 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photophoretic separation of single-walled carbon nanotubes: a novel approach to selective chiral sorting†
David
Smith
*ab,
Christopher
Woods
c,
Annela
Seddon
*ab and
Heinrich
Hoerber
b
aBristol Centre for Functional Nanomaterials, Centre for Nanoscience and Quantum Information, University of Bristol, Bristol, BS8 1FD, UK. E-mail: annela.seddon@bristol.ac.uk; Tel: +44 (0)117 3940015
bHH Wills Physics Laboratory, University of Bristol, Bristol, BS8 1TL, UK. E-mail: dave.smith@bristol.ac.uk; h.hoerber@bristol.ac.uk; Tel: +44 (0)117 3317787
cCentre for Computational Chemistry, School of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: Christopher.woods@bristol.ac.uk
First published on 28th January 2014
Abstract
For over two decades single-walled carbon nanotubes (SWCNTs) have been used in a broad range of electronic and optical applications, however the selective chiral sorting of SWCNTs with guaranteed optoelectronics characteristics is imperative to the industrial realization of such applications. In this paper we provide the results of modeling an optical sorting method that utilizes the inherent opto-electronic properties of the SWCNTs, thus guaranteeing the properties of the extracted populations. Utilizing the resonant transfer of photonic momentum, we simulate chiral sorting of two chiral populations in an aqueous environment based on the frequency dependent optical absorption properties of the nanotubes. We show that photonic sorting is not only feasible, but may be up to faster than density gradient centrifugation techniques. Our simulations investigate the effects of laser power, temperature and orientation. We find that 96% purity can be achieved in less than 12 minutes by operating at 9 × 107 W m−2 (20 mW in a 20 μm chamber) at elevated temperatures.
Introduction
Since they were first observed in 1991,1 many different applications have been proposed for carbon nanotubes including energy conversion,2–4 chemical and optical sensors,5–7 hydrogen storage,8–11 high-speed electronics12–16 and computing,17 nanometer wires and high strength composites.18 In recent years some applications have been commercially realized but these are mainly composite materials which have little dependence upon diameter or chirality. Although advanced optoelectronic applications have been demonstrated2 the lack of bulk chiral selectivity with guaranteed optoelectronic properties means these are, generally, still confined to the laboratory. The electronic and optical properties of single walled carbon nanotubes (SWCNTs) intrinsically depend upon their chirality due the graphene wrapping.19 The chirality of a carbon nanotube is described by two integer values (m and n), its chiral vector (the dashed line in Fig. 1) that describes how it is rolled from a graphene sheet.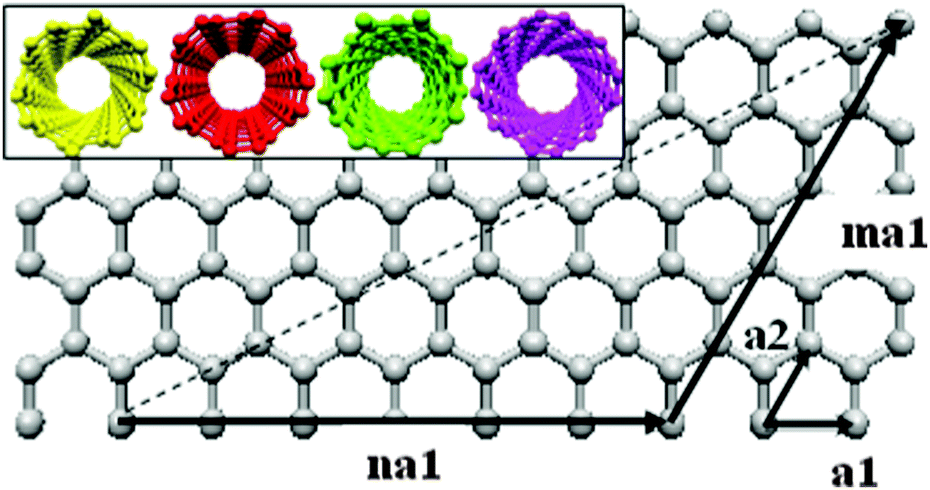 | ||
| Fig. 1 Graphene sheet showing how the chiral vector (dashed arrow) of a (6,5) carbon nanotube is described by two integer values m and n. Inset: end-on-view of various chirality carbon nanotubes; from left to right, (6,4), (5,5), (7,1), (8,4). | ||
For many advanced applications mono-chiral dispersions are essential20 and a range of post-synthetic sorting techniques have been developed to achieve this. These techniques include density gradient ultra-centrifugation,21–30 ion exchange chromatography,31 gel chromatography32 and conjugated polymer extraction.33 However, density gradient ultracentrifugation is the most commonly used. The density gradient ultracentrifugation of nanotubes was first demonstrated by Arnold et al.21 in 2006 who showed diameter control the of ∼0.02 nm. Significant effort was subsequently invested in developing the technique. Fagan et al.25 demonstrated that density gradient ultracentrifugation could be used for SWCNT length fractionation whilst numerous other groups have demonstrated that the technique can be used two separate metallic and semiconductor SWCNT.21,23,24 More recently chiral purity exceeding 97% have now been obtained via DGU22 and some groups have even demonstrated enantiomeric selectivity.26,28 However processing times limit the scalability of density gradient ultracentrifugation. For example the procedure originally used by Arnold et al.21 in 2006 required 12 hours of DGU processing time whereas the more recent enantiomeric selectivity demonstrated by Ghosh et al.28 required 18 hours of processing.
Chiral recognition and chromatographic techniques have also been vigorously investigated. In 2007 Nish et al.33 demonstrated that specific aromatic polymers would selectively disperse particular SWCNT species. Whilst this is a relatively simple method the resulting dispersions still only demonstrated a chiral purity of ∼60%. In 2009 Tu et al.31 used custom fabricated DNA sequences that preferentially encapsulate specific chiralities depending upon the DNA sequence. The encapsulation allowed for the selective chemical modification and separation using ion-exchange chromatography. However, such an approach may not commercially viable due to the prohibiting cost of DNA strand synthesis and (if required) the subsequent difficulties with DNA removal. More recently, a simple gel chromatographic method of sorting was demonstrated by Liu et al.32 The procedure is able to refine SWCNT's based upon their interaction with the allyl dextran-based size-exclusion gel. The technique is clearly very powerful and yet careful study of the photoluminescence result still demonstrates the presence of other chiralities within the refined samples.
Although both of the aforementioned techniques are very powerful tools for producing mono-chiral dispersions they cannot guarantee the optoelectronics characteristics of the purified SWCNTs (merely their chirality) due to the indirect approaches they adopt for chiral recognition. To sort high quality, defect free SWCNTs a direct method based upon optoelectronic interactions would be desirable. The simplest method used to characterize poly-dispersed carbon nanotube suspensions is UV-Vis-NIR absorption spectroscopy. This optical technique gives an indication to the chiralities of SWCNTs present in a sample, based on the exact frequency or wavelength of their absorption peaks. In the following work, we demonstrate using a computer simulation how it is possible to use absorption as a tool for separation rather than just for characterization.
The potential to use the absorption of photons for separation is due to photons possessing an amount of momentum based upon their frequency, and hence their energy. Therefore, if one chirality of SWCNTs absorbs more photons at a given energy than a different chirality, the preferentially absorbing sample will experience a greater photon pressure. Examination of the density of states (DOS) of SWCNTs shows sharp spikes, which are Van Hove singularities34 and occur at energies dependent upon the chirality of the carbon nanotubes. The peaks in the absorption spectra are due to optically excited transitions preferentially occurring at energies coinciding with transitions between Van Hove singularities. Hence, by tuning a photonic source, for example a laser, to the transition energy between corresponding Van Hove singularities, it should be possible to preferentially impart photonic momentum upon a single chirality of SWCNTs.
A fundamental question to ask of such a separation strategy would be whether sufficient photonic momentum can be imparted on the SWCNT populations to overcome Brownian based diffusive re-mixing without optically saturating the transitions. The work presented here demonstrates that this is possible and has been tested by simulations run under a number of possible conditions to elucidate the required separation times of two populations of SWCNTs. We have chosen to use a simulations rather than an analytical approach to allow for the SWCNTs reduced freedom of rotation due to interaction with the channel sidewalls.
Theory
Brownian movement (i.e. diffusion) of SWCNTs has both rotational and translational components and here we base our simulation upon previously published work.35 Brownian translation was considered as a Gaussian distribution of random steps in the x, y and z directions. Such a distribution would have a mean of zero and a standard deviation of σ. σ can be found using eqn (1).| σ2 = 〈(Bx,y,z)(Bx,y,z)〉 = 2Dx,y,zΔt | (1) |
| Dx,y,z = kBTξx,y,z−1 | (2) |
 | (3) |
 | (4) |
| ξ⊥ = 2ξ= | (5) |
Brownian rotation was modeled by finding the standard deviation in two orthogonal planes normal to the SWCNT axis. The rotational standard deviation is given by:
| σ2 = 〈(BUV,UW)(BUV,UW)〉 = 2DRΔt | (6) |
In the eqn (6), BUV and BUW are the mean absolute rotational steps in the two planes normal to the SWCNTs' axis. Eqn (7) shows how DR is the rotational diffusivity and which can be found from the ratio of the thermal energy to the rotational hydrodynamic frictional coefficient ξR, where ξR is obtained from eqn (8):
| DR = kBT/ξR | (7) |
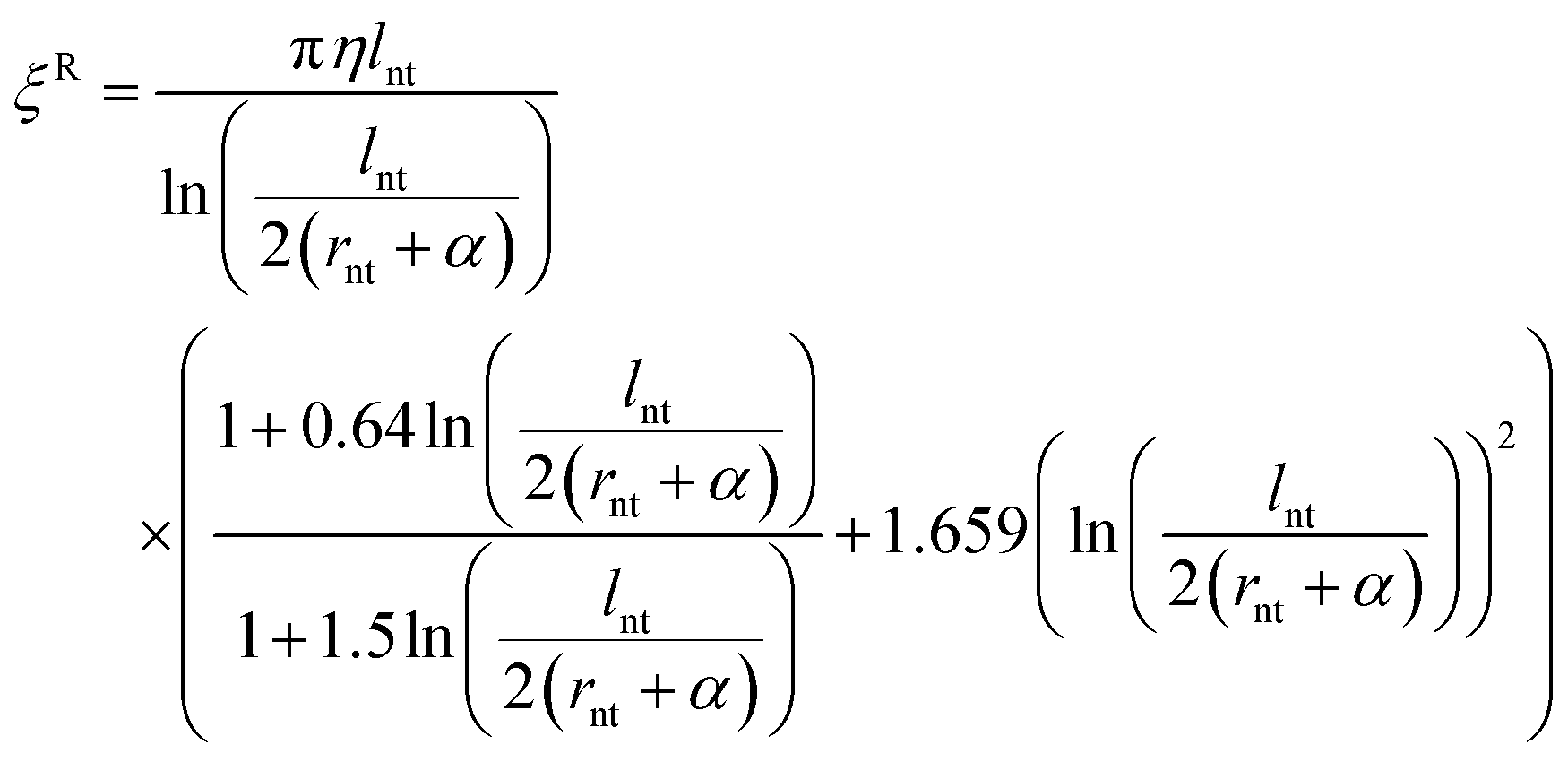 | (8) |
Many studies have been conducted both experimentally36–38 and theoretically39,40 into the behaviour of asymmetric objects in optical traps. However, optically traps typically exhibit strong degrees of polarisation and intensity gradients producing forces that both confine and orient objects. Conversely, in our case such forces could be a hindrance. Therefore, we assume an unpolarised, uniform optical field (i.e. no optical gradients) of relatively low intensity. The photonic translation caused by the momentum transfer from absorbed photons is assumed to be along the direction of the Poynting vector (i.e. along the direction of propagation) and for convenience this is chosen to be the z-direction with no components in x or y. Absorption in SWCNTs arises due to many phenomena, including plasmon absorption, lattice scattering and photoelectric absorption. The first two effects are non-specific and non-saturable, creating a universal background absorption irrespective of chirality. However, the degree of photoelectric absorption at a given energy depends upon the SWCNT chirality. Further to this, it has been shown that the photoelectric absorption of SWCNTs is a saturable process with a continuous wave saturation intensity of ∼5 × 107 W m−2.41
As such, the energy absorbed (ψ) by a partially saturable absorbing SWCNT in a time period Δt is given by:
 | (9) |
Once the total absorbed energy has been deduced the associated momentum change in the z-direction (Pz) can be found from eqn (10) where c is the speed of light.
 | (10) |
This momentum acts against the hydrodynamic frictional drag coefficient in the z-direction obtained in the Brownian translation calculations. The resulting photonic step is given by eqn (11):
| Bz = Pz·ξz | (11) |
Methods
All simulations were run on the Bluecrystal HPC facility at the University of Bristol. Parallel implementation was realized by utilizing the Python MPI4Py module. A typical simulation utilized 3 nodes with 8 cores per node such that there were 24 cores in total. The general procedure for these simulations can be found in the ESI† in Fig. S1. The SWCNTs are distributed evenly across all cores and are allowed to evolve for 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles (54 seconds) before being re-gathered and their positions stored. This improved efficiency of the simulation by minimizing the amount of MPI communication required. Consequently the efficiency scaling is essentially linear; doubling the number of cores halves the processing time. 54 seconds is chosen between storage steps producing 200 data points during a 180 minute simulation.
000 cycles (54 seconds) before being re-gathered and their positions stored. This improved efficiency of the simulation by minimizing the amount of MPI communication required. Consequently the efficiency scaling is essentially linear; doubling the number of cores halves the processing time. 54 seconds is chosen between storage steps producing 200 data points during a 180 minute simulation.
The general experimental parameters can also be found in the ESI† in Table S1. Briefly, a 1 ms time step is chosen because during this time SWCNTs typically rotate less than 10 degrees allowing for a stable simulation. A simulated ‘Run Time’ of 180 minutes is needed to observe chiral separation. SWCNT lengths and diameters were based on SG65i SWCNTs which are commercially available from both Sigma-Aldrich and Southwest Nanotechnologies. The additional radial value is chosen so that hydrodynamic radius (which may be significantly larger than the SWCNT radius due to the presence of dispersion agents) is equal that of Sodium-dodecylbenzenesulfonate (SDBS) encapsulated SWCNTs. SDBS which has been shown to be one of the best surfactants for SWCNT dispersion.44 Surfactant encapsulated SWCNTs have a larger hydrodynamic radius than SWCNTs dispersed via DNA or polymer encapsulation. Therefore, we allow for the ‘worst case scenario’. The absorption-ratio was chosen by studying absorption spectra.42,43 Low SWCNT concentrations were assumed and therefore SWCNT–SWCNT hydrodynamic coupling effects were considered negligible and ignored. We also model our samples as individual SWCNTs; as such they are not bundled. Assuming individual SWCNTs present at low concentrations means that any inter-SWCNT interactions, such as Exciton Energy Transfer (EET) or Fluorescence resonance energy transfer (FRET) can be disregarded. The potential experimental implication of EET or FRET within small bundles is considered in the discussion below.
We investigated a photophoretic45,46 system due to the simple setup geometry and success of other phoretic approaches in chiral processing.47 Before conducting these investigations we verified the validity of our program by running Brownian evolution simulations of randomly orientated SWCNTs that can translate & rotate, and of aligned SWCNTs that can only undergo translation. We then analysed the results to obtain diffusion coefficients of aligned and randomly orientated SWCNTs and compared our findings with published results.
The simulated photophoretic system used a single resonantly tuned laser lightly focused into a 20 × 20 × 1500 μm channel. To assess channel sidewall interactions limiting the SWCNTs' freedom of rotation and subsequent absorption cross section we initially simulated the Brownian motion of 10000 SWCNTs over a 250 second period. We then recorded their absorption cross section as a function of distance from the channel sidewall.
To assess the effectiveness of photophoretic chiral sorting a mixed population of SWCNTs (50% on-resonance:50% off-resonance) was then introduced at one end of the a similar channel so that the photonic pressure drove the suspended SWCNTs along the channel.
When considering the following simulations, it is more useful to consider photonic intensity rather than absolute laser power. As such, laser powers are measured in terms of their relative photonic intensity (RPI); the ratio of photonic intensity in the sample chamber to a previously measured saturation intensity41 of 5 × 107 W m−2. Therefore, an RPI of 1.0 equates to a photonic intensity which is the same as the saturation intensity.
Accordingly an RPI of 2.0 represent a photonic intensity which is double the saturation intensity. In this way an RPI of 1.0 corresponds to a laser power of 20 mW uniformly distributed across the modelled channel. Initially we verified that photophoretic separation of SWCNTs is viable and compared the separation times of aligned and randomly orientated dispersions. Alignment perpendicular to the z-axis should increase the SWCNT absorption cross-section and therefore may improve the separation efficiency. These simulations were conducted with an RPI of 1.0 (i.e. the saturation intensity) and at room temperature (20 °C).
Subsequently, we conducted a power sweep at a constant temperature of 20 °C, and a temperature sweep at a constant RPI of 1.0. For the power sweep the RPI was increased in steps of 0.25 between 0.25 and 2.5, whilst for the temperature sweep the solution temperature was increased from 0 °C to 100 °C in 10 °C steps. We also allowed for the decrease in the hydrodynamic viscosity with increasing temperature via an empirical relationship. This relationship and a corresponding table of values can be found in the ESI† in eqn (S1) and Table S2.
At elevated temperatures surfactant encapsulation may not be stable method of SWCNT dispersal. However other dispersal methods are valid at elevated temperatures. For example it has been shown48 that 12-base single-stranded DNA homopolymers of Guanine can disperse SWCNTs at temperature exceeding 90 °C.
Finally, we conducted a series of simulations to ascertain the optimum separation conditions in terms of the RPI and temperature. This variable space assessment was conducted by setting the RPI and temperature to all possible combinations. The respective ranges and increments were the same as for the RPI and temperature sweeps discussed above. Thus the full assessment consists of 110 simulations.
Results
Our approach was validated by simulating diffusion caused by the translation of SWCNTs due to their Brownian motion. Fig. 2a and b shows the output of the simulation of randomly orientated SWCNTs before and after 5 seconds of free diffusion, with their time dependent mean square displacement (MSD) along the x, y and z axes plotted in Fig. 2c.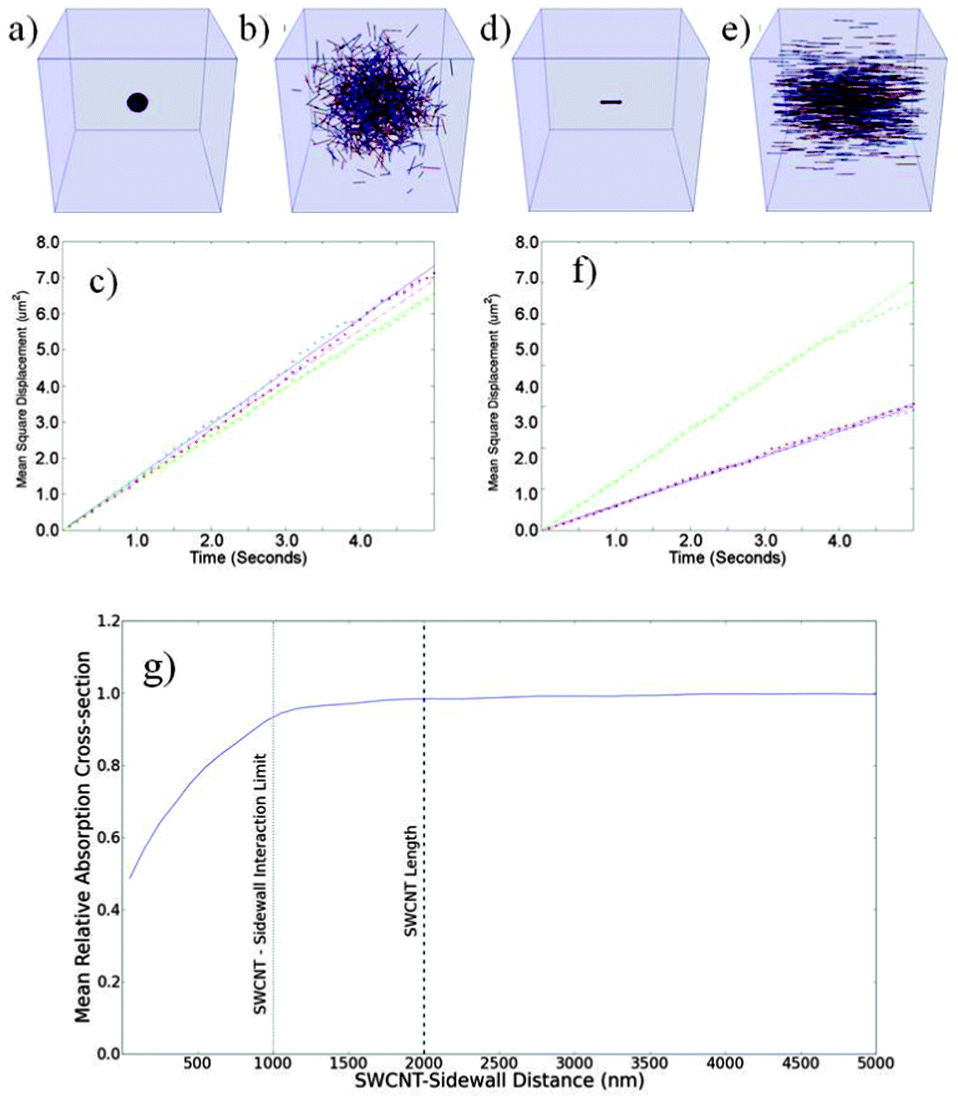 | ||
| Fig. 2 Verification of translational Brownian simulations (a–f) and sidewall effects (g). The box dimensions (a–d) are 16 μm × 16 μm × 16 μm. Randomly orientated SWCNTs (a) before and (b) after 5 seconds of evolution. (c) Mean square displacement (MSD) as a function of time for randomly orientated, rotationally free SWCNTs in the x (blue), y (red) and z (green) directions, (d–e) aligned SWCNTs (d) before and (e) after 5 seconds of evolution. (f) MSD as a function of time for each axis when non-rotating SWCNTs are aligned parallel to the z-axis, with x (blue), y (red) and z (green). The linear fits shown in (c) and (f) are used to obtain 1-dimensional diffusion constants. (g) Mean SWCNT absorption cross-section as a function of SWCNT (centre point)-sidewall separation. | ||
Fig. 2d and e show aligned SWCNTs before and after 5 seconds of free diffusion with their time dependent MSD shown in Fig. 2f. The related linear fits for each axis x, y and z gave diffusion constants of 0.732, 0.693 and 0.657 μm2 s−1 for randomly orientated SWCNTs. For the SWCNTs that are aligned parallel to the z-axis and not allowed to rotate, the diffusion constants are 0.599, 0.614 and 1.21 μm2 s−1 along the respective x, y and z axes. These results are in good agreement with the literature.22Fig. 2g shows the how the effect of the channel sidewall limits the SWCNT's freedom of rotation and the mean hence absorbing cross section. On the average the sidewall effects reduced the absorbing cross section by over a range of ∼1.2 μm.
Fig. 3a shows photophoretic chiral separations of randomly orientated SWCNTs and Fig. 3b shows analogous data for aligned SWCNTs. Randomly orientated SWCNTs achieved 2σ-separation after ∼84 minutes of processing at a distance of ∼400 μm from their starting location. Aligned SWCNT achieved 2σ-separation after ∼109 minutes at a distance of 480 μm.
 | ||
| Fig. 3 Mean position as a function of time of on-resonance (red) and off-resonance (blue) SWCNT populations undergoing photophoretic separation. (a) Distribution of randomly orientated SWCNT dispersions; (b) distribution of aligned SWCNT dispersions. The error bars are 2σ and thus encompass 96% of each population. The 2σ-separation times are 84 minutes and 109 minutes respectively. | ||
Fig. 4a shows stacked distribution plots of randomly oriented SWCNTs as a function of increasing RPI; the stacked distribution plots of randomly oriented SWCNTs as a function of temperature are shown in Fig. 4b. The increasing angular separation shown between the off-resonance SWCNTs (blue) and the on-resonance SWCNT (red) regions indicates an increased rate of separation both with increasing RPI or increasing temperature.
 | ||
| Fig. 4 Stacked distribution plots showing: (a) increasing separation rates (at a constant solution temperature of 20 °C) with increasing relative photonic intensity (RPI) and (b) increasing separation rates (at constant RPI of 1.0) with increasing temperature. The RPI is a measure of the photonic intensity relative to the saturation intensity. Thus an RPI of one represents the saturation intensity. | ||
Finally, Fig. 5 shows the variable space simulations surface plot of RPI against temperature of predicted 2σ-separation times for randomly aligned SWCNTs obtained via a ‘best-fit’ 2nd order polynomial surface. This found a minimum 2σ-separation time of ∼9 minutes at a distance of 280 μm with an RPI of 1.75 and a solution temperature of 100 °C. As a general trend the 2σ-separation times decrease with increasing temperature and RPI. At low temperatures and RPI some simulations never achieve 2σ-separation within the 180 minutes simulated period.
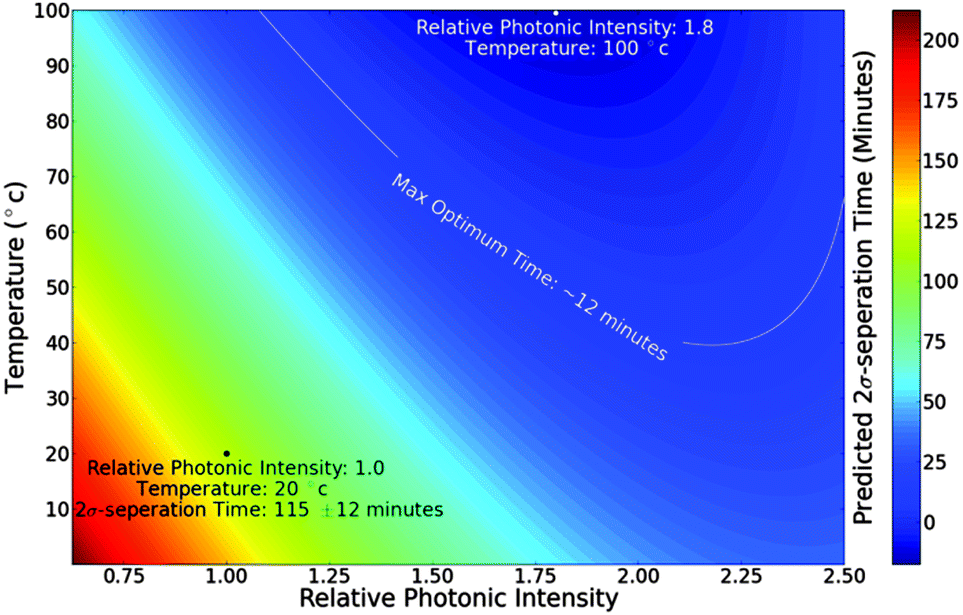 | ||
| Fig. 5 Relative photonic intensity (RPI) – temperature surface plot of predicted 2σ-separation times for randomly aligned SWCNTs obtained via a ‘best-fit’ 2nd order polynomial surface. The error of ±12 minutes was found from the standard deviation of simulated and predicated values. The black dot corresponds to the result shown in Fig. 3b. The white dot as at the surface minima indicates the conditions for optimum separation. The white contour line represents the 12 minute error bound of the optimum conditions and is the suggested experimental region. | ||
Discussion
The validity of our simulation was checked by: (i) estimating the diffusion constant of randomly aligned SWCNTs over a 5 second period compared to published values49 and (ii) checking that aligned SWCNT diffuse twice as readily along their axis of alignment as they do perpendicular to it.20 As the x, y and z axes can be considered equivalent, one would expect the 1-dimensional diffusion constants should be the same along all axes for the randomly orientated SWCNTs and similarly along the x and y directions for the aligned SWCNTs. The randomly orientated SWCNTs diffusion constant varies from 0.657 to 0.732 μm2 s−1 with a mean of 0.694 μm2 s−1. For the aligned SWCNTs the diffusion constants are 0.599 and 0.614 with a mean of 0.607 μm2 s−1. We therefore estimate that our uncertainty of the 1-dimensional diffusion constant is less than 0.05 μm2 s−1. The 3-dimensional diffusion constant is then calculated for randomly aligned SWCNTs from the Euclidian norm of the 1-dimensional diffusion constants. This is found to be 1.21 ± 0.08 μm2 s−1, which falls within the range of previously measured values.49 For the SWCNTs aligned to the z-axis, the z-axis diffusion constant (1.2 μm2 s−1) is twice that of the x-axis (0.599 μm2 s−1) and y-axis (0.614 μm2 s−1) diffusion constants. This agrees with theoretical results,50 further reassuring us as to the validity of our simulation. Our investigations into the effect of the sidewalls limiting the absorption cross-section found that the mean cross section fell gradually from ∼95% (at 1200 nm separation) to ∼50% (at 100 nm separation) of the bulk value. This is interesting as the separation at which the SWCNTs no longer interact with the sidewall is 1000 nm (i.e. the 1/2 length). However, the absorption cross-section doesn't recover to within 5% of the bulk value for a further 200 nm. Suggesting a degree of orientation memory. Considering how the sidewall effect may alter the photophoretic behaviour we calculate that over the 20 μm × 20 μm channel the mean SWCNT absorption cross-section is only reduced by 6% and compared to no sidewall interaction and should therefore have a limited effect on the sorting efficiency.Initial testing on the photophoretic separation gave rise to the results shown in Fig. 3a and b, which confirm that SWCNT chiral separation can be achieved in less than 2 hours for both aligned and randomly distributed populations. Aligned SWCNT displayed shorter 2σ separation times due to their increased cross-section in the xy plane and were found to achieve 2σ separation in approximately three quarters of the time that it took for randomly orientated SWCNTs to achieve the same degree of separation. Whilst this effect was expected, the enhancement is significantly less than had been originally envisaged and suggests that alignment procedures would only offer marginal gains to separation. Consequently, potential future separation devices do not need to include alignment mechanisms and therefore allow for simpler and more elegant approaches to be considered.
Fig. 4 shows increasing rates of separation when both the RPI (Fig. 4a) and the temperature (Fig. 4b) are increased. The separation rates increase with RPI because the photonic ‘pushing’ effect becomes stronger at higher RPIs. However, at RPIs above 1.0 the absorption band will saturate. Therefore it should be more difficult to discriminate between the on-resonance and off-resonance SWCNTs. The reduced discrimination will result in slower separation rates. A possible explanation for not observing slower separation rates at higher RPIs, is that the effect of saturation limiting the separation may only become apparent at RPIs greater 2.5 which was the limit of what was simulated here.
The other interesting observation is that separation rates increase with temperature. As the diffusivity is proportional to temperature (see eqn (2)), one would expect the diffusion driven remixing of the SWCNTs to be greater at higher temperatures and hence slow the overall rate of separation. However, this is not the case. One also has to consider the change in viscosity with temperature to understand this observation. Between 0 °C and 100 °C the hydrodynamic viscosity of water decreases by 85% from 1.79 mN s m−2 to 0.27 mN s m−2 whilst the absolute temperature only increases by 37% from 273 K to 373 K. Therefore, even though the SWCNTs diffuse slightly faster, the significantly lower viscosities allow them to propagate far further for a given photonic momentum and thus to separate faster.
Fig. 5 shows the variable space assessment of the effect of RPI and temperature on 2σ separation times and found that an RPI of 1.75 at 100 °C results in the shortest separation time of ∼9 minutes. This is 12× improvement over the ∼109 minute result for an RPI of 1.0 at room temperature (20 °C) shown in the bottom of Fig. 3b. As discussed above, the elevated temperature for optimum separation can be explained due to the decreasing viscosity with increased temperature. However, the optimum RPI of 1.75 is more surprising. Once again, one would expect the shortest separation times to occur at an RPI of 1.0 where the photonic intensity is greatest without saturating the absorption band. While RPIs above 1.0 lead to faster separation because of the increased photonic intensity, the associated saturation causes slower separation because of the decreasing chiral discrimination. An optimum RPI above 1.0 suggests that there is a balance between the faster separation due to the increased intensity and the slower separation due to the increased saturation. Our results show that this balance occurs at an RPI of 1.75.
Finally we fit a 2nd order polynomial surface to our variable space simulation results. This approach is chosen to impartially identify optimum separation conditions considering all simulations results. If no optimum conditions exist then a 2nd order polynomial fit has sufficient freedom to reduce to a linear fit and merely indicate a general trend. However, if optimum conditions do exist then these conditions are found at the surface minima. Any results that did not achieve 2σ separation in the 180 minute simulated period are excluded prior to fitting.
The resulting 2nd order polynomial surface that predicts 2σ-separation times as a function of temperature and RPI is shown in Fig. 5. A surface minimum is observed at an RPI of 1.8 and a temperature of 100 °C. While this agrees well with the previously observed conditions for optimum separation (RPI: 1.75, temperature: 100 °C) it predicts a near zero optimum 2σ separation time. With respect to a ±12 minute fitting error this is within acceptable limits and agrees with our observation that 2σ separation occurs after 9 minutes under optimum conditions. We also take ±12 minutes to be the error on our observed 2σ separation times.
The ±12 minute uncertainty is utilized to identify a suggested experimental operating region. This is indicated by the white contour line in Fig. 5. By studying the region constrained within this contour we suggest that elevated temperatures (for example above 40 °C) at an RPI of 1.8 should be initially investigated. An RPI of 1.8 corresponds to a photonic intensity of 9 × 107 W m−2. However, the maximum temperature that would be of use experimentally would have to consider the temperature stability of the dispersal method. This temperature will be dependent on the dispersal agent and there will be a balance between the improved separation rates afforded by higher operating temperatures and the potential instability of the sample. This would be a parameter which would necessitate an experimental investigation.
Also one should consider the experimental implications of absorption by small SWCNT bundles that may have implications on the photophoretic sorting efficiency. As mentioned previously bundles that contain many SWCNTs do not typically exhibit absorption bands whereas isolated SWCNTs do. However, it has been shown51,52via Exciton Energy Transfer (EET) that bundles that contain a small number of SWCNTs can exhibit preferential resonant absorption. Such bundles preferentially absorb at the resonant energy wavelength due to the presence of an on-resonance SWCNT in the bundle. The energy then couples into an adjacent SWCNT within the bundle before decaying either radiatively or non-radiatively. Fortunately, the energy transfer and potential re-emission at a lower energy should not have an effect on the sorting efficiency because the averaged re-emission is omni-directional and photophoresis is an absorption driven process. However, the off-resonance SWCNTs in the bundle would be extracted along with the preferentially absorbing on-resonance SWCNT and thus could contaminate the refined population. Such bundles will have a larger hydrodynamic radius than isolated SWCNTs and therefore propagate more slowly. We hope that the larger hydrodynamic radius (and hence frictional coefficient) will therefore limit the photonic drift velocities and mitigate the problems of small bundle absorption. We have not yet simulated it but we will conduct these studies soon.
It is also worth considering alternative experimental configurations as our simulation only considers a simple linear channel as an initial approach. However, the continuous introduction and extraction of SWCNT samples could compromise the sorting quality; for example in the device discussed here, all chiralities would travel in the same direction and are separated because they propagate at different speeds. Whilst it would be possible to use a photophoretic process to sort a single sample it would be more difficult to add a second sample. If a second sample were to be added at the start of the channel then the on-resonance SWCNTs of the second sample could catch up and mix with the off-resonance SWCNTs separated from the first sample. However, this may be easily overcome by implementing timed introduction and extraction cycles.
With this in mind, future work will simulate modified devices that utilize counter-propagation forces so that the separated SWCNT populations propagate in opposing directions. If the magnitude of a counter-propagating force could be correctly balanced relative to the (propagating) photonic pressure forces then it could be possible to ‘drive’ the two populations to opposite ends of the sample chamber. Two such counter-propagating forces have been identified: fluidic counter-flow & optical counter-propagation. Fluidic counter-flow simulations will be used to investigate the fluidic conditions required for flowing the SWCNT solution towards the resonant optical source. Simulations of optical counter-propagation will investigate the use of an off-resonance optical source which acts equally upon all chiralities.
Conclusion
Although procedures for chirality sorting SWCNT have emerged in recent years they are unable to guarantee the opto-electronic properties of the sorted samples. We have proposed using a resonance based optical sorting technique that by its very nature will guarantee the opto-electronic properties. To this end we have introduced and modeled the concept of photophoretic chiral sorting of SWCNT to investigate the effect of orientation, temperature and optical power on separation times. Whilst fixing the SWCNT orientation reduces the room temperature separation times from 109 ± 12 minutes to 84 ± 12 minutes, elevated temperatures and powers offer far greater enhancements. The 2σ separation time can be reduced to less than 12 minutes by operating at 9 × 107 W m−2 (1.8× the saturation intensity) at 100 °C. These simulations show that optical sorting of SWCNTs is not only feasible but could be over 100 times faster than current approaches whilst guaranteeing high quality SWCNT separation. Although we appreciate the pressing need for experimental verification we propose that this work suggests an exciting avenue for chiral sorting of SWCNTs.DS is funded by an EPSRC Doctoral Training Centre Grant EP/G036780/1.
Notes and references
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- C. X. Chen, W. Zhang, E. S. W. Kong and Y. F. Zhang, Appl. Phys. Lett., 2009, 94, 3 Search PubMed.
- Y. Jia, A. Y. Cao, X. Bai, Z. Li, L. H. Zhang, N. Guo, J. Q. Wei, K. L. Wang, H. W. Zhu, D. H. Wu and P. M. Ajayan, Nano Lett., 2011, 11, 1901–1905 CrossRef CAS PubMed.
- D. D. Tune, B. S. Flavel, R. Krupke and J. G. Shapter, Adv. Energy Mater., 2012, 2, 1043–1055 CrossRef CAS.
- A. R. Rocha, M. Rossi, A. Fazzio and A. J. R. da Silva, Phys. Rev. Lett., 2008, 100, 176083 Search PubMed.
- T. Ueda, M. M. H. Bhulyan, H. Norimatsu, S. Katsuki, T. Ikegami and F. Mitsugi, Phys. E, 2008, 40, 2272–2277 CrossRef CAS PubMed.
- Z. Zanolli, R. Leghrib, A. Felten, J. J. Pireaux, E. Llobet and J. C. Charlier, ACS Nano, 2011, 5, 4592–4599 CrossRef CAS PubMed.
- E. Poirier, R. Chahine, P. Benard, D. Cossement, L. Lafi, E. Melancon, T. K. Bose and S. Desilets, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 961–967 CrossRef CAS.
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209–2214 CrossRef CAS PubMed.
- K. Spyrou, D. Gournis and P. Rudolf, ECS J. Solid State Sci. Technol., 2013, 2, M3160–M3169 CrossRef CAS PubMed.
- P.-J. Tsai, C.-H. Yang, W.-C. Hsu, W.-T. Tsai and J.-K. Chang, Int. J. Hydrogen Energy, 2012, 37, 6714–6720 CrossRef CAS PubMed.
- P. Avouris, Z. H. Chen and V. Perebeinos, Nat. Nanotechnol., 2007, 2, 605–615 CrossRef CAS PubMed.
- M. P. Anantram and F. Leonard, Rep. Prog. Phys., 2006, 69, 507–561 CrossRef CAS.
- M. L. Geier, P. L. Prabhumirashi, J. J. McMorrow, W. C. Xu, J. W. T. Seo, K. Everaerts, C. H. Kim, T. J. Marks and M. C. Hersam, Nano Lett., 2013, 13, 4810–4814 CrossRef CAS PubMed.
- D. M. Sun, C. Liu, W. C. Ren and H. M. Cheng, Small, 2013, 9, 1188–1205 CrossRef CAS PubMed.
- C. Wang, K. Takei, T. Takahashi and A. Javey, Chem. Soc. Rev., 2013, 42, 2592–2609 RSC.
- M. M. Shulaker, G. Hills, N. Patil, H. Wei, H.-Y. Chen, H. S. P. Wong and S. Mitra, Nature, 2013, 501, 526–530 CrossRef CAS PubMed.
- M. M. Shokrieh and R. Rafiee, Mech. Compos. Mater., 2010, 46, 155–172 CrossRef CAS.
- T. W. Odom, J. L. Huang, P. Kim and C. M. Lieber, J. Phys. Chem. B, 2000, 104, 2794–2809 CrossRef CAS.
- A. Vijayaraghavan, J. Mater. Chem., 2012, 22, 7083–7087 RSC.
- M. S. Arnold, A. A. Green, J. F. Hulvat, S. I. Stupp and M. C. Hersam, Nat. Nanotechnol., 2006, 1, 60–65 CrossRef CAS PubMed.
- A. A. Green and M. C. Hersam, Adv. Mater., 2011, 23, 2185–2190 CrossRef CAS PubMed.
- A. L. Antaris, J. W. T. Seo, A. A. Green and M. C. Hersam, ACS Nano, 2010, 4, 4725–4732 CrossRef CAS PubMed.
- A. L. Antaris, J. W. T. Seo, R. E. Brock, J. E. Herriman, M. J. Born, A. A. Green and M. C. Hersam, J. Phys. Chem. C, 2012, 116, 20103–20108 CAS.
- J. A. Fagan, M. L. Becker, J. Chun and E. K. Hobbie, Adv. Mater., 2008, 20, 1609–1613 CrossRef CAS.
- A. A. Green, M. C. Duch and M. C. Hersam, Nano Res., 2009, 2, 69–77 CrossRef CAS.
- M. S. Arnold, S. I. Stupp and M. C. Hersam, Nano Lett., 2005, 5, 713–718 CrossRef CAS PubMed.
- S. Ghosh, S. M. Bachilo and R. B. Weisman, Nat. Nanotechnol., 2010, 5, 443–450 CrossRef CAS PubMed.
- N. Berton, F. Lemasson, J. Tittmann, N. Sturzl, F. Hennrich, M. M. Kappes and M. Mayor, Chem. Mater., 2011, 23, 2237–2249 CrossRef CAS.
- M. Kawai, H. Kyakuno, T. Suzuki, T. Igarashi, H. Suzuki, T. Okazaki, H. Kataura, Y. Maniwa and K. Yanagi, J. Am. Chem. Soc., 2012, 134, 9545–9548 CrossRef CAS PubMed.
- X. Tu, S. Manohar, A. Jagota and M. Zheng, Nature, 2009, 460, 250–253 CrossRef CAS PubMed.
- H. P. Liu, D. Nishide, T. Tanaka and H. Kataura, Nat. Commun., 2011, 2, 8 Search PubMed.
- A. Nish, J. Y. Hwang, J. Doig and R. J. Nicholas, Nat. Nanotechnol., 2007, 2, 640–646 CrossRef CAS PubMed.
- L. Van Hove, Phys. Rev., 1953, 89, 1189–1193 CrossRef CAS.
- M. J. Mendes, H. K. Schmidt and M. Pasquali, J. Phys. Chem. B, 2008, 112, 7467–7477 CrossRef CAS PubMed.
- O. M. Marago, P. H. Jones, F. Bonaccorso, V. Scardaci, P. G. Gucciardi, A. G. Rozhin and A. C. Ferrari, Nano Lett., 2008, 8, 3211–3216 CrossRef CAS PubMed.
- D. M. Carberry, S. H. Simpson, J. A. Grieve, Y. Wang, H. Schafer, M. Steinhart, R. Bowman, G. M. Gibson, M. J. Padgett, S. Hanna and M. J. Miles, Nanotechnology, 2010, 21, 7 CrossRef PubMed.
- D. B. Phillips, D. M. Carberry, S. H. Simpson, H. Schafer, M. Steinhart, R. Bowman, G. M. Gibson, M. J. Padgett, S. Hanna and M. J. Miles, J. Opt., 2011, 13, 8 CrossRef.
- S. H. Simpson and S. Hanna, J. Opt. Soc. Am. A, 2011, 28, 850–858 CrossRef PubMed.
- S. H. Simpson and S. Hanna, Nanotechnology, 2012, 23, 205502 CrossRef CAS PubMed.
- M. S. Arnold, J. E. Sharping, S. I. Stupp, P. Kumar and M. C. Hersam, Nano Lett., 2003, 3, 1549–1554 CrossRef CAS.
- Y. Q. Tan and D. E. Resasco, J. Phys. Chem. B, 2005, 109, 14454–14460 CrossRef CAS PubMed.
- M. E. Itkis, D. E. Perea, S. Niyogi, S. M. Rickard, M. A. Hamon, B. Zhao and R. C. Haddon, Nano Lett., 2003, 3, 309–314 CrossRef CAS.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. P. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593–596 CrossRef CAS PubMed.
- H. J. Keh and H. J. Tu, Colloids Surf., A, 2001, 176, 213–223 CrossRef CAS.
- N. T. Tong, J. Colloid Interface Sci., 1975, 51, 143–151 CrossRef.
- R. Kuhn and S. Hoffstetterkuhn, Chromatographia, 1992, 34, 505–512 CAS.
- F. Albertorio, M. E. Hughes, J. A. Golovchenko and D. Branton, Nanotechnology, 2009, 20, 9 CrossRef PubMed.
- D. A. Tsyboulski, S. M. Bachilo, A. B. Kolomeisky and R. B. Weisman, ACS Nano, 2008, 2, 1770–1776 CrossRef CAS PubMed.
- M. M. Tirado, C. L. Martinez and J. G. Delatorre, J. Chem. Phys., 1984, 81, 2047–2052 CrossRef PubMed.
- P. H. Tan, A. G. Rozhin, T. Hasan, P. Hu, V. Scardaci, W. I. Milne and A. C. Ferrari, Phys. Rev. Lett., 2007, 99, 4 Search PubMed.
- O. N. Torrens, D. E. Milkie, M. Zheng and J. M. Kikkawa, Nano Lett., 2006, 6, 2864–2867 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Simulation details and parameters; calculated viscosities. See DOI: 10.1039/c3cp54812k |
| This journal is © the Owner Societies 2014 |