 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-physical properties of 2-(1-ethynylpyrene)-adenosine: influence of hydrogen bonding on excited state properties†
P.
Trojanowski
a,
J.
Plötner
a,
C.
Grünewald
b,
F. F.
Graupner
c,
C.
Slavov
a,
A. J.
Reuss
a,
M.
Braun
a,
J. W.
Engels
b and
J.
Wachtveitl
*a
aInstitute for Physical and Theoretical Chemistry, Goethe University Frankfurt, Max-von-Laue-Straße 7, 60438 Frankfurt/Main, Germany. E-mail: wveitl@theochem.uni-frankfurt.de; Fax: +496979829709; Tel: +496979829351
bInstitute for Organic Chemistry and Chemical Biology, Goethe University Frankfurt, Max-von-Laue-Straße 7, 60438 Frankfurt/Main, Germany
cFaculty of Physics, Center for Integrative Protein Science, Ludwig Maximilians University Munich, Oettingenstrasse 67, 80538 Munich, Germany
First published on 14th May 2014
Abstract
The photo-physical properties of 2-(1-ethynylpyrene)-adenosine (PyA), a fluorescent probe for RNA dynamics, were examined by solvation studies. The excited-state dynamics display the influence of the vicinity on the spectral features. Combining improved transient absorption and streak camera measurements along with a new analysis method provide a detailed molecular picture of the photophysics. After intramolecular vibrational energy redistribution (IVR), two distinct states are observed. Solvent class (protic/aprotic) and permittivity strongly affect the properties of these states and their population ratio. As a result their emission spectrum is altered, while the fluorescence quantum yield and the overall lifetime remain nearly unchanged. Consequently, the hitherto existing model of the photophysics is herein refined and extended. The findings can serve as basis for improving the information content of measurements with PyA as a label in RNA.
Introduction
Monitoring dynamics of proteins, membranes or DNA/RNA depends on specific and versatile probes. Molecular fluorescence can be very sensitive depending on the molecule and its surroundings covering a great variety of lifetimes or spectral characteristics.1 Therefore fluorophores are perfectly suited and thus ubiquitously applied as molecular probes. Pyrene is used often among the large number of fluorophores. The basic spectral characteristics and photo-dynamics of the monomer2–8 as well as its excimer characteristics2,9,10 are well studied. Numerous derivatives of pyrene were synthesized to examine the influence of substitutions,8,11 artificial donor–acceptor systems,12–15 its aggregation behaviour and ISC/IC dynamics.16,17 A number of groups introduced pyrene into DNA to investigate electron transfer into nucleobases via photo-excitation of pyrene,15,18–24 specific base/structure recognition,25–30 FRET-pair studies,30–32 molecular beacons,33–35 hybridization probes36–42etc. and accomplished a remarkable understanding of the photo-physics and dynamics of pyrene coupled to deoxyribonucleic acid nucleosides. A great variety of fluorophores as probes for RNA were also synthesized, aiming for a structural and functional probe with minimal structural perturbation and optimal photo-physical properties,43 which is challenging due to the greater structural flexibility and complexity of RNA.44–47 Pyrene as a label has a small share of this variety in RNA. It was introduced with different linkers at different positions to the RNA nucleosides. One favoured position is the C2′ of ribose to detect ribonucleic acid48–53 as well as to monitor hybridization42,53,54 and folding.55 C2′ was chosen because the label should point out of a typical A-helix structure motif. Another approach is the direct modification of the nucleobase and was applied to DNA nucleosides by groups like Wagenknecht,15,18–24,36,39 Netzel,56,57 Kim37 and Berlin.58We previously presented the 2-(1-ethynylpyrene)-adenosine (PyA, Fig. 1), a pyrene modified RNA nucleoside, where pyrene is attached at the 2-position of adenine.59 This position was chosen in order to locate the pyrene in the minor groove of a RNA A-helix.
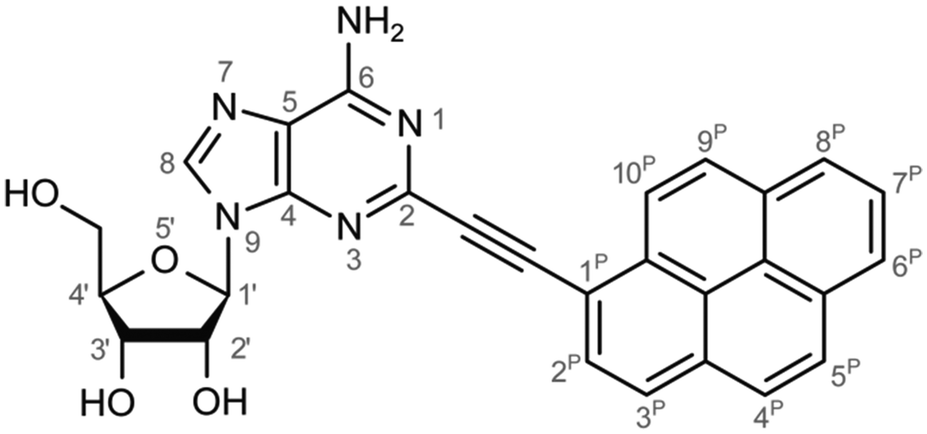 | ||
| Fig. 1 Lewis structure of the pyrene modified adenosine 2-(1-ethynylpyrene)-adenosine (PyA). | ||
The photo-physics of a protected nucleoside form60 as well as the applicability and the characteristics of PyA in modified 15-mer oligonucleotides61 as a RNA folding probe were investigated.62 Based on spectroscopic characteristics of CT states12,63 and findings of other groups, which observed ICT states in similar systems24,64 the proposed model of the photophysics involved an intramolecular charge transfer process (ICT).61 This intramolecular charge transfer was suggested to take place from the pyrene to the adenine64 and to be followed by a proton transfer to adenine.60 However, it remained unclear whether a proton transfer indeed takes place and how the surrounding contributes to the spectral properties and dynamics of 2-(1-ethynylpyrene)-adenosine.
This study reports on the solvent dependence of the photo-induced dynamics by using steady-state, transient absorption and streak camera measurements as well as quantum chemical calculations and leads to a more complete picture of the photo-induced processes of 2-(1-ethynylpyrene)-adenosine.
Material and methods
Sample preparation
2-(1-Ethynylpyrene)-adenosine (PyA) was synthesized using methodologies described earlier for the synthesis of protected PyA.59In summary, a solvent mixture of 60 ml of dimethylformamide and 5 ml of triethylamine was degassed thoroughly to exclude oxygen in the reaction mixture. 1.00 g (2.55 mmol, 1.00 eq.) of 2-iodoadenosine, 0.751 g (3.32 mmol, 1.30 eq.) of 1-ethynylpyrene, 84 mg (0.44 mmol, 17 mol%) of copper(I)iodide and 176 mg of bis(triphenylphosphine)palladium(II)chloride (0.25 mmol, 10 mol%) were added to the solvent mixture. The orange reaction mixture was stirred under argon for 19 h at room temperature. The mixture was filtered off to remove copper(I)iodide, the solution was adsorbed on silica gel and purified by column chromatography. Elution with a solvent mix of dichloromethane and methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and subsequent crystallization from methanol afforded 2-(1-ethynylpyrene)-adenosine as yellow powder in 80% yield.
:1) and subsequent crystallization from methanol afforded 2-(1-ethynylpyrene)-adenosine as yellow powder in 80% yield.
A DMSO stock solution was diluted to prepare the samples in other solvents and therefore a remaining amount of 1%-DMSO is present in all solvents. As a side effect tBuOH remains liquid at room temperature (∼23 °C).
Steady-state spectroscopy
Absorption spectra were recorded on a JASCO V670 absorption spectrometer between 300 nm and 600 nm using a fused silica cuvette with 1 mm optical path length. All spectra were background corrected for solvent absorption. In acetone, the spectrum was limited to 340 nm due to strong solvent absorption. The concentration of PyA for all absorption measurements varied between 150–200 μM.Fluorescence spectra were taken on a JASCO FP-8500 fluorimeter between 360 nm and 700 nm using a fused silica cuvette with the dimensions 3 × 3 × 3 mm3. The excitation wavelength varied between 371 nm and 376 nm depending on the exact position of the absorption maximum. The concentration was adjusted to approximately 0.1 OD/3 mm (∼10 μM) to minimize reabsorption effects. The spectra were corrected for remaining reabsorption and the spectral properties of the detection system. Fluorescence quantum yields (Φfl) were obtained by a relative method with quinine bisulfate (10 μM, Φfl = 0.60 ± 0.02 at 350 nm in 0.5 M H2SO4 (ref. 65 and 66)). A cross reference with quinine bisulfate (10 μM) in 0.05 M H2SO4 at 366 nm was done to account for excitation wavelength and H2SO4 concentration specific variations.
Vis-pump–probe spectroscopy
The time resolved transient absorption (TA) measurements were performed using a self-built pump–probe setup.67 Ultra short laser pulses (150 fs) were provided by a laser–amplifier system (Clark, MXR-CPA-2001) operating at a repetition rate of 1 kHz at a central wavelength of 775 nm. For the probe pulses, a single filament white light (WL) was generated by focusing the laser fundamental in a CaF2 crystal of 5 mm thickness. The WL covered a spectral range between 350 nm and 700 nm and was focused to a diameter of ∼100 μm at the sample position. The pump pulses at 388 nm were produced by second harmonic generation (SHG) in a 0.1 mm BBO crystal (200 fs pulse duration) and focused to a diameter of ∼200 μm at the sample position. The pump pulse energy was adjusted to 100 nJ to avoid two-photon absorption processes (total amount of excited molecules <6%), even though no changes in dynamics were observed for pulse energies up to 250 nJ. Difference spectra of excited and unexcited samples were recorded by blocking every second laser pulse in the excitation pulse pathway using a chopper. Probe pulses were dispersed using a spectrometer resulting in a spectral resolution of 4 nm and recorded by a gated detection system. This detection system is composed of a 128 channel photodiode array (PDA) combined with a signal processing chip (Hamamatsu Photonics, S8865-128), a driver circuit (Hamamatsu Photonics, C9118) and a data acquisition card (National Instruments, NI-6120), which digitizes the analog PDA signals at 16 bits. The experiments were performed under magic angle conditions (54.7° pump–probe polarization angle difference) and continuous movement of the sample in the plane perpendicular to the direction of pulse propagation. The signal to noise ratio was 103 for an average of 6 scans (2.5 × 103 pulses per scan).Streak camera spectroscopy
A Hamamatsu streak camera system (C5680-24 C) was employed to measure time-resolved fluorescence. For the femtosecond excitation pulses a CPA laser system (Clark, MXR-CPA-2001) at 780 nm was frequency doubled (390 nm) by second harmonic generation in a BBO crystal. The excitation energy varied between 40 nJ and 200 nJ depending on the sample and the time frame. It was focused onto the sample and detected under an angle of 15° to avoid excitation light to be collected. The excitation beam diameter was about 15 μm at the sample position. The induced fluorescence light was collected by an air-gap achromat (f = 80 mm, 20 mm diameter, Type OUV 4.20, Bernhard Halle Nachfl. GmbH). The collimated fluorescence light passed a wire-grid polarizer (Moxtek UBB01C, diameter 36 mm) positioned at the magic angle before being focused with a second achromatic lens (f = 75 mm NUV, diameter 25 mm, Edmund Optics) onto the entrance slit of the spectrograph (Princeton Instruments, Acton Series SP2356, f = 300 mm, 50 lines per mm grating blazed at 600 nm). The slit widths were held to a minimum in order to achieve best temporal and spectral resolution. For further details of the setup see ref. 68. The spectrometer slit width was set between 5 μm and 15 μm corresponding to an overall wavelength resolution of 1.3 nm to 2 nm, respectively. The focal plane of the spectrograph coincided with the photocathode of the streak camera system. The time window was set by a fast single-sweep unit (Hamamatsu, M5677-01) with sweep velocities chosen accordingly to the investigated process. In the nanosecond regime, data were acquired in continuous sweeping mode. For detailed observation of the spectral shift in the picosecond regime time resolution was improved by deploying single sweeping combined with jitter correction using a fraction of excitation light as a temporal fix point in each frame. Therefore an overall time resolution of <5 ps, 6–20 ps, 40–50 ps and 80–100 ps was achieved for the corresponding time windows of 0.12 ns, 0.48 ns, 4.27 ns and 10.33 ns, respectively. For a more detailed explanation of the streak camera operation see ref. 69. All data were collected in photon-counting mode, typically yielding between 104 and 103 counts for one detection bin at the emission peak (bin defined by: wavelength width = spectral resolution, temporal width = effective width of instrumental response function). To collect sufficient fluorescence intensity for an adequate signal and reasonable statistics, excitation energy was chosen individually. However, in all experiments the photon flux was kept low enough to avoid pile-up artefacts. The total acquisition time for each measurement amounted to ∼15 min for the continuous streaking and ∼1 h for single sweep measurements. The sample solutions were pumped through a flow cell (fused silica, 0.5 mm light path length) to exchange the excited sample volume between consecutive laser pulses. All samples were prepared to yield less than 0.15 OD/mm absorbance required to match linear conditions avoiding reabsorption. The same sample preparation was used for all time frames. Therefore the total number of excitation shots accumulated for ∼2 ml of sample preparation with concentrations ∼70 μM typically amounted to 5 × 105. To prove intactness of the sample throughout the whole streaking experiment stationary absorption spectra were recorded using a spectrophotometer (LAMBDA 750, Perkin Elmer) and compared before and after the time resolved measurements.Data analysis
The LDA was performed with the help of OPTIMUS – a newly developed time-resolved data analysis program in our group. The details about the program and the analysis methods used will be presented in detail elsewhere. Here we give only a short description of the LDA method to facilitate the discussion of the results.
The time-resolved data are quantitatively described by a spectrum of lifetimes, τ:
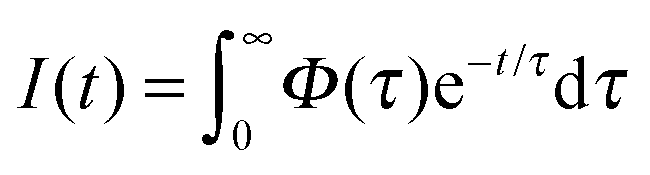
where S and Sfit are the experimental data and the fit, P is the penalty function which takes a specific form depending on the type of regularization used and λ is a Lagrange multiplier defining the relative importance of the two constraints.
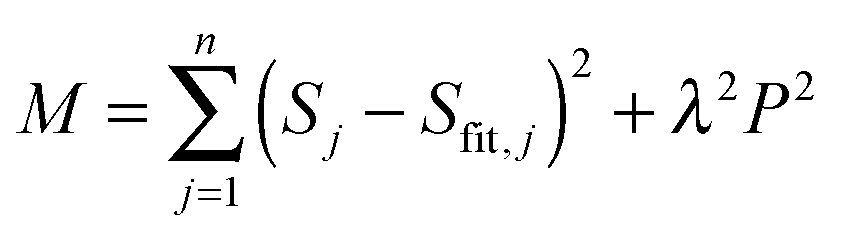
Most commonly, in time-resolved spectroscopy (see ref. 72–77 and references therein) variants of the maximum entropy method78 and Tikhonov regularization79 are used. Currently OPTIMUS performs Tikhonov regularization and allows selection between several penalty functions. The search of the Lagrange multiplier can be performed either manually or with the help of dedicated search methods – the L-curve criterion,80,81 the minimal product method82,83 and generalized cross-validation.75,84,85 The results shown here were obtained using the identity matrix as a smoothing norm and an optimal value for the regularization parameter, determination based on the L-curve and generalized cross-validation methods.
The LDA recovers the lifetime distribution at each detection wavelength and thus the results can be presented in the form of a contour map, which, similarly to ref. 72, we call the lifetime density map (LDM). The reading of the maps is straightforward following the convention for DAS from GLA of transient absorption – positive pre-exponential amplitudes (red) describe either decay of absorption or the increase of bleaching/stimulated emission, while negative pre-exponential amplitudes (blue) reflect either decay of bleaching/stimulated emission or the increase of absorption. LDM allows easier comparison between different samples and different experimental conditions.
We should mention here that the de-convolution of the instrument response function and the fitting of the coherent oscillations at time zero caused by the interaction of the temporarily overlapping pump and probe beams are performed simultaneously using the numerical inverse Laplace transform. In OPTIMUS, the deconvolution of the IRF is implemented similarly to the procedure described in ref. 70, taking into account the wavelength dependent chirp. The coherent contributions at time zero are modelled as a sum of a Gaussian function and its first and second derivatives as in ref. 86.
Quantum-chemical calculations
All quantum-chemical calculations have been performed using the Turbomole88 program package and the Q-Chem program.89In this study, we use time-dependent density-functional theory (TDDFT)90,91 and configuration-interaction singles methods (CIS)92 (for an overview see e.g.ref. 93 and references therein). Both single reference methods are often used to study excited states of large molecules due to their relatively low computational cost.
TDDFT suffers from the electron-transfer self-interaction (SI) error, which results in an underestimation of the excitation energies of charge-transfer (CT) and Rydberg states.94–99 The SI error is influenced by the amount of Hartree–Fock (HF) exchange in the used exchange correlation (xc) functional. This was pointed out by Dreuw et al.94 To evaluate the influence of the SI error for the PyA system an exchange correlation (xc) functional study applying xc-functionals with different amounts of HF exchange has been performed (B-LYP: pure DFT functional, 0% HF exchange; B3-LYP: hybrid functional, 20% HF exchange; BH-LYP: hybrid functional, 50% HF exchange).
The outcome has been compared with the results of a CIS calculation, which does not suffer from the SI error and therefore gives the correct ordering of the excited states in the framework of the single-reference methods and does not underestimate the excitation energy of CT states.
Single-reference methods neglect doubly or higher excited configurations of the wave function, which are needed for a correct description of electron correlation energy. It is known that for conjugated systems like pyrene, the influence of doubly excited configurations is significant. To clarify the influence of the higher excited configurations, we used a variant of configuration interaction singles doubles (CISD) named ‘scaled opposite spin CIS(D)′ (SOS-CIS(D)) methods100 as implemented in Q-Chem.
Experimental results
Steady-state characterization
Absorption and fluorescence spectra (Fig. 2) were taken for the pyrene-modified adenosine (PyA, Fig. 1) in eight different solvents, subdivided into two classes, protic (MeOH, EtOH, iPrOH and tBuOH) and aprotic (DMSO, DMF, acetone and EtOAc) solvents. The solvents of each class were chosen to cover a broad permittivity range (Table 1).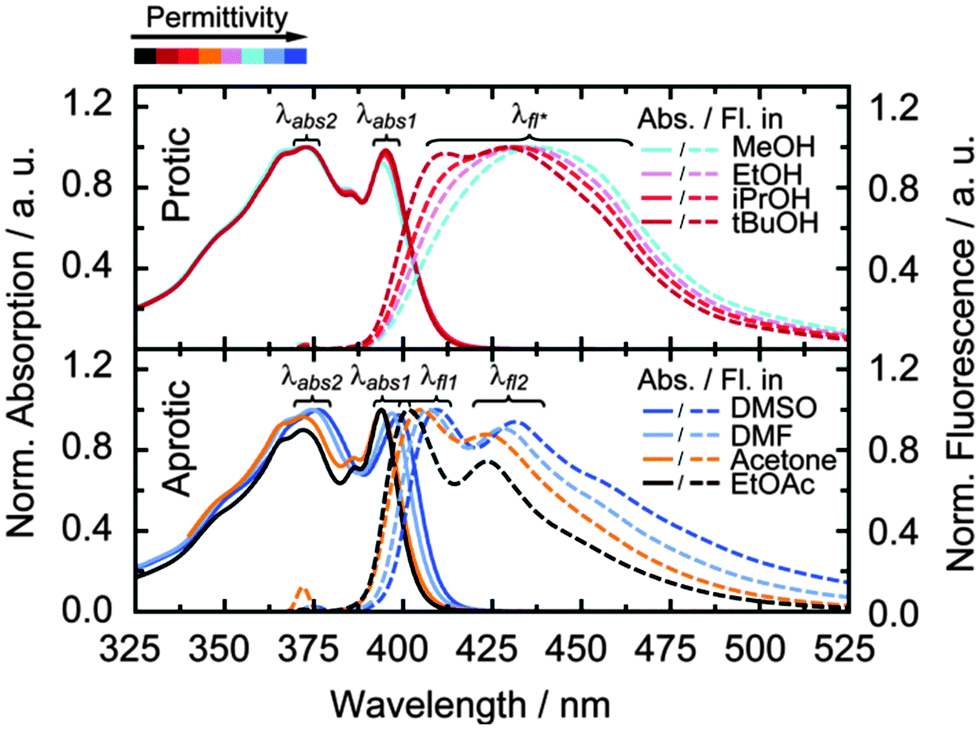 | ||
| Fig. 2 Normalized absorption (solid lines) and normalized fluorescence (dashed lines) spectra for pyrene modified adenosine (150 μM and 10 μM, respectively) in four protic (upper panel) and four aprotic (lower panel) solvents. For the fluorescence spectra PyA was excited at λabs2: MeOH – 372 nm, EtOH, iPrOH and tBuOH – 373 nm, DMSO – 376 nm, DMF – 374 nm, Acetone and EtOAc – 372 nm. | ||
| PyA in | ε r 101 | Abs./nm | Fluor./nm | Φ fl | |||
|---|---|---|---|---|---|---|---|
| λ abs2 | λ abs1 | λ fl1 | λ fl2 | ||||
| Aprotic | DMSO | 47.2 | 376 | 399 | 410 | 432 | 0.79 |
| DMF | 38.3 | 374 | 397 | 407 | 428 | 0.76 | |
| Acetone | 21.0 | 371 | 394 | 405 | 424 | 0.84 | |
| EtOAc | 6.1 | 371 | 394 | 399 | 422 | 0.78 | |
| Protic | MeOH | 33.0 | 371 | 394 | 440* | 0.79 | |
| EtOH | 25.3 | 372 | 395 | 437* | 0.79 | ||
| iPrOH | 20.2 | 372 | 395 | 434* | 0.76 | ||
| tBuOH | 12.5 | 372 | 395 | 431* | 0.72 | ||
In protic solvents, the fluorescence is in general broad and unstructured with weak shoulders, which get more pronounced with decreasing permittivity. Accordingly, the fluorescence splits into two clearly distinguishable maxima at 412 nm and 426 nm in tBuOH.
In aprotic solvents, a fine structure is visible in the fluorescence spectra, which undergoes a bathochromic shift by ∼10 nm with increasing permittivity.
Fluorescence quantum yields are around 0.8 for both solvent classes (Table 1, Φfl). In aprotic solvents, the fluorescence quantum yield shows no clear tendency with respect to the solvent permittivity, whereas protic solvents cause a slight increase of the fluorescence quantum yield with increasing permittivity.
Transient absorption of PyA in MeOH
TA measurements were performed to further elucidate the solvent and permittivity dependence of PyA in all eight solvents. They revealed a complex time-dependent behaviour with the same number of components, which differ only in their amplitude and temporal progression in each solvent. Therefore, we will point out our course of analysis exemplary for the methanol dissolved sample. The spectral characteristics and lifetimes in the other solvents are described thereafter and are summarized in Table 2.| PyA in | Time resolved data | Lifetimes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transient absorption (TA) | Streak camera (SC) | TA/ps | SC/ns | |||||||||
| Excited state abs./nm | Stimulated emission/nm | Fluorescence/nm | ||||||||||
| λ ESA1 | λ ESA2 | λ ESA2S | λ SE1 | λ SE2 | λ trfl1 | λ trfl2 | τ 1 | τ 2 | τ 3 | τ 4 * | ||
| Aprotic | DMSO | 666 | 575–n.d. | 403 | 450–575 | 410 | 432 | 0.2 | 3.7 | 2.1 | ||
| DMF | 661 | 545–n.d. | 401 | 430–545 | n.d. | n.d. | 0.3 | 3.7 | n.d. | |||
| Acetone | 664 | 515–660 | 398 | 420–515 | n.d. | n.d. | 0.3 | 3.7 | n.d. | |||
| EtOAc | 662 | 515–670 | 398 | 420–515 | 399 | 422 | 0.2 | 3.7 | 1.8 | |||
| Protic | MeOH | 663 | 515–630 | 608 | 399 | 435–515 | 432 | 440 | 0.3 | 2–10 | 10–20 | 2.4 |
| EtOH | 663 | 515–650 | 611 | 399 | 425–515 | n.d. | n.d. | 0.2 | 9.6 | 76 | n.d. | |
| iPrOH | 663 | 520–655 | 613 | 398 | 420–520 | n.d. | n.d. | 0.2 | 17 | 130 | n.d. | |
| tBuOH | 662 | 520–655 | 614 | 398 | 420–520 | 427 | 430 | 0.2 | 11 | 170 | 1.8 | |
In MeOH, the TA spectrum (Fig. 3B) shows three different main signals that cannot be separated clearly. The ground-state bleach (GSB, negative amplitude) is visible from 371 nm to ∼410 nm. The GSB signal partially overlaps with a structured stimulated emission (SE, negative amplitude), which dominates the spectrum between 400 nm and 500 nm. At longer wavelengths (>500 nm), broad and structured excited-state absorption (ESA, positive amplitude) is present, which extends beyond the spectral window of the measurements (∼690 nm).
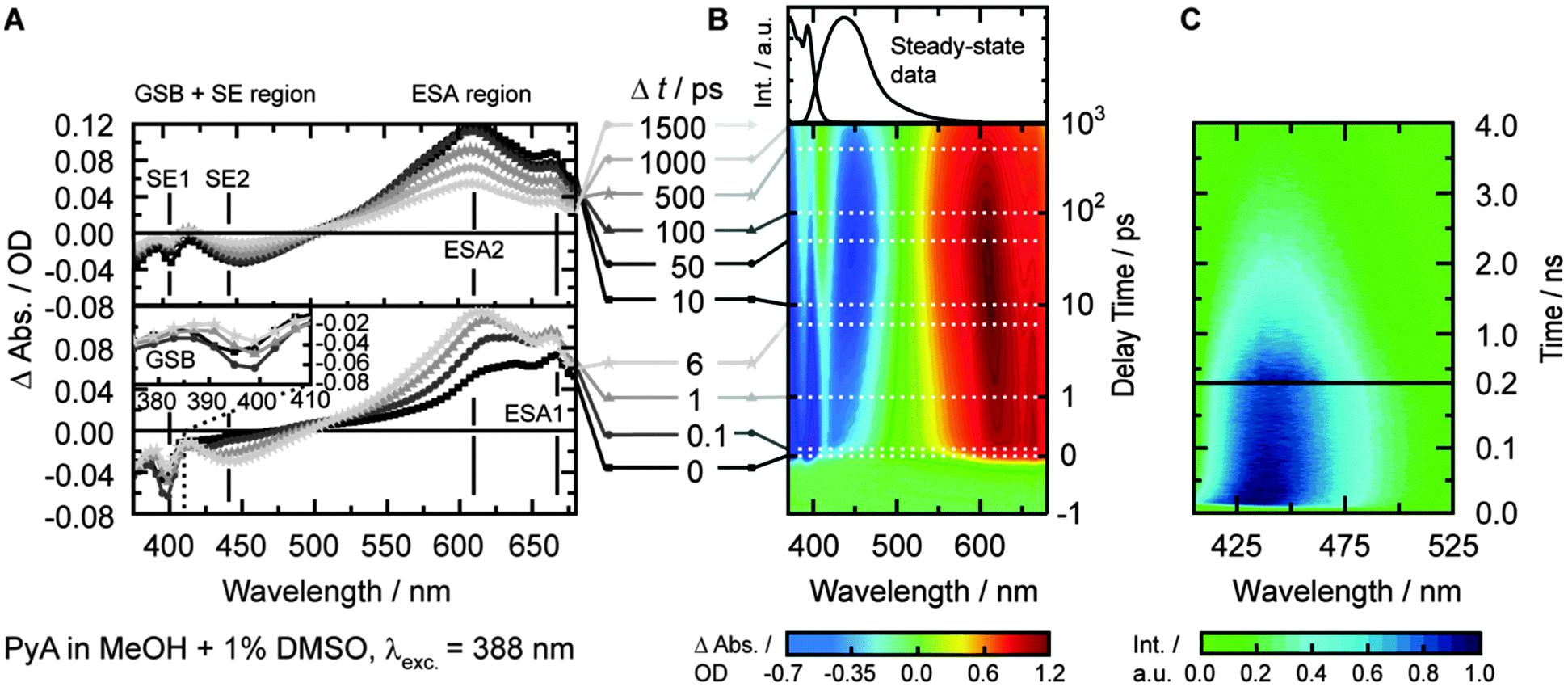 | ||
| Fig. 3 Dataset for PyA in MeOH and 1% DMSO excited at 388 nm. (A) Time slices between 0 ps and 6 ps (lower panel) as well as 10 ps and 1500 ps (upper panel). Inset shows GSB with superimposed SE. (B) Contour plot between −1 ps and 1000 ps, with highlighted positions of time slices (dashed lines). (C) Streak camera measurements with a ∼5 ns decay window (upper panel) and a ∼0.5 ns decay window (lower panel). | ||
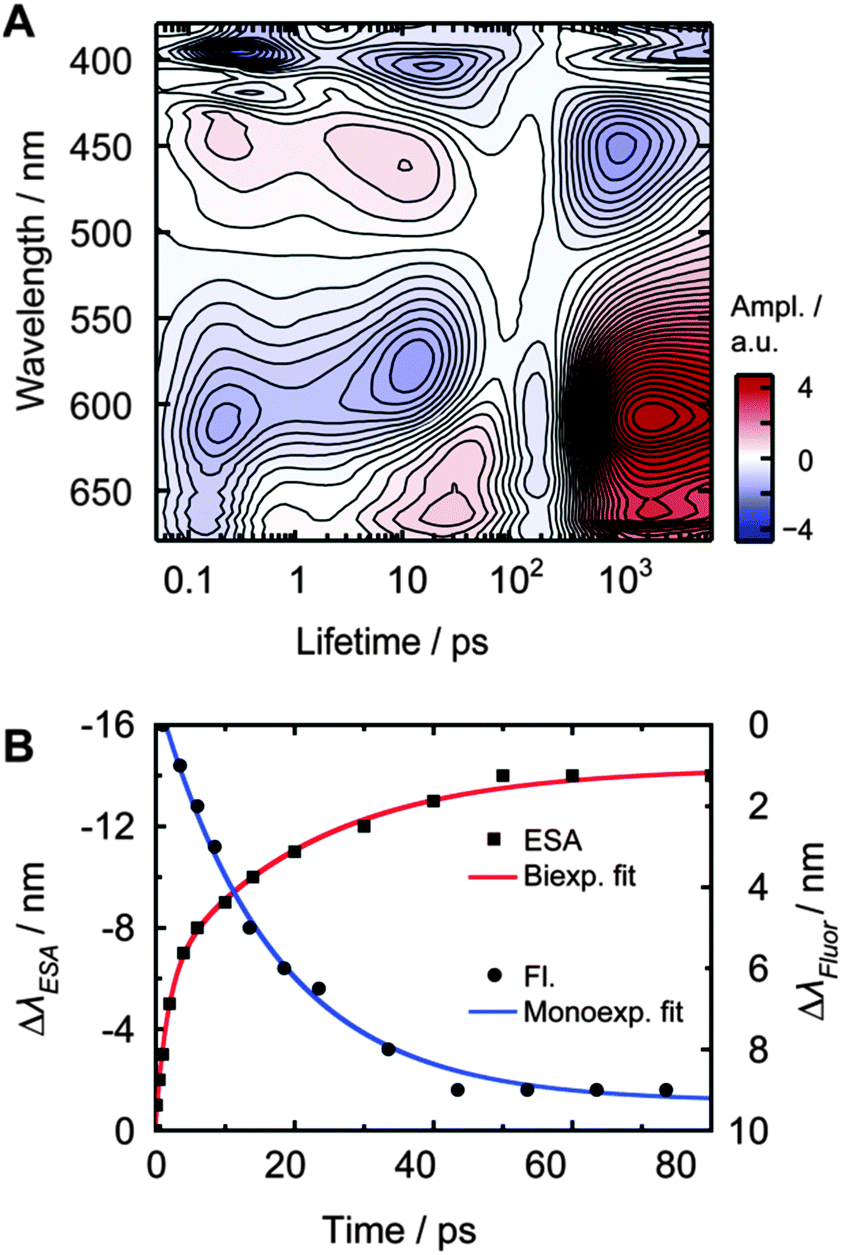 | ||
| Fig. 4 Analysis of PyA in MeOH and 1% DMSO excited at 388 nm. (A) Lifetime density amplitude map. (B) Spectral shift of the unstructured ESA2 maximum (cubes) and biexponential fit (red line, τESA2a = 1.8 ± 0.3 ps and τESA2b = 22 ± 3 ps), as well as spectral shift of the fluorescence maximum (circles) measured with the streak camera and monoexponential fit (blue line, τSE2b = 17 ± 1 ps). | ||
Since the SE signal is negative in TA, the slightly positive signal after 50 ps at 411 nm (Fig. 3A) means that we observe a broad ESA1 band. A broad ESA in this region is typical for pyrene and its derivatives.6,8,16,19,23,56 This ESA, which is superimposed by the SE, prevents a proper quantification of the spectral shift and the determination of the exact spectral position of the SE2 band.
Streak camera measurements
Streak camera measurements were performed to obtain the fluorescence dynamics without the disturbing contributions of ESA and GSB (Fig. 3C). DMSO, EtOAc, MeOH and tBuOH were chosen as representative solvents with the largest change in permittivity for each solvent class.The time frame of ∼0.5 ns revealed a transient spectral shift of ∼8 nm (∼420 cm−1) from 432 nm to 440 nm with a time constant of 17 ± 1 ps (Fig. 4B, τSE2b, blue curve). The spectral shift occurs with a similar time constant like the slow component of the ESA2 shift (τESA2b = 22 ps), so these two time constants should describe the same process. The lack of the fast time constant obtained for the ESA2 shift (τESA2a = 1.8 ps) can be explained by the limited time resolution of the streak camera.
Streak camera measurements with a time frame of ∼5 ns revealed an overall lifetime (τ4) of 2.0 ± 0.1 ns (Fig. 3C, upper plot).
Summary of the time-resolved behaviour
After photo-excitation, the appearance of structured bands ESA1 and SE1 during the first 500 fs is observed. Within less than 6 ps, ESA1 and SE1 slightly lose amplitude. At the same time, the unstructured ESA2 and SE2 bands gain amplitude. The ESA1 band is correlated with the SE1 band and the ESA2 band with the SE2 band, respectively. Both ESA bands and SE bands are energetically close. ESA2 and SE2 spectrally shift towards each other within 10–20 ps. The spectral shift is also visible in the time-resolved fluorescence measurements and could be analysed in detail only in MeOH, because of the spectroscopically hardly separable emitting states. All signals decay with a lifetime of approximately 2 ns, as determined by the streak camera measurements.LDM of PyA in MeOH
In methanol, the LDM for PyA (Fig. 4A) shows a first time constant (τ1) around 200 to 300 fs, which is within our time resolution. Minima at 395 nm, 419 nm, 611 nm and 662 nm and a maximum at 445 nm are observable. These extremes represent a change in amplitude due to IVR, observable in the SE1 and ESA1 bands.Between ∼2 ps and ∼30 ps, elongated extremes at 445 nm and 611 nm are visible. The maximal intensity of these extremes shifts towards each other with increasing lifetime. They are composed of two separate lifetimes, which are hardly distinguishable in MeOH. This conclusion is supported by the LDM of PyA in other solvents, where the separation is clearly visible (Fig. 5). Furthermore, the fit of the spectral ESA shift (Fig. 4B) also shows two lifetimes. The shorter of these two lifetimes (τ2, ∼2–10 ps) describes the increase of the ESA2. The longer one (τ3, ∼10–20 ps) dominates the signals and describes the spectral shift. The signals around 20 ps at 404 nm and 663 nm are a consequence of the spectral shift (Fig. 4).
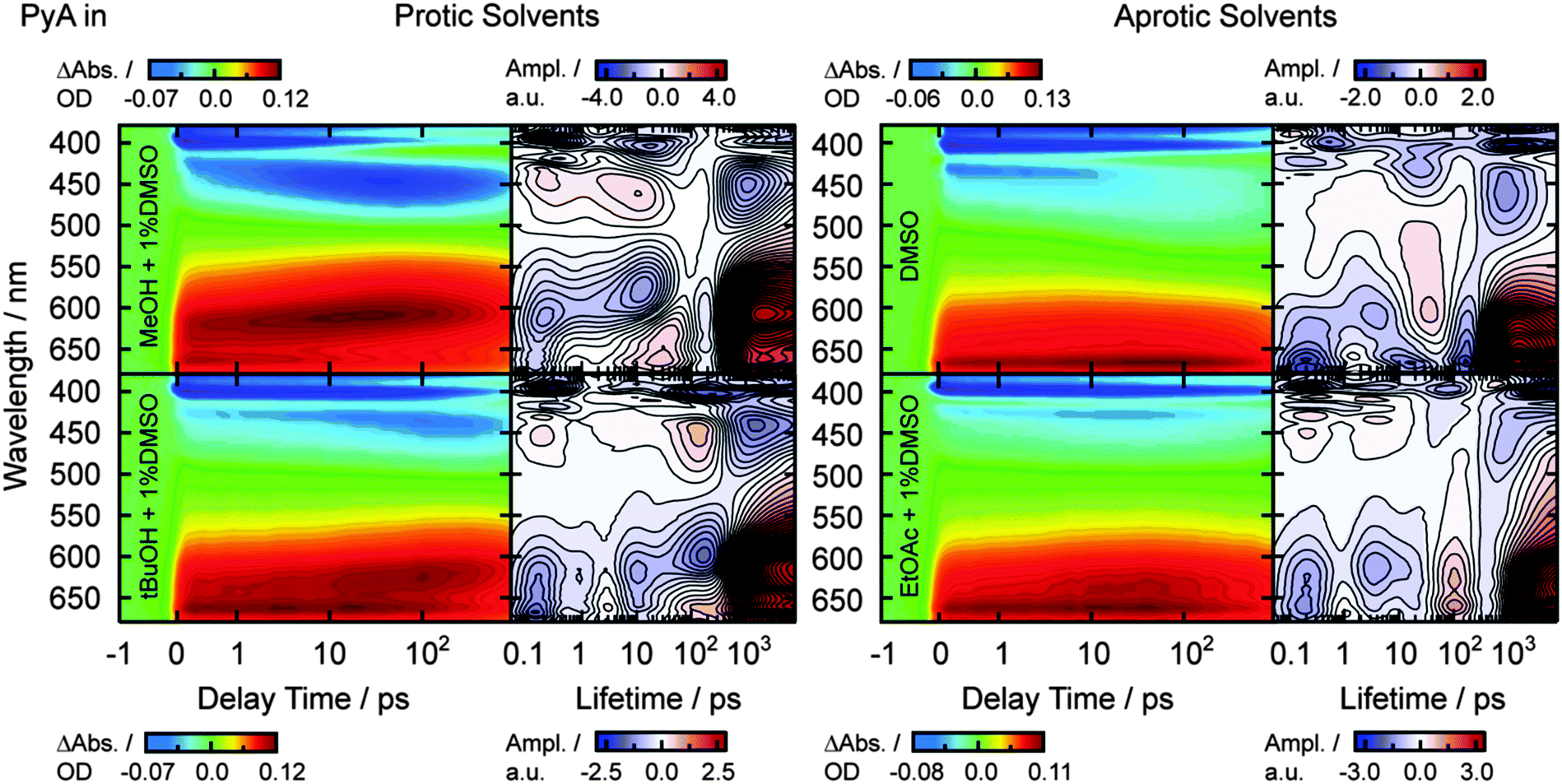 | ||
| Fig. 5 Transient absorption spectra (λexc = 388 nm) and LDM of PyA in protic solvents (MeOH and tBuOH, left side) and in aprotic solvents (DMSO and EtOAc, right side). MeOH and DMSO as the upper limit of the measured permittivity range of each solvent class and tBuOH and EtOAc as the lower limit. | ||
Lifetimes between 1 ns and 3 ns show a broad distribution at 452 nm, 606 nm and 626 nm, which describe the decay of both, SE and ESA bands. The longest lifetimes cannot be completely recovered from the LDMs, due to the limited amount of data points and limited time range. Therefore, the overall decay lifetime τ4 was determined by fluorescence measurements (Table 2).
Comparison of the spectral behavior of PyA in different solvents
After the fast spectral shift of the ESA1 band (Fig. 5 and Table 2, τ1 = 0.2–0.3 ps), strong solvent class and permittivity dependencies are visible. The spectral position of ESA1 changes slightly to longer wavelengths with increasing permittivity in aprotic solvents (Table 2, λESA1). This is also the case for the ground state absorption (Fig. 2 and Table 1, λabs1).In protic solvents, strong mostly unstructured ESA2 and SE2 bands appear within 20 ps (Table 2, τ2). A higher solvent permittivity leads to slightly larger amplitudes of ESA2 and SE2 bands. Additionally, the following hypsochromic shift of ESA2 and bathochromic shift of SE2 is stronger and becomes linearly faster with increasing permittivity (Table 2, τ3).
In aprotic solvents, a weak unstructured ESA2 (SE2) band appears with a time constant of ∼3.7 ps, which is slightly faster than in protic solvents (Table 2, τ2). In DMSO and DMF, the ESA2 band is largely superimposed by the ESA1 band, while spectrally separated in EtOAc and acetone. Like in protic solvents, the ESA2 (SE2) amplitude increases with increasing permittivity, but is in general low when compared to protic solvents (Fig. 5). A spectral shift of the SE2 band is observable only in DMSO and DMF, but not visible in their ESA2 bands. This can be explained by the spectrally close and strong ESA1 band, which is superimposed on the much weaker ESA2 in aprotic solvents. However, a spectral shift of the SE2 is assumed for all solvents due to the spectral shift observed in the steady-state emission spectra.
The overall lifetime was determined via streak camera measurements for representatives of both solvent classes with the highest and lowest permittivity. In both solvent classes, τ4 is in the range of 2 ns and increases with the solvent permittivity (Table 2, Δτprotic4 ≈ 0.6 ns, Δτaprotic4 ≈ 0.3 ns).
Results of quantum chemical calculations of PyA
Ground state geometry optimization
For this study the PyA molecule has been fully optimized in the electronic ground-state using DFT with the above mentioned xc-functionals (B-LYP, B3-LYP and BH-LYP) and Dunnnings double zeta basis set (DZP) as implemented in Turbomole. Calculations were done in vacuum neglecting solvent interactions.Optimized ground state structures show a planar arrangement of the base and the pyrene moiety independent of the applied method. The ribose is coordinated to the adenosine by an intramolecular H-bond between the 2′ OH of the ribose and the N3 atom of the base (Fig. 6).
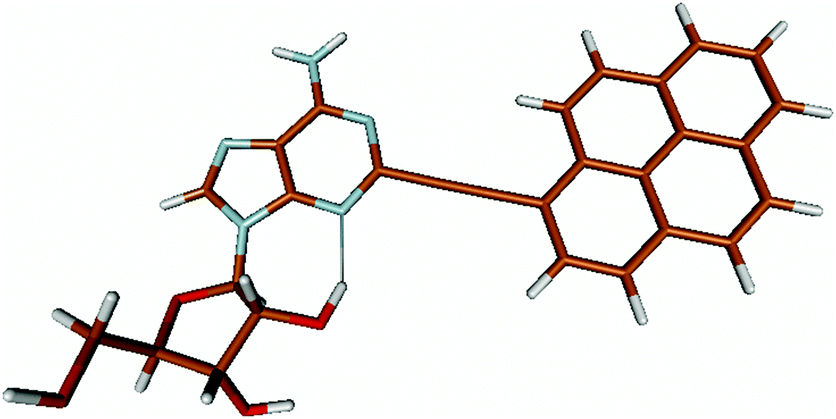 | ||
| Fig. 6 Ground state optimized structure of PyA (DFT/BH-LYP). | ||
Method evaluation
The vertically excited states have been calculated from the ground state optimized structures using the corresponding methods (Table 3) and the DZP basis set. For the SOS-CIS(D) method the BH-LYP optimized structure has been used along with the DZP basis set and the SVP auxiliary fitting basis set.| Transition: S0 → | E exc/nm | Character | f OSC | ||
|---|---|---|---|---|---|
| B-LYP | |||||
| S1 | 475.3 | HOMO | −LUMO | (78%) | 0.67 |
| S2 | 447.0 | HOMO − 1 | −LUMO | (62%) | 0.108 |
| S3 | 426.3 | HOMO − 2 | −LUMO | (96%) | 0.0011 |
| B3-LYP | |||||
| S1 | 403.5 | HOMO | −LUMO | (95%) | 0.95 |
| S2 | 355.1 | HOMO − 1 | −LUMO | (40%) | 0.011 |
| HOMO | −LUMO + 1 | (28%) | |||
| HOMO − 2 | −LUMO | (20%) | |||
| S3 | 335.5 | HOMO − 1 | −LUMO | (42%) | 0.0064 |
| HOMO − 2 | −LUMO + 1 | (35%) | |||
| HOMO | −LUMO + 1 | (18%) | |||
| BH-LYP | |||||
| S1 | 348.2 | HOMO | −LUMO | (92%) | 1.043 |
| S2 | 309.0 | HOMO − 2 | −LUMO | (28%) | 0.006 |
| HOMO | −LUMO + 1 | (25%) | |||
| HOMO − 1 | −LUMO | (21%) | |||
| S3 | 270.1 | HOMO − 1 | −LUMO | (46%) | 0.421 |
| HOMO | −LUMO + 2 | (26%) | |||
| CIS | |||||
| S1 | 334.5 | HOMO | −LUMO | (79%) | 0.92 |
| S2 | 288.3 | HOMO − 1 | −LUMO | (34%) | 0.0017 |
| HOMO | −LUMO + 1 | (26%) | |||
| HOMO − 2 | −LUMO | (12%) | |||
| S3 | 254.0 | HOMO | −LUMO + 2 | (22%) | 0.438 |
| HOMO − 1 | −LUMO | (14%) | |||
| SOS-CIS(D) | |||||
| S1 | 330.9 | HOMO − 1 | −LUMO | (40%) | 0.0045 |
| HOMO | −LUMO + 1 | (18%) | |||
| S2 | 287.5 | HOMO | −LUMO | (79%) | 1.3 |
| S3 | 226.5 | HOMO | −LUMO + 2 | (26%) | 0.58 |
| HOMO − 1 | −LUMO | (13%) | |||
As one can see from Table 3, for the TDDFT methods the excited states show a functional dependent blue shift of the excitation wavelengths of the transitions (Eexc). This is due to the admixture of increasing amounts of HF-exchange (B-LYP < B3-LYP < BH LYP) in the different functionals.
B3-LYP performs better than the B-LYP functional, but still the ordering of states differs from the CIS results. For example the third excited state (S3) can be identified as an intruder state, which has been calculated at much too low energy compared to the CIS method. This is a result of its high CT character and the SI error of TDDFT.
The increased amount of HF exchange in the BH-LYP functional corrects for this error and so the state ordering is in agreement with the CIS results.
Comparison with experimental data
All DFT calculations and the CIS method show an optically allowed S0 → S1 transition with a high oscillator strength of 0.7–1 (Table 3), which corresponds to a HOMO–LUMO transition.The second excited state (S2) is a dark state. For the BH-LYP method, the S0 → S2 transition has a complex character and possesses three dominant contributions, namely a HOMO − 1 to LUMO, a HOMO to LUMO + 1 and a HOMO − 2 to LUMO transition.
The optically allowed transition to the third excited state (S3) corresponds to a HOMO − 1 to LUMO and a HOMO to LUMO + 2 transition.
It should be noted that the energy difference between the two allowed transitions (S0 → S1 and S0 → S3) calculated with the BH-LYP and CIS methods is in reasonable agreement with the experimental data. A comparison between the calculated and measured absorption spectra can be found in the ESI† (Fig. S4).
In contrast to this the PyA S0 → S1 transition has high oscillator strength (Table 3) and corresponds to a HOMO to LUMO transition. Comparing the molecular orbitals (MOs) of PyA with the ones of pyrene (Table 4) this transition is similar to the bright S0 → S2 transition of pyrene. The contributing molecular orbitals for the transition to the S1 state in PyA and S2 state in pyrene are only slightly influenced by substitution on the pyrene 1 position, Table 4.
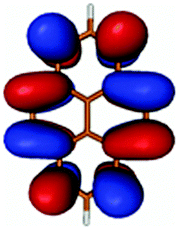
| PyA | Pyrene | |
|---|---|---|
| LUMO + 1 |
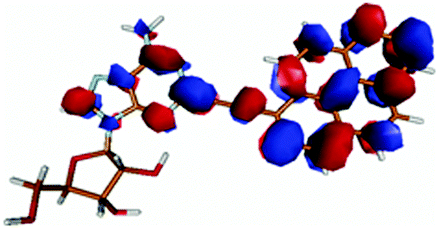
|
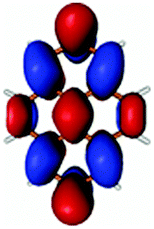
|
| LUMO |
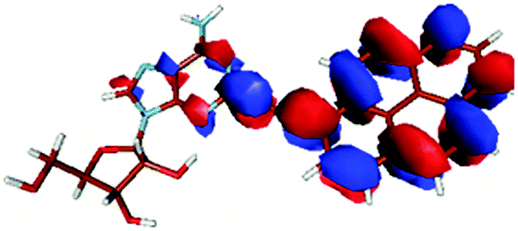
|
|
| HOMO |
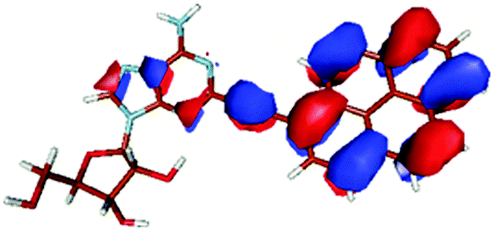
|
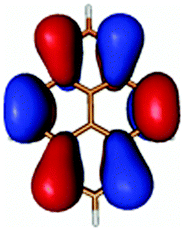
|
| HOMO − 1 |
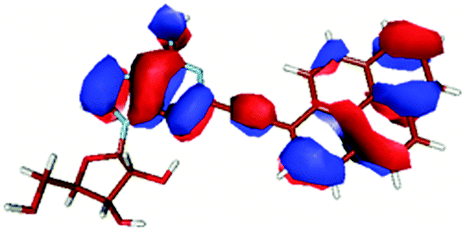
|
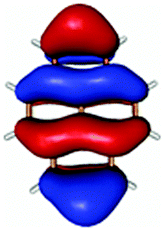
|
| HOMO − 2 |
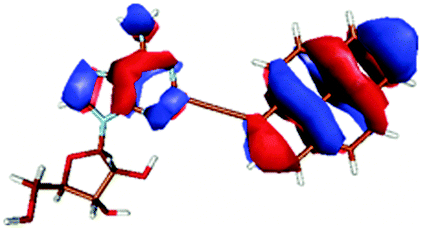
|
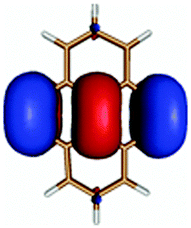
|
The second excited state (S2) of PyA is a dark state like the S1 state of pyrene. The S0 → S2 transition has a complex character like the S0 → S1 transition of pyrene, but possesses an additional third dominant contribution, namely a HOMO − 2 to LUMO transition. The PyA HOMO − 2 is similar to the pyrene HOMO − 1 orbital, but the PyA HOMO − 1 orbital is not comparable to any of the pyrene orbitals. The additional transition shows the strong influence of the substitution on the transition character. Thus in the case of PyA, the original pyrene S1 state gets destabilized and becomes the PyA S2 state.
Summing up these findings, we can conclude that within the framework of the single reference methods the ordering of the excited states of PyA is reversed with respect to the excited states of pyrene. Accordingly, the fluorescent state of PyA is the S1 state which explains the observed small Stokes shift of the PyA fluorescence, compared to the large Stokes shift for pyrene.
Discussion
The model of the photophysics shown in Fig. 7 summarizes, extends and adjusts the originally proposed one61 with new insights into the nature of the states and the photo-dynamics.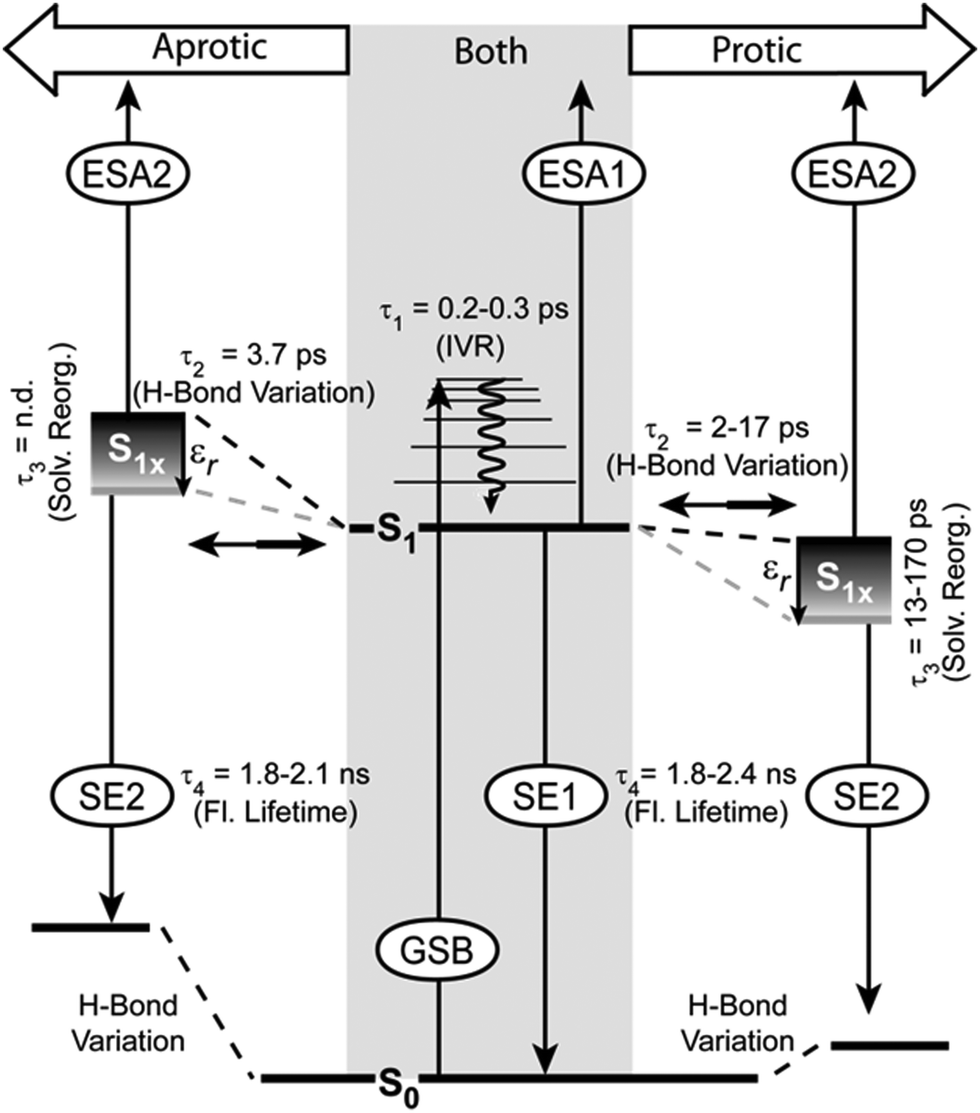 | ||
| Fig. 7 Model of relaxation pathways and observed signals in the TA for PyA in aprotic (left side) and protic (right side) solvents. | ||
Ground state properties
In general, the substitution pattern of pyrene strongly influences the spectral position as well as the oscillator strength for S0 → S1 and S0 → S2 transitions in absorption and fluorescence spectra depending on the position11,15,103 and the electrochemical nature of the substituent.103,104 Especially, the 1-position of pyrene (Fig. 1) shows strong electronic coupling with its substituents.11,15,23 Therefore, the absorption spectrum of PyA strongly differs from that of pyrene.Quantum chemical calculations revealed one optically allowed and one forbidden transition in the spectral range between 325 nm to 420 nm, but were not able to describe a clear order of the states. In the case of pyrene, fluorescence (according to Kasha's rule) occurs from the S1 state, which has very low oscillator strength. Therefore, pyrene fluorescence shows a pronounced Stokes shift (∼40 nm) and is long-lived (∼238 ns in DMSO).3 PyA shows a weak Stokes shift (∼10 nm in DMSO, Table 2) of the emission and is short-lived (τ4, ∼2 ns). In addition with the structural similarity to the pyrene emission, this indicates that we observe a S0 → S1 transition. The high fluorescence quantum yield (∼0.8) implies that this transition has high oscillator strength. This rules out excitation into higher energetic states at 388 nm. Therefore, the fine structure of the absorption band between 325 nm and 420 nm (Fig. 2) is the result of a superimposed vibrational fine structure like in the case of unsubstituted pyrene.
A change of the absorption ratio from the first (λabs1) towards the second (λabs2) maximum, which is accompanied by a bathochromic shift, is observed in aprotic solvents with increasing permittivity. No spectral shift and a much weaker change of the absorption ratio is observed in protic solvents, this indicates a intermolecular hydrogen-bond based alteration of the ground-state electron-density distribution, which reduces the influence of the permittivity on PyA. In aprotic solvents, the changes in the absorption spectra are a result of the changing solvent permittivity, which is due to the lack of solvent hydrogen bonding. In particular, the hydrogen bond between N3 and the 2′OH group observed in the quantum chemical calculations should influence the photo-dynamics.
Excited state dynamics
In agreement with our previous findings,61 two excited states and four time constants describe the photo-physics of PyA in MeOH sufficiently. The values of τ1, τ2 and τ4 remain in the same time range. The time constant τ3 (10–20 ps), which describes the spectral shift, differs by one order of magnitude (τ3 = 470 ps).60 This difference in the new datasets is a result of the higher spectral resolution and the improved analysis approach. By using lifetime density maps, the shortcoming of an exponential global fit analysis for spectral shifts, which are influenced linearly by time, was overcome.Development of S1 substates
The S1 state is spectrally represented by the ESA1 and SE1 signals. Between 325 nm to 420 nm higher vibronic S1 states are excited, therefore the first time constant (τ1) can be attributed to intramolecular vibrational energy redistribution (IVR). The IVR is not influenced by the varying solvents and lies around 0.2 ps to 0.3 ps in all solvents, which is a typical value for pyrene.6,8,12 The spectral positions of the ESA1 and the ground state absorption bands shift weakly (∼10 nm) to longer wavelengths with increasing solvent permittivity. This indicates that the S0 and the S1 state differ only slightly in their dipole moment. This explains why the S1 state and therefore the ESA1 and the SE1 bands do not show significant solvent dependencies. The molecule in the S1 state can partially adopt another conformation within τ2, which belongs to the same potential energy surface. The conformation of this substate is unknown, but energetically close to the initial conformation. This conformational substate is further on called the S1x state. The S1x state manifests itself in the TA by the ESA2 and the SE2 bands.In aprotic solvents, the ESA1 signal is dominant and therefore the S1 state is mostly populated. Streak camera and steady-state spectra are dominated by a structured emission in aprotic solvents, showing that the S1 state is the origin of the structured emission.
In protic solvents, both bands possess high amplitudes and therefore a significantly populated S1x state can be assumed. A high S1x state population is accompanied by an unstructured red-shifted emission (Fig. 2).
Emission wavelengths which represent either mostly the S1 state (410 nm) or the S1x state (450 nm) do not differ in their excitation spectra (Fig. S2, ESI†). This finding supports the model that the relaxed S1 state populates the S1x state (Fig. 7).
S1x state properties
Either the S1 state or the S1x state emission dominate the spectra, while no significant differences in the fluorescence quantum yields are noticeable in the different solvent classes. This implies that S1 and S1x exhibit similar fluorescence quantum yields. This rules out a strong intramolecular charge transfer character of the S1x state, because this would result in a strong reduction of the fluorescence quantum yield and a much stronger bathochromic shift of the emission maximum with increasing solvent permittivity.14,22,105,106 Then again, a small transfer of electron density, which is not localized on a functional group, does not strongly alter the fluorescence properties.106 The HOMO and LUMO (S0 → S1) show partial electron density contributions on the nucleobase. A weak delocalized CT character of the S1x state also explains the spectral shift of the SE2 band (τ3). This bathochromic shift reflects a solvent-permittivity dependent stabilization of the S1x state due to solvent reorganization. Higher solvent permittivity leads to a stronger spectral shift of the S1x state (Fig. 5). The acceleration of the spectral shift with increasing solvent permittivity (Table 2, τ3) is a result of faster solvent reorganization, due to the stronger electrostatic interactions between PyA and the solvents with a high permittivity.Thus, a slight electron density transfer, which leads to a slightly more polar S1x state, is sufficient to explain the experimental findings. The reaction coordinate, which describes the transition to the S1x state, could not be determined by quantum chemical calculations due to the size of the system.
Solvent dependence of equilibrium population
As mentioned above, a high solvent permittivity and protic solvents, which are able to form hydrogen bonds, lead to a higher S1x population. This is visible in the reduced share of structured emission. The exact population ratio between the S1 state and the S1x state is not quantifiable, due to the strong overlap of the SE with the ESA bands and because of the (most probably) different transition dipole-moments of the ESA1 and the ESA2 transitions. Nevertheless, the solvent class and the solvent permittivity allow the control of the population ratio. Increased S1x population results from an energetic stabilization of the S1x state (Fig. 7). The influence of the solvent class on the population ratio is much stronger than the permittivity. In aprotic solvents, the S1 state is favoured even for solvents with high permittivity. In protic solvents, the situation is reversed and the S1x state is more highly populated even in solvents with a low permittivity (Fig. 5). Therefore, the S1x state has to be energetically more stable than the S1 state (Fig. 7). Even though the energetic position of the S1x state varies, the spectral position of the SE2 band is only weakly affected. This means that the ground state has to be equally affected in its energetic position.Interestingly, only one decay time for S1 and S1x is observed in the streak camera measurements, which is in the same range for both solvent classes and varies only slightly with permittivity (Table 2, τ4). In tBuOH for example, S1 and S1x are nearly equally populated, but do not show an additional decay lifetime in the streak camera measurements. This indicates, in combination with the energetic proximity of both states, that S1 and S1x undergo fast population exchange (τ2), which leads to an equilibrium at the nanosecond timescale.
Hydrogen bond variation in the excited states
The intramolecular hydrogen bond between N3 and the 2'OH group in the ground state (S0) restricts the glycosidic torsion and is also present in the S1 state. The above mentioned solvent dependent behaviour leads us to the assumption that by reaching the S1x state this bond is weakened by forming the conformation, which is connected to the S1x state and influences the electron density on the adenine.In the case of aprotic solvents (the S1x state energetically higher than the S1 state), the intramolecular hydrogen bond hinders the formation and makes this conformation unfavourable. The corresponding ground state is also destabilized with respect to the original S0 state.
In protic solvents, the S1x state is now energetically favoured relative to the S1 state. This means that the surrounding solvent molecules probably form intermolecular hydrogen bonds with the N3 of adenine instead of the 2'OH group. The result is that the conformation of the S1x state is now able to form and therefore energetically favoured.
Conclusions
Our systematic spectroscopic study provides detailed evidence that hydrogen bonding as well as the solvent permittivity strongly influence the photo-dynamics of PyA. Compared to a slightly different derivative of PyA60 and PyA modified RNA,61 the ultrafast dynamics vary in the excited state characteristics, assignments of time constants and provide new insights into the role of intramolecular hydrogen bonds (Fig. 7).Higher vibronic states of the S1 are excited between 325 nm to 420 nm and no contributions of other states are detectable. After IVR (τ1), the locally excited S1 state populates the conformational substate S1x within τ2. This S1x state does not have a strong charge transfer character, but is slightly more polar than the S1 state and hence gets stabilized by solvents with a high permittivity. The polar character of the S1x state has to be the result of a restricted delocalized electron density distribution over the nucleobase. Due to that, τ3 describes the solvent permittivity dependent stabilization of the S1x state, rather than the (previously assumed) decay of the S1 state.61 Therefore, the difference in magnitude of τ3 to previous data is a result of the better resolved datasets and the improved analysis approach. Both emitting states (S1 and S1x) are in equilibrium and decay with the same time constant (τ4).
However, the ground state and population ratio between the S1 state and the S1x state are strongly influenced by hydrogen bonds. A hydrogen bond between the 2'OH of the ribose and the N3 of the adenine for PyA was revealed by the quantum chemical calculations. This hydrogen bond influences the energetic position of the S1x state depending on the solvent class. In aprotic solvents, the S1x state is energetically destabilized relative to the S1 state. This leads to the population ratio in favour of the S1 state and therefore a structured fluorescence band. Protic solvents disturb this hydrogen bond by interfering via intermolecular hydrogen bonds. As a result, the S1x state is energetically stabilized relative to the S1 state and an unstructured fluorescence band is observed. In both solvent classes, a high permittivity further stabilizes the S1x state independent of the S0 state due to solvent reorganization, which results in a spectral shift and less structured fluorescence. No additional proton transfer could be observed.
These findings allow addressing even slight changes in the emission structure to permittivity changes in the surroundings of PyA upon e.g. ligand binding in RNA or to solvent inaccessible PyA positions.
Funding
The authors thank the Deutsche Forschungsgemeinschaft for funding via the Collaborative Research Center 902 ‘Molecular Principles of RNA-based Regulation'.Acknowledgements
The authors thank Prof. Dr Wolfgang Zinth for the possibility to measure streak camera spectra. The authors would also like to thank Prof. Irene Burghardt and Prof. Andreas Dreuw for providing computational resources and financial support.References
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2010 Search PubMed.
- M. F. M. Post, J. Langelaar and J. D. W. van Voorst, Chem. Phys. Lett., 1971, 10, 468–472 CrossRef CAS.
- A. Nakajima, Bull. Chem. Soc. Jpn., 1973, 46, 2602–2604 CrossRef CAS.
- T. Kubota, K. Kano, B. Uno and T. Konse, Bull. Chem. Soc. Jpn., 1987, 60, 3865–3877 CrossRef CAS.
- P. Foggi, L. Pettini, I. Santa, R. Righini and S. Califano, J. Phys. Chem., 1995, 99, 7439–7445 CrossRef CAS.
- F. V. R. Neuwahl and P. Foggi, Laser Chem., 1999, 19, 375–379 CrossRef CAS PubMed.
- D. S. Karpovich and G. J. Blanchard, J. Phys. Chem., 1995, 99, 3951–3958 CrossRef CAS.
- M. Raytchev, E. Pandurski, I. Buchvarov, C. Modrakowski and T. Fiebig, J. Phys. Chem. A, 2003, 107, 4592–4600 CrossRef CAS.
- T. Förster, Angew. Chem., 1969, 81, 364–374 CrossRef.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, California, 1991 Search PubMed.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L. O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS PubMed.
- T. Fiebig, K. Stock, S. Lochbrunner and E. Riedle, Chem. Phys. Lett., 2001, 345, 81–88 CrossRef CAS.
- E. Pandurski and T. Fiebig, Chem. Phys. Lett., 2002, 357, 272–278 CrossRef CAS.
- J. Sung, P. Kim, Y. O. Lee, J. S. Kim and D. Kim, J. Phys. Chem. Lett., 2011, 2, 818–823 CrossRef CAS.
- C. Wanninger-Weiss and H.-A. Wagenknecht, Eur. J. Org. Chem., 2008, 64–69 CrossRef CAS.
- C. E. Crespo-Hernandez, G. Burdzinski and R. Arce, J. Phys. Chem. A, 2008, 112, 6313–6319 CrossRef CAS PubMed.
- Y. Niko, Y. Hiroshige, S. Kawauchi and G. Konishi, Tetrahedron, 2012, 68, 6177–6185 CrossRef CAS PubMed.
- N. Amann, E. Pandurski, T. Fiebig and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2002, 41, 2978–2980 CrossRef CAS.
- N. Amann, E. Pandurski, T. Fiebig and H.-A. Wagenknecht, Chem. – Eur. J., 2002, 8, 4877–4883 CrossRef CAS.
- A. Trifonov, I. Buchvarov, H.-A. Wagenknecht and T. Fiebig, Chem. Phys. Lett., 2005, 409, 277–280 CrossRef CAS PubMed.
- A. Trifonov, M. Raytchev, I. Buchvarov, M. Rist, J. Barbaric, H.-A. Wagenknecht and T. Fiebig, J. Phys. Chem. B, 2005, 109, 19490–19495 CrossRef CAS PubMed.
- C. Wagner and H.-A. Wagenknecht, Chem. – Eur. J., 2005, 11, 1871–1876 CrossRef CAS PubMed.
- P. Kaden, E. Mayer-Enthart, A. Trifonov, T. Fiebig and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2005, 44, 1637–1639 CrossRef PubMed.
- C. Wanninger-Weiß, L. Valis and H.-A. Wagenknecht, Bioorg. Med. Chem., 2008, 16, 100–106 CrossRef PubMed.
- A. Okamoto, K. Kanatani and I. Saito, J. Am. Chem. Soc., 2004, 126, 4820–4827 CrossRef CAS PubMed.
- K. Kawai, H. Yoshida, T. Takada, S. Tojo and T. Majima, J. Phys. Chem. B, 2004, 108, 13547–13550 CrossRef CAS.
- Y. J. Seo, J. H. Ryu and B. H. Kim, Org. Lett., 2005, 7, 4931–4933 CrossRef CAS PubMed.
- Y. J. Seo, I. J. Lee, J. W. Yi and B. H. Kim, Chem. Commun., 2007, 2817–2819 RSC.
- M. Nakamura, Y. Fukunaga, K. Sasa, Y. Ohtoshi, K. Kanaori, H. Hayashi, H. Nakano and K. Yamana, Nucleic Acids Res., 2005, 33, 5887–5895 CrossRef CAS PubMed.
- H. Kashida, T. Takatsu, K. Sekiguchi and H. Asanuma, Chem. – Eur. J., 2010, 16, 2479–2486 CrossRef CAS PubMed.
- T. Kato, H. Kashida, H. Kishida, H. Yada, H. Okamoto and H. Asanuma, J. Am. Chem. Soc., 2013, 135, 741–750 CrossRef CAS PubMed.
- I. K. Astakhova, T. Santhosh Kumar, M. A. Campbell, A. V. Ustinov, V. A. Korshun and J. Wengel, Chem. Commun., 2013, 49, 511–513 RSC.
- N. Venkatesan, Y. Jun Seo and B. Hyean Kim, Chem. Soc. Rev., 2008, 37, 648–663 RSC.
- K. Huang and A. Martí, Anal. Bioanal. Chem., 2012, 402, 3091–3102 CrossRef CAS PubMed.
- R. Zhang, L. Yang, M. Zhao, J. Dong, H. Dong, Y. Wen, X. Zhan, H. Yang and G. Wang, Polymer, 2013, 54, 1289–1294 CrossRef CAS PubMed.
- E. Mayer, L. Valis, C. Wagner, M. Rist, N. Amann and H.-A. Wagenknecht, ChemBioChem, 2004, 5, 865–868 CrossRef CAS PubMed.
- G. T. Hwang, Y. J. Seo, S. J. Kim and B. H. Kim, Tetrahedron Lett., 2004, 45, 3543–3546 CrossRef CAS PubMed.
- G. T. Hwang, Y. J. Seo and B. H. Kim, Tetrahedron Lett., 2005, 46, 1475–1477 CrossRef CAS PubMed.
- L. Valis, E. Mayer-Enthart and H.-A. Wagenknecht, Bioorg. Med. Chem., 2006, 16, 3184–3187 CrossRef CAS PubMed.
- I. V. Astakhova, A. V. Ustinov, V. A. Korshun and J. Wengel, Bioconjugate Chem., 2011, 22, 533–539 CrossRef CAS PubMed.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS PubMed.
- S. P. Sau and P. J. Hrdlicka, J. Org. Chem., 2012, 77, 5–16 CrossRef CAS PubMed.
- S. Srivatsan and A. Sawant, Pure Appl. Chem., 2010, 83, 213–232 CrossRef.
- I. Tinoco Jr and C. Bustamante, J. Mol. Biol., 1999, 293, 271–281 CrossRef CAS PubMed.
- J. A. Cruz and E. Westhof, Cell, 2009, 136, 604–609 CrossRef CAS PubMed.
- K. S. Karunatilaka and D. Rueda, Chem. Phys. Lett., 2009, 476, 1–10 CrossRef CAS PubMed.
- A. D. Garst, A. L. Edwards and R. T. Batey, Cold Spring Harbor Perspect. Biol., 2010, 1–13 Search PubMed.
- A. Mahara, R. Iwase, T. Sakamoto, K. Yamana, T. Yamaoka and A. Murakami, Angew. Chem., Int. Ed., 2002, 41, 3648–3650 CrossRef CAS.
- A. Mahara, R. Iwase, T. Sakamoto, T. Yamaoka, K. Yamana and A. Murakami, Bioorg. Med. Chem., 2003, 11, 2783–2790 CrossRef CAS.
- O. A. Krasheninina, D. S. Novopashina and A. G. Venyaminova, Russ. J. Bioorg. Chem., 2011, 37, 244–248 CrossRef CAS.
- R. Waki, A. Yamayoshi, A. Kobori and A. Murakami, Chem. Commun., 2011, 47, 4204–4206 RSC.
- K. K. Karlsen, A. Pasternak, T. B. Jensen and J. Wengel, ChemBioChem, 2012, 13, 590–601 CrossRef CAS PubMed.
- T. Ueda, A. Kobori, A. Yamayoshi, H. Yoshida, M. Yamaguchi and A. Murakami, Bioorg. Med. Chem., 2012, 20, 6034–6039 CrossRef CAS PubMed.
- G. Wang, G. V. Bobkov, S. N. Mikhailov, G. Schepers, A. Van Aerschot, J. Rozenski, M. Van der Auweraer, P. Herdewijn and S. De Feyter, ChemBioChem, 2009, 10, 1175–1185 CrossRef CAS PubMed.
- S. K. Silverman and T. R. Cech, Biochemistry, 1999, 38, 14224–14237 CrossRef CAS PubMed.
- T. L. Netzel, K. Nafisi, J. Headrick and B. E. Eaton, J. Phys. Chem., 1995, 99, 17948–17955 CrossRef CAS.
- T. L. Netzel, M. Zhao, K. Nafisi, J. Headrick, M. S. Sigman and B. E. Eaton, J. Am. Chem. Soc., 1995, 117, 9119–9128 CrossRef CAS.
- A. Malakhov, E. Malakhova, S. Kuznitsova, I. Grechishnikova, I. Prokhorenko, M. Skorobogatyi, V. Korshun and Y. Berlin, Russ. J. Bioorg. Chem., 2000, 26, 34–44 CrossRef CAS.
- C. Grünewald, T. Kwon, N. Piton, U. Förster, J. Wachtveitl and J. W. Engels, Bioorg. Med. Chem., 2008, 16, 19–26 CrossRef PubMed.
- U. Förster, N. Gildenhoff, C. Grünewald, J. W. Engels and J. Wachtveitl, J. Lumin., 2009, 129, 1454–1458 CrossRef PubMed.
- U. Förster, C. Grünewald, J. W. Engels and J. Wachtveitl, J. Phys. Chem. B, 2010, 114, 11638–11645 CrossRef PubMed.
- U. Förster, K. Lommel, D. Sauter, C. Grünewald, J. W. Engels and J. Wachtveitl, ChemBioChem, 2010, 11, 664–672 CrossRef PubMed.
- S. Techert, S. Schmatz, A. Wiessner and H. Staerk, J. Phys. Chem. A, 2000, 104, 5700–5710 CrossRef CAS.
- R. Huber, T. Fiebig and H.-A. Wagenknecht, Chem. Commun., 2003, 1878–1879 RSC.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- L. Dworak, V. V. Matylitsky and J. Wachtveitl, ChemPhysChem, 2009, 10, 384–391 CrossRef CAS PubMed.
- F. J. Lederer, F. F. Graupner, B. Maerz, M. Braun and W. Zinth, Chem. Phys., 2014, 428, 82–89 CrossRef CAS PubMed.
- G. Ryseck, T. Schmierer, K. Haiser, W. Schreier, W. Zinth and P. Gilch, ChemPhysChem, 2011, 12, 1880–1888 CrossRef CAS PubMed.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- A. A. Istratov and O. F. Vyvenko, Rev. Sci. Instrum., 1999, 70, 1233–1257 CrossRef CAS PubMed.
- R. Croce, M. G. Müller, R. Bassi and A. R. Holzwarth, Biophys. J., 2001, 80, 901–915 CrossRef CAS.
- Z. Ablonczy, A. Lukács and E. Papp, Biophys. Chem., 2003, 104, 249–258 CrossRef CAS.
- V. A. Lórenz-Fonfría and H. Kandori, Appl. Spectrosc., 2007, 61, 74–84 CrossRef PubMed.
- V. K. Mulligan, K. C. Hadley and A. Chakrabartty, Anal. Biochem., 2012, 421, 181–190 CrossRef CAS PubMed.
- V. A. Voelz and V. S. Pande, Proteins: Struct., Funct., Bioinf., 2012, 80, 342–351 CrossRef CAS PubMed.
- R.-J. Kutta, T. Langenbacher, U. Kensy and B. Dick, Appl. Phys. B: Lasers Opt., 2013, 111, 203–216 CrossRef CAS.
- E. T. Jaynes, Phys. Rev., 1957, 106, 620–630 CrossRef.
- A. N. Tikhonov, Dokl. Akad. Nauk SSSR, 1963, 151, 501–504 Search PubMed.
- K. Miller, SIAM J. Math. Anal., 1970, 1, 52–74 CrossRef PubMed.
- P. Hansen, BIT Numer. Math., 1990, 30, 658–672 CrossRef.
- J. Lian, D. Yao and B. He, in Engineering in Medicine and Biology Society, 1998. Proceedings of the 20th Annual International Conference of the IEEE, 1998, vol. 4, pp. 2155–2158.
- J. Lian and B. He, Brain Topogr., 2001, 13, 209–217 CrossRef CAS.
- G. H. Golub, M. Heath and G. Wahba, Technometrics, 1979, 21, 215–223 CrossRef.
- V. A. Lórenz-Fonfría and H. Kandori, Appl. Spectrosc., 2007, 61, 428–443 CrossRef PubMed.
- A. L. Dobryakov, S. A. Kovalenko, A. Weigel, J. L. Pérez-Lustres, J. Lange, A. Müller and N. P. Ernsting, Rev. Sci. Instrum., 2010, 81, 113106 CrossRef CAS PubMed.
- H. Satzger and W. Zinth, Chem. Phys., 2003, 295, 287–295 CrossRef CAS PubMed.
- R. Ahlrichs, TurboMole V 5.10, 2007 Search PubMed.
- J. Kong, C. White, A. Krylov, D. Sherrill, R. Adamson, T. Furlani, M. Lee, A. Lee, S. Gwaltney, T. Adams, C. Ochsenfeld, A. Gilbert, G. Kedziora, V. Rassolov, D. Maurice, N. Nair, Y. Shao, N. Besley, P. Maslen, J. Dombroski, H. Daschel, W. Zhang, P. Korambath, J. Baker, E. Byrd, T. Van Voorhis, M. Oumi, S. Hirata, C. Hsu, N. Ishikawa, J. Florian, A. Warshel, B. Johnson, P. Gill, M. Head-Gordon and J. Pople, J. Comput. Chem., 2000, 21, 1532–1548 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. E. Casida, Recent Advances in Density Functional Methods, Part I, World Scientific, Singapore, 1995 Search PubMed.
- J. E. D. Bene, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1971, 55, 2236–2241 CrossRef PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- A. Dreuw, J. L. Weisman and M. Head-Gordon, J. Chem. Phys., 2003, 119, 2943–2946 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef CAS PubMed.
- J. Plötner and A. Dreuw, Chem. Phys., 2008, 347, 472–482 CrossRef PubMed.
- D. J. Tozer, J. Chem. Phys., 2003, 119, 12697–12699 CrossRef CAS PubMed.
- N. T. Maitra, J. Chem. Phys., 2005, 122, 234104 CrossRef PubMed.
- J. Neugebauer, O. Gritsenko and E. J. Baerends, J. Chem. Phys., 2006, 124, 214102 CrossRef PubMed.
- D. Casanova, Y. M. Rhee and M. Head-Gordon, J. Chem. Phys., 2008, 128, 164106 CrossRef PubMed.
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, Boca Ranton, New York, London, Tokyo, 81st edn, 2000 Search PubMed.
- I. S. K. Kerkines, I. D. Petsalakis, G. Theodorakopoulos and W. Klopper, J. Chem. Phys., 2009, 131, 224315 CrossRef PubMed.
- H. Maeda, T. Maeda, K. Mizuno, K. Fujimoto, H. Shimizu and M. Inouye, Chem. – Eur. J., 2006, 12, 824–831 CrossRef CAS PubMed.
- C. Yao, H.-B. Kraatz and R. P. Steer, Photochem. Photobiol. Sci., 2005, 4, 191–199 CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- A. Morimoito, T. Yatsuhashi, T. Shimada, L. Biczók, D. A. Tryk and H. Inoue, J. Phys. Chem. A, 2001, 105, 10488–10496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Em. spectra of PyA at 376 nm and 421 nm (Fig. S1), exc. spectra in MeOH and DMSO (Fig. S2), TA spectra of PyA in all solvents (Fig. S3) and comparison of experimental absorption spectra of PyA in DMSO with QC calculations (Fig. S4). See DOI: 10.1039/c4cp01148a |
| This journal is © the Owner Societies 2014 |