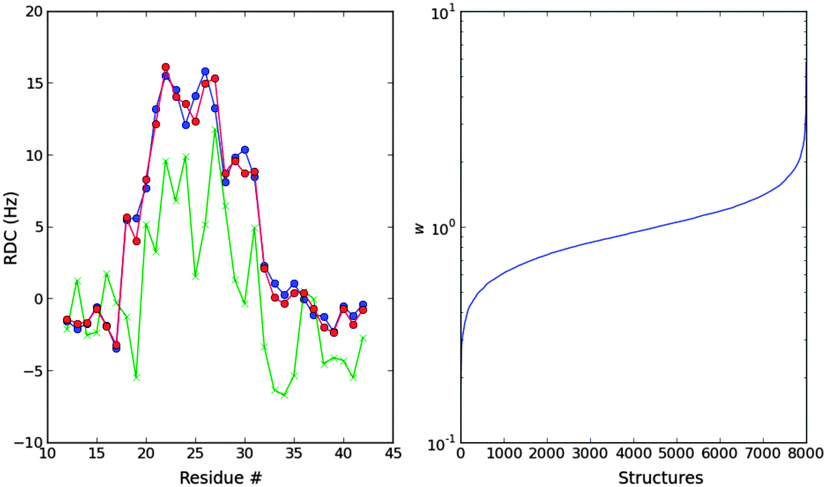 | ||
| Fig. 7 Left: ME fit (red) of the Campari ensemble to the experimental RDCs (blue). The unweighted ensemble is shown in green. Right: distribution of the weights after the optimization. | ||
We now turn to the comparison with the Campari ensemble. This comparison is illustrative because it allows disentangling the fitting procedure with prior distribution of the ensemble. Indeed, the comparison we did with Blackledge and co-workers was comparing a different ensemble and a different fitting procedure. This is a common practice in this field: different groups have developed sampling force fields and fitting procedures and the results contain information of both. For example, Forman-Kay group results are based on their ENSEMBLE selection procedure15,17 from a TRaDES force field16,20 generated structures. The present comparison will shed light on the information RDCs provide giving two different ensembles and the same fitting procedure.
The temperature of the Campari force field is better defined than that of Profasi, because the best fitting temperature corresponds to the experimental temperature. However, the initial ensemble has a worse agreement with the experimental N–H RDCs and therefore it needs a larger re-weighting (Fig. 7).
The secondary structure of this ensemble is considerably different from that of Profasi. It lacks the very abundant short helices of the Profasi ensemble and contains mainly helical fragments in the region of residues 22–32. This is, indeed, the region that the RDCs suggest should have helical fragments, and the region where Blackledge and co-workers deduced the helices were. There is a quantitative difference because the amount of helices in the Campari ensemble is lower than that obtained by Blackledge11 (see also Fig. 4 in ref. 59). However, it is true that both convey a similar ensemble, whereas the Profasi one is qualitatively different. Despite the differences, the Campari and the Profasi ensemble to fit N–H RDCs have similar scaling factors (α = 3.97 and 3.67, respectively).
As before, the initial ensemble is similar to the optimized one, so that because the original Profasi and the Campari ensemble differ, the optimized ensembles still differ, even qualitatively. Even using the same fitting procedure, the starting ensemble has a pervasive influence in the optimized one. This is because the MaxEnt principle minimizes the modifications to the original ensemble, but this is a positive quality because it avoids overfitting or biasing the optimization procedure.
Again, we can introduce the Cα–Hα RDCs to increase the number of experimental data. As with Profasi, the reweighting increases, but the final ensemble is qualitatively very similar to the original. The cross-validation with Cα–Hα RDCs shows that the Campari predicted values are closer to the experimental ones. In spite of being closer, the N–H RDC reweighted ensemble does not improve the Cα–Hα (Fig. S7, ESI†) in agreement with the results of Profasi, and suggesting that the Cα–Hα are independent of the N–H RDCs.
Despite the difference between the Campari and Profasi ensemble, it is worth emphasizing that both are able to reproduce the positive N–H RDCs in the central region, and that the MaxEnt re-weighted ensemble does not differ significantly from the original ones. This may seem disappointing – if we expected them to collapse to the same final ensemble – but it also shows that the initially generated ensembles are physically reasonable. Based on the relation between energy and probability ΔEi = −RTlog(wi/w0i), where w0i = 1/N, the energy difference for a reweighting of 0.5 is only 0.4 kcal mol−1. Unfortunately, if we want to predict secondary structure elements we need these force fields to do better, and the RDC data can be used to improve them. The weight distribution of IDP structures is not peaked as with folded proteins, and thus can be easily reweighted to fit experimental data. Therefore agreement with experimental data does not guarantee a real structural ensemble. If we expect insights from the simulated ensembles we need force fields to have more predictive power. Campari seems to be more successful in this respect.
The Campari ensemble is “simpler” to interpret, but this does not seem to us a valid reason to favour it. In contrast, the Profasi ensemble needs less re-weighting and thus has more predictive power. It is true, however, that the use of an artificially high temperature in the Profasi ensemble is introducing a parameter that the Campari force field predicts to a good accuracy and this can also be the cause for the higher errors of the Cα–Hα in the Profasi ensemble. The Profasi temperature was originally defined as the correct scaling parameter of the energy to reproduce the melting temperature of the Trp cage peptide.45 For IDPs maybe this parameter can be slightly scaled and it is then transferable to other sequences or maybe rescaling some of the energy terms results in a shifted temperature. Further systems need to be tested but our preliminary results suggest that the higher temperature is transferable among IDPs.
If, as before, we remove the structures that have w < 0.75 and leave the remaining unweighted, the fit of the Campari ensemble is very good (Fig. S12, ESI†). In this case, the number of structures removed is larger, 2074 out of 8000 (Fig. S13, ESI†). As with the Profasi ensemble, the structures that get a larger re-weighting are the ones that have larger RDC norm. The consistency of the re-weighting starting from different ensembles with different RDCs strengthens our confidence in the validity of the MaxEnt algorithm that we present.
Ideally, one wishes to start with a large pool of structures and let the data select the ones that agree with the ensemble. Different initial distributions should swamp to the same re-weighted distribution. Unfortunately, this is not the case: not even for folded proteins!41 RDCs do not convey enough information to make the initial distribution irrelevant. Our perspective is that the biophysical community has made heroic efforts in developing experimental techniques to probe IDPs, and then has hoped the data to speak by themselves, overlooking the influence of the prior distribution that the force fields produce.
Profasi and Campari can predict secondary structure elements in IDP ensembles based only on first principles, i.e. they can go beyond random coil force fields. But the ensembles they generate are different, and the RDC fitting cannot make them equal, not even similar. They do have an influence on the final ensemble that can fit the RDC data. This is not to say that the RDCs are not informative, but that the ensembles that fit the data combine the information from the RDCs with that of the force fields. Efforts should be made to improve both experimental methods and force fields. Indeed, we believe that the efforts in the latter field lag behind the experimental developments attained in the IDP world.
Comparison with FitEnsemble
The recent publication of FitEnsemble,38 a method to reweight calculated ensembles to experimental data, prompted us to compare this approach with ours. The advantage of FitEnsemble is that it is a fully Bayesian approach. It is one order of magnitude slower than MaxEnt, but that involves times of a bit more than a minute, which is still very competitive. The problem is that it cannot work with scale invariant quantities such as RDCs. Here we take the scaling factor of the optimized ensemble with MaxEnt to compare both methods.The agreement with both methods is very high (Fig. 8). We also see that the uncertainty in the weights is low compared to its dispersion. That confirms our assumption that this is not a key parameter. We found that the resulting FitEnsemble fit has much lower errors than the introduced experimental uncertainty. In particular, for an uncertainty of 1 Hz, the fit has a root-mean-square error of 0.2. Therefore we optimized our MaxEnt to a threshold of 0.1. For the FitEnsemble, we used a regularization strength of 3, as suggested by the authors but we checked that values of 0.3 and 30 essentially produced the same average results and the same dispersion.
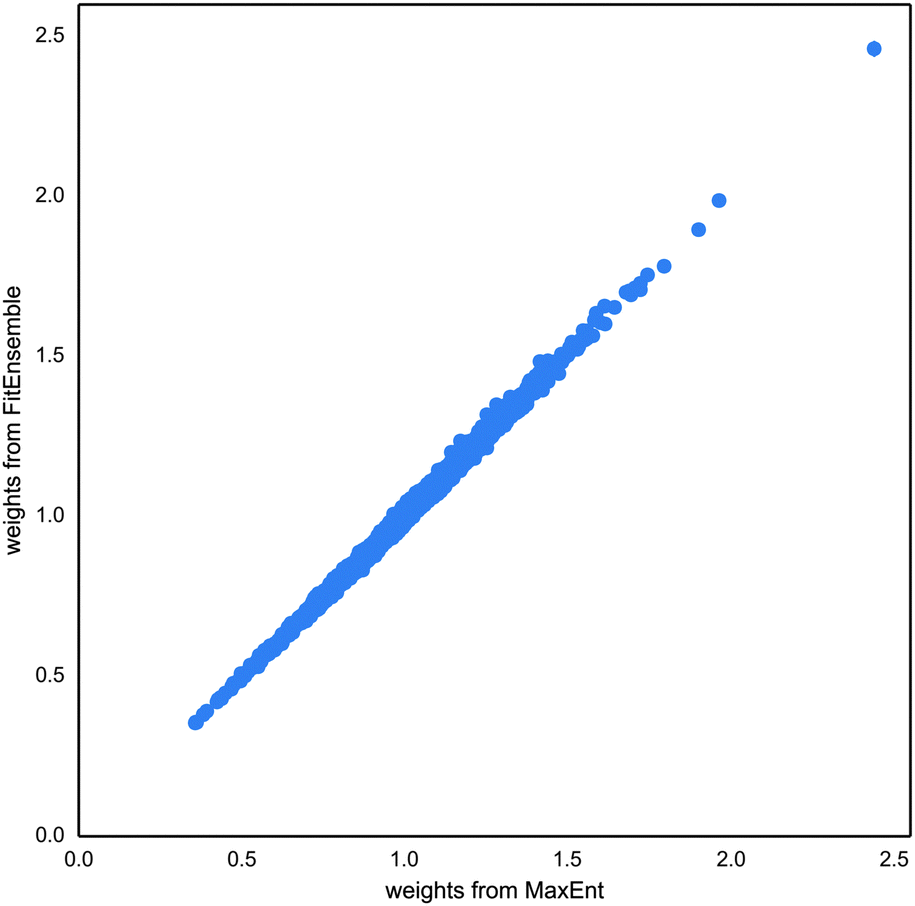 | ||
| Fig. 8 Comparison of the fitting of MaxEnt and FitEnsemble.38 FitEnsemble results include the estimated error from the Bayesian procedure, but it is of the order of the point size. | ||
The extension of FitEnsemble to include a scale parameter seems to be an interesting approach. Still, questions about the convergence of MCMC for RDC ensembles need to be addressed, as well as ensuring that it remains a computationally affordable method.
Conclusions
We present an algorithm based on the maximum entropy principle, which minimizes the information introduced in the fitting of experimental data to a given ensemble. We adapted the algorithm to work with scale invariant measures, such as RDCs. The algorithm is implemented in an open source code freely available.27 The advantage of our method is that it can be used by different experimental groups using different ensembles, as it can use any given set of structures. It can use thousands of structures and converges in a few seconds. It also avoids the risk of overfitting, as the number of parameters depends only on the number of experimental data, and not on the number of structures in the ensemble. Cross-validation shows that more than 7000 structures need to be used to get errors close to the experimental errors of 1 Hz.It has been claimed that RDCs are one of the best probes of IDPs' residual secondary structure,12 but other studies have questioned the relevance of RDCs in IDP modelling.17 Our results, with both a synthetic and an experimental data set, suggest that RDCs can shift the ensembles' secondary structure composition, but only to a limited extent. Different sets of RDCs – N–H and Cα–Hα – give complementary information and improve the reweighting; however the vast conformational space that IDPs can sample makes it a complex case of inferential structure determination,22 so that even with the large number of RDC experimental data, the amount of data is sparse compared to the size of the ensemble.40
Neither all-atom nor coarse-grained force fields have the precision to describe an IDP ensemble,61 as errors of 1 or 2 kcal mol−1 can significantly shift the populations of helices or other secondary or tertiary structure elements. Therefore the need to use experimental data to improve these ensembles is mandatory. But the experimental data is insufficient to fully determine this ensemble, and the pervasive influence of the force field cannot be overlooked, if we wish to have consistent representations of IDP ensembles.
Even though both Campari and Profasi predict certain secondary structure elements, their ensembles are qualitatively different. That determines the composition of the MaxEnt reweighted ensembles. The combination of Cα–Hα and N–H RDCs suggests that Campari is more suitable to describe IDPs than Profasi. We still need further work to test other force fields, improve them, and check other complementary sources of data that help up further select the ensembles. One of our future goals is to include SAXS and chemical shifts in our maximum entropy code.
Appendix
Here we derive the expression of the gradient of f1 and f2, needed for their optimization.For the sake of simplicity we will derive the gradient of f1 piecewise. We only consider when the argument in eqn (3) is larger than the threshold; otherwise the gradient is the null vector. The gradient of the average RDC is
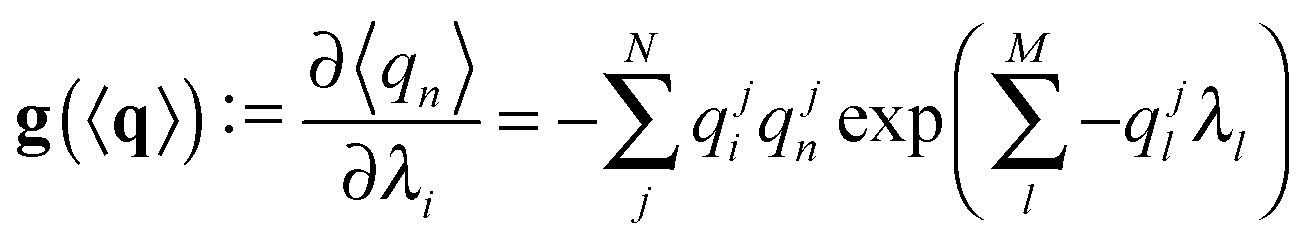
where s is the sign function of 〈q〉·Q. Finally,
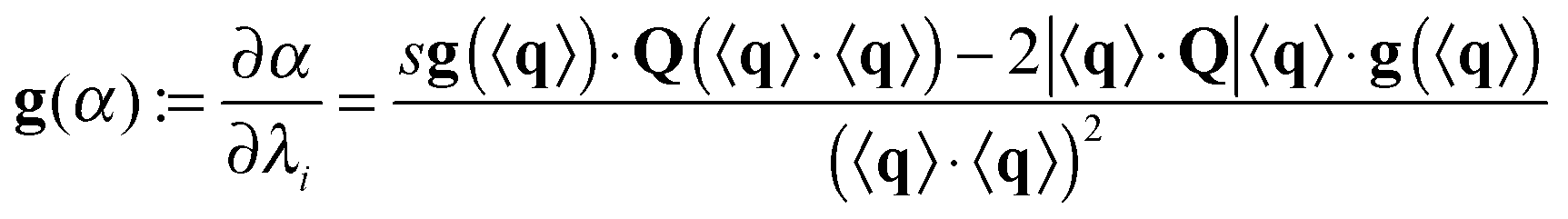
where × represents the outer product. The gradient for f2 is trivial:
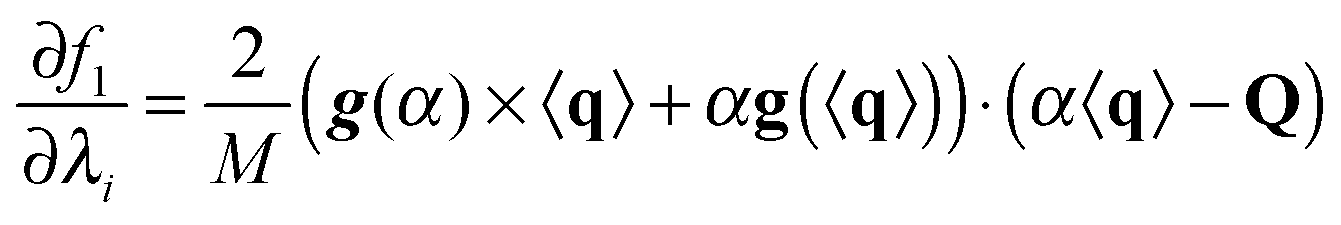
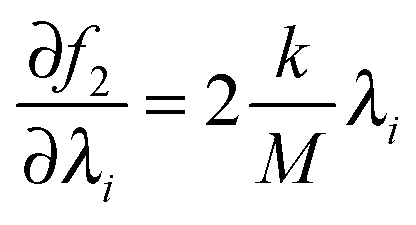
Acknowledgements
We would like to thank X. Salvatella and P. Bernadó for critically reading the manuscript. We acknowledge financial support from the Ministerio de Economía y Competitividad (CTQ2012-33324) and the Generalitat de Catalunya (2009SGR01472). MS-M thanks the Ministerio de Economia y Competitividad for a predoctoral fellowship. We thank the CCUC and the RES (BCV-2013-3-0015) for computational resources.Notes and references
- M. M. Babu, R. van der Lee, N. S. de Groot and J. Gsponer, Curr. Opin. Struct. Biol., 2011, 21, 432–440 CrossRef CAS PubMed.
- V. N. Uversky, C. J. Oldfield and a. K. Dunker, Annu. Rev. Biophys., 2008, 37, 215–246 CrossRef CAS PubMed.
- J. Wang, Z. Cao, L. Zhao and S. Li, Int. J. Mol. Sci., 2011, 12, 3205–3219 CrossRef CAS PubMed.
- C. K. Fisher and C. M. Stultz, Curr. Opin. Struct. Biol., 2011, 21, 426–431 CrossRef CAS PubMed.
- M. Fuxreiter, Mol. BioSyst., 2012, 8, 168–177 RSC.
- V. Iešmantavičius, J. Dogan, P. Jemth, K. Teilum and M. Kjaergaard, Angew. Chem., Int. Ed., 2014, 53, 1548–1551 CrossRef PubMed.
- A. Mohan, C. J. Oldfield, P. Radivojac, V. Vacic, M. S. Cortese, A. K. Dunker and V. N. Uversky, J. Mol. Biol., 2006, 362, 1043–1059 CrossRef CAS PubMed.
- W.-L. Hsu, C. J. Oldfield, B. Xue, J. Meng, F. Huang, P. Romero, V. N. Uversky and a. K. Dunker, Protein Sci., 2013, 22, 258–273 CrossRef CAS PubMed.
- K. Chen and N. Tjandra, Top. Curr. Chem., 2012, 326, 47–67 CrossRef CAS.
- L. Salmon, M. R. Jensen, P. Bernadó and M. Blackledge, Methods Mol. Biol., 2012, 895, 115–125 CAS.
- M. R. Jensen, K. Houben, E. Lescop, L. Blanchard, R. W. H. Ruigrok and M. Blackledge, J. Am. Chem. Soc., 2008, 130, 8055–8061 CrossRef CAS PubMed.
- M. R. Jensen, P. R. L. Markwick, S. Meier, C. Griesinger, M. Zweckstetter, S. Grzesiek, P. Bernadó and M. Blackledge, Structure, 2009, 17, 1169–1185 CrossRef CAS PubMed.
- R. Schneider, J. Huang, M. Yao, G. Communie, V. Ozenne, L. Mollica, L. Salmon, M. R. Jensen and M. Blackledge, Mol. BioSyst., 2012, 8, 58–68 RSC.
- W. Y. Choy and J. D. Forman-Kay, J. Mol. Biol., 2001, 308, 1011–1032 CrossRef CAS PubMed.
- J. A. Marsh, C. Neale, F. E. Jack, W.-Y. Choy, A. Y. Lee, K. A. Crowhurst and J. D. Forman-Kay, J. Mol. Biol., 2007, 367, 1494–1510 CrossRef CAS PubMed.
- J. A. Marsh, J. M. R. Baker, M. Tollinger and J. D. Forman-Kay, J. Am. Chem. Soc., 2008, 130, 7804–7805 CrossRef CAS PubMed.
- J. A. Marsh and J. D. Forman-Kay, Proteins, 2012, 80, 556–572 CrossRef CAS PubMed.
- A. F. Ángyán and Z. Gáspári, Molecules, 2013, 18, 10548–10567 CrossRef PubMed.
- V. Ozenne, F. Bauer, L. Salmon, J.-R. Huang, M. R. Jensen, S. Segard, P. Bernadó, C. Charavay and M. Blackledge, Bioinformatics, 2012, 28, 1463–1470 CrossRef CAS PubMed.
- H. J. Feldman and C. W. V. Hogue, Proteins, 2000, 131, 112–131 CrossRef.
- G. W. Daughdrill, S. Kashtanov, A. Stancik, S. E. Hill, G. Helms, M. Muschol, V. Receveur-Bréchot and F. M. Ytreberg, Mol. BioSyst., 2012, 8, 308–319 RSC.
- W. Rieping, M. Habeck and M. Nilges, Science, 2005, 309, 303–306 CrossRef CAS PubMed.
- E. Jaynes, Phys. Rev., 1957, 106, 620–630 CrossRef.
- S. Pressé, K. Ghosh, J. Lee and K. A. Dill, Rev. Mod. Phys., 2013, 85, 1115–1141 CrossRef.
- W. Boomsma, J. Ferkinghoff-Borg and K. Lindorff-Larsen, PLoS Comput. Biol., 2014, 10, e1003406 Search PubMed.
- B. Róycki, Y. C. Kim and G. Hummer, Structure, 2011, 19, 109–116 CrossRef PubMed.
- http://https://github.com/MelchorSanchez/MaxEnt .
- J. W. Pitera and J. D. Chodera, J. Chem. Theory Comput., 2012, 8, 3445–3451 CrossRef CAS.
- A. D. White and G. A. Voth, J. Chem. Theory Comput., 2014, 10, 3023–3030 CrossRef CAS.
- B. Roux and J. Weare, J. Chem. Phys., 2013, 138, 084107 CrossRef PubMed.
- A. Cavalli, C. Camilloni and M. Vendruscolo, J. Chem. Phys., 2013, 138, 094112 CrossRef PubMed.
- S. Esteban-Martín, R. B. Fenwick and X. Salvatella, J. Am. Chem. Soc., 2010, 132, 4626–4632 CrossRef PubMed.
- R. B. Fenwick, S. Esteban-Martín and X. Salvatella, Eur. Biophys. J., 2011, 40, 1339–1355 CrossRef CAS PubMed.
- S. Meier, S. Grzesiek and M. Blackledge, J. Am. Chem. Soc., 2007, 129, 9799–9807 CrossRef CAS PubMed.
- E. Jones, E. Oliphant, P. Peterson, et al., SciPy: Open Source Scientific Tools for Python, 2001, see http://www.scipy.org/scipylib/citing.html, accessed 28th October 2014.
- C. K. Fisher, A. Huang and C. M. Stultz, J. Am. Chem. Soc., 2010, 132, 14919–14927 CrossRef CAS PubMed.
- C. K. Fisher, O. Ullman and C. M. Stultz, Pac. Symp. Biocomput., 2012, 82–93 Search PubMed.
- K. a. Beauchamp, V. S. Pande and R. Das, Biophys. J., 2014, 106, 1381–1390 CrossRef CAS PubMed.
- A. Patil, D. Huard and C. J. Fonnesbeck, J. Stat. Softw., 2010, 35, 1–81 Search PubMed.
- S. Olsson, J. Frellsen, W. Boomsma, K. V. Mardia and T. Hamelryck, PLoS One, 2013, 8, e79439 CAS.
- S. Olsson, W. Boomsma, J. Frellsen, S. Bottaro, T. Harder, J. Ferkinghoff-Borg and T. Hamelryck, J. Magn. Reson., 2011, 213, 182–186 CrossRef CAS PubMed.
- S. Olsson, B. R. Vögeli, A. Cavalli, W. Boomsma, J. Ferkinghoff-Borg, K. Lindorff-Larsen and T. Hamelryck, J. Chem. Theory Comput., 2014, 10, 3483–3491 CrossRef.
- K. Berlin, C. A. Castañeda, D. Schneidman-Duhovny, A. Sali, A. Nava-Tudela and D. Fushman, J. Am. Chem. Soc., 2013, 135, 16595–16609 CrossRef CAS PubMed.
- P. Bernadó, L. Blanchard, P. Timmins, D. Marion, R. W. H. Ruigrok and M. Blackledge, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17002–17007 CrossRef PubMed.
- A. Irbäck and S. Mohanty, Biophys. J., 2005, 88, 1560–1569 CrossRef PubMed.
- A. Irbäck, S. Mitternacht and S. Mohanty, PMC Biophys., 2009, 2, 2 Search PubMed.
- A. Irbäck and S. Mohanty, J. Comput. Chem., 2006, 27, 1548–1555 CrossRef PubMed.
- M. Zweckstetter, Nat. Protoc., 2008, 3, 679–690 CrossRef CAS PubMed.
- G. Nodet, L. Salmon, V. Ozenne, S. Meier, M. R. Jensen and M. Blackledge, J. Am. Chem. Soc., 2009, 131, 17908–17918 CrossRef CAS PubMed.
- O. I. Obolensky, K. Schlepckow, H. Schwalbe and A. V. Solov'yov, J. Biomol. NMR, 2007, 39, 1–16 CrossRef CAS PubMed.
- A. Vitalis and R. V. Pappu, J. Comput. Chem., 2009, 30, 673–699 CrossRef CAS PubMed.
- X. Cong, N. Casiraghi, G. Rossetti, S. Mohanty, G. Giachin, G. Legname and P. Carloni, J. Chem. Theory Comput., 2013, 9, 5158–5167 CrossRef CAS.
- S. A. Jónsson, S. Mohanty and A. Irbäck, Proteins, 2012, 80, 2169–2177 CrossRef PubMed.
- A. H. Mao, S. L. Crick, A. Vitalis, C. L. Chicoine and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8183–8188 CrossRef CAS PubMed.
- M. Varadi, S. Kosol, P. Lebrun, E. Valentini, M. Blackledge, A. K. Dunker, I. C. Felli, J. D. Forman-Kay, R. W. Kriwacki, R. Pierattelli, J. Sussman, D. I. Svergun, V. N. Uversky, M. Vendruscolo, D. Wishart, P. E. Wright and P. Tompa, Nucleic Acids Res., 2014, 42, D326–D335 CrossRef CAS PubMed.
- R. Bürgi, J. Pitera and W. F. van Gunsteren, J. Biomol. NMR, 2001, 19, 305–320 CrossRef.
- D. S. Weinstock, C. Narayanan, A. K. Felts, M. Andrec, R. M. Levy, K.-P. Wu and J. Baum, J. Am. Chem. Soc., 2007, 129, 4858–4859 CrossRef CAS PubMed.
- B. Richter, J. Gsponer, P. Várnai, X. Salvatella and M. Vendruscolo, J. Biomol. NMR, 2007, 37, 117–135 CrossRef CAS PubMed.
- J. Iglesias, M. Sanchez-Martínez and R. Crehuet, Intrinsically Disord. Proteins, 2013, 1, e25323 CrossRef.
- S. Mohanty, J. H. Meinke and O. Zimmermann, Proteins, 2013, 1–11 Search PubMed.
- M. R. Jensen and M. Blackledge, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1557–E1558 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the PALES calculation and Fig. S1 to S9. See DOI: 10.1039/c4cp03114h |
| This journal is © the Owner Societies 2014 |