
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous imaging of both product ions: exploring gateway states for HCl as a benchmark molecule
M.
Poretskiy
*a,
A. I.
Chichinin
bc,
C.
Maul
a and
K.-H.
Gericke
a
aInstitut für Physikalische und Theoretische Chemie, Technische Universität Braunschweig, Hans-Sommer-Straße 10, 38106 Braunschweig, Germany. E-mail: mikhail.poretskiy@gmail.com
bInstitute of Chemical Kinetics and Combustion, Institutskaya, 3, Novosibirsk, 630090, Russia
cNovosibirsk State University, Pirogova Str., 2, Novosibirsk, 630090, Russia
First published on 5th August 2014
Abstract
Simultaneous imaging of both positive and negative product ions is used to exclusively study photoion pair formation free from interference of competing fragmentation channels. Resonance enhanced multi-photon excitation allows us to interrogate potential energy surfaces for vastly differing molecular geometries. 3D imaging provides complete fragment information. We applied the technique to HCl as a benchmark and identified the gateway state leading to photoion pairs. The approach can easily be applied to any molecule exhibiting a potential with an attractive part at large internuclear distances.
Complete characterization of elementary chemical processes is a major challenge in reaction dynamics. Ideally, it would require determination of the fully quantum state resolved joint reaction probability for product pairs coincidently formed in the same elementary process. However, true coincidence measurements for chemical reactions are extremely difficult to realize, and lifting the requirement for observed products to result from the same elementary process, the next best strategy is the fully quantum state resolved simultaneous observation of all product species formed in the process under investigation. In this context, ion pair production is particularly interesting. It is one of several competing processes occurring in many molecules and has been studied for ground state geometries by photoexcitation in the vacuum ultraviolet (VUV). Based on these studies, we report a novel technique of resonance enhanced multi-photon excitation relying on readily available UV light in conjunction with three-dimensional (3D) imaging for simultaneously monitoring of both oppositely charged ionic products. Thus we can interrogate a wide range of molecular geometries, exclusively address the photoion pair channel in a complex environment of competing processes, and simultaneously observe both products. 3D fragment momentum vector distributions are determined containing complete photofragment information, subsets of which are conventional quantities as kinetic energy distributions, spatial fragment distributions, and anisotropy parameters. We demonstrate power and potential of our method for HCl fragmentation as prototype because its fragmentation and ionization dynamics have intensively been studied and the energy partitioned onto the ionic fragments is discrete and fixed by conservation of energy and linear momentum. From our data, the existence of a Rydberg gateway state on the route to photoion pair generation in HCl is verified. Experimental approach and excitation scheme are easily applicable to any molecule exhibiting potential energy surfaces with attractive parts at large internuclear distances, paving the way for determining joint reaction probabilities of coincidently formed products.
Photodissociating neutral molecules AB either produces neutral fragments A + B or oppositely charged ionic species A+ + B−, as was first observed in 1932.1 Today over 50 molecules are known to fragment into ionic pairs upon photoabsorption. Numerous studies were performed employing vacuum ultraviolet (VUV) single photon absorption,2,3 nevertheless, the issue of whether the formation mechanism is a direct transition from the ground state into the ion pair state or rather a Rydberg state excitation followed by a non adiabatic transition to the ion pair state is still being discussed controversially.4,5
Long range Coulomb attraction between oppositely charged ionic fragments is reflected by shallow slopes of ion pair state potential energy surfaces, and noticeable attractive forces act at large inter-nuclear distances. In contrast, potential energy surfaces of valence states correlating to neutral fragments are characterized by steep slopes at small inter-nuclear distances with significant interaction not extending beyond a few hundred picometers. The energetically lowest ion pair states result from excitation within the valence electron shell. Therefore ion pair states represent a sub-class of valence states. However, ion pair states result from “high energy” excitation within the valence shell and often exhibit similar energies as Rydberg states which result from electronic excitation “outside” the valence shell. High energies and long range attractions of ion pair states make result in numerous avoided crossings between ion pair and Rydberg states of identical symmetry, yielding several double minima potential energy surfaces with quite peculiar dissociation and/or ionization dynamics. As an example, we recently reviewed the intriguing dynamics evolving on an (incomplete) set of HCl Rydberg, valence, and ion pair potentials.6
Shedding light on these complex dynamics is challenging and requires obtaining as much information about a fragmentation process as possible. Ideally, one should monitor all fragments simultaneously or, rather, in coincidence. Also, one should obtain as much information about each single fragment as possible, i.e. one should monitor each fragment's quantum state and three-dimensional momentum vector. Three-dimensional (3D) imaging techniques with the time-resolving delay line anode detector are at hand nowadays that can provide the desired complete data for a single laser shot excitation7–9 or by superposition of sequentially obtained slices.10,11 From the experimental observables, common quantities as e.g. kinetic energy distributions, quantum state populations and spatial anisotropies for particle ensembles are easily obtained by suitable averaging and/or projection.
To this end three-dimensional imaging is combined with resonance enhanced multi-photon ionization. Tuning the resonant excitation laser lets one address single quantum levels of an electronic state (IS1) under investigation while another photon (or several photons) subsequently interrogates the selected state yielding detectable fragments.
 | (1a) |
 | (1b) |
 | (1c) |
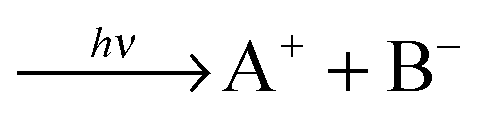 | (1d) |
 | (1e) |
The scope of this letter is to present simultaneous 3D photoion pair imaging as experimental method for detailed studying of intramolecular excitation dynamics among Rydberg and ion-pair moieties of highly excited electronic states of small molecules with focussing on coincident ion pair formation. Data analysis strategies are presented and implications are discussed for the example of ionization and fragmentation of the HCl molecule following a specific vibrational excitation of its double minimum B1Σ+ state. Experimental approach and data analysis can be transferred to any molecule exhibiting suitable ion pair potential energy. A much wider range of the relevant potential energy surfaces can be examined than with conventional one-photon VUV absorption. Simultaneous detection of both anionic and cationic species in ion pair production makes use of the previously demonstrated analytic power of resonance enhanced multi photon imaging letting one explore regions of potential energy surfaces inaccessible for conventional techniques.
The 3D imaging experimental setup has been described in detail elsewhere.8,9 We modified our setup with a second time-of-flight (TOF) detector such that both positive and negative ions can be observed, both separately and simultaneously. The second detector is currently being upgraded to a full 3D detector. The improved set-up differs from former versions primarily by the presence of two TOF spectrometers, each equipped with its own detector. The two spectrometers share the symmetry axis (see Fig. 1) and are the mirror reflection of one another. A pulsed nozzle generated a molecular beam of the precursor substance between the two spectrometers perpendicular to their symmetry axis. For the data presented here the nozzle was operated with pure HCl. The molecular beam was intersected by one laser beam propagating perpendicularly to both the spectrometer symmetry axis and to the molecular beam. Laser radiation at wavelengths around 235 nm was produced by frequency doubling the output of a tunable Nd:YAG laser pumped dye laser and resonantly excited HCl by two-photon absorption at a total energy around 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 cm−1. A third photon from the same laser fragmented the excited HCl molecule to either neutral or ion pair fragments. For ion pair formation the absorption of a third photon was essential since the ion pair threshold at 1390.9 kJ mol−1 (ref. 23) (or 116255 cm−1). Lies far above the two photon energy level. The linear laser polarization could be adjusted between 0° and 90° with respect to the TOF spectrometer axis. Laser beam (X), molecular beam (Y), and spectrometer axis (Z) define the Cartesian coordinate system in the laboratory. Negative ions were registered by a delay line detector (Roentdek DLD-80), after passing one side of the TOF spectrometer. Interference from photoelectrons is avoided by installing permanent magnets providing a homogeneous magnetic field, not influencing the trajectories of heavy chlorine negative ions. Simultaneously, positive ions passed the other side of the spectrometer and were registered by a homebuilt 1D detector (double stage multichannel plate (MCP) assembly + anode). For the time being, three-dimensional images of positive ions were obtained by reversing spectrometer voltages while currently three-dimensionality is being installed in both arms of the spectrometer.
100 cm−1. A third photon from the same laser fragmented the excited HCl molecule to either neutral or ion pair fragments. For ion pair formation the absorption of a third photon was essential since the ion pair threshold at 1390.9 kJ mol−1 (ref. 23) (or 116255 cm−1). Lies far above the two photon energy level. The linear laser polarization could be adjusted between 0° and 90° with respect to the TOF spectrometer axis. Laser beam (X), molecular beam (Y), and spectrometer axis (Z) define the Cartesian coordinate system in the laboratory. Negative ions were registered by a delay line detector (Roentdek DLD-80), after passing one side of the TOF spectrometer. Interference from photoelectrons is avoided by installing permanent magnets providing a homogeneous magnetic field, not influencing the trajectories of heavy chlorine negative ions. Simultaneously, positive ions passed the other side of the spectrometer and were registered by a homebuilt 1D detector (double stage multichannel plate (MCP) assembly + anode). For the time being, three-dimensional images of positive ions were obtained by reversing spectrometer voltages while currently three-dimensionality is being installed in both arms of the spectrometer.
 | ||
| Fig. 1 Double arm ion pair three-dimensional (3D) imaging for simultaneous detection of positive and negative photoions. | ||
The complete information about all three components of the velocity vectors of Cl− and H+ is directly obtained from the experiment (Fig. 2). Thus, the ion-pair photodissociation channel is directly analyzable from the 3D raw data without need for mathematical reconstruction relying on inversion algorithms as it is necessary for a 2D experiment. However, displaying data in two dimensions requires suitable projection. Here, two kinds of data presentation are used: (1) conventional 2D projection into the XY plane (Fig. 2A) corresponding to experimentally observed images in 2D imaging, and (2) meridian projection (Fig. 2B and C) described in detail elsewhere.20 Mathematical transformations describing the two methods are given in Table 1. For comparison, 1D projection onto a single (spectrometer) axis is given corresponding to measured data in TOF spectrometry. Only the r conserving meridian projection copes with the spherical symmetry of the investigated fragmentation process while the other two projection methods rather obey a Cartesian way of thinking unsuited for instructive presentation of experimental 3D imaging data. In any case, one must bear in mind that for a 3D imaging experiment any 2D display projection necessarily goes along with a loss of information, whereas the complete information is always maintained in the original data.
 | ||
| Fig. 2 Two-dimensional projections of 3D images for HCl ion pair dissociation via resonant two-photon excitation of the V1Σ+ (v = 12, J = 0) intermediate state. Laser polarization is vertical in all panels. (A) Cl− conventional 2D plot. (B) Cl− meridian 2D plot. (C) H+ meridian 2D plot. Only the outer intensive ring results from ion pair dissociation. Other features result from dissociation into neutral, electronically excited fragments which are subsequently photoionized or from photoionization of the parent molecule which is subsequently dissociated. | ||

For informative 2D projections, the plane of projection must contain the electric field vector E. In Fig. 2 where IS1 = V1Σ+ (v = 12, J = 0) the electric field vector E is oriented along the y axis. The full 3D fragment distribution is symmetric about the axis defined by the electric field vector, provided all photons have identical polarization 2D projections of Cl− (Fig. 2A and B) and H+ ions (Fig. 2C) resulting from the multiphoton excitation of HCl demonstrate significant angular anisotropy, with HCl excitation and dissociation being of parallel character, i.e. electric field vector, transition dipole moment of HCl, and fragment recoil direction are all oriented parallel to each other. The parallel mechanism is strictly proved by mathematical analysis of the 3D velocity vector distribution giving a precise result of 2.0 for the β anisotropy parameter in eqn (2):
| P(θE) ∼ 1 + βP2(cosθE) | (2) |
Here, θE is the angle between electric field vector of the excitation light and fragment momentum vector and P2 is the second Legendre polynomial. Speed characteristics of photonegative ions are best obtained from meridian projection20 of the 3D distribution (Fig. 2B and C). For Cl− meridian projection yields only one ring with a radius corresponding to the most probable speed. In contrast, the H+ speed distribution exhibits a richer structure resulting from various pathways accessible for production of H+ upon HCl multi-photon ionization. Only one H+ ring results from ion pair dissociation, all others are evidence of competing fragmentation channels initially producing neutral, electronically excited fragments subsequently being photoionized or ionized parent molecules subsequently being dissociated. The Cl− ring is in perfect agreement with the H+ ion pair ring obtained in this and in previous investigations,6,21 considering linear momentum conservation of an initially resting HCl molecule:
| p(H+) + p(Cl−) = p(HCl) = 0 | (3) |
The β parameter for Cl− in Fig. 2B was determined to be 1.92 ± 0.12, and the β parameter for H+ in the outer intensive ring in Fig. 2C is 1.59 ± 0.43. Again, both values agree very well with each other within the experimental error. The experimental error in H+ is slightly larger because (1) the H+ ion pair signal cannot cleanly be separated from the interfering channels mentioned above, and (2) the fast H+ ions travel for the most part of their trajectory in the spectrometer far away from the spectrometer axis, such that off-axis inhomogeneities in the electric fields start to become noticeable.
One dimensional representations of both H+ and Cl− speed distributions are shown in Fig. 3. Speed axes are scaled by the 35Cl to 1H mass ratio of 34.96885/1.0078324 accounting for linear momentum conservation. The conservation of linear momentum together with a β parameter of 2 is proof of the ion-pair channel being the only source of negative chlorine ions. This proof is critical because theoretically a different mechanism of negative ion formation were possible: HCl− decomposition after dissociative photoelectron attachment to HCl.25
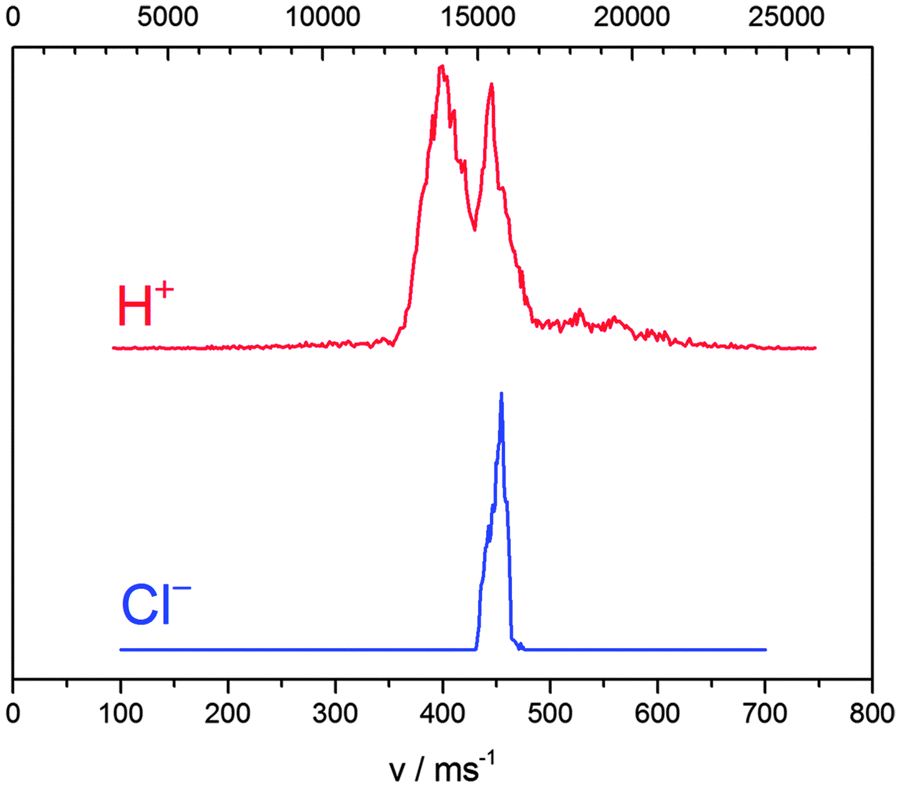 | ||
| Fig. 3 Speed distributions for H+ and Cl− photoions obtained after resonant two-photon excitation of the V1Σ+ (v = 12, J = 0) intermediate state of HCl. The horizontal axis is scaled according to fragment masses: equal positions correspond to equal linear momenta. Only the H+ peak at 15500 m s−1 results from ion pair dissociation. Other features result from dissociation into neutral, electronically excited fragments which are subsequently photoionized or from photoionization of the parent molecule which is subsequently dissociated. | ||
The general picture of HCl ion pair formation is as follows. From the Cl− speed distribution we know, that HCl absorbed exactly 3 photons. The H+ + Cl− term is singular and correlates to the V1Σ+ molecular state, i.e. there is no other way to produce H+ + Cl− except the V1Σ+ molecular state. These two statements make clear that HCl* (V1Σ+) absorbs a third photon and subsequently returns to the V state in a radiationless manner as described in detail below.
An intriguing result proving the outlined mechanism is shown in Fig. 4. We observed a pronounced variation of the total Cl− yield with the vibrational quantum number v′ = 7–14 of the resonantly excited intermediate state V1Σ+. The largest yield occurs for v′ = 12. The yield decreases for adjacent vibrational quantum numbers to zero, whereas a small secondary maximum is observed for v′ = 8. While not shown here, we also observed ion pair formation following initial resonant excitation of the E1Σ+ and g3Σ−(0+) states. We interpret these features as being due to strongly varying Franck–Condon factors between the initially excited V1Σ+ state and a secondary intermediate Rydberg state IS2 acting as gateway state to ion pair fragmentation. We modeled this behavior for the strongest transition X1Σ+ → V1Σ+ (v′ = 12) → IS2 by calculating wavefunctions for all involved states of HCl, which have been reproduced from the references.26,27 We assumed that the intermediate state IS2 acting as gateway state to photoion pair production is formed from the same potential energy surfaces (E1Σ+ and V1Σ+) that also combine to form the double minimum B1Σ+ state. Then the inner wall of IS2 will essentially be contributed to by the ion pair state itself, and an adiabatic transition through the avoided crossing with the Rydberg state forming the IS2 outer part will occur with large probability and result in ion pair dissociation. By tuning the laser, we do not only change the wavelength and the vibrational quantum number v′ of IS1 (the V1Σ+ state), but also the position of the outer turning point of vibrationally excited HCl. The ion pair yield is a most sensitive gauge for the resulting overlap of the IS1 (V1Σ+) wavefunction with the IS2 gateway state wavefunction. Calculation results are depicted in Fig. 5 for v′ = 12. The intermediate state IS1 (V1Σ+, v′ = 12) is resonantly ionized by tuning the excitation laser, while the gateway state IS2 is predominantly accessed from the molecular geometry with maximum overlap of IS1 and IS2 wavefunctions. Ion pair fragmentation occurs after a non-adiabatic transition back to the V1Σ+ state. The return of the molecule from the originally excited V1Σ+ state back to the V1Σ+ state via IS2 is peculiar, but purely incidental because the V1Σ+ state is easily accessible owing to well-characterized and strong transitions from the electronic ground state. It must be noted, however, that the same mechanism occurred for initial excitation of the E1Σ+ and g3Σ− states as well.
 | ||
| Fig. 4 Dependence of the ion yield on the vibrational quantum number v′ of the resonantly excited V1Σ+ (v′) state. | ||
 | ||
| Fig. 5 Dependence of Cl− yield on the third power of the laser intensity measured for IS1 = V1Σ+ (v′ = 12). The inset shows the laser intensity range for which a linear dependence of the ion signal on the third power of the laser intensity is experimentally observed. For larger laser intensities saturation effects are responsible for the deviation of the cubic dependence. | ||
Several more features of the complex fragmentation mechanism of HCl following resonant multi-photon excitation were observed, most importantly the dependence of the ion pair yield (Fig. 5) on laser intensity. This dependence from the third power of laser light intensity is linear only at reduced intensities, reflecting the three photon origin of ion pair formation. At higher intensities the process becomes proportional to lesser powers of the laser intensity owing to saturation. The discussion of the more complicated effects is beyond the scope of this letter. Similar calculations of the nuclear wavefunction overlap integrals as shown in Fig. 6 were performed for all experimentally monitored vibrational levels (cf.Table 2), and the pronounced intensity variation around v′ = 12 as well as the bimodal intensity distribution of Fig. 4 are in accordance with the proposed decay mechanism. Moreover, wavefunction calculations predict the outcome of experiments on hitherto unstudied transitions.
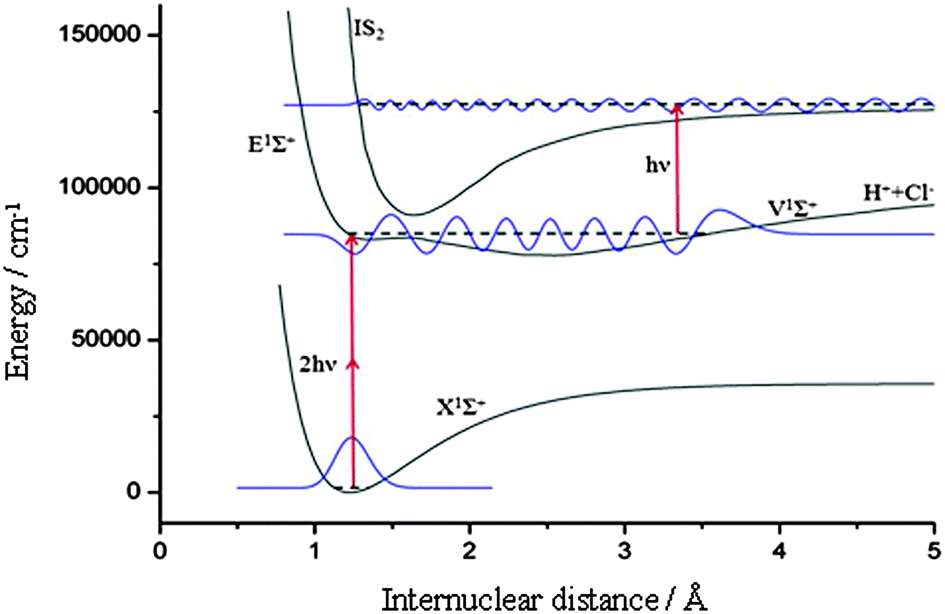 | ||
| Fig. 6 Representation of the photoion pair production process in HCl. The first intermediate state IS1 is resonantly ionized by tuning the excitation laser through the second intermediate state IS2 – the gateway state to photoion pair production. The latter is predominantly accessed from the molecular geometry with maximum overlap of IS1 and IS2 wavefunctions, shown here for the case IS1 = V1Σ+ (v′ = 12). Fragmentation into ion pairs occurs after a non-adiabatic transition back to the V1Σ+ state. | ||
| v | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|
| I v /I12 | 0.0025 | 0.2116 | 0.0576 | 0.0184 | 0.5086 | 1 | 0.2931 | 0.0039 |
As conclusion, investigating ion pair dissociation by studying both positive and negative photoions simultaneously allows one to experimentally select photoion pair production as the reaction of interest among a variety of competing processes generating chemically identical products. Employing resonant multi-photon excitation allows one to enlarge the portions of relevant potential energy surfaces amenable to experimental interrogation. Using 3D imaging provides complete information about fragment quantum states, three-dimensional momentum vector distributions and spatial fragment anisotropies. Thus, simultaneous detection of positive and negative photoions has a very large potential to facilitate the data analysis of ion pair fragmentation processes. Similar studies are feasible for larger molecules, provided similar excitation schemes for intermediate states with large equilibrium internuclear distances exist. More specifically, our results show that intermediate Rydberg gateway states play a decisive role in the formation of photoion pairs in HCl.
References
- A. Terenin and B. Popov, Phys. Z. Sowjetunion, 1932, 2299 Search PubMed.
- J. Berkowitz, in VUV and Soft X-Ray Photoionization, ed. U. Becker and D. A. Shirley, Plenum Press, New York, 1996 Search PubMed.
- A. G. Suits and J. W. Hepburn, Annu. Rev. Phys. Chem., 2006, 57, 431 CrossRef CAS PubMed.
- D. A. Shaw, D. M. P. Holland and I. C. Walker, J. Phys. B: At., Mol. Opt. Phys., 2006, 39, 3549 CrossRef CAS.
- T. Ridley, J. T. Hennessy, R. J. Donovan, K. P. Lawley, S. Wang, P. Brint and E. Lane, J. Phys. Chem. A, 2008, 112, 7170 CrossRef CAS PubMed.
- C. Maul, A. I. Chichinin and K.-H. Gericke, J. Phys. B: At., Mol. Opt. Phys., 2011, 410108 Search PubMed.
- A. I. Chichinin, T. Einfeld, C. Maul and K.-H. Gericke, Rev. Sci. Instrum., 2002, 73, 1856 CrossRef CAS PubMed.
- A. I. Chichinin, T. Einfeld, K.-H. Gericke and C. Maul, in Imaging in Molecular Dynamics: Technology and Applications, ed. B. Whitaker, Cambridge University Press, Cambridge, 2003 Search PubMed.
- A. I. Chichinin, S. Kauczok, K.-H. Gericke and C. Maul, Int. Rev. Phys. Chem., 2009, 28, 607 CrossRef CAS.
- C. R. Gebhardt, T. P. Rakitzis, P. C. Samartzis, V. Ladopoulos and T. N. Kitsopoulos, Rev. Sci. Instrum., 2001, 72, 3848 CrossRef CAS PubMed.
- D. Townsend, M. P. Minitti and A. G. Suits, Rev. Sci. Instrum., 2003, 74, 2530 CrossRef CAS PubMed.
- C. Romanescu, S. Manzhos, D. Boldovsky, J. Clarke and H.-P. Loock, J. Chem. Phys., 2004, 120, 767 CrossRef CAS PubMed.
- S. Manzhos, C. Romanescu, H.-P. Loock and J. G. Underwood, J. Chem. Phys., 2004, 121, 11802 CrossRef CAS PubMed.
- C. Romanescu and H.-P. Loock, J. Chem. Phys., 2007, 127, 124304 CrossRef PubMed.
- A. Kvaran, H. Wang, K. Matthiasson, A. Bodi and E. Jonsson, J. Chem. Phys., 2008, 129, 164313 CrossRef PubMed.
- K. Matthiasson, H. Wang and A. Kvaran, J. Mol. Spectrosc., 2009, 255, 1 CrossRef CAS PubMed.
- A. Kvaran, K. Matthiasson and H. Wang, J. Chem. Phys., 2009, 131, 044324 CrossRef PubMed.
- K. Matthiasson, J. Long, H. Wang and A. Kvaran, J. Chem. Phys., 2011, 134, 164302 CrossRef PubMed.
- J. Long, H. Wang and A. Kvaran, J. Chem. Phys., 2013, 138, 044308 CrossRef PubMed.
- A. I. Chichinin, C. Maul and K.-H. Gericke, J. Chem. Phys., 2006, 124, 224324 CrossRef CAS PubMed.
- A. I. Chichinin, P. S. Shternin, N. Gödecke, S. Kauczok, C. Maul, O. S. Vasyutinskii and K.-H. Gericke, J. Chem. Phys., 2006, 125, 034310 CrossRef CAS PubMed.
- S. Kauczok, C. Maul, A. I. Chichinin and K.-H. Gericke, J. Chem. Phys., 2010, 133, 024301 CrossRef CAS PubMed.
- NIST-JANAF Thermochemical Tables, ed. by M. W. Chase Jr., 4th ed., J. Phys. Chem. Ref. Data 1998, Monograph No. 9.
- G. Audi and A. H. Wapstra, Nucl. Phys. A, 1995, 595, 409 CrossRef.
- Q.-B. Lu and L. Sanche, J. Chem. Phys., 2001, 115, 5711 CrossRef CAS PubMed.
- P. J. Bruna and S. D. Peyerimhoff, in Ab initio Methods in Quantum Chemistry, ed. K. P. Lawley, Wiley, New York, 1987 Search PubMed.
- H. Lefebvre-Brion, H. P. Liebermann and G. J. Vázquez, J. Chem. Phys., 2011, 134, 204104 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2014 |