 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular simulation of gas adsorption and diffusion in a breathing MOF using a rigid force field†
E.
García-Pérez
*a,
P.
Serra-Crespo
a,
S.
Hamad
b,
F.
Kapteijn
a and
J.
Gascon
a
aCatalysis Engineering-Chemical Engineering Department, Delft University of Technology, Julianalaan, 136, 2628 BL Delft, The Netherlands. E-mail: e.garciaperez81@gmail.com; Fax: +31 152785006; Tel: +31 152786976
bDepartment of Chemical, Physical, and Natural System, University Pablo de Olavide, Ctra. Utrera km. 1, 41013, Seville, Spain
First published on 11th June 2014
Abstract
Simulation of gas adsorption in flexible porous materials is still limited by the slow progress in the development of flexible force fields. Moreover, the high computational cost of such flexible force fields may be a drawback even when they are fully developed. In this work, molecular simulations of gas adsorption and diffusion of carbon dioxide and methane in NH2-MIL-53(Al) are carried out using a linear combination of two crystallographic structures with rigid force fields. Once the interactions of carbon dioxide molecules and the bridging hydroxyls groups of the framework are optimized, an excellent match is found for simulations and experimental data for the adsorption of methane and carbon dioxide, including the stepwise uptake due to the breathing effect. In addition, diffusivities of pure components are calculated. The pore expansion by the breathing effect influences the self-diffusion mechanism and much higher diffusivities are observed at relatively high adsorbate loadings. This work demonstrates that using a rigid force field combined with a minimum number of experiments, reproduces adsorption and simulates diffusion of carbon dioxide and methane in the flexible metal–organic framework NH2-MIL-53(Al).
A Introduction
Metal–organic frameworks (MOFs) consist of metal or metal clusters (“connectors”) linked by organic ligands (“linkers”)1–6 forming nanoporous crystalline solid materials. The ligands act as spacers, creating an open porous structure with very high pore volume and surface area. Due to their unusual variety in terms of chemical composition, accessibility and pore dimensions as well as to their low densities (0.2–1 g cm−3) and high surface areas (500–4500 m2 g−1), MOFs are considered promising candidates in applications involving adsorption and separation of strategic gases such as carbon dioxide, methane, and hydrogen.3,4,6–10The first report on gas adsorption using MOFs was published in 199711 and since then, MOFs have become an active field of research, resulting in numerous publications.12–16 In case of carbon dioxide adsorption, several approaches have been followed to improve selectivity: adsorbate–surface interactions, flexibility/gate opening mechanisms, and cooperative effects14 (i.e. flexibility and specific interactions) have been reported as very effective ways of improving selectivity. In view of the high selectivities, relatively low enthalpies of adsorption, and fast kinetics, MOFs are among the most promising solids for carbon dioxide capture and separation.
Several authors have applied molecular simulations to study the adsorption and separation of carbon dioxide in metal–organic frameworks, i.e. Wang et al.,17 Babarao et al.,18 Keskin and Sholl,19 Walton et al.,20 Yang and Zhong,21–23 Calero et al.,24 Chen and Jiang,25 Liu et al.,26 and Wells and Chaffee.27 Also combined experiment and modeling techniques are used in work by Pera-Titus et al.,28 Lescouet et al.,29 Serra-Crespo et al.,30 Chen et al.,25 Stavitski et al.,31 Boutin et al.,32 and Montoro et al.33 on functionalized MOFs. Although the origin of the improved separation performance in functionalized MOFs is not yet fully understood, it is clear from the above mentioned works that a delicate interplay of weak dispersion forces with framework polarity plays an important role.
A special class of MOFs is formed by those whose pore dimensions may change without breaking chemical bonds within the framework. This results in special properties like the breathing effect8,34 and the gate phenomenon35,36 where pores contract or open during molecule adsorption. An example of a breathing type material is the MIL-53 series. MIL-53 is built from MO6 octahedra (where M can be three-valent ions Fe3+, Cr3+, Al3+, Ga3+, In3+ or Sc3+) formed from trans bridging OH ions and the oxygens of coordinate, bridging 1,4-benzene-dicarboxylate linkers.34 In this way a crystalline material with 1-D diamond shaped pores is formed. Amino-MIL-53(Al)37 is a material with the topology of MIL-53. During the synthesis of amino-MIL-53(Al), 2-amino terephthalic acid is used as the linker molecule instead of terephthalic acid. The isoreticular material obtained has the following formula: Al(OH)[O2C–C6H3NH2–CO2].
During the last few years, we have extensively worked on NH2-MIL-53(Al), demonstrating the high potential of this functionalized MOF in carbon dioxide separation and its thrilling optical properties.16,31,32,38–41 Both, the high carbon dioxide selectivity and the optical switch behavior of this structure are due to its unique breathing behavior. In the presence of carbon dioxide, two different pore configurations were found. The narrow-pore (np) has a smaller cell and pore volume and it is the dominant phase at lower pressures. Once certain pressure is reached, there is a pore expansion to the large-pore configuration (lp).16 The chemical composition remains the same, and these configurations only differ in their pore dimensions.
The practical implementation of transition between phases has proved challenging from a simulation point of view. The development of specific force fields for every different structure and the intensive computational efforts needed to run simulations using flexible force fields certainly play against the goal of simulations taking over experimental efforts for predictive purposes.42–45 In this work, we demonstrate that by using a rigid force field and with a minimum number of experiments, it is possible to reproduce adsorption and to simulate diffusion of carbon dioxide and methane in the flexible metal–organic framework NH2-MIL-53(Al).
The remainder of this paper is structured as follows. In Section B, the details of the simulation methodology and experiments are presented, including descriptions of the simulation techniques, force fields, and models used for the molecules and adsorbents. In Section C, the computed adsorption isotherms for carbon dioxide, Henry coefficients, and enthalpies of adsorption at zero coverage of both carbon dioxide and methane, the adsorption of the binary CO2:CH4 mixture, and the diffusion of carbon dioxide molecules inside the pores are analyzed. Finally we give some concluding remarks.
B Simulation and experimental section
Atomic charges are used for all the atoms in the structure. In order to calculate the atomic charges for the narrow and large-pore structures of NH2-MIL-53(Al), we used the Mulliken charge partitioning scheme46 from all electron GGA DFT calculations employing the PW91 functional47 and a double numerical plus polarisation (dnp) basis set.48 The calculations were carried out with the DMol3 code.49 We did not use the cluster approach to calculate the charges, because breaking the bonds can lead to spurious effects. We took into account the whole unit cell in the process of calculation of the charges. Ramsahye et al.50 showed that the charges obtained with this method are less dependent on the geometry than charges obtained with electrostatic potential fitting methods of cluster models of the periodic structures, and they employed these charges to model successfully the adsorption of various molecules in MIL-47 and MIL-53.50–52Guest molecules are considered as small rigid molecules. Carbon dioxide is modeled as a linear molecule with a C–O bond-length of 1.149 Å, and partial charges are distributed along each molecule to reproduce the experimental quadrupole moment.53,54 Methane molecules are described with a united atom model, in which each molecule is treated as a single interaction centre.55 The energy of each gas atomic interactions is described by Lennard-Jones and Coulombic potentials. Lorentz–Berthelot mixing rules were used to calculate mixed Lennard-Jones parameters.
Monte Carlo (MC) simulations were performed to study the adsorption of carbon dioxide and methane in the Grand Canonical ensemble. To compute the Henry coefficients and enthalpies of adsorption at zero coverage of the studied molecules simulations were performed in the NVT ensemble using the narrow-pore structure. Simulations were performed using the in-house RASPA code.56 The simulations were performed in cycles, and in each cycle, a MC move was chosen at random with a fixed probability: translation, regrowth, rotation, and insertion/deletion of molecules (in the Grand Canonical ensemble). We used at least 107 cycles and charge interactions were computed using Ewald sums with a relative precision of 10−6. Absolute adsorption was converted to excess adsorption57 for comparison with experimental values.
Molecular dynamics (MD) simulations were carried out to compute the self-diffusivities of the carbon dioxide molecules inside the pores. The self-diffusivities were obtained by calculating the slope of the mean-square displacement at long times.58 In these simulations the velocity-Verlet algorithm was used to integrate Newton's law of motion. MD simulations of 107 cycles in the NVT ensemble (Nóse–Hoover chain thermostat) were performed using a time step of 0.5 fs. 104 equilibration cycles were used and we took the initial positions of the molecules from previous MC simulations. The Lennard-Jones potential is cut and shifted with the cutoff distance set to 12 Å. NH2-MIL-53(Al) structures are modeled as rigid structures with Lennard-Jones parameters taken from the universal DREIDING force field.59 Simulations were performed at 257, 263, 273, and 298 K using 32 unit cells (a = 19.7939 Å, b = 7.8496 Å, c = 6.5934 Å, α = γ = 90°, β = 73.5673°) for the narrow-pore structure and 16 unit cells for the large-pore structure (a = 17.4426 Å, b = 12.0429 Å, c = 6.8447 Å, α = γ = 90°, β = 89.99°). The crystal structures previously reported by our group16 were used for the simulations. The pore volumes of the structures during the simulations are 0.07 cc g−1 and 0.52 cc g−1, for the np and lp forms respectively.
Adsorption isotherms of carbon dioxide (purity of 99.995%) and methane (purity of 99.95%) were determined using the volumetric technique with an apparatus from BEL Japan (Belsorp HP). Around 0.5 g of NH2-MIL-53(Al) was placed in the sample container. Before every measurement, the adsorbent was pre-treated by increasing the temperature to 473 K at a rate of 10 K min−1 under vacuum and maintaining the temperature for two hours. The measurements for carbon dioxide and methane adsorption were carried out at 257, 263, 273, and 298 K.
C Results
The experimental adsorption isotherm for carbon dioxide in NH2-MIL-53(Al) at 257 K was compared with the adsorption isotherms obtained from simulation using both narrow-pore and large-pore crystallographic structures. The experimental isotherm shows a pronounced step, attributed to the transition in the framework structure due to the breathing mechanism. Carbon dioxide adsorption isotherms display the typical s-shape of breathing materials like regular MIL-53. The first plateau is reached at 3 mol kg−1 and pressures below 700 kPa. At higher pressures a drastic increase in the amount adsorbed occurred, results in a saturation loading of 10 mol kg−1. The same behavior is found over a wide range of temperatures. The experimental saturation loading (∼10 mol kg−1) is reproduced using simulations for the large pore structure. However, experimental adsorption in the low pressure range is underestimated. In order to address this issue, we increased the cell volume of the np configuration16 by a 0.65% (increase of the cell volume of 0.65%), resulting in a slight increase in loading in the simulated adsorption isotherm.In order to obtain a better match between experimental and simulated data in the np configuration, the framework–adsorbate interactions were optimized using a universal force field. Although universal force fields are usually expected to provide realistic predictions of molecular structures, when dealing with complex adsorbents like MOFs, these generic force fields usually deliver poor results. Previous studies pointed at a strong interaction between carbon dioxide molecules and the framework hydroxyls.60 Using experimental data, we could quantify the extent of this interaction by direct fitting of the data on the isotherm (Fig. 1(a)). CCO2–HOH Lennard-Jones parameters were obtained by calibrating the force field through explicitly fitting one point (10 kPa) of the experimental isotherm at 257 K. After calculating the appropriate Lennard-Jones interaction at this single temperature, the full isotherm could be reproduced and predicted over a wide range of temperatures (see Fig. 1 (b) 263 K, (c) 273 K, and (d) 298 K) using the np structure, resulting in an excellent agreement with experimental data (full green symbols).
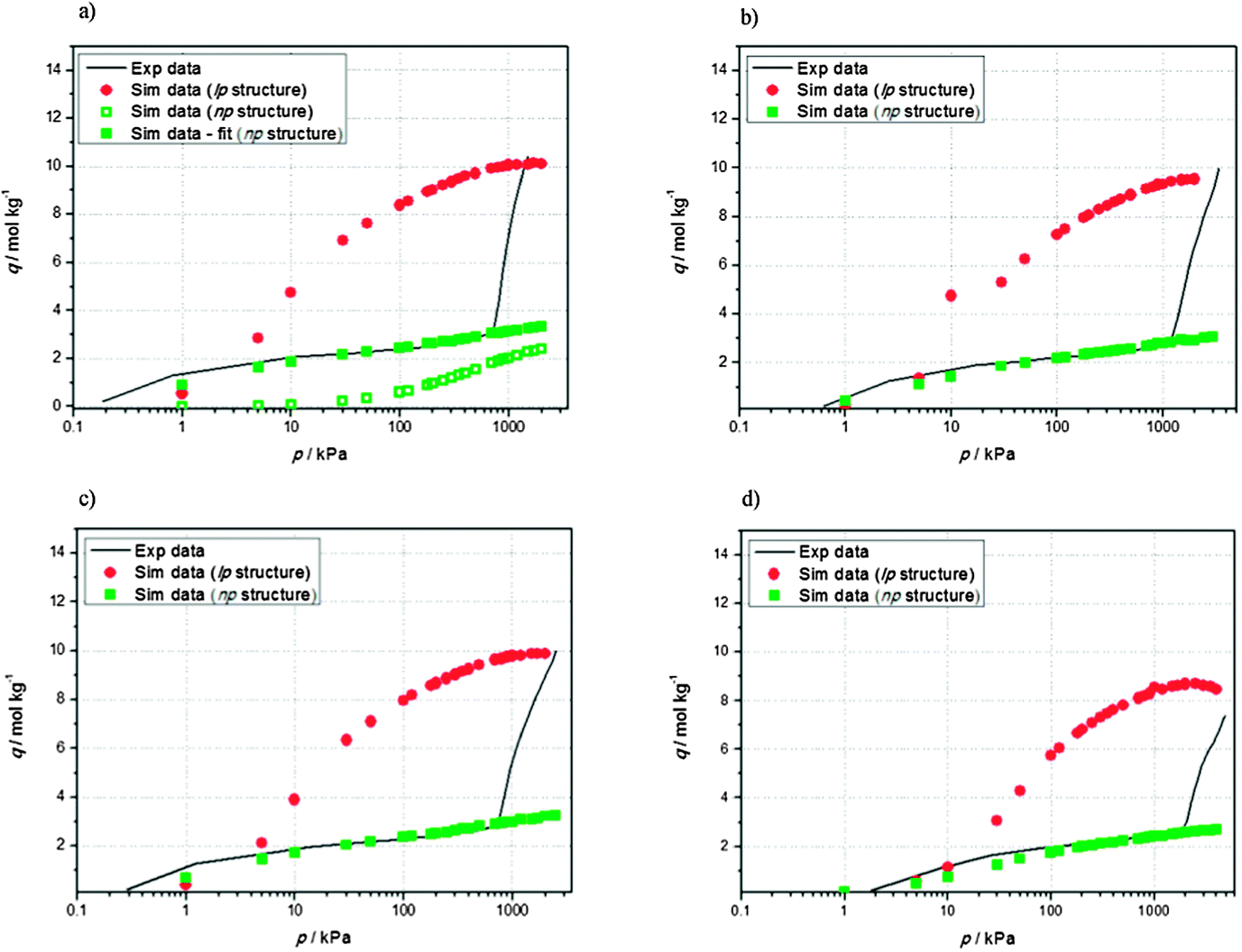 | ||
| Fig. 1 Comparison of simulated and experimental carbon dioxide adsorption isotherms in NH2-MIL-53(Al) at (a) 257 K, (b) 263 K, (c) 263 K, and (d) 298 K. Experimental isotherm is represented by a black line and simulation results are represented by open symbols (adsorption isotherm in the np structure before the fitting) and full symbols (green and red symbols for the np and lp structures, respectively). | ||
Simulated zero-coverage adsorption enthalpies and Henry coefficients for carbon dioxide and methane are compared with previous experimental data39 in Table 1. Simulated adsorption values are very close to the experimental data. Results for carbon dioxide confirm a strong interaction between the molecule and the structure. In case of methane, a much larger difference between experimental and simulated KH is evident. We attribute this difference to the large error in the calculation of the experimental value due to the very low affinity of the framework for methane.
| ΔH (kJ mol−1) | K H (mol kg−1 Pa−1) | |||
|---|---|---|---|---|
| Exp. data | Sim. | Exp. Data | Sim. | |
| Carbon dioxide | 38.4 | 37.8 | 1.16 × 10−4 | 1.20 × 10−4 |
| Methane | <20 | 5.5 | 1.4 × 10−6 | 1.98 × 10−11 |
The main goal of this work is to capture the np → lp transition in NH2-MIL-53(Al) at a low computational cost. In Fig. 2, experimental adsorption isotherms of carbon dioxide at different temperatures are shown as a function of the relative pressures (p/p0). The adsorption isotherms present a similar behavior. In the studied temperature range, the np → lp phase transition occurs at the same relative pressure of about p/p0 = 0.3, where p0 is the pressure of the vapour in equilibrium with its non-vapour phases. This fact clearly infers that the phase transition is related to the chemical potential of the adsorbate. Based on our previous crystallographic results,16 a first estimation could be made of the relative population of the np and lp phases during framework expansion. Based on that estimation of the relative population of each phase, a linear combination of the simulated isotherms using the rigid structures as function of p/p0 was constructed. An excellent match between the isotherm obtained by the linear combination of the rigid simulated isotherms and the experimental data was obtained (Fig. 3(a), blue asterisks). By extrapolating the obtained results to higher temperatures, the transition between the two crystallographic phases of NH2-MIL-53(Al) can be well reproduced at different temperatures from only one experimental isotherm and a linear combination of the percentages of each coexisting structure at a given p/p0. Following this approach, both the experimental and the computational costs are minimized, and flexible force fields are not needed to capture the breathing effects due to the interaction with guest molecules. Fig. 3 shows the experimental adsorption isotherms (black line) at (b) 263 K, (c) 273 K, and (d) 298 K compared with the results obtained via linear combination of the two coexisting structures (blue asterisks). For the whole temperature range studied, agreement between simulation and experimental data is excellent.
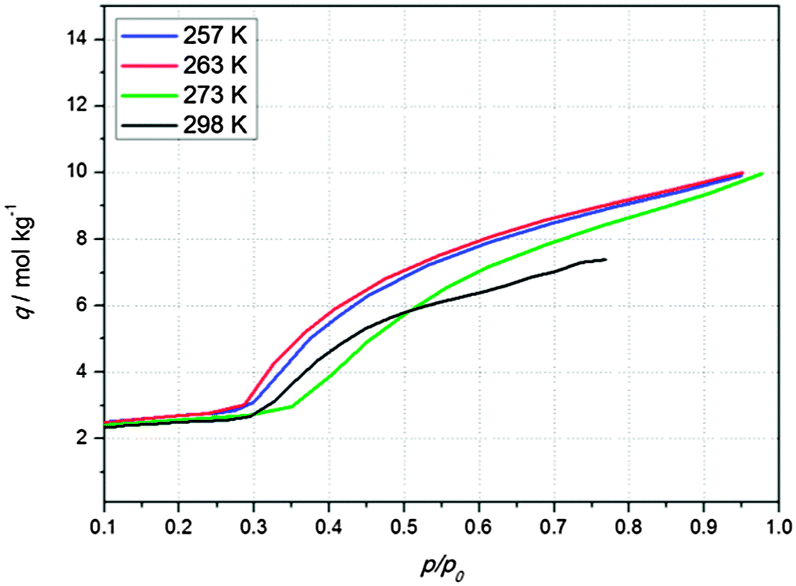 | ||
| Fig. 2 Experimental adsorption isotherms of carbon dioxide at different temperatures are shown as a function of the relative pressures (p/p0). The adsorption isotherms present a similar behavior. Independently of the temperature, the np → lp phase transition occurs at circa p/p0 = 0.3. | ||
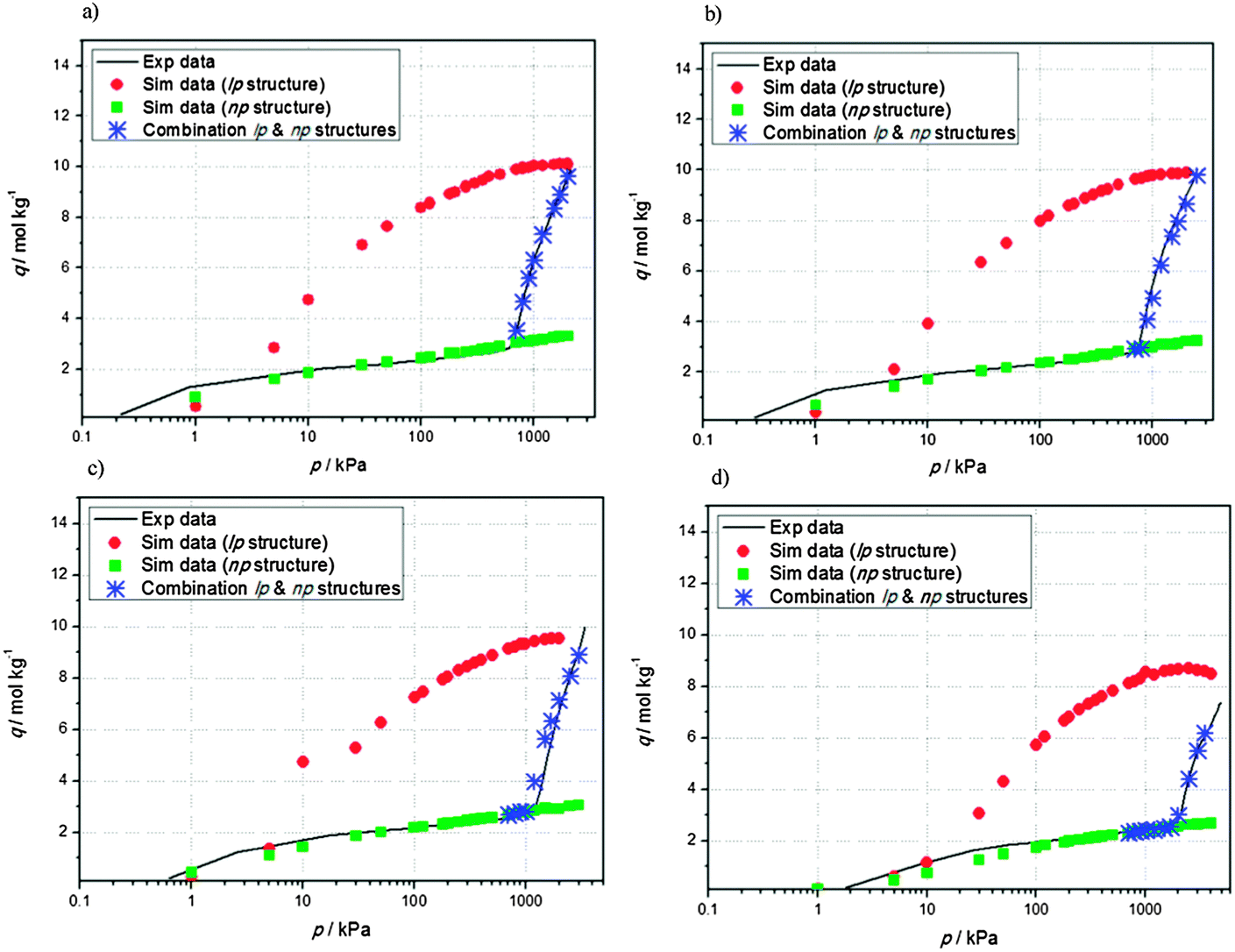 | ||
| Fig. 3 Experimental adsorption isotherms (black line) at (a) 257 K, (b) 263 K, (c) 273 K, and (d) 298 K compared to the results obtained via linear combination of the two coexisting structures (blue asterisks). | ||
Adsorption isotherms of individual components and equimolar carbon dioxide and methane mixtures as a function of the partial pressures at 298 K computed using MC simulations are shown in Fig. 4. The adsorption of (top) carbon dioxide and (bottom) methane is presented for the single component (full symbols) and as a part of the binary mixture (empty symbols) in the narrow pore form (circles and diamonds, respectively), in the large pore form (squares and triangles, respectively) and in the linear combination of the two coexisting structures (asterisks). Carbon dioxide starts adsorbing at lower pressures than methane as single component and has a 33% higher saturation loading. The adsorption of carbon dioxide is almost independent of the presence of methane.
 | ||
| Fig. 4 Simulated adsorption isotherms of (top) carbon dioxide and (bottom) methane as single components (full symbols) and as a part of a binary equimolar mixture (empty symbols) in the np and lp structures at 298 K. Partial pressures are used for comparison. | ||
In contrast, when methane is part of the binary equimolar mixture, its equilibrium loading is much smaller than when it adsorbs as single component. The strong decrease in methane adsorption in the presence of carbon dioxide is due to two different reasons: (i) on one hand, carbon dioxide molecules are already confined in the pores before methane can be adsorbed due to the constrained pore dimensions of the np form and (ii) the different interaction strengths between the two adsorbates and the hydroxyl groups of the metal–organic framework. Due to the significant quadrupole moment of carbon dioxide, the interaction with the bridging hydroxyls is much higher than for the non-polar methane molecule. As a result, NH2-MIL-53(Al) is a highly selective adsorbent.
MD simulations are a powerful tool to obtain information concerning gas diffusion for single components in MOFs. Up to now, only a few computational studies based on MD simulations have been reported so far on the diffusion of carbon dioxide in MOFs.61–65 As we have recently shown,38 the interaction between amino groups of the linker and the hydroxyl groups of the structure prevents rotation of the linker, in contrast to the unfunctionalized MIL-53 structure. Therefore, diffusion of molecules from one channel to another is highly unlikely, making directional diffusion along the channels the most likely diffusion mechanism in NH2-MIL-53(Al).
In Fig. 5, calculated self-diffusivities as a function of the number of adsorbed molecules per unit cell at 273 K are presented. The very slow diffusivity for less than 3 molecules per unit cell, the maximum loading for the close configuration of NH2-MIL-53(Al), is related to the fact that molecules are not able to pass each other due to their orientation into the one dimensional channels. Diffusivity is faster once the structure is open, even when a larger number of molecules are adsorbed per unit cell. The latter is due to the larger available space in the pore and the weaker interaction with the pore walls. Fig. 6 shows two snapshots of carbon dioxide in the np (left) and lp (right) structures of NH2-MIL-53(Al) taken directly from our simulations at 273 K. Although we are not able to provide a comparison with experimental data on NH2-MIL-53(Al), on the basis of the excellent agreement found between experiments and simulations for equilibrium adsorption data, we are confident that the diffusivities here reported are qualitatively correct, as it has been shown previously for zeolites using similar molecular simulation methods.66,67
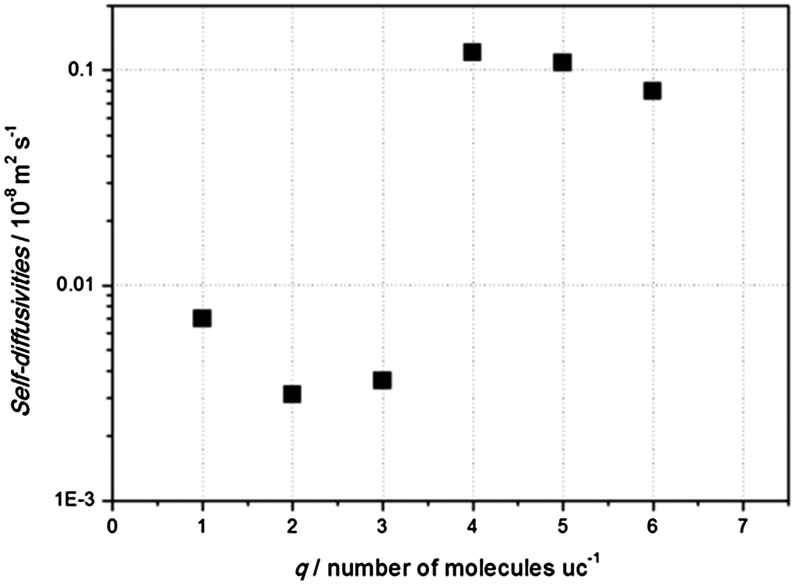 | ||
| Fig. 5 Computed self-diffusivity of carbon dioxide molecules as a function of the number of adsorbed molecules per unit cell in NH2-MIL-53(Al) at 273 K. | ||
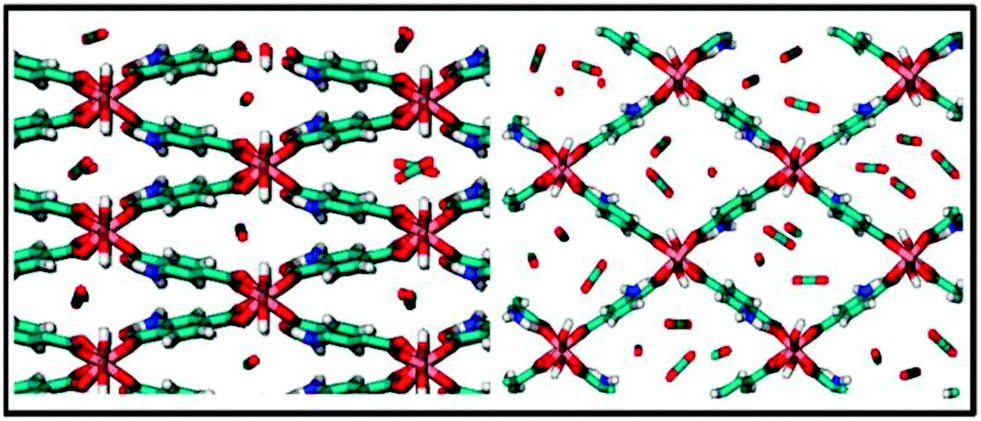 | ||
| Fig. 6 Snapshots of carbon dioxide in the np (left) and lp (right) structures of NH2-MIL-53(Al) taken directly from our simulations at 273 K. | ||
D Conclusions
Adsorption of carbon dioxide and methane in the flexible metal–organic framework NH2-MIL-53(Al) has been simulated, both for pure component and equimolar binary mixtures, using rigid force fields for the framework. The use of flexible force fields was avoided following a two-steps process, with which we obtained a very good agreement between the available experimental data and simulations at different temperatures. The first step is the optimization of the interactions of carbon dioxide molecules and the bridging hydroxyls groups of the framework followed by the use of a linear combination of the np and lp forms based on experimental results. The step found in the adsorption isotherms of carbon dioxide due to the breathing effect is well reproduced by our simulations, and the high selectivity towards carbon dioxide in the presence of methane is explained on the basis of size exclusion and affinity. Computed enthalpies of adsorption and Henry coefficients for both gases are also in agreement with experimental data. Computed self-diffusivities of carbon dioxide in NH2-MIL-53(Al) show two different regions. The pore expansion by the breathing effect influences the self-diffusion mechanism and much higher diffusivities are observed at relatively high adsorbate loadings.Summarizing, in this work we have shown that adsorption and diffusion of gases can be simulated in flexible porous materials without employing flexible force fields and with very limited experimental efforts.
Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. 335746, CrystEng-MOF-MMM (J.G.). E.G.P. thanks the Spanish MEC for her postdoctoral fellowship.Notes and references
- B. L. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS PubMed.
- A. K. Cheetham and C. N. R. Rao, Science, 2007, 318, 58–59 CrossRef CAS PubMed.
- J. L. C. Rowsell, E. C. Spencer, J. Eckert, J. A. K. Howard and O. M. Yaghi, Science, 2005, 309, 1350–1354 CrossRef CAS PubMed.
- X. B. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keefe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS PubMed.
- P. L. Llewellyn, S. Bourrelly, C. Serre, Y. Filinchuk and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 7751–7754 CrossRef CAS PubMed.
- C. Serre, S. Bourrelly, A. Vimont, N. A. Ramsahye, G. Maurin, P. L. Llewellyn, M. Daturi, Y. Filinchuk, O. Leynaud, P. Barnes and G. Ferey, Adv. Mater., 2007, 19, 2246–2251 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J. S. Chang, D. Y. Hong, Y. K. Hwang, S. H. Jhung and G. Ferey, Langmuir, 2008, 24, 7245–7250 CrossRef CAS PubMed.
- M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725–1727 CrossRef CAS.
- G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- S. Natarajan and P. Mahata, Chem. Soc. Rev., 2009, 38, 2304–2318 RSC.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- S. Couck, E. Gobechiya, C. E. A. Kirschhock, P. Serra-Crespo, J. Juan-Alcaniz, A. M. Joaristi, E. Stavitski, J. Gascon, F. Kapteijn, G. V. Baron and J. F. M. Denayer, ChemSusChem, 2012, 5, 740–750 CrossRef CAS PubMed.
- S. Y. Wang, Q. Y. Yang and C. L. Zhong, Sep. Purif. Technol., 2008, 60, 30–35 CrossRef CAS PubMed.
- R. Babarao, Z. Q. Hu, J. W. Jiang, S. Chempath and S. I. Sandler, Langmuir, 2007, 23, 659–666 CrossRef CAS PubMed.
- S. Keskin and D. S. Sholl, J. Phys. Chem. C, 2007, 111, 14055–14059 CAS.
- K. S. Walton, A. R. Millward, D. Dubbeldam, H. Frost, J. J. Low, O. M. Yaghi and R. Q. Snurr, J. Am. Chem. Soc., 2008, 130, 406–407 CrossRef CAS PubMed.
- Q. Y. Yang and C. L. Zhong, ChemPhysChem, 2006, 7, 1417–1421 CrossRef CAS PubMed.
- Q. Y. Yang and C. L. Zhong, J. Phys. Chem. B, 2006, 110, 655–658 CrossRef CAS PubMed.
- Q. Y. Yang, C. Y. Xue, C. L. Zhong and J. F. Chen, AIChE J., 2007, 53, 2832–2840 CrossRef CAS.
- A. Martin-Calvo, E. Garcia-Perez, J. Manuel Castillo and S. Calero, Phys. Chem. Chem. Phys., 2008, 10, 7085–7091 RSC.
- Y. F. Chen and J. W. Jiang, ChemSusChem, 2010, 3, 982–988 CrossRef CAS PubMed.
- Y. H. Liu, D. H. Liu, Q. Y. Yang, C. L. Zhong and J. G. Mi, Ind. Eng. Chem. Res., 2010, 49, 2902–2906 CrossRef CAS.
- B. A. Wells and A. L. Chaffee, Adsorption, 2011, 17, 255–264 CrossRef CAS.
- M. Pera-Titus, T. Lescouet, S. Aguado and D. Farrusseng, J. Phys. Chem. C, 2012, 116, 9507–9516 CAS.
- T. Lescouet, E. Kockrick, G. Bergeret, M. Pera-Titus, S. Aguado and D. Farrusseng, J. Mater. Chem., 2012, 22, 10287–10293 RSC.
- P. Serra-Crespo, E. V. Ramos-Fernandez, J. Gascon and F. Kapteijn, Chem. Mater., 2011, 23, 2565–2572 CrossRef CAS.
- E. Stavitski, E. A. Pidko, S. Couck, T. Remy, E. J. M. Hensen, B. M. Weckhuysen, J. Denayer, J. Gascon and F. Kapteijn, Langmuir, 2011, 27, 3970–3976 CrossRef CAS PubMed.
- A. Boutin, S. Couck, F. X. Coudert, P. Serra-Crespo, J. Gascon, F. Kapteijn, A. H. Fuchs and J. F. M. Denayer, Microporous Mesoporous Mater., 2011, 140, 108–113 CrossRef CAS PubMed.
- C. Montoro, E. Garcia, S. Calero, M. A. Perez-Fernandez, A. L. Lopez, E. Barea and J. A. R. Navarro, J. Mater. Chem., 2012, 22, 10155–10158 RSC.
- C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louer and G. Ferey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- C. Gucuyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef PubMed.
- J. van den Bergh, C. Gucuyener, E. A. Pidko, E. J. M. Hensen, J. Gascon and F. Kapteijn, Chem. – Eur. J., 2011, 17, 8832–8840 CrossRef CAS PubMed.
- T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Ferey and N. Stock, Inorg. Chem., 2009, 48, 3057–3064 CrossRef CAS PubMed.
- P. Serra-Crespo, M. A. van der Veen, E. Gobechiya, K. Houthoofd, Y. Filinchuk, C. E. A. Kirschhock, J. A. Martens, B. F. Sels, D. E. De Vos, F. Kapteijn and J. Gascon, J. Am. Chem. Soc., 2012, 134, 8314–8317 CrossRef CAS PubMed.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Remy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- S. Couck, T. Remy, G. V. Baron, J. Gascon, F. Kapteijn and J. F. M. Denayer, Phys. Chem. Chem. Phys., 2010, 12, 9413–9418 RSC.
- P. Serra-Crespo, E. Stavitski, F. Kapteijn and J. Gascon, RSC Adv., 2012, 2, 5051–5053 RSC.
- F. Salles, A. Ghoufi, G. Maurin, R. G. Bell, C. Mellot-Draznieks and G. Ferey, Angew. Chem., Int. Ed., 2008, 47, 8487–8491 CrossRef CAS PubMed.
- D. S. Coombes, F. Cora, C. Mellot-Draznieks and R. G. Bell, J. Phys. Chem. C, 2009, 113, 544–552 CAS.
- A. Ghoufi, A. Subercaze, Q. Ma, P. G. Yot, Y. Ke, I. Puente-Orench, T. Devic, V. Guillerm, C. Zhong, C. Serre, G. Ferey and G. Maurin, J. Phys. Chem. C, 2012, 116, 13289–13295 CAS.
- L. Sarkisov, R. L. Martin, M. Haranczyk and B. Smit, J. Am. Chem. Soc., 2014, 136, 2228–2231 CrossRef CAS PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- B. Delley, J. Phys. Chem., 1996, 100, 6107–6110 CrossRef CAS.
- DMol3, San Diego, v. 4.0, Accelrys Inc., 2005 Search PubMed.
- N. A. Ramsahye, G. Maurin, S. Bourrelly, P. Llewellyn, T. Loiseau and G. Ferey, Phys. Chem. Chem. Phys., 2007, 9, 1059–1063 RSC.
- N. A. Ramsahye, G. Maurin, S. Bourrelly, P. L. Llewellyn, T. Devic, C. Serre, T. Loiseau and G. Ferey, Adsorption, 2007, 13, 461–467 CrossRef CAS PubMed.
- L. Hamon, H. Leclerc, A. Ghoufi, L. Oliviero, A. Travert, J.-C. Lavalley, T. Devic, C. Serre, G. Ferey, G. De Weireld, A. Vimont and G. Maurin, J. Phys. Chem. C, 2011, 115, 2047–2056 CAS.
- A. Garcia-Sanchez, C. O. Ania, J. B. Parra, D. Dubbeldam, T. J. H. Vlugt, R. Krishna and S. Calero, J. Phys. Chem. C, 2009, 113, 8814–8820 CAS.
- J. G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021–12024 CrossRef CAS.
- J. P. Ryckaert and A. Bellemans, Faraday Discuss., 1978, 95–106 RSC.
- D. Dubbeldam, S. Calero, D. E. Ellis and R. Q. Snurr, Northwestern University, Evanston, IL, version 1.0 edn, 2008.
- T. Duren, L. Sarkisov, O. M. Yaghi and R. Q. Snurr, Langmuir, 2004, 20, 2683–2689 CrossRef.
- D. Dubbeldam, D. C. Ford, D. E. Ellis and R. Q. Snurr, Mol. Simul., 2009, 35, 1084–1097 CrossRef CAS.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- P. Serra-Crespo, E. Gobechiya, E. V. Ramos-Fernandez, J. Juan-Alcaniz, A. Martinez-Joaristi, E. Stavitski, C. E. A. Kirschhock, J. A. Martens, F. Kapteijn and J. Gascon, Langmuir, 2012, 28, 12916–12922 CrossRef CAS PubMed.
- A. I. Skoulidas and D. S. Sholl, J. Phys. Chem. B, 2005, 109, 15760–15768 CrossRef CAS PubMed.
- R. Babarao and J. W. Jiang, Langmuir, 2008, 24, 6270–6278 CrossRef CAS PubMed.
- F. Salles, H. Jobic, A. Ghoufi, P. L. Llewellyn, C. Serre, S. Bourrelly, G. Ferey and G. Maurin, Angew. Chem., Int. Ed., 2009, 48, 8335–8339 CrossRef CAS PubMed.
- F. Salles, H. Jobic, T. Devic, V. Guillerm, C. Serre, M. M. Koza, G. Ferey and G. Maurin, J. Phys. Chem. C, 2013, 117, 11275–11284 CAS.
- Q. Y. Yang, C. L. Zhong and J. F. Chen, J. Phys. Chem. C, 2008, 112, 1562–1569 CAS.
- R. Krishna, J. M. van Baten, E. Garcia-Perez and S. Calero, Chem. Phys. Lett., 2006, 429, 219–224 CrossRef CAS PubMed.
- R. Krishna, J. M. van Baten, E. Garcia-Perez and S. Calero, Ind. Eng. Chem. Res., 2007, 46, 2974–2986 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The definitions of the crystallographically different atoms for the (a) np form and (b) lp form of NH2-MIL-53(Al). Complete set of parameters and charges for adsorbates and adsorbents. See DOI: 10.1039/c3cp55416c |
| This journal is © the Owner Societies 2014 |