 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Energy transfer properties of Rhodobacter sphaeroides chromatophores during adaptation to low light intensity†
B.
Driscoll‡
a,
C.
Lunceford‡
a,
S.
Lin
*b,
K.
Woronowicz
c,
R. A.
Niederman
c and
N. W.
Woodbury
*b
aDepartment of Physics, Arizona State University, Tempe, AZ 85287, USA
bDepartment of Chemistry and Biochemistry and The Biodesign Institute at Arizona State University, Tempe, AZ 85287, USA. E-mail: nwoodbury@asu.edu
cDepartment of Molecular Biology and Biochemistry, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA
First published on 26th June 2014
Abstract
Time-resolved fluorescence spectroscopy was used to explore the pathway and kinetics of energy transfer in photosynthetic membrane vesicles (chromatophores) isolated from Rhodobacter (Rba.) sphaeroides cells harvested 2, 4, 6 or 24 hours after a transition from growth in high to low level illumination. As previously observed, this light intensity transition initiates the remodeling of the photosynthetic apparatus and an increase in the number of light harvesting 2 (LH2) complexes relative to light harvesting 1 (LH1) and reaction center (RC) complexes. It has generally been thought that the increase in LH2 complexes served the purpose of increasing the overall energy transmission to the RC. However, fluorescence lifetime measurements and analysis in terms of energy transfer within LH2 and between LH2 and LH1 indicate that, during the remodeling time period measured, only a portion of the additional LH2 generated are well connected to LH1 and the reaction center. The majority of the additional LH2 fluorescence decays with a lifetime comparable to that of free, unconnected LH2 complexes. The presence of large LH2-only domains has been observed by atomic force microscopy in Rba. sphaeroides chromatophores (Bahatyrova et al., Nature, 2004, 430, 1058), providing structural support for the existence of pools of partially connected LH2 complexes. These LH2-only domains represent the light-responsive antenna complement formed after a switch in growth conditions from high to low illumination, while the remaining LH2 complexes occupy membrane regions containing mixtures of LH2 and LH1–RC core complexes. The current study utilized a multi-parameter approach to explore the fluorescence spectroscopic properties related to the remodeling process, shedding light on the structure-function relationship of the photosynthetic assembles. Possible reasons for the accumulation of these largely disconnected LH2-only pools are discussed.
Introduction
The photosynthetic apparatus of Rhodobacter (Rba.) sphaeroides is composed of light harvesting complexes 1 and 2 (LH1 and LH2), and the reaction center (RC). In the membrane, the RC and LH1 form a tightly associated core complex (LH1–RC) found mainly in a dimeric form in which each RC is surrounded by an open LH1 ring-like structure, also containing an integral PufX protein.1–3 Atomic force microscopy (AFM) studies have shown that the LH1–RC dimers generally align in arrays within the photosynthetic membrane and are surrounded by LH2 complexes4 (Fig. 1a). The antenna (LH1 and LH2) and the RC protein complexes are thought to assemble in the cytoplasmic membrane, which then invaginates inward, forming chromatophore membrane vesicles that contain the mature apparatus.5–7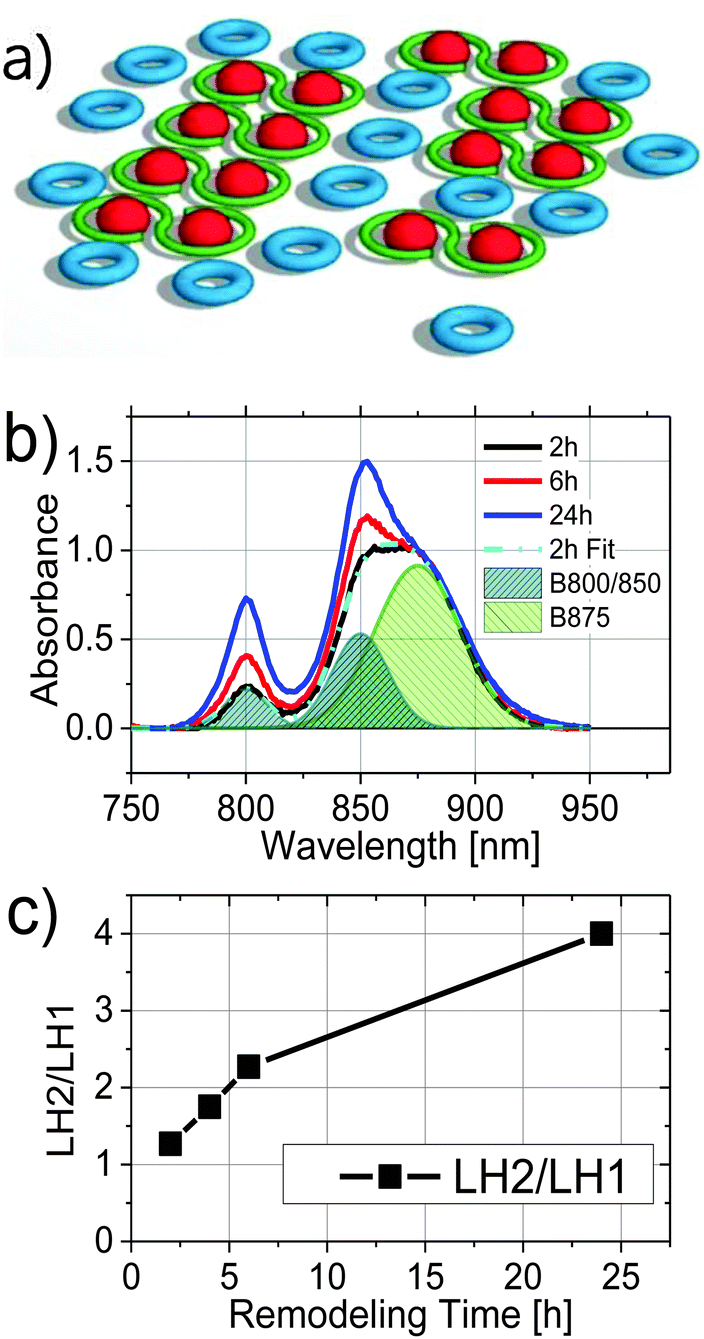 | ||
| Fig. 1 (a) schematic illustration of photosynthetic units, LH2 (blue), LH1 (green) and RC (red), as arranged in the chromatophore structure; (b) absorption spectra of chromatophores as a function of remodeling time after shifting from high-light to low-light growth conditions, 2 h (black), 4 h (red), and 24 h (blue) normalized to the LH1 absorption maximum at 875 nm. The spectrum at 4 h remodeling time (not shown) is similar to that of the 6 h spectrum. The shaded curves are three Gaussians centered at 800, 850, and 875 nm, used in the fitting of the 2 h absorption spectrum; (c) the relative amplitude ratio of LH2 and LH1 at different remodeling times. | ||
The structures of the individual LH1, LH2, and RC complexes are known at atomic resolution and their spectroscopic and kinetic properties have been extensively studied (for recent reviews, see ref. 8–10). Each LH1–RC complex in the LH1–RC dimer is composed of 14 pairs of α/β subunits and 28 closely associated bacteriochlorophyll a (Bchl a) molecules surrounding the RC. The LH1 BChl molecules have a QY absorption band centered at 875 nm (B875) and a fluorescence spectrum with a maximum at 885 nm. The LH2 complex, in contrast, has two spectroscopically distinct types of BChl molecules. There are 9 pairs of α/β subunits in this case containing 9 BChl molecules that are well separated from the others and absorb at 800 nm (B800) and 18 much more closely associated BChl molecules that absorb at 850 nm (B850). Excitation energy transfer takes place very rapidly from the B800 ring to the B850 ring of LH2 and therefore the fluorescence from this complex is emitted almost entirely by the B850 BChls (fluorescing with a maximum at 860 nm).11 The fluorescence quantum yield of the RC is much smaller than that from either LH1 or LH2 due to the picosecond quenching of the excited state via electron transfer,12,13 and thus there is essentially no observable RC fluorescence from these membranes.
The ratio of LH2 to LH1–RC complexes in mature intracytoplasmic membrane vesicles (chromatophores) depends on both the light intensity and the time the cells have grown at that intensity. When grown under continuous low-light intensity, the LH2/LH1 ratio is larger than when grown under continuous high-light intensity. This is due to an increase in the number of LH2 complexes at low light rather than a change in LH1. When cells are grown anaerobically under high-light and are then shifted to low-light conditions, the photosynthetic apparatus adapts via a process referred to as antenna remodelling.6,14 During low-light remodeling, additional LH2 complexes are produced. The increase in LH2 complexes results in an increase in the total antenna absorption cross-section. In principle, this is consistent with that idea that additional absorbance cross section results in additional energy transfer to the RC, thus partially compensating for the lower light intensity.
If the LH2 that accumulates during low-light adaptation does so to increase the functional absorbance cross section, one might think they would simply add to the existing LH2 pool, increasing its size. However, AFM images of chromatophores showed a heterogeneous distribution of the LH2 complexes with two types of organization.4 Ten to twenty LH2 units are seen to closely associate with the LH1–RC complexes, and this group appears to increase only moderately as remodeling occurs. Most of the accumulation of LH2 appears to result in the formation of LH2-only domains that are more distal from the LH1–RC arrays. It was suggested that these more distal LH2 domains may form a light-responsive complement of LH2 antenna.4 A radiolabeling study following the assembly of the photosynthetic apparatus has also shown that mature membranes assemble new LH2 rings during the remodeling process in chromatophores in response to the lowered light intensity.6 Once again, it was found that the newly assembled LH2 complexes mainly group in areas not directly associated with existing arrays of LH1–RC complexes and the proximal LH2 complexes that are present under high light. The functionality of these distal LH2 only pools is thus far poorly understood. Recent studies have suggested that this arrangement may slow the turnover rate of the RC14 and/or that under unnaturally high excitation light these extra pigments are involved in photoprotection.15
These recent observations suggest that further exploration of the light-harvesting capabilities of the additional pools of LH2 would be enlightening. Here, the energy transfer properties of the proximal LH2 pools are investigated by measuring the picosecond time-resolved fluorescence of photosynthetic membranes at various points during the remodeling process, as well as under various excitation intensities.
Experimental
Sample preparation
Whole cells were grown to OD680 of at least 0.2 anaerobically at high-light intensity (1100 W m−2) and then switched to a lower intensity (100 W m−2), initiating the remodeling process. The cells collected at 2, 4, 6, and 24 hours after initiation of low light conditions were then passed through a French press and chromatophores were isolated from the lysate using rate-zone ultracentrifugation.14 Absorption spectra of these samples were obtained using a Varian UV-visible spectrophotometer (Cary 5, Varian).Time-resolved fluorescence spectroscopy
Fluorescence emission spectra were recorded as a function of delay time using a streak camera system described previously.16 The excitation pulse was generated from a Nd:YVO4-pumped, model-locked titanium:sapphire laser producing 100 fs pulses at a repetition rate of 76 MHz and a wavelength of 800 nm (Coherent Inc., Verdi-18 and Mira-900). The excitation intensity was controlled using neutral density filters, and was kept at 1 mW (5.3 × 107 photons per pulse or 7.6 × 108 photons per pulse per cm2) for the study of fluorescence kinetics as a function of remodeling time, but was varied between 0.1 mW and 20 mW for the excitation intensity dependence study. A beam diameter of 3 mm was set to excite the maximal volume of the sample in the 3 × 3 mm2 cuvette while minimizing artefacts resulting from scattering. A long-pass filter, with 50% transmittance at 825 nm was placed between the sample and the detector to reduce scattered excitation light, and an IR sheet polarizer was set to 54.7° to collect fluorescence at the “magic angle” relative to the excitation polarization. The fluorescence was then focused onto the entrance slit of a spectrograph (Chomex 250IS) coupled to a streak camera and a CCD (Hamamatsu C5680). Individual fluorescence measurements involved integration times between 1 and 6 h, depending on the sample. Time-resolved fluorescence spectra were recorded over a wavelength range of 830 to 940 nm, and an 800 ps time window. The full-width-at-half-maximum of the instrument response function was ∼10 ps.Data analysis and modeling
Time resolved fluorescence emission spectra as a function of decay time were corrected for the wavelength sensitivity of the detecting system, and globally fit to a sum of exponential decays (eqn (1)) using a locally written software package, ASUFIT. The quality of the fit was judged by the reduced chi squared value. Fitting was performed with the least number of exponential decay components statistically required and all fits yielded reduced chi squared values ≤1.7 (additional components did not reduce the chi squared value further).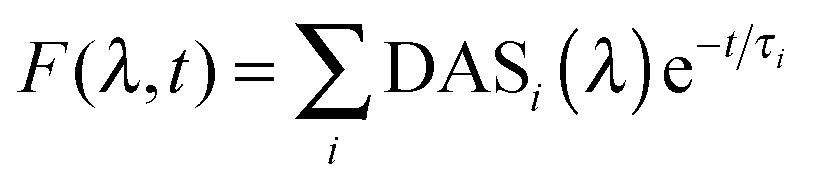 | (1) |
In eqn (1), DASi(λ) is the wavelength dependent amplitude of the ith decay component (Decay Associated Spectra), and τi is the ith exponential time constant.
Results
Absorption spectroscopic properties of chromatophores as a function of remodeling time
Fig. 1b compares the absorption spectra of 2, 6, and 24 h samples normalized at 875 nm, the wavelength of LH1 absorption maximum. The rise in the B850 peak was only slightly greater in the 6 h relative to 4 h chromatophores (4 h spectrum not shown). It is clear that as the remodeling proceeded, relative intensities of the LH2 absorption bands at 800 nm and 850 nm increased. In order to determine the amount of LH2 relative to LH1 as a function of the remodeling process, each absorption spectrum was fit by a sum of three Gaussians centered at 800, 850, and 875 nm, corresponding to absorption in LH2 (B800 and B850) and LH1 (B875), respectively. The three Gaussian bands obtained from fitting of the 2 h sample are illustrated in Fig. 1b (shaded areas). The ratio of the amplitudes at 800 nm and 875 nm is plotted as a function of remodeling time and shown in Fig. 1c. Consistent with prior studies,14 the amount of LH2 increased relative to LH1 with remodeling time, with the 24 h sample showing ∼3 fold more LH2 than the 2 h sample.Fluorescence kinetics of LH2 and LH1 from chromatophores at different stages of remodeling
To explore energy transfer in chromatophores as a function of the remodeling process, time-resolved fluorescence spectra from LH2 and LH1 were analyzed kinetically following selective excitation of the higher energy band of LH2 (B800) at 800 nm. The optical density of each sample was adjusted to 0.2 at the excitation wavelength to ensure that the same number of photons were absorbed by each at a given excitation intensity.All the experiments were performed at room temperature and samples were not stirred during data collection. Under the conditions described above, the reaction centers were in the closed state during the measurements (i.e., the majority of the reaction centers in the sample had the primary electron donor, P, oxidized). Global fitting of the fluorescence decays to a series of exponential decay terms (see Methods) required three components for an adequate fit. The resulting DAS for each remodeling time sample are shown in Fig. 2, and the decay lifetimes as a function of remodeling time are plotted in Fig. 3. Because the excitation energy transfer from the 800 nm to the 850 nm absorbing BChls of LH2 takes place on the subpicosecond time scale, the fluorescence signal from the 800 nm BChls of LH2 is very weak and no attempt was made to measure it or to include that process in the kinetic analysis. The amplitude spectrum of the first kinetic component (DAS1, black curves in Fig. 2), has a positive band peaking at 859 nm (LH2) and a negative band at 885 nm (LH1) at all times during remodeling, consistent with energy transfer from LH2 to LH1. This kinetic component has been previously observed in mature photosynthetic membranes.8 The time constant associated with this process is between 20–40 ps as shown in Fig. 3(a). The amplitude spectrum of the second component (DAS2, red curves in Fig. 2) is again largely similar at all times during remodeling and has a positive band centered at 880 nm, which corresponds primarily to fluorescence from LH1. The decay time constant associated with this component is consistent with fluorescence kinetics previously recorded for LH1–RC complexes;17 it rises rapidly from ∼70 to ∼130 ps after the 2 h time point and then remains nearly constant (Fig. 3b). The amplitude spectrum of the third component, DAS3 (blue curves in Fig. 2), is blue-shifted relative to DAS2, and the extent of the blue shift increases with the remodeling time. Furthermore, a nearly 6-fold increase in the lifetime of this component, from 170 to 970 ps, is observed as remodeling progresses (Fig. 3c). The 970 ps time constant seen at 24 h for DAS3 is similar to the longest-lived lifetime seen for chromatophores made from an LH2-only mutant (∼1.2 ns),18 suggesting that the LH2 giving rise to this component does not undergo efficient energy transfer to LH1 (note that at the 2 h time point, the 20, 70 and 190 ps components are probably mixed in the fitting, a possibility supported by the observed gradual blue shift in the DAS spectrum of the slow component at longer remodeling times). This likely explains the apparent ∼2-fold increase in lifetime of the first and second components between 2 and 4 h.
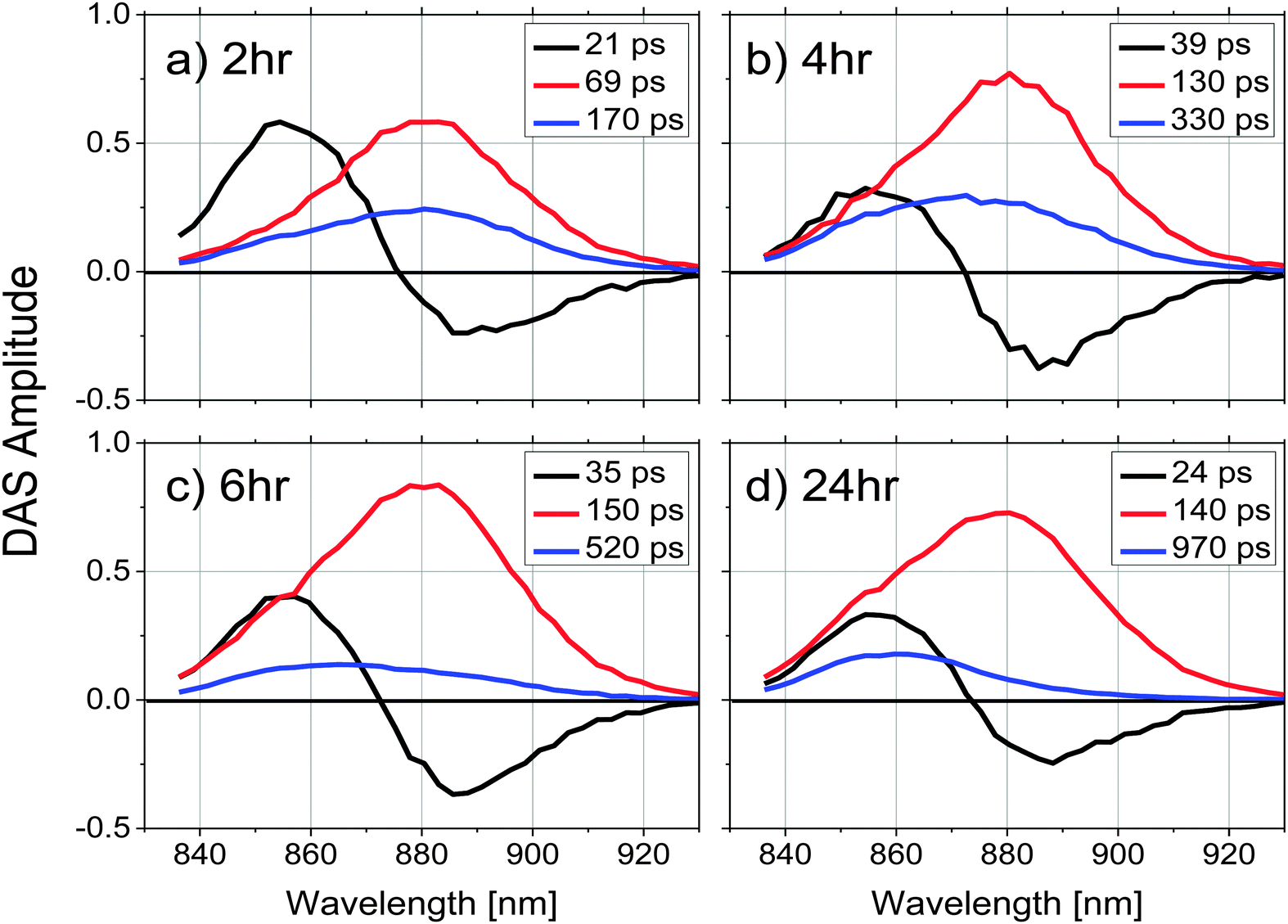 | ||
| Fig. 2 Decay associated spectra (DAS) for chromatophores after a remodeling time of (a) 2, (b) 4, (c) 6 and (d) 24 hours, obtained from a 3-exponential global fitting of the time-resolved fluorescence emission spectra. Samples were adjusted to have the same absorbance at 800 nm, the laser excitation wavelength. A 3D plot of a data set with residuals (Fig. S3, ESI†) illustrates the fitting quality. | ||
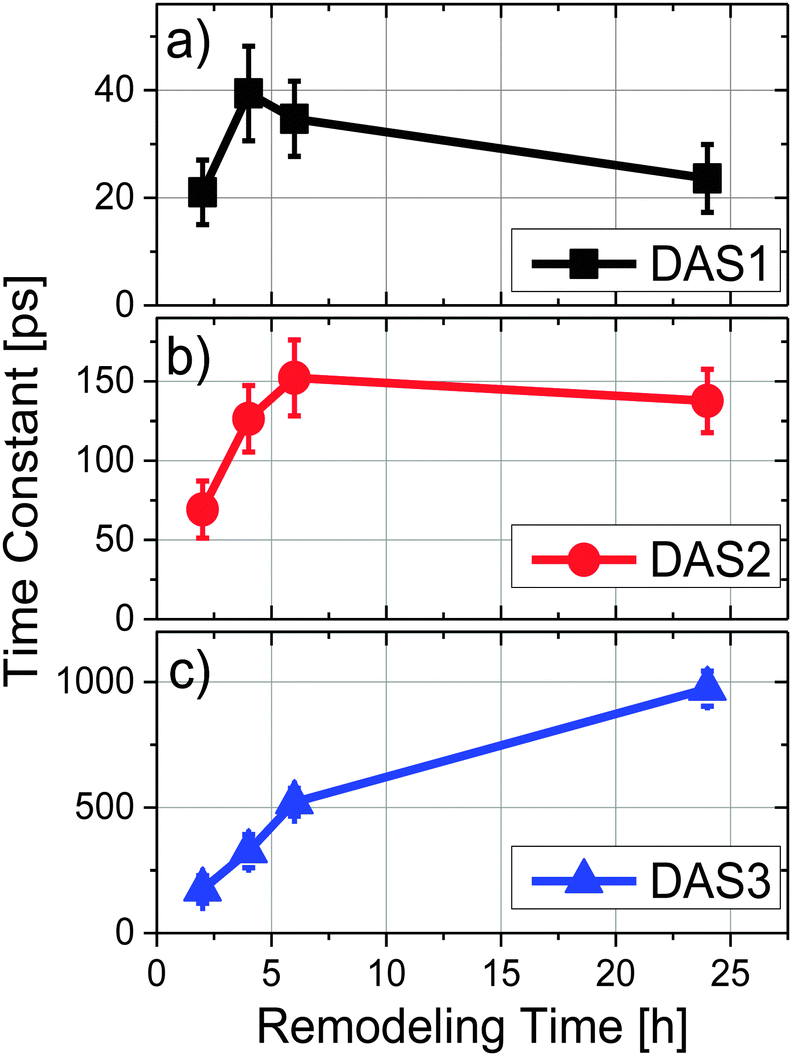 | ||
| Fig. 3 Time constants as a function of remodeling time where (a) DAS1, (b) DAS2 and (c) DAS3 correspond to the short (20–30 ps), middle (70–150 ps) and long (170 ps–1 ns) terms, respectively. | ||
To evaluate the contributions of LH1 and LH2 to each DAS independently, the three amplitude spectra associated with each sample were analyzed by fitting the wavelength region between 830 nm and 930 nm of each spectrum to 2 Gaussian functions with peaks centered at 860 and 890 nm. Both the peak wavelength and the width of each Gaussian were held constant during the fitting, allowing only the amplitudes of the two spectral components to vary. An example of such a spectral decomposition is illustrated in Fig. 4d. The DAS of different samples were normalized to the initial fluorescence intensity recorded close to time zero. The relative amplitudes of the resulting fits are shown in Fig. 4a and b for DAS1 and DAS3 respectively, and a ratio of the peak amplitudes of the Gaussian functions centered at 860 nm and 890 nm (corresponding to LH2 and LH1, respectively) as a function of remodeling time are given for the longest-lived component (DAS3) in Fig. 4c. Because DAS2 does not significantly change spectrally during remodeling, it is not shown.
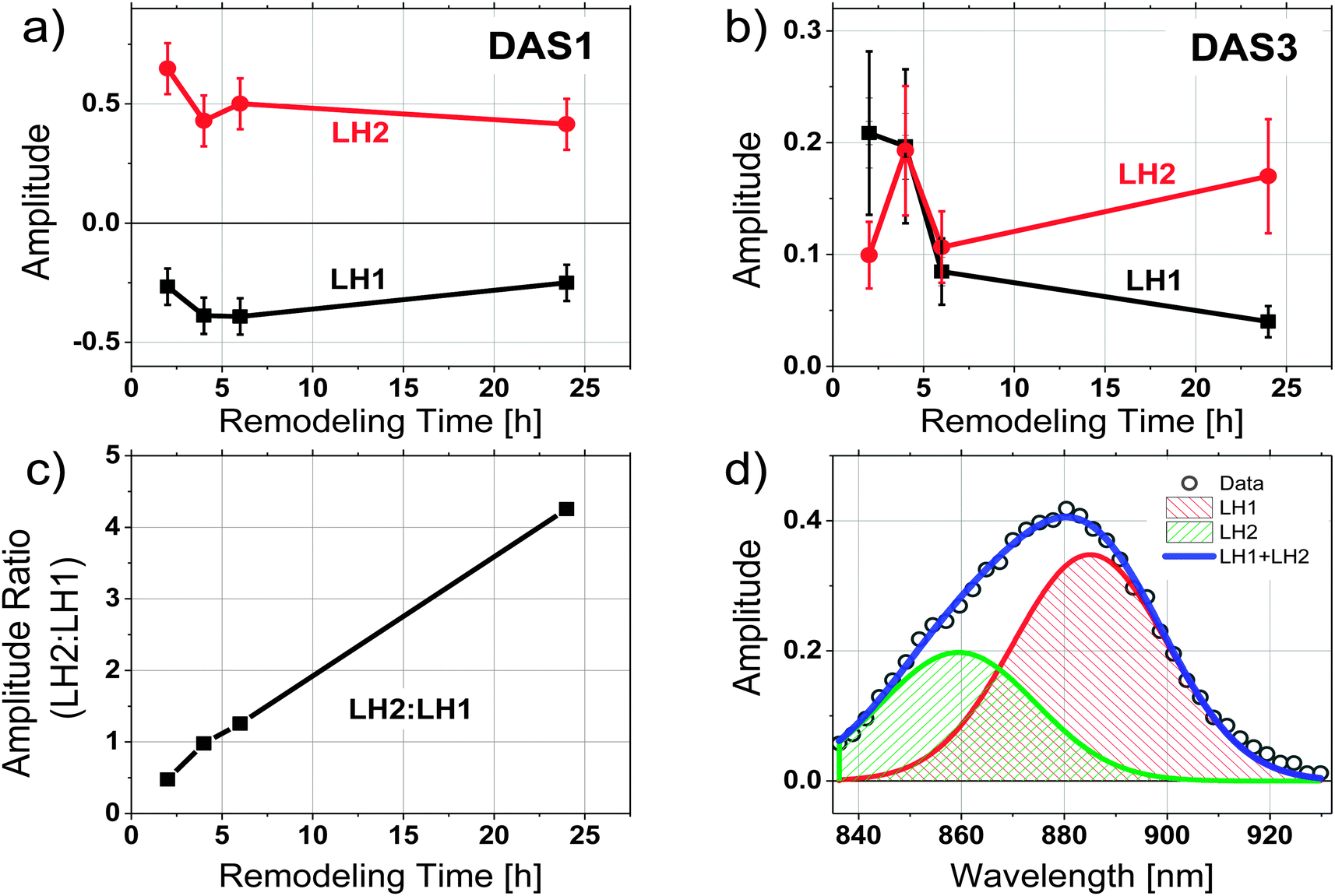 | ||
Fig. 4 (a) Amplitudes of Gaussian curves used to characterize the trends of fluorescence contributions from LH2 and LH1 as a function of remodeling time. (a) The short time component (DAS1). Amplitudes of the LH1 wavelengths (∼885 nm) are negative as a result of the DAS fitting, which indicates a rise in fluorescence from LH1, (b) identical analysis on the long-lived time component (DAS3), (c) ratio of the amplitudes LH2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LH1 (860 nm:885 nm). (d) Illustration of spectral deconvolution into two Gaussians of DAS3 from the 2 h sample at 1 mW excitation intensity. :LH1 (860 nm:885 nm). (d) Illustration of spectral deconvolution into two Gaussians of DAS3 from the 2 h sample at 1 mW excitation intensity. | ||
The relative contributions of LH2 and LH1 in each DAS illustrated in Fig. 4a–c show that: (1) when LH2 is excited, excitation energy is transferred to LH1 with a time constant of ∼40 ps, (2) the LH1 excited state formed decays with a time constant of ∼150 ps, and (3) the longest decay process, which takes place on the order of hundreds of ps, shows the most pronounced change as a function of antenna remodeling time and is mainly due to fluorescence from LH2. The DAS associated with this decay also indicates an increased fluorescence contribution from LH2 as remodeling proceeds (it shifts to the blue, Fig. 2). These results are consistent with the idea that as remodeling progresses, there is a larger and larger amount of LH2 generated that is not well connected to the remainder of the photosynthetic apparatus in terms of excitation energy transfer. Apparently, there are at least two pools of LH2 complexes present with distinct kinetic profiles.
Excitation intensity dependence study
In order to further explore the origins of the different fluorescence components, the excitation intensity dependence of the time resolved fluorescence spectra was measured as a function of remodeling time for the 2 and 24 h samples. Excitation intensities ranging from 0.1–20 mW were used, (note, 1 mW = 7.6 × 108 photons per pulse per cm2 at the excitation wavelength) and both global fitting and Gaussian deconvolution of the resulting amplitude spectra were performed, as described above.Similar to the results described above, 3 kinetic components with lifetimes in the range of 20–30 ps, 100–150 ps, and 600–1000 ps were resolved from each data set. The spectral profile of the middle component, dominated by the decay of LH1, remains relatively unchanged under all excitation intensities for both the 2 h and 24 h samples. Significant spectral changes in the DAS as a function of excitation intensity were observed in DAS1 and DAS3 (Fig. S1 and S2, ESI†). Two Gaussian functions, representing the fluorescence from LH2 and LH1, were used to fit the DAS1 and DAS3 of the 2 and 24 h samples at each excitation intensity. Fig. 5 compares the amplitudes of the resulting two Gaussian functions plotted as a function of excitation intensity, after normalizing to the amplitude of DAS2. The positive amplitude of DAS1, representing the maximum of LH2 emission at 860 nm, increases with the excitation intensity (Fig. 5a and c), while the negative amplitude at 890 nm (LH1) reduces with the excitation intensity. Because of the nature of the spectrum associated with energy transfer between LH2 and LH1, it is not clear whether the observed changes represent an increase in contribution from LH2, a decrease in contribution from LH1, or both. DAS3, in contrast, shows a decrease in the relative contribution from LH2 for the 24 h sample and a decrease in both LH2 and LH1 for the 2 h sample (Fig. 5b and d). These results are most easily explained in terms of annihilation that occurs in both LH1 and LH2 at the 2 h time point and primarily takes place in LH2 at the 24 h time point.
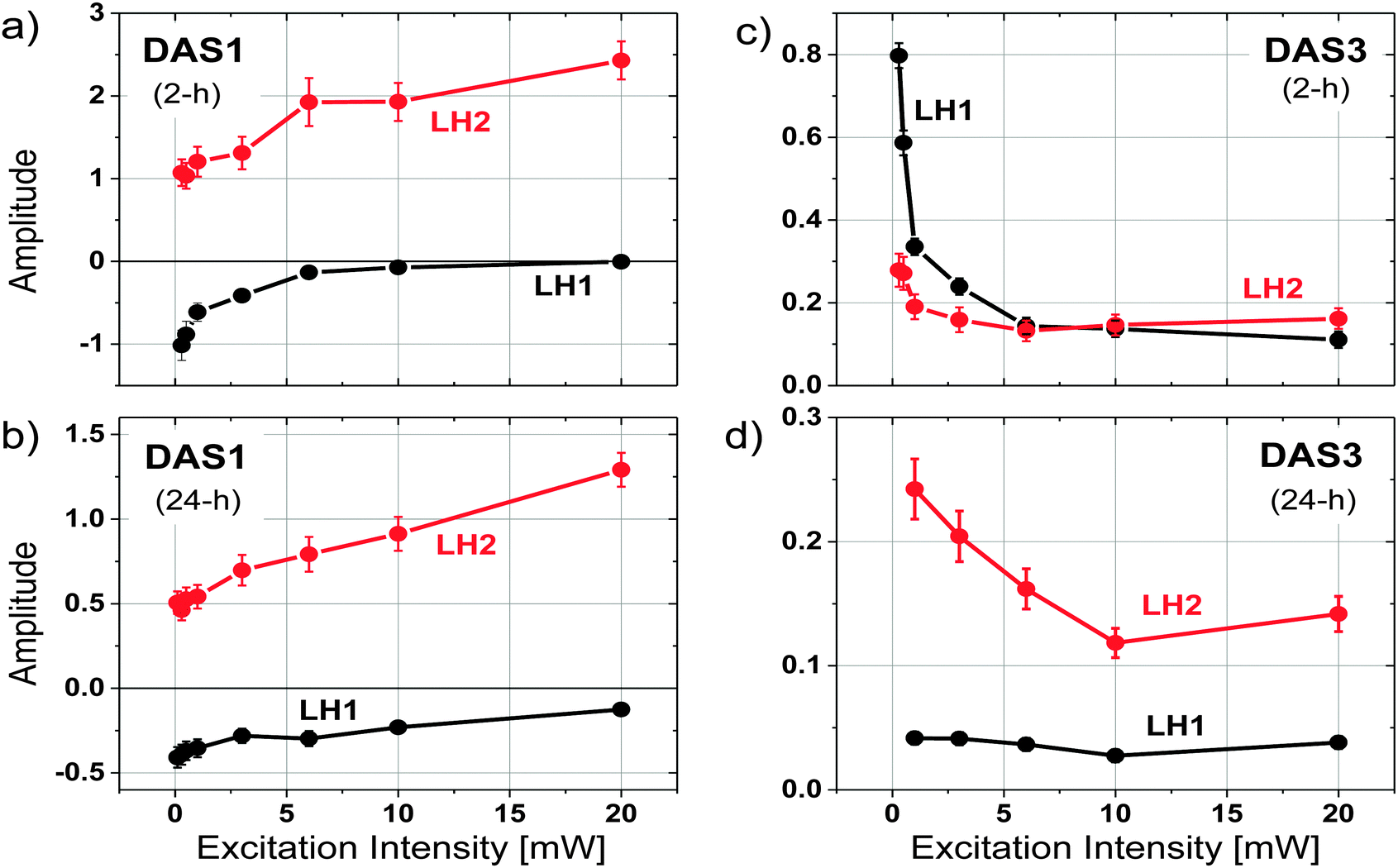 | ||
| Fig. 5 Amplitudes of Gaussian bands used to characterize the trends of fluorescence contributions from LH2 and LH1 as a function of excitation intensity. (a) The short time component (DAS1) of the 2 h sample. (b) DAS1 for the 24 h sample. (c) The long-time component (DAS3) of the 2 h sample. (d) DAS3 for the 24 h sample as a function of excitation intensity. | ||
Discussion
Previous work has shown that when cell growth conditions are shifted from high to low light, the amount of LH2 in the chromatophores increases. Logically, one would assume this larger absorbance cross section per reaction center would result in increased light harvesting at low light. Structural studies of the remodeling process have begun to provide a picture of the evolving organization of the photosynthetic apparatus as additional LH2 is added.6 The observation of two types of organization for LH2 complexes, one type of LH2 closely associated with the linear arrays of dimeric LH1–RC complexes and the other type located outside these arrays in larger clusters, raises questions regarding how this architectural design is related to light-harvesting function. The goal of the current study is to explore the functional evolution of the light harvesting apparatus during the remodeling transition, with particular attention paid to the function of LH2 as an extended antenna.Changes in the distribution of excited states between LH1 and LH2 pools during remodeling
The most significant result from the above analysis is that the increase in LH2 with remodeling does not correspond to an increase in energy transfer from the LH2 to LH1–RC complexes. The additional LH2 complexes assembled under low-light conditions seem to form LH2 pools, which are poorly connected or completely disconnected from the LH1–RC arrays. A qualitative scheme depicting possible organizational changes as LH2 complexes assemble in the remodeled photosynthetic unit is shown in Fig. 6. At early remodeling times, an array of core complexes is surrounded by a small number of LH2 complexes, as has been suggested in previous studies.4,6 As remodeling of the photosynthetic apparatus proceeds, more LH2 complexes are added but at least a portion of these newly-added LH2 complexes are functionally disconnected or poorly connected to the LH1–RC dominated domains. This results in a low efficiency of excitation energy transfer from the distal pool, and an increasingly long decay time approaching the inherent decay time of isolated LH2 complexes.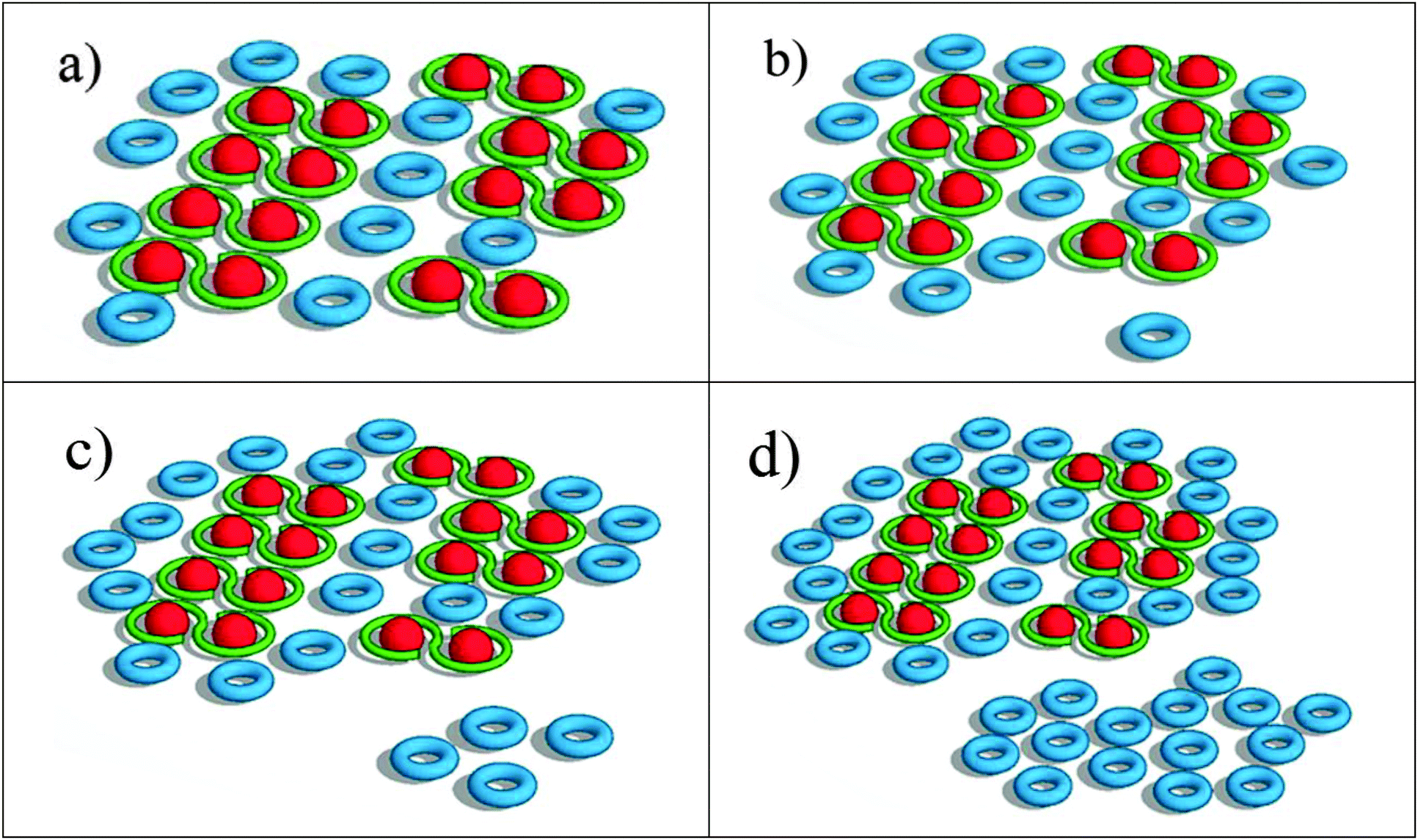 | ||
| Fig. 6 Schematic of LH2 accumulation as remodeling time increases. Representations are of (a) 2 h, (b) 4 h, (c) 6 h and (d) 24 h. The ratio of LH2:LH1–RC, as well as the ratio of the two different pools of LH2 complexes matches that used in the simulation. | ||
Compelling evidence for this scheme is seen in Fig. 4c, where the ratios of contributions from LH2 and LH1 to the longest-lived amplitude spectrum, DAS3, is given. The plot indicates a significant increase in fluorescence contribution from LH2 relative to LH1. This is of particular interest when combined with the observation that as remodeling time increases, the lifetime of DAS3 approaches the lifetime of isolated LH2, 1.2 ns (see Fig. 3c). This suggests that the increase in LH2 between 2 and 24 h results in a population of LH2 complexes that are poorly connected to LH1 complexes, and thus RCs, at least at the level of excitation intensity used in the measurement. The kinetic evidence presented here for the existence of disconnected, or poorly connected, LH2 pools matches very well with the AFM observations of LH2-only clusters formed during growth under low-light.4
Different mechanisms of the two types of LH2 pools in handling excess excitation
Exciton redistribution between LH2 and LH1 complexes due to the accumulation of LH2 during remodeling were further explored by measuring the excitation intensity dependence of the time resolved fluorescence. Interestingly, the DAS3 of the 2 h sample additionally exhibits a decrease in LH1 fluorescence, providing evidence for annihilation in LH1 (Fig. 5c and Fig. S1c, ESI†). The similar trend of the excitation intensity dependent decrease in the fluorescence of the LH1 and LH2 components of the longer-lived fluorescence (Fig. 5b) indicates that there exists a strong coupling between LH2 and LH1 complexes in the 2 h chromatophores. In contrast, the kinetic behavior of the 24 h sample shows a decrease in the part of the long DAS3 component attributed to LH2 as the excitation intensity is increased, while the LH1 portion of DAS3 remains largely unchanged (Fig. 5d). This indicates that in the 24 h sample, excitation annihilation occurs predominantly in the LH2 only pools, again, consistent with the proposed notion of functionally disconnected pools of LH2.Kinetic simulation
The observed fluorescence kinetics as a function of remodeling time were reproduced using a simple model for the physical relationship between the photosynthetic constituents and were found to be consistent with the above interpretation. In order to put the scheme in Fig. 6 on a more quantitative footing, the system was simulated in terms of a random walk energy transfer network. For these simulations, all reaction centers were assumed to be closed (note that closed reaction centers still quench excitations from LH1). Each exciton carried a probability to hop between different LH2 complexes with a rate constant of 100 ns−1, corresponding roughly to the published time constant for energy transfer between LH2 complexes.8 Each LH2 and LH1 complex is considered as an individual point on a 2D grid. The model allowed for radiative decay (fluorescence) and singlet–singlet excited state annihilation within both LH2 and LH1 pools. Annihilation was assumed to occur whenever two excitons met in a single complex, and fluorescence was assumed to occur with rate constants of 0.83 ns−1 and 10 ns−1 for LH2 and LH1, respectively, which are the fluorescence decay rates from LH2-only and LH1-only mutants.8 At any boundary between LH2 and LH1, the probability of transfer from LH2 → LH1 was set at 1/10, and back transfer from LH1 → LH2 at 1/50. These values were estimated from previously reported characteristic times of energy transfer and the ratio verified by Boltzmann energy statistics. In the 4, 6, and 24 h simulations, a portion of the LH2 complexes were assumed to reside in LH2-only pools that were not as well connected to LH1 as other LH2, which was done by reducing the transfer rate into and out of the pools from 100 ns−1 to 0.67 ns−1. Additionally, the ratio of LH2/LH1 used in the simulations was obtained from the ratios calculated from the absorption spectra as given in Fig. 1b. These ratios were then combined with known extinction coefficients to reveal relative concentrations of LH2 and LH1.Output from the simulation provided fluorescence counts as a function of time (one time interval corresponded to 1 ps). Attempts to fit this output globally, as was done with the experiment data, proved unsuccessful presumably due to the bimodal nature of the output (LH1 fluorescence and LH2 fluorescence). Time constants associated with each kinetic process were thus obtained by individually fitting each fluorescence vs. time data set with a minimum set of exponential decay components. Fits readily provided time constants consistent with the experimental results; offering a fast component of ∼30 ps, seen as a decay in the LH2 fluorescence and a rise in LH1, a component of 100–150 ps, dominated by LH1 fluorescence, and a long-lived component dominated by LH2 which ranged from 170–200 ps before the inclusion of LH2 only pools and 200–1000 ps after.
The results of these simulations are given in Fig. 7. Fig. 7a shows the simulation as a function of remodeling time without the addition of a functionally weakly connected or disconnected pool of LH2. Using the literature-value rate constants given above and assuming homogeneity in rate constants across like constituents, it was not possible to obtain simulation data approximating the long component time constant (DAS3).
 | ||
| Fig. 7 Comparison of simulation (open symbols with solid lines) and streak camera derived long-lived time-constants (DAS3, solid symbols with dashed lines) (a) without and (b) with a functionally disconnected pool for samples at different remodeling times under low excitation intensity. | ||
Instead, much faster decay time constants were obtained. Inclusion of a portion of LH2 in a disconnected pool, as described above, yielded results (Fig. 7b) that were much more consistent with the data seen in Fig. 3, supporting the concept that much of the added LH2 absorbance is due to unconnected or poorly connected LH2 complexes.
Possible functions of the additional LH2 pools formed during the remodeling
Previous studies have shown an increasing amount of LH2 during remodeling, when cell growth shifted from high to low light intensity.6,14 This expansion of LH2 pools would be expected to enhance the light-harvesting capability of the antenna network to maintain photosynthetic activity in the system. However, the studies described above imply that the added LH2 is largely unconnected to the RC, a concept also supported by previous radiolabelling studies.6The role of light level in modulating the connectivity of energy transfer in antennas in vitro has been studied in the past,15 suggesting that there may be an inherent response even in the absence of changes in the number of LH2 complexes. The in vivo remodeling process has been observed in other species of purple bacteria and it has been suggested that it may play a role in photoprotection19 or in modulating the rate at which reaction centers turnover,14,20 but the fundamental question of why it would be advantageous to generate large amounts of unconnected LH2 upon decreasing the light level remains unanswered.
In considering this question, it is important to remember that laboratory growth conditions are quite different from those experienced in the organism's natural habitat. The “high” level of light used for bacterial growth in these studies is in the range of the maximum solar intensity level that would hit the surface of the earth. The “low” level is one tenth of that. In the natural environment, diurnal light–dark cycles and clouds as well as the level of shading from surrounding bacteria or other organisms in a liquid environment, mean that the dynamic range of light exposure any bacterium sees may be much greater than ten-fold (and probably shifted significantly downward from the intensities used here). Furthermore, the duration and frequency of exposure to high and low light levels is likely very different in the natural environment compared to the laboratory. This makes it difficult to extrapolate the current results to natural function. What one can say is that decreasing the average light intensity seen by the bacterium over hours initiates the production of LH2 complexes that are not all initially involved in functional light harvesting. It is possible, for example, that production of LH2 and its functional incorporation into the photosynthetic apparatus are controlled by separate environmental factors that are not appropriately mimicked in the growth regime used in this study.
Acknowledgements
This work was supported by National Science Foundation award No. PHY-1057827 (NWW, SL, BD, and CL) and subaward No. 12-764 (RAN), and by U. S. Department of Energy (DE-FG02-08ER15957) from the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science (RAN). We thank Joseph M. Kay and Arpit A. Patel for assistance in membrane preparation, and Dr A. Freiberg for helpful discussions.Notes and references
- T. Walz, S. J. Jamieson, C. M. Bowers, P. A. Bullough and C. N. Hunter, J. Mol. Biol., 1998, 282, 833–845 CrossRef CAS PubMed.
- R. Niederman, Structure, function and formation of bacterial intracytoplasmic membranes, Springer-Verlag, Heidelberg, Berlin, 2006 Search PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Iowa State University Press, Blackwell Science Ltd., Ames, Iowa, 2002 Search PubMed.
- S. Bahatyrova, R. N. Frese, C. A. Siebert, J. D. Olsen, K. O. van der Werf, R. van Grondelle, R. A. Niederman, P. A. Bullough, C. Otto and C. N. Hunter, Nature, 2004, 430, 1058–1062 CrossRef CAS PubMed.
- R. A. Niederman, D. E. Mallon and J. J. Langan, Biochim. Biophys. Acta, 1976, 440, 429–447 CrossRef.
- C. N. Hunter, J. D. Tucker and R. A. Niederman, Photochem. Photobiol. Sci., 2005, 4, 1023–1027 CAS.
- M. Koblizek, J. D. Shih, S. I. Breitbart, E. C. Ratcliffe, Z. S. Kolber, C. N. Hunter and R. A. Niederman, Biochim. Biophys. Acta, 2005, 1706, 220–231 CrossRef CAS PubMed.
- R. J. Cogdell, A. Gall and J. Kohler, Q. Rev. Biophys., 2006, 39, 227–324 CrossRef CAS PubMed.
- C. L. Tavano and T. J. Donohue, Curr. Opin. Microbiol., 2006, 9, 625–631 CrossRef CAS PubMed.
- J. N. Sturgis and R. A. Niederman, Photosynth. Res., 2008, 95, 269–278 CrossRef CAS PubMed.
- V. Sundstrom, T. Pullerits and R. van Grondelle, J. Phys. Chem. B, 1999, 103, 2327–2346 CrossRef.
- N. W. Woodbury and J. P. Allen, in Anoxygenic Photosynthetic Bacteria, ed. R. E. Blankenship, M. T. Madigan and C. E. Bauer, Kluwer Academic Publishers, Dordrecht, 1995, vol. 2, pp. 527–557 Search PubMed.
- W. Zinth and J. Wachtveitl, ChemPhysChem, 2005, 6, 871–880 CrossRef CAS PubMed.
- K. Woronowicz, D. Sha, R. N. Frese and R. A. Niederman, Biochemistry, 2011, 50, 4819–4829 CrossRef CAS PubMed.
- G. J. Magis, M. J. den Hollander, W. G. Onderwaater, J. D. Olsen, C. N. Hunter, T. J. Aartsma and R. N. Frese, Biochim. Biophys. Acta, 2010, 1798, 637–645 CrossRef CAS PubMed.
- B. Thangaraj, C. C. Jolley, I. Sarrou, J. B. Bultema, J. Greyslak, J. P. Whitelegge, S. Lin, R. Kouril, R. Subramanyam, E. J. Boekema and P. Fromme, Biophys. J., 2011, 100, 135–143 CrossRef CAS PubMed.
- A. Freiberg, J. P. Allen, J. C. Williams and N. W. Woodbury, Photosynth. Res., 1996, 48, 309–319 CrossRef CAS PubMed.
- H. Bergstrom, V. Sundstrom, R. Vangrondelle, T. Gillbro and R. Cogdell, Biochim. Biophys. Acta, 1988, 936, 90–98 CrossRef.
- S. Scheuring and J. N. Sturgis, Science, 2005, 309, 484–487 CrossRef CAS PubMed.
- K. Woronowicz and R. A. Niederman, Adv. Exp. Med. Biol., 2010, 675, 161–178 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01981d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2014 |