 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen activation on Pt–Sn nanoalloys supported on mixed Sn–Ce oxide films
Armin
Neitzel
a,
Yaroslava
Lykhach
*a,
Tomáš
Skála
b,
Nataliya
Tsud
b,
Viktor
Johánek
b,
Mykhailo
Vorokhta
b,
Kevin C.
Prince
cd,
Vladimír
Matolín
b and
Jörg
Libuda
ae
aLehrstuhl für Physikalische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstrasse 3, 91058 Erlangen, Germany. E-mail: yaroslava.lykhach@fau.de; Fax: +49 9131 8528867; Tel: +49 9131 8520944
bDepartment of Surface and Plasma Science, Charles University, Faculty of Mathematics and Physics, V Holešovičkách 2, 18000 Prague, Czech Republic
cElettra-Sincrotrone Trieste SCpA, Strada Statale 14, km 163.5, 34149 Basovizza-Trieste, Italy
dIOM, Strada Statale 14, km 163.5, 34149 Basovizza-Trieste, Italy
eErlangen Catalysis Resource Center, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstrasse 3, 91058 Erlangen, Germany
First published on 19th May 2014
Abstract
We have studied the interaction of H2 with Pt–Sn nanoalloys supported on Sn–Ce mixed oxide films of different composition by means of synchrotron radiation photoelectron spectroscopy and resonant photoemission spectroscopy. The model catalysts are prepared in a three step procedure that involves (i) the preparation of well-ordered CeO2(111) films on Cu(111) followed by subsequent physical vapor deposition of (ii) metallic Sn and (iii) metallic Pt. The formation of mixed Sn–Ce oxide is accompanied by partial reduction of Ce4+ cations to Ce3+. Pt deposition leads to the formation of Pt–Sn nanoalloys accompanied by the partial re-oxidation of Ce3+ to Ce4+. Subsequent annealing promotes further Pt–Sn alloy formation at expense of the Sn content in the Sn–Ce mixed oxide. Adsorption of H2 on Pt–Sn/Sn–Ce–O at 150 K followed by stepwise annealing results in reversible reduction of Ce cations caused by spillover of dissociated hydrogen between 150 and 300 K. Above 500 K, annealing of Pt–Sn/Sn–Ce–O in a hydrogen atmosphere results in irreversible reduction of Ce cations. This reduction is caused by the reaction of hydrogen with oxygen provided by the mixed oxide substrate via the reverse spillover to Pt–Sn nanoalloy. The extent of the hydrogen and oxygen spillover strongly depends on the amount of Sn in the Sn–Ce mixed-oxide. We observe an enhancement of hydrogen spillover on Pt–Sn/Sn–Ce–O at low Sn concentration as compared to Sn-free Pt/CeO2. Although the extent of hydrogen spillover on Pt–Sn/Sn–Ce–O with high Sn concentration is comparable to Pt/CeO2, the reverse oxygen spillover is substantially suppressed on these samples.
1. Introduction
Bimetallic nanoparticles, nanoalloys,1–5 represent a new class of nanomaterials with the potential to tailor catalytic properties. The latter may be tuned via the particle's electronic structure or ensemble effects. Bimetallic nanoalloys consisting of Pt alloyed with other metals, e.g. Ag,4 Ni,6 Pd,7 Ru,8,9 or Sn,9–12 have a great potential for application in direct methanol or ethanol fuel cells. Another benefit from alloying Pt with less expensive, non-noble metals is to reduce the costs of the anode fuel cell material.13 Pt–Sn nanoalloys are among the most attractive candidates, also because of their high tolerance towards CO.14High dispersion of the bimetallic nanoparticles is an important parameter for effective fuel cell operation.13 This can be achieved through the careful control of the composition and size of the bimetallic nanoalloys.5 Alternatively, an oxide support can be employed which can prevent agglomeration of metallic nanoparticles. For instance, the addition of cerium oxide to Pt/C catalysts significantly improves the durability of the catalyst.15 Furthermore, co-deposition of Pt and cerium oxide allows to stabilize very small Pt particles16 or even yields atomically dispersed Pt.17–19 Due to their low Pt content, fuel cell catalysts containing highly dispersed Pt in cerium oxide matrices yield very high specific power, i.e. the ratio of the power density and the weight of the noble metal, when compared to commercial PtRu electrodes.18
Recently, Pt–Sn nanoalloys dispersed in porous cerium oxide have been proposed as perspective materials for fuel cell applications.20 Operating under hydrogen feed, these materials showed excellent specific power values, which considerably exceed even those of Pt–CeO2 catalyst with similar Pt concentration.
The microscopic origins of these effects and the role of Sn remain under discussion. It is speculated that the high activity of the catalyst maybe related to higher probability of hydrogen dissociation at Pt–Sn sites or to the higher degree of reduction of cerium oxide upon interaction with Sn.21 Previous studies have shown that Sn interacts strongly with both components, Pt22,23 and cerium oxide.24,25 There are several bulk Pt–Sn alloy phases with defined composition, such as Pt3Sn, PtSn, PtSn2.5,23 In those cases where Sn has been deposited on Pt(111)26–28 and Pt(110),22,29 formation of different ordered surface alloys has been observed. For nanoparticles the situation is of course more complex, due to the possible formation of core–shell structures, segregation patterns or random alloy structures due to entropic and kinetics effects.1–6 Interestingly, none of the before mentioned surface alloys on Sn/Pt(111) turned out to be active for dissociation of molecular hydrogen.28 In contrast, a moderate increase in hydrogen uptake has been observed at small Sn concentration on bimetallic Pt–Sn supported on alumina.30
Deposition of Sn on stoichiometric cerium oxide results in the formation of mixed Sn–Ce oxide which is accompanied by reduction of a part of the cerium cations to Ce3+.24,25 Similar to cerium oxide, the mixed Sn–Ce oxide shows moderate activity towards hydrogen dissociation above 550 K.31,32 Therefore, activation of hydrogen at room temperature is not possible on Pt-free mixed Sn–Ce oxide. Surprisingly, fuel cells containing Pt–Sn nanoalloys dispersed on cerium oxide yield high power densities already at room temperature.20 Therefore, the extraordinary reactivity of the Pt–Sn nanoalloy dispersed on cerium oxide must result from a synergistic effect between the intermetallic and the oxide phase. In the following, we explore the origin of this synergistic interplay between the Pt–Sn nanoalloy and the oxide support during reaction with molecular hydrogen under well-controlled conditions. Our study is based on the preparation of model systems consisting of Pt–Sn nanoalloys supported on mixed Sn–Ce oxide films with different Sn concentration and different degree of reduction. We employ synchrotron radiation photoelectron spectroscopy (SRPES) and resonant photoemission spectroscopy (RPES) to follow both the changes in the composition of the Pt–Sn nanoalloy and the degree of reduction of the Sn–Ce mixed oxide, both during preparation and during reaction with molecular hydrogen.
2. Materials and methods
2.1 Sample preparation
Three types of model Pt–Sn/Sn–Ce–O catalyst samples have been investigated. Their preparation followed a three steps procedure as shown in Fig. 1. First, epitaxial CeO2(111) films were grown on clean Cu(111) (MaTecK GmbH, 99.999%) by physical vapor deposition (PVD) of Ce metal (goodfellow, 99.99%) from Mo crucible in oxygen atmosphere (5 × 10−7 mbar, Linde, 99.999%) at 523 K, followed by annealing of the films at 523 K in oxygen atmosphere at the same pressure for 5 min. The preparation method33,34 yielded a continuous and stoichiometric CeO2(111) film with a thickness in the range of 1.7–2.0 nm. Low energy (LEED) and reflection high energy (RHEED) electron diffraction study of the prepared films confirmed the epitaxial growth of CeO2(111) with the characteristic (1.5 × 1.5) superstructure relative to the Cu(111) substrate.34,35 According to previous scanning tunneling microscopy (STM) studies, flat CeO2(111) terraces are separated by steps showing defect structures similar to those on single-crystalline CeO2(111).36,37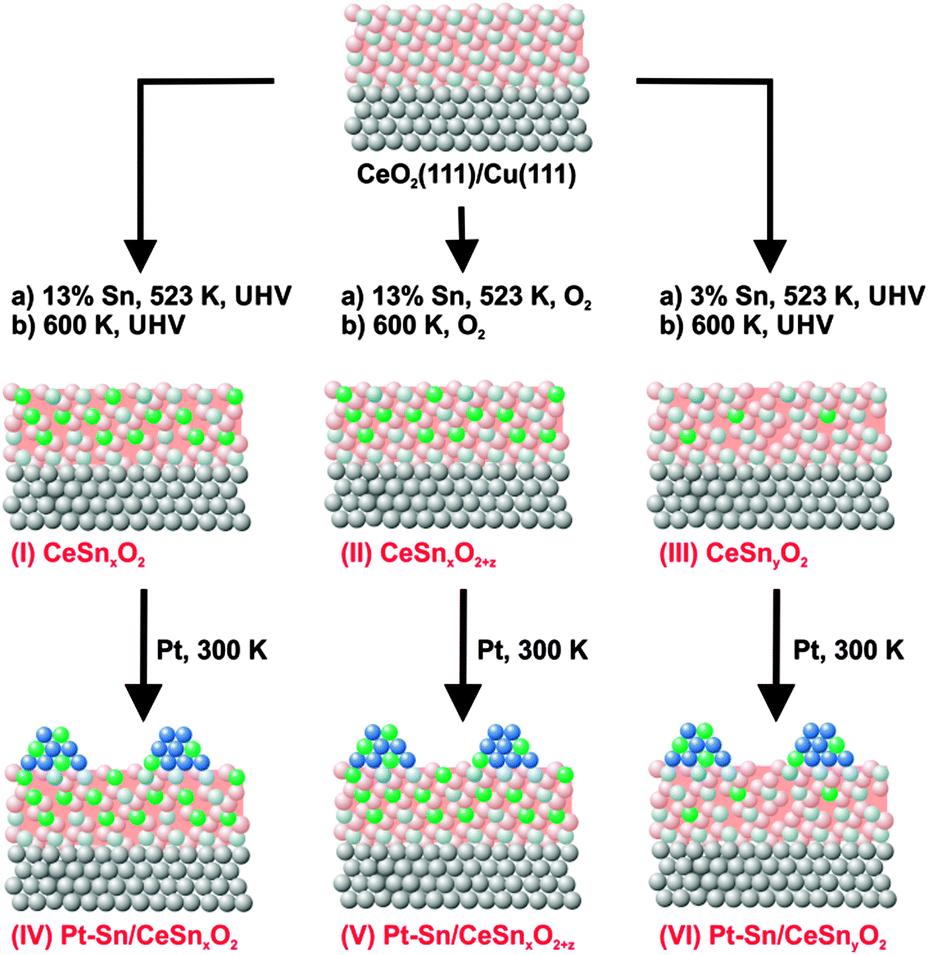 | ||
| Fig. 1 Schematic representation of three step preparation of model catalytic systems. | ||
In the following step, three types of Sn–Ce–O substrates denoted as SnxCeO2 (I, high Sn concentration, strongly reduced), SnxCeO2+z (II, high Sn concentration, oxidized), SnyCeO2 (III, low Sn concentration, reduced) were prepared. All samples were prepared by PVD of Sn from a Mo crucible onto the CeO2(111) films, either in ultrahigh vacuum (UHV) (I, III) or in an oxygen atmosphere (II) (5 × 10−7 mbar) at 523 K. Subsequently, the substrates were annealed either in UHV (I, III) or in an oxygen atmosphere (II) (5 × 10−7 mbar) at 600 K for 3 min. The subscripts “x” and “y” denote different Sn concentration in CeO2. The subscript “z”, where z > 0, indicates oxidizing preparation conditions of SnxCeO2+z (II). There, SnxCeO2 (I) and SnxCeO2+z (II) contain about 13% of Sn and SnyCeO2 (III) contains about 3% of Sn with respect to the total thicknesses of the mixed Sn–Ce oxide. The Sn concentration was calibrated by means of X-ray photoelectron spectroscopy (XPS).
Similar amounts of Pt were deposited by means of PVD from a Pt wire (goodfellow, 99.99%) onto the three samples SnxCeO2 (I), SnxCeO2+z (II), and SnyCeO2 (III) at 300 K in UHV. The nominal thickness of the deposited Pt layers was 0.3–0.4 nm as determined by XPS. The corresponding samples are denoted Pt–Sn/SnxCeO2 (IV), Pt–Sn/SnxCeO2+z (V), and Pt–Sn/SnyCeO2 (VI).
The interaction of hydrogen with the samples (I–VI) was studied in two regimes, denoted as “desorption” and “reaction”. The first regime (desorption) involved exposure of the samples to 50 L (1 L = 1.33 × 10−6 mbar s) of hydrogen (4 × 10−7 mbar, 165 s) at 150 K followed by stepwise annealing in UHV up to 750 K. The annealing was performed such that the samples were briefly annealed to the stated temperatures and cooled down after each annealing step. In the second regime (reaction), the samples were repeatedly exposed to 50 L of H2 at constant temperatures ranging from 150 K to 750 K. Hydrogen (Linde 99.999%) was dosed by backfilling the UHV chamber.
2.2 Synchrotron radiation photoelectron spectroscopy (SRPES) and resonant photoemission spectroscopy (RPES)
High-resolution synchrotron radiation photoelectron spectroscopy (SRPES) was performed at the Materials Science Beamline, Elettra synchrotron light facility in Trieste, Italy. The bending magnet source provides synchrotron light in the energy range of 21–1000 eV. The UHV end station (base pressure 1 × 10−10 mbar) is equipped with a multichannel electron energy analyzer (Specs Phoibos 150), a rear view LEED optics, an argon sputter gun, and a gas inlet system. The basic setup of the chamber includes a dual Mg/Al X-ray source.The core level spectra of O 1s, Pt 4f, and Sn 4d were acquired at photon energies of 650, 180, and 60 eV, respectively. The binding energies (BEs) in the spectra were calibrated with respect to the Fermi level. Additionally, Al Kα radiation (1486.6 eV) was used to measure O 1s, C 1s, Ce 3d, Sn 3d, Pt 4f, and Cu 2p3/2 core levels. All spectra were acquired at constant pass energy and at an emission angle for the photoelectrons of 20° or 0° with respect to the sample normal, while using the X-ray source or synchrotron radiation, respectively. The values of total spectral resolution were 1 eV (Al Kα), 150 meV (hν = 60 eV), 200 meV (hν = 115–180 eV), and 650 meV (hν = 650 eV). Fitting of the Sn 4d spectra revealed multiple spectral components. The components associated with Sn2+ and Sn4+ states were fitted with a Voigt profile and those associated with the Pt–Sn alloy were fitted with a Doniach–Šunjić convoluted with a Gaussian profile with fixed asymmetry parameter of 0.05 after subtraction of a composite background. The composite background consisted of a baseline spectrum obtained prior to Sn deposition and a Shirley background. The use of the composite background was necessary to compensate for the shape of the background in the Sn 4d region. The widths and the branching ratios for the Sn 4d components associated with Sn–Ce–O were determined prior to Pt deposition and kept fixed thereafter. For the Sn components that emerged after Pt deposition the widths of the peaks and their relative binding energies were fixed and the branching ration was set to 1.5. Additional details regarding the fitting procedure will be given in the next section.
Valence band spectra were acquired at three different photon energies, 121.4, 124.8, and 115.0 eV, that correspond to the resonant enhancements in Ce3+, Ce4+ ions, and to off-resonance conditions, respectively. Analysis of the spectra obtained with these photon energies forms the basis of RPES.38,39 The Ce3+ resonance at a photon energy of 121.4 eV is caused by a Super-Coster–Kronig decay involving electron emission from Ce 4f states located about 1.4 eV below the Fermi edge. The Ce4+ resonance at a photon energy of 124.8 eV involves emission of O 2p electrons (hybridized with Ce states) from the valence band around 4.0 eV. The valence band spectrum measured at a photon energy of 115 eV is used as a background for the calculation of the intensity difference of the features on- and off-resonance, denoted as the resonant enhancements for Ce3+ (D(Ce3+)) and for Ce4+ (D(Ce4+)), respectively. The resonant enhancement ratio (RER), calculated as D(Ce3+)/D(Ce4+), is the direct measure of the degree of reduction of the mixed Sn–Ce oxide. All SRPES data were processed using KolXPD fitting software.40 During the experiment, the sample temperature was controlled by a DC power supply passing a current through Ta wires holding the sample. Temperatures were monitored by a K-type thermocouple attached to the back of the sample.
3. Results and discussion
3.1 Preparation and thermal stability of the Pt–Sn nanoalloy on Sn–Ce oxide: influence of the Sn concentration and the degree of reduction
Sn 4d spectra obtained from SnxCeO2 (I, high Sn concentration, reduced), SnxCeO2+z (II, high Sn concentration, oxidized), and SnyCeO2 (III, low Sn concentration) substrates are shown in Fig. 2a–c (top traces), respectively. All three spectra resemble a single spin–orbit-split doublet with the 4d5/2 component located at binding energies of 25.5 eV on samples (I–II) and 25.3 eV on sample (III). The corresponding doublets are labeled (A). Fitting of the Sn 4d spectra revealed a similar spin–orbit splitting (1.04 eV), but different widths and branching ratios between the 4d5/2 and 4d3/2 components on the three substrates (I–III). The spin–orbit branching ratio in Sn 4d spectra are between 1.26 and 1.33, deviating strongly from the statistical value of 1.5.41 Such deviations from the statistical branching ratios have been observed experimentally in photoelectron spectra of several shallow Sn 4d,42–44 In 4d,44 Pb 5d,44 Bi 5d,44 Hg 5d,45 Xe 5d46 orbitals obtained at low photon energies. The varying spin–orbit branching ratio results from resonances caused by spin–orbit interaction-activated interchannel coupling between final states with different j value46 that leads to variation of the partial cross sections in the doublet.46–48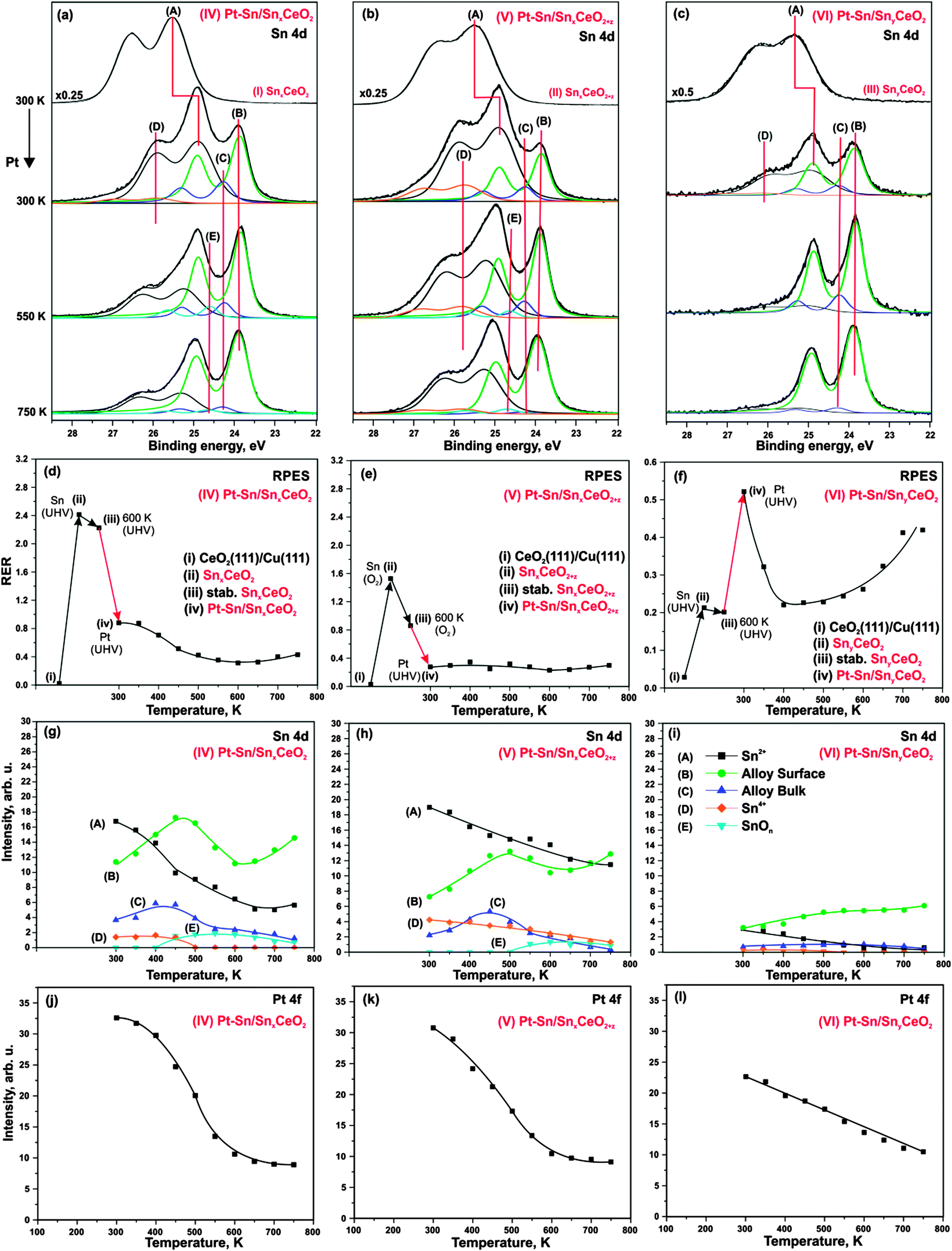 | ||
| Fig. 2 Sn 4d spectra obtained from (IV) Pt–Sn/SnxCeO2 (a), (V) Pt–Sn/SnxCeO2+z (b), and (VI) Pt–Sn/SnyCeO2 (c) before and after Pt deposition onto the corresponding (I) SnxCeO2 (top, a), (II) SnxCeO2+z (top, b), and (III) SnyCeO2 (top, c) followed by annealing under UHV. Evolution of RERs (d–f), integrated Sn 4d (g–i), and Pt 4f (j–l) on Pt–Sn/SnxCeO2 (d, g, j), Pt–Sn/SnxCeO2+z (e, h, k), Pt–Sn/SnyCeO2 (f, i, l), respectively, as a function of temperature. Sn 4d and Pt 4f spectra were obtained with photon energies 60 and 180 eV, respectively. | ||
According to the binding energies of the Sn 4d5/2 components observed on the samples I–II (25.5 eV) and III (25.3 eV) we assign the oxidation state of Sn cations to 2+.49 Note that the structure and the oxidation state of Sn–Ce mixed oxides strongly depend on the Sn concentration and the preparation conditions.25,50–53 Many Sn–Ce–O phases have been reported with different oxidation states of Sn and Ce.50 In some particular cases, this may cause ambiguities in identification of the oxidation state of Sn. De Padova et al.49 suggested to use binding energy shifts between coexisting oxide and metallic Sn 4d components to identify the oxidation state. Binding energy differences of 1.4–1.5 eV were associated with Sn2+ while larger differences (up to 2 eV) were attributed to Sn4+. In additional experiments (data not shown) we prepared the sample with higher Sn content that show well-resolved oxide and metal components in Sn 4d spectrum. The binding energy difference between the two components was 1.4 eV which further supports the assignment of the doublets (A) on samples (I–III) to Sn2+.
Deposition of Sn results in a partial reduction of cerium oxide. This effect is reflected by the increase of the RER on all three samples, as shown in Fig. 2d–f. On SnxCeO2 (I) and SnyCeO2 (III), the degree of reduction of the Sn–Ce oxide (RER) scales linearly with the amount of Sn deposited in UHV.24 For deposition of Sn in oxygen (sample II) a lower degree of reduction of the mixed oxide is observed. Whereas deposition of Sn in UHV (SnxCeO2 (I)) yields a RER of 2.4, the deposition of the same amount of Sn under oxygen atmosphere (SnxCeO2+z (II)) yields a RER of 1.6. The following annealing of samples SnxCeO2 (I) and SnyCeO2 (III) in UHV up to 600 K causes a weak re-oxidation of the ceria. In sharp contrast, strong re-oxidation of Sn–Ce mixed oxide is observed only upon annealing of SnxCeO2+z (II) in oxygen up to 600 K. Interestingly, no new spectral features at higher binding energies were found in Sn 4d spectrum on this sample (Fig. 2b, top spectrum). This shows that neither Sn deposition nor the following annealing procedure in oxygen leads to oxidation of Sn2+ to Sn4+.
Deposition of Pt on the substrates (I–III) at 300 K in UHV leads to significant changes in Sn 4d spectra shown in Fig. 2a–c (second spectrum from the top). The corresponding samples are labeled Pt–Sn/SnxCeO2 (IV), Pt–Sn/SnxCeO2+z (V), and Pt–Sn/SnyCeO2 (VI). First of all, Pt deposition resulted in a decrease of the intensity of component (A) and a simultaneous shift to lower binding energy, yielding a value of 24.9 eV on samples IV–V and 25.0 eV on sample VI. The corresponding shifts of the Sn 4d components (A) are 0.6 eV (samples IV–V) and 0.3 eV (sample VI). These shifts are accompanied by the concomitant shifts of the O 1s peak by 0.4 eV (sample IV), 0.2 eV (sample V) and 0.0 eV (sample VI). We will discuss the origin of these shifts later. Additionally, three new components (B–D) emerged at 23.9 eV (B), 24.3 eV (C), and 25.9 eV (D) in Sn 4d spectra on all samples (IV–VI). The binding energies of components (B) and (C) are similar to that of metallic Sn (24.0 eV).22,49,54 However, formation of Pt–Sn nanoalloys, rather than metallic Sn, is energetically favorable.23 Therefore, we associate both component (B) and (C) with Pt–Sn nanoalloys. The components (B) and (C) can be attributed to a surface and a bulk contribution from the Pt–Sn alloy, respectively.22,27,55
The formation of Pt–Sn nanoalloy induces changes in the oxidation states of both tin and cerium in the samples (IV–VI). Specifically, emergence of a weak component (D) in the spectra of the samples (IV–VI) is associated with partial oxidation of Sn2+ to Sn4+. The binding energy difference between these two states (approximately 1.0 eV) is close to the values 0.7 eV reported in the literature.49,56 Considering the small intensity of the peak (D), we assume that this effect is minor. The most prominent effect is, however, the partial re-oxidation of Ce3+ to Ce4+, which is evident from the decrease in RER on samples (IV) and (V) (see Fig. 2d and e, respectively). In contrast to the samples (IV) and (V), we observe a reduction of cerium cations upon Pt deposition in case of sample (VI), i.e. at low Sn concentration (Fig. 2f). The partial reduction of cerium cations upon deposition of metals (Pt, Pd) on CeO2(111) films has been reported earlier.38,57 It was assigned to a partial charge transfer from the metal particles to cerium oxide.
Therefore, we conclude that there are two competing effects which give rise to changes of the cerium oxidation state in mixed Sn–Ce oxide upon deposition of Pt (IV–VI). On the one hand, Sn migrates from the mixed Sn–Ce oxide into the Pt particles and, thereby, changes its oxidation state from 2+ to 0. The charge required for the reduction of Sn could in part be provided by the remaining Sn2+, which is oxidized to Sn4+ but, primarily, by the oxidation of Ce3+ to Ce4+. This charge transfer channel prevails at high Sn concentration, i.e. on samples (IV) and (V). On the other hand, there is a partial charge transfer from Pt to the mixed oxide (resulting in Ce3+ centers), which also occurs upon deposition of Pt on CeO2(111).38 This channel prevails at low Sn concentration, i.e. on sample (VI). Similar behavior has been observed upon Pd deposition on Sn–Ce mixed oxide.57 Therefore, the final oxidation state of cerium cations is the sum of counteracting effects and must be discussed carefully.
Several phenomena may contribute to the slightly different binding energy shifts of the Sn 4d (A) and O 1s components on samples (IV–VI), including band bending, chemical shifts and final state effects. Empirically we have found a concomitant shift of both Sn 4d (A) and O 1s to lower binding energy with decreasing Sn concentration in Sn–Ce mixed oxides (data not shown), and the same trend would be expected upon Pt–Sn alloying. On the other hand, Pt deposition on CeO2(111) causes a shift of the O 1s levels by about 0.2 eV to higher binding energy (data not shown), which is associated with charge transfer from Pt to the oxide support. In addition, local differences in electronic screening are expected upon formation of supported alloy particles, which will give rise to different final state shifts. These opposing contributions may explain the different binding energy shifts observed in the Sn 4d and O 1s levels, but cannot be resolved on the basis of the present experiments. The stability of the prepared samples (IV–VI) has been studied upon annealing in UHV. The corresponding Sn 4d spectra and evolution of the integrated intensities of all Sn 4d spectral components are shown in Fig. 2a–c and g–i, respectively. Annealing of the samples (IV–VI) resulted in a substantial decrease of component (A) (Sn2+ in mixed oxide) which was accompanied by an increase of both components (B) and (C) associated with the Pt–Sn alloys. This behavior suggests a thermally induced Pt–Sn alloying which is accompanied by migration of Sn from the mixed Sn–Ce oxide into the alloy.
During the annealing, the intensity of the component (D) (Sn4+) gradually decreases on all samples until it vanishes on samples (IV) and (VI) at 500 K. Above 450 K, the new weak component (E) emerges at 24.5 eV on samples (IV) and (V), i.e. at high Sn concentration, but not on the sample (VI) where the Sn concentration is low. The binding energy of this component is in between the typical values for Sn0 and Sn2+. A likely explanation would be associated with the formation of sub-stoichiometric SnOn species, possibly in contact with the Pt–Sn nanoalloy. The binding energy of the component (E) is close to the value of 24.44–24.46 eV associated with SnO species formed upon oxidation of Pt–Sn alloys.22 In this temperature regime, the oxygen can be provided to Pt–Sn nanoalloys by reverse spillover from Sn–Ce mixed oxide.38 We may speculate that simultaneous spillover of oxygen and Sn may result in partial encapsulation of Pt–Sn nanoalloys by sub-stoichiometric SnOn species. However, taking into account low intensity of the corresponding component (E), we assume that this effect is minor. The absence of the component (E) on Pt–Sn/SnyCeO2 (VI) suggests that encapsulation of Pt–Sn does not occur at low Sn concentration.
With respect to the thermal stability and morphology of the prepared layers, the concentration of Sn in the samples appears to be a key parameter. This is evident from the comparison of the integrated Pt 4f intensities on samples (IV–VI) and their development upon annealing (Fig. 2j–l). The lower Pt 4f intensities at 300 K suggest formation of larger Pt–Sn particles on sample (VI, low Sn concentration) as compared to samples (IV–V, high Sn concentration).
Thermally induced coalescence and growth of the Pt–Sn particles occurs on all samples (IV–VI). However, the decrease of the integrated Pt 4f intensities is more pronounced on samples (IV–V) than on sample (VI) (Fig. 2j–l). Partial encapsulation of Pt–Sn nanoalloy by SnOn species may contribute to the attenuation of the Pt 4f intensity on samples (IV–V) above 500 K.
As mentioned above, the evolution of RER on samples (IV–VI) upon annealing has to be discussed carefully taking into account multiple processes. In addition to the two processes discussed above (charge transfer from Pt to the oxide and migration of Sn from the mixed oxide into Pt–Sn nanoalloy), oxygen vacancies become mobile at elevated temperature and may migrate from the interface into the bulk. For Sn-free Pt/CeO2(111) system, we have shown that vacancies are removed from the surface above 300 K.38 Therefore, the decrease of RER on samples (IV) and (VI) may result from both processes, Sn diffusion and vacancy migration. The decrease of RER is not well resolved on sample (V) due to the strong oxidation during sample preparation.
Above 500 K, the evolution of RER differs considerably on samples (IV–VI). Only RER on sample (VI) (Fig. 2f) is similar to those observed for Sn-free Pt/CeO2(111).38 Based on a comparison with the latter, we associate the increase of RER above 500 K with the reverse oxygen spillover from the oxide substrate to the Pt–Sn nanoalloy. Reverse oxygen spillover is a general phenomenon on ceria-based catalysts38,58 and has been thoroughly studied on Pt/CeO2(111).38 Note that reverse oxygen spillover is not accompanied by migration of Sn to the Pt–Sn nanoalloy on sample (VI). According to the development of Sn 4d components (A) and (D) (Fig. 2i), the Sn is nearly completely removed from the cerium oxide phase below 500 K. Also, the SnOx component (E) in the Sn 4d spectra (Fig. 2c) is absent suggesting that migration of Sn from the oxide support to Pt–Sn nanoalloy stops before the reverse oxygen spillover sets in.
Unlike on sample (VI), annealing above 500 K does not cause any significant changes of the RER on samples (IV–V). We attribute this to the counteracting processes of Sn migration to the Pt–Sn nanoalloy (leading to partial re-oxidation) and oxygen reverse spillover (leading to partial reduction) which occur in parallel.
3.2 Stability of Pt-free Sn–Ce oxide under hydrogen treatment
First, we studied interaction of hydrogen with Pt-free Sn–Ce mixed oxides. Selected Sn 4d core level spectra obtained from SnxCeO2 (I) after annealing in hydrogen are shown in Fig. 3a. The Sn 4d spectrum obtained prior to hydrogen exposure is shown at the top.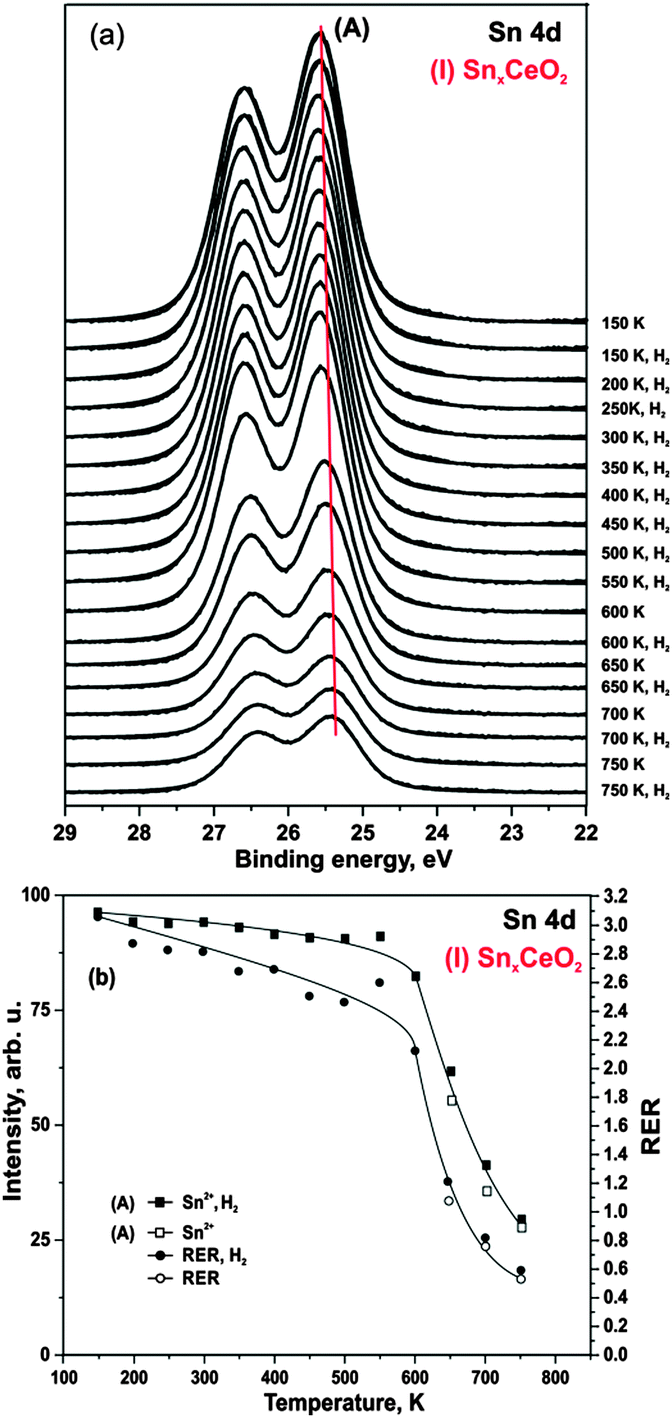 | ||
| Fig. 3 Sn 4d spectra (a), and evolutions of integrated Sn 4d intensities (squares) and RERs (circles) (b) on (I) SnxCeO2 during annealing under 50 L H2 exposure (filled symbols) at constant temperatures between 150 to 750 K and occasional annealing in UHV (empty symbols). | ||
We find that exposure of the SnxCeO2 (I) to hydrogen between 150 and 750 K does not cause any measurable reduction of Sn2+ to metallic Sn. The integrated intensity of the Sn2+ component remains unchanged up to 600 K (Fig. 3b). However, annealing above 600 K leads to a significant decrease of the Sn 4d intensity of the component (A). We assign this effect to thermally induced diffusion of Sn2+ ions into deeper layers.24 The fact that this effect is induced thermally and not by hydrogen is evident from the comparison of the Sn 4d intensity in which we first annealed in UHV and then in hydrogen at the same temperature (see Fig. 3b).
As evident from Fig. 3b, the integrated intensity of Sn 4d corresponds to the evolution of RER throughout the whole temperature range. This is consistent with migration of Sn2+ into the bulk, thus, leading to a partial re-oxidation of cerium cations at the surface. Further, no loss of oxygen was observed as can be concluded from the unchanged O 1s signal (data not shown).
3.3 Stability of the Pt–Sn/Sn–Ce–O model catalysts under hydrogen treatment
Next, we investigated the interaction of H2 with three Pt–Sn/Sn–Ce–O samples (IV–VI). Two sets of experiments are explored. In the first set of experiments we expose the samples to 50 L H2 at 150 K followed by stepwise annealing in UHV. We refer to this procedure as hydrogen “desorption” experiment. In the second set of experiments we anneal the samples in H2 (exposure 50 L) at temperatures between 150 and 750 K. We refer to this procedure as a hydrogen “reaction” experiment. The corresponding intensities in the Sn 4d (component (A) and sum of components B, C, and E) and Pt 4f regions are shown in Fig. 4. In addition to the “desorption” and “reaction” experiment we show a “reference” experiment that corresponds to annealing in UHV (i.e. in absence of hydrogen).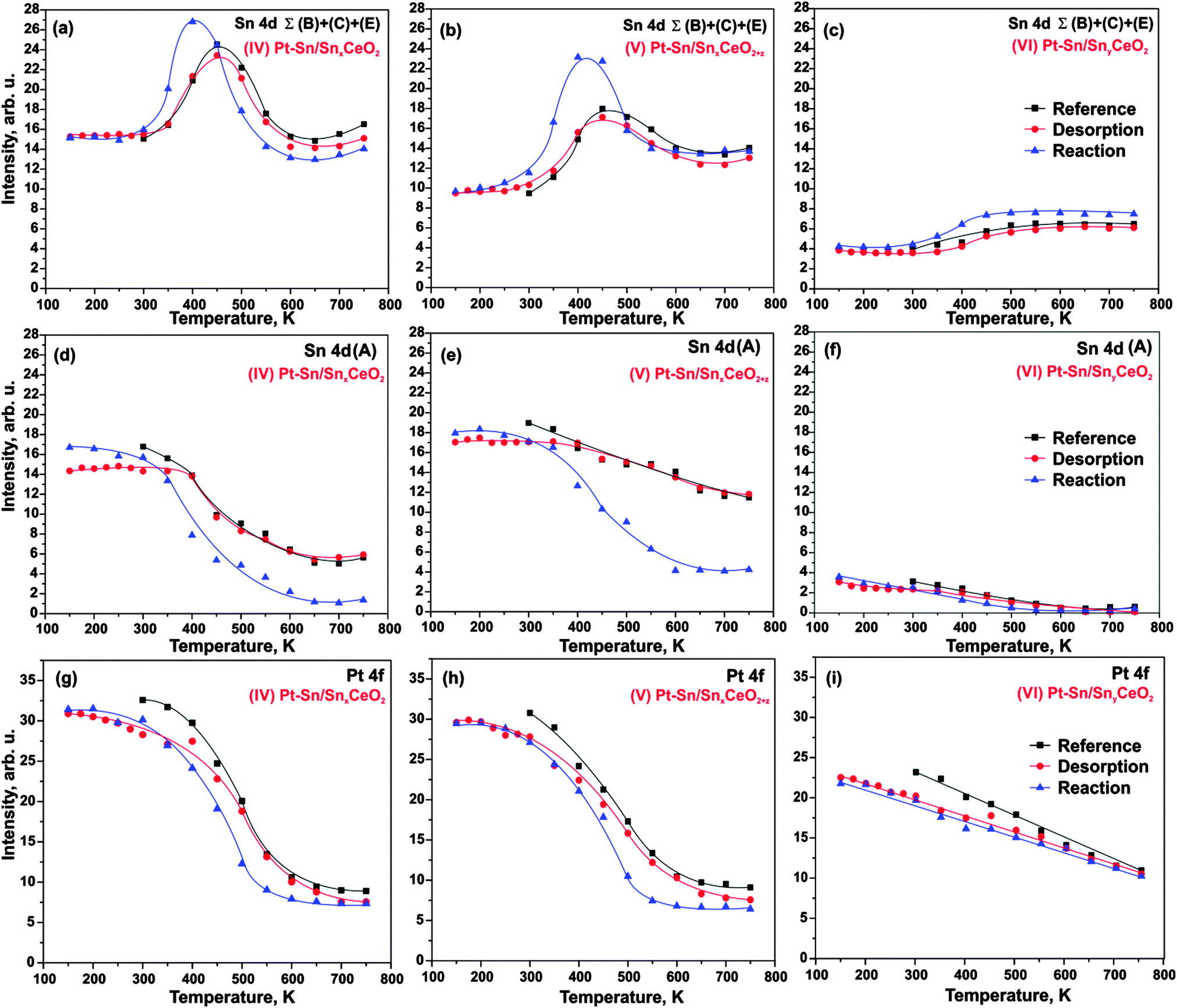 | ||
| Fig. 4 Integrated Sn 4d of summed (Σ(B) + (C) + (E)) (a–c), (A) (d–f) spectral components, and Pt 4f intensities (g–i) on a (IV) Pt–Sn/SnxCeO2 (a, d, g), (V) Pt–Sn/SnxCeO2+z (b, e, h), and (VI) Pt–Sn/SnyCeO2 (c, f, i) as functions of temperature. | ||
Note that exposure to 50 L H2 at 150 K does not cause any detectable changes in the Sn 4d and Pt 4f regions on any of the three samples (IV–VI), nor do the signals show significant deviations from the reference experiment during the annealing in UHV. Small intensity variations may be caused by traces of contaminants, primarily water, adsorbed from the background gas at low temperature. Therefore, we conclude that the changes of morphology and composition induced by the adsorption and desorption experiment are minor. In brief, we identify the same temperature regions as without H2 treatment: the sharp increase of the reduced Sn components (B, C, E) between 300 and 450 K is caused by onset of Sn diffusion into the Pt particles. At temperatures above 450 K sintering of the Pt–Sn nanoalloys lead to a decrease of both signals, Pt 4f and Sn 4d (B–D).
In contrast to the H2 “adsorption” experiment, a clear effect is observed for the “reaction” experiment (in which we expose the sample to H2 at different temperatures) on the samples with high Sn concentration (IV–V). We find that the decrease of the oxide component (A) is shifted to lower temperatures (onset around 300 K). A similar shift is observed for the increase of the “reduced” Sn (B, C, E), which, in addition, is more pronounced between 300 K and 400 K (see Fig. 4a, b, d and e). These changes are accompanied by a concomitant shift of the decrease in the Pt 4f intensity (Fig. 4a, b, g and h). Our observations suggest that the decomposition of the mixed Sn–Ce oxide is accelerated by the reaction with H2. As a result, diffusion of Sn to the Pt particles and Pt–Sn alloy formation is facilitated at relatively lower temperature, i.e. in the temperature region between 300 and 400 K. At temperatures above 400 K, particle sintering dominates and leads to decrease of both alloy components, Pt 4f and Sn 4d (B, C, E). Interestingly, the decrease in intensity exceeds that of the reference experiment, suggesting that sintering is also facilitated under H2 treatment.
3.4 Redox behavior and spillover on Pt–Sn/Sn–Ce–O model catalysts under hydrogen treatment
Finally, we investigated the cerium oxidation state during H2 activation on Pt–Sn/Sn–Ce–O model catalysts as a function of the Sn concentration. The developments of the RER on samples (IV–VI) during annealing in UHV and upon interaction with H2 are shown in Fig. 5a–c. The experimental conditions are the same as described in Section 3.3 and, accordingly, we label the experiments as “reference”, “desorption”, and “reaction”. In addition, we added the RER on the Sn-free Pt/CeO2(111) for comparison.59,60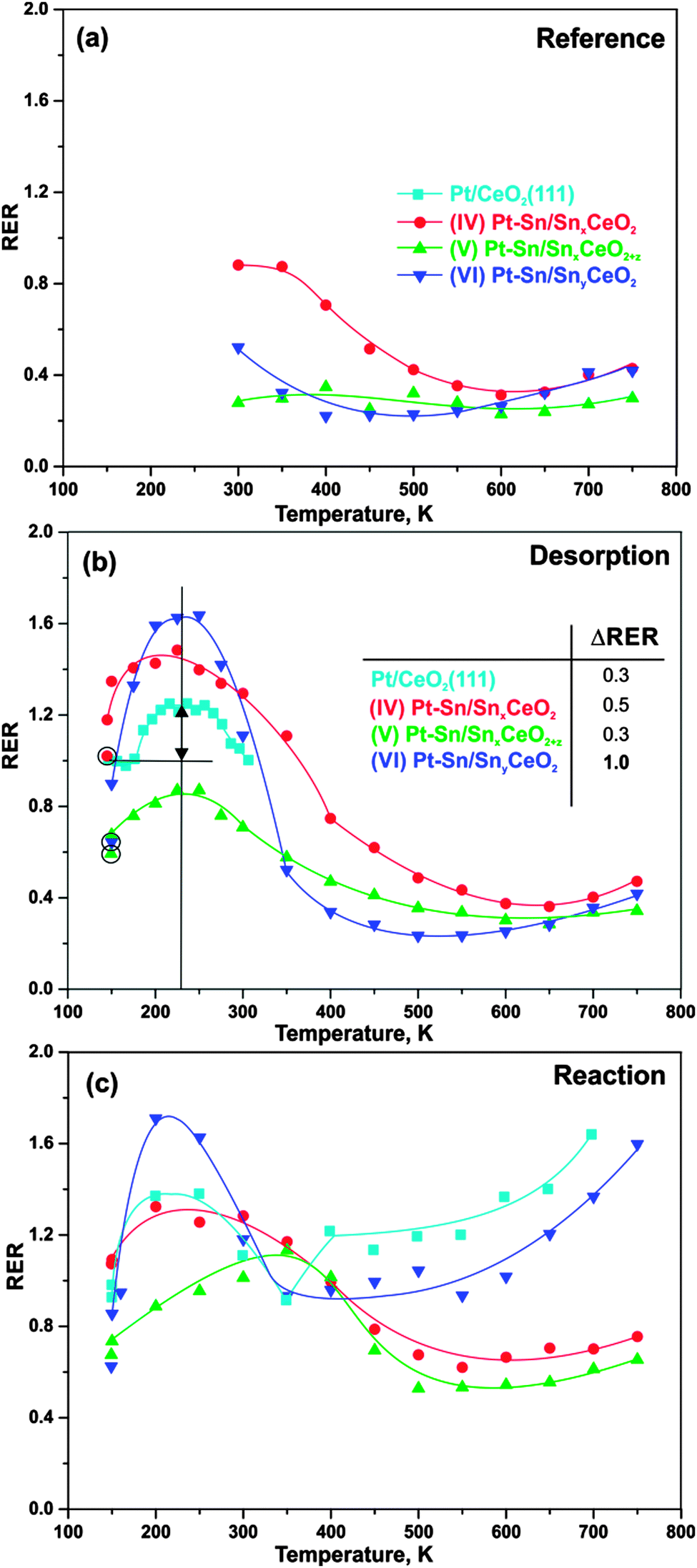 | ||
| Fig. 5 Evolutions of RERs on Pt/CeO2(111), (IV) Pt–Sn/SnxCeO2, (V) Pt–Sn/SnxCeO2+z, and (VI) Pt–Sn/SnyCeO2 as functions of temperature during stepwise annealing in UHV (a, reference), adsorption of 50 L H2 at 150 K followed by stepwise annealing in UHV (b, desorption), annealing under exposure to 50 L H2 at constant temperatures between 150 and 750 K (c, reaction). The circled symbols in (b) indicate RERs prior to H2 adsorption. Differences between the initial and the maximum RERs achieved at 225 K specify an extent of hydrogen spillover from Pt–Sn nanoalloys to the support. | ||
The evolution of RER on samples (IV–VI) in UHV, i.e. under “reference” condition, has been discussed in Section 3.1. Briefly, the decrease of the RER between 300 and 500 K is associated with re-oxidation of Ce3+ caused by the reduction of Sn2+ upon Pt–Sn alloy formation. An additional contribution arises from migration of oxygen vacancies from the surface into deeper layers.38 Above 500 K, reverse oxygen spillover from the oxide support to the nanoalloys leads to partial reduction of mixed Sn–Ce oxide, counteracting the above oxidation channels.
Earlier we reported that adsorption of hydrogen on Pt/CeO2(111) at 150 K followed by annealing in UHV results in reversible reduction of cerium oxide between 190 and 310 K.59 The corresponding RER evolution is shown in Fig. 5b. Here, the increase of RER between 190 and 210 K is associated with spillover of hydrogen from the Pt particles to CeO2 followed by formation of hydroxyl groups that are stable up to 260 K. Reverse hydrogen spillover and desorption of hydrogen from Pt particles is activated between 260 and 310 K and leads to re-oxidation of cerium oxide. As can be seen from the data in Fig. 5b, the above mentioned reversible reduction of cerium cations occurs also on samples (IV–VI). The extent of the reversible reduction can be estimated from the difference between RERs before hydrogen adsorption at 150 K (circled symbols in Fig. 5b) and the value of the RER at 225 K, where hydrogen spillover from the particles to the oxide has reached its maximum. The table inset in Fig. 5b shows the estimated extent of hydrogen spillover on all samples. Apparently, the extent of hydrogen spillover on samples (IV) and (V), i.e. at high Sn concentration, is comparable to the Sn-free Pt/CeO2(111). Surprisingly, the extent of hydrogen spillover is considerably larger on sample (VI), i.e. at low Sn concentration. There are several possible explanations for this observation. First, the effect may be caused by the modified hydrogen adsorption on the nanoalloy. Secondly, the stability and mobility of the hydroxyl groups on cerium oxide could be modified by Sn doping. In this respect it is noteworthy that dissociation of molecular hydrogen was found to be suppressed on Sn/Pt(111).28 A moderate destabilization of hydrogen adsorption on the Pt–Sn nanoalloy, i.e. at low Sn concentration, may, on the other hand, provide an additional driving force for hydrogen spillover to the oxide. Such effect would be consistent with the experimental observation that the onset of hydrogen spillover is shifted to a lower temperature in the presence of tin. On the Sn-free samples the onset is observed between 190 and 200 K (see Fig. 5 and ref. 59) whereas on all Sn-containing samples hydrogen spillover is already observed at temperatures as low as 150 K. Theoretical studies on alloy nanoparticles may help to clarify this point. Above 350 K, the RER returns to similar values as obtained under “reference” conditions (Fig. 5a). As described in the previous section, the interaction of hydrogen with the samples (IV–VI) does not cause changes of the Sn diffusion and sintering behavior under “desorption” conditions. Therefore, we assume that similarly to Pt/CeO2(111), the hydrogen spillover is reversed on samples (IV–VI) between 250 and 350 K leading to desorption of molecular hydrogen. This process reestablishes the original oxidation state of the cerium cations.
Irreversible reduction of cerium oxide occurs upon reaction of hydrogen with reversely spilled-over oxygen on the Pt particles for Pt/CeO2(111).59 The corresponding development of the RER is shown in Fig. 5c. The evolution of RER under the “reaction” condition shows irreversible reduction in the temperature region between approximately 450 and 700 K. The region of irreversible reduction on sample (VI) with low Sn concentration is very similar to those observed on the Sn-free Pt/CeO2(111).59 However, the degree of reduction on sample (VI) is lower than it is on Sn-free Pt/CeO2 (Fig. 5c). The effect is even more pronounced on the samples with high Sn concentration (IV–V). We attribute this observation to the fact that oxygen reverse spillover and Sn diffusion from the oxide into the Pt–Sn nanoalloy occurs in the same temperature region. As discussed above, both effects lead to opposite changes of the oxidation state of cerium cations and thus may obscure or even cancel each other. However, the accelerated decomposition of the mixed Sn–Ce oxide followed by Pt–Sn alloying discussed in Section 3.1 proved the existence of oxygen reverse spillover also for the samples with high Sn concentration (IV–V).
Finally, comparison of the Sn 4d spectra obtained from samples (IV–VI) after annealing to 750 K under “reference”, “desorption”, and “reaction” conditions is shown in Fig. 6a–c. It is seen that the component (A) associated with Sn2+ in the mixed Sn–Ce oxide decreased considerably after the annealing of the samples (IV–V) under the “reaction” conditions as compared to annealing under UHV (“reference” conditions) and with H2 exposure at low temperature (“desorption” conditions). Thus, the extent of the decomposition of mixed Sn–Ce oxide could be used as a measure of the oxygen spillover on these samples under the “reaction” conditions. The differences observed between the samples (IV) and (V) suggests that the initial degree of reduction of the mixed oxide has a major influence on reverse oxygen spillover. In particular, the sample (V) prepared with lower degree of reduction of the mixed oxide shows lower extent of the mixed oxide decomposition under “reaction” conditions and therefore points to less facile oxygen reverse spillover on this sample as compared to the sample (IV).
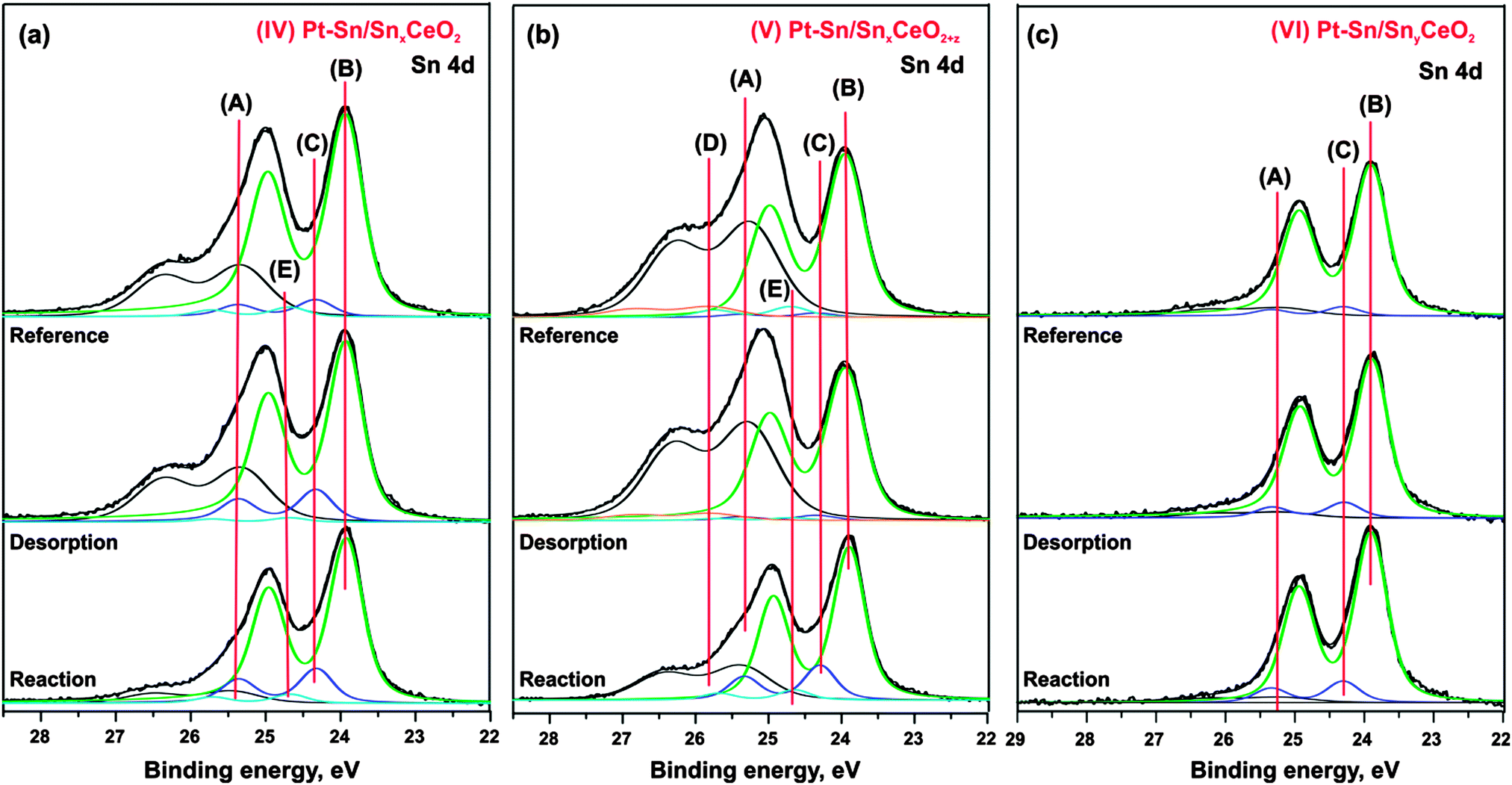 | ||
| Fig. 6 Sn 4d spectra obtained from (IV) Pt–Sn/SnxCeO2 (a), Pt–Sn/SnxCeO2+z (b), and Pt–Sn/SnyCeO2 (c) after annealing at 750 K in UHV (reference), after exposure to H2 (50 L) at 150 K followed by annealing to 750 K (desorption), after annealing at 750 K under exposure to 50 L H2 (reaction). | ||
4. Conclusion
Hydrogen activation on Pt–Sn nanoalloys supported on mixed Sn–Ce oxides has been studied as a function of the Sn concentration and the degree of reduction of mixed Sn–Ce oxide by means of SRPES and RPES.(1) The model catalysts are prepared in three steps that involve: (i) preparation of stoichiometric CeO2(111)/Cu(111) films; (ii) Sn deposition under UHV or oxygen atmosphere; (iii) Pt deposition in UHV at 300 K. In this way, Pt–Sn/Sn–Ce–O systems are obtained in which the Pt coverage, the Sn concentration, and the degree of reduction of the Sn–Ce mixed oxide can be controlled independently.
(2) During the sample preparation formation of mixed Sn–Ce oxides occurs upon deposition of metallic Sn on well-ordered CeO2(111) films at 523 K. Sn is formed exclusively in the oxidation state 2+. The formation of the mixed oxide is accompanied by a strong reduction of cerium cations, i.e. transformation of Ce4+ to Ce3+. A lower degree of reduction can be established by deposition of Sn in oxygen atmosphere. Upon Pt deposition at 300 K, a Pt–Sn nanoalloy is formed, regardless of the Sn concentration and the degree of reduction of the mixed oxide. Formation of the Pt–Sn alloy is accompanied by partial re-oxidation of cerium cations and by formation of a small fraction of Sn4+. The resulting degree of reduction of mixed Sn–Ce oxide depends on the Sn concentration and is further modified by charge transfer between the Pt–Sn nanoalloys and the oxide support.
(3) At temperatures above 400 K, the Pt–Sn nanoalloy on the mixed Sn–Ce oxides undergoes sintering. This process is accompanied by enhanced diffusion of Sn from Sn–Ce mixed oxide into the Pt–Sn nanoalloys.
(4) Oxygen reverse spillover occurs on all Sn-containing systems irrespective of the Sn concentration. At high Sn concentration, however, oxygen reverse spillover is accompanied by migration of Sn which may result in partial encapsulation of the Pt–Sn nanoalloys by SnOn species. At low Sn concentration, the encapsulation of Pt–Sn does not occur due to the decomposition of the mixed oxide below the onset temperature of oxygen reverse spillover.
(5) Spillover of hydrogen between the Sn–Ce mixed oxide and the Pt–Sn nanoalloy occurs on all samples, irrespective of the Sn concentration and degree of reduction of the Sn–Ce oxide. However, the highest extent of hydrogen spillover was observed at low Sn concentration. The onset temperature for hydrogen spillover is lowered by Sn doping (150 K). Above 250 K hydrogen reverse spillover is followed by H2 desorption and leads to re-oxidation in the temperature range up to 350 K.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
“chipCAT” (FP7-NMP-2012-SMALL-6, Grant Agreement no. 310191), DFG, Ministry of Education of the Czech Republic (LG12003, LD13054), COST Action CM1104, Czech Science Foundation 204/11/1183, (FP7/2007–2013, no. 312284)Abbreviations
| SRPES | Synchrotron radiation photoelectron spectroscopy |
| RPES | Resonant photoemission spectroscopy |
| LEED | Low energy electron diffraction |
| UHV | Ultrahigh vacuum |
| RER | Resonant enhancement ratio |
Acknowledgements
The authors gratefully acknowledge financial support by the European Commission in the framework of FP7 Project “chipCAT” (FP7-NMP-2012-SMALL-6, Grant Agreement no. 310191) and by the Ministry of Education of the Czech Republic (LG12003 and LD13054). We acknowledge additional support from the DFG within the excellence cluster “Engineering of Advanced Materials” in the framework of the Excellence Initiative as well as support from the COST Action CM1104 “Reducible oxide chemistry, structure and functions”. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 312284. V.J. acknowledges support by Czech Science Foundation 204/11/1183. Y.L., A.N., V.J., N.T., and K.C.P. thank Elettra for excellent working conditions and support.References
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- D. Bochicchio and R. Ferrando, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 165435 CrossRef.
- U. Banin, Nat. Mater., 2007, 6, 625–626 CrossRef CAS PubMed.
- H. Liu, F. Ye, Q. Yao, H. Cao, J. Xie, J. Y. Lee and J. Yang, Sci. Rep., 2014, 4, 3969 Search PubMed.
- X. Wang, L. Altmann, J. Stöver, V. Zielasek, M. Bäumer, K. Al-Shamery, H. Borchert, J. Parisi and J. Kolny-Olesiak, Chem. Mater., 2013, 25, 1400–1407 CrossRef CAS.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed.
- Y.-J. Wang, D. P. Wilkinson, A. Guest, V. Neburchilov, R. Baker, F. Nan, G. A. Botton and J. Zhang, J. Power Sources, 2013, 221, 232–241 CrossRef CAS PubMed.
- Z. Liu, F. Su, X. Zhang and S. W. Tay, ACS Appl. Mater. Interfaces, 2011, 3, 3824–3830 CAS.
- E. Antolini, J. Power Sources, 2007, 170, 1–12 CrossRef CAS PubMed.
- F. Han, X. Wang, J. Lian and Y. Wang, Carbon, 2012, 50, 5498–5504 CrossRef CAS PubMed.
- C.-T. Hsieh, Y.-S. Chang and K.-M. Yin, J. Phys. Chem. C, 2013, 117, 15478–15486 CAS.
- G. Samjeske, S.-i. Nagamatsu, S. Takao, K. Nagasawa, Y. Imaizumi, O. Sekizawa, T. Yamamoto, Y. Uemura, T. Uruga and Y. Iwasawa, Phys. Chem. Chem. Phys., 2013, 15, 17208–17218 RSC.
- H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS PubMed.
- D.-H. Lim, D.-H. Choi, W.-D. Lee and H.-I. Lee, Appl. Catal., B, 2009, 89, 484–493 CrossRef CAS PubMed.
- D.-H. Lim, W.-D. Lee, D.-H. Choi and H.-I. Lee, Appl. Catal., B, 2010, 94, 85–96 CrossRef CAS PubMed.
- E. You, R. Guzmán-Blas, E. Nicolau, M. Aulice Scibioh, C. F. Karanikas, J. J. Watkins and C. R. Cabrera, Electrochim. Acta, 2012, 75, 191–200 CrossRef CAS PubMed.
- V. Matolín, I. Matolínová, M. Václavů, I. Khalakhan, M. Vorokhta, R. Fiala, I. Piš, Z. Sofer, J. Poltierová-Vejpravová, T. Mori, V. Potin, H. Yoshikawa, S. Ueda and K. Kobayashi, Langmuir, 2010, 26, 12824–12831 CrossRef PubMed.
- R. Fiala, I. Khalakhan, I. Matolínová, M. Václavů, M. Vorokhta, Z. Sofer, S. Huber, V. Potin and V. Matolín, J. Nanosci. Nanotechnol., 2011, 11, 5062–5067 CrossRef CAS PubMed.
- M. Vorokhta, I. Khalakhan, I. Matolínová, M. Kobata, H. Yoshikawa, K. Kobayashi and V. Matolín, Appl. Surf. Sci., 2013, 267, 119–123 CrossRef CAS PubMed.
- V. Matolín, M. Cabala, I. Matolínová, M. Škoda, M. Václavů, K. C. Prince, T. Skála, T. Mori, H. Yoshikawa, Y. Yamashita, S. Ueda and K. Kobayashi, Fuel Cells, 2010, 10, 139–144 Search PubMed.
- M. Vaclavu, I. Matolinova, J. Myslivecek, R. Fiala and V. Matolin, J. Electrochem. Soc., 2009, 156, B938–B942 CrossRef CAS PubMed.
- N. Tsud, T. Skála, K. Veltruská, M. Škoda, K. C. Prince and V. Matolín, J. Phys.: Condens. Matter, 2011, 23, 215002 CrossRef CAS PubMed.
- X. Su, F. Yin, M. Huang, Z. Li and C. Chen, J. Alloys Compd., 2001, 325, 109–112 CrossRef CAS.
- T. Skála, F. Šutara, K. C. Prince and V. Matolín, J. Electron Spectrosc. Relat. Phenom., 2009, 169, 20–25 CrossRef PubMed.
- V. Matolín, M. Cabala, V. Cháb, I. Matolínová, K. C. Prince, M. Škoda, F. Šutara, T. Skála and K. Veltruská, Surf. Interface Anal., 2008, 40, 225–230 CrossRef.
- Y. L. Tsai, C. Xu and B. E. Koel, Surf. Sci., 1997, 385, 37–59 CrossRef CAS.
- E. Janin, M. Björkqvist, T. M. Grehk, M. Göthelid, C. M. Pradier, U. O. Karlsson and A. Rosengren, Appl. Surf. Sci., 1996, 99, 371–378 CrossRef CAS.
- M. T. Paffett, S. C. Gebhard, R. G. Windham and B. E. Koel, J. Phys. Chem., 1990, 94, 6831–6839 CrossRef CAS.
- E. Janin, H. von Schenck, S. Helldén, O. Tjernberg, U. O. Karlsson and M. Göthelid, Surf. Sci., 2002, 515, 462–470 CrossRef CAS.
- K. Balakrishnan and J. Schwank, J. Catal., 1991, 127, 287–306 CrossRef CAS.
- Y.-Z. Chen, B.-J. Liaw and C.-W. Huang, Appl. Catal., A, 2006, 302, 168–176 CrossRef CAS PubMed.
- S. Bernal, J. J. Calvino, G. A. Gifredo, J. M. Rodriguez-Izquiero, V. Perrichon and A. Laachir, J. Catal., 1992, 137, 1–11 CrossRef CAS.
- V. Matolín, J. Libra, I. Matolínová, V. Nehasil, L. Sedláček and F. Šutara, Appl. Surf. Sci., 2007, 254, 153–155 CrossRef PubMed.
- F. Šutara, M. Cabala, L. Sedláček, T. Skála, M. Škoda, V. Matolín, K. C. Prince and V. Cháb, Thin Solid Films, 2008, 516, 6120–6124 CrossRef PubMed.
- K. Mašek, J. Beran and V. Matolín, Appl. Surf. Sci., 2012, 259, 34–38 CrossRef PubMed.
- T. Staudt, Y. Lykhach, L. Hammer, M. A. Schneider, V. Matolín and J. Libuda, Surf. Sci., 2009, 603, 3382–3388 CrossRef CAS PubMed.
- F. Dvořák, O. Stetsovych, M. Steger, E. Cherradi, I. Matolínová, N. Tsud, M. Škoda, T. Skála, J. Mysliveček and V. Matolín, J. Phys. Chem. C, 2011, 115, 7496–7503 Search PubMed.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- V. Matolin, I. Matolinova, L. Sedlacek, K. C. Prince and T. Skala, Nanotechnology, 2009, 20, 215706 CrossRef CAS PubMed.
- J. Libra, KolXPD, version 1.8.0.26, 2003–2012 Search PubMed.
- D. Briggs and P. Seah, Practical Surface Analysis: Auger and X-ray photoelectron spectroscopy, Wiley, 1990 Search PubMed.
- H. T. Anyele, C. L. Griffiths, A. A. Cafolla, C. C. Matthai and R. H. Williams, Appl. Surf. Sci., 1998, 123–124, 480–484 CrossRef.
- R. A. Pollak, S. Kowalczyk, L. Ley and D. A. Shirley, Phys. Rev. Lett., 1972, 29, 274–277 CrossRef CAS.
- G. Margaritondo, J. E. Rowe and S. B. Christman, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 19, 2850–2855 CrossRef CAS.
- T. E. H. Walker, J. Berkowitz, J. L. Dehmer and J. T. Waber, Phys. Rev. Lett., 1973, 31, 678–681 CrossRef CAS.
- S. S. Kumar, T. Banerjee, P. C. Deshmukh and S. T. Manson, Phys. Rev. A: At., Mol., Opt. Phys., 2009, 79, 043401 CrossRef.
- N. G. Stoffel and G. Margaritondo, Phys. Lett. A, 1980, 75, 319–320 CrossRef.
- G. Margaritondo, R. Rosei, J. H. Weaver and W. M. Becker, Solid State Commun., 1980, 34, 401–404 CrossRef CAS.
- P. De Padova, M. Fanfoni, R. Larciprete, M. Mangiantini, S. Priori and P. Perfetti, Surf. Sci., 1994, 313, 379–391 CrossRef CAS.
- R. Lin, Y.-J. Zhong, M.-F. Luo and W.-P. Liu, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2001, 40, 36–40 Search PubMed.
- S. Mihaiu, A. Brăileanu, M. Bán, J. Madarász and G. Pokol, J. Optoelectron. Adv. Mater., 2006, 8, 572–575 CAS.
- B. Tolla, A. Demourgues, O. Isnard, M. Menetrier, M. Pouchard, L. Rabardel and T. Seguelong, J. Mater. Chem., 1999, 9, 3131–3136 RSC.
- N. Tsud, T. Skála, K. Mašek, P. Hanyš, M. Takahashi, H. Suga, T. Mori, H. Yoshikawa, M. Yoshitake, K. Kobayashi and V. Matolín, Thin Solid Films, 2010, 518, 2206–2209 CrossRef CAS PubMed.
- M. Škoda, M. Cabala, V. Cháb, K. C. Prince, L. Sedláček, T. Skála, F. Šutara and V. Matolín, Appl. Surf. Sci., 2008, 254, 4375–4379 CrossRef PubMed.
- Y. Jugnet, D. Loffreda, C. Dupont, F. Delbecq, E. Ehret, F. J. Cadete Santos Aires, B. S. Mun, F. Aksoy Akgul and Z. Liu, J. Phys. Chem. Lett., 2012, 3, 3707–3714 CrossRef CAS.
- J.-M. Themlin, M. Chtaïb, L. Henrard, P. Lambin, J. Darville and J.-M. Gilles, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 2460–2466 CrossRef CAS.
- T. Skála, F. Šutara, M. Škoda, K. C. Prince and V. Matolín, J. Phys.: Condens. Matter, 2009, 21, 0550051–0550059 CrossRef PubMed.
- G. S. Zafiris and R. J. Gorte, J. Catal., 1993, 139, 561–567 CrossRef CAS.
- Y. Lykhach, T. Staudt, M. Vorokhta, T. Skála, V. Johánek, K. C. Prince, V. Matolín and J. Libuda, J. Catal., 2012, 285, 6–9 CrossRef CAS PubMed.
- Reprinted from J. Catal., 285, Y. Lykhach, T. Staudt, M. Vorokhta, T. Skála, V. Johánek, K. C. Prince, V. Matolín and J. Libuda, Hydrogen spillover monitored by resonant photoemission spectroscopy, 6–9, Copyright (2014), with permission from Elsevier.
| This journal is © the Owner Societies 2014 |