 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DFT and ONIOM study of C–H hydroxylation catalyzed by nitrobenzene 1,2-dioxygenase†
Inacrist
Geronimo
and
Piotr
Paneth
*
Institute of Applied Radiation Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland. E-mail: paneth@p.lodz.pl
First published on 19th May 2014
Abstract
A detailed description of the mechanism of C–H hydroxylation by Rieske non-heme iron dioxygenases remains elusive, as the nature of the oxidizing species is not definitively known. DFT calculations on cluster models of nitrobenzene 1,2-dioxygenase were done to explore possible mechanisms arising from oxidation by either the experimentally observed FeIII–OOH complex or the putative high-valent HO–FeV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate formed through a heterolytic O–O bond cleavage. Hydrogen abstraction by HO–FeVO, followed by oxygen rebound, was found to be consistent with experimental studies. The findings from the quantum mechanical cluster approach were verified by accounting for the effect of the protein environment on transition state geometries and reaction barriers through ONIOM calculations.
O intermediate formed through a heterolytic O–O bond cleavage. Hydrogen abstraction by HO–FeVO, followed by oxygen rebound, was found to be consistent with experimental studies. The findings from the quantum mechanical cluster approach were verified by accounting for the effect of the protein environment on transition state geometries and reaction barriers through ONIOM calculations.
Introduction
Monooxygenation is among the wide range of reactions catalyzed by Rieske non-heme iron dioxygenases (RDOs),1,2 which include aromatic cis-dihydroxylation, desaturation, sulfoxidation, O- and N-dealkylation and amine oxidation.3–6 The reactivity of RDOs toward stable aromatic compounds has made them important to the development of biodegradation measures for recalcitrant environmental pollutants.7 The challenges of selective functionalization of alkanes in the chemical industry have also driven research on biomimetic iron catalysts, which benefit from an understanding of the mechanism of these enzymes.8 The active site of RDOs is a high-spin mononuclear FeII bound to two histidines and a bidentate aspartate residue, forming the recurring 2-His-1-carboxylate facial triad motif of non-heme iron-containing oxygenases. Two external electrons supplied by NAD(P)H are transferred to the active site during the catalytic cycle via a Rieske [2Fe–2S] cluster located within 12 Å of FeII in an adjacent α subunit. Formation of the reduced enzyme–substrate complex induces conformational changes that allow O2 binding and activation.1,2A ferric (hydro)peroxo complex is believed to be a key reaction intermediate based on extensive studies on naphthalene 1,2-dioxygenase (NDO), including crystallographic data,9 single turnover10 and peroxide shunt experiments,11 and theoretical calculations.12 However, there is no consensus on whether this species reacts directly with the substrate or initially undergoes O–O bond cleavage to form a high-valent iron–oxo intermediate. It was recently found that a high-spin (S = 5/2) biomimetic complex, [FeIII(OOH)(TMC)]2+ [TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane], is capable of C–H hydroxylation of alkylaromatics.13 The low-spin (S = 1/2) FeIII–OOH intermediate of the non-heme antitumor drug bleomycin (activated BLM) is also believed to initiate DNA cleavage by directly abstracting hydrogen.14 In both cases, O–O bond cleavage prior to H abstraction was found to have a higher activation energy.
On the other hand, hydroxylation catalyzed by enzymes such as 2-oxo acid dioxygenases and aromatic amino acid hydroxylases, which also have a 2-His-1-carboxylate facial triad motif, involves electron donation from a cofactor or cosubstrate allowing the formation of an FeIVO intermediate.1,15 Direct spectroscopic evidence of this species has been reported for taurine-α-ketoglutarate dioxygenase,16 tyrosine hydroxylase17 and phenylalanine hydroxylase.18 In contrast, only one electron is provided by the Rieske center in RDOs,10,19 which would lead to a HO–FeVO species as in heme enzymes and methane monooxygenase.15 However, the non-heme ligands in RDOs do not stabilize high oxidation states (through spin delocalization and ligand oxidation) as well as porphyrin and thiolate ligands do for heme enzymes.20 This is supported by cluster model calculations showing that O–O bond cleavage in high-spin FeIII–OOH is endothermic with a prohibitively high energy barrier (26.5 kcal mol−1).12 Nevertheless, experimental studies of monooxygenase reactions of RDOs suggest the involvement of a HO–FeVO intermediate. The latter is capable of solvent exchange which would explain the 18O-labeled products obtained from toluene dioxygenase (TDO)-catalyzed oxidation of indan.21 As in the case of a FeIII–OOH oxidant,13,14 H abstraction by HO–FeVO would lead to the formation of radical intermediates, which were inferred from the rearranged products observed in the reactions of NDO with norcarane and bicyclohexane19 and TDO with indene.21,22
Aliphatic hydroxylation catalyzed by cytochrome P450 enzymes, which share the chemistry of RDOs (with the exception of cis-dihyroxylation),23 proceeds through a rebound mechanism. The reactive intermediate is [(Por˙)FeIVO]+1, also known as compound I (Cpd I), where the oxidizing species is formally FeVO. Hydrogen is initially abstracted by the oxo ligand, and in the rebound step, the resulting hydroxyl group recombines with the radical carbon to yield the alcohol product (Scheme 1).24 Low-spin biomimetic non-heme iron complexes catalyze substrate hydroxylation in a similar manner,25 with HO–FeVO as the proposed reactive species.26–29 HO–FeVO has been detected in a Fe–Me,HPytacn [Me,HPytacn = 1-(2′-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane] complex using variable-temperature mass spectrometry.26 The O–O bond cleavage is facilitated by solvent water as evidenced by incorporation of 18O from H218O in the product.26,27 Previous calculations on the Fe–TPA [TPA = tris(2-pyridylmethyl)amine] complex show that this mechanism is energetically feasible due to the weak O–O bond of low-spin FeIII–OOH.30 The Fe–Pytacn and Fe–TPA complexes can be considered as functional models of RDOs unlike the Fe–TMC complex since these contain two cis-exchangeable sites.25
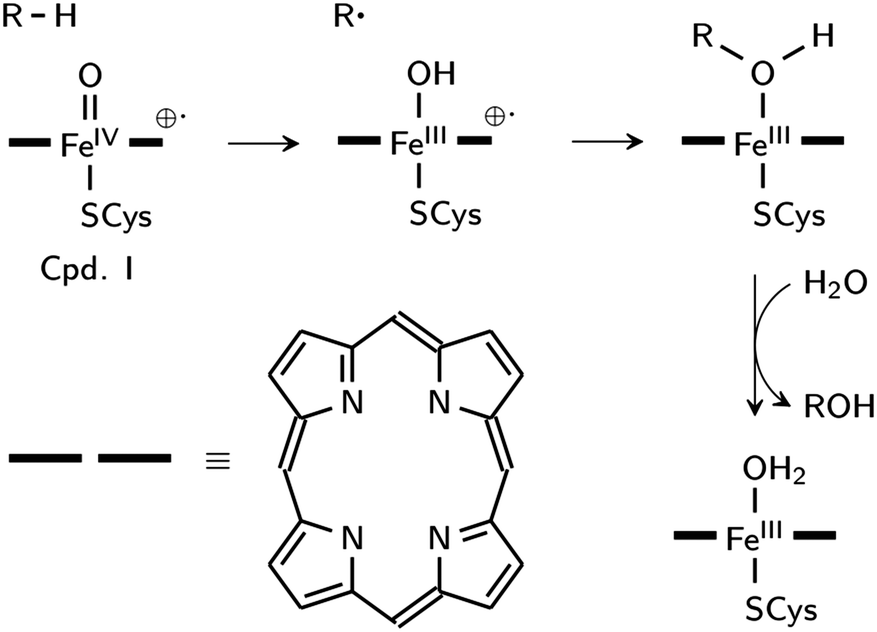 | ||
| Scheme 1 Mechanism of aliphatic hydroxylation catalyzed by cytochrome P450. | ||
The present work examines the C–H hydroxylation of 2-nitrotoluene (2NT) catalyzed by nitrobenzene 1,2-dioxygenase (NBDO), a member of the naphthalene family of RDOs that include NDO.31 This reaction, along with the competing aromatic cis-dihydroxylation, is an important biodegradation pathway of nitroaromatic pollutants. cis-Dihydroxylation has been shown to involve oxidation by HO–FeVO, in which the hydroxo group initially attacks the aromatic ring followed by the oxo ligand.32 Direct oxidation by FeIII–OOH and reaction through a HO–FeVO intermediate were investigated using density functional theory (DFT) calculations on cluster models. Quantum mechanics/molecular mechanics (QM/MM) calculations, specifically the ONIOM (Our own N-layered Integrated molecular Orbital and molecular Mechanics) method, were then performed with the most plausible mechanism to elucidate the effect of the protein environment on geometry, electronic structure and energetics.
Computational methods
Preparation of the initial structure of the NBDO–2NT complex, as well as details of the molecular dynamics (MD) simulation, are discussed in the ESI.† The root-mean-square deviation (RMSD) with respect to the crystal structure (Fig. S1, ESI†) and hydrogen bonding interactions formed during simulation (Table S1, ESI†) are also reported.DFT calculations on cluster models
A cluster model consisting of mononuclear Fe, the sidechains of histidine and aspartate residues (imidazole and acetate, respectively), and a hydroperoxo ligand (M1 model) was derived from the initial structure and used to explore the possible mechanisms of C–H hydroxylation. Geometries were optimized using the B3LYP hybrid density functional33–36 and the double-ζ-quality LACVP* basis set, which incorporates the effective core potential of Hay and Wadt37 for Fe, and a 6-31G* basis set for the other atoms. The solvent effects were included using the Polarizable Continuum Model (PCM)38,39 with a dielectric constant of ε = 5.62 (corresponding to chlorobenzene) to mimic the hydrophobic environment of the substrate pocket in RDOs.40 Stationary points were confirmed by frequency analysis and the validity of transition states was established by intrinsic reaction coordinate (IRC) calculations at the same level. Single-point energies were determined using the LACV3P+* basis set for Fe and 6-311+G* for the rest of the atoms. The larger basis set gives the correct ground state and inclusion of polarization functions was shown to be important when dealing with O–O bonds.41 Spin contamination in the doublet state was corrected using the spin projection scheme,42,43 although the difference from uncorrected values is less than 0.5 kcal mol−1. These methods have been found to give satisfactory results for both heme and non-heme iron systems.41,44,45The O–O bond cleavage and H abstraction steps were also modeled with the inclusion of carboxamide (M2 model) to represent Asn-258, which positions the substrate for oxidation through the H bonding interaction with the nitro O atom.31 Optimization of M2 model geometries was performed with the Cβ atoms of the ligands frozen to reproduce positions in the crystal structure and to prevent carboxamide from forming artificial hydrogen bonds with active site ligands. DFT calculations were done using Gaussian 09.46 Molecular orbitals of the reactant complex and the H abstraction transition state were analyzed using QMForge.47
ONIOM calculations on the enzymatic model
The lowest energy structure from the 2.5 ns molecular dynamics simulation, in which the initial methyl H–O bond is 2.50 Å, was chosen as the starting point for the ONIOM calculations. The residues located outside the 20 Å radius of the active site region were removed to reduce the system size. Solvent waters within this region were also kept as they may play a role in the O–O bond cleavage step. The truncated protein was capped with acetyl and N-methyl amide groups and neutralized with counterions. The final structure used to model the reaction is illustrated in Fig. 1 and consists of 9522 atoms. | ||
| Fig. 1 Truncated enzyme model. The QM region composed of Fe–OOH, His-206, His-211 and Asp-360 sidechains, and 2NT is shown as sticks. | ||
The QM region composed of Fe, the hydroperoxo ligand, the sidechains of His-206, His-211 and Asp-360, and 2NT (49 atoms) was treated at the B3LYP/LACVP* level while the MM region was described using the AMBER ff99SB force field.48 The active site ligands were truncated at the Cα–Cβ bond and hydrogen link atoms were added to saturate the dangling bonds of the QM atoms at the QM/MM boundary. The charge of the link atom is equal to the total charge of the atoms left out (N, H, CA, HA, C, and O) while the charge of Cα was adjusted so that the total charge of the backbone atoms is zero. This was done to maintain integer charges for the QM (+1) and MM (0) regions.49
The ONIOM energy is expressed as
| EONIOM = EMM(QM,MM) + EQM(QM) − EMM(QM) | (1) |
Results and discussion
Possible oxidizing species
The crystal structure of the NDO–O2–indole complex shows side-on binding of O2 with Fe–O bond distances of 1.7 and 2.0 Å.9 The presumed reactive species is the protonated FeIII–OOH (1),11,12 for which the calculated ground state is a sextet as magnetic circular dichroism data for NDO confirm.2 No 41 was found while the doublet state is 14.4 kcal mol−1 higher in energy. The end-on adduct (1′) is lower in energy by 1.0 kcal mol−1 and it has been observed in a high-spin biomimetic non-heme complex52 unlike 1. 41′ and 21′ lie 4.4 and 17.2 kcal mol−1, respectively, above the ground state.Oxidation of the methyl sidechain of 2NT can proceed through either direct reaction with FeIII–OOH (1 or 1′) or via a HO–FeVO intermediate (2) (Fig. 2). The latter is formed through heterolytic O–O bond cleavage, which requires 27.8 kcal mol−1 in the sextet ground state. However, 2 has a quartet ground state, as verified by CCSD(T) calculations on model FeVO complexes showing preference for the high-spin state when a π-donor ligand such as OH is cis to FeVO.42 The sextet state, which is 3.2 kcal mol−1 higher in energy, can be considered as an excited state FeIV–O˙ species53 based on the spin densities of Fe (3.06) and the oxo ligand (1.01). The doublet state, on the other hand, lies 20.4 kcal mol−1 above the ground state.
 | ||
| Fig. 2 Optimized geometries and spin populations for side-on (1) and end-on (1′) S = 5/2 FeIII–OOH and S = 3/2 HO–FeVO (2). Distances are given in Å. | ||
O–O bond homolysis, which yields FeIVO and OH˙, was not considered as it would be inconsistent with the regio- and stereospecific products observed in monooxygenase reactions catalyzed by RDOs.3,21,22,54 Solvent exchange, as inferred from the 18O-labeled products, would also not be possible with this intermediate. The failure of scavengers to inhibit monohydroxylation confirms that the hydroxyl radical is not involved in the enzymatic reaction.21
The possible reaction mechanisms for oxidation by 1′ (mechanism I), 1 (mechanism II) and 2 (mechanisms III and IV) are summarized in Scheme 2. Optimized M1 geometries for the H abstraction transition state in mechanisms I–III are shown in Fig. 3 while those for the other stationary points and for mechanism IV can be found in the ESI† (Fig. S2–S5). Potential energy surfaces (PES) of mechanisms I–III at different spin states are illustrated in Fig. 4–6. The energies were calculated relative to separated reactants as it was not possible to optimize an appropriate geometry of the reactant complex due to the tendency of the substrate to either form a spurious hydrogen bond with the hydroperoxo ligand or dissociate completely.
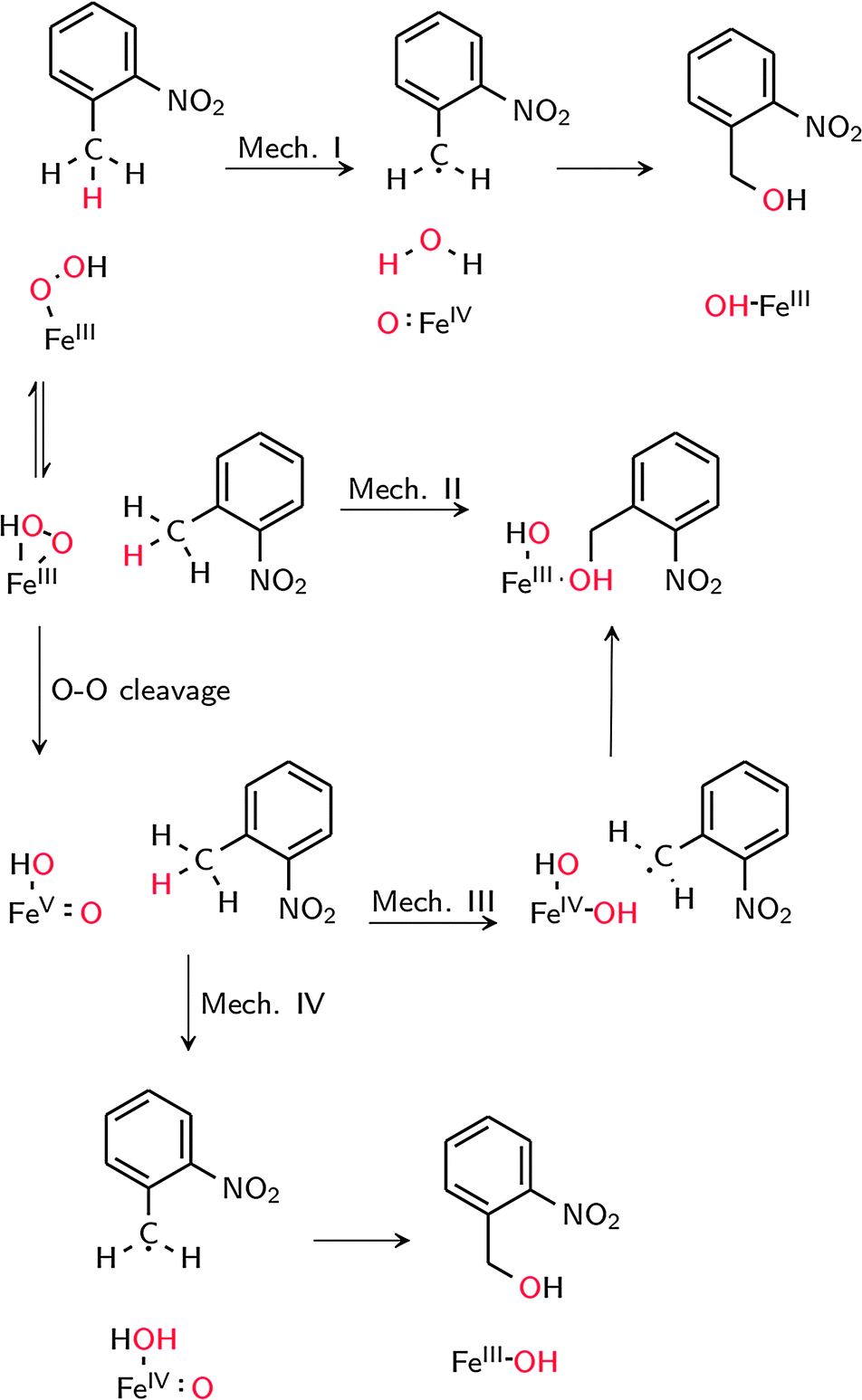 | ||
| Scheme 2 Possible mechanisms for C–H hydroxylation. | ||
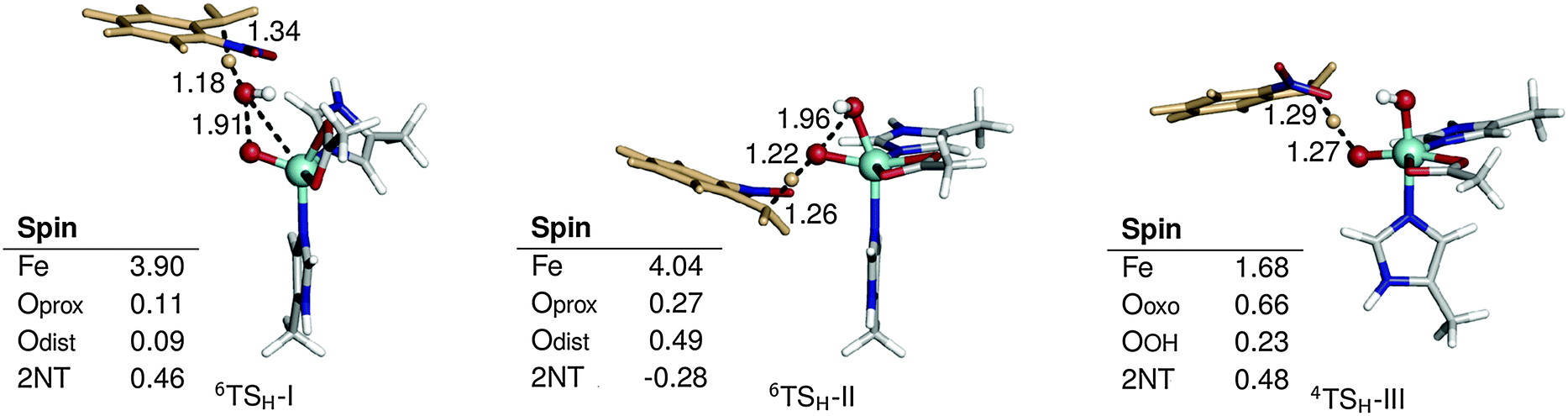 | ||
| Fig. 3 Optimized geometries and spin populations for H abstraction transition states (TSH) in mechanisms I–III. Distances are given in Å. | ||
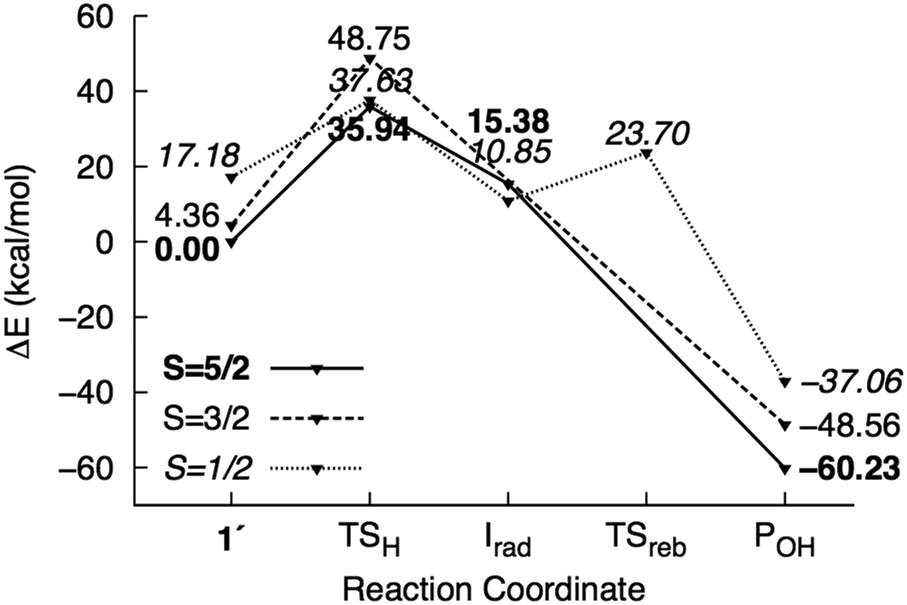 | ||
| Fig. 4 Potential energy surface of mechanism I calculated using the M1 model. Energies are reported relative to 61′ + 2NT. | ||
 | ||
| Fig. 5 Potential energy surface of mechanism II calculated using the M1 model. Energies are reported relative to 61 + 2NT. | ||
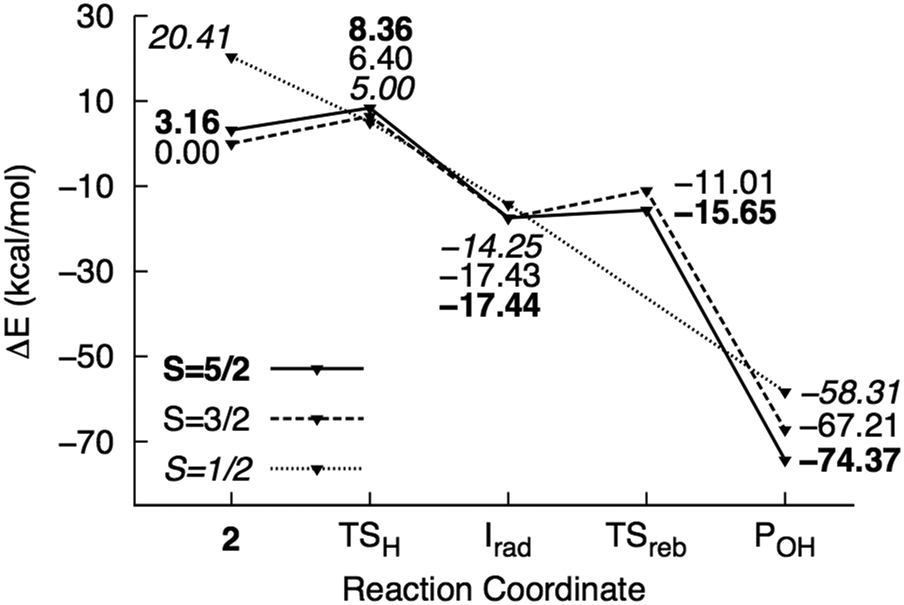 | ||
| Fig. 6 Potential energy surface of mechanism III calculated using the M1 model. Energies are reported relative to 42 + 2NT. | ||
Concerted O–O bond cleavage and H abstraction by FeIII–OOH
H abstraction by the distal O atom (Odist) of 1′ (mechanism I) requires 35.9 kcal mol−1 in the sextet ground state. Activation energies in the quartet and doublet states are 44.4 and 20.4 kcal mol−1, respectively (Fig. 4). 6TSH-I is characterized as a late transition state (Wiberg bond index rC–H = 0.38), as verified by the significant spin density on 2NT (0.46). Moreover, the spin density on OdistH (0.09) is consistent with the fact that H2O is nearly formed in the transition state. On the other hand, the decreased total spin density of Fe–Oprox (from 4.42 to 4.01) and shortening of the bond from 1.86 to 1.75 Å indicate oxidation to FeIVO (Fig. 3). Formation of the radical 6Irad-I is an endothermic process (15.4 kcal mol−1). 6Irad-I is a shoulder-type energy minimum that transforms to the product 6POH-I in a barrierless process. This involves the transfer of the abstracted H to Oprox upon recombination of OdistH˙ and the methyl C radical. 6POH-I is located outside the first coordination shell of the metal center (dFe–O = 3.6 Å) and is exothermic by −60.2 kcal mol−1. The doublet state follows the same mechanism with the exception of a barrier (12.8 kcal mol−1) for the rebound step (2TSreb-I). On the other hand, no intermediate was found in the quartet state as H abstraction leads directly to the alcohol product.
Direct reaction with 1 (mechanism II) involves concerted O–O bond cleavage and H abstraction by the proximal O atom (Oprox), with an activation energy of 23.9 kcal mol−1 in the sextet state. The transition states in the quartet and doublet states lie at higher energies compared to 6TSH-II, and the corresponding barriers are 32.6 and 22.1 kcal mol−1, respectively (Fig. 5). 6TSH-II can be characterized as central (rC–H= 0.46) with a nearly linear O–O–H angle (172°). O–O bond cleavage is reflected in the increased spin density of OdistH from 0.09 to 0.48. However, the negligible change in the Fe spin density (∼4) suggests that the metal center is not involved in the reaction. 2NT, on the other hand, has a negative spin density of −0.28 indicating the transfer of an α-spin electron to the O–O σ* orbital (Fig. 3). No intermediate is formed as oxygen rebound occurs spontaneously after additional electron donation from 2NT completes O–O bond cleavage. 6POH-II is coordinated to the ferric center (dFe–O = 2.3 Å) and is 3.0 kcal mol−1 higher in energy than 6POH-I.
Stepwise mechanism via a HO–FeV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O intermediate
O intermediate
H abstraction by the oxo ligand of 2 (mechanism III) has a barrier of 6.4 kcal mol−1 in the ground state. However, the energy of the doublet transition state is slightly lower by 1.4 kcal mol−1 (Fig. 6). 4TSH-III is central as in mechanism II with rC–H = 0.47. Spin densities on 2NT (0.48) and Fe (1.68) indicate radical formation and reduction, respectively (Fig. 3). H abstraction yields S = 1 FeIV–OH ferromagnetically coupled to the nitrobenzyl radical giving a total spin of S = 3/2 in an exothermic process (−17.4 kcal mol−1). However, the sextet state, wherein S = 2 FeIV–OH is ferromagnetically coupled to the radical, is isoenergetic. 6Irad-III is achieved through a spin flip to an unoccupied α-spin MO.55 On the other hand, 2Irad-III is 3.2 kcal mol−1 higher in energy. The rebound step involving C–O bond formation has an early transition state (dC–O = 2.7–2.8 Å) with a low energy barrier in the sextet (1.8 kcal mol−1) and a higher one (6.4 kcal mol−1) in the quartet state. As in previous mechanisms, alcohol formation is exothermic (−77.5 and −67.2 kcal mol−1 for the sextet and quartet states, respectively) and the product is still bound to the active site (dFe–O = 2.2 Å). There is no barrier to the rebound step in the doublet state and the resulting alcohol product lies 16.0 kcal mol−1 above 6POH-III.
H abstraction by the OH ligand of 2 (mechanism IV) was also investigated and found to be similar to mechanism I, in which the abstracted H atom is transferred from the bound H2O to the oxo ligand in the rebound step (Fig. S5, ESI†). However, the barrier to H abstraction in the quartet state was found to be much higher at 12.8 kcal mol−1. While the activation energy is low in the sextet state (3.0 kcal mol−1), the rebound step requires 13.3 kcal mol−1 and the alcohol product is dissociated from the active site.
Although the fact that only one electron is provided by the Rieske center in RDOs implies that the oxidant is most likely either 1(1′) or 2,10,11,19 H abstraction by HO–FeIVO was also examined for comparison with 2. Calculations at the quintet (ground) state show that HO–FeIVO is less oxidizing than 2, with an activation energy of 21.0 kcal mol−1 for H abstraction by the oxo ligand.
Comparison of different mechanisms and experimental evidence
To summarize, the sextet PES is the lowest energy pathway for mechanisms I, II and IV while in the case of mechanism III, the sextet and quartet PES are close in energy. The reactant complexes and H abstraction transition states were then recalculated using the M2 model (Fig. S6, ESI†) and activation energies are summarized in Table 1. Mechanism I has the highest barrier, which is in contrast to the similarly high-spin Fe–TMC complex with a reported activation enthalpy of only 16.1 kcal mol−1 (B3LYP/6-311+G**). The high reduction potential of the complex was attributed to minimal ligand charge donation and lowered energy of the redox-active molecular orbital (RAMO) resulting from a configuration interaction with the hydroperoxo σ* orbital.13 In comparison, the RAMO of 1′, which is a β-spin dx2−y2–σ* bonding orbital (Fig. 7), is increased in energy, presumably due to antibonding interactions with the π orbital of the acetate ligand, which contributes 6.0%. The activation energy for O–O bond homolysis in 1′ is 29.8 kcal mol−1, which indicates that oxidation of Fe and formation of OdistH˙ dominate contributions to the barrier for mechanism I.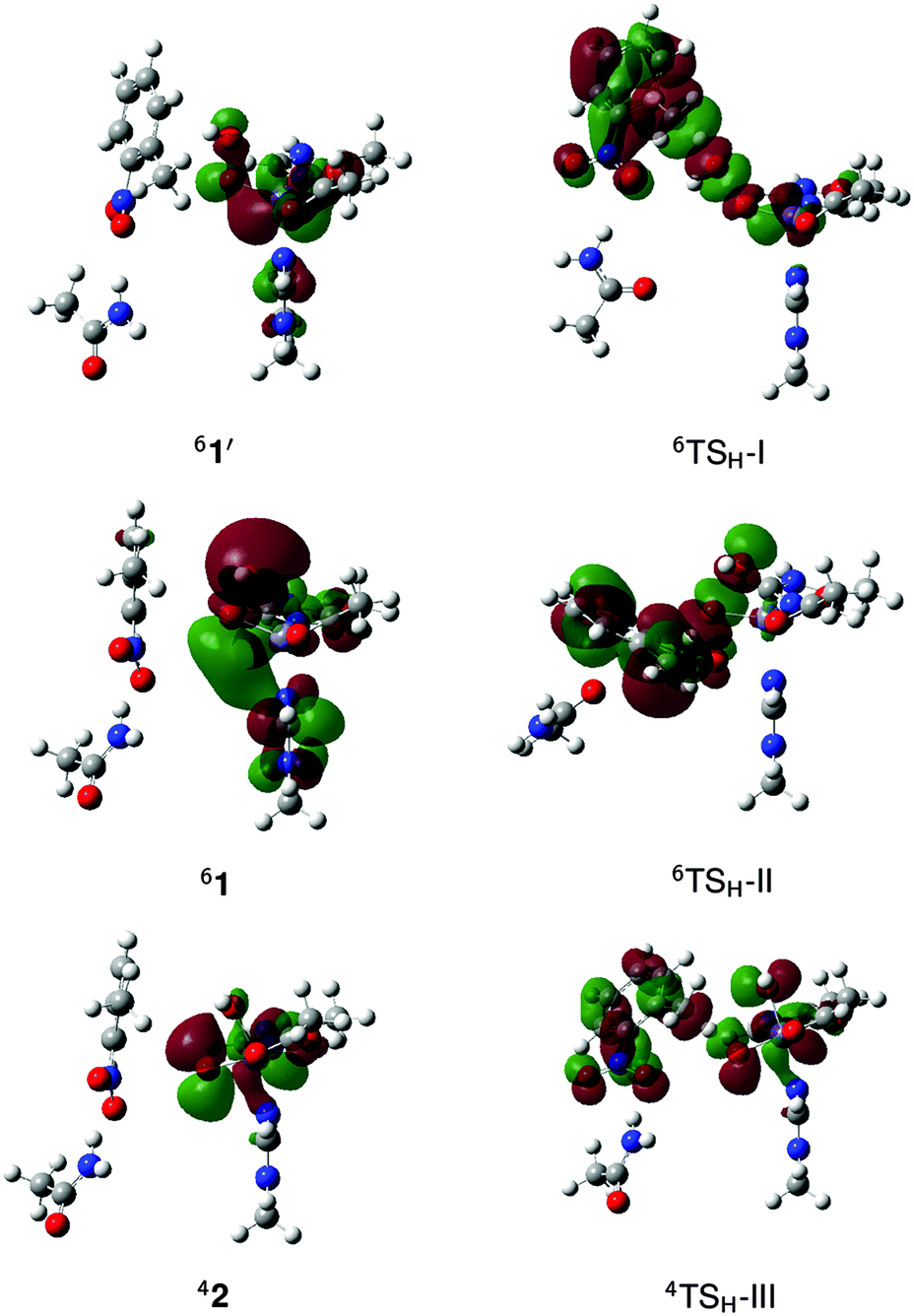 | ||
| Fig. 7 Redox-active molecular orbitals of the reactant complex (1′, 1 or 2) and the H abstraction transition state (TSH) in mechanisms I–III. | ||
The activation energy for mechanism II is lower than that for mechanism I by about 10 kcal mol−1. Unlike the latter, O–O bond cleavage is heterolytic and Fe remains in the ferric state throughout the process. Thus, Fe is not involved in the redox reaction and the electrons that must be transferred to the O–O σ* orbital for bond cleavage instead comes from the substrate. This preserves favorable exchange interactions in the metal center.56 Moreover, the large Oprox character of the O–O σ* orbital (18.9%) makes it a better electrophile55 than the RAMO involved in mechanism I (2.4% contribution from OdistH) (Fig. 7). While direct reaction with 1 has been implicated in the cis-dihydroxylation of naphthalene,12 aliphatic hydroxylation through this concerted mechanism has no precedent.
Mechanisms III and IV have the lowest activation energies. However, the rebound barrier for mechanism IV is more than 10 kcal mol−1 higher than that for mechanism III as this involves concerted O–H bond breaking and C–O bond formation. In the case of mechanism III, the H abstraction barrier is lower in the quartet state by 1.7 kcal mol−1. 2 is a better electrophile as indicated by the large coefficient of the oxo ligand in the RAMO (39.7%),55 which is a β-spin dxy–π* antibonding orbital (Fig. 7). The stepwise mechanism (TSH-III) allows the substrate to attack sideways since the RAMO is a Fe–O π* orbital (Fig. 7). In contrast, the concerted mechanism requires the C–H bond of the substrate to be linearly aligned with the O–O bond for good overlap between the substrate π orbital and the Fe–O σ* (TSH-I) or O–O σ* (TSH-II) orbital (Fig. 7).
Radical clock experiments on the monooxygenase reaction of NDO with norcarane and bicyclohexane yield a significant fraction of the radical rearrangement product, with a radical lifetime of the order 10 ns.19 Further evidence of a radical intermediate is inferred from the TDO-catalyzed oxidation of deuterated indene to inden-1-ol. Deuterium was found in either the C1 or C3 position, which is consistent with the formation of an allylic or benzylic radical intermediate that can have different resonance forms.21,22 Mechanism III is thus the most consistent with experimental studies. Although the activation energy for direct reaction with 1 (mechanism II) is slightly lower by 1.6 kcal mol−1 compared to formation of 2, the mechanism does not involve a radical intermediate, contrary to experiment. On the other hand, the radical intermediate in mechanism I forms the alcohol product in a barrierless process, which would not allow rearrangement to occur. Moreover, the formation of the radical itself is endothermic and requires a high activation energy. Finally, mechanism IV has a large rebound barrier and the alcohol product is dissociated from the active site (as in mechanism I), which is not consistent with the proposed catalytic cycle wherein the product is not released until the ferric active site is reduced back to FeII.10,11
The involvement of 2 as the oxidant is also in agreement with the recent theoretical study on aromatic cis-dihydroxylation of nitrobenzene and 2NT by NBDO.32 It was found that substrate oxidation by 1 has a higher barrier than formation of 2 and do not lead to cis-diol formation. Initial attack of the hydroxo ligand of 2 on the ring has an activation energy of 2.4 kcal mol−1 (S = 5/2) at the B97-D/LACV3P+* level for both substrates. The corresponding barrier for H abstraction by 2 (6TSH-III) is higher at 6.1 kcal mol−1.
Effect of the protein environment
C–H hydroxylation via mechanism III was further studied using the ONIOM method to incorporate steric and electronic effects from the protein environment. Energies in the sextet and quartet states were calculated using the ME and EE schemes and compared in Table 2, with higher values from the latter due to polarization of the QM region induced by H bonding interactions with the MM environment.| ONIOM | Protein effectb | |||||
|---|---|---|---|---|---|---|
| ME | EEa | E MM(QM,MM) − EMM(QM) | E QM(QM)c | E(M2 model)c | E QM(QM) − E(M2 model) | |
| a B3LYP-D2 energies in square brackets, B97-D energies in parenthesis. b Based on decomposition of ONIOM-EE energies into QM and MM contributions. c Without zero-point energy correction. | ||||||
| S = 5/2 | ||||||
| 1 | 0.00 | 0.00 [0.00] (0.00) | 0.00 | 0.00 | 0.00 | 0.00 |
| TSO–O | 22.46 | 24.79 [23.60] (13.37) | −2.76 | 29.47 | 29.55 | −0.08 |
| 2 | 12.52 | 14.67 [12.63] (1.65) | −3.00 | 19.54 | 21.72 | −2.18 |
| TSH | 16.66 | 25.54 [20.74] (6.82) | −2.66 | 33.48 | 32.89 | 0.59 |
| Irad | −10.63 | 3.31 [0.28] (−9.46) | −1.09 | 7.45 | 7.17 | 0.28 |
| TSreb | −10.77 | 11.07 [7.58] (−3.07) | −0.90 | 14.07 | 8.69 | 5.38 |
| POH | −64.59 | −44.90 [−50.24] (−44.67) | −0.33 | −46.18 | −56.36 | 10.17 |
| S = 3/2 | ||||||
| 2 | 8.42 | 11.12 [7.97] (0.43) | −1.76 | 13.51 | 17.97 | −4.47 |
| TSH | 15.65 | 24.55 [18.62] (7.92) | −0.68 | 29.23 | 31.16 | −1.93 |
| Irad | −4.98 | 3.48 [−0.88] (−4.31) | 0.47 | 3.86 | 8.17 | −4.31 |
| TSreb | −0.73 | 13.95 [9.29] (3.55) | −0.10 | 14.92 | 11.57 | 3.36 |
| POH | −53.69 | −32.94 [−39.36] (34.64) | −0.36 | −35.09 | −48.11 | 13.02 |
Heterolytic O–O bond cleavage (TSO–O) is the rate-limiting step and has activation energies of 22.5 and 24.8 kcal mol−1 in the ME and EE schemes, respectively. The barrier was recalculated in the ONIOM-ME scheme using different initial structures and results are summarized in Table S2 (ESI†). The values ranged from 22.5 to 25.7 kcal mol−1, suggesting that some protein conformations are more favorable toward O–O bond cleavage than others. It has been previously shown through free-energy perturbation corrections to ONIOM calculations on isopenicillin N synthase that dynamical contributions, specifically statistical effects due to fluctuations about the average protein geometry, lower the O–O bond heterolysis barrier obtained from the static method by about 3 kcal mol−1.57 Thus, it is possible that the barrier is overestimated in the present study due to negligence of geometric polarization of the MM region. ONIOM-ME and ONIOM-EE geometries for the stationary points in the O–O bond cleavage step are similar. The spin population of 1 is also comparable in both schemes, but ONIOM-EE predicts a more significant electron transfer from Fe to the antibonding O–O σ* orbital at the transition state. There is also a difference of about 0.1 in the spin density of the oxo ligand in 42 (Fig. S7, ESI†).
Concerted O–O bond cleavage and H abstraction by 1 was also investigated given that the activation energy obtained using the M2 model was lower compared to that for formation of 2. To locate the transition state, O–O–H and O–H–C angles were incrementally brought to 170° and 175°, respectively, to position the substrate for overlap between its π orbital and the O–O σ* orbital (see TSH-II, Fig. 7). A potential energy scan was then generated along the H2NT–Oprox reaction coordinate. The highest point (dH–O = 1.2 Å, dO–O = 1.8 Å) has an energy of 40.5 kcal mol−1 at the ONIOM-ME(B3LYP/LACVP*:AMBER) level (Fig. S8, ESI†) and optimization after release of restraints led to the TSO–O geometry. The calculations were repeated for two other structures from the simulation but the transition state was also not located. Moreover, the energy was already over 40 kcal mol−1 at dH–O = 1.9 Å. Steric hindrance from second-shell ligands, particularly Val-207 (Fig. S9, ESI†), prevents the substrate from relaxing to the transition state geometry TSH-II (Fig. 3) and leads to a repulsive MM energy contribution (∼5 kcal mol−1 in the first structure calculated).
H abstraction by 2 (TSH) in the ONIOM-ME PES has a lower energy in the quartet state, corresponding to a barrier of 7.2 kcal mol−1. 4TSH was obtained using three other structures from the simulation and results calculated by the ONIOM-ME method are summarized in Table S3 (ESI†). C–H and O–H bond distances and spin densities did not vary significantly (with the exception of one structure) and activation energies were within 1–2 kcal mol−1. Irad is much more stable in the sextet state by 5.6 kcal mol−1. The rebound step to form POH is essentially barrierless in the sextet PES. On the other hand, the ONIOM-EE activation energy for H abstraction is higher at 13.4 kcal mol−1 due to the change in the charge distribution of the substrate as it reacts with 2, which is unaccounted for in the ONIOM-ME method. Another notable difference from the ONIOM-ME method is that formation of Irad is much more endothermic, with the quartet and sextet states becoming nearly isoenergetic. The rebound barrier in the sextet PES also increased to 7.8 kcal mol−1.
The ONIOM-ME method fails to give a correct description of the electronic structure of the stationary points along this reaction. This can be most clearly seen by comparing the spin population of 4Irad from the two methods (Fig. 8). A β-spin electron is transferred from the substrate to the active site during H abstraction and the ONIOM-EE method consistently yields a spin density of 1.0 on 2NT. In contrast, incomplete electron transfer was obtained from the ONIOM-ME method, as indicated by the smaller spin density on 2NT (0.39). Moreover, the spin density became more localized on Fe, as the value increased from 1.5 in 4TSH to 2.7 in 4Irad. Based on ONIOM-EE calculations, the spin-flip in Irad to the sextet state results in a spin density of 3.3 on Fe. This increased to 3.8 in 6TSreb while the spin density of 2NT decreased to 0.7. In 6POH, the ONIOM-ME method shows a residual spin density on the oxo ligand and 2NT, unlike in the ONIOM-EE method, where the spin density is concentrated on Fe and the hydroxo ligand. The Fe–O bond lengths are comparable in the two methods except in the case of 6Irad, where bonds are 0.4–0.9 Å longer in the ONIOM-ME geometries. The ONIOM-ME method also predicts earlier transition states, as can be seen from the relatively long H–O (1.39 Å) and C–O (2.85 Å) bonds in 4TSH and 6TSreb, respectively.
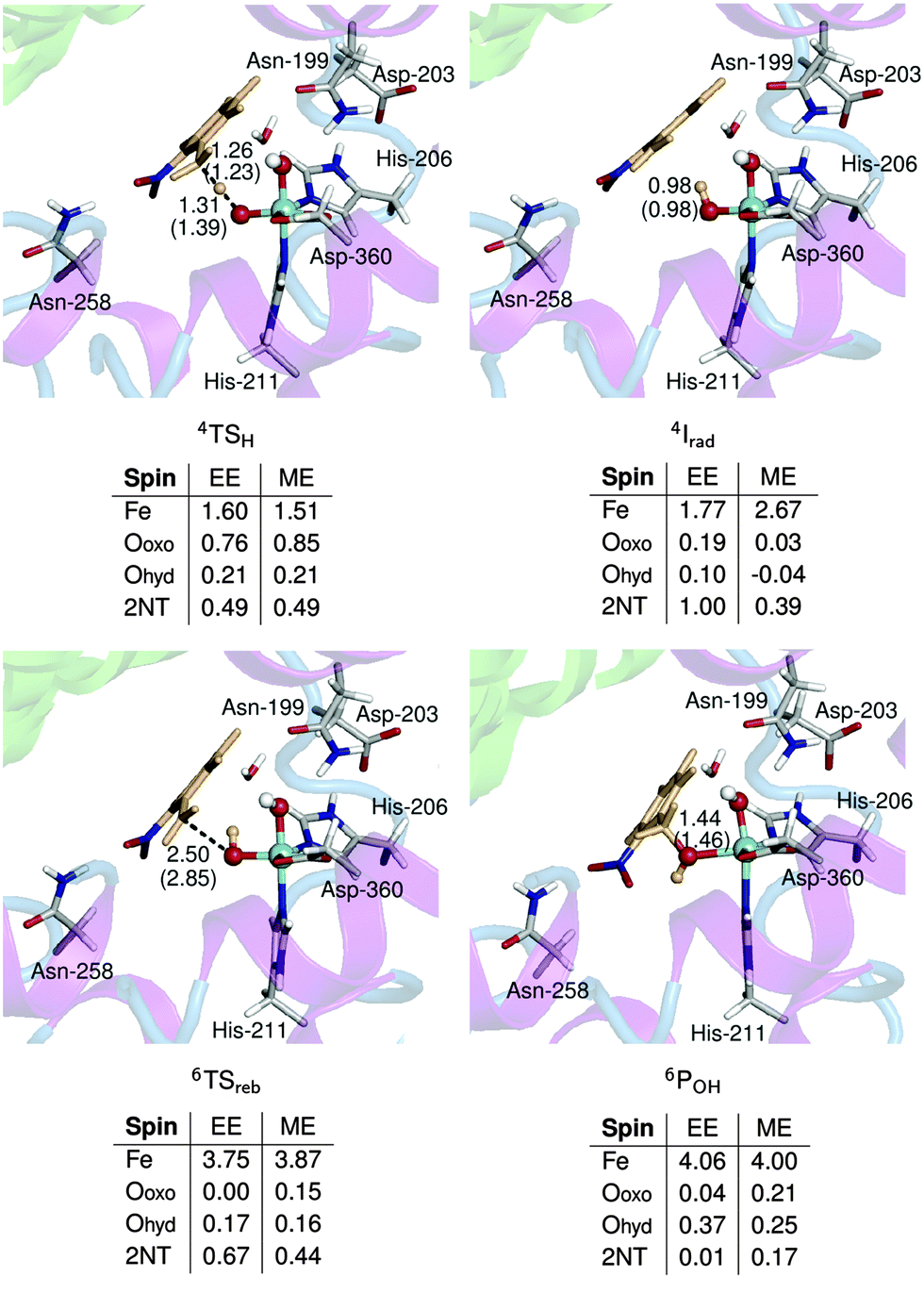 | ||
| Fig. 8 Optimized geometries and spin populations of stationary points in the C–H hydroxylation step calculated using mechanical (ME) and electronic (EE) embedding schemes. Distances (Å) in parenthesis are obtained using the ME scheme. Residues and a water molecule forming hydrogen bonds with the QM region are also shown. | ||
A comparison between the QM energy (EQM(QM)) and the energy calculated using the cluster model (M2) gives a qualitative idea of the effect of the protein environment on the geometry and the electronic structure of the active site (high-level effect). The protein effect on the energetics of the reaction can also be evaluated in terms of EMM(QM,MM) − EMM(QM), which includes the non-bonded interactions between the QM and MM regions (low-level effect).58 The decomposition of ONIOM-EE energies into high- and low-level effects is summarized in Table 2.
The high-level effect for TSO–O is negligible but it stabilizes 42 by about 4 kcal mol−1. The spin density of 42 in the two models is similar but ONIOM-EE shows a slightly larger (∼3°) O–Fe–O angle. On the other hand, the low-level effect for TSO–O is 3 kcal mol−1 while that for 42 is 2 kcal mol−1. Thus, the protein environment makes heterolytic O–O bond cleavage slightly more favorable.
QM energies of 4TSH and 4Irad are about 2 and 4 kcal mol−1 lower than those in the M2 model, while QM energies are higher for 6TSreb and 6POH by about 5 and 10 kcal mol−1. For 6TSreb, the difference can be attributed to the much earlier transition state predicted by the M2 model based on the longer C–O bond length (by 0.7 Å) and the higher spin density on 2NT (0.81 compared to 0.67 in ONIOM-EE) (Fig. S10, ESI†). In the case of 6POH, the difference in energy is possibly an artifact of including Asn-258 (which moved closer to the active site, see Fig. S10, ESI†) in the M2 model since the geometry and spin population are similar for the two models. In contrast to the O–O bond cleavage step, the low-level effects for the C–H hydroxylation step only ranged from −0.9 (6TSreb) to 0.5 (4Irad) kcal mol−1 since the substrate essentially remained in the same position during the reaction.
Effect of dispersion interactions
The effect of dispersion interactions on ONIOM-EE energies was also investigated in light of its possible importance in obtaining accurate activation energies for enzymatic reactions, as shown in a QM/MM study of H abstraction by P450cam.59Table 2 includes dispersion-corrected ONIOM-EE energies calculated using B3LYP-D2, which was used by Lonsdale et al.,59 and B97-D, which was used in the study on cis-dihydroxylation by NBDO mentioned above.32 A comparison of the energy profiles for the overall reaction is shown in Fig. 9. It can be seen that the O–O bond cleavage barrier is lowered by dispersion, and in the case of B97-D, the barrier is only 13.4 kcal mol−1. This is slightly lower than those reported by Pabis et al.32 (14.6–16.7 kcal mol−1), which were obtained using various cluster models of NBDO. Formation of ground state 2 is also essentially thermoneutral (0.4 kcal mol−1) using the B97-D functional. The H abstraction barrier in the quartet state decreased to 10.7 kcal mol−1 and the resulting 4Irad is stabilized when the D2 correction is added to B3LYP energies. In contrast, the B97-D functional yields a slightly lower energy for 6TSH, resulting in a barrier of only 6.4 kcal mol−1 relative to 42. 6Irad is also more stable than the corresponding quartet state by 5.2 kcal mol−1, which would be consistent with the notion that a higher number of identical-spin unpaired electrons results in more favorable exchange interactions.56 On the other hand, all methods consistently indicate that the rebound step is more favorable in the sextet PES, with B97-D giving the lowest barrier (6.4 kcal mol−1) and B3LYP-D2 giving the most exothermic reaction energy (−50.5 kcal mol−1).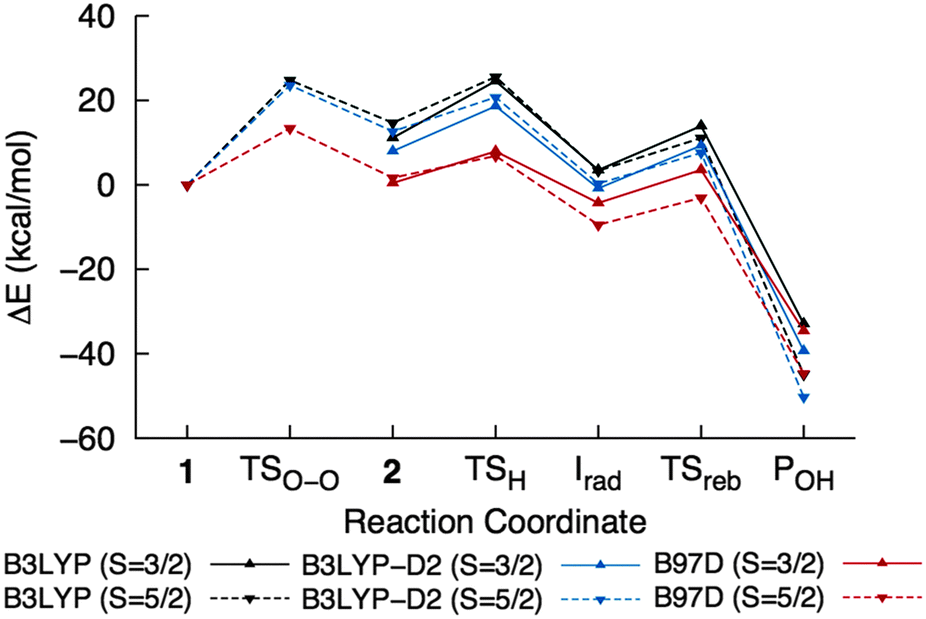 | ||
| Fig. 9 ONIOM-EE energy profiles for the overall reaction calculated using different functionals. The values are listed in Table 2. | ||
Possible role of other residues in the substrate pocket
The substrate pocket of NBDO is composed of mostly hydrophobic residues, some of which have been shown to influence stereo- or regioselectivity. In particular, Asn-258 may be crucial to aromatic cis-dihydroxylation of mononitrotoluenes by suitably positioning the ring for oxygen attack through hydrogen bonding.60 This is indicated by the predominance of the competing reaction, C–H hydroxylation, in the N258V mutant of NBDO60 and in NDO,31 which has valine at the equivalent position. On the other hand, there is no experimental evidence for the direct involvement of neighbouring residues in the reaction itself. Several hydrogen bonding interactions with the active site were observed during simulation and have also been reported in a recent MD study of NBDO.61 These include interactions between (a) the substrate and Asn-258, (b) His-206 and Asp-203, which bridges the active site and the adjacent Rieske cluster, (c) the hydroperoxo ligand and a water molecule, (d) the same water molecule and Asn-199, which is believed to be connected to a water channel facilitating proton transfer,9 (e) the hydroperoxo ligand and Asn-199 and (f) Asp-203 and Asn-199 (Fig. 8, Table S1, ESI†).ONIOM-EE geometries of 6TSO–O and 4TSH obtained with the QM region consisting only of Fe, the hydroperoxo ligand, the sidechains of His-206, His-211 and Asp-360, and 2NT were re-optimized with inclusion of these residues to investigate the effect on energy, geometry and electronic structure. In the case of O–O bond cleavage, the barrier increased with inclusion of other residues in the QM region although the difference is less than 1 kcal mol−1 (Table 3). On the other hand, the H atom abstraction barrier decreased by about 1 kcal mol−1 with addition of Asn-258 in the QM region (Table 4). However, 6TSO–O and 4TSH geometries and spin densities did not change significantly with the QM region.
| QM region | Atoms | ΔE‡a (kcal mol−1) | O–O bond distance (Å) | Spin densities | ||
|---|---|---|---|---|---|---|
| Fe | Oprox | Odist | ||||
| a Without zero-point correction. b Fe–OOH + His-206 + His-211 + Asp-360 + 2NT. | ||||||
| Coreb | 49 | 26.71 | 1.88 | 3.70 | 0.49 | 0.31 |
| + H2O | 52 | 26.93 | 1.88 | 3.70 | 0.50 | 0.32 |
| + Asp-203 | 55 | 27.02 | 1.88 | 3.69 | 0.49 | 0.32 |
| + H2O + Asp-203 | 58 | 27.28 | 1.88 | 3.69 | 0.51 | 0.31 |
| + H2O + Asp-203 + Asn-199 | 66 | 27.19 | 1.88 | 3.69 | 0.52 | 0.30 |
| QM region | Atoms | ΔE‡a (kcal mol−1) | Bond distances (Å) | Spin densities | ||||
|---|---|---|---|---|---|---|---|---|
| C–H | O–H | Fe | Ooxo | Ohyd | 2NT | |||
| a Without zero-point correction. b Fe–OOH + His-206 + His-211 + Asp-360 + 2NT. | ||||||||
| Coreb | 49 | 16.81 | 1.26 | 1.31 | 1.60 | 0.76 | 0.21 | 0.49 |
| + Asp-203 | 55 | 16.91 | 1.26 | 1.30 | 1.59 | 0.74 | 0.21 | 0.48 |
| + Asn-258 | 57 | 15.68 | 1.25 | 1.32 | 1.59 | 0.77 | 0.21 | 0.49 |
| + Asn-199 | 57 | 17.49 | 1.27 | 1.30 | 1.61 | 0.75 | 0.20 | 0.49 |
Conclusions
DFT calculations on cluster models of nitrobenzene 1,2-dioxygenase reveal the mechanistic differences between C–H hydroxylation by FeIII–OOH and HO–FeVO. Direct reaction with FeIII–OOH involves concerted H abstraction and O–O bond cleavage to form the alcohol product. On the other hand, H abstraction by HO–FeVO, which is formed through O–O bond heterolysis in FeIII–OOH, leads to a radical intermediate. The latter mechanism is consistent with deuterium labeling studies and radical clock experiments on other Rieske non-heme iron dioxygenases. However, the activation energy for formation of the HO–FeVO oxidant calculated using cluster models is higher compared to that for direct reaction with FeIII–OOH. ONIOM calculations gave a better description of the energetics of the two possible mechanisms through explicit inclusion of the steric effects of the protein environment. The concerted mechanism with FeIII–OOH requires the C–H bond to be aligned with the O–O bond for good orbital overlap, but formation of this transition state was hindered by the surrounding residues. In comparison, H abstraction by HO–FeVO involves a sideways attack of the substrate, which was slightly stabilized by the protein environment. Formation of HO–FeVO also became more favorable and it is possible that dispersion and free-energy perturbation corrections would lower the calculated activation energy further. On the other hand, no evidence was found to indicate the involvement of other residues in the substrate pocket on the reaction itself. Inclusion of Asn-199, Asp-203 and Asn-258, which form hydrogen bonding interactions with the active site, in the QM region of the ONIOM model also did not affect O–O bond cleavage and H abstraction barriers and transition state geometries significantly. The results of the study can provide an insight into the nature of the oxidant and the mechanism of aliphatic hydroxylation in Rieske non-heme iron dioxygenases in general.
Acknowledgements
This work was supported by the grants FP7-264329 from the 7th Framework Programme, Marie Curie Action ITN “CSI:Environment” and PSRP-025/2010 from the Polish-Swiss Research Program. Access to computing facilities at the Lodz University of Technology Computer Center (under the PLATON project) is gratefully acknowledged.Notes and references
- P. C. A. Bruijnincx, G. van Koten and R. J. M. K. Gebbink, Chem. Soc. Rev., 2008, 37, 2716–2744 RSC.
- T. Ohta, S. Chakrabarty, J. D. Lipscomb and E. I. Solomon, J. Am. Chem. Soc., 2008, 130, 1601–1610 CrossRef CAS PubMed.
- D. T. Gibson, S. M. Resnick, K. Lee, J. M. Brand, D. S. Torok, L. P. Wackett, M. J. Schocken and B. E. Haigler, J. Bacteriol., 1995, 177, 2615–2621 CAS.
- J. Lee and H. Zhao, Angew. Chem., Int. Ed., 2006, 45, 622–625 CrossRef CAS PubMed.
- D. R. Boyd, N. D. Sharma, B. E. Byrne, S. A. Haughey, M. A. Kennedy and C. C. R. Allen, Org. Biomol. Chem., 2004, 2, 2530–2537 CAS.
- S. M. Resnick, K. Lee and D. T. Gibson, J. Ind. Microbiol. Biotechnol., 1996, 17, 438–457 CrossRef CAS.
- L. P. Wackett, Enzyme Microb. Technol., 2002, 31, 577–587 CrossRef CAS.
- M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., Int. Ed., 2012, 51, 10712–10723 CrossRef CAS PubMed.
- A. Karlsson, J. V. Parales, R. E. Parales, D. T. Gibson, H. Eklund and S. Ramaswamy, Science, 2003, 299, 1039–1042 CrossRef CAS PubMed.
- M. D. Wolfe, J. V. Parales, D. T. Gibson and J. D. Lipscomb, J. Biol. Chem., 2001, 276, 1945–1953 CrossRef CAS PubMed.
- M. D. Wolfe and J. D. Lipscomb, J. Biol. Chem., 2003, 278, 829–835 CrossRef CAS PubMed.
- A. Bassan, M. R. A. Blomberg and P. E. M. Siegbahn, JBIC, J. Biol. Inorg. Chem., 2004, 9, 439–452 CrossRef CAS PubMed.
- L. V. Liu, S. Hong, J. Cho, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2013, 135, 3286–3299 CrossRef CAS PubMed.
- M. S. Chow, L. V. Liu and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13241–13245 CrossRef CAS PubMed.
- E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef CAS PubMed.
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger, Jr. and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef CAS PubMed.
- B. E. Eser, E. W. Barr, P. A. Frantom, L. Saleh, J. M. Bollinger, Jr., C. Krebs and P. F. Fitzpatrick, J. Am. Chem. Soc., 2007, 129, 11334–11335 CrossRef CAS PubMed.
- A. J. Panay, M. Lee, C. Krebs, J. M. Bollinger, Jr. and P. F. Fitzpatrick, Biochemistry, 2011, 50, 1928–1933 CrossRef CAS PubMed.
- S. Chakrabarty, R. N. Austin, D. Deng, J. T. Groves and J. D. Lipscomb, J. Am. Chem. Soc., 2007, 129, 3514–3515 CrossRef CAS PubMed.
- A. Decker and E. I. Solomon, Curr. Opin. Chem. Biol., 2005, 9, 152–163 CrossRef CAS PubMed.
- L. P. Wackett, L. D. Kwart and D. T. Gibson, Biochemistry, 1988, 27, 1360–1367 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, N. I. Bowers, R. Boyle, J. S. Harrison, K. Lee, T. D. H. Bugg and D. T. Gibson, Org. Biomol. Chem., 2003, 1, 1298–1307 CAS.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- D. Kumar, S. P. de Visser, P. K. Sharma, S. Cohen and S. Shaik, J. Am. Chem. Soc., 2004, 126, 1907–1920 CrossRef CAS PubMed.
- I. Prat, A. Company, V. Postils, X. Ribas, L. Que, Jr., J. M. Luis and M. Costas, Chem. – Eur. J., 2013, 19, 6724–6738 CrossRef CAS PubMed.
- I. Prat, J. S. Mathieson, M. Güell, X. Ribas, J. M. Luis, L. Cronin and M. Costas, Nat. Chem., 2011, 3, 788–793 CrossRef CAS PubMed.
- K. Chen and L. Que, Jr., J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed.
- Y. He, J. D. Gorden and C. R. Goldsmith, Inorg. Chem., 2011, 50, 12651–12660 CrossRef CAS PubMed.
- J. England, R. Gondhia, L. Bigorra-Lopez, A. R. Petersen, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2009, 5319–5334 RSC.
- A. Bassan, M. R. A. Blomberg, P. E. M. Siegbahn and L. Que, Jr., J. Am. Chem. Soc., 2002, 124, 11056–11063 CrossRef CAS PubMed.
- R. Friemann, M. M. Ivkovic-Jensen, D. J. Lessner, C.-L. Yu, D. T. Gibson, R. E. Parales, H. Eklund and S. Ramaswamy, J. Mol. Biol., 2005, 348, 1139–1151 CrossRef CAS PubMed.
- A. Pabis, I. Geronimo and P. Paneth, J. Phys. Chem. B, 2014, 118, 3245–3256 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 97, 9173–9177 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef PubMed.
- B. Mennucci, E. Cancès and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS.
- D. J. Heyes, M. Sakuma, S. P. de Visser and N. S. Scrutton, J. Biol. Chem., 2009, 284, 3762–3767 CrossRef CAS PubMed.
- E. Derat, D. Kumar, H. Hirao and S. Shaik, J. Am. Chem. Soc., 2006, 128, 473–484 CrossRef CAS PubMed.
- H. Chen, W. Lai, J. Yao and S. Shaik, J. Chem. Theory Comput., 2011, 7, 3049–3053 CAS.
- K. Yamaguchi, F. Jensen, A. Dorigo and K. N. Houk, Chem. Phys. Lett., 1988, 149, 537–542 CrossRef CAS.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS PubMed.
- S. P. de Visser, J. Am. Chem. Soc., 2006, 128, 9813–9824 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revisions A.02 and D.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. L. Tenderholt, QMForge Version 2.1, Stanford University, Stanford, CA, 2007 Search PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- M. Lundberg, T. Kawatsu, T. Vreven, M. J. Frisch and K. Morokuma, J. Chem. Theory Comput., 2009, 5, 222–234 CrossRef CAS.
- T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. Montgomery, Jr., K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815–826 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- F. Li, K. K. Meier, M. A. Cranswick, M. Chakrabarti, K. M. V. Heuvelen, E. Münck and L. Que, Jr., J. Am. Chem. Soc., 2011, 133, 7256–7259 CrossRef CAS PubMed.
- A. Miłaczewska, E. Broclawik and T. Borowski, Chem. – Eur. J., 2013, 19, 771–781 CrossRef PubMed.
- K. Lee and D. T. Gibson, Appl. Environ. Microbiol., 1996, 62, 3101–3106 CAS.
- M. L. Neidig, A. Decker, O. W. Choroba, F. Huang, M. Kavana, G. R. Moran, J. B. Spencer and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12966–12973 CrossRef CAS PubMed.
- D. Usharani, D. Janardanan, C. Li and S. Shaik, Acc. Chem. Res., 2013, 46, 471–482 CrossRef CAS PubMed.
- T. Kawatsu, M. Lundberg and K. Morokuma, J. Chem. Theory Comput., 2011, 7, 390–401 CrossRef CAS.
- M. Lundberg and K. Morokuma, J. Phys. Chem. B, 2007, 111, 9380–9389 CrossRef CAS PubMed.
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Chem. Theory Comput., 2012, 8, 4637–4645 CrossRef CAS.
- K.-S. Ju and R. E. Parales, Appl. Environ. Microbiol., 2006, 72, 1817–1824 CrossRef CAS PubMed.
- A. Pabis, I. Geronimo, D. M. York and P. Paneth, J. Chem. Theory Comput., 2014 DOI:10.1021/ct500205z.
Footnote |
| † Electronic supplementary information (ESI) available: Protein structure preparation, MD simulation methodology and analysis, figures of optimized stationary points and additional ONIOM calculations using other initial structures from the simulation. See DOI: 10.1039/c4cp01030b |
| This journal is © the Owner Societies 2014 |