 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development and understanding of cobaloxime activity through electrochemical molecular catalyst screening†
David W.
Wakerley
and
Erwin
Reisner
*
Christian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 6th February 2014
Abstract
Electrochemical molecular catalyst screening (EMoCS) has been developed. This technique allows fast analysis and identification of homogeneous catalytic species through tandem catalyst assembly and electrochemistry. EMoCS has been used to study molecular proton reduction catalysts made from earth abundant materials to improve their viability for water splitting systems. The efficacy of EMoCS is proven through investigation of cobaloxime proton reduction activity with respect to the axial ligand in aqueous solution. Over 20 axial ligands were analysed, allowing rapid identification of the most active catalysts. Structure–activity relationships showed that more electron donating pyridine ligands result in enhanced catalytic currents due to the formation of a more basic Co–H species. The EMoCS results were validated by isolating and assaying the most electroactive cobaloximes identified during screening. The most active catalyst, [CoIIICl(dimethyl glyoximato)2(4-methoxypyridine)], showed high electro- and photoactivity in both anaerobic and aerobic conditions in pH neutral aqueous solution.
Introduction
As oil reserves draw closer to depletion,1 alternative infrastructures must be installed to accommodate increasing global energy demands. Hydrogen is an attractive substitute for fossil fuels that oxidises without producing harmful emissions. Water stands as a cheap and convenient hydrogen source, however a disconcerting 48 million tonnes of hydrogen are derived each year from non-renewable hydrocarbon feedstocks.2 Efficient and sustainable generation of hydrogen from water is therefore a top priority of contemporary catalyst research.3–5Molecular proton reduction catalysts are capable of evolving hydrogen through electro- and photocatalysis.6,7 Progressive research has focused on fabricating proton reduction catalysts based on earth abundant and therefore scalable elements, rather than precious metals.8 This effort has produced catalysts that generate hydrogen with high turnover frequencies,9 low overpotentials,10 from water11 and even under aerobic conditions.12 However, synthetic catalyst activity still falls short of the more industrially relevant catalytic rates and turnover numbers attained by Pt13,14 and H2 evolving enzymes called hydrogenases.15
A number of techniques have recently emerged that allow heterogeneous surfaces to be screened for catalytic activity, quickly yielding new, highly active materials.16–20 In contrast, the synthesis of novel molecular catalysts remains a very slow and often fruitless practice and as such a molecular screening procedure would dramatically accelerate progress in this field.
This work describes the development of electrochemical molecular catalyst screening (EMoCS). This concept extends upon combinatorial approaches21,22 and is specifically tailored to discover molecular redox catalysts for renewable fuel generation. EMoCS is achieved through tandem in situ assembly and electrochemistry of a complex and is carried out in aqueous solution, which ensures all catalysts identified are active in conditions relevant for large scale application.23
EMoCS has been used here to investigate cobaloximes (Fig. 1),24–29 an O2-tolerant12 catalyst family that display proton reduction activity at low overpotentials30 in neutral aqueous solution.31–33 Substantial improvements are required before these catalysts can be considered useful for generating hydrogen outside of the laboratory environment.
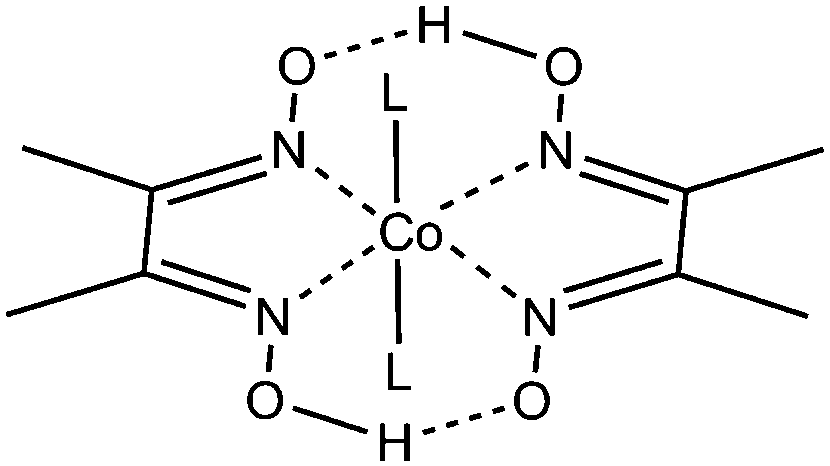 | ||
| Fig. 1 General chemical structure of a cobaloxime with a cobalt ion and two dimethyl glyoximato ligands in the equatorial coordination sphere. The axial ligands (L) are typically a pyridine, halide or solvent molecule. | ||
Enhancing cobaloxime activity has thus been the subject of considerable scrutiny. Proton reduction proceeds through several key oxidation states and requires protonation of a Co–H species (Scheme 1). The hydride intermediate can be made more basic by increasing electron density on the Co center34 but this also leads to more negative reduction potentials, thereby increasing the catalytic overpotential. A number of studies have explored modification of the complex to exploit this relationship, however this has focused predominantly on the equatorial macrocycles,30,34 leaving the axial ligand relatively unexplored in aqueous solution.
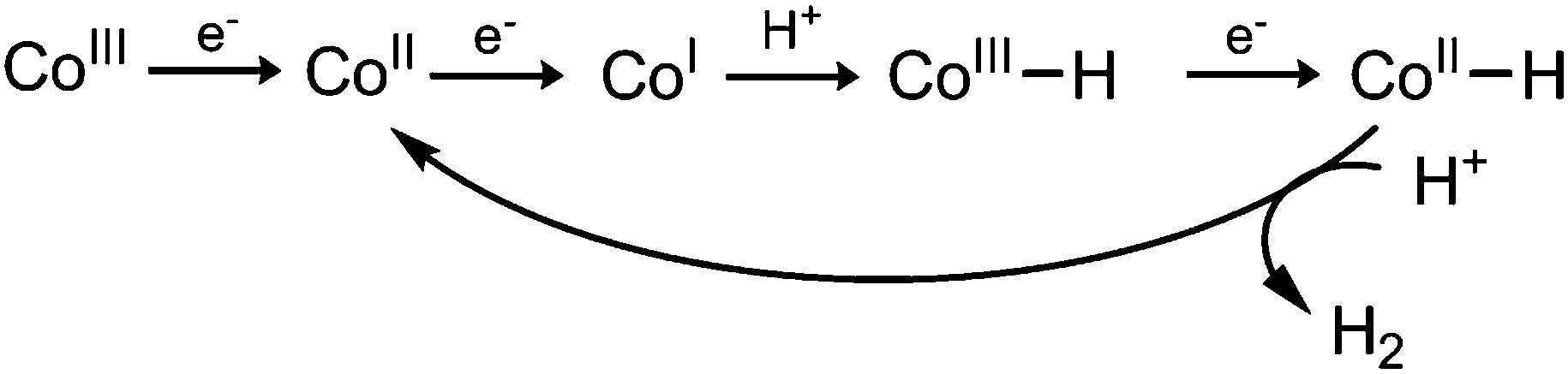 | ||
| Scheme 1 Proposed mechanism of H2 formation.26 | ||
Although the native CoIII state of the cobaloxime has two axial substituents, upon reduction to lower oxidation states one dissociates, leaving a free coordination site for catalysis.35,36 The remaining axial ligand is trans to the intermediate hydride formed during proton reduction (Scheme 1) and it is well documented that the presence of an axial aromatic N-binding ligand, such as pyridine, enhances the reactivity with H2 and the reduction of protons in organic media.35,37 Attempts to augment this effect are limited in number and have drawn conflicting conclusions concerning the effect of more electron donating axial ligands on cobaloxime activity.24,35
EMoCS is capable of clarifying the role of the cobaloxime axial ligand in aqueous solution at a rate unattainable by synthetic procedures. This was used to elucidate structure–activity relationships (SARs) and subsequently the most active cobaloximes have been isolated and tested to confirm the efficacy of the EMoCS technique.
Results and discussion
In situ assembly of cobaloximes
The EMoCS process is simple; starting from a solution of a labile metal ion, a defined amount of ligand is added and the electrochemical response of the assembled complex is recorded. Analysis of catalytic currents then indicates the most promising metal–ligand combinations to investigate further.Initial experiments explored this premise by monitoring the in situ formation of cobaloximes. Starting from a solution of [Co(H2O)6]Cl2 in aqueous phosphate buffer (Pi, 0.1 M, pH 7), cyclic voltammograms (CVs) were recorded using a glassy carbon (GC) working electrode as constituent ligands were added (Fig. 2).
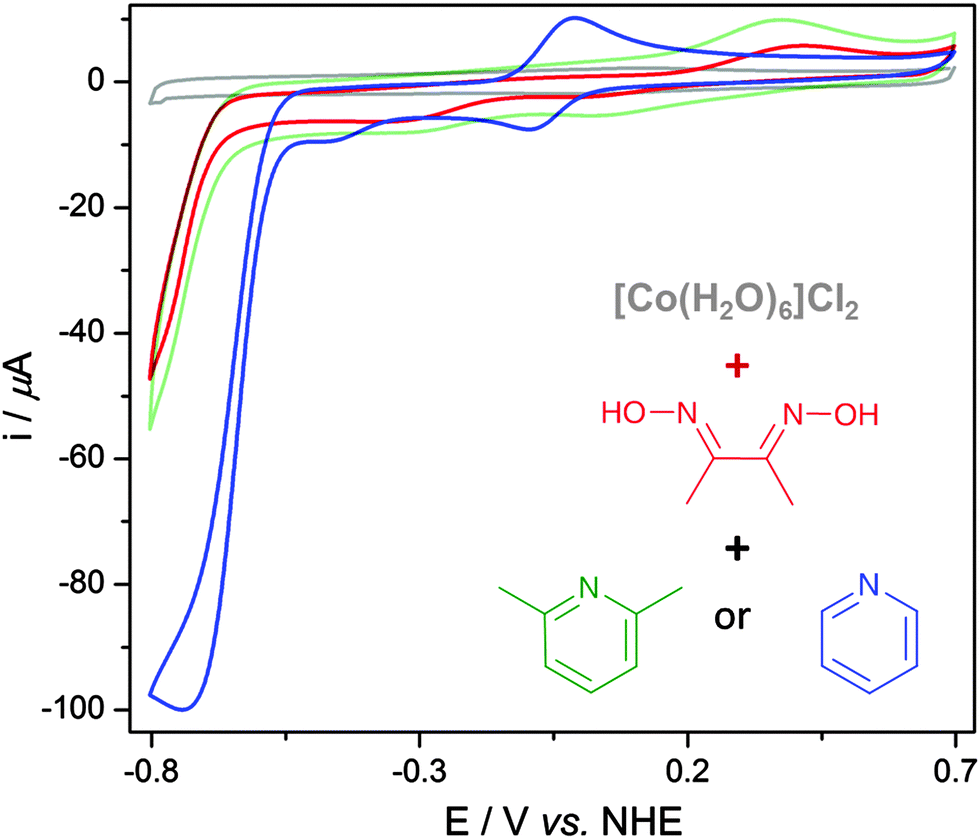 | ||
| Fig. 2 CVs of [Co(H2O)6]Cl2 (2 mM) (grey) after addition of 2 equiv. of dmgH2 (red), followed by addition of 4 equiv. of 2,6-dimethylpyridine (green) or pyridine (blue trace). Measurements were recorded in Pi (0.1 M, pH 7) at 100 mV s−1. | ||
The grey trace in Fig. 2 is a CV of [Co(H2O)6]Cl2 (2 mM), which shows no pronounced electrochemical features between −0.8 and +0.7 V vs. NHE. Sonication of [Co(H2O)6]Cl2 with two equiv. of dimethylglyoxime (dmgH2) results in the spontaneous formation of a brown solution, which corresponds to dissolved [Co(dmgH)2(H2O)2] as confirmed by UV-vis spectroscopy and mass spectrometry (Fig. S1 and S2, ESI†). This compound displays a small proton reduction wave with an onset potential of approximately −0.65 V vs. NHE (Fig. 2, red trace), consistent with previous studies.30
Addition of pyridine to the solution of [Co(dmgH)2(H2O)2] formed [Co(dmgH)2(pyridine)2], which was confirmed by 1H-NMR spectroscopy and mass spectrometry (Fig. S3 and S4, ESI†). Introducing pyridine resulted in the appearance of a reversible CoIII/CoII couple at approximately −0.03 V vs. NHE and a catalytic wave at a lower overpotential (onset at −0.54 V vs. NHE) in the CV (Fig. 2, blue trace). The catalytic wave occurs upon reduction of CoII to CoI and its size correlates to catalytic activity.38 The wave height was found to increase until approximately four equivalents of pyridine were added (Fig. S5, ESI†), at this point it was assumed all water ligands were substituted.
Addition of four equiv. of 2,6-dimethylpyridine to [Co(dmgH)2(H2O)2] produced no significant change in the CV (Fig. 2, green trace) compared to [Co(dmgH)2(H2O)2] (red trace). 2,6-Dimethylpyridine is sterically hindered and cannot coordinate to the Co ion, confirming that the observed increase in activity upon addition of pyridine was a result of ligation to the cobaloxime to form a more active catalyst. Similar accounts of in situ formation have been documented for other catalysts,39–43 however this assembly has not been exploited to methodically screen for catalytic activity.
Electrochemical Molecular Catalyst Screening (EMoCS)
EMoCS was achieved by repeating the tandem assembly and electrochemistry across a large range of axial ligands. To screen each cobaloxime, 4 equiv. of potential ligand were injected into a vial of [Co(dmgH)2(H2O)2] (2 mM) in Pi (0.1 M, pH 7). This solution was carefully degassed for 5 min with N2, after which CVs were recorded.CVs generated from EMoCS are shown in Fig. 3 and Fig. S6 (ESI†) and the data is summarised in Table 1. The screening procedure was highly reproducible with a small standard deviation in all experiments (typically <5%). It is apparent that pyridines with electron donating groups, such as 4-methoxypyridine (2) and 4-methylpyridine (3), form highly effective catalysts that surpass the activity of the pyridine (4) substituted cobaloxime. 3,5-Dimethylpyridine (1) (in contrary to 2,6-dimethylpyridine) can coordinate to the Co center and also forms a cobaloxime that displays a high level of activity. The CV of this species decays rapidly upon consecutive scans due to poor electrostability, which was not observed for the other cobaloximes (Fig. S7, ESI†).
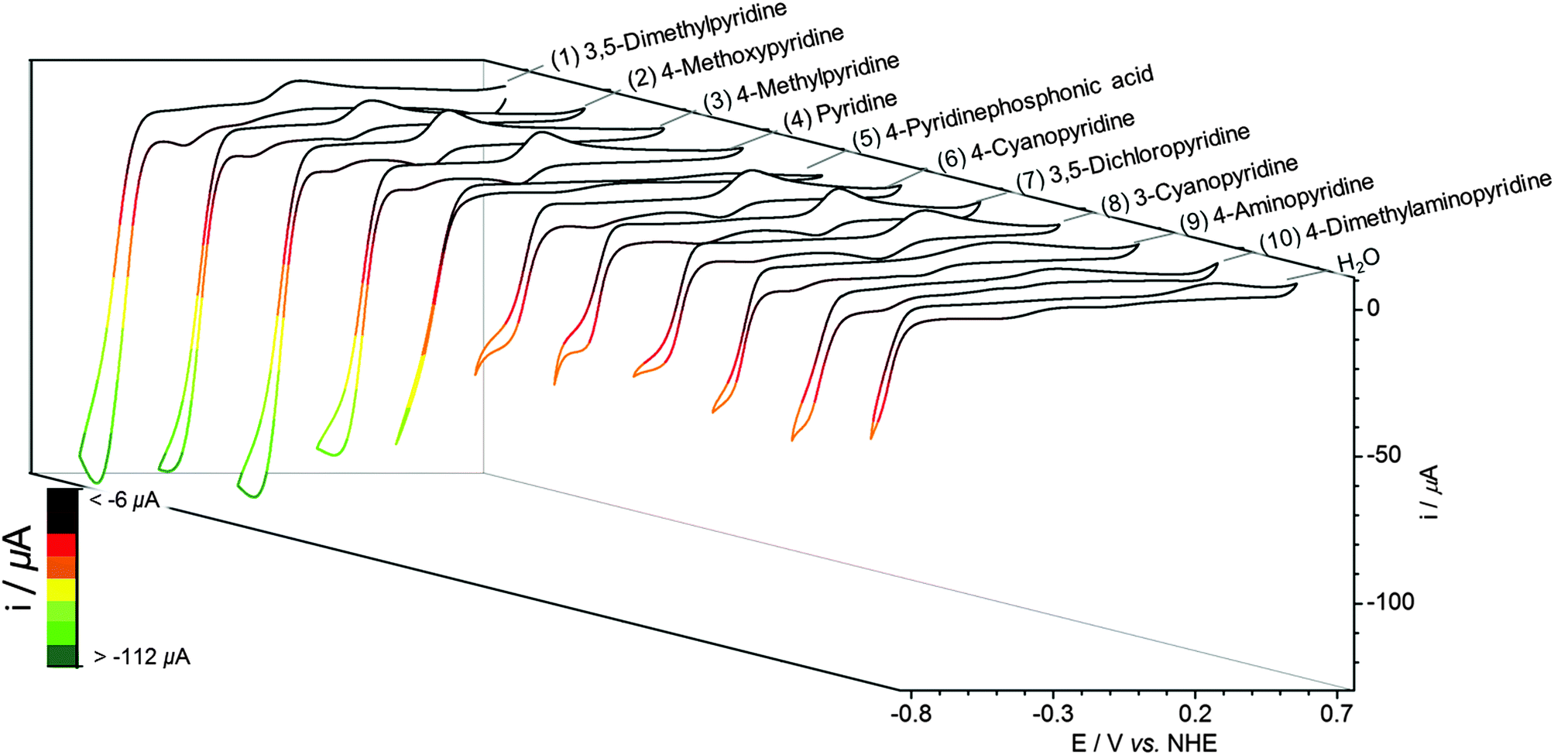 | ||
| Fig. 3 CVs of each screened cobaloxime solution after addition of 4 equiv. of axial pyridine derivative to [Co(dmgH)2(H2O)2] precursor (2 mM) in Pi (0.1 M, pH 7) at a scan rate of 100 mV s−1, listed in order of increasing electrocatalytic activity from right to left. CVs shown are the second consecutive scan with the exception of 3,5-dimethylpyridine (first scan shown), which displays poor stability during catalysis. | ||
| No. | Axial ligand | E 1/2 CoIII/CoII/V vs. NHE | Catalytic onset/V vs. NHE | Catalytic current/μA | E L/V |
|---|---|---|---|---|---|
| a No well-defined reversible CoIII/CoII couple was observable, preventing the accurate determination of E1/2 CoIII/CoII. b This value rapidly decayed upon consecutive scans. c See ref. 46. d Values determined in this study from Fig. S8 (ESI) and eqn (2). e E L values converted from corresponding Hammett parameters (σp) (ref. 50, R. Museo and O. Sciacovelli, J. Org. Chem., 1997, 62, 9031–9033 and C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195.). f E L value calculated based on the deprotonated phosphonic acid (hydrogen phosphonate anion) or protonated form of the amine substituent (ammonium cation). | |||||
| 1 | 3,5-Dimethylpyridine | −0.06 | −0.52 | −116b ± 4.3 | 0.21c |
| 2 | 4-Methoxypyridine | −0.07 | −0.54 | −105 ± 2.6 | 0.22d |
| 3 | 4-Methylpyridine | −0.07 | −0.54 | −103 ± 1.8 | 0.23c |
| 4 | Pyridine | −0.03 | −0.54 | −90.7 ± 1.1 | 0.25c |
| 5 | 4-Pyridinephosphonic acid | —a | −0.52 | −79.7 ± 3.3 | 0.27e,f |
| 6 | 4-Cyanopyridine | 0.14 | −0.52 | −50.5 ± 1.1 | 0.32c |
| 7 | 3,5-Dichloropyridine | 0.19 | −0.53 | −45.8 ± 4.4 | 0.33c |
| 8 | 3-Cyanopyridine | 0.15 | −0.53 | −39.4 ± 0.8 | 0.33d |
| 9 | 4-Aminopyridine | —a | −0.60 | −42.0 ± 0.8 | 0.32e,f |
| 10 | 4-Dimethylaminopyridine | —a | −0.61 | −36.9 ± 5.1 | 0.33e,f |
Other noteworthy ligands include the more withdrawing cyanopyridines (6, 8) and 3,5-dichloropyridine (7), which produce catalysts that function at lower overpotentials, albeit with relatively low catalytic currents (Table 1). The 4-pyridinephosphonic acid (5) substituted complex also shows an appreciable catalytic current.
As for the less effective catalysts, pyridines bearing donating 4-aminopyridine (9) and 4-dimethylaminopyridine (10) substituents show surprisingly low activity. This observation was attributed to the amines being protonated at pH 7 (pKa of 4-aminopyridine = 9.12 and dimethylaminopyridine = 9.6044) to form electron withdrawing ammonium-substituted pyridines. Compounds containing non-aromatic nitrogen atoms did not appear to bind to the cobaloxime and displayed nearly identical CVs to the [Co(dmgH)2(H2O)2] precursor upon addition (Fig. S6, ESI†).27 A similarly small catalytic influence was observed when adding 4-hydroxypyridine, which may be due to the formation of a 4-pyridone tautomer.45
An imidazole substituted catalyst showed high catalytic currents (Fig. S6, ESI†), but the electron-rich cobaloxime makes reduction of CoII more difficult,35 consequently producing a less attractive catalytic overpotential for this species. The cobaloxime substituted with a withdrawing pyrazine ligand similarly showed a notable catalytic current (Fig. S6, ESI†).
Structure–Activity Relationships (SARs) from EMoCS
The data from EMoCS showed clear trends that were used to elucidate SARs between axial pyridine and cobaloxime performance (Table 1). The same trends were also seen when using different equivalents of axial ligand.Lever's parameterisation approach was employed to rationalise the EMoCS data.46 This electrochemical parameterisation was originally developed to predict an Mn/Mn−1 redox potential by assuming that all electronic contributions from ligand to metal are additive (eqn (1)).
| E1/2(Mn/Mn−1)/V vs. NHE = SM·(ΣEL) + IM | (1) |
The slope, SM, and intercept, IM, are dependent on the metal, oxidation state, geometry of the complex, the spin state, stereochemistry and complex net charge in aqueous solution.46–49 The electrochemical ligand parameter, EL, reflects the net electron-donating character of the ligands.46 The unique advantage of using EL values for each ligand over other parameters, such as pKa values, is that they reflect both the σ and π interactions of a given metal–ligand bond. An EL value defines how these interactions will change the electronic properties of a complex. More electron donating ligands have more negative EL values, as the complex to which they are ligated has a more negative redox potential.
E L values for each axial ligand used in EMoCS were taken from the literature46 or converted from respective Hammett parameters50 (Table 1). A plot of ΣEL against the CoIII/CoII reduction potential then allowed analysis of the underlying electronic change introduced by each axial ligand to the catalyst (Fig. S8, ESI†). The observed correlation produced eqn (2), with SM and IM values similar to those found for RuIII/RuII (SM = 1.14 and IM = −0.35) in aqueous solution.46Eqn (2) can be employed to predict the CoIII/CoII couple of any related aqueous cobalt complex and was subsequently used to generate unreported EL values for 4-methoxypyridine and 3-cyanopyridine (Table 1).
| E1/2(CoIII/CoII)/V vs. NHE = 1.08·(ΣEL) − 0.60 | (2) |
E L values could not be used to predict the onset potential of the catalytic wave as it does not shift significantly between the most active cobaloximes (Fig. 4a and Table 1). This observation suggests that there is only a weak interaction between the low valent Co center and the pyridine at this stage of the mechanism (Fig. 5).35,51
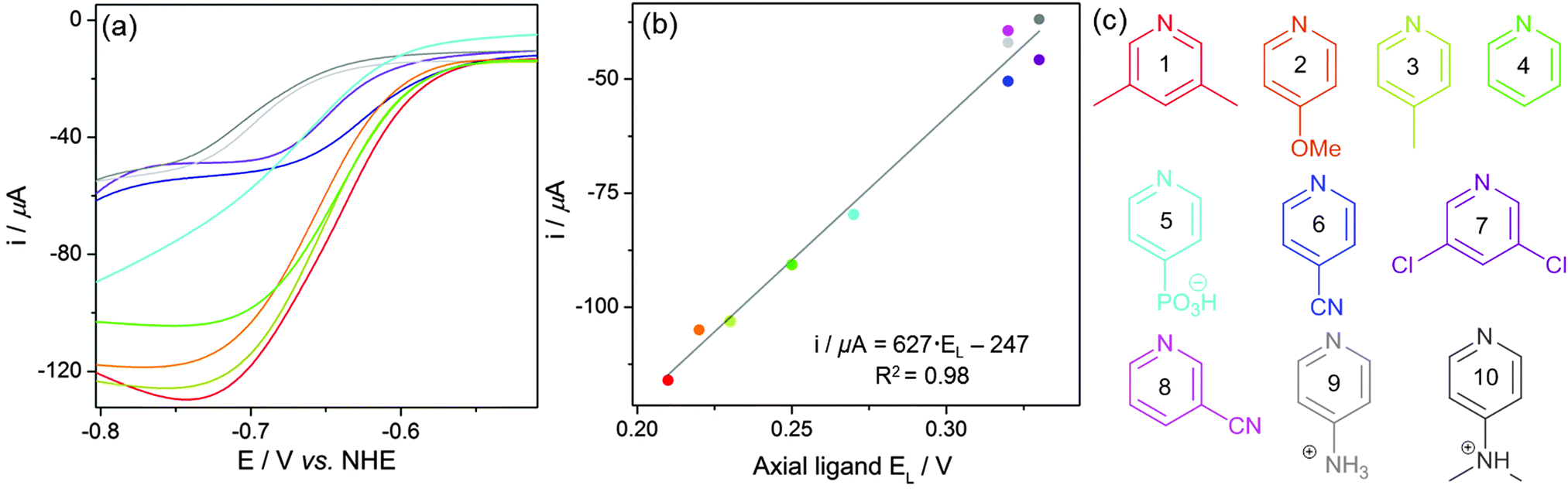 | ||
| Fig. 4 (a) Overlaid catalytic waves from CVs of pyridine-substituted cobaloximes shown in Fig. 3. (b) Correlation between the ligand parameter, EL, of each axial ligand with the baseline-corrected catalytic currents from Table 1. (c) Summary of the analysed pyridine ligands. | ||
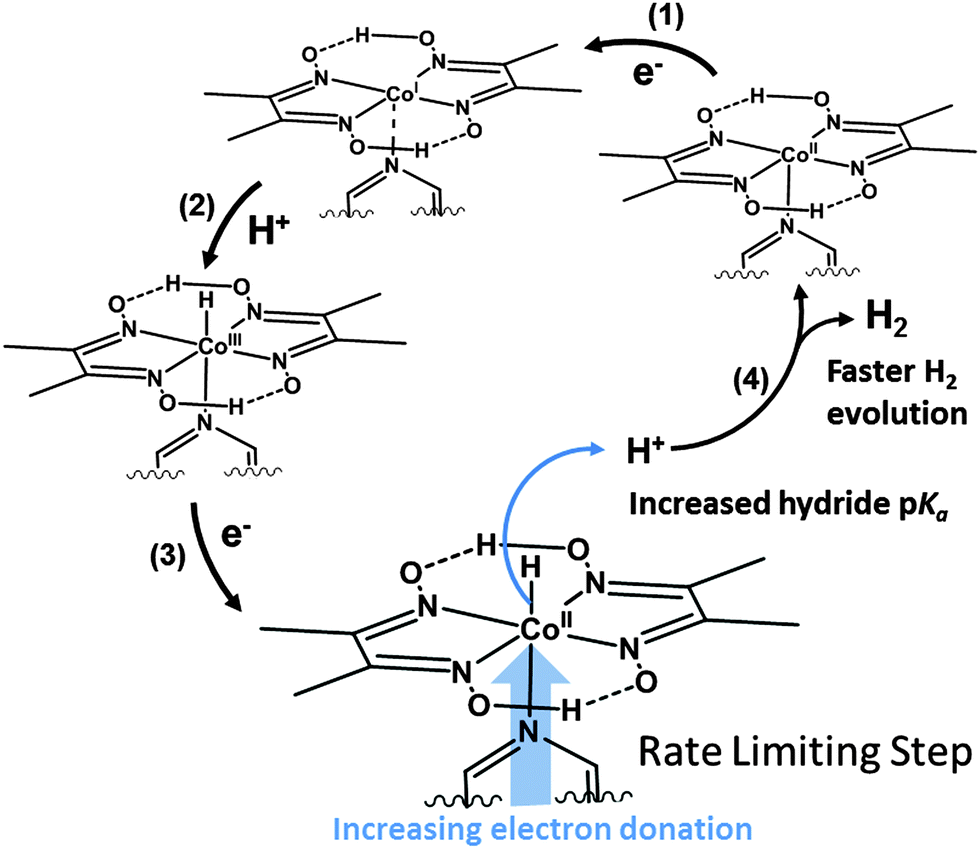 | ||
| Fig. 5 A mechanistic overview of hydrogen generation from cobaloximes illustrating the proposed effect of the axial ligand on the rate of proton reduction. | ||
Conversely, EL values do correlate well with the size of the catalytic wave, as more electron donating pyridines produce higher catalytic currents (Fig. 4). This trend is in line with previous mechanistic studies (Fig. 5).52 The reduction of CoII yields a highly nucleophilic CoI species that is quickly protonated to form CoIII–H. CoIII–H is subsequently reduced to a highly basic CoII–H, which is protonated to form H2. Protonation of a Co–H and release of H2 has been proposed to be rate limiting,53 therefore catalytic activity can be tuned by adjusting the basicity of this species. Adding more electron donating substituents to the cobaloxime yields a more basic CoII–H, explaining the linear trend in Fig. 4b.
Eqn (3) was extracted from the plot in Fig. 4b and provides an effective means to predict the activity of a substituted cobaloxime from the respective EL value, which will be a valuable tool for future cobaloxime design. The correlation also demonstrates that activities found in non-aqueous solvents are not directly applicable to an aqueous system. The 4-dimethylaminopyridine substituted Co catalyst was previously described as the most active cobaloxime in organic solvents with Et3NH+ as a proton source.35 However, protonation of the amine residue in water produces the opposite effect and forms an inactive cobaloxime.
| Catalytic current/μA = 627·EL − 247 | (3) |
Comparison to isolated cobaloximes
Cobaloximes bearing the more activating axial ligands were subsequently isolated to confirm the efficacy of EMoCS. This was achieved through synthesis of [CoIIICl(dmgH)2(4-methoxypyridine)] (A), [CoIIICl(dmgH)2(4-methylpyridine)] (B),54 [CoIIICl(dmgH)2(pyridine)] (C),55 and [CoIIICl(dmgH)2(4-pyridine-phosphonic acid)] (D).31 Catalyst A is a novel complex and has been characterised by 1H-NMR spectroscopy, mass spectrometry, elemental analysis and single crystal X-ray structure analysis (Fig. 6). B has been synthesised previously, but has not been studied with respect to proton reduction activity. The activity of C24,35 and D56 is known.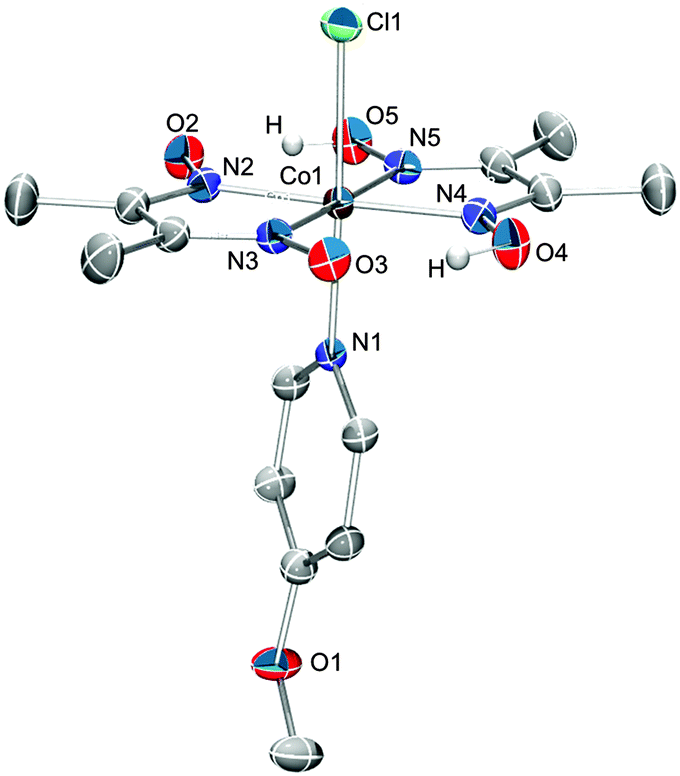 | ||
| Fig. 6 Single-crystal X-ray structure of compound A with atom ellipsoids of 50% probability showing the atom-labeling scheme for all non-C atoms. Solvent molecules and hydrogen atoms (except O–H) have been omitted for clarity. Monoclinic P21/n, a = 8.000(1), b = 13.5322(2), c = 19.2803(3) Å, β = 92.183(1)°. R1 = 3.87%, wR2 = 11.64%. More information can be found in Table S1 (ESI†). | ||
Electrochemical analysis was undertaken of each isolated complex. An additional advantage of the EMoCS procedure is that it produced water-soluble cobaloximes, whereas complexes A to C were isolated as complexes with a chlorido-ligand and required dissolution in organic solvents before adding water. All electrochemical experiments of isolated complexes consequently contain 10% acetone for solubility.
Linear sweep voltammograms (LSVs) of each cobaloxime (Fig. S9, ESI†) demonstrated the exact trend in catalytic current seen in Fig. 3, which fully authenticated the EMoCS procedure. Subsequent controlled potential electrolysis (CPE) experiments generated measurable quantities of H2, confirming that the electrochemical currents were a result of prolonged proton reduction. The electrocatalytic activity was quantified through CPE on a GC electrode held at −0.7 V vs. NHE in a degassed 1 mM solution of each complex. The H2 produced per hour was deduced via gas chromatography after electrolysis (Table 2). The amounts of H2 generated again agree with the EMoCS; compounds A and B produced more hydrogen per hour than the less active catalysts, C and D.
| Catalyst | H2 μmol h−1 in 0% O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
H2 μmol h−1 in air |
|---|---|---|
| a CPE performed at −0.7 V vs. NHE in 9 mL TEOA/Na2SO4 buffer (0.1 M each, pH 7) and 1 mL acetone. b Measured in 2% CH4 in N2. c Percentage in brackets shows Faradaic efficiency. | ||
| A | 0.92 ± 0.05 (77 ± 2%)c | 0.89 ± 0.07 (65 ± 6%)c |
| B | 0.84 ± 0.06 (75 ± 7%)c | 0.69 ± 0.05 (64 ± 10%)c |
| C | 0.78 ± 0.09 (68 ± 4%)c | 0.76 ± 0.03 (59 ± 6%)c |
| D | 0.65 ± 0.04 (62 ± 4%)c | 0.62 ± 0.02 (43 ± 2%)c |
CPE was also performed on a Hg-pool electrode for compounds A to D to confirm that a molecular catalyst is responsible for H2 formation. Hg forms amalgams with metallic deposits, thereby preventing the formation of a heterogeneous electroactive deposit on the electrode.57 Hg-pool electrode CPE at −0.7 V vs. NHE yielded at least 10 turnovers from each cobaloxime in the bulk solution, confirming that hydrogen evolution was a result of homogenous cobaloxime catalysis.
Proton reduction activity in the presence of O2
Oxygen inhibition has been identified as a key obstacle in producing viable molecular proton reduction catalysts.3,58 Compound D has recently shown remarkable stability in the presence of O2 and retains the majority of its electrocatalytic activity in pH 7 electrolyte solution,12 a feature that sets cobaloximes apart from other catalytic molecules.CPE was consequently repeated with each catalyst in air. The H2 evolution activity of A–D decreased by typically less than 5% in each case compared to anaerobic conditions (Table 2). The tolerance of cobaloximes towards O2 is outstanding considering the potential competition between proton and oxygen reduction at a low valent Co site,59 which suggests that competing oxygen reduction at the cobaloxime is low under the employed conditions.
On the other hand, the Faradaic efficiency (FE) undergoes a noticeable decrease between inert and aerobic atmospheres (Table 2). The observed decrease is a result of oxygen reduction at the GC electrode, which competes for electrons with the cobaloxime. It appears that the limiting factor in the efficiency of cobaloximes under aerobic conditions is therefore not the catalyst, but the electrode material.
The general decrease in FE from cobaloxime A to D (Table 2) in the presence of air was assigned to their respective activities. The rate of underlying oxygen reduction by the GC remains constant in all experiments, consequently using a complex that operates at a faster rate can lessen the relative loss of the FE. The only other FE reported in air was 52% by a Co corrole catalyst60 and as such, at 65% compound A sets a new benchmark for a discrete synthetic catalyst under this demanding environment.
We also tested the most active catalyst, compound A, (0.2 μmol) for photocatalytic H2 generation in a homogenous, noble-metal free system with an organic dye, Eosin Y (0.1 μmol), and triethanolamine (4.49 mL, pH 7, 0.1 M) as a hole scavenger.12,61,62 Visible light irradiation (λ > 420 nm, 100 mW cm−2, AM 1.5 G) of the solution in a photoreactor (3.4 mL headspace) resulted in the formation of 12.3 ± 0.6 (TONCo = 62 ± 3) and 3.9 ± 0.4 (TONCo = 19 ± 2) μmol H2 after two hours of irradiation at 25 °C under anaerobic and aerobic conditions, respectively (Fig. 7). EMoCS therefore produced a complex that acts as a very active proton reduction catalyst in both electrocatalytic and photocatalytic systems under both anaerobic and aerobic atmospheres.
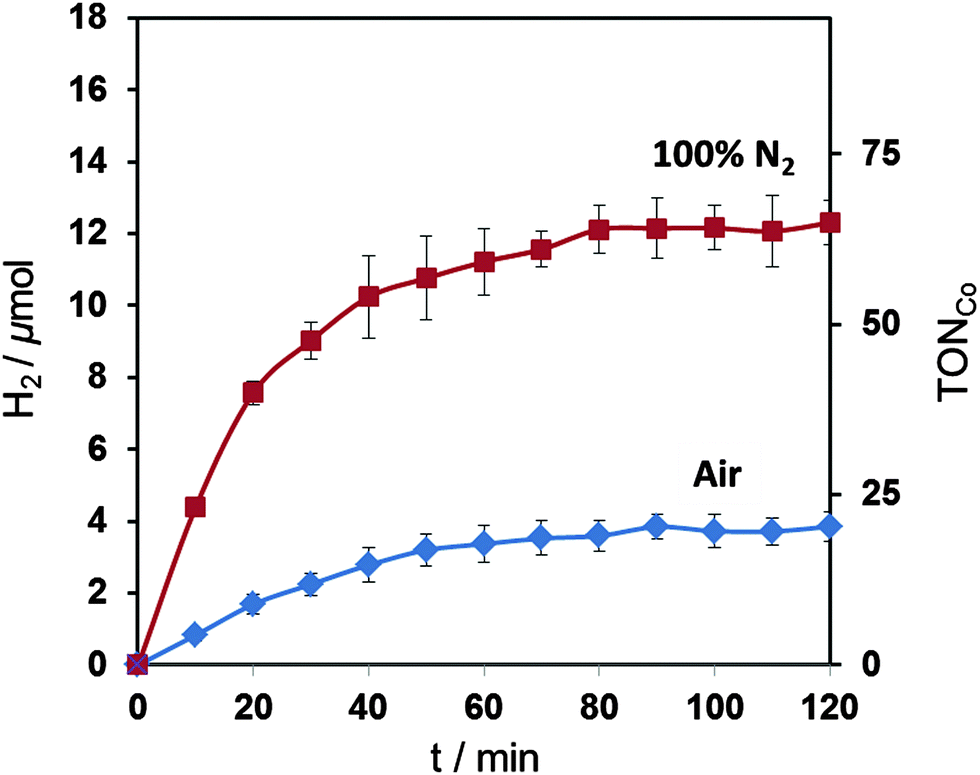 | ||
| Fig. 7 Visible light driven (λ > 420 nm, 100 mW cm−2, AM 1.5 G) H2 production by cobaloxime A (0.2 μmol) with Eosin Y (0.1 μmol) in TEOA (4.49 mL, pH 7, 0.1 M) and acetone (10 μL). | ||
Conclusions
EMoCS is a simple and fast approach to molecular catalyst design that can develop accurate SARs and identify more active species. Data from over 20 potential ligands were collected from the electrochemical screening within hours and provided a full analysis of the axial ligand's influence on cobaloxime activity in pH neutral aqueous solution. Undertaking a study of this magnitude by isolating and analysing each individual catalyst in the solid state would take considerably more time.63Utilisation of electrochemical parameterisation to differentiate the ligands based on net electron donating character provided a rational analysis of the EMoCS data. A more electron donating ligand increases the rate of proton reduction catalysis due to the formation of a more basic Co–H species. Importantly, this parameterisation allows activity to be predicted in the future.
EMoCS led to the fast identification of a novel catalyst A, which performs better than previously known cobaloximes. Cobaloxime A was isolated and fully characterised and was found to operate electrocatalytically under aerobic conditions with negligible decrease in proton reduction activity compared to an inert atmosphere. The same catalyst also functioned with high efficiency in a homogeneous, noble metal free, photocatalytic scheme.
EMoCS is particularly attractive for labile 3d transition metal ions, such as Co, Ni and Fe, and automating this technique will allow high-throughput combinatorial screening to be undertaken across a library of ligands. The fundamentals of EMoCS are applicable to any molecular electrocatalytic process and it is therefore foreseeable that other attractive catalytic reactions, such as oxygen reduction, CO2 reduction or water oxidation, will benefit from this technique in the near future.
Experimental
Reagents
All chemical reagents were obtained from commercial suppliers at the highest available purity for analytical measurements. All solvents were HPLC grade and Millipore water was used in all electrochemical analysis.Methods and instrumentation
1H NMR spectra were recorded on a Bruker 400 MHz spectrometer at room temperature. Electro-spray ionisation mass spectrometry (ESI-MS) was carried out on a Quattro LC electrospray mass spectrometer in H2O (assembled cobaloximes) or MeOH (isolated complex A). UV-visible spectroscopy was undertaken on a Varian Cary 50 UV-vis spectrometer using a quartz cuvette. Elemental analysis was carried out by the University of Cambridge microanalysis service.Assembly of cobaloximes for EMoCS
[Co(H2O)6]Cl2 (238 mg, 2 mmol) and dmgH2 (232 mg, 4 mmol) were added to a solution of Pi (500 mL, 0.1 M, pH 7) and sonicated at 40 °C for 30 min. The resultant dark brown solution was neutralised to pH 7 with aqueous NaOH and [Co(dmgH)2(H2O)2] was characterised by UV-vis spectroscopy and mass spectrometry (Fig. S1 and S2, ESI†).To assemble each cobaloxime, [Co(dmgH)2(H2O)2] (5 mL of 2 mM) in Pi (0.1 M, pH 7) was pipetted into a sample vial and 40 μL of a 1 M stock solution of an axial ligand was injected into the solution.
Synthesis of [CoCl(dmgH)2(4-methoxypyridine)] (A)
[CoCl2(dmgH)(dmgH2)] (0.50 g, 1.4 mmol), prepared as described previously,55 was dissolved in CHCl3 (25 mL) with 4-methoxypyridine (0.7 mL, 6.9 mmol) and stirred for 10 min. H2O (10 mL) was added and the resultant solution was stirred vigorously for one hour. The CHCl3 layer was washed three times with water and concentrated in vacuo to approximately 5 mL. Precipitation of the crude product occurred upon addition of EtOH. Brown crystals of [CoCl(dmgH)2(4-methoxypyridine)] (A, 0.39 g, 49%) were obtained after recrystallisation from hot acetone. 1H NMR (400 MHz, CDCl3, Me4Si) δ (ppm): 2.43 (12H, s, Me), 3.84 (3H, s, OMe), 6.73 (2H, d, J 7 Hz, 2H Pyr-m), 8.04 (2H, d, J 7 Hz, Pyr-o). ESI-MS: m/z 433.9 ([M + H]+, 13%), 397.9 (21), 310.8 (10), 288.9 (100). Elemental analysis found C 41.35; H 5.47; N 14.67. Calc. for CoC14H19N5O5Cl·C3H6O: C 41.52; H 5.53; N 14.24.Single X-ray data collection and structure solution refinement for cobaloxime A
Single crystals suitable for X-ray diffraction studies were grown by slow vapour diffusion of diethyl ether into a saturated solution of A in CHCl3. X-ray crystallography was carried out with Mo Kα radiation (λ = 0.71073 Å) and a Nonius Kappa CCD diffractometer fitted with an Oxford Cryosystems Cryostream cooling apparatus. The single crystal was mounted on the tip of a glass fibre and the data was collected under a stream of N2 at 180 K and refined against all reflections. The weighted R-factor wR and goodness of fit (GOF) are based on F2. The structure of A contains a disordered diethyl ether solvent molecule at a special position, which was assigned with bond length constraints and a common isotropic displacement parameter for the C and O atoms. The glyoximato –OH hydrogen atoms were located in the difference Fourier map.Electrochemical measurements
Electrochemical measurements were carried out on an Ivium CompactStat potentiostat in a three-electrode cell using a 3 mm diameter GC disk working electrode (IJ Cambria Scientific Ltd), Pt-mesh counter electrode and Ag/AgCl (KCl sat.) reference electrode (BASi). Solutions were purged with N2 and measurements were taken at room temperature in buffered electrolyte solutions of triethanolamine (TEOA) containing Na2SO4 (0.1 M each, pH 7) or aqueous Pi buffer (0.1 M, pH 7). Potentials were converted to NHE according to the relationship E(Ag/AgCl (KCl sat.)) + 0.197 V = E(NHE).64E 1/2 of CoIII/CoII was taken as the potential at the midpoint of the reduction wave. Standard deviation was calculated using data from at least three fresh solutions. Catalytic current was taken as the difference between the peak and baseline of each catalytic wave.
Controlled Potential Electrolysis (CPE)
CPE was performed with a GC disk (0.07 cm2; 10 mL electrolyte solution) or a Hg-pool (∼1 cm2; 1 mL solution) working electrode in a tailor-made, air-tight CPE cell containing the cobaloxime (1 mM) in aqueous TEOA–Na2SO4 (0.1 M each, pH 7) and acetone in 9:1 volume ratio. Hydrogen was quantified by gas chromatography and the Faradaic efficiency was calculated according to eqn (4):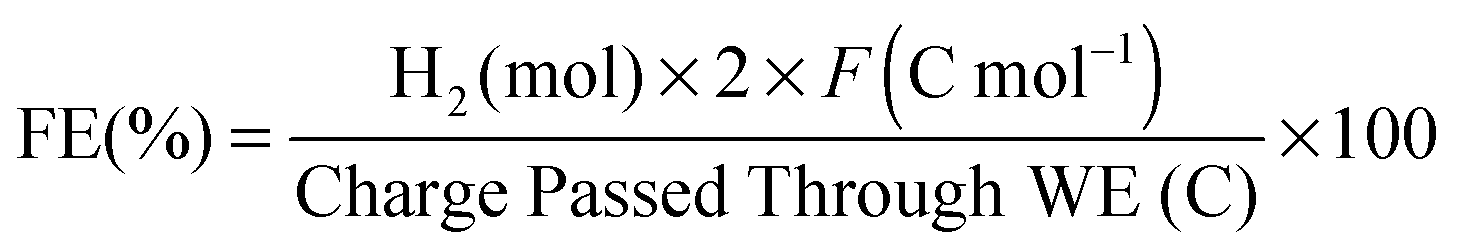 | (4) |
Gas chromatography analysis
Gas chromatography was carried out on an Agilent 7890A gas chromatograph with a 5 Å molecular sieve column at 45 °C and N2 carrier gas with a flow rate of approximately 3 mL min−1. Methane (2% CH4 in N2) was used as internal or external standard.Photocatalytic generation of H2
Cobaloxime A (0.2 μmol in 0.1 mL) and Eosin Y (0.1 μmol in 0.1 mL) were added to an aqueous TEOA solution (4.3 mL, 0.1 M, pH 7) and purged with N2 or left under air. The photocatalytic system was sealed with a rubber septum, thermostated with a water circulator at 25 °C and irradiated by a solar light simulator (Newport Oriel, 150 W, 100 mW cm−2) equipped with an air mass (AM) 1.5 global filter. IR irradiation was filtered with a water filter and UV irradiation with a 420 nm cut-off filter (UQG Optics). H2 was quantified every ten minutes by analysing 20 μL of headspace gas by gas chromatography until catalytic activity had ceased. The O2 level remained constant in the headspace during photocatalytic experiments.Acknowledgements
This work was supported by the EPSRC (EP/H00338X/2 to E.R. and a DTA studentship for D.W.W.), Christian Doppler Research Association and the OMV Group. We thank Dr John E. Davies for acquiring the crystallographic data. Further thanks are extended to Fezile Lakadamyali for providing samples of compounds C and D and Dr Moritz Kuehnel for fruitful discussions.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS PubMed.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- W. T. Eckenhoff, W. R. McNamara, P. Du and R. Eisenberg, Biochim. Biophys. Acta, 2013, 1827, 958–973 CrossRef CAS PubMed.
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268–9274 RSC.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 CAS.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. Rakowski DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- N. P. Dasgupta, C. Liu, S. Andrews, F. B. Prinz and P. Yang, J. Am. Chem. Soc., 2013, 135, 12932–12935 CrossRef CAS PubMed.
- D. Wang, T. Hisatomi, T. Takata, C. Pan, M. Katayama, J. Kubota and K. Domen, Angew. Chem., Int. Ed., 2013, 52, 11252–11256 CrossRef CAS PubMed.
- E. Reisner, Eur. J. Inorg. Chem., 2011, 1005–1016 CrossRef CAS.
- H. Ye, H. S. Park and A. J. Bard, J. Phys. Chem. C, 2011, 115, 12464–12470 CAS.
- P. N. Anunson, G. R. Winkler, J. R. Winkler, B. A. Parkinson and J. D. Schuttlefield Christus, J. Chem. Educ., 2013, 90, 1333–1340 CrossRef CAS.
- N. Na, S. Zhang, X. Wang and X. Zhang, Anal. Chem., 2009, 81, 2092–2097 CrossRef CAS PubMed.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res., 2011, 25, 3–16 CrossRef.
- K. C. Leonard and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 15885–15889 CrossRef CAS PubMed.
- E. Danielson, J. H. Golden, E. W. McFarland, C. M. Reaves, W. H. Weinberg and X. D. Wu, Nature, 1997, 389, 944–948 CrossRef CAS PubMed.
- C. Hoffmann, A. Wolf and F. Schüth, Angew. Chem., Int. Ed., 1999, 38, 2800–2803 CrossRef CAS.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952–4962 CrossRef CAS PubMed.
- T. M. McCormick, Z. Han, D. J. Weinberg, W. W. Brennessel, P. L. Holland and R. Eisenberg, Inorg. Chem., 2011, 50, 10660–10666 CrossRef CAS PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- G. N. Schrauzer, Angew. Chem., Int. Ed., 1976, 15, 417–426 CrossRef CAS PubMed.
- C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817–1824 CrossRef CAS PubMed.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS PubMed.
- C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS PubMed.
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695–1697 RSC.
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed.
- L. A. Berben and J. C. Peters, Chem. Commun., 2010, 46, 398–400 RSC.
- B. H. Solis and S. Hammes-Schiffer, J. Am. Chem. Soc., 2011, 133, 19036–19039 CrossRef CAS PubMed.
- M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS PubMed.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2011, 50, 11252–11262 CrossRef CAS PubMed.
- L. I. Simándi, É. Budó-Záhonyi and Z. Szeverényi, Inorg. Nucl. Chem. Lett., 1976, 12, 237–241 CrossRef.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- W. Zhang, J. Hong, J. Zheng, Z. Huang, J. Zhou and R. Xu, J. Am. Chem. Soc., 2011, 133, 20680–20683 CrossRef CAS PubMed.
- Z.-J. Li, X.-B. Li, J.-J. Wang, S. Yu, C.-B. Li, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2013, 6, 465–469 CAS.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed.
- Z. Chen and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 700–703 CrossRef CAS PubMed.
- L. Chen, M. Wang, K. Han, P. Zhang, F. Gloaguen and L. Sun, Energy Environ. Sci., 2014, 7, 329–334 CAS.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- C. Chen, M. Wu, A. Yeh and T. Y. R. Tsai, Inorg. Chim. Acta, 1998, 267, 81–86 CrossRef CAS.
- A. B. P. Lever, Inorg. Chem., 1990, 29, 1271–1285 CrossRef CAS.
- A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2007, 1473–1482 CrossRef CAS.
- E. Reisner, V. B. Arion, A. Eichinger, N. Kandler, G. Giester, A. J. L. Pombeiro and B. K. Keppler, Inorg. Chem., 2005, 44, 6704–6716 CrossRef CAS PubMed.
- E. Reisner, V. B. Arion, B. K. Keppler and A. J. L. Pombeiro, Inorg. Chim. Acta, 2008, 361, 1569–1583 CrossRef CAS PubMed.
- H. Masui and A. B. P. Lever, Inorg. Chem., 1993, 32, 2199–2201 CrossRef CAS.
- K. A. Lance, K. A. Goldsby and D. H. Busch, Inorg. Chem., 1990, 29, 4537–4544 CrossRef CAS.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 16774–16776 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 1060–1065 CrossRef CAS PubMed.
- V. R. Vijayaraghavan and M. Santappa, J. Inorg. Nucl. Chem., 1974, 36, 163–168 CrossRef CAS.
- W. C. Trogler, R. C. Stewart, L. A. Epps and L. G. Marzilli, Inorg. Chem., 1974, 13, 1564–1570 CrossRef CAS.
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem.–Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- S. Losse, J. G. Vos and S. Rau, Coord. Chem. Rev., 2010, 254, 2492–2504 CrossRef CAS PubMed.
- M. Shamsipur, A. Salimi, H. Haddadzadeh and M. F. Mousavi, J. Electroanal. Chem., 2001, 517, 37–44 CrossRef CAS.
- B. Mondal, K. Sengupta, A. Rana, A. Mahammed, M. Botoshansky, S. G. Dey, Z. Gross and A. Dey, Inorg. Chem., 2013, 52, 3381–3387 CrossRef CAS PubMed.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS PubMed.
- Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659–14669 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 974386. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cp00453a |
| This journal is © the Owner Societies 2014 |