 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing molecular interaction in ionic liquids by low frequency spectroscopy: Coulomb energy, hydrogen bonding and dispersion forces
Koichi
Fumino
a,
Sebastian
Reimann
a and
Ralf
Ludwig
*ab
aUniversität Rostock, Institut für Chemie, Abteilung für Physikalische Chemie, Dr.-Lorenz-Weg 1, 18059 Rostock, Germany
bLeibniz-Institut für Katalyse an der Universität Rostock e.V., Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: ralf.ludwig@uni-rostock.de; Fax: +49 381 498-6524; Tel: +49 381 498-6517
First published on 23rd May 2014
Abstract
Ionic liquids are defined as salts composed solely of ions with melting points below 100 °C. These remarkable liquids have unique and fascinating properties and offer new opportunities for science and technology. New combinations of ions provide changing physical properties and thus novel potential applications for this class of liquid materials. To a large extent, the structure and properties of ionic liquids are determined by the intermolecular interaction between anions and cations. In this perspective we show that far infrared and terahertz spectroscopy are suitable methods for studying the cation–anion interaction in these Coulomb fluids. The interpretation of the measured low frequency spectra is supported by density functional theory calculations and molecular dynamics simulations. We present results for selected aprotic and protic ionic liquids and their mixtures with molecular solvents. In particular, we focus on the strength and type of intermolecular interaction and how both parameters are influenced by the character of the ions and their combinations. We show that the total interaction between cations and anions is a result of a subtle balance between Coulomb forces, hydrogen bonds and dispersion forces. For protic ionic liquids we could measure distinct vibrational modes in the low frequency spectra indicating clearly the cation–anion interaction characterized by linear and medium to strong hydrogen bonds. Using isotopic substitution we have been able to dissect frequency shifts related to pure interaction strength between cations and anions and to different reduced masses only. In this context we also show how these different types of interaction may influence the physical properties of ionic liquids such as the melting point, viscosity or enthalpy of vaporization. Furthermore we demonstrate that low frequency spectroscopy can also be used for studying ion speciation. Low vibrational features can be assigned to contact ion pairs and solvent separated ion pairs. In conclusion we showed how detailed knowledge of the low frequency spectra can be used to understand the change in interaction strength and structure by variation of temperature, solvent polarity and solvent concentration in ionic liquids and their mixtures with molecular solvents. In principle the used combination of methods is suitable for studying intermolecular interaction in pure molecular liquids and their solutions including additive materials such as nanoparticles.
 Koichi Fumino | Koichi Fumino received his BSc and MEng degrees in chemistry from Ritsumeikan University, Japan. He also earned his PhD degree in physical chemistry in 1998. He was then a postdoctoral fellow at Department of Chemistry, University of Virginia, USA. He is currently working as a postdoctoral research fellow in the research group of Ralf Ludwig at the Institute for Chemistry, University of Rostock, Germany. His current research interests are focused on physicochemical properties of ionic liquids and their mixtures. |
 Sebastian Reimann | Sebastian Reimann studied Chemistry at the University of Rostock, Germany, where he received the PhD in Organic Chemistry in 2013 under the mentorship of Prof. Peter Langer and Prof. Oliver Kühn. Meanwhile, he joined the group of Prof. Mukund P. Sibi. at the North Dakota State University, USA. In 2013 he was postdoc with Prof. Ralf Ludwig at the University of Rostock working in the field of ionic liquids. In July 2013 he became the manager for research and development at Evonik Industries AG. |
 Ralf Ludwig | Ralf Ludwig studied Physics at the Rheinisch-Westfälische Technische Hochschule Aachen, Germany, where he received his PhD in Physical Chemistry in 1991 under the mentorship of Prof. Manfred Zeidler. From 1993 to 1995 he was postdoc with Prof. Tom Farrar and Prof. Frank Weinhold at the University of Wisconsin in Madison, USA. Shortly after, he joined the research group of Prof. Alfons Geiger at the University of Dortmund, Germany, where he obtained his habilitation in Physical Chemistry in 1999. Five years later he was appointed professor for Physical Chemistry at the University of Rostock, Germany, where he became chair of Physical Chemistry in 2012. His major scientific activities are in the field of clusters, liquids and solutions. |
1. Introduction
1.1 The role of this perspective
The pioneers of infrared spectroscopy and hydrogen bonding, George C. Pimentel and Aubrey L. McClellan, have stated fifty years ago that “Of all the vibrational degrees of freedom of hydrogen bonded polymers, νσ and νβ are the most interesting.”1 The vibrational modes νσ and νβ describe intermolecular stretching and bending motions between two hydrogen bonded molecules along the bond A–H⋯B, respectively (Scheme 1).2,3 The force constants obtained from these frequencies provide information about the hydrogen bonding potential function as well as being a measure of the bond strength. Moreover, the extent to which these low-energy vibrations affect the band shape and intensity of the bonded A–H stretch (νs) may be determined, and their very important contribution to thermodynamic functions can be calculated. When Pimentel and McClellan issued their statement they thought about intermolecular H-bonds between two or more molecules resulting in chains, rings or three dimensional networks of molecular liquids such as water, alcohols, polypeptides or proteins.1–3 Of course, it should also be possible to observe spectral features which can be related to intermolecular interaction between cations and anions or between ions and solvent molecules, respectively. In principle, low vibrational spectroscopy should provide information about interaction in ionic liquids, ionic liquid mixtures and ionic liquid mixtures with molecular solvents.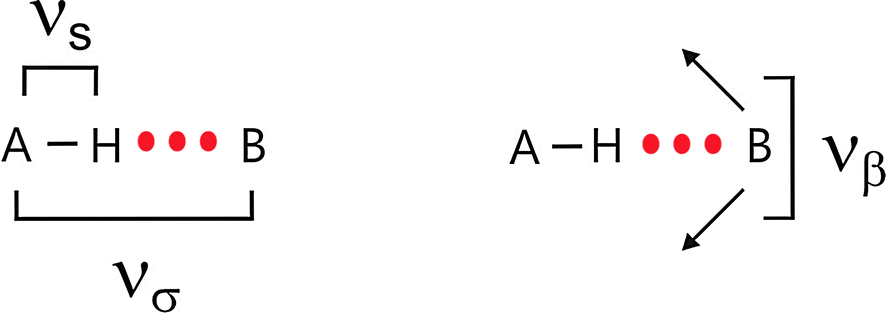 | ||
| Scheme 1 Stretching (νσ) and bending (νβ) vibrational modes of two hydrogen bonded molecules along the bond A–H⋯B. | ||
A couple of years ago we started to study intermolecular interaction in ionic liquids by far infrared (FIR) spectroscopy and later on by terahertz (THz) spectroscopy as well. At the beginning this attempt did not seem to be very promising. Primarily, the low frequency spectra are surprisingly rich in information. Even for light molecules, low-energy intramolecular vibrations as torsions and certain skeletal motions are located in the far infrared region. This was probably the reason why researchers gave up exploring the low frequency range without any reasonable support by theory and simulation. Secondly, ionic liquids (ILs) solely consist of ions and hydrogen bonding was assumed to be negligible. Furthermore, a mélange of Coulomb interaction, hydrogen bonding and dispersion forces further hampers the interpretation of far infrared spectra as well.
Within the framework of this perspective article we show that these problems can be overcome, emphasizing that low frequency vibrational spectroscopy provides important information about interaction strength, the type of interaction and their influence on the properties of ionic liquids. For this reason we have initiated a program for direct spectroscopic observation of cation–anion interaction and in particular of hydrogen bonding in these Coulomb fluids. Unambiguous assignment of the low frequency vibrational bands could be achieved due to the synthesis of ionic liquids including specific interaction sites and supporting density functional theory (DFT) calculated frequencies of related ionic liquid clusters. In a few cases we also performed molecular dynamics simulations (MD) for analyzing the far infrared spectra. Of course, reasonable radiation sources such as high-pressure mercury lamps, better beam-splitters and more sensitive detectors provide strongly improved access to the far infrared spectra these days.4
1.2 The outline of this perspective
The outline of this perspective article is as follows. First we would like to introduce briefly ionic liquids and some of their unique properties. Then we show that far infrared (FIR) and terahertz (THz) spectroscopy are suitable methods for studying the cation–anion interaction in these Coulomb fluids. Mostly the interpretation of the experimental low frequency spectra requires support from theoretical and simulation methods which are therefore introduced as well. It is not the aim of this perspective article to cover the whole families of aprotic ionic liquids (APILs) and protic ionic liquids (PILs) including combinations of all commonly used cations and anions. Instead we picked out special combinations which allow the study of the anion–cation interaction in detail. We present the results for chosen APILs and PILs. We focus on the strength and type of intermolecular interaction and how both parameters are influenced by the character of the ions and their combinations. In particular we would like to show that the total interaction between cations and anions is a result of a subtle balance among Coulomb forces, hydrogen bonds and dispersion forces. This can be nicely demonstrated by switching from APILs to PILs. For PILs we could measure distinct vibrational modes in the FIR indicating clearly the cation–anion interaction characterized by linear and medium to strong hydrogen bonds. Using isotopic substitution we have been able to dissect frequency shifts related to pure interaction strength between cations and anions and to different reduced masses only. In this context we show how these different types of interaction may influence the physico-chemical properties of ILs such as the melting point, viscosity or enthalpy of vaporization. Finally, we demonstrate that FIR spectroscopy can also be used for studying ion speciation. Low vibrational features can be assigned to contact ion pairs (CIPs) and solvent separated ion pairs (SIPs). We were successful in evaluating the spectra quantitatively resulting in equilibrium constants and free energies of the ion-pair species. Ion speciation can now be studied as a function of solvent polarity, solvent concentration and temperature. In conclusion we demonstrate how detailed knowledge of the low frequency spectra can be used to understand the change in interaction strength and structure by variation of temperature, solvent polarity and solvent concentration in IL mixtures.1.3 Low frequency vibrational spectroscopy
Knowledge of intermolecular forces is a requisite for understanding material properties. They determine whether matter sticks together, gases condense to liquids or liquids freeze to solids. To study these forces is in particular interesting for ILs.5–8 Although the structure and properties of these fluid materials are determined to a large extent by the Coulomb forces, hydrogen bonding and dispersion forces can play a crucial role in the understanding of their unique properties. The strong anion–cation interaction in these Coulomb fluids is characterized by extremely low vapour pressures as well as high enthalpy of vaporization.9–16 These unique properties make them attractive for science and technology. As discussed above, the measurement of the interaction between cations and anions in ILs is still a challenge. In principle these interactions can be studied by experimental techniques which cover the frequency range of interest. Meanwhile there is a substantial number of spectra available covering the frequency range of interest between 10 and 300 cm−1, equivalent to 0.3 and 9 THz or 1 and 36 meV. The variety of spectroscopic methods includes optical femtosecond Raman-induced Kerr-effect spectroscopy (RIKES),17–40 terahertz time-domaine spectroscopy (THz-TDS),40–43 as well as conventional steady-state spectroscopic methods such as Raman spectroscopy,44–47 and far infrared spectroscopy (FIR).48–55 Experimental work has been accompanied by numerous theoretical studies including ab initio calculations on ion-pairs or ion-pair clusters, classical molecular dynamics simulations and Car–Parrinello molecular dynamics simulations.56–79 However, in most cases only a combination of experimental and theoretical methods provides a reasonable spectral assignment and reliable interpretation of the low frequency spectra. In this perspective article we discuss the use of conventional steady-state spectroscopic methods such as FIR and Raman spectroscopy for the investigation of intermolecular interaction in ILs including hydrogen bonding. Also TD-THz is applied which does not measure dynamics. Although the measurement is made in the time domain, only equilibrium properties of the sample are probed.80As discussed above, we are mainly interested in the low frequency range between 10 and 300 cm−1 covering nicely the vibrational modes due to intermolecular interaction. For imidazolium-based ILs the cation–anion interaction is characterized by the stretching (νσ) and bending (νβ) modes as illustrated in Scheme 2.
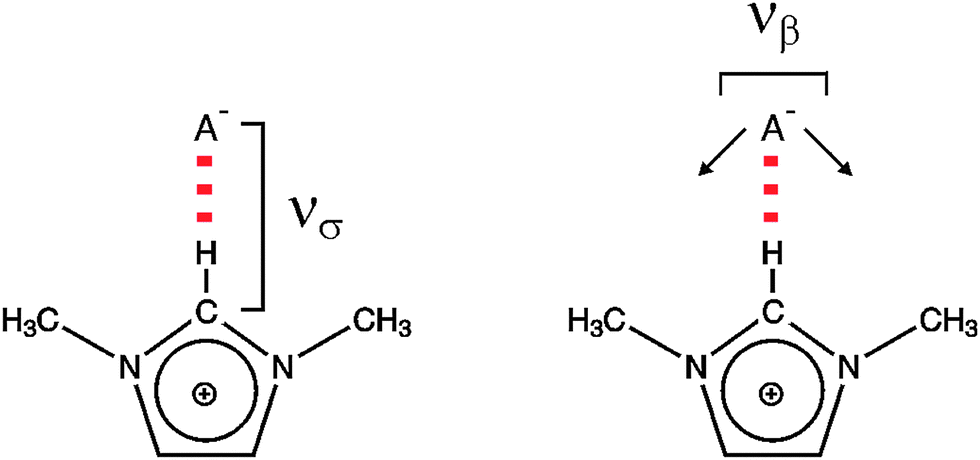 | ||
| Scheme 2 The stretching (νσ) and bending (νβ) modes of the cation–anion interaction for the +C(2)–H⋯A− interaction within 1,3-dimethyl-imidazolium cation. | ||
To which extent this cation–anion interaction in APILs is determined by hydrogen bonding is one of the issues of this perspective article. The situation becomes clearer for PILs, in particular for those which have only one strong interaction site at the cation. As an example the trimethylammonium cation is shown in Scheme 3.
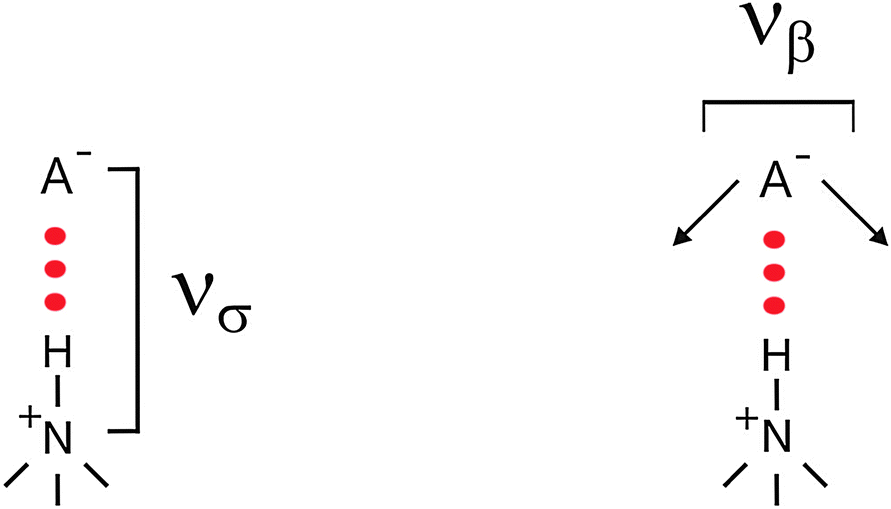 | ||
| Scheme 3 The stretching (νσ) and bending (νβ) frequencies of the cation–anion interaction for the +N–H⋯A− interaction including a trimethyl ammonium cation. | ||
The THz spectroscopy measurements were performed using a TeraView TPS spectra 3000 transmission spectrometer. For all measurements the sample compartment of the spectrometer was purged with dry nitrogen gas. Transmission terahertz spectra were collected from 2 cm−1 to 120 cm−1 at a resolution of 1.2 cm−1. Each rapid-scan spectrum, the average of 1800 co-added scans, took 60 s to record. Three spectra of each sample were recorded. A liquid cell consisting of z-cut quartz windows and a 300 μm spacer was used for all measurements.
The pulsed terahertz attenuated total reflection (ATR) spectra were recorded using the same THz pulsed spectrometer and a silicon ATR module. The silicon ATR crystal is cut at an angle of 35°. These THz ATR spectra could be recorded from 15 to 100 cm−1 (0.2 to 3 THz) at a resolution of 1.2 cm−1.
In all cases the sample temperature was maintained by an external Haake DC 30/K 20 bath chiller and recorded using a NiCrNi thermocouple attached directly to the cell.
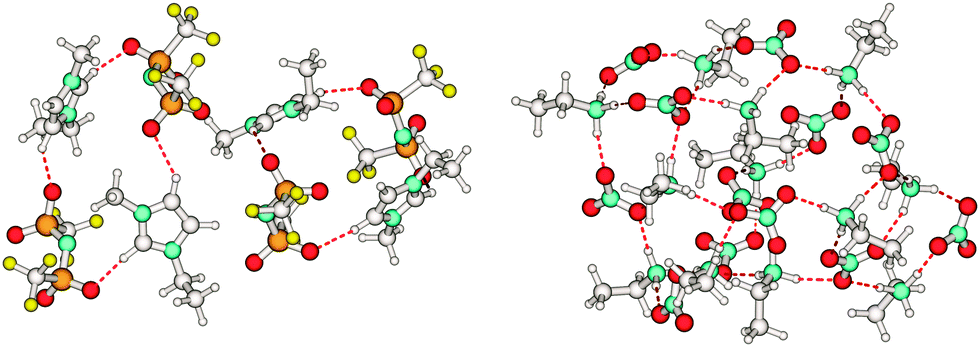 | ||
| Scheme 4 The cation–anion interaction in a tetramer of the aprotic ionic liquid 1-ethyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide [C2mim][NTf2] indicated by the +C(2/4/5)–H⋯O− bonds (left) and in a 12-mer of the protic ionic liquid ethyl ammonium nitrate [EtNH3][NO3] indicated by the +N–H⋯O− bond (right). | ||
For calculating the APIL and PIL clusters between four and six conformers of each cluster were taken into account. No conformational search was undertaken. The starting geometries of the conformers were chosen this way that a maximum of donor and acceptor abilities at the ions were saturated. Using this approach we could minimize surface effects which could lead to different properties compared to those in the bulk phase. Recently it could be shown by the group of Johnson that the main features of the bulk spectrum are already developed in small IL clusters indicating similar binding and interaction in the gas and the bulk phases. This finding strongly supports our procedure. In some of our studies we could also show that from a particular cluster size on (in most of the cases tetramers) the energetic and spectroscopic properties did not change anymore indicating that cooperative effects are sufficiently saturated.81 Finally for each size the lowest energy conformers were used for calculating spectroscopic properties.
For the studies before 2010, ab initio calculations were performed at the Hartree–Fock level with the Gaussian 98 program82 using the internal stored 3-21G basis set. All optimized clusters were found to have only positive frequencies, demonstrating that they are all true equilibrium species on the RHF/3-21G potential-energy surface. The basis set superposition error (BSSE) corrected binding energies and average binding energies per ion of IL clusters usually comprising between two and six ion-pairs have been calculated.83
The zero point energies (ZPE) were also considered. However, both the BSSE and the ZPE corrected energies were negligible for comparison of binding energies of different sized clusters.
Studies after 2010 could calculate geometries and frequencies of all IL clusters at the density functional theory (DFT) level B3LYP, using the internal stored 6-31+G* basis set of the Gaussian 09 program.84 In some cases Grimme's DFT-D3 method was applied for the calculation of dispersion forces.85
It is an ongoing debate whether gas phase like configurations as calculated here are able to reflect bulk phase behaviour as measured here. In our case this approach is justified because the low vibrational modes are mainly determined by local and directional interaction. If we enlarge the calculated clusters by adding further ion-pairs we observe some slight cooperative effects but no significant change in the spectroscopic properties of the configurations. Thus the calculations provide nearly quantitative information.
The molecular volumes discussed in Chapter 5.2 were computed by a Monte-Carlo integration as implemented in the Gaussian programs.82,84 The volume is defined inside a contour of 0.001 electrons Bohr−3 density. The computed volume is only accurate to about two significant figures, but this is sufficient for our purposes. We were interested in comparing volumes of different cations rather than determining accurate absolute values.
There are several ways to calculate the FIR spectra from theory and simulation. Mostly, the vibrational spectra in the low frequency range are obtained using normal-mode analysis within the harmonic approximation as well as from velocity autocorrelation functions in a molecular dynamics trajectory generated from empirical force fields or from ab initio MD simulation (AIMD).62,63 We used empirical force field for calculating the FIR spectra of ILs from the Fourier transform of the velocity autocorrelation function, using eqn (1):
 | (1) |
2. Aprotic (APILs) und protic (PILs) ionic liquids
Ionic liquids are salts in the liquid state. The physical properties of these Coulomb fluids can be simply tuned by varying the size, shape and symmetry of cations and anions. Such a ‘design’ provides novel potential applications for this class of new liquid materials. The structure and properties of ILs are determined by the intermolecular interaction between anions and cations. The molecular interaction is described by a subtle balance among Coulomb forces, hydrogen bonds, and dispersion forces and is thus crucial for understanding the properties of ILs. All the important properties such as structure, diffusion, viscosity, conductivity or melting point depend on these interactions between cations and anions in ILs. Thus, the development of a fundamental understanding of their chemical and physical properties at the molecular level is of current interest. Furthermore, reliable structure–property relationships can help us to synthesize IL compositions possessing a desired set of properties.In addition, ILs have changed from a lab curiosity to materials of tremendous academic and industrial interest. This new and remarkable liquid substance with unique and fascinating properties offers a phenomenal opportunity for new science and technology.5–10 Ionic liquids are of particular interest due to their wide range of possible applications as solvents for reaction and material processing, as extraction media or as working fluids in mechanical applications. Starting in the field of electrochemistry and based upon their wide electrochemical windows and good conductivities, ILs found useful applications in sensors, solar cells, solid-state photocells, batteries, separation devices and as thermal fluids, lubricants, hydraulic fluids, ionogels and fuels.5–9 Hereby, two types of ionic liquids gained increasing interest in science and technology: aprotic (APILs) and protic (PILs) ionic liquids.
2.1 Aprotic ionic liquids (APILs)
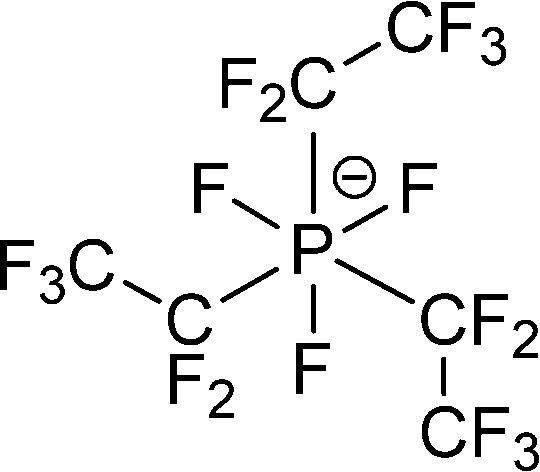
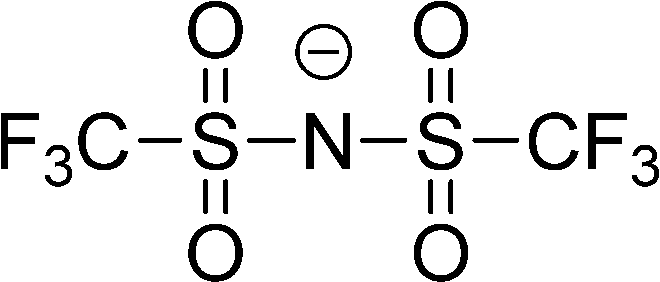
The preferred aprotic ionic liquids (APILs) are those with pyrrolidium, pyridinium, tetraalkylammonium, or tetraalkylphosphonium ions. However, the most widely employed cation is the 1-alkyl-3-methylimidazolium. The imidazolium-based salts exhibit unique properties which stem from the electronic structure of the aromatic cations. This electronic structure can be described by a delocalized 3-center-4-electron configuration across the N(1)–C(2)–N(3) moiety, a double bond between C(4) and C(5) at the opposite side of the ring, and a weak delocalization in the central region. The hydrogen atoms C(2)–H, C(4)–H, and C(5)–H have almost the same charge, but carbon C(2) is positively charged due to the electron deficit in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, whereas C(4) and C(5) are practically neutral. Notably, the resulting acidity of the hydrogen atoms is the key feature to understand the properties of these salts. Thus the imidazolium-based ILs are a good choice for measuring molecular interactions such as hydrogen bonding. The anions range from mostly hydrophobic to strongly hydrophilic one including tris(pentafluoroethyl)trifluorophosphate [FAP]−, tetrafluoroborate [BF4]−, hexafluorophosphate [PF6]−, trifluoromethylsulfonate ([TfO]−), bis(trifluoromethylsulfonyl)imide ([NTf2]−), dicyanamide ([N(CN)2]−), thiocyanate ([SCN]−), ethylsulfate ([EtSO4]−), diethylphosphate ([Et2PO4]−) or acetate ([OAc]−). To find a reasonable sequence in anion interaction strength and its influence on IL properties was a significant part of this work (Tables 1 and 2).
N bond, whereas C(4) and C(5) are practically neutral. Notably, the resulting acidity of the hydrogen atoms is the key feature to understand the properties of these salts. Thus the imidazolium-based ILs are a good choice for measuring molecular interactions such as hydrogen bonding. The anions range from mostly hydrophobic to strongly hydrophilic one including tris(pentafluoroethyl)trifluorophosphate [FAP]−, tetrafluoroborate [BF4]−, hexafluorophosphate [PF6]−, trifluoromethylsulfonate ([TfO]−), bis(trifluoromethylsulfonyl)imide ([NTf2]−), dicyanamide ([N(CN)2]−), thiocyanate ([SCN]−), ethylsulfate ([EtSO4]−), diethylphosphate ([Et2PO4]−) or acetate ([OAc]−). To find a reasonable sequence in anion interaction strength and its influence on IL properties was a significant part of this work (Tables 1 and 2).
| Cations | Abbreviation | Anions | Name |
|---|---|---|---|
| Structure | |||

|
[PF3(C2F5)3]− or [FAP]− |
|
Tris(pentafluoroethyl)-trifluorophosphate |
| [BF4]− |
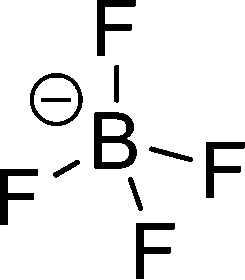
|
Tetrafluoroborate | |
| [PF6]− |

|
Hexafluorophosphate | |
| [(CF3SO2)2N]− or [NTf2]− |
|
Bis(trifluoromethylsulfonyl)imide | |
| [SCN]− |
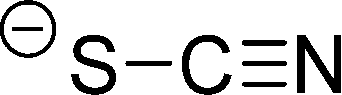
|
Thiocyanate | |
| [N(CN)2]− |
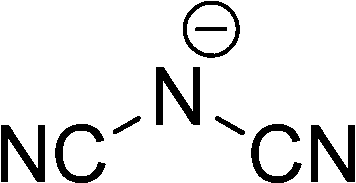
|
Dicyanamide | |
| [EtOSO3]− |
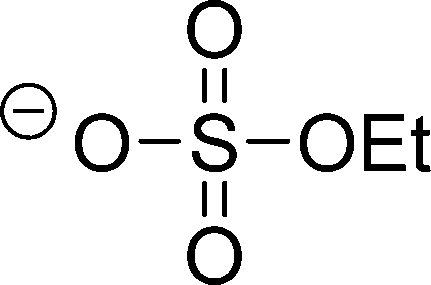
|
Ethylsulfate | |
| [(EtO)2PO2]− |
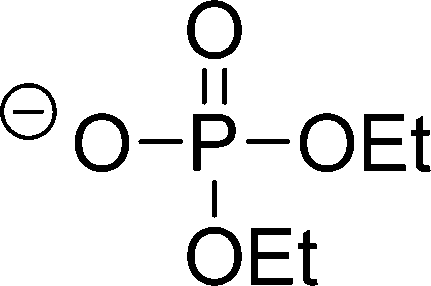
|
Diethylphosphate | |
| [OAc]− |

|
Acetate |
| Cations | Abbreviation | Anions | Name |
|---|---|---|---|
| Structure | |||

|
[NO3]− |
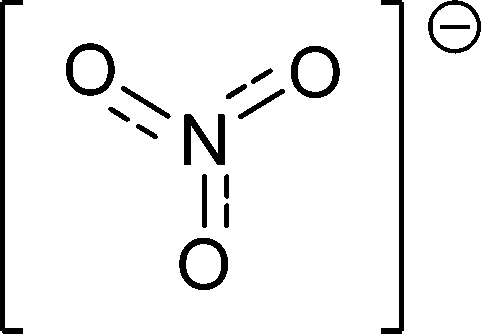
|
Nitrate |
| [MeSO3]− |
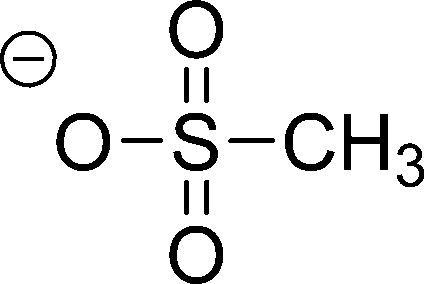
|
Methylsulfonate | |
| [OTf]− |
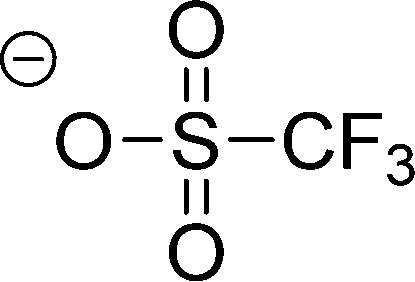
|
Trifluoromethylsulfonate | |
| [NTf2]− |
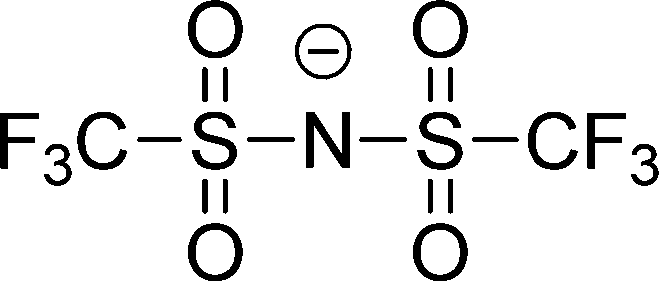
|
Bis(trifluoromethylsulfonyl)imide | |
| I− | I− | Iodide |
2.2 Protic ionic liquids (PILs)
Protic ionic liquids (PILs) are an important subgroup of ionic liquids formed by a combination of equimolar amounts of a Brønsted acid and a Brønsted base.10 The key feature that distinguishes PILs from other ILs is the proton transfer from the acid to the base, leading to the presence of proton-donor and proton-acceptor sites, which can be used to build a hydrogen-bond network. PILs have a number of unique properties in contrast to other ILs, just as water which is different from “normal” molecular liquids. Thus, PILs are a perfect system to study the presence and strength of hydrogen bonding. The number of H-bond donors, the interaction strength, and the network formation can be varied as desired. For our studies we have mainly used ammonium-based PILs including anions with variable interaction potential ranging from weakly interacting iodide to strongly interacting nitrate (Table 2).3. Studies of aprotic ionic liquids
3.1 Cation–anion interaction detected by far infrared spectroscopy
In the first study we could show that the cation–anion interaction in APILs can be detected by FIR spectroscopy. For that purpose we measured FIR spectra of these imidazolium-based ILs [C2mim][SCN], [C2mim][N(CN)2], [C2mim][EtSO4] and [C2mim][NTf2].49,50 The interpretation of the measured spectra was supported by ab initio calculated frequencies of IL clusters. The low frequency vibrational bands between 50 and 120 cm−1 could be assigned to bending and stretching modes of the +C–H⋯A− cation–anion interaction in the ILs. Characteristic bands shift in frequency and change intensity corresponding to the strength of the calculated interaction energies caused by varying the anion. Thus we present a direct probe for studying the strength of interaction energies between cations and anions in ILs. The FIR spectra of the neat ILs [C2mim][SCN], [C2mim][N(CN)2], [C2mim][EtSO4], and [C2mim][NTf2] are shown for the frequency range between 30 and 300 cm−1 (Fig. 1). The observed differences in the spectra can only stem from weak intramolecular vibrations of various anions and/or specific cation–anion interactions because the imidazolium cation (C2mim+) is kept fixed. Vibrational frequencies and intensities change significantly with the anion interaction strength. Strong support for the interpretation of the low vibrational frequencies came from ab initio calculations of IL clusters ([C2mim][Anion])n where n is the number of ion pairs. The assumption is that the largest clusters give the most liquid-like frequency spectra. In large clusters C(2)–H as well as C(4/5)–H are involved in hydrogen bonding with different strengths leading to slightly different intermolecular frequencies and intensities (see also Scheme 2).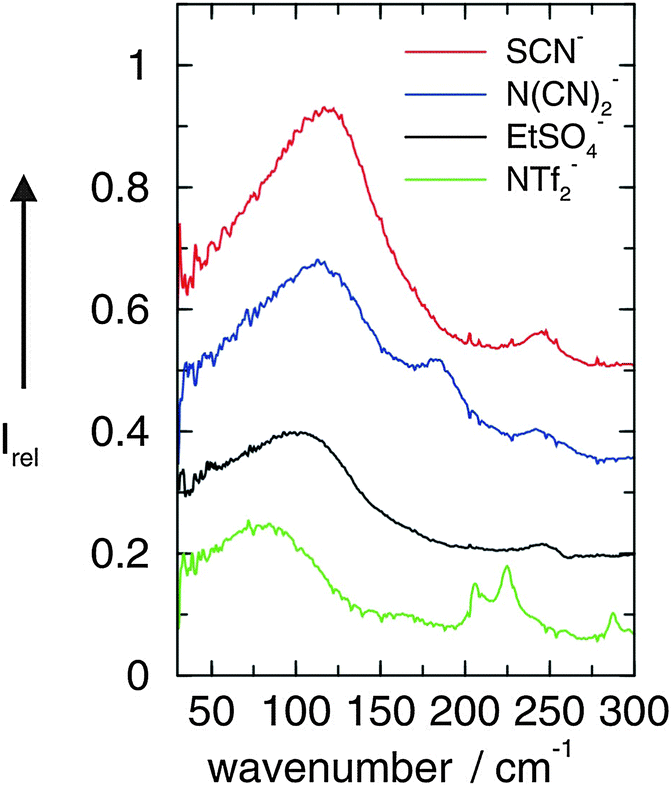 | ||
| Fig. 1 FIR spectra of [C2mim][SCN], [C2mim][N(CN)2], [C2mim][EtSO4], and [C2mim][NTf2] ionic liquids measured at 273 K. Reprinted from ref. 49 with permission. Copyright 2008 Wiley VCH. | ||
A detailed interpretation is given for the low frequency spectra of [C2mim][N(CN)2]. Fig. 2 shows the measured FIR spectrum of [C2mim][N(CN)2] along with the ab initio calculated low frequencies of the IL clusters (n = 2, 4, 6, 8). All vibrational modes could be assigned to weak intramolecular modes of the ions. However, the most interesting bands are found below 150 cm−1. The calculated frequencies of IL clusters with different size suggest that the main intensity at about 120 cm−1 can be clearly attributed to the stretching modes of the hydrogen bonds +C–H⋯A−, where C–H can be C(2)–H and/or C(4/5)–H (Scheme 2). The vibrational bands at about 50–60 cm−1 stem from the corresponding bending modes of these hydrogen bonds. These modes are significantly weaker in intensity. Obviously, the stretching mode of the hydrogen bond provides a sensitive probe for the cation–anion interaction.
 | ||
| Fig. 2 FIR spectrum of [C2mim][N(CN)2] at 273 K compared with ab initio calculated vibrational modes of corresponding IL clusters ([C2mim][N(CN)2])n with n = 2, 4, 6, 8. It is observed that the major vibrational bands are reflected by the calculated frequencies which are corrected for the harmonic approximation. Reprinted from ref. 49 with permission. Copyright 2008 Wiley VCH. | ||
3.2 Frequency shifts due to increasing anion interaction strength
We could show so far that vibrational bands in the FIR provide important information about the anion–cation interaction in APILs. However, there were still some difficulties in identifying the anion–cation interaction precisely. As discussed in the introduction this frequency range can be strongly populated. Besides the pure anion–cation interaction, other contributions can stem from weak intramolecular vibrational modes of anions and cations or unspecific librational motions or lattice modes. But even if the vibrational band of the anion–cation interaction can be clearly assigned, its position in the spectral range is still determined by two parameters. The equation of the simple harmonic oscillator![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = (1/2πc)(k/μ)1/2 shows that the frequency (wavenumber) is determined by the square root of the ratio between the force constant k and the reduced mass μ. Thus frequency shifts in the spectrum can be referred to changing force constants or to different reduced masses or both.
= (1/2πc)(k/μ)1/2 shows that the frequency (wavenumber) is determined by the square root of the ratio between the force constant k and the reduced mass μ. Thus frequency shifts in the spectrum can be referred to changing force constants or to different reduced masses or both.
Stronger frequency shifts can be expected by using the same imidazolium cation but anions with significantly different interaction potential. In principle this has been shown in the foregoing study, where we used ILs including the same C2mim+ imidazolium cation but diverse anions NTf2−, EtSO4−, N(CN)2− and SCN−. As shown in Fig. 1, the most intense bands shifted from 82 to 116 cm−1 in this order and could be related linearly to the DFT calculated binding energies.49 Although there has been some evidence that not the full masses of the anions and cations take part in the intermolecular vibrational motion and that the interaction is local in nature, the shift to higher frequencies could also be explained by the decreasing reduced masses of the ILs instead of increasing force constants due to stronger interaction. It is obvious to use anions of similar mass, but this approach is difficult to realize. Voluminous and heavy anions only weakly interact, whereas small and light anions provide strong interaction potential due to their higher surface charge density. Both the reduced mass as well as the force constant lead to shifts into the same direction, namely to lower or higher frequencies, respectively.
For this purpose we used imidazolium-based ILs with C2mim+ cation fixed and anions which are close in molecular mass as shown in Fig. 3.90 Additionally to the ILs including anions N(CN)2− and SCN−, which we recorded again for a broader frequency range down to 10 cm−1, we measured FIR spectra of 1-ethyl-3-methylimidazolium nitrate [C2mim][NO3] and 1-ethyl-3-methylimidazolium acetate [C2mim][CH3COO].
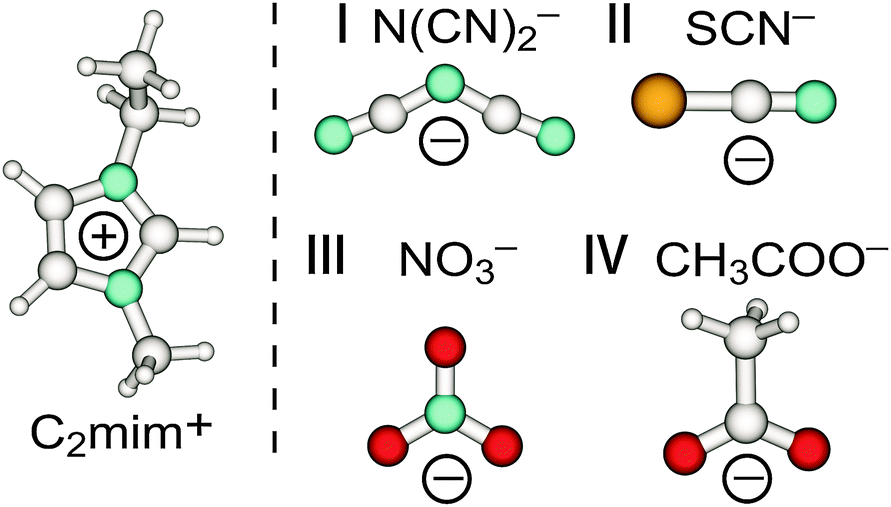 | ||
| Fig. 3 Structures of anions dicyanamide (N(CN)2−) (I), thiocyanate (SCN−) (II), nitrate (NO3−) (III) and acetate (CH3COO−) (IV) in C2mim+ imidazolium-based ionic liquids. Reprinted from ref. 90 with permission. Copyright 2012 American Chemical Society. | ||
The low-frequency FTIR spectra of [C2mim][N(CN)2] (I), [C2mim][SCN] (II), [C2mim][NO3] (III) and [C2mim][CH3COO] (IV) in the range between 20 and 300 cm−1 are shown in Fig. 4. Beside similarities the spectra also show significant differences.
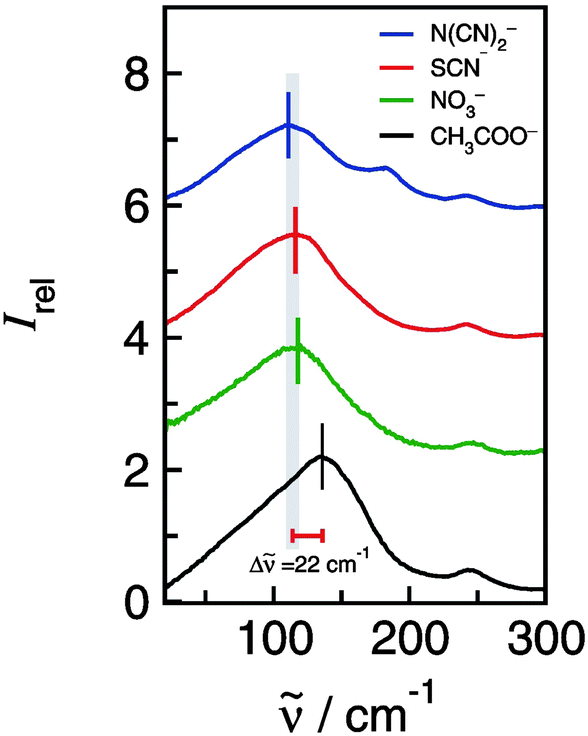 | ||
| Fig. 4 Far infrared spectra of [C2mim][N(CN)2], [C2mim][SCN], [C2mim][NO3] and [C2mim][CH3COO]. The maxima in the spectra indicate the anion–cation interaction as supported by combined experimental and theoretical analysis. Reprinted from ref. 90 with permission. Copyright 2012 American Chemical Society. | ||
As discussed before the frequencies below 150 cm−1 can be assigned to the stretching modes +C–H⋯A− describing the anion–cation interaction.49 In Fig. 4 it is shown that the FIR spectra of ILs I–III are similar in the anion–cation stretching region between 100 and 150 cm−1. But one IL is different. For [C2mim][CH3COO] (IV) the vibrational mode that describes the +C–H⋯A− interaction occurs at 136 cm−1. This mode is strongly shifted to higher wavenumbers although the reduced mass is only the second lowest among all ILs. Such a strong increase in frequency (22 cm−1) can be only referred to increasing interaction strength between cations and anions. This finding is supported by DFT calculations. The energies per ion pair are up to 35 kJ mol−1 higher for [C2mim][CH3COO] than those for ILs I–III.89
The observed interaction strength of CH3COO− is well known.91–93 The acetate anion is even able to overcome the strong hydrogen bond network in cellulose.94–97 Imidazolium-based ILs including the weaker interacting anions (I–III) are not capable of competing with the cellulose–cellulose interaction. Recently, we could show that the dissolution power of ILs largely depends on the ability of the anion to disrupt the H-bond network in a well-defined model compound such as pentaerythritol.96,97 Moreover we could show that the frequency shifts in the OH stretching region of the model compounds support the development of effective cellulose solvents. Strong redshifted OH stretching modes of the model compound are related to high wavenumber shifts in the FIR spectra. Both experimental findings support stronger solute–solvent or anion–cation interaction, in agreement with our results obtained for the FIR spectra of [C2mim][CH3COO]. Here we would like to point out that the use and studies of acetate containing ILs is a rapidly growing field with a particular focus on hydrogen bonding.98–101
3.3 Frequency shift due to the variation of the cation interaction sites
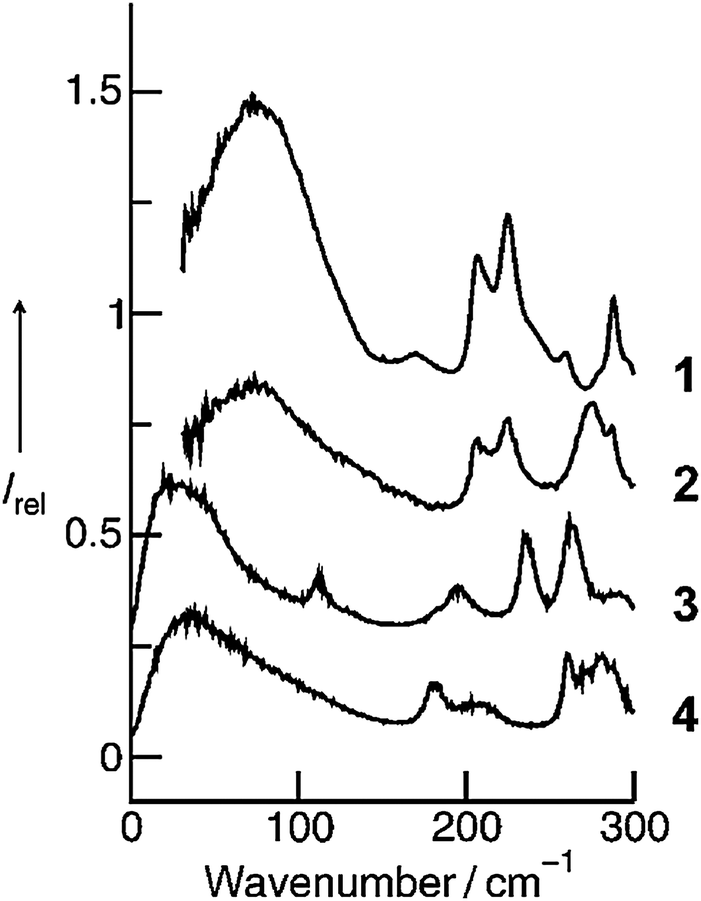
By varying the number and strength of the cation interaction sites we hoped to obtain some information about the importance of hydrogen bonds. The strategy here is to vary the cation. Instead of switching from imidazolium to other types of cations we modified the interaction sites at the imidazolium cation. Overall, it is assumed that hydrogen bonding plays an important role in the properties and reaction dynamics of these Coulomb systems. The presence of hydrogen bonding in 1-alkyl-3-methylimidazolium salts was first reported by Seddon et al.102 Evidence for hydrogen bonding stems from X-ray diffraction, mid-infrared and NMR spectroscopy. Local and directional interactions such as H-bonds in imidazolium-based ILs are indicated by shorter C–H⋯anion distances, redshifted C–H frequencies and downfield shifted C–H proton chemical shifts.48,70,102–110 Support for hydrogen bonding in this type of APILs also comes from theoretical studies.56,70,111–113 From time to time the presence of hydrogen bonding in ILs is strongly challenged at all. Some authors claim that hydrogen bonding is not invoked for explaining IL's properties.114–116It was the purpose of our work to address this important issue by providing direct spectroscopic evidence for hydrogen bonding in imidazolium-based ILs.52 First, we show that the different reduced masses have negligible effects on the frequency shifts because the interaction is local in nature. Second, we enhance the anion–cation interaction due to the increasing number and strength of H-bond abilities supporting that we certainly observe H-bond stretching frequencies in ILs. These experimental findings are supported by ab initio calculations on larger IL clusters. We measured FIR spectra of imidazolium-based ILs containing the same anion bis(trifluoromethylsulfonyl)imide (NTf2−) but various cations 1,2,3-trimethyl-imidazolium (1,2,3-trimethyl-im+, 1), 1,3-dimethyl-imidazolium (1,3-dimethyl-im+, 2), 1,2-dimethyl-imidazolium (1,2-dimethyl-im+, 3) and 1-methyl-imidazolium (1-methyl-im+, 4), respectively.49,50,52 We expected to find similar vibrational modes for the anions but different contributions stemming from the modified cations of these ILs. For the direct spectroscopic observation of H-bonds in molecular liquids, some criteria have been established for making more positive assignments. The most convincing identification can be obtained when a group incapable of hydrogen bonding is substituted for the hydrogen atom. Consequently, the vibrational bands associated with hydrogen-bond stretches or bends disappear completely. In our examples the formation of hydrogen bonds is possible via C(4)–H and C(5)–H of the cation in all ILs (Fig. 5). We have gradually substituted the methyl (CH3) group for the hydrogen (H) at the C(2)–H and N–H positions. In 1 both interactions via C(2)–H and N–H are eliminated, in 2 and 3 additional H-bonds are possible either via the C(2)–H or N–H, whereas in 4 both interactions are possible. In the sequence 1–4 the potential H-bond capabilities increase.
 | ||
| Fig. 5 Structures of cations of [1,2,3-trimethyl-im][NTf2] (1), [1,3-dimethyl-im][NTf2] (2), [1,2-dimethyl-im][NTf2] (3), and [1-methyl-im][NTf2] (4). Different position and numbers of H-bond abilities are indicated by the dotted lines. Reprinted from ref. 52 with permission. Copyright 2010 Wiley VCH. | ||
The FIR spectra were measured in the frequency range between 30 and 300 cm−1 (see Fig. 6). The main focus was put on the maximum intensities of the FIR modes below 150 cm−1. The vibrational bands of interest can be observed at 62.3 cm−1 for 1, 85.7 cm−1 for 2, 96.7 cm−1 for 3 and 100.7 cm−1 for 4, respectively. They all can be assigned to stretching vibrational bands of hydrogen bonds +C–H⋯A− and/or +N–H⋯A−. In Fig. 6 it is observed that the interaction strength between the cation and anion is significantly different. The frequencies of the band maxima shift to higher wavenumbers in the order of ILs from 1 to 4. Obviously, the interaction energy is enhanced for this sequence of ILs. Increasing hydrogen bonding leads to shorter intermolecular bond lengths and larger force constants. Enhanced H-bonds result in higher wavenumbers and increasing intensities of the vibrational bands. Such a behaviour is reflected in the FIR spectra. Consequently, we could relate the low vibrational frequencies to the average binding energies from ab initio calculated IL clusters. The binding energies per ion pair of IL tetramers could be plotted against the measured frequencies νσ (see Fig. 7, filled symbols). The almost linear plot suggests that the measured low vibrational bands reflect the strength of intermolecular forces. Moreover, the relation to hydrogen bonding is obvious.
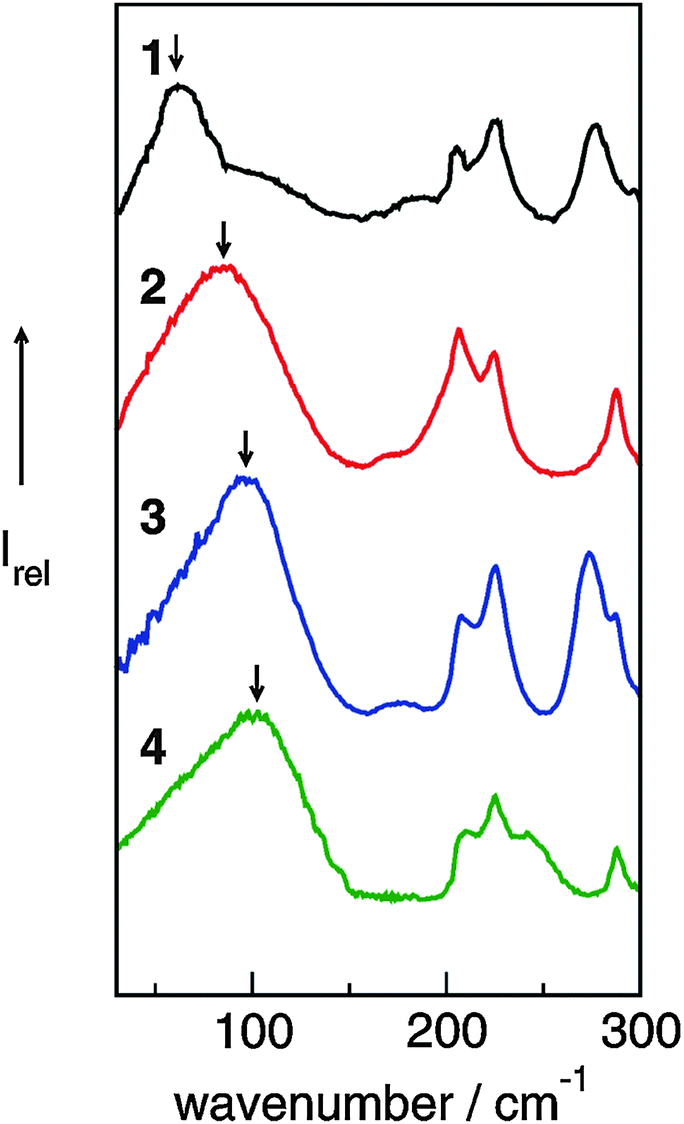 | ||
| Fig. 6 Far infrared spectra of [1,2,3-trimethyl-im][NTf2] (1), [1,3-dimethyl-im][NTf2] (2), [1,2-dimethyl-im][NTf2] (3), [1-methyl-im][NTf2] (4) at 323 K for 2–4 and 383 K for 1. Reprinted from ref. 52 with permission. Copyright 2010 Wiley VCH. | ||
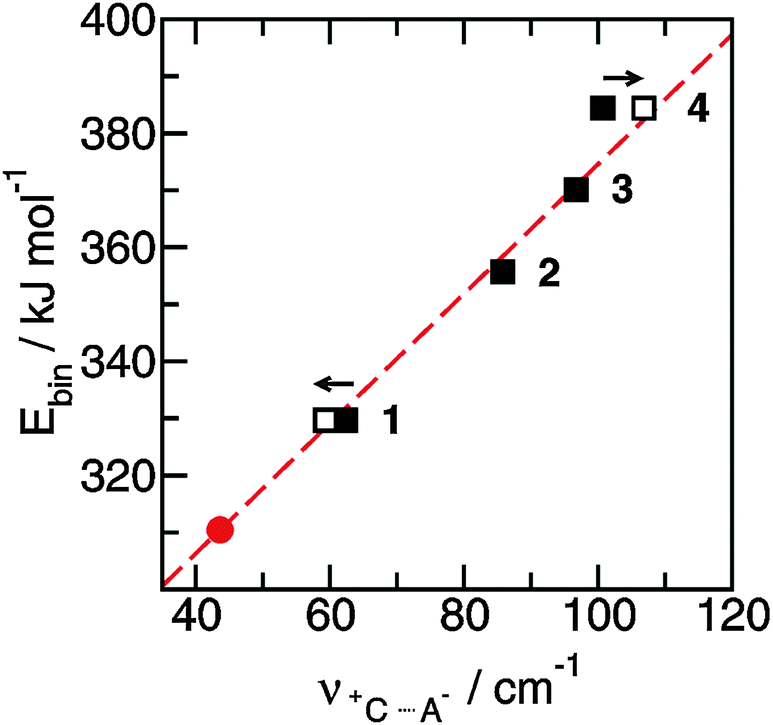 | ||
| Fig. 7 Average binding energies Ebin per ion pair in tetramers of the ionic liquids 1–4 plotted versus the measured H-bond frequencies +C⋯A−. Using the obtained linear relationship we can predict the H-bond frequency for ionic liquid ([1,2,3,4,5-pentamethyl-im][NTf2]) (filled circle). The filled symbols represent the measured frequencies and the open symbols give the frequencies corrected for the reduced masses. Reprinted from ref. 52 with permission. Copyright 2010 Wiley VCH. | ||
Overall, we reported the direct observation of H-bond stretching frequencies in pure imidazolium-based ILs from FIR spectroscopy. Nearly excluding reduced mass effects, the frequency shifts could be related to increasing force constants indicating stronger cation–anion interactions. Ab initio calculations suggest a linear relationship between the interaction energies and the intermolecular stretching frequencies. Both properties are related to the increasing H-bond capabilities in the varying imidazolium cations. This finding clearly indicates that the stretching frequencies are a direct measure of hydrogen bonding in the ILs.
3.4 Hydrogen bonding in aprotic ionic liquids
In the foregoing work we could show that the cation–anion interaction is at least enhanced by hydrogen bonding at the C(2)–H position. That this specific interaction can influence the physical properties of ILs was first noticed by Bonhôte et al.106 His group discovered that methylation of the C(2) position of 1,3-dialkylimidazolium containing ILs disrupts the predominant hydrogen-bonding interaction between cations and anions leading to surprising changes in the physicochemical properties. Elimination the C(2)–H⋯A− H-bond interaction by alkyl substitution was expected to lower the melting point and to decrease the viscosity. Instead, the opposite behaviour was observed. C(2) methylation increased the melting point from 50 to 120 °C, whereas methylation at the C(5)-position only moderately increased the melting point. Similar characteristics were found for the viscosities. Altogether, the ionic liquids protonated at the C(2) position exhibit the lowest melting points and viscosity.There have been several efforts to explain the unusual property behaviour of imidazolium-based ILs. Hunt117 studied 1-butyl-2,3-dimethyl-imidazolium and 1-butyl-3-methyl-imidazolium based ILs by DFT methods for understanding these effects at the molecular level. Also here less hydrogen bonding and slight reduction in Coulomb attraction between the cation and the anion in [1-butyl-3-methyl-im]Cl suggest that phase transition temperatures and viscosities decrease compared to those of the [1-butyl-2,3-dimethyl-imidazolium]Cl in agreement with experimental findings. It was argued that the effect due to a loss in hydrogen bonding is outweighed by those due to a loss in entropy. The amount of disorder in the system is reduced by the elimination of ion-pair conformers which are stable for [1-butyl-3-methyl-imidazolium]Cl but not for [1-butyl-2,3-dimethyl-im]Cl. An increase in the rotational barrier of the butyl chain limits free rotation and facilitates alkyl chain association. In the so-called “entropy theory” by Hunt117 it is still presumed that hydrogen bonding stabilizes imidazolium-based ILs.
The C(2) methylation effects for [1-butyl-3-methyl-imidazolium]X and [1-butyl-2,3-dimethyl-imidazolium]X, where X− is Cl−, Br−, I−, BF4−, and PF6− were also studied by Endo et al.118 These authors reported that the melting points and freezing points of these ILs increase with methylation at the C(2) position. They argued that the overcompensation of the phase transition entropy decreases with the transition enthalpy decrease. Their findings are in agreement with Hunt's results although quantum chemical calculations of isolated ion-pairs should be regarded with suspicion.
We suggested another explanation for understanding the property behavior of imidazolium-based ILs (see Chapter 3.1–3.3). We proposed that local and directional hydrogen bonds formed between cations and anions destroy the charge symmetry resulting in fluidized ILs. Hydrogen bonds can be regarded as “defects” in the Coulomb network of ILs. These defects increase the dynamics of the cations and anions, leading to lower melting points and viscosities. Such a behaviour could be observed for 1-ethyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide and 1-ethyl-2,3-dimethyl-imidazolium bis(trifluoromethylsulfonyl)imide based on FIR measurements and DFT calculations on IL clusters. In additional studies we could show that the cation–anion interaction in imidazolium-based ILs is enhanced due to hydrogen bonding as indicated by frequency shifts to higher wavenumbers in FIR and terahertz (THz) spectra.50,52,119,120
Noack et al.120 showed for similar ILs 1-ethyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide, 1-ethyl-2,3-dimethyl-imidazolium bis(trifluoromethylsulfonyl)imide, and 1-butyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide and 1-butyl-2,3-dimethyl-imidazolium bis(trifluoromethylsulfonyl)imide that electron density changes can alter the position and strength of interionic interactions and lead to reduced configurational variations. From their spectroscopic work they concluded that both factors can alter the melting points and viscosities. They summarized that neither the “entropy theory” of Hunt117 nor the “defect hypothesis” of our earlier studies50,52 alone are capable of explaining the changes in the physicochemical properties but complement each other.
There are other ideas discussing the relevance of hydrogen bonding to the properties of ILs.121–123 Spohr and Patey studied the influence of charge location on IL properties by MD simulations. Charge displacement from the center of mass of the ions shows the formation of directional ion-pairs leading to low conductivities. If the hydrogen bonding strength exceeds a critical value melting points and viscosities increase again.121 Zahn et al. showed that hydrogen bonding at the C(2) position enhances the mobility of the anion resulting in lower melting points. Low melting points could be referred to flat energy landscapes of ion-pairs.122
Izgorodina et al. investigated two possible structural or energetic sources of the increased viscosity due to the methylation at the C(2) position: firstly, ion association, as probed by the Walden rule, and secondly differences in the potential energy surface profiles that favor ionic transport in the non-C2-methylated imidazolium-based ILs.123 They showed that the increased viscosity could be a result of higher potential energy barriers between the energetically preferred conformations on the potential energy surface inhibiting the overall ion transport.
For supporting the “defect hypothesis”, we used the same bis(trifluoromethylsulfonyl)-imide anion (NTf2−) for all ILs to avoid any effects from changing negative charge distributions. Only the imidazolium cations are different: 1,2,3,4,5-pentamethyl-imidazolium (I), 1,2,3,4-tetramethyl-imidazolium (II), 1,3,4,5-tetramethyl-imidazolium (III) and 1,2,4,5-tetramethyl-imidazolium (IV) (see Fig. 8). To reduce the conformational flexibility of the cations, only methyl groups and hydrogen atoms at the imidazolium ring, but no longer alkyl chains are taken into consideration. Additionally, we tried to avoid mass or volume effects by synthesizing ILs with imidazolium cations II, III and IV, all having four methyl groups as ring substituents. Now, the focus can be put on effects resulting from anion–cation interactions only.
 | ||
| Fig. 8 Structures of cations for imidazolium-based ionic liquids I–IV. The positions of possible H-bonds are indicated by the dotted lines. Reprinted from ref. 119 with permission. Copyright 2011 The Royal Society of Chemistry. | ||
For better understanding of our concept, interaction sites of the imidazolium cations are given as red dotted lines (Fig. 8). They show possible local and directional interactions via C(5)–H, C(2)–H or N–H, which should be important for the structure and properties of ILs.
We measured FIR spectra of ILs I–IV between 10 and 150 cm−1 or 0.3 and 4.5 THz. The vibrational bands are relatively broad but not featureless. Without deconvolution of the vibrational spectra and supporting DFT calculated frequencies, vibrational bands of enhanced intensity for each IL can be observed as shown by the arrows (Fig. 9). For IL I we could demonstrate before that the ATR as well as absorption FIR and THz spectra could be deconvoluted into three vibrational bands. DFT calculations on an cluster of an ion-pair trimer supported the number of vibrational bands in this frequency range. Moreover, DFT calculated frequencies suggested that the vibrational band at about 60 cm−1 describes the anion–cation interaction. Because in I no specific interaction site for hydrogen bonding is present, this interaction mainly stems from Coulomb forces and to a minor extent from non-directional dispersion forces. The arrow for II at about 59 cm−1 indicates a similar type of interaction. Although there is possible interaction with C(5)–H, there is no specific interaction and thus no enhancement by hydrogen bonding resulting in increasing frequencies. Such a behavior is well known from earlier studies.52 Interaction via C(4/5)–H is negligible and does not cause significant frequency shifts. That is completely different for ILs III and IV as shown in Fig. 9. Here, the maxima of the low frequency vibrational bands are shifted to 77 and 102 cm−1 caused by hydrogen bond enhanced anion–cation interaction. However, although supported by DFT calculations the deconvolution procedure of the spectra is not unique in any case. The spectra show characteristic features, but the relative halfwidth and the intensities are difficult to identify.
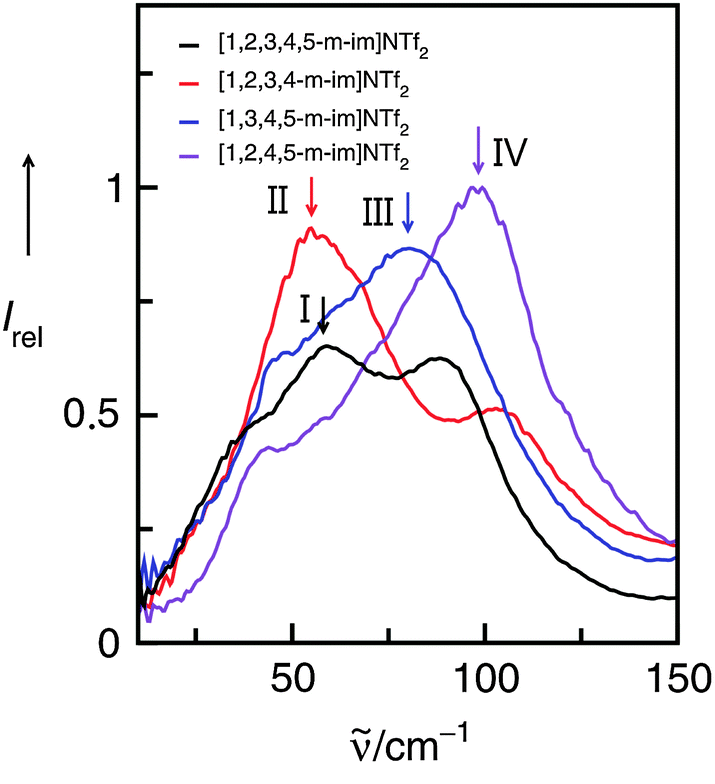 | ||
| Fig. 9 Far infrared spectra of imidazolium-based ionic liquids I–IV. The maxima in the spectra indicate the anion–cation interaction as supported by the subsequent experimental and theoretical analysis. Reprinted from ref. 119 with permission. Copyright 2011 The Royal Society of Chemistry. | ||
For better assignment of the vibrational bands we additionally measured the FIR spectra of 50 mol% mixtures of the ILs I/II, I/III and I/IV, versus the background spectra of the pure ILs I and II, III, IV, respectively. The resulting difference spectra give positive and/or negative contributions for the intra- and intermolecular vibrational bands present or missing in these mixtures. If we now subtract II, III, IV from the 50 mol% mixtures with I, we expect positive contributions from I, but negative contributions from II, III or IV, respectively.
The spectra and difference spectra of the I/III mixtures are shown in Fig. 10. If IL I is subtracted three negative bands are observed in the difference spectrum. The contributions at about 30, 60 and 90 cm−1 can be well assigned to the frequencies we derived from the deconvolution procedure.
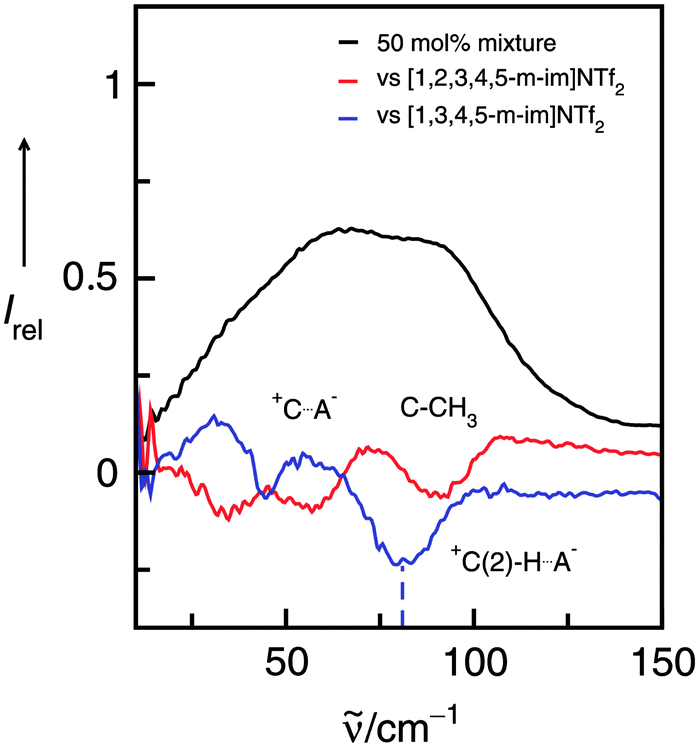 | ||
| Fig. 10 Far infrared spectra of a 50 mol% mixture of the ILs I and III (black line) along with the difference spectra obtained from measurements of this mixture versus the background spectrum of IL I (red line) and IL III (blue line), respectively. The negative contribution at about 77 cm−1 can be assigned to the +C(2)–H⋯A− interaction in IL III and is indicated by the dotted line. Reprinted from ref. 120 with permission. Copyright 2011 The Royal Society of Chemistry. | ||
3.5 Substituting the hydrogen by a methyl group at the C(2) carbon
In a consecutive study we measured the low-frequency spectra of [C2mim][NTf2] and [C2C1mim][NTf2].50,124 Although we used the same path length for the IR cell, all intensities of [C2C1mim][NTf2] are substantially lower, except those of the cations above 250 cm−1. The intramolecular bending and wagging modes of the anion NTf2− are affected by suppressing the important H-bonds via C(2)–H (see Scheme 2). However, the intensities of the vibrational bands which we referred to the bending and stretching modes of the C(2)–H⋯A− and C(4/5)–H⋯A− interactions are now significantly reduced. Furthermore, the remaining low-intensity band is red-shifted from 83.5 cm−1 to 79.0 cm−1. Although the shift is small relative to the rather broad bands, this behaviour could be detected for spectra at all temperatures (see Fig. 11).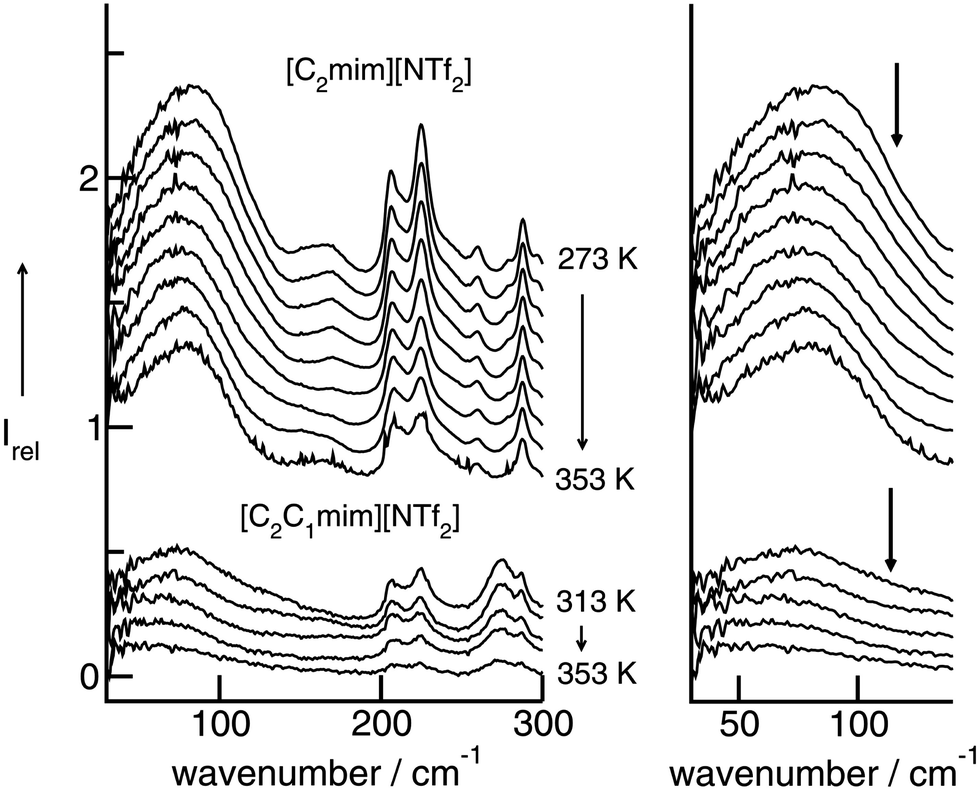 | ||
| Fig. 11 Far infrared spectra of [C2mim][NTf2] (top) and [C2C1mim][NTf2] (bottom) as a function of temperature. To the right, enlarged spectra are given for the intermolecular vibrational modes between 30 and 150 cm−1. Reprinted from ref. 124 with permission. Copyright 2009 Wiley VCH. | ||
Switching off the C(2)–H interaction leads to H-bonds via C(4/5)–H. These interactions are weaker, leading to a slight redshift of the intermolecular vibrational band. The overall H-bond contribution is significantly reduced in favor of increasing Coulomb interactions in [C2C1mim][NTf2]. This can be observed in FIR spectra by increasing contributions above 100 cm−1, represented by a long tail up to 200 cm−1. The Coulomb interactions are stronger than the H-bonds and occur at higher wavenumbers. Substituting the C(2)–H hydrogen in [C2mim][NTf2] by the C(2)–CH3 methyl group in [C2C1mim][NTf2] we replace a localized and highly directional H-bond in favour of a non-localized and smeared-out Coulomb interaction.
These characteristic features in the FIR spectra can be used to evaluate force fields of ILs which have been developed for molecular dynamics (MD) and Monte Carlo (MC) simulations. For the simulation of FIR spectra we used two different force fields.71,86 In the CLaP force field86 the same charges and Lennard-Jones parameters for all the ring hydrogen HA/C(2)–H, HW/C(4/5)–H are used throughout. Instead, in the refined force field by Köddermann et al.,71 the Lennard-Jones parameters σ and ε were changed in such a way that the C(2) interactions were significantly favoured over the C(4)–C(5) interactions with the anion. This was achieved by decreasing σ and increasing ε for these hydrogen atoms. The parameters were parameterized to reproduce NMR rotational correlation times for cations and water molecules in [C2mim][NTf2].
The experimental and simulated FIR spectra of the pure ILs are shown in Fig. 12. It is not expected that all inter- and intramolecular vibrational modes are given quantitatively in frequency and intensities. Despite the shortcomings, the power spectrum should reflect motions present in the ILs. The simulations reproduce the positions of the absorption maxima fairly well, although in the simulations the absorption peaks occur at lower frequencies than in the measured spectra.
 | ||
| Fig. 12 Measured far infrared spectra of [C2mim][NTf2] (1) and [C2C1mim][NTf2] (2) as well as simulated far infrared spectra of the [C2mim][NTf2] using the force field by Köddermann et al.71 (3) and the CLaP force field86 (4), respectively. Reprinted from ref. 124 with permission. Copyright 2009 Wiley VCH. | ||
We now discuss the profiles of the low vibrational bands below 150 cm−1, which can be assigned to the cation–anion interactions. Whereas the shape of the spectral band obtained from the MD simulation using the Köddermann's force field resembles that from the measured spectrum of [C2mim][NTf2], the spectral feature resulting from the MD simulations based on the CLaP force field looks significantly different. The latter resembles the characteristics of the spectral band observed experimentally for [C2C1mim][NTf2]. Both spectra, the MD simulated spectrum for [C2mim][NTf2] as well as the measured spectrum for [C2C1mim][NTf2] show long tails towards higher wavenumbers, whereas the low vibrational bands are less pronounced. In our opinion this feature does not surprise. The CLaP force field suggests no specific preference for the C(2)–H interaction and treats all ring hydrogen atoms equally, as indicated by identical Lennard-Jones parameters. Consequently, the power spectra resemble those of the measured spectra of [C2C1mim][NTf2] where this specific interaction is deleted by methylation of the C(2) position.
4. Studies of protic ionic liquids
4.1 Stronger H-bond interaction: shift from aprotic to protic ionic liquids
Aprotic (APILs) and protic ionic liquids (PILs) differ significantly in their ability to form hydrogen bonds. Therefore characteristic differences in FIR spectra describing the cation–anion interactions are expected. For observing changes in the spectral features from APILs to PILs, we measured the FIR spectra of the imidazolium-based APILs [C4C1mim][BF4] (1), [C4mim][BF4] (2) and [C4mim][NO3] (3) as well as PIL [PrAm][NO3] (4) (see Fig. 13). The consideration is as follows: at first we increase the interaction strength in the APILs by replacing the methyl group at the C(2) position of [C4C1mim] by a hydrogen atom getting [C4mim] (1 → 2). Thereby the anion BF4− remains the same. For IL 2 hydrogen bonding via C(2)⋯H⋯A− is possible. Then we replace the weakly interacting anion BF4− by the strongly interacting anion NO3− in the imidazolium-based APIL without replacing the cation [C4mim] (2 → 3). Finally we substitute the imidazolium cation [C4mim] by the propylammonium cation [PrAm] with still the same anion NO3− (3 → 4). In Fig. 14 FIR spectra of all four ILs (1–4) are shown for the frequency range between 30 and 600 cm−1. It is observed that the spectra show significant differences but also share some common features. The anions BF4− and NO3− have been chosen for contributing to this frequency range by intramolecular frequencies. Thus differences can only be referred to weak intramolecular vibrations of the different cations and/or specific cation–anion interactions. | ||
| Fig. 13 The imidazolium-based aprotic ionic liquids [C4C1mim][BF4] (1), [C4mim][BF4] (2), [C4mim][NO3] (3) and the protic ionic liquid [PrAm][NO3] (4). Similar colors indicate like cations and anions, respectively. Reprinted from ref. 76 with permission. Copyright 2009 The Royal Society of Chemistry. | ||
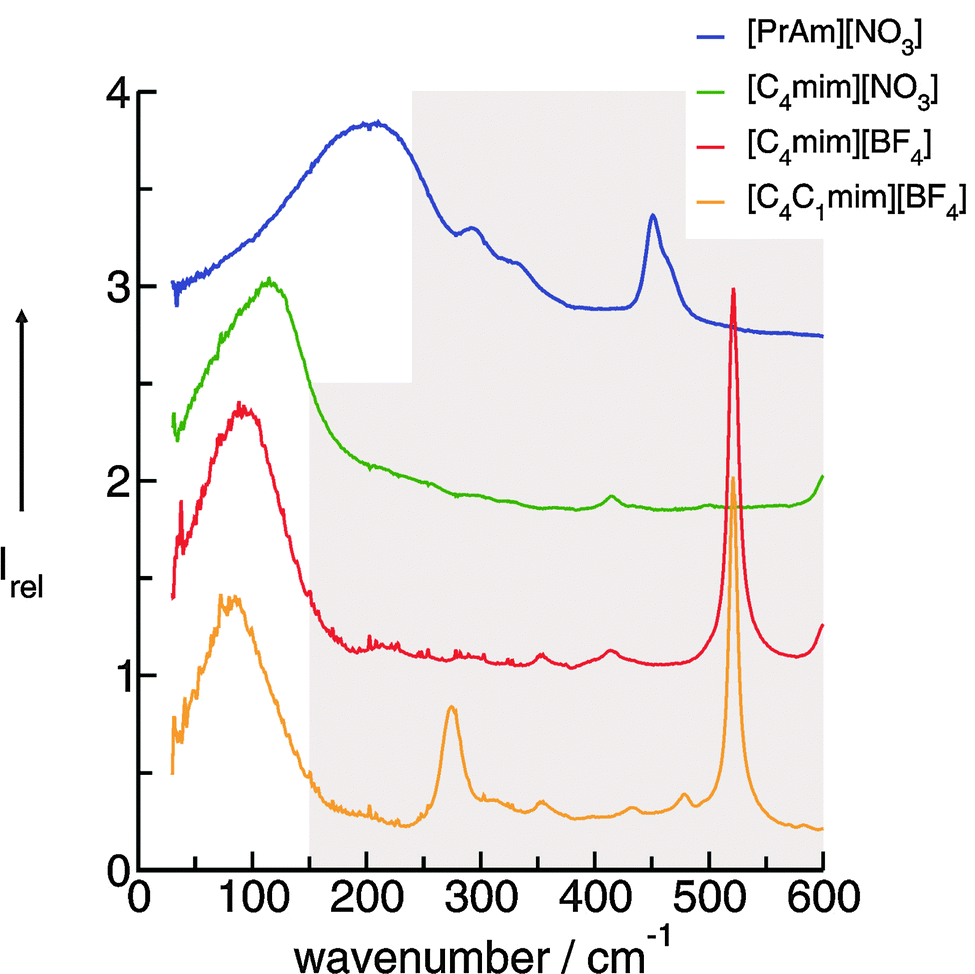 | ||
| Fig. 14 FIR spectra of [C4C1mim][BF4], [C4mim][BF4], [C4mim][NO3] and [PrAm][NO3] at 353 K. Reprinted from ref. 76 with permission. Copyright 2009 The Royal Society of Chemistry. | ||
In PILs, the proton transfer from the acid to the base leads to the presence of proton-donor and proton-acceptor sites resulting in hydrogen-bond networks. This is observed in the FIR spectra. Again, the most interesting bands show up below 250 cm−1. The DFT calculated frequencies of the PIL clusters suggest that the frequencies at about 224 cm−1 and 159 cm−1 can be assigned to the asymmetric and symmetric stretching modes of the hydrogen bonds +N⋯H–O− (νas(HBs) and νs(HBs)). The vibrational bands at around 78 cm−1 can be attributed to corresponding bending modes of these hydrogen bonds (δ(HBs)). As for the intramolecular modes, the asymmetric stretches exhibit higher intensities than the symmetric stretches. The asymmetric and symmetric stretching modes as well as the bending modes of the hydrogen bonds can be related to those reported for liquid water and ice.
Overall it can be summarized that the low vibrational modes of all ILs reflect cation–anion interactions. Again the shift to higher wavenumbers for these bands can be related to increasing DFT calculated binding energies. The correlation of intermolecular frequencies and calculated NBO delocalization energies is even more plausible, suggesting that the increase in interaction energy from (1) to (4) results from increasing hydrogen bonding as supported by Weinhold's NBO analysis.125,126 The NBO stabilization energy is expected to be the dominant attractive contribution to H-bond formation. It is closely related to the cooperative strengthening and shortening of H-bonds, because intermolecular charge delocalization enhances the Lewis base (donor) strength of the anion and the Lewis acid (acceptor) strength of the cation. In Fig. 15 the 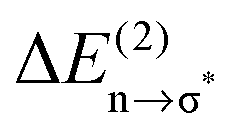 delocalization energies of ion-pair monomers and dimers of ILs (1–4) are shown. The energies of the dimeric ion pair are only slightly higher than those for the single ion pair. The reason is the larger number of H-bonds in the dimer rather than cooperative effects. The intermolecular stabilization energies as well as the total binding energies per ion were plotted versus the measured intermolecular frequencies. The obtained relations suggest that the measured low-frequency vibrational bands reliably describe the intermolecular forces. In Fig. 15 it is shown that the intermolecular stabilization energies more strongly increase in the order from (1) to (4) than the corresponding binding energies. The ratios of
delocalization energies of ion-pair monomers and dimers of ILs (1–4) are shown. The energies of the dimeric ion pair are only slightly higher than those for the single ion pair. The reason is the larger number of H-bonds in the dimer rather than cooperative effects. The intermolecular stabilization energies as well as the total binding energies per ion were plotted versus the measured intermolecular frequencies. The obtained relations suggest that the measured low-frequency vibrational bands reliably describe the intermolecular forces. In Fig. 15 it is shown that the intermolecular stabilization energies more strongly increase in the order from (1) to (4) than the corresponding binding energies. The ratios of 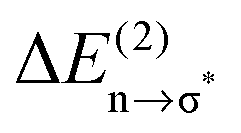 and Ebin increase in this sequence, suggesting that the H-bond contribution to the total interaction energy increases from 0.06 for [C4C1mim][BF4] (1), via 0.15 for [C4mim][BF4] (2), 0.33 for [C4mim][NO3] (3) to 0.53 for PIL [PrAm][NO3] (4), respectively. By choosing specific cation–anion combinations it is obviously possible to tune the ratio from Coulomb forces to hydrogen bonding by switching from APILs to PILs.
and Ebin increase in this sequence, suggesting that the H-bond contribution to the total interaction energy increases from 0.06 for [C4C1mim][BF4] (1), via 0.15 for [C4mim][BF4] (2), 0.33 for [C4mim][NO3] (3) to 0.53 for PIL [PrAm][NO3] (4), respectively. By choosing specific cation–anion combinations it is obviously possible to tune the ratio from Coulomb forces to hydrogen bonding by switching from APILs to PILs.
 | ||
Fig. 15 B3LYP/6-31+G* calculated binding energies Ebin (circles) and NBO delocalization energies 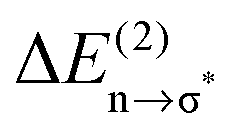 (squares) of ionic liquid clusters x [1 = monomers (open symbols)], [2 = dimers (filled symbols)] for [C4C1mim][BF4] (1), [C4mim][BF4] (2), [C4mim][NO3] (3) and [PrAm][NO3] (4). The inlay shows the ratios between both energies ( (squares) of ionic liquid clusters x [1 = monomers (open symbols)], [2 = dimers (filled symbols)] for [C4C1mim][BF4] (1), [C4mim][BF4] (2), [C4mim][NO3] (3) and [PrAm][NO3] (4). The inlay shows the ratios between both energies (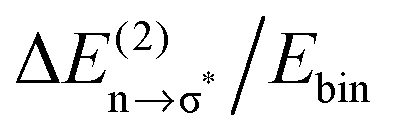 ) indicating increasing contributions of hydrogen bonding for the given order of ionic liquids. Reprinted from ref. 76 with permission. Copyright 2009 The Royal Society of Chemistry. ) indicating increasing contributions of hydrogen bonding for the given order of ionic liquids. Reprinted from ref. 76 with permission. Copyright 2009 The Royal Society of Chemistry. | ||
4.2 Detecting and quantifying H-bonds
For quantifying hydrogen bonding, we studied a well chosen set of PILs. Room temperature PILs were measured in the liquid state, PILs in the solid state were studied as pellets. Thus, some of the chosen systems may be called protic molten salts because they have melting points above 100 °C. However, all PILs considered here include trialkylammonium cations as Brønsted base possessing only a single H-bond donor possibility via N–H. We expected to observe a distinguished vibrational band that can be clearly assigned to N–H⋯anion hydrogen bonds.127 For some methyl ammonium nitrates Raman and IR spectra in the low frequency range have been measured some time ago.128,129 Due to the lack of systematic variation of the number of possible H-bond and unavailable DFT calculated frequencies, the vibrational modes could be only roughly referred to “translational and librational motions of cations and anions”.First, we measured the FIR spectra of tetramethylammonium nitrate [(CH3)4N][NO3] and trimethylammonium nitrate [(CH3)3NH][NO3] as shown in Fig. 16a. For [(CH3)4N][NO3] a broad vibrational band is observed which is generally attributed to librational contributions of interacting ions. Intramolecular vibrational motions of the ammonium cation or nitrate anion are not observed in this frequency range. The spectrum of [(CH3)3NH][NO3] shows a similar band at around 100 cm−1, but also a distinct vibrational band at around 170 cm−1. Because no intra molecular modes can be referred to the ions, this vibrational band is associated with anion–cation interaction and can be only assigned to the +N–H⋯NO3− hydrogen bond. This interpretation is supported by DFT calculated frequencies of clusters ([(CH3)3NH][NO3])n with n = 2, 4, 6, 8, 10. For comparison with the measured spectrum, the calculated FIR spectra were also simulated by applying a width at half maximum of 10 cm−1 for the Lorentzian functions for each vibrational mode (see Fig. 16b). Although the frequencies are calculated in the harmonic approximation and no correction is applied, the calculated spectra represent the measured spectra. The vibrational band observed at around 170 cm−1 is also found in the calculated spectra for each cluster. There is no additional frequency shift of this band with increasing cluster size beyond the tetramer. Caused by the matrix effect in the pellets, vibrational bands are better separated in the calculated than in the measured spectra. At that time we can realize that the distinct vibrational band at around 170 cm−1 is well isolated and that it can be clearly attributed to the +N–H⋯NO3− hydrogen bond.
 | ||
| Fig. 16 (a) FIR spectra of trimethylammonium nitrate [(CH3)3NH][NO3] (top) and tetramethylammonium nitrate [(CH3)4N][NO3] (bottom) in the pellet state. The additional vibrational band at 170 cm−1 in the spectrum can be assigned to +N–H⋯NO3− intermolecular interaction (indicated by the arrow) and is consequently missing in the spectrum for [(CH3)3N][NO3]. (b) Calculated FIR spectra of clusters of ion-pairs for ([(CH3)3NH][NO3])n with n = 2, 4, 6, 8, 10. The calculated spectra were also simulated by using a width at half maximum of 10 cm−1 for each vibrational mode. No cluster size dependence beyond the ion-pair tetramer is observed. The calculated spectra perfectly reproduce the measured spectrum (top), in particular the H-bond vibrational mode. Reprinted from ref. 127 with permission. Copyright 2012 Wiley VCH. | ||
We then tried to quantify this interaction strength. Steiner pointed out in his review that hydrogen bonds possessing mainly ionic as well as little covalent nature are not classified as “strong” despite high dissociation energies.130 Instead, these interactions should be termed ionic with a moderate hydrogen bond formed on top. We show here that this “on top” H-bond energy can be calculated. For clusters of ([(CH3)4][NO3])n and ([(CH3)3NH][NO3])n up to n = 6 the binding energies per ion pair were calculated and corrected for the basis set superposition error. Similar to the calculated FIR spectra the binding energies do not change for clusters beyond trimers (Fig. 16b). Mostly three types of energies contribute to the overall interaction: Coulomb forces, hydrogen bonding and dispersion forces. In principle, Coulomb and dispersion forces should be similar for both PILs. However, the additional methyl group in [(CH3)4N][NO3] leads to slightly larger dispersion forces compared to [(CH3)3NH][NO3]. For confirmation we applied Grimme's DFT-D3 method and calculated the non-covalent interactions.85 The dispersion forces per ion-pair for both PILs only differ by about 2 kJ mol−1. Obviously the difference in energy can be mainly referred to the presence of hydrogen bonding in [(CH3)3NH][NO3]. The energy difference of both PIL clusters now provides a reliable measure of the H-bond strength which is estimated to be 49.4 kJ mol−1 (see the inset of Fig. 17). If the dispersion correction is taken into account, the H-bond energy only slightly decreased to 47.3 kJ mol−1. These H-bonds are usually classified to be located between moderate and strong.130
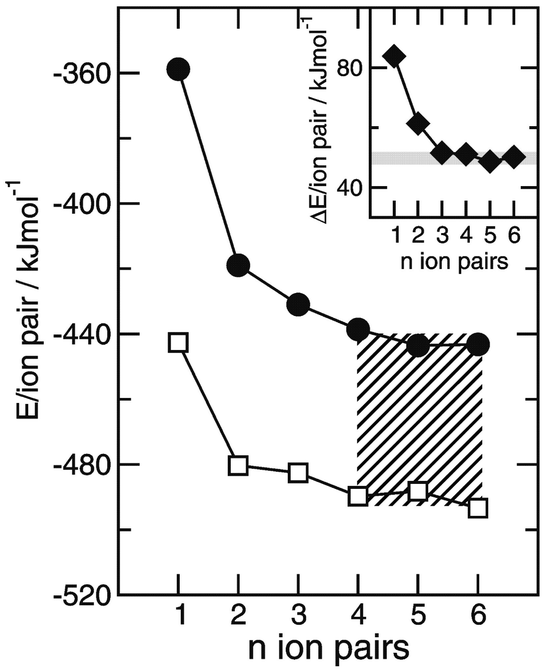 | ||
| Fig. 17 DFT calculated cluster energies per ion-pair for ([(CH3)4N][NO3])n (filled circles) and ([(CH3)3NH][NO3])n (open squares) with n = 1–6. The energy differences between clusters of similar size of these protic ionic liquids are shown in the inlay and represent the H-bond strength of the +N–H⋯NO3− interaction in [(CH3)3NH][NO3]. The H-bond energy is quantified to be 49.4 kJ mol−1. The slightly higher dispersion energies in clusters ([(CH3)4N][NO3])n due to the additional methyl group of the cation reduce the H-bond energy by only about 2 kJ mol−1. Reprinted from ref. 127 with permission. Copyright 2012 Wiley VCH. | ||
We additionally considered several anions and alkyl group substitution at the cation.127 Overall we could conclude that the interaction between the trialkyl ammonium cation and the respective anion is local and directional in nature. The observed frequency shifts can be related to different interaction strengths caused by changing anions or inductive effects resulting from alkyl group substitution at the cation. For all PILs taken into account the H-bond vibrational modes were found in the range of 140–180 cm−1. This is close to the observed vibrational band of the water H-bond network stemming from O–H⋯O interaction.131–134 It seems thus surprising that the H-bonds in PILs (47 kJ mol−1) are about two times stronger than those in water (22 kJ mol−1). However, the intermolecular interaction in water strongly depends on the reduced mass as a result of its low molecular weight. We calculated the force constants and the corresponding reduced masses of the low vibrational frequencies of a water dimer and compared them to those of the PIL ion-pair [(CH3)3NH][NO3]. The ratios [(CH3)3NH][NO3]/H2O for the force constants (2.16) and the reduced masses (2.12) are similar. Obviously, the significantly lower reduced mass of water compensates for its small force constants leading to intermolecular vibrational modes for water and PILs in the same frequency range.
4.3 Pure interaction, corrected for reduced mass effects
The interaction between cations and anions in ILs can be controlled by increasing the molecular volume of the ions. Simultaneously the reduced mass increases and the frequency shift to lower wavenumbers results from both parameters. In this context, further work was needed to derive the pure intermolecular interaction energy corrected for reduced mass effects from low frequency vibrational bands.We could show that combining synthesis of a well chosen set of ILs, systematic far infrared measurements as well as DFT calculated frequencies, reduced masses and force constants of IL clusters allows the complete analysis of the FIR spectral region. In particular we could demonstrate that frequency shifts describing the intermolecular interactions between anions and cations in ILs can be dissected into reduced mass and pure interaction effects.
Cations and anions in well chosen APILs and PILs have specific features and functions to overcome the typical difficulties in analyzing the FIR spectra and assigning the interaction contributions. Six PILs have been synthesized, one APIL has been purchased and already measured in earlier studies.49,50,135 The full set of ILs included triethylammonium methylsulfonate [(C2H5)3NH][CH3SO3] (Ia), triethylammonium trifluoromethylsulfonate [(C2H5)3NH][CF3SO3] (IIa), triethylammonium bis(trifluoromethylsulfonyl)imide [(C2H5)3NH][NTf2] (IIIa) and 1-ethyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide [C2mim][NTf2] (IV). The PILs Ib–IIIb have also been synthesized including deuterated ethyl groups in the triethyl ammonium cations resulting in [(C2D5)3NH][CH3SO3] (Ib), [(C2D5)3NH][CF3SO3] (IIb) and [(C2D5)3NH][NTf2] (IIIb), respectively.
The PILs included the same triethylammonium cation throughout. Therefore changes in the interaction strength can result only from varying anions. Three mobile ethyl groups in the ammonium cations result in room temperature PILs as well. Highly flexible ethyl groups lead to a larger variety of states and are entropically favored. Another important feature of this cation is the single proton donor function N–H. The only possible directional interaction +N–H⋯A− yields isolated vibrational bands in the FIR spectra which can be clearly attributed to the H-bond enhanced anion–cation interaction.127 The chosen anions show decreasing interaction strength in the order [NTf2−] < [CF3SO3−] < [CH3SO3−] due to fluorination of the methyl groups. The problem here is that the decreasing interaction strength is accompanied by increasing reduced mass of the anions. Consequently both properties result in shifts to lower vibrational frequencies and thus do not allow the discrimination between reduced mass and force constants effects. To overcome this problem we deuterated the three ethyl groups in the ammonium cations for the PILs Ia–IIIa. Without changing the anion–cation interaction strength we are now able to measure the frequency shifts stemming from the reduced masses only.
The measured FIR spectra cover the frequency range from 20 to 220 cm−1 (see Fig. 18). The frequencies above 180 cm−1 can be clearly assigned to intramolecular vibrational modes mainly of the anion as described earlier.127,135 In ILs II and III including CF3SO3− and NTf2− the vibrational modes above 200 cm−1 result from wagging modes of the OSO groups within the anions.
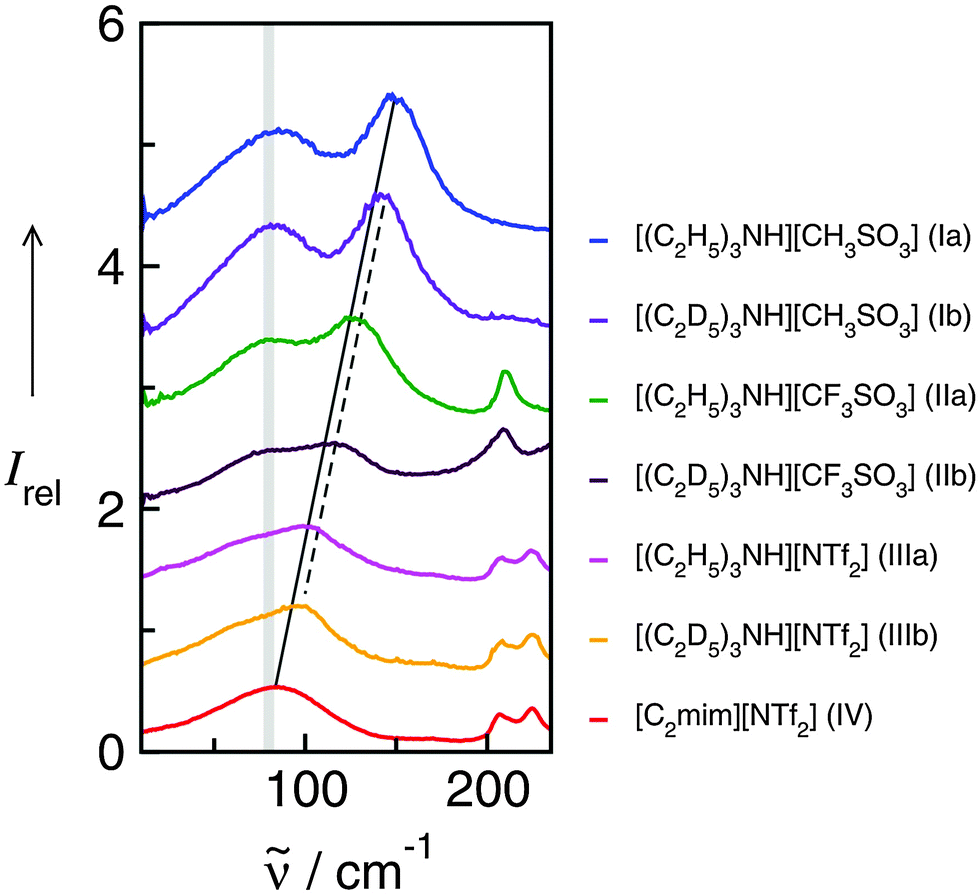 | ||
| Fig. 18 Far infrared spectra of [(C2H5)3NH][CH3SO3] (Ia), [(C2H5)3NH][CF3SO3] (IIa), [(C2H5)3NH][NTf2] (IIIa), and [C2mim][NTf2] (IV) and the corresponding protic ionic liquids for the fully deuterated ethyl groups of the ammonium cation (Ib–IIIb). The vibrational band at 149 cm−1 for Ia can be assigned to the +N–H⋯CH3SO3− intermolecular interaction. This interaction strength is decreasing with the series of Ia–IIIa as indicated by the straight line. For the deuterated PILs the reduced mass effect is shown by the dotted line. Reprinted from ref. 135 with permission. Copyright 2013 Wiley VCH. | ||
In the low frequency range only contributions from intermolecular interaction can be observed. The intramolecular bending and rotational contributions of the cation ethyl groups are relatively weak compared to the distinct local interactions +N–H⋯A−. Two main features characterize the low frequency spectra of the ILs. A broad and unspecific vibrational mode at about 80 cm−1 is attributed to librational modes overlapped by bending modes of H-bonds. This interpretation is supported by calculated frequencies of IL clusters. The most important vibrational mode is observed at 149.4 cm−1 for Ia and shifts down to 105.2 cm−1 for IIIa. The corresponding mode of APIL IV is detected at 83.2 cm−1, wherein this contribution cannot be separated from the librational modes. Some time ago this interpretation raised the controversy whether the contribution at 83 cm−1 in APILs such as [C2mim][NTf2] can be assigned to anion–cation interaction and H-bonding at all.55 In this work we could clearly address and answer this question. By increasing the anion–cation interaction due to enhanced H-bonding this vibrational mode continuously moves to higher frequencies as displayed by the straightline in Fig. 18. However, not the entire frequency shift from 149.4 cm−1 to 83.2 cm−1 for Ia to IV can be explained by decreasing interaction strength between anions and cations. Following the simple equation of the harmonic oscillator = (1/2πc)(k/μ)1/2 frequency shifts can result from both different reduced masses and different force constants indicating the interaction strength between anions and cations.
For frequencies fully determined by increasing reduced masses, a frequency shift from 149.4 cm−1 for Ia to 117.6 cm−1 for IV with Δ = 31.8 cm−1 is expected. Instead a larger frequency shift Δ = 66.2 cm−1 is observed. Consequently the additional frequency shift of about Δ = 34.4 cm−1 can be exclusively attributed to decreasing force constants due to decreasing interaction strength. Now the deuterated PILs Ib–IIIb come into play. The measured frequencies for the deuterated PILs are about 6–12 cm−1 lower than those of the protonated species in accord with the calculated frequency shifts. The trend of the frequency shifts for the deuterated PILs is shown by the dotted red line which is nearly parallel to that of the protonated species. The slope of the red dashed line is somewhat steeper than that of the solid line, because the additional mass becomes less significant for the heavier, deuterated PILs. The frequency shifts given by the straight, solid line include both increasing reduced masses and decreasing force constants (ILs Ia, IIa, IIIa and IV). The shift of the dotted line for the deuterated PILs (Ib, IIb and IIIb) versus the straightline for the protonated species displays the effect of the reduced masses only. Overall, the measurements show that frequency shifts result to the same extent from changing force constants and reduced masses.
We find perfect linear behaviour between the measured frequencies and the calculated binding energies per ion of the IL tetramers (see Fig. 19).49,52,135 The resulting linear dependency still includes reduced mass effects (filled symbols), but the expected frequency shifts due to increasing reduced masses only are given by the steeper slope (open symbols). The area between both lines presents the frequency shifts stemming from changing interaction strength only. The data analysis results in a total frequency shift of Δ = 67.8 cm−1 between PIL Ia and IV where Δ = 33.1 cm−1 and Δ = 34.7 cm−1 can be related to decreasing force constants and increasing reduced masses, respectively. Obviously, changing force constants and reduced masses contribute in an equal way to the measured frequency shift.
 | ||
| Fig. 19 Density functional calculated energies per ion of tetramers for ILs Ia–IV plotted versus the corresponding vibrational modes of the intermolecular interaction +N–H⋯anion (filled symbol). The open symbol indicates the frequencies which are expected if only the reduced mass contributes to the frequency shift. PIL Ia (149.4 cm−1) is chosen as a reference system. The shaded area gives the frequency shifts due to increasing force constants indicating the interaction strength between anion and cation. The bar for Ia indicates the accuracy of the calculated binding energies per ion by taking different conformers into account for Ia. Reprinted from ref. 135 with permission. Copyright 2013 Wiley VCH. | ||
Finally we could relate the frequency shifts to changing interaction strength between anions and cations in the ILs. A redshift of about 1 cm−1 for the vibrational band corresponds to lower interaction energy between cations and anions of about 1.69 ± 0.06 kJ mol−1. Although this relation was derived mainly from a set of PILs, it should also hold for APILs.
4.4 Frequency shifts due to increasing temperature and phase transition and their relation to changing interaction energies
In the foregoing study we presented a reliable estimate for changing interaction strength between anions and cations in APILs and PILs. Frequency shifts in FIR spectra could be referred to stronger or weaker interactions in these Coulomb fluids. This relation opens a new path for studying the variation of interaction strength due to the changing temperature or the physical environment.131 Frequency shifts observed for the solid–liquid phase transition could give some estimate of enthalpies of melting. Another possibility is to analyze changing interaction energies of ILs in solution by using solvents of different polarity. In Fig. 20 FIR spectra of PIL triethylammonium methylsulfonate [(C2H5)3NH][CH3SO3] (TEAMS) are shown as a function of temperature. The maxima of the vibrational bands between 147 and 158 cm−1 can be assigned to the +N–H⋯CH3SO3− intermolecular interaction. With increasing temperature from 273 to 303 K for the solid phase and from 313 to 353 K for the liquid phase, both vibrational bands are shifted by about 2 cm−1 to lower frequencies. Following the above given relationship between the wavenumber and energy such a shift indicates weaker interaction in the order of 3.4 kJ mol−1. The solid to liquid phase transition taking place between 303 and 313 K results in a frequency shift of about 7 cm−1 corresponding to a change in the energy of about 12 kJ mol−1. Unfortunately, the enthalpy of melting is not available for TEAMS. However, the measured enthalpies of melting for similar ammonium containing ILs range between 6.4 kJ mol−1 for trimethyl-butyl-ammonium bis(trifluoromethylsulfonyl)imide and 15.2 kJ mol−1 for dimethyl-propyl-butyl-ammonium bis(trifluoromethylsulfonyl)imide, respectively.137 These data for related compounds are close to the predicted value for TEAMS obtained from FIR spectroscopy.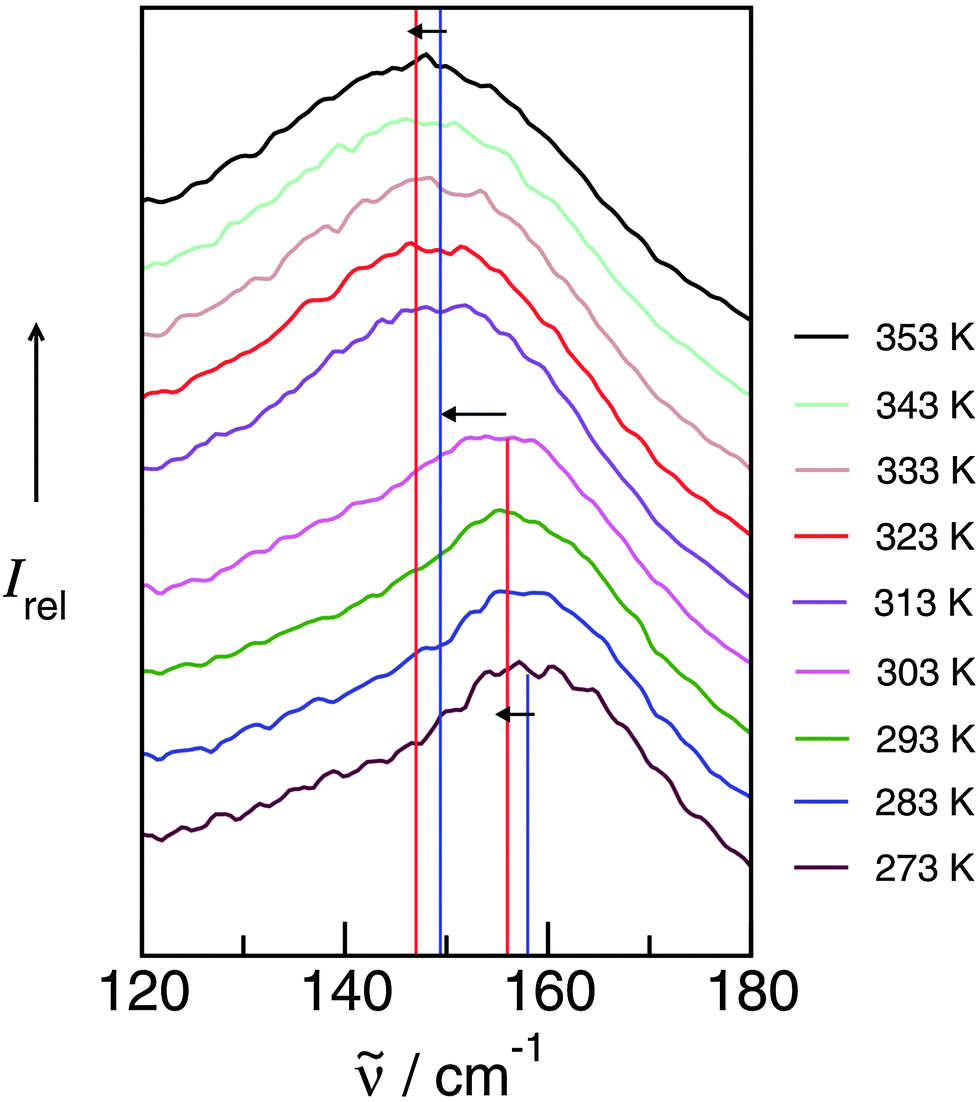 | ||
| Fig. 20 FIR spectra of triethylammonium methylsulfonate [(C2H5)3NH][CH3SO3] as a function of temperature. The maxima of the vibrational bands between 147 and 158 cm−1 can be assigned to the +N–H⋯CH3SO3− intermolecular interaction. This interaction strength is decreasing with increasing temperature and with the solid to liquid phase transition as indicated by the arrows. Reprinted from ref. 136 with permission. Copyright 2013 Elsevier. | ||
4.5 Ion speciation
An important feature affecting the properties of ILs is ion pair formation. Ion pairing in solution results from the subtle balance between ion–solvent interaction and cation–anion interaction.138–142 This in particular holds for ILs where a mélange of Coulomb forces, hydrogen bonds and dispersion forces between cations and anions results in characteristic macroscopic properties of these Coulomb fluids.143–145 For sufficiently strong anion–cation interaction contact ion pairs (CIPs) are the dominant structures. If the ion–solvent interaction dominates, solvent shared and solvent-separated ion pairs (SIPs) prevail.138For PILs including trialkylammonium as the cation, ‘quasi’ CIPs can already exist in the neat Coulomb fluid. Additional to Coulomb interaction a strong and directional hydrogen bond between the NH of the cation and the oxygen of the anion results in strongly bound CIPs for the pure PIL.127 First, we show qualitatively that FIR spectroscopy in combination with DFT methods allows us to study ion speciation of PILs in aqueous solutions. With this combined approach we expected to observe the transfer from CIPs to SIPs in aqueous solutions of triethylammonium methylsulfonate (TEAMS). The water concentrations were 50, 70, 80 and 90 mol%. The background spectra of both the PIL and water were measured for eliminating the contributions of the pure ionic and the pure molecular liquids, respectively. As discussed before for TEAMS, the vibrational band at 150 cm−1 in the FIR spectrum can be referred to the cation–anion interaction along the +N–H⋯O− bond.127,146 The well known feature of the low frequency water spectrum is the broad vibrational band at around 200 cm−1 which is assigned to the intermolecular interaction within the extended hydrogen bonding network of water.48,131–134 In Fig. 21 the difference spectra of the TEAMS–water mixtures measured against the pure TEAMS and the pure water samples are shown. The clue of these measurements is discussed for the difference spectra of the 50 mol% PIL–water mixture. If the pure water spectrum is subtracted from the mixture spectrum, positive contributions can stem from vibrational modes of the remaining internal TEAMS or arising TEAMS–water interactions. Negative intensities can be referred to missing pure water contributions. Contrarily, if the pure TEAMS spectrum is subtracted from the mixture spectrum, positive contributions result from water–water or TEAMS–water interactions, whereas negative intensities can be assigned to decreasing TEAMS concentration.
 | ||
| Fig. 21 Far infrared difference spectra of the mixtures of [(C2H5)3NH][CH3SO3] (TEAMS) with water. The spectra were obtained by subtracting the pure water (red line) and the pure TEAMS (blue line) contributions as background. The vibrational band at about 150 cm−1 in both difference spectra could be assigned to remaining or missing contributions from +N–H⋯CH3SO3− intermolecular interaction. With increasing water concentration this vibrational mode disappears and a new maximum occurs at about 180 cm−1 above 80 mol% of water (indicated by the grey bar). Reprinted from ref. 146 with permission. Copyright 2013 Wiley VCH. | ||
So far we have reported that both difference spectra of the 50 mol% mixture show the vibrational mode at 150 cm−1 supporting that CIPs are fully present. The TEAMS–water vibrational modes can be found in the same frequency range as those of water. By constantly increasing the water concentration from 50 to 90 mol%, the difference spectra change typically. The characteristic vibrational mode at 150 cm−1 describing the cation–anion interaction of the CIPs strongly loses intensities and finally cannot be observed as distinct bands anymore. However, an additional vibrational band occurs at around 180 cm−1. In the other difference spectra the negative bands of the TEAMS are better structured. It can be concluded that CIPs are strongly reduced in the mixtures with increasing water concentration. At the same time the TEAMS–water contributions increase in intensity. The cross-over point for the difference spectra is observed at the same frequency at about 180 cm−1. Notably its intensity increases with increasing water concentration. However, these contributions cannot stem from anion–water interactions which are observed at 200 cm−1. Furthermore, at these concentrations all proton acceptor sites of the anion are saturated. We claim that due to the transfer from CIPs to SIPs in particular above 80 mol% water, cation–water interaction via N–H⋯O (water) occurs and changes the difference spectra. Our interpretation of the measured difference spectra is assisted by DFT calculations of an isolated contact ion pair and hydrated CIPs and SIPs of TEAMS (see Fig. 22).
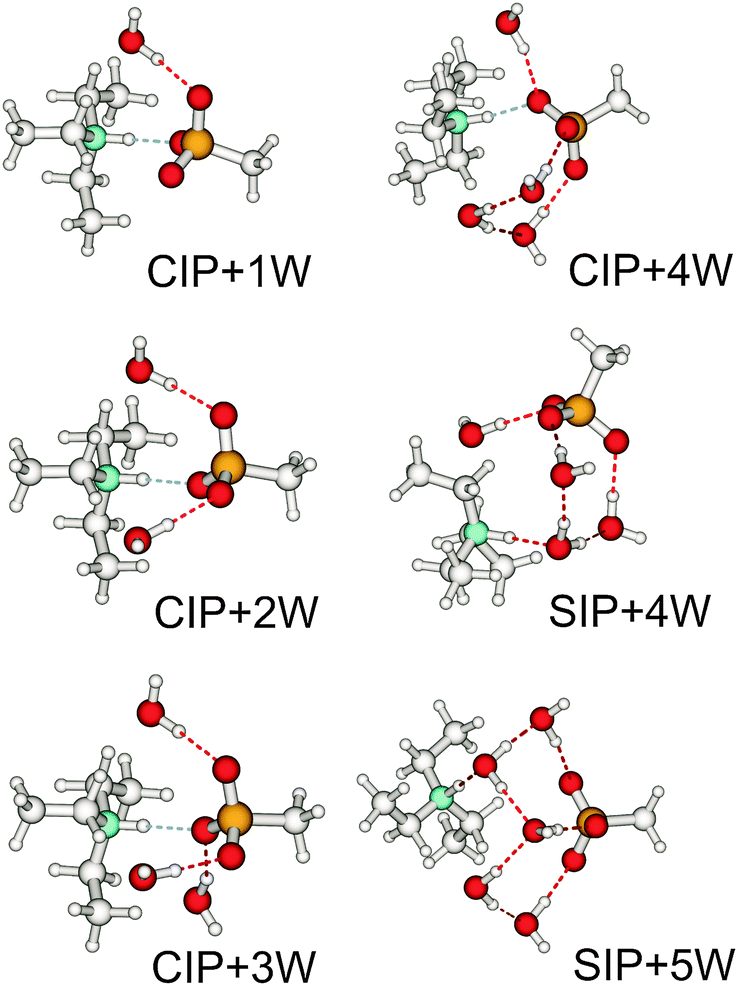 | ||
| Fig. 22 Density functional calculated clusters of [(C2H5)3NH][CH3SO3] (TEAMS) ion pairs and different numbers of water molecules representing 50, 66, 75, 80 and 83 mol% water concentration. Starting with the configurations including four water molecules (4 W) the solvent-separated ion pairs (SIP) are enthalpically favoured over the contact ion pairs (CIP). The intermolecular interaction between cation and anion (+N–H⋯anion, blue dotted line) is replaced by the intermolecular interaction between the cation and water (+N–H⋯water, red dotted line). In these SIP configurations (e.g. SIP + 5 W) one additional H-bond can be formed due to the coordination ability of the water molecules. Reprinted from ref. 146 with permission. Copyright 2013 Wiley VCH. | ||
The calculated binding energies of all CIP and SIP configurations are shown in Fig. 23. For the configuration with four and more water molecules SIPs are better in energy than CIPs. The reason is that SIP configurations provide increased potential for H-bond formation. The OH proton donors of water can be integrated in H-bonded structural motifs. The enthalpic preference of SIPs between 20 and 30 kJ mol−1 holds for all configurations including four or more water molecules.
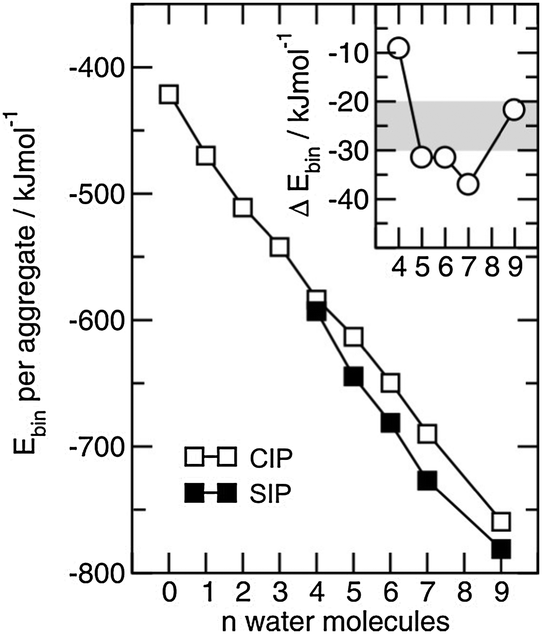 | ||
| Fig. 23 Total binding energies for clusters of [(C2H5)3NH][CH3SO3] (TEAMS) ion pairs and different numbers of water molecules representing water concentrations between 50 and 90 mol%. It is shown that the solvent-separated ion pairs (SIP) are energetically favored over the contact ion pair (CIP) configurations including four or more water molecules (filled symbols). The energies are lower by about 20–30 kJ mol−1 due to the formation of one additional H-bond in the SIP (see inlay). Reprinted from ref. 146 with permission. Copyright 2013 Wiley VCH. | ||
Here we measured the FIR difference spectra of PIL–water mixtures for studying ion speciation. Vibrational bands representing the cation–anion interaction can be distinguished from those stemming from anion–water or cation–water interactions. Changing difference spectra can be attributed to the transfer from CIPs to SIPs with increasing water concentration. Minimum four water molecules are needed to activate this process. The SIPs are enthalpically favored over the CIPs due to increasing H-bond possibilities and cooperative effects. This interpretation is supported by calculated structures, energies and frequencies of hydrated PIL ion pairs. The transfer from contact to solvent separated configurations strongly depends on the interaction power of the PILs anion. If the anion CH3SO3− is substituted by CF3SO3− the cation–anion interaction is reduced and the transfer process may begin at lower water concentration.
4.6 Network formation
For the PILs in the foregoing studies only linear hydrogen bonds but no two- or threedimensional H-bond networks could be formed. This situation is different for mono- or dialkyl ammonium cations providing two or three proton donor functions. The resulting network structures for PILs may be comparable to those of water.51 We checked that by measuring the FIR spectra of the neat PILs ethylammonium nitrate (EAN), propylammonium nitrate (PAN), and dimethylammonium nitrate (DMAN) in the range between 30 and 600 cm−1 (see Fig. 24). The spectra show some common features but also significant differences. Because the anion (NO3−) is the same for all PILs under consideration, the differences can only stem from weak intramolecular vibrations of the various cations and/or from specific cation–anion interactions.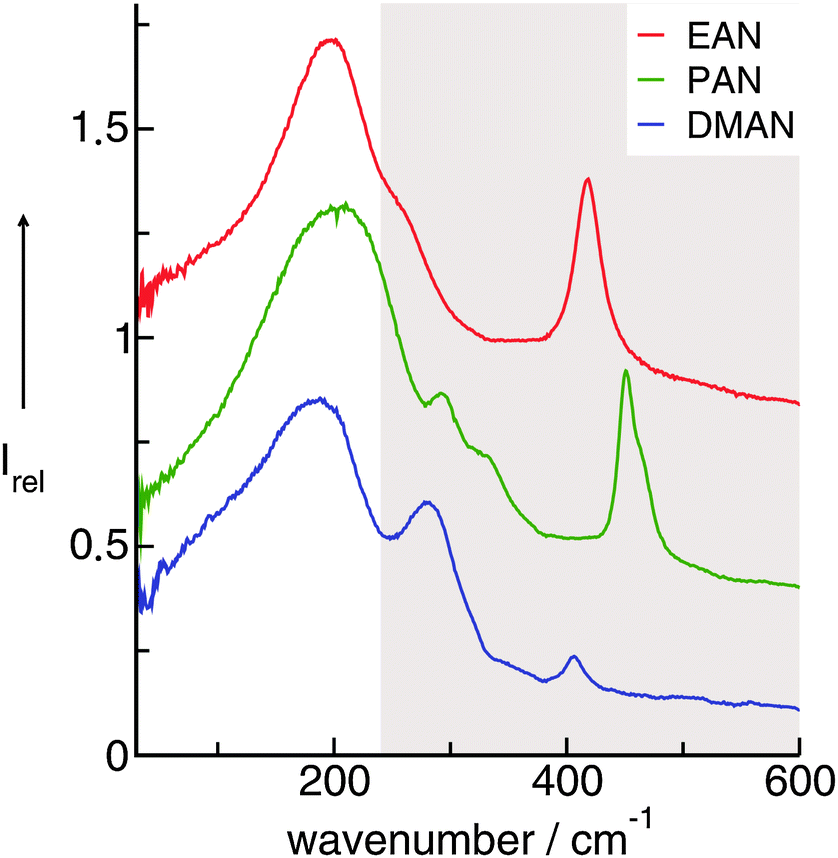 | ||
| Fig. 24 FIR spectra of ethylammonium nitrate (EAN), propylammonium nitrate (PAN), and dimethylammonium nitrate (DMAN) measured at 353 K. Reprinted from ref. 51 with permission. Copyright 2009 Wiley VCH. | ||
The calculated frequencies of the PIL clusters suggest that the bands between 199 and 224 cm−1 as well as between 134 and 159 cm−1 can be attributed to the asymmetric and symmetric stretching modes of the hydrogen bonds NH⋯O (νas(HBs) and νs(HBs)). The vibrational bands between 60 and 78 cm−1 can be attributed to corresponding bending modes of these hydrogen bonds (δ(HBs)) (see Fig. 25). Similar to the intramolecular case the asymmetric stretches show higher intensities than the symmetric stretches. The frequency difference for the asymmetric and symmetric stretches for all alkyl ammonium nitrates is observed to be 65 cm−1, suggesting comparable interaction strengths between cations and anions for all PILs. Both stretching modes as well as the corresponding bending mode of the hydrogen bonds can be compared to those measured for liquid water and ice.
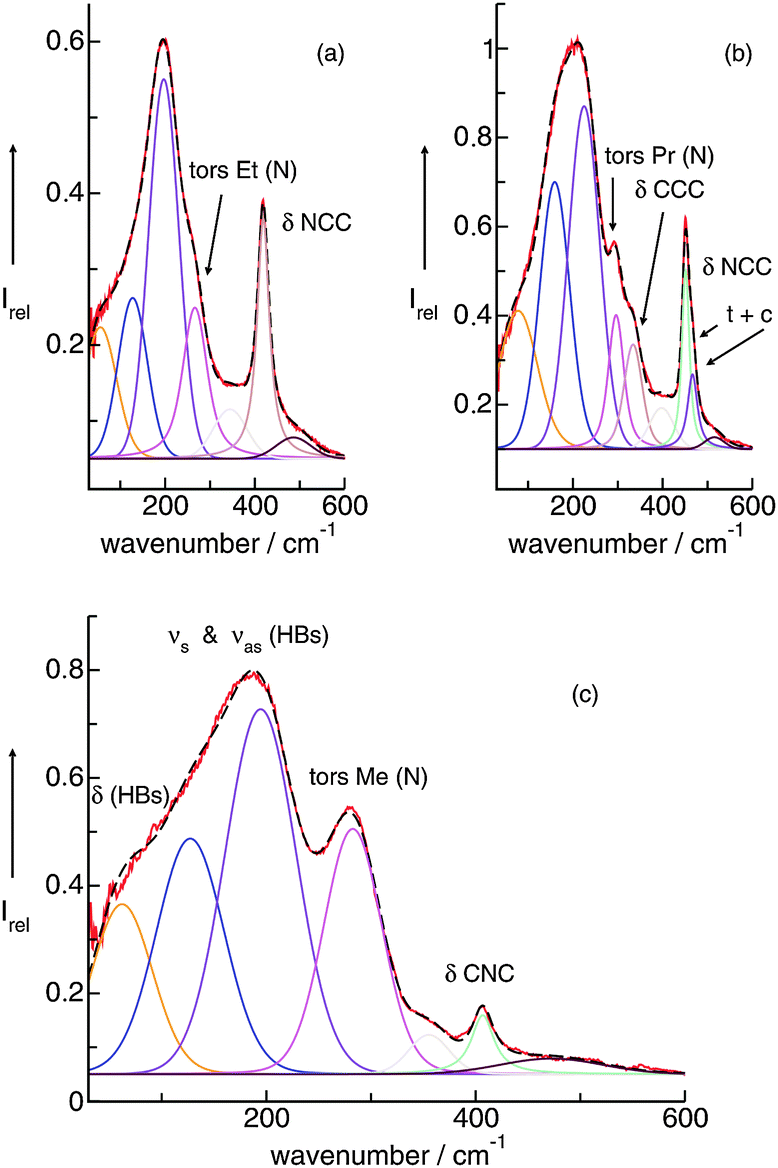 | ||
| Fig. 25 Measured FIR spectra of ethylammonium nitrate (EAN) (a), propylammonium nitrate (PAN) (b), and dimethylammonium nitrate (DMAN) (c), deconvoluted into distinct vibrational bands, which can all be assigned with the help of the frequencies calculated by DFT. For DMAN, the detailed assignment to intramolecular bending modes and intermolecular stretching and bending modes is shown. Reprinted from ref. 51 with permission. Copyright 2009 Wiley VCH. | ||
The characteristic vibrational bands of liquid water in the low-frequency range up to 250 cm−1 are well known. Cluster calculations support that stretching modes of the OH⋯O hydrogen bonding stretching modes occur at 200 cm−1.131 In ice Ih at 263 K, a major sharp band is observed at 215 cm−1 and a minor sharp band is found at 155 cm−1.132 Bending vibrational modes of the hydrogen bonds are out-of-alignment translations relative to the hydrogen bonds between 40–60 cm−1.133 Recently, Brubach et al.134 matched the structure of the connectivity band of water to a sum of four Gaussians. For all temperatures they obtained constant bands positioned at 88, 130, 195, and 251 cm−1. The intensities of the hydrogen bond stretches increased with decreasing temperature.
Overall, the vibrational bands of ice and liquid water in the FIR spectra can be referred to the herein measured bands at 199–224, 134–159, and 60–78 cm−1 in the PILs. This analogy indicates comparable strength of hydrogen bonding and related network structures. Obviously both water and the alkyl ammonium nitrates can form three-dimensional H-bond networks of similar strength (see Fig. 26).
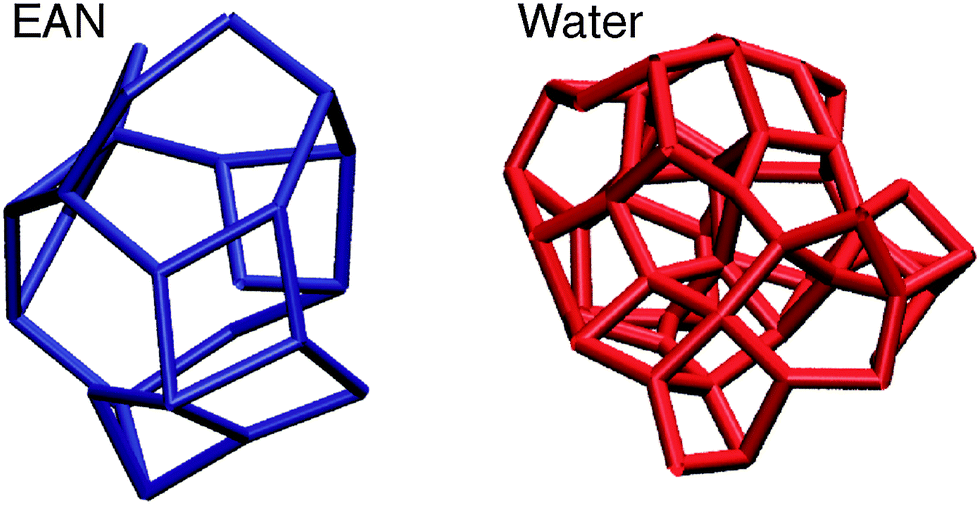 | ||
| Fig. 26 Network structures of ethylammonium nitrate (EAN) and water obtained from DFT calculations on larger clusters. For EAN the connections between the nitrogen atoms of the cation and anion are shown, whereas for water the oxygen–oxygen distances are displayed. Both liquids have three-dimensional networks, but a tetrahedral network is only present for water. Reprinted from ref. 51 with permission. Copyright 2009 Wiley VCH. | ||
In this work we presented FIR spectra of PILs EAN, PAN and DMAN. Supported by DFT calculations, the deconvoluted vibrational bands could be assigned to intermolecular bending and stretching modes of the hydrogen bonds +NH⋯O−. The characteristic features of the low-frequency spectra of PILs resemble those of water because both liquids form three-dimensional network structures. However, only water exhibits a tetrahedral H-bond network. Such a network is impossible for EAN because of different donor–acceptor abilities of the ions.147–150
5. Influence of H-bonding on APIL and PIL properties
As discussed in Chapter 3.4 several strategies have been proposed to fluidize ILs. The most promising strategy is the use of weakly polar anions which reduce the interaction energy between cations and anions with the result that melting points and viscosities decrease.151 Another opportunity is the increase of asymmetry for the imidazolium cation by adding different substituents at the N(1) and N(3) positions. Substituting the methyl group by an ethyl group at the C(1) position in [C1mim][NTf2] leads to lower melting points,106,152–154 lower viscosities106,155,156 and a small reduction of the enthalpies of vaporization73 for [C2mim][NTf2]. We reported a third possibility for tuning IL properties. We could show that strong, directional and localized hydrogen bonds (H-bonds) fluidize imidazolium-based ILs (see also Chapter 3.4).50,52 Thus deleting the interaction via C(2)–H due to substitution of the hydrogen atom by a methyl group increases melting points and viscosities significantly.Similar arguments concerning the preformation of ion-pairs were thoroughly discussed by Tokuda et al.155,157 and Widegren et al.158 Meanwhile there is more experimental evidence that H-bonds increase the asymmetry in the well-organized Coulomb fluids leading to more fluid ILs. In Fig. 27 measured melting points, viscosities and enthalpies of vaporization are shown for imidazolium-based ILs with [C(2)–H] and without [C(2)–CH3] H-bonds at the C(2)–H position. If the methyl group at the C(2) position is replaced by a hydrogen atom allowing the formation of H-bonds between cations and anions, the melting points and the viscosities are lowered throughout, independent of the chosen anion. It is expected that the addition of a methyl group increases the molecular volume resulting in higher melting points and viscosities.159 However, these effects do account only partially for the significant property changes.
 | ||
| Fig. 27 Melting points, viscosities and enthalpies of vaporization for imidazolium-based ionic liquids showing the differences between protonated and methylated C(2) positions in the imidazolium cation: (a) melting points of C2mim+ (squares) and C2C1mim+ (circles) containing several anions, (b) viscosities of C2mim+ (squares) and C2C1mim+ (circles) with anions NTf2− and OTf−, (diamonds) viscosities for C2mim+ cations which are methylated at the C(5) position, (c) enthalpies of vaporization of C4mim+ (squares) and C3C1mim+ (circles) with anions NTf2− and OTf−. Reprinted from ref. 79 with permission. Copyright 2009 Wiley VCH. | ||
Bonhôte et al.106 could show that methylation at the C(2)–H position in [C2mim][NTf2] increases the viscosity substantially from 34 MPa s to 88 MPa s, whereas it is only less affected (η = 37 MPa s) for methylation at the C(5)–H position. This behaviour was also found for melting points.106 Even stronger evidence stems from recent isothermographic determination of vaporization of 1-alkyl-3-methyl-imidazolium ILs. Luo et al. reported that in the case of the [C3C1mim][NTf2] cation bearing a methyl group at the C(2) position, distinctly higher enthalpies of vaporization relative to that expected for this side chain length are observed.160 This is surprising based on the fact that the molecular volume Vm is nearly identical for [C3C1mim] and [C4mim] cations having the same anion. The authors interpreted this result by considering the fact that the C(2) proton of [C4mim] can engage in strong hydrogen bonding with the anion of the IL, supporting the formation of stronger bound ion pairs. In contrast, if this hydrogen bond is removed, ΔvapH increases as shown in Fig. 27. These experimental results are in complete agreement with our findings that directional and localized H-bonds liquefy ILs.50–52
5.1 Enthalpies of vaporization
Here we show that measuring the strength of anion–cation interaction by FIR spectroscopy opens a new path for estimating enthalpies of vaporization of ILs. Instead of relating transport and thermodynamic properties it is more reliable to focus on properties which describe the cohesive energies of ILs and may quantify the energetics involved in the vaporization process.53,161 We measured FIR spectra of 11 ILs including the same 1-ethyl-3-methyl-imidazolium cation (C2mim+) and various anions bis(trifluoromethylsulfonyl)imide (NTf2−), ethylsulfate (EtSO4−), dicyanamide (N(CN)2−), tricyanomethide (C(CN)3−), tetracyanoborate (B(CN)4−), butylsulfate (BuSO4−), nitrate (NO3−), tetrafluoroborate (BF4−), tris(pentafluoroethyl)trifluorophosphate (PF3(C2F5)3− or FAP), trifluoromethylsulfonate (CF3SO3− or OTf−), and thiocyanate (SCN−). All spectra are shown in Fig. 28.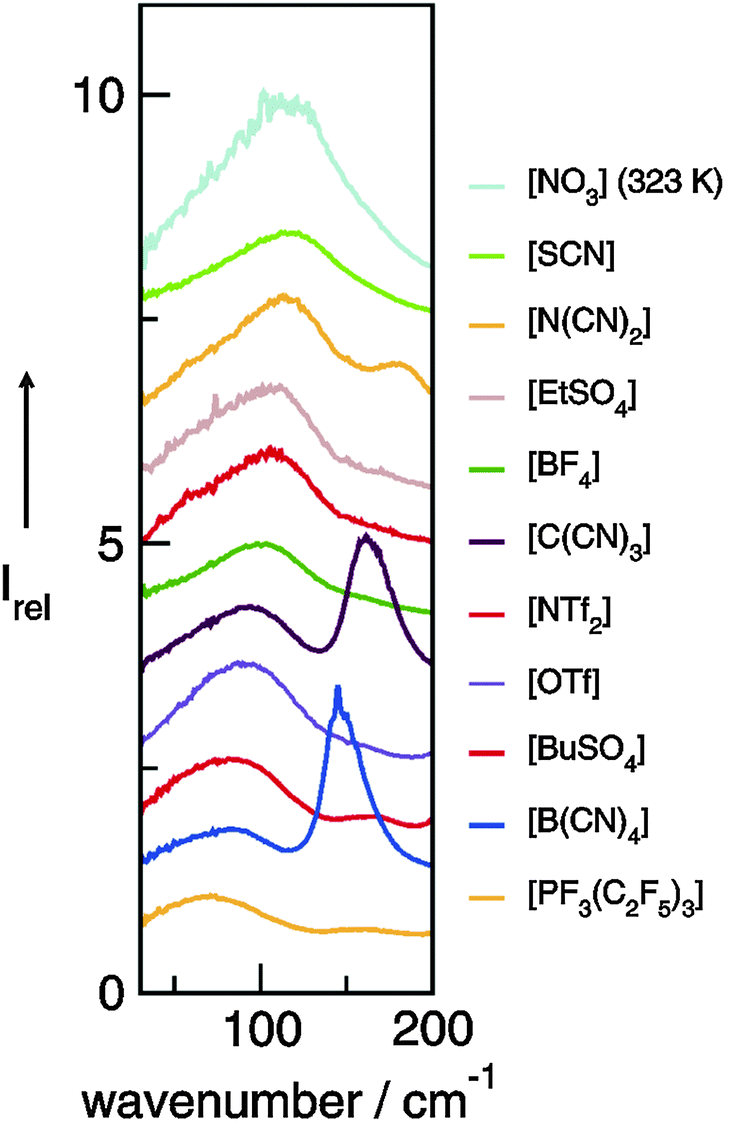 | ||
| Fig. 28 Far infrared spectra of ionic liquids including the same 1-ethyl-3-methylimidazolium cation (C2mim) but various anions bis(trifluoromethylsulfonyl)imide (NTf2−), ethylsulfate (EtSO4−), dicyanamide (N(CN)2−), tricyanomethide (C(CN)3−), tetracyanoborate (B(CN)4−), butylsulfate (BuSO4−), nitrate (NO3−), tetrafluoroborate (BF4−), tris(pentafluoroethyl) trifluorophosphate (PF3(C2F5)3−, FAP), trifluoromethanesulfonate (CF3SO3−, OTf−), and thiocyanate (SCN−), respectively. Reprinted from ref. 53 with permission. Copyright 2009 Wiley VCH. | ||
The observed maxima in the measured FIR spectra mainly coincide with those obtained from the deconvolution procedure into Voigt functions. The most accepted values of the enthalpies of vaporization of some imidazolium-based ILs are then plotted versus wavenumbers of the vibrational bands obtained from the deconvolution procedure (Fig. 29). We obtained a linear relationship between enthalpies of vaporization and the intermolecular vibrational frequencies. Lower enthalpies of vaporization correspond to lower wavenumbers and higher ΔvapH values can be assigned to higher frequencies. By fitting the experimental data we obtained a linear equation [eqn (2)]:
| ΔvapH = 75.1 kJ mol−1 + 0.75 kJ mol−1 cm−1 × | (2) |
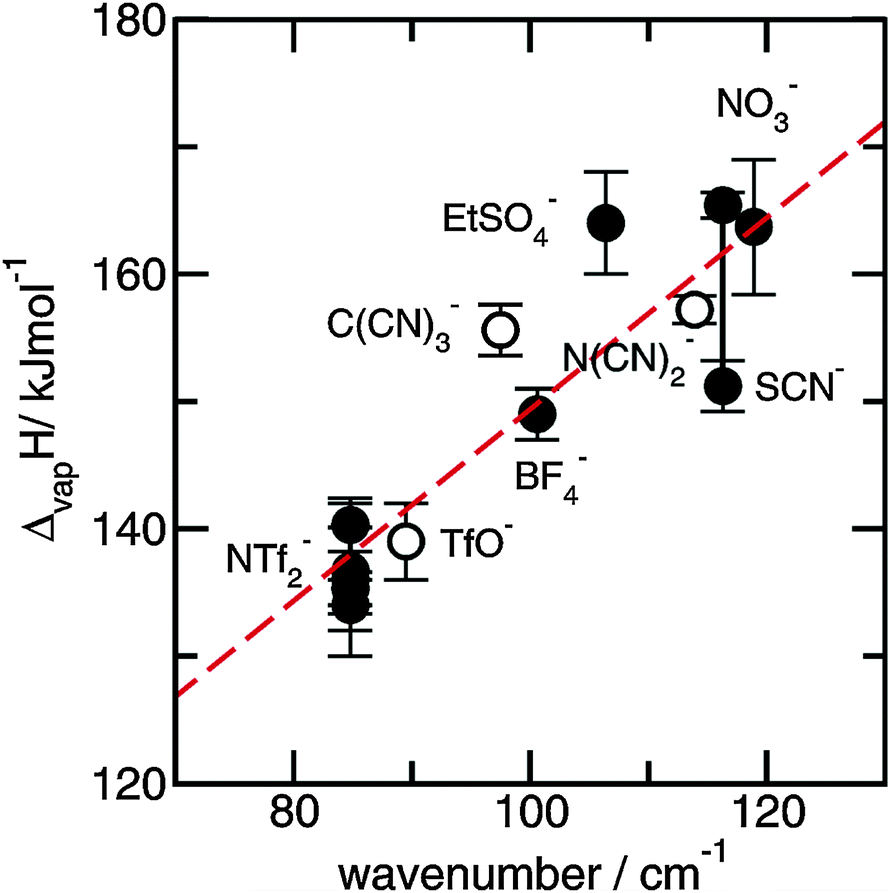 | ||
| Fig. 29 Measured maxima of the low frequency vibrational bands from far infrared spectra plotted versus enthalpies of vaporization taken from literature. A linear relation could be achieved. Deviations are smaller than those assumed for the experimental ΔvapH values. Reprinted from ref. 53 with permission. Copyright 2010 Wiley VCH. | ||
5.2 Melting points
We could show in the two foregoing chapters that ILs are salts with surprisingly low melting temperatures which is of importance for any application.162 The working temperature range of ILs is set by the melting and boiling or decomposition temperature. In particular the melting points (Tm) vary substantially for reasons presently not fully understood.117–123 We can show here that the melting points of imidazolium-based ILs are decreased by about 100 K if an extended ionic and hydrogen bond network is disrupted by localized interactions, which might be hydrogen bonds as well.To exclude any effects from varying anions, we have chosen a set of 8 ILs, all with the same bis(trifluoromethylsulfonyl)imide anion. Only the cation is different: imidazolium (I), 1-methyl-imidazolium (II), 1,3-dimethyl-imidazolium (III), 1,2-dimethyl-imidazolium (IV), 2,4,5-trimethyl-imidazolium (V), 1,2,3-trimethyl-imidazolium (VI), 1,2,4,5-tetramethyl-imidazolium (VII), and 1,2,3,4,5-pentamethyl-imidazolium (VIII) (see Fig. 30).
 | ||
| Fig. 30 Structures of imidazolium-based cations of imidazolium (I), 1-methyl-imidazolium (II), 1,3-dimethyl-imidazolium (III), 1,2-dimethyl-imidazolium (IV), 2,4,5-trimethyl-imidazolium (V), 1,2,3-trimethyl-imidazolium (VI), 1,2,4,5-tetramethyl-imidazolium (VII), and 1,2,3,4,5-pentamethyl-imidazolium (VIII). The different positions of H-bond abilities are indicated by bold dots and the remaining interaction sites as dotted lines. Reprinted from ref. 144 with permission. Copyright 2011 Wiley VCH. | ||
All melting points for ILs I–VIII are now plotted versus ab initio computed volumes (A− + C+) of anions and cations (Fig. 31). It was shown by Krossing et al. and Markusson et al. that the calculated volumes are in good agreement with those obtained from crystal structure or density measurements.151,163 As shown in Fig. 31 no linear trend for Tmversus volume can be observed. The highest melting points are found for ILs I and VIII. Both of these cations, imidazolium and 1,2,3,4,5-penta-methyl-imidazolium do not exhibit specific interaction sites. In principle this is not true for IL I which owns five interaction sites via C–H or N–H bonds as indicated by the black dotted lines in Fig. 30. However, none of these interaction sites is favored over others and therefore not specific. In Fig. 31 it is shown by the dashed line that Tm increases with increasing volume. However, Tm of IL VI can be found on that line too. This is surprising because the 1,2,3-trimethylimidazolium cation exhibits interaction sites for the anion via C(4)–H and C(5)–H. However, it is known from recent studies that these interactions are weak compared to those via C(2)–H which is known to be strongly acidic.49,106 The major decrease in Tm of nearly 100 K is found for the ILs III, IV and VII. These ILs all provide single interaction sites via C(2)–H in III or via N–H in IV and VII. The local and directional interaction via hydrogen bonding results in preformation of ion-pairs characterized by lower “lattice energies” compared to the energy of isolated ion-pairs. Obviously, the preformation of ion-pairs increases the quasi-molecular character of the pure ILs. Dupont suggested a similar idea for adding solvent molecules and macromolecules to ILs.139 He argued that the degradation of the H-bond network results in nanostructures with polar and non-polar regimes.164,165
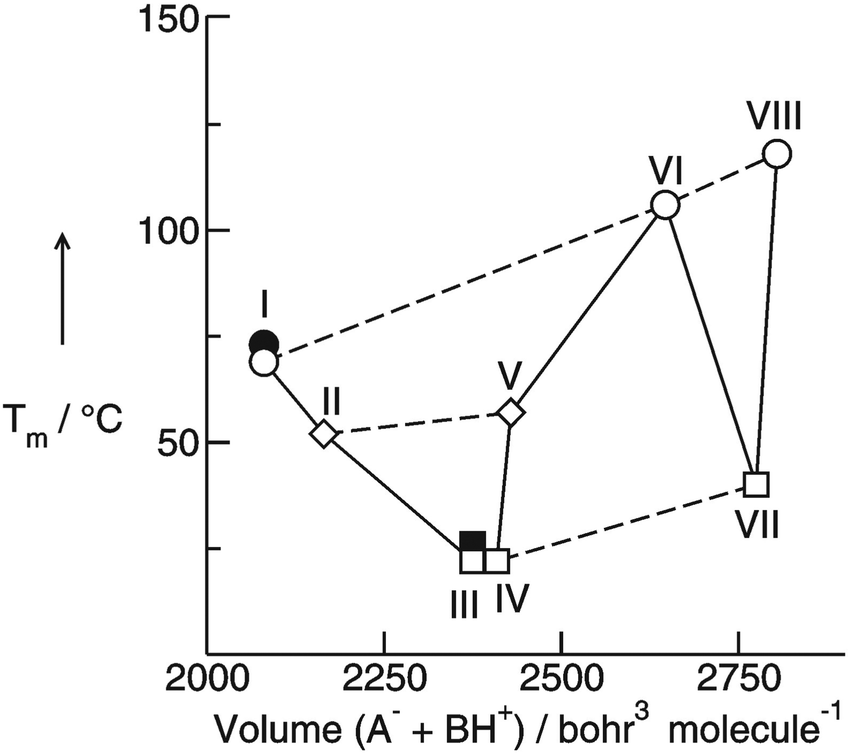 | ||
| Fig. 31 T m versus volume (A− + C+) for the ionic liquids I–VIII including various imidazolium cations and the same bis(trifluoromethylsulfonyl)imide anion (NTf2−). As indicated by the dotted lines, there is an increase in Tm with increasing volume for the ionic liquids with no specific interaction site (circles), with one interaction site (squares) and with two interaction sites (diamonds). Overall it is shown that the presence of single interaction sites at the cation leads to significant decrease in Tm as given for ILs III, IV and VII, respectively. The filled symbols indicate further Tm value from the literature. Reprinted from ref. 144 with permission. Copyright 2011 Wiley VCH. | ||
Obviously, there is strong evidence that Tm can be lowered significantly if single local and directional interactions such as H-bonding are present. For the imidazolium-based ILs under investigation these effects can add up to 100 K. Such a decrease in the melting point significantly expands the working temperature range of ILs. This phenomenon has been observed already by Bonhôte et al. some time ago. His group synthesized ILs including imidazolium cations and perfluorinated hydrophobic anions.106 They recognized significant H-bonding via C(2)–H and hoped for lower melting points by replacing the proton at the C(2) position by alkyl groups. The opposite behavior was observed. Alkylation increased the melting points and viscosities. However, Bonhôte et al. put their focus on synthesizing low melting and highly conductive ILs for solar cell applications and did not consider this phenomenon in detail. Here we could show that hydrogen bonding in ILs can have opposite effects on properties compared to that expected for H-bonded molecular liquids.
6. Conclusions and perspectives
Summarizing some of our results from FIR and THz spectroscopy combined with ab initio and DFT calculations on larger clusters of aprotic and protic ionic liquids we can conclude the following: FIR and THz are suitable methods for studying the anion–cation interaction in ionic liquids. By varying anions and cations in a systematic way it could be shown that increasing interaction strength leads to frequency shifts to higher wavenumbers due to increasing force constants indicating stronger interaction between the ion species. Further on it is demonstrated that although hydrogen bonding contributes only about 10% of the overall interaction energy in aprotic ionic liquids, the local and directional nature of this type of interaction significantly influences the unique properties of this liquid material. For protic ionic liquids it could be shown that the vibrational band assigned to the anion–cation interaction can be well separated from other low-frequency vibrational modes due to librational and rattling motions. Stronger H-bonds further shift the frequencies to higher wavenumbers and result in distinguished vibrational bands which can be used for studying the interaction strength as a function of temperature and phase transition. By using isotopic substitution we could dissect the frequency shifts into contributions stemming from changing reduced masses and force constants. The resulting relationships between frequencies and energies open a new path for studying the variation of interaction strength due to the changing temperature or physical environment. Overall also access to thermophysical properties of ionic liquids such as heat capacities and enthalpies of vaporization is provided. The linear behavior between the FIR frequencies and enthalpies of vaporization as introduced recently still holds, but it is not corrected for reduced mass effects. Overall, the relation between FIR frequency shifts and changing interaction strengths provides further options. Frequency shifts observed for the solid–liquid phase transition could give some estimate for enthalpies of melting. Another possibility is to analyze changing interaction energies for ionic liquids in solution by using solvents of different polarity. In diluted ionic liquids it could be studied whether ion-pair or cluster formation leads to stronger anion–cation interaction compared to that in the neat liquid. Furthermore we could demonstrate that FIR spectroscopy is an interesting method to investigate ion speciation at least in protic ionic liquids. Spectral features can be clearly assigned to contact and solvent separated ion pairs. In principle, FIR spectroscopy in combination with DFT calculations and/or MD simulations can be used for studying not only ionic liquids but all H-bonded molecular liquids and their mixtures with different polar solvents.166–170 Another perspective is the study of dissolution and precipitation of molecules and nanoparticles in ionic liquids. Our approach could be used for a better understanding of the role of these ‘composite’ ionic liquids as templates, stabilizers and immobilizing agents. Little is known about how the properties of ionic liquids and ionic liquids mixtures depend on the amount or chemical identity of additives such as solvents, salts or nanoparticles. Some of these investigations are currently on the way in our laboratories.Acknowledgements
This work was supported by the DFG priority programme SPP 1191. Financial support was also provided by the DFG collaborative research center SFB 652.References
- G. P. Pimentel and A. L. McClellan, The hydrogen bond, W. H. Freeman and Co., San Francisco, 1960 Search PubMed.
- W. J. Hurley, I. D. Kuntz and G. E. Leroi, J. Am. Chem. Soc., 1966, 88, 3199–3202 CrossRef CAS.
- R. F. Lake and H. W. Thompson, Proc. R. Soc. London, Ser. A, 1966, 291, 469–477 CrossRef.
- P. R. Griffiths and C. Homes, Instrumentation for Far-infrared in Spectroscopy in Handbook of Vibrational Spectroscopy, John Wiley & Son Ltd., 2006 Search PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- F. Endres and S. Z. El Abedin, Phys. Chem. Chem. Phys., 2006, 8, 2101–2116 RSC.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2077–2084 CrossRef PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CAS.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- P. Wasserscheid, Nature, 2006, 439, 797 CrossRef CAS PubMed.
- D. H. Zaitsau, G. J. Kabo, A. A. Strechan, Y. U. Paulechka, A. Tschersich, S. P. Verevkin and A. Heintz, J. Phys. Chem. A, 2006, 110, 7303–7306 CrossRef CAS PubMed.
- R. Ludwig and U. Kragl, Angew. Chem., 2007, 119, 6702–6704 CrossRef CAS; R. Ludwig and U. Kragl, Angew. Chem., Int. Ed., 2007, 46, 6582–6584 CrossRef PubMed.
- J. M. S. S. Esperança, J. N. Canongia Lopes, M. Tariq, L. M. N. B. F. Santos, J. W. Magee and L. P. N. Rebelo, J. Chem. Eng. Data, 2010, 55, 3–12 CrossRef.
- S. P. Verevkin, Angew. Chem., 2008, 120, 5149–5152 CrossRef CAS; S. P. Verevkin, Angew. Chem., Int. Ed., 2008, 47, 5071–5074 CrossRef PubMed.
- H. Shirota, ChemPhysChem, 2012, 13, 1638–1648 CrossRef CAS PubMed.
- E. W. Castner Jr., J. F. Wishart and H. Shirota, Acc. Chem. Res., 2007, 40, 1217–1227 CrossRef PubMed.
- B. R. Hyun, S. V. Dzyuba, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. A, 2002, 106, 7579–7585 CrossRef CAS.
- J. R. Rajian, S. F. Li, R. A. Bartsch and E. L. Quitevis, Chem. Phys. Lett., 2004, 393, 372–377 CrossRef PubMed.
- D. Xiao, J. R. Rajian, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. B, 2006, 110, 16174–16178 CrossRef CAS PubMed.
- D. Xiao, J. R. Rajian, A. Cady, S. Li, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. B, 2007, 111, 4669–4677 CrossRef CAS PubMed.
- D. Xiao, J. R. Rajian, L. G. Hines Jr., S. Li, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. B, 2008, 112, 13316–13325 CrossRef CAS PubMed.
- D. Xiao, L. G. Hines Jr., R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. B, 2009, 113, 4544–4548 CrossRef CAS PubMed.
- D. Xiao, L. G. Hines Jr., S. Li, R. A. Bartsch, E. L. Quitevis, O. Russina and A. Triolo, J. Phys. Chem. B, 2009, 113, 6426–6433 CrossRef CAS PubMed.
- O. Russina, A. Triolo, L. Gontrani, R. Caminiti, D. Xiao, L. G. Hines Jr., R. A. Bartsch, E. L. Quitevis, N. Plechkova and K. R. Seddon, J. Phys.: Condens. Matter, 2009, 21, 424121 CrossRef.
- D. Xiao, L. G. Hines Jr., M. W. Holtz, K. Song, R. A. Bartsch and E. L. Quitevis, Chem. Phys. Lett., 2010, 497, 37–42 CrossRef CAS PubMed.
- P. Yang, G. A. Voth, D. Xiao, L. G. Hines Jr., R. A. Bartsch and E. L. Quitevis, J. Chem. Phys., 2011, 135, 034502 CrossRef PubMed.
- G. Giraud, C. M. Gordon, I. R. Dunkin and K. Wynne, J. Chem. Phys., 2003, 119, 464–477 CrossRef CAS PubMed.
- D. A. Turton, J. Hunger, A. Stoppa, G. Hefter, A. Thoman, M. Walther, R. Buchner and K. Wynne, J. Am. Chem. Soc., 2009, 131, 11140–11146 CrossRef CAS PubMed.
- H. Shirota, A. M. Funston, J. F. Wishart and E. W. Castner Jr., J. Chem. Phys., 2005, 122, 184512 CrossRef PubMed.
- H. Shirota and E. W. Castner Jr., J. Phys. Chem. A, 2005, 109, 9388–9392 CrossRef CAS PubMed.
- H. Shirota and E. W. Castner Jr., J. Phys. Chem. B, 2005, 109, 21576–21585 CrossRef CAS PubMed.
- H. Shirota, J. F. Wishart and E. W. Castner Jr., J. Phys. Chem. B, 2007, 111, 4819–4829 CrossRef CAS PubMed.
- H. Shirota, K. Nishikawa and T. Ishida, J. Phys. Chem. B, 2009, 113, 9831–9839 CrossRef CAS PubMed.
- T. Ishida, K. Nishikawa and H. Shirota, J. Phys. Chem. B, 2009, 113, 9840–9851 CrossRef CAS PubMed.
- T. Fujisawa, K. Nishikawa and H. Shirota, J. Chem. Phys., 2009, 131, 244519 CrossRef PubMed.
- H. Shirota, H. Fukazawa, T. Fujisawa and J. F. Wishart, J. Phys. Chem. B, 2010, 114, 9400–9412 CrossRef CAS PubMed.
- H. Fukazawa, T. Ishida and H. Shirota, J. Phys. Chem. B, 2011, 115, 4621–4631 CrossRef CAS PubMed.
- H. Shirota and T. Ishida, J. Phys. Chem. B, 2011, 115, 10860–10870 CrossRef CAS PubMed.
- K. Yamamoto, M. Tani and M. Hangyo, J. Phys. Chem. B, 2007, 111, 4854–4859 CrossRef CAS PubMed.
- M. Koeberga, C.-C. Wu, D. Kimc and M. Bonn, Chem. Phys. Lett., 2007, 439, 60–64 CrossRef PubMed.
- A. Chakraborty, T. Inagaki, M. Banno, T. Mochida and K. Tominaga, J. Phys. Chem. A, 2011, 115, 1313–1319 CrossRef CAS PubMed.
- K. Iwata, H. Okajima, S. Saha and H. Hamaguchi, Acc. Chem. Res., 2007, 40, 1174–1181 CrossRef CAS PubMed.
- M. C. C. Ribeiro, J. Chem. Phys., 2010, 133, 024503 CrossRef PubMed.
- M. C. C. Ribeiro, J. Chem. Phys., 2011, 134, 244507 CrossRef PubMed.
- A. Wulf, K. Fumino, R. Ludwig and P. F. Taday, ChemPhysChem, 2010, 11, 349–353 CrossRef CAS PubMed.
- A. Dominguez-Vidal, N. Kaun, M. J. Ayora-Canada and B. Lendl, J. Phys. Chem. B, 2007, 111, 4446–4452 CrossRef CAS PubMed.
- K. Fumino, A. Wulf and R. Ludwig, Angew. Chem., 2008, 120, 3890–3894 CrossRef CAS; K. Fumino, A. Wulf and R. Ludwig, Angew. Chem., Int. Ed., 2008, 47, 3830–3834 CrossRef PubMed.
- K. Fumino, A. Wulf and R. Ludwig, Angew. Chem., 2008, 120, 8859–8862 CrossRef CAS; K. Fumino, A. Wulf and R. Ludwig, Angew. Chem., Int. Ed., 2008, 47, 8731–8734 CrossRef PubMed.
- K. Fumino, A. Wulf and R. Ludwig, Angew. Chem., 2009, 121, 3230–3233 CrossRef CAS; K. Fumino, A. Wulf and R. Ludwig, Angew. Chem., Int. Ed., 2009, 48, 3184–3186 CrossRef PubMed.
- A. Wulf, K. Fumino and R. Ludwig, Angew. Chem., 2010, 122, 459–463 CrossRef CAS; A. Wulf, K. Fumino and R. Ludwig, Angew. Chem., Int. Ed., 2010, 49, 449–453 CrossRef PubMed.
- K. Fumino, A. Wulf, S. P. Verevkin, A. Heintz and R. Ludwig, ChemPhysChem, 2010, 11, 1623–1626 CrossRef CAS PubMed.
- C. Roth, T. Peppel, K. Fumino, M. Köckerling and R. Ludwig, Angew. Chem., 2010, 122, 10419–10423 CrossRef CAS; C. Roth, T. Peppel, K. Fumino, M. Köckerling and R. Ludwig, Angew. Chem., Int. Ed., 2010, 49, 10221–10224 CrossRef PubMed.
- T. Buffeteau, J. Grondin, Y. Danten and J.-C. Lassegues, J. Phys. Chem. B, 2010, 114, 7587–7592 CrossRef CAS PubMed.
- P. A. Hunt, B. Kirchner and T. Welton, Chem. – Eur. J., 2006, 12, 6762–6775 CrossRef CAS PubMed.
- P. A. Hunt, I. R. Gould and B. Kirchner, Aust. J. Chem., 2007, 60, 9–14 CrossRef CAS.
- F. Dommert, J. Schmidt, B. Qiao, Y. Zhao, C. Krekeler, L. D. Site, R. Berger and C. Holm, J. Chem. Phys., 2008, 129, 224501 CrossRef PubMed.
- S. Zahn, J. Thar and B. Kirchner, J. Chem. Phys., 2010, 132, 124506 CrossRef PubMed.
- E. Izgorodina, Phys. Chem. Chem. Phys., 2010, 13, 4189–4207 RSC.
- H. V. Spohr and G. N. Patey, J. Chem. Phys., 2010, 132, 154504 CrossRef PubMed.
- S. S. Sarangi, S. K. Reddy and S. Balasubramanian, J. Phys. Chem. B, 2011, 115, 1874–1880 CrossRef CAS PubMed.
- K. Wendler, M. Brehm, F. Malberg, B. Kirchner and L. D. Site, J. Chem. Theory Comput., 2012, 8, 1570–1579 CrossRef CAS.
- S. M. Urahata and M. C. C. Ribeiro, J. Chem. Phys., 2005, 122, 024511 CrossRef PubMed.
- S. M. Urahata and M. C. C. Ribeiro, J. Chem. Phys., 2006, 124, 074513 CrossRef PubMed.
- Z. Hu, X. Huang, H. V. R. Annapureddy and C. J. Margulis, J. Phys. Chem. B, 2008, 112, 7837–7849 CrossRef CAS PubMed.
- T. Ishida, J. Non-Cryst. Solids, 2011, 357, 454–462 CrossRef CAS PubMed.
- T. Y. Yan, C. J. Burnham, M. G. Del Popolo and G. A. Voth, J. Phys. Chem. B, 2004, 108, 11877–11881 CrossRef CAS.
- A. Wulf, R. Ludwig, P. Sasisanker and H. Weingärtner, Chem. Phys. Lett., 2007, 439, 323–326 CrossRef CAS PubMed.
- A. Wulf, K. Fumino, D. Michalik and R. Ludwig, ChemPhysChem, 2007, 8, 2265–2269 CrossRef CAS PubMed.
- T. Köddermann, D. Paschek and R. Ludwig, ChemPhysChem, 2007, 8, 2464–2470 CrossRef PubMed.
- R. Ludwig, Phys. Chem. Chem. Phys., 2008, 10, 4333–4339 RSC.
- T. Köddermann, D. Paschek and R. Ludwig, ChemPhysChem, 2008, 9, 549–555 CrossRef PubMed.
- T. Köddermann, R. Ludwig and D. Paschek, ChemPhysChem, 2008, 9, 1851–1858 CrossRef PubMed.
- D. Paschek, T. Köddermann and R. Ludwig, Phys. Rev. Lett., 2008, 100, 115901 CrossRef.
- K. Fumino, A. Wulf and R. Ludwig, Phys. Chem. Chem. Phys., 2009, 11, 8790–8794 RSC.
- R. Ludwig, J. Phys. Chem. B, 2009, 113, 15419–15422 CrossRef CAS PubMed.
- D. Kerlé, R. Ludwig, A. Geiger and D. Paschek, J. Phys. Chem. B, 2009, 113, 12727–12735 CrossRef PubMed.
- R. Ludwig and D. Paschek, ChemPhysChem, 2009, 10, 516–519 CrossRef CAS PubMed.
- C. A. Schmuttenmaer, Chem. Rev., 2004, 104, 1759–1779 CrossRef CAS PubMed.
- C. J. Johnson, J. A. Fournier, C. T. Wolke and M. A. Johnson, J. Chem. Phys., 2013, 139, 224305 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98 (Revision A.10), Gaussian, Inc., Pittsburgh, PA, 2001 Search PubMed.
- S. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, 03, Revision, C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. N. Lopes, J. Deschamps and A. A. H. Pádua, J. Phys. Chem. B, 2004, 108, 2038–2047 CrossRef CAS.
- H. W. Horn, W. C. Swope, J. W. Pitera, J. D. Madura, T. J. Dick, G. L. Hura and T. Head-Gordon, J. Chem. Phys., 2004, 120, 9665–9678 CrossRef CAS PubMed.
- T. Köddermann, C. Wertz, A. Heintz and R. Ludwig, Angew. Chem., 2006, 118, 3780–3785 CrossRef; T. Köddermann, C. Wertz, A. Heintz and R. Ludwig, Angew. Chem., Int. Ed., 2006, 45, 3697–3702 CrossRef PubMed.
- F. Dommert, K. Wendler, B. Qiao, L. Delle Site and C. Holm, J. Mol. Liq., 2014, 192, 32–37 CrossRef CAS PubMed.
- K. Fumino, K. Wittler and R. Ludwig, J. Phys. Chem. B, 2012, 116, 9507–9511 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- H. Zhang, J. Wu, J. Zhang and J. He, Macromolecules, 2005, 38, 8272–8277 CrossRef CAS.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staigeer, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- T. Torimoto, T. Tsuda, K. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef CAS PubMed.
- B. Kosan, C. Michels and F. Meister, Cellulose, 2008, 15, 59–66 CrossRef CAS.
- Z. Papanyan, C. Roth, D. Paschek and R. Ludwig, ChemPhysChem, 2011, 12, 2400–2404 CrossRef CAS PubMed.
- Z. Papanyan, C. Roth, S. Reimann and R. Ludwig, ChemPhysChem, 2013, 14, 3667–3671 CrossRef CAS PubMed.
- N. R. Dhumal, H. J. Kim and J. Kiefer, J. Phys. Chem. A, 2009, 113, 10397–10404 CrossRef CAS PubMed.
- V. Kempter and B. Kirchner, J. Mol. Struct., 2010, 972, 22–34 CrossRef CAS PubMed.
- S. B. C. Lehmann, M. Roatsch, M. Schöppke and B. Kirchner, Phys. Chem. Chem. Phys., 2010, 12, 7473–7486 RSC.
- J. Kiefer, M. M. Molina and K. Noack, ChemPhysChem, 2012, 13, 1213–1220 CrossRef CAS PubMed.
- A. K. Abdul-Sada, A. M. Greenway, P. B. Hitchcock, T. J. Mohammed, K. R. Seddon and J. A. Zora, Chem. Commun., 1986, 1753–1754 RSC.
- P. B. Hitchcock, K. R. Seddon and T. J. Welton, J. Chem. Soc., Dalton Trans., 1993, 2639–2643 RSC.
- J. D. Holbrey, W. M. Reichert, M. Nieuwenhuyzen, S. Johnston, K. R. Seddon and R. D. Rogers, Chem. Commun., 2003, 1636–1637 RSC.
- J. D. Holbrey, W. M. Reichert, M. Nieuwenhuyzen, O. Sheppard, C. Hardacre and R. D. Rogers, Chem. Commun., 2003, 476–477 RSC.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalynasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- J. D. Tubbs and M. M. Hoffmann, J. Solution Chem., 2004, 33, 381–394 CrossRef CAS.
- R. W. Berg, M. Deetlefs, K. R. Seddon, I. Shim and J. M. Thompson, J. Phys. Chem. B, 2005, 109, 19018–19025 CrossRef CAS PubMed.
- S. A. Katsyuba, E. E. Zvereva, A. Vidiš and P. J. Dyson, J. Phys. Chem. B, 2007, 111, 352–370 CrossRef CAS PubMed.
- T. Köddermann, C. Wertz, A. Heintz and R. Ludwig, ChemPhysChem, 2006, 7, 1944–1949 CrossRef PubMed.
- A. Yokozeki, D. J. Kasprzak and M. B. Shiflett, Phys. Chem. Chem. Phys., 2007, 9, 5018–5026 RSC.
- S. Kossmann, J. Thar, B. Kirchner, P. A. Hunt and T. Welton, J. Chem. Phys., 2006, 124, 174506 CrossRef PubMed.
- B. L. Bhargava and S. J. Balasubramanian, J. Chem. Phys., 2007, 127, 114510 CrossRef CAS PubMed.
- J.-C. Lassègues, J. Gronding, D. Cavagnat and P. Johansson, J. Phys. Chem. A, 2009, 113, 6419–6421 CrossRef PubMed.
- S. Tsuzuki, H. Tokuda and M. Mikami, Phys. Chem. Chem. Phys., 2007, 9, 4780–4784 RSC.
- Y. Jeon, J. Sung, C. Seo, H. Lim, H. Cheong, M. Kang, B. Ouchi and D. Kim, J. Phys. Chem. B, 2008, 112, 4735–4740 CrossRef CAS PubMed.
- P. A. Hunt, J. Phys. Chem. B, 2007, 111, 4844–4853 CrossRef CAS PubMed.
- T. Endo, T. Kato and K. Nishikawa, J. Phys. Chem. B, 2010, 114, 9201–9208 CrossRef CAS PubMed.
- K. Fumino, T. Peppel, M. Geppert-Rybczyńska, D. H. Zaitsau, J. K. Lehmann, S. P. Verevkin, M. Köckerling and R. Ludwig, Phys. Chem. Chem. Phys., 2011, 13, 14064–14075 RSC.
- K. Noack, P. S. Schulz, N. Paape, J. Kiefer, P. Wasserscheid and A. Leipertz, Phys. Chem. Chem. Phys., 2010, 12, 14153–14161 RSC.
- H. V. Spohr and G. N. Patey, J. Chem. Phys., 2009, 130, 104506 CrossRef CAS PubMed.
- S. Zahn, G. Bruns, J. Thar and B. Kirchner, Phys. Chem. Chem. Phys., 2008, 10, 6921–6924 RSC.
- E. I. Izgorodina, R. Maganti, V. Armel, P. M. Dean, J. M. Prigle, K. R. Seddon and D. R. MacFarlane, J. Phys. Chem. B, 2011, 115, 14688–14697 CrossRef CAS PubMed.
- T. Köddermann, K. Fumino, R. Ludwig, J. N. C. Lopes and A. A. H. Pádua, ChemPhysChem, 2009, 10, 1181–1186 CrossRef PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge, University Press, Cambridge, 2005 Search PubMed.
- K. Fumino, E. Reichert, K. Wittler, R. Hempelmann and R. Ludwig, Angew. Chem., 2012, 124, 6340–6344 CrossRef CAS; K. Fumino, E. Reichert, K. Wittler, R. Hempelmann and R. Ludwig, Angew. Chem., Int. Ed., 2012, 51, 6236–6240 CrossRef PubMed.
- M. Mylrahan and T. K. K. Srinivasan, J. Chem. Phys., 1988, 89, 1634–1641 CrossRef PubMed.
- M. Mylrahan and T. K. K. Srinivasan, J. Phys. C: Solid State Phys., 1988, 21, 1673–1690 CrossRef.
- T. Steiner, Angew. Chem., 2002, 114, 50–80 CrossRef CAS; T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef.
- V. I. Gaiduk and J. K. Vij, Phys. Chem. Chem. Phys., 2001, 3, 5173–5181 RSC.
- N. Miura, T. Kitagawa, K. I. Nishikawa and A. Moon, Joint 30th Int. Conf. in Infrared and Millimeter Waves and 13th Int. Conf. on Terahertz Electronics, 2005.
- V. I. Gaiduk and D. S. F. Crothers, J. Mol. Liq., 2006, 128, 145–160 CrossRef CAS PubMed.
- J.-B. Brubach, A. Mermet, A. Filabozzi, A. Gerschel and P. Roy, J. Chem. Phys., 2005, 122, 184509 CrossRef PubMed.
- K. Fumino, V. Fossog, K. Wittler, R. Hempelmann and R. Ludwig, Angew. Chem., 2013, 125, 2425–2429 CrossRef CAS; K. Fumino, V. Fossog, K. Wittler, R. Hempelmann and R. Ludwig, Angew. Chem., Int. Ed., 2013, 52, 2368–2372 CrossRef PubMed.
- K. Fumino and R. Ludwig, J. Mol. Liq., 2014, 192, 94–102 CrossRef CAS PubMed.
- D. R. McFarlane, P. Meakin, N. Amini and M. Forsyth, J. Phys.: Condens. Matter, 2001, 13, 8257–8267 CrossRef.
- Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585–4621 CrossRef CAS PubMed.
- J. Dupont, J. Braz. Chem. Soc., 2004, 15, 341–350 CrossRef CAS PubMed.
- M.-M. Huang, K. Schneiders, P. S. Schulz, P. Wasserscheid and H. Weingärtner, Phys. Chem. Chem. Phys., 2011, 13, 4126–4131 RSC.
- J. Hunger, A. Stoppa, R. Buchner and G. Hefter, J. Phys. Chem. B, 2008, 112, 12913–12919 CrossRef CAS PubMed.
- Y. Jiang, H. Nadolny, S. Käshammer, S. Weibels, W. Schröer and H. Weingärtner, Faraday Discuss., 2012, 154, 391–407 RSC.
- T. Köddermann, S. Klembt, D. Klasen, D. Paschek, U. Kragl and R. Ludwig, ChemPhysChem, 2012, 13, 1748–1752 CrossRef PubMed.
- T. Peppel, C. Roth, K. Fumino, D. Paschek, M. Köckerling and R. Ludwig, Angew. Chem., 2011, 123, 6791–6795 CrossRef CAS; T. Peppel, C. Roth, K. Fumino, D. Paschek, M. Köckerling and R. Ludwig, Angew. Chem., Int. Ed., 2011, 50, 6661–6665 CrossRef PubMed.
- Y. Zhang and E. J. Maginn, Phys. Chem. Chem. Phys., 2012, 14, 12157–12164 RSC.
- P. Stange, K. Fumino and R. Ludwig, Angew. Chem., 2013, 125, 3064–3068 CrossRef CAS; P. Stange, K. Fumino and R. Ludwig, Angew. Chem., Int. Ed., 2013, 52, 2990–2994 CrossRef PubMed.
- H. Weingärtner, A. Knocks, W. Schrader and U. Kaatze, J. Phys. Chem., 2001, 105, 8646–8650 CrossRef.
- D. F. Evans, S.-H. Chen, G. W. Schriver and E. M. Arnett, J. Am. Chem. Soc., 1981, 103, 481–482 CrossRef CAS.
- D. F. Evans, E. W. Kaler and W. J. Benton, J. Phys. Chem., 1983, 87, 533–535 CrossRef CAS.
- D. F. Evans, A. Yamauchi, G. J. Wei and V. A. Bloomfield, J. Phys. Chem., 1983, 87, 3537–3541 CrossRef CAS.
- I. Krossing, J. M. Slattery, C. Gaguenet, P. J. Dyson, A. Oleinikova and H. Weingärtner, J. Am. Chem. Soc., 2006, 128, 13427–13434 CrossRef CAS PubMed.
- H. Every, A. G. Bishop, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2000, 45, 1279–1284 CrossRef CAS.
- C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. N. V. K. Aki and J. F. Brennecke, J. Chem. Eng. Data, 2004, 49, 954–964 CrossRef CAS.
- H. L. Ngo, K. LeCompte, L. Hargens and A. B. McEwen, Thermochim. Acta, 2000, 357, 97–102 CrossRef.
- H. Tokuda, K. Hayamizu, K. Ishii, A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2005, 109, 6103–6110 CrossRef CAS PubMed.
- J. M. Crosthwaite, M. J. Muldoon, J. K. Dixon, J. L. Anderson and J. F. Brennecke, J. Chem. Thermodyn., 2005, 37, 559–568 CrossRef CAS PubMed.
- H. Tokuda, S. Tsuzuki, M. A. B. H. Susan, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2006, 110, 19593–19600 CrossRef CAS PubMed.
- J. A. Widegren, Y.-M. Wang, W. A. Henderson and J. W. Magee, J. Phys. Chem. B, 2007, 111, 8959–8964 CrossRef CAS PubMed.
- J. M. Slattery, C. Daguenet, P. J. Dyson, T. J. S. Schubert and I. Krossing, Angew. Chem., 2007, 119, 5480–5484 CrossRef CAS; J. M. Slattery, C. Daguenet, P. J. Dyson, T. J. S. Schubert and I. Krossing, Angew. Chem., Int. Ed., 2007, 46, 5384–5388 CrossRef PubMed.
- H. Luo, G. A. Baker and S. Dai, J. Phys. Chem. B, 2008, 112, 10077–10081 CrossRef CAS PubMed.
- O. Borodin, J. Phys. Chem. B, 2009, 113, 11463–11478 CrossRef CAS PubMed.
- H. Weingärtner, Angew. Chem., 2008, 120, 664–682 CrossRef; H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef PubMed.
- H. Markusson, J.-P. Belière, P. Johansson, C. A. Angell and P. Jacobson, J. Phys. Chem. A, 2007, 111, 8717–8723 CrossRef CAS PubMed.
- P. A. Z. Suarez, S. Einloft, J. E. L. Dullius, R. F. de Souza and J. Dupont, J. Chim. Phys. Phys.-Chim. Biol., 1998, 95, 1626–1639 CrossRef CAS.
- M. Antonietti, D. B. Kuang, B. Smarsly and Z. Yong, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS PubMed.
- A. Heintz, R. Ludwig and E. Schmidt, Phys. Chem. Chem. Phys., 2011, 13, 3268–3273 RSC.
- R. Ludwig, E. Maginn and S. Balasubramanian, ChemPhysChem, 2012, 13, 1603–1605 CrossRef CAS PubMed.
- C. Roth, A. Appelhagen, N. Jobst and R. Ludwig, ChemPhysChem, 2012, 13, 1708–1717 CrossRef CAS PubMed.
- C. Roth, A. Rose and R. Ludwig, ChemPhysChem, 2012, 13, 3102–3105 CrossRef CAS PubMed.
- C. Roth, S. Chatzipapadopoulos, D. Kerlé, F. Friedriszik, M. Lütgens, S. Lochbrunner, O. Kühn and R. Ludwig, New J. Phys., 2012, 14, 105026 CrossRef CAS.
| This journal is © the Owner Societies 2014 |