
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SERS spectroscopic evidence for the integrity of surface-deposited self-assembled coordination cages†
Marina
Frank
a,
Sebastian
Funke
b,
Hainer
Wackerbarth
b and
Guido H.
Clever
*a
aInstitute of Inorganic Chemistry, Georg-August University Göttingen, Tammannstr. 4, 37077 Göttingen, Germany. E-mail: gclever@gwdg.de; Web: http://www.clever-lab.de
bPhotonic Sensor Technology, Laser-Laboratorium Göttingen e.V., Hans-Adolf-Krebs-Weg 1, 37077 Göttingen, Germany
First published on 29th August 2014
Abstract
A series of self-assembled coordination cages [Pd4Ln8] based on a phenothiazine backbone has been investigated by means of Raman spectroscopy in solution and by Surface Enhanced Raman Scattering (SERS) on a nanostructured Au surface. The experiments demonstrate that the cages can be clearly distinguished from their constituting ligands by their Raman spectroscopic signatures. Furthermore, the structural integrity of the interpenetrated coordination cages upon deposition on the Au surface was demonstrated for the first time. The signal assignment of the experimental vibrational spectra was supported by Density Functional Theory (DFT) calculations on suitable molecular models.
The study of self-assembled coordination cages1 is gaining in interest because of their potential for application in selective guest binding,2 stabilization of reactive compounds,3 catalysis4 and the synthesis of redoxactive5 and light-switchable materials.6 Although self-assembled systems are routinely studied in solution or as bulk materials, the examination of surface-confined supramolecular structures has become a popular alternative.7
Phenothiazine and its derivatives have been widely investigated because of their attractive properties in pharmacology8 and their use in charge separation devices.9
Applications in the latter field are based on their good electron donor abilities. For example, phenothiazine derivatives have been used as surface-mounted photosensitizers on semiconducting supports in dye-sensitized and organic polymer solar cells.10 Traditionally, these applications make use of discrete organic building blocks or covalent polymers. When self-assemblies are considered to be used in such a context, however, the surface stability of the supramolecules becomes an important question.
In the present paper we show a detailed experimental study by means of Raman spectroscopy in solution and on nanostructured Au surfaces for a series of recently reported self-assembled coordination cages [Pd4L1–38].11 These closely related interpenetrated double cages contain eight bis-monodentate ligands L1–3, which are based on the heterocycle phenothiazine and differ in their oxygenation state at the central sulfur atom. In total, four square-planar coordinated Pd(II) metal ions are coordinated by these ligands (Fig. 1a). As we showed before, these double cages are capable of binding small guest molecules such as BF4− and halide anions in their three pockets.12 In addition, we showed that S-oxidation of the cages can be achieved, both, in solution or in the solid state by the action of oxidants such as Fe(III) or Cu(II) salts, organic peroxides or molecular oxygen.11
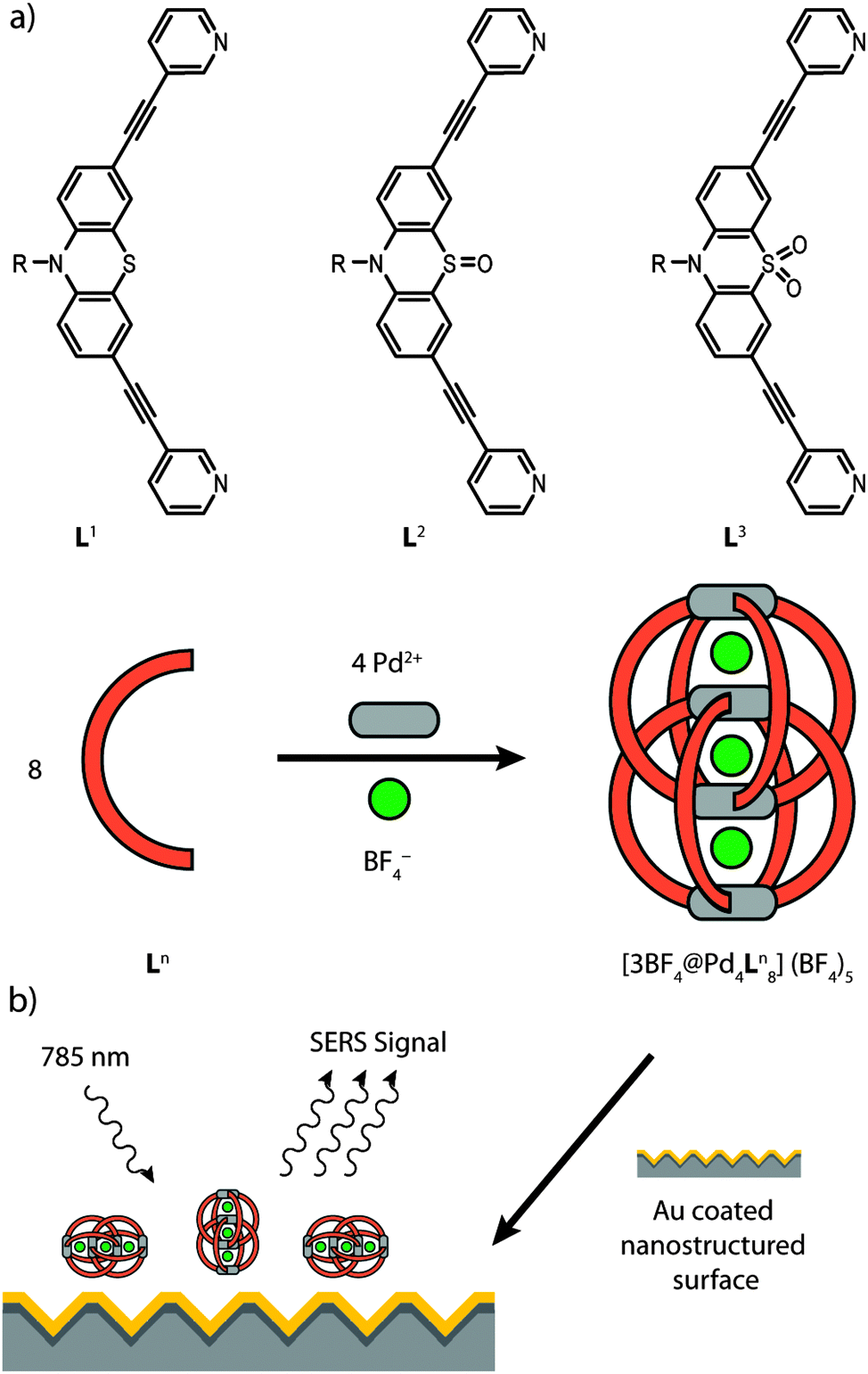 | ||
| Fig. 1 (a) Investigated ligands Ln and their corresponding self-assembled double cages [3BF4@Pd4Ln8](BF4)5 with n = 1–3 (R = hexyl; not all BF4− counter anions are shown). (b) Schematic representation of the SERS (Surface Enhanced Raman Scattering) experimental setup. The size of the double cages (length 2.5–2.6 nm) is exaggerated compared to the size of the nanoscopic surface features (1.4 × 1.4 μm) of the metal substrate. | ||
Surface Enhanced Raman Scattering (SERS) is an invaluable and straightforward technique to approach the question of surface stability of supramolecular structures. SERS allows to overcome some limitations of conventional Raman spectroscopy.13 The enhancement of the signal intensity can be attributed to two effects: an increase in the electromagnetic field intensity near the metal surface induced by the resonant excitation of the surface plasmons of the nanostructured metal and/or a chemical effect induced by a modulation of the electronic polarizability of the bound molecule. In the first case the enhancement is not dependent on the specific interactions between the molecule and the metal, but is strongly related to the characteristics of the metal surface. The contribution from the electromagnetic enhancement can be up to ten orders of magnitude while the chemical enhancement is between 10 and 100.14 Furthermore, redox-driven and electric field induced structural changes on metallic surfaces can be directly monitored by SERS under electrochemical control.15
In the context of macrocyclic and self-assembled supramolecular structures, SERS methods have been used only scarcely so far.16 In order to examine the surface stability of the interpenetrated coordination cages we have compared their vibrational spectra after deposition on a gold surface by using SERS with the ones in solution by using Raman spectroscopy. Care was taken that the characteristic bands of the ligands and the cages are not overlapped by solvent bands, therefore the solution measurements were carried out in two solvents (acetonitrile and acetone, see Fig. S1 in the ESI†). The SERS measurements were carried out using Au coated nanostructured silicon chips. The analytes were deposited as acetonitrile solutions on the surface and the solvent was allowed to evaporate yielding a thin film (Fig. 1b). The complete evaporation of the solvent was indicated by the lack of any acetonitrile bands in the SERS spectra. The plasmonic effect of the nanostructured surface was verified by a comparison with a planar Au surface (see Fig. S2 in the ESI†). Density functional theory (DFT) computations were employed to achieve a partial assignment of the experimentally obtained vibrations for the electronic ground state of the examined molecules.
Interestingly, a comparison of the double cage spectra with the spectra of their constituting ligands reveals a significant difference not only in the intensity distribution of the vibrational bands, but also in the frequencies of some characteristic signals. For example, the vibrational bands in the normal Raman spectrum of [3BF4@Pd4L18](BF4)5 at 2210 cm−1, 1163 cm−1 and 1031 cm−1 are shifted to higher wavenumbers by Δω ∼ 6–9 cm−1 with respect to the free ligand L1 (see Fig. 2). In contrast, the three intense Raman bands at 1568 cm−1, 1578 cm−1 and 1595 cm−1 are displaced to lower wavelengths by 2–8 cm−1. Since these significant differences between the ligand and cage spectra are conserved in the results of the SERS measurements, we conclude that the supramolecular assemblies do not decompose/disassemble on the Au surface under release of the free ligands. Similar results have been obtained for the oxygenated ligand derivatives L2 and L3 and their corresponding double cages (see Fig. 3 and the ESI†). The frequency shifts in the spectra of double cage [3BF4@Pd4L38](BF4)5 are rather small compared to the free ligand L3. Still, the cage spectrum can be unambiguously differentiated from the ligand spectrum.
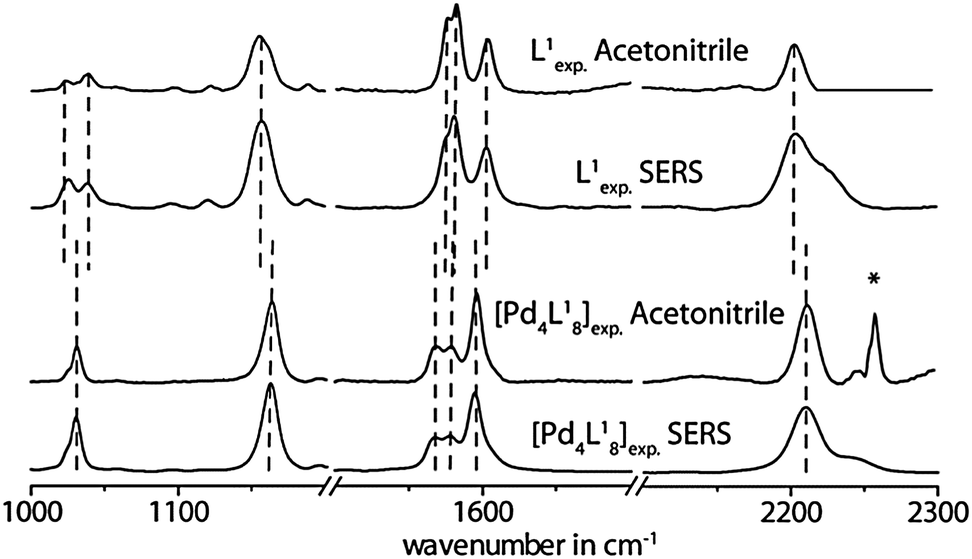 | ||
| Fig. 2 Comparison of experimental Raman spectra measured in solution (acetonitrile) and on the Au surface for the free ligand L1 and the double cage [3BF4@Pd4L18](BF4)5. *Artefact. | ||
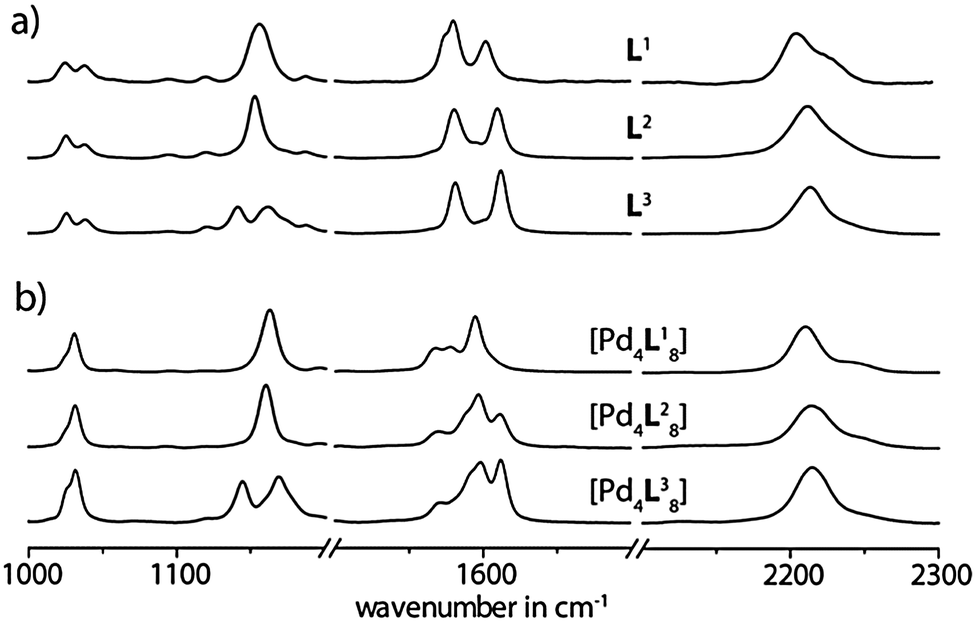 | ||
| Fig. 3 Comparison of experimental SERS spectra for (a) free ligands Ln (n = 1–3) and (b) corresponding double cages [3BF4@Pd4Ln8](BF4)5. | ||
In principal, the SERS effect can cause a shift in the Raman frequencies. This is usually the case if strong interactions between a non-bonding orbital of the molecules and the surface are present. Then even new bands can appear due the formation of charge transfer complexes and the chemical enhancement can be observed. In the absence of such effects the frequencies of the bands in the SERS spectrum are hardly shifted with respect to the Raman spectrum in solution.17 Moreover, the comparison of the band intensities of Raman and SERS spectra provide information about the orientation of molecules on the surface. Modes involving changes in molecular polarizability with a component normal to the surface are the most enhanced. The herein observed similarities of the spectral parameters of the solid analyte (ligand or cage), the analyte in solution and the analyte adsorbed on the nanostructured Au surface indicate weak intermolecular interactions and weak interactions between the analyte and the surface (see Fig. S3 and S4 in the ESI†). Weak interactions between the molecules and the surface are not expected to lead to specific orientations of the adsorbed molecules. This assumption agrees quite well with the fact that even the intensity distribution of the bands of the Raman and SERS spectrum of the cages (ligands) are almost identical, as the similar intensity distribution of the bands points toward a random orientation of the cages (ligands) on the surface as it is illustrated in Fig. 1b.
Although a comparison of the ligand spectra among each others shows a substantial similarity in the intensity distribution patterns of the most prominent bands, some of the signals appear at significantly different frequencies. For example, the band at 2203 cm−1 in the normal Raman spectrum of L1 shifts to 2209 cm−1 for L2 and up to 2216 cm−1 for L3. The resonance around 1142 cm−1 is split in to two signals for the dioxygenated derivative L3, which is not the case for the other ligands. Likewise, the spectra of the cages are distinguishable. In particular, the band assigned to the alkyne group vibration is shifted from 2210 cm−1 for [3BF4@Pd4L18](BF4)5 over 2214 cm−1 for [3BF4@Pd4L28](BF4)5 up to 2215 cm−1 for [3BF4@Pd4L38](BF4)5 in the normal Raman spectrum. As in the case of the ligand L3, the double cage [3BF4@Pd4L38](BF4)5 shows an additional band at 1145 cm−1. Moreover, the distinction between the cages is obvious for the Raman bands around 1600 cm−1. In this area, three bands appear for all three cages. In the case of [3BF4@Pd4L18](BF4)5 these bands are shifted to lower wavenumbers, while the bands for the oxygenated derivatives come at higher wavenumbers. The oxygenated double cages can be distinguished by the intensity pattern of the bands around 1600 cm−1.
For the assignment of the vibrational bands (see Fig. S7–S9 in the ESI†), the spectra were calculated by DFT methods on the B3LYP/6-31G* level of theory as implemented in Gaussian 09.18Fig. 4 compares the experimental SERS spectra of the ligands L1–3 with the results from the calculations. Due to a systematic overestimation of the vibrational frequencies by the used computational method, the calculated spectra had to be corrected by using an appropriate scaling factor or a scaling equation. For this reason, some scaling methods have been tested, such as scaling with a functional- and basis-set-specific constant factor (0.9614 × ωcalc = ωcorr),19 a reported scaling equation for phenothiazines (0.9519 × ωcalc + 23.3 = ωcorr)20 and a self-developed scaling equation (0.9377 × ωcalc + 45.2 = ωcorr), which gave the best fit. Although the first two scaling approaches predicted the frequencies for the signals in the middle parts of the spectra quite well, in the ranges with smaller and larger wavenumbers, however, the deviation of the shifts from the experimental values was more pronounced. The best correlation was achieved by applying the self-derived linear scaling equation and this was finally used for the uniform scaling of all further calculated values. The obtained errors for the most intensive bands were less than 1% compared to the experimental spectra (see Table 1). The intensity patterns were satisfyingly reflected by the calculation method, although the intensities of the bands around 2200–2220 cm−1 were overestimated by the calculations.
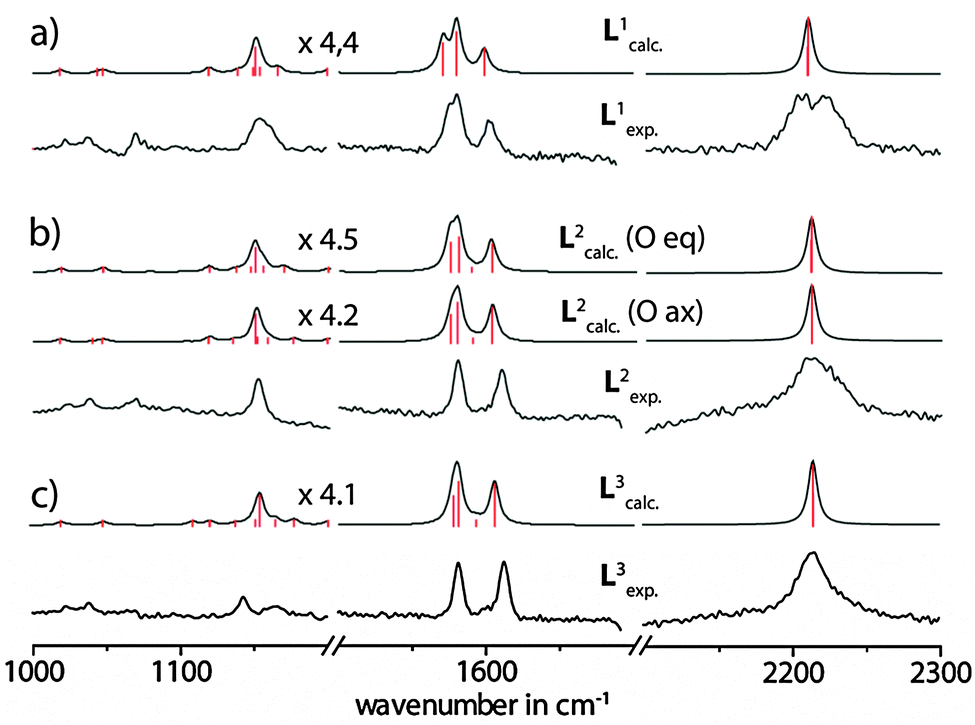 | ||
| Fig. 4 Comparison of the experimental Raman spectra in solution (acetone) and the calculated vibrational Raman spectra for the free ligands (a) L1, (b) L2 and (c) L3. Calculated spectra were subjected to a gaussian peak broadening of 4 cm−1. The intensities of the calculated spectra below 1800 cm−1 were scaled by the factors printed in the figure. | ||
| Ligand L1 | Ligand L2 | Ligand L3 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Experimental frequenciesa in cm−1 | Raman calculated frequenciesb in cm−1 | Experimental frequenciesa in cm−1 | Raman calculated frequenciesb in cm−1 for Oax | Raman calculated frequenciesb in cm−1 for Oeq | Experimental frequenciesa in cm−1 | Raman calculated frequenciesb in cm−1 | |||||||
| Raman | SERS | ω corr | Relative intensity | Raman | SERS | ω corr | Relative intensity | ω corr | Relative intensity | Raman | SERS | ω corr | Relative intensity |
| Experimental intensity description: vs = very strong; s = strong; m = medium; w = weak; vw = very weak; sh = shoulder.a This work.b Scaled frequencies with the scaling equation ωcorr = 45.2 + 0.9377ωcalc. | |||||||||||||
| 1022 m | 1025 m | 1018 | 1 | 1024 w | 1025 m | 1018 | 1 | 750 | 1 | 1022 w | 1025 m | 1018 | 1 |
| 1037 m | 1038 m | 1043 | 1 | 1038 m | 1037 m | 1046 | 1 | 1018 | 1 | 1038 w | 1038 m | 1046 | 1 |
| 1057 sh | 1047 | 1 | 1120 w | 1118 | 1 | 1046 | 2 | 1107 | 2 | ||||
| 1120 vw | 1118 | 1 | 1119 | 2 | 1118 | 1 | 1121 w | 1119 | 1 | ||||
| 1119 | 2 | 1135 | 1 | 1118 | 2 | 1136 | 1 | ||||||
| 1138 | 1 | 1152 s | 1153 vs | 1150 | 15 | 1136 | 1 | 1150 | 2 | ||||
| 1148 | 2 | 1152 | 3 | 1146 | 1 | 1142 m | 1142 m | 1153 | 13 | ||||
| 1154 s | 1156 vs | 1150 | 13 | 1158 | 1 | 1149 | 13 | 1166 w | 1162 m | 1164 | 2 | ||
| 1153 | 3 | 1172 w | 1176 | 2 | 1155 | 4 | 1172 sh | 1176 | 2 | ||||
| 1165 | 3 | 1187 w | 1199 | 1 | 1169 | 3 | 1187 w | 1199 | 1 | ||||
| 1187 vw | 1199 | 1 | 1199 | 1 | 1199 | 1 | 1199 | 1 | |||||
| 1248 w | 1249 | 2 | 1247 | 1 | 1249 | 2 | 1276 vw | 1242 | 1 | ||||
| 1500 vw | 1497 | 1 | 1302 vw | 1303 | 1 | 1321 | 1 | 1298 w | 1302 | 1 | |||
| 1576 sh | 1575 vs | 1570 | 16 | 1326 vw | 1326 | 2 | 1470 | 1 | 1330 w | 1326 | 2 | ||
| 1580 vs | 1580 vs | 1579 | 25 | 1494 | 1 | 1493 | 1 | 1473 vw | 1470 | 1 | |||
| 1601 s | 1602 s | 1598 | 12 | 1581 vs | 1584 vs | 1575 | 14 | 1574 | 18 | 1500 vw | 1494 | 1 | |
| 2203 vs | 2204 vs | 2209 | 44 | 1580 | 25 | 1580 | 23 | 1577 | 13 | ||||
| 2209 | 100 | 1595 w | 1590 | 1 | 1589 | 1 | 1581 vs | 1581 vs | 1580 | 23 | |||
| 3071 | 1 | 1610 vs | 1610 vs | 1603 | 2 | 1602 | 16 | 1592 | 2 | ||||
| 2209 vs | 2212 vs | 2212 | 55 | 2211 | 53 | 1612 vs | 1612 vs | 1605 | 21 | ||||
| 2212 | 100 | 2211 | 100 | 2216 vs | 2213 vs | 2214 | 100 | ||||||
| 3071 | 1 | 2214 | 50 | ||||||||||
| SO frequencies | SO frequencies | ||||||||||||
| 1035 | <1 | 1038 | <1 | 1046 | <1 | ||||||||
| 1060 | <1 | 1069 | <1 | 1254 | <1 | ||||||||
Fig. 5 shows the assignment of the most prominent signals in the spectrum of ligand L1 to the calculated vibrational modes. Obviously, the stretching band of the alkyne linkers is degenerated and is represented by two frequencies at 2209.0 and 2209.4 cm−1. Due to line broadening effects only one signal is observed in the experimental spectra. The next three less intensive bands are found at 1597.9 cm−1, 1569.9 cm−1 and 1150.0 cm−1. These modes consist of group vibrations comprising predominantly the phenothiazine backbone or the pyridine units which is in good agreement with literature reported data for phenothiazine and ethynylpyridine derivatives.21 Similar vibrational modes were obtained in the calculations of the oxygenated ligand derivatives L2 and L3 (see the ESI†). The sulfoxide stretching bands were predicted to be found at 1034 cm−1 and 1060 cm−1 for the axial isomer of ligand L2 and 1038 cm−1 and 1069 cm−1 for the equatorial isomer with respect to the position of the oxygen substituent. The question arose whether the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations alone can be used for the unambiguous discrimination between the three ligand (or cage) isomers? Unfortunately, the SO stretching resonances of L2 are a part of group vibrations together with numerous backbone deformations. For ligand L3, the most prominent sulfoxide vibrations were found at 1046 cm−1 and 1254 cm−1. This is in good agreement with the reported vibrational bands for other oxygenated phenothiazine derivatives.22 Nevertheless, all sulfoxide vibrations are of very low intensity and therefore not very well suited to serve as characteristic and outstanding markers of the ligand systems. Therefore, the assignment of the samples to the ligands and cages in their different oxidation states was rather based on a comprehensive comparison of the spectral features in the range between ∼1000 and 2300 cm−1 as described above.
O vibrations alone can be used for the unambiguous discrimination between the three ligand (or cage) isomers? Unfortunately, the SO stretching resonances of L2 are a part of group vibrations together with numerous backbone deformations. For ligand L3, the most prominent sulfoxide vibrations were found at 1046 cm−1 and 1254 cm−1. This is in good agreement with the reported vibrational bands for other oxygenated phenothiazine derivatives.22 Nevertheless, all sulfoxide vibrations are of very low intensity and therefore not very well suited to serve as characteristic and outstanding markers of the ligand systems. Therefore, the assignment of the samples to the ligands and cages in their different oxidation states was rather based on a comprehensive comparison of the spectral features in the range between ∼1000 and 2300 cm−1 as described above.
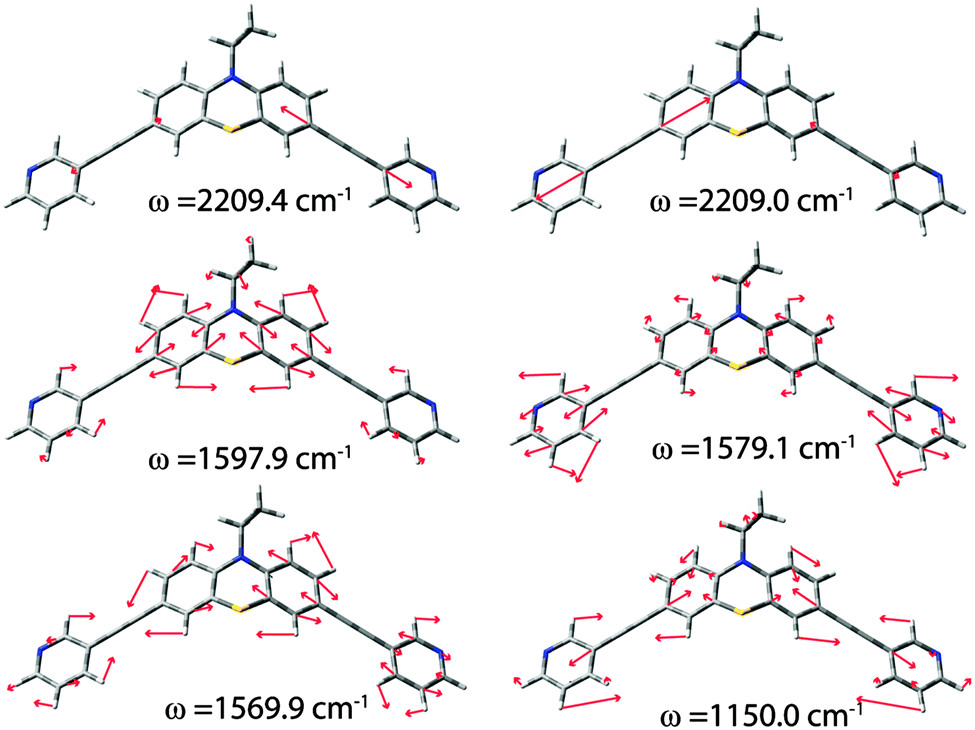 | ||
| Fig. 5 Calculated vibrational modes with the highest intensities for ligand L1. | ||
Materials and experimental setup
Calculations
Density functional theory (DFT) calculations were performed using the Gaussian '09 program with the Becke 3-Parameter Lee–Yang–Parr correlation functional (B3LYP) and the 6-31G* split-valence polarization basis set. No imaginary frequencies were observed for the optimized structures. The applied scaling equation for correcting the computed vibrational frequencies was found empirically. In order to save computation time the hexyl residues were truncated and replaced by ethyl groups.Raman spectroscopy
The Raman and SERS spectra were collected with a standard system (Kaiser Optical System Inc., Ann Arbor, MI, USA); the 785 nm (line width 0.06 nm) GaAlAs diode laser (Invictus, Kaiser Optical Systems, Inc.) light was focused onto the sample. The incident power of the laser emission was ca. 50 mW at the probe head by a recording time of 10 and 5 s with 1 and 20 accumulations for SERS and Raman experiments, respectively. The diffracted light was recorded with a CCD camera (iDus, Andor Technology plc.) The spectral resolution was 5 cm−1. The most important prerequisite when comparing Raman shifts is the reproducibility (or repeatability) of the experiment. Kaiser Optical Systems provide a Raman shift tolerance between ±0.5 and ±1.0 cm−1. Individual system performance will not vary to this extent. Upon calibration, a system should yield Raman shift values repeatable to ±0.1 cm−1.For the normal Raman measurements the concentration was 2.80 mM for the ligand and 0.35 mM for the double cage. The normal Raman spectra have been corrected by subtraction of the solvent bands. Two different solvents (acetonitrile and acetone) were employed to check for overlapping bands in various regions of the spectra. Due to subtraction artefacts only regions with no significant overlap with the solvent bands are depicted and discussed here. For the full spectra and the overlapping regions of the solvents and the analytes see ESI.† In the case of the SERS measurements 1 μL of the solutions were used to coat the Au substrate. The solvent was let to evaporate before the measurements.
All SERS spectra were recorded on a commercially available nanostructured gold surface (Klarite, Renishaw Diagnostics Ltd). This SERS substrate consists of gold-coated periodic square lattice of inverted pyramid pits (∼1.5 μm wide and ∼1 μm deep).17a,23
Due to the background signal, the Raman and surface enhanced Raman spectra have to be corrected by a baseline correction. We have developed an algorithm for baseline correction by connecting cubic splines.24
Conclusions
In this paper we could show that a series of self-assembled, redox-active double cages remains structurally stable upon adsorption on an Au surface. This observation will be helpful for building devices such as flow sensors or organic electronic circuits based on surface-confined functional supramolecular systems. Both, the previously reported host–guest chemistry and the unique electrochemical features of our interpenetrated cages promise to serve as exploitable functionalities in this context. We could show that it is possible to differentiate between the free ligands and the ligands embedded in the supramolecular structures by means of Raman and SERS spectroscopy. In addition, we could demonstrate that vibrational spectroscopy allows for an unambiguous differentiation of the species even within the series of the three structurally very similar cage structures that only differ by the oxygenation state of the ligand's sulfur atoms.Acknowledgements
M.F. thanks the Evonik Foundation for a PhD fellowship. We thank the DFG (CL 489/2-1) and the Fonds der Chemischen Industrie for generous financial support.Notes and references
- (a) M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161 RSC; (b) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825 CrossRef CAS PubMed; (c) D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136 CrossRef CAS PubMed; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (e) T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476 RSC.
- (a) N. Gimeno and R. Vilar, Coord. Chem. Rev., 2006, 250, 3161 CrossRef CAS PubMed; (b) J. L. Sessler, P. Gale, W.-S. Cho and S. J. Rowan, Anion Receptor Chemistry, Monographs in Supramolecular Chemistry, Royal Society of Chemistry, Cambridge, 2006 Search PubMed; (c) S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980 RSC; (d) S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191 CrossRef CAS PubMed; (e) S. Freye, D. M. Engelhard, M. John and G. H. Clever, Chem. – Eur. J., 2013, 19, 2114 CrossRef CAS PubMed; (f) J. M. Dieterich, G. H. Clever and R. A. Mata, Phys. Chem. Chem. Phys., 2012, 14, 12746 RSC.
- (a) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed; (b) D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745 CrossRef CAS PubMed; (c) M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2006, 128, 6558 CrossRef CAS PubMed.
- (a) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed; (b) in Molecular Encapsulation: Organic Reactions in Constrained Systems, ed. U. H. Brinker and J. MieussetWiley, Hoboken, 2010 Search PubMed.
- S. Bivaud, J.-Y. Balandier, M. Chas, M. Allain, S. Goeb and M. Sallé, J. Am. Chem. Soc., 2012, 134, 11968 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319 CrossRef CAS PubMed.
- (a) E. Menozzi, R. Pinalli, E. A. Speets, B. J. Ravoo, E. Dalcanale and D. N. Reinhoudt, Chem. – Eur. J., 2004, 10, 2199 CrossRef CAS PubMed; (b) T. D. Nguyen, H. R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029 CrossRef CAS PubMed; (c) T. Kudernac, S. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402 RSC; (d) N. Miyashita and D. G. Kurth, J. Mater. Chem., 2008, 18, 2636 RSC; (e) A. G. Slater, P. H. Beton and N. R. Champness, Chem. Sci., 2011, 2, 1440 RSC.
- M. J. Ohlow and B. Moosmann, Drug Discovery Today, 2011, 16, 119 CrossRef CAS PubMed.
- (a) D. G. McCafferty, D. A. Friesen, E. Danielson, C. G. Wall, M. J. Saderholm, B. W. Erickson and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8200 CrossRef CAS; (b) M. Borgström, O. Johansson, R. Lomoth, H. B. Baudin, S. Wallin, L. Sun, B. Akermark and L. Hammarström, Inorg. Chem., 2003, 42, 5173 CrossRef PubMed; (c) E. A. Weiss, M. J. Ahrens, L. E. Sinks, A. V. Gusev, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 5577 CrossRef CAS PubMed; (d) H. Tian, X. Yang, R. Chen, Y. Pan, L. Li, A. Hagfeldt and L. Sun, Chem. Commun., 2007, 3741 RSC; (e) K. Kawai, Y. Osakada, M. Fujitsuka and T. Majima, J. Phys. Chem. B, 2008, 112, 2144 CrossRef CAS PubMed; (f) M. Marszalek, S. Nagane, A. Ichake, R. Humphry-Baker, V. Paul, S. M. Zakeeruddin and M. Grätzel, J. Mater. Chem., 2011, 22, 889 RSC; (g) P. K. Poddutoori, A. S. D. Sandanayaka, N. Zarrabi, T. Hasobe, O. Ito and A. van der Est, J. Phys. Chem. A, 2011, 115, 709 CrossRef CAS PubMed; (h) S. Bay, T. Villnow, G. Ryseck, V. Rai-Constapel, P. Gilch and T. J. J. Müller, ChemPlusChem, 2013, 78, 137 CrossRef CAS PubMed.
- (a) R. Argazzi, C. A. Bignozzi, T. A. Heimer, F. N. Castellano and G. J. Meyer, J. Am. Chem. Soc., 1995, 117, 11815 CrossRef CAS; (b) H. Tian, X. Yang, R. Chen, Y. Pan, L. Li, A. Hagfeldt and L. Sun, Chem. Commun., 2007, 3741 RSC; (c) G. Sang, Y. Zou and Y. Li, J. Phys. Chem. C, 2008, 112, 12058 CrossRef CAS; (d) P. Rajakumar, C. Satheeshkumar, M. Ravivarma, S. Ganesan and P. Maruthamuthu, J. Mater. Chem. A, 2013, 1, 13941 RSC.
- M. Frank, J. Hey, I. Balcioglu, Y.-S. Chen, D. Stalke, T. Suenobu, S. Fukuzumi, H. Frauendorf and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 10102 CrossRef CAS PubMed.
- M. Frank, J. M. Dieterich, S. Freye, R. A. Mata and G. H. Clever, Dalton Trans., 2013, 42, 15906 RSC.
- For reviews see: (a) K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, J. Phys.: Condens. Matter, 2003, 14, R597–R624 CrossRef; (b) M. Moskovits, J. Raman Spectrosc., 2005, 36, 485 CrossRef CAS PubMed; (c) W. E. Smith, Chem. Soc. Rev., 2008, 37, 955 RSC; (d) L. Guerrini and D. Graham, Chem. Soc. Rev., 2012, 41, 7085 RSC.
- (a) P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 60 Search PubMed; (b) S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756 CrossRef PubMed.
- (a) H. Wackerbarth and P. Hildebrandt, ChemPhysChem, 2003, 4, 714 CrossRef CAS PubMed; (b) H. Wackerbarth, U. Klar, W. Günther and P. Hildebrandt, Appl. Spectrosc., 1999, 53, 283 CrossRef CAS.
- (a) A. McNally, R. J. Forster and T. E. Keyes, Phys. Chem. Chem. Phys., 2009, 11, 848 RSC; (b) S. Mahajan, T.-C. Lee, F. Biedermann, J. T. Hugall, J. J. Baumberg and O. A. Scherman, Phys. Chem. Chem. Phys., 2010, 12, 10429 RSC; (c) E. H. Witlicki, C. Johnsen, S. W. Hansen, D. W. Silverstein, V. J. Bottomley, J. O. Jeppesen, E. W. Wong, L. Jensen and A. H. Flood, J. Am. Chem. Soc., 2011, 133, 7288 CrossRef CAS PubMed.
- (a) H. Wackerbarth, C. Salb, L. Gundrum, M. Niederkrüger, K. Christou, V. Beushausen and W. Viöl, Appl. Opt., 2010, 49, 4362 CrossRef CAS PubMed; (b) H. Wackerbarth, L. Gundrum, C. Salb, K. Christou and W. Viöl, Appl. Opt., 2010, 49, 4367 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502 CrossRef CAS.
- M. Alcolea Palafox, M. Gil, J. L. Núñez and G. Tardajos, Int. J. Quantum Chem., 2002, 89, 147 CrossRef PubMed.
- (a) D. Pan and D. L. Phillips, J. Phys. Chem. A, 1999, 103, 4737 CrossRef CAS; (b) D. Pan, L. C. T. Shoute and D. L. Phillips, J. Phys. Chem. A, 1999, 103, 6851 CrossRef CAS; (c) H. Jian, J. Xiang, K. Sun, J. Sun, C. Chen, B. Zhou, Y. Liu and G. Xu, J. Colloid Interface Sci., 2000, 229, 212 CrossRef CAS PubMed; (d) R. Millen, D. de Faria and M. Temperini, Vib. Spectrosc., 2001, 27, 89 CrossRef CAS.
- (a) M. Bolboaca, T. Iliescu and W. Kiefer, Chem. Phys., 2004, 298, 87 CrossRef CAS PubMed; (b) M. Bolboaca, T. Iliescu, C. Paizs, F. D. Irimie and W. Kiefer, J. Phys. Chem. A, 2003, 107, 1811 CrossRef CAS.
- N. M. B. Perney, J. J. Baumberg, M. E. Zoorob, M. D. B. Charlton, S. Mahnkopf and C. M. Netti, Opt. Express, 2006, 14, 847 CrossRef CAS.
- H. Wackerbarth, C. Lenth, S. Funke, L. Gundrum, F. Rotter, F. Büttner, J. Hagemann, M. Wellhausen, U. Plachetka, C. Moormann, M. Strube and A. Walte, Proc. SPIE, 2013, 8896, 889609 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further Raman, SERS and calculated spectra. See DOI: 10.1039/c4cp02188f |
| This journal is © the Owner Societies 2014 |