An ammonia-stabilized mixed-cation borohydride: synthesis, structure and thermal decomposition behavior†
Yanjing
Yang
a,
Yongfeng
Liu
*a,
Hui
Wu
bc,
Wei
Zhou
bc,
Mingxia
Gao
a and
Hongge
Pan
*a
aState Key Laboratory of Silicon Materials, Key Laboratory of Advanced Materials and Applications for Batteries of Zhejiang Province & Department of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: mselyf@zju.edu.cn; hgpan@zju.edu.cn; Fax: +86 571 87952615; Tel: +86 571 87952615
bNIST Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, Maryland 20899-6102, USA
cDepartment of Materials Science and Engineering, University of Maryland, College Park, Maryland 20742-2115, USA
First published on 14th November 2013
Abstract
We demonstrate the synthesis, crystal structure and thermal decomposition behavior of a novel ammonia-stabilized mixed-cation borohydride where the NH3 groups enable the coexistence of Li and Mg cations as an “assistant”. Li2Mg(BH4)4·6NH3, which is comprised of orderly arranged Mg[NH3]62+ ammine complexes and Li2[BH4]42− complex anions, was synthesized by the mechanochemical reaction between Mg(BH4)2·6NH3 and LiBH4. This novel compound crystallizes in a tetragonal P43212 (No. 96) structure with lattice parameters a = b = 10.7656(8) Å and c = 13.843(1) Å with very short dihydrogen bonds, which determine a very low onset temperature of 80 °C for hydrogen release and are also responsible for the nucleation of Li2Mg(BH4)4·3NH3 as a decomposition intermediate. Mechanistic investigations on the thermal decomposition showed that the Hδ+–Hδ− combination in the ammonia-stabilized mixed-cation borohydride was significantly enhanced due to the strengthened Mg–N bonds. Upon heating, 11.02 moles of H2 (equivalent to 11.1 wt%) and 3.07 moles of NH3 are evolved from one mole of Li2Mg(BH4)4·6NH3 with a three-step reaction. The insights into the formation mechanism of ammonia-stabilized mixed-cation borohydride and the role played by NH3 group are very useful as a guideline for the design and synthesis of novel B–N-based materials with high hydrogen content.
Introduction
The accelerating depletion of fossil fuels and the growing concerns on the environmental crises necessitate utilization of green and renewable sources of energy. Due to its high abundance on earth, high energy content and pollution-free oxidation product, hydrogen is regarded as a primary candidate for future energy sources.1 However, safe, efficient and economical hydrogen storage is still a huge challenge.2 Hydrogen storage in solid materials, which is often preferred over traditional compressed gas and liquefied hydrogen storage due to its safe operating conditions and high volumetric capacity, is under extensive investigation.2 Among the materials developed for hydrogen storage,3 metal borohydrides containing [BH4]− anions have been attracting intense interest as one of the most promising hydrogen storage materials due to their quite high gravimetric hydrogen content.4 Unfortunately, the high thermal stability, slow dehydrogenation–hydrogenation kinetics and poor hydrogen storage reversibility often hamper their practical use for mobile applications.5–7 Considerable efforts have been devoted to addressing these thermodynamic and kinetic limitations, including catalyst doping,8 reactive composites,9,10 nanoconfinement,11 and cation/anion substitution,12,13etc. In particular, forming dual- or multiple-cation borohydrides was demonstrated recently as an effective way to manipulate the thermodynamic properties since the thermal stability of metal borohydrides signified by the dehydrogenation temperature is determined by their compositions and structures at the molecular level. A series of mixed-cation borohydrides with completely novel structures were synthesized and characterized, such as LiK(BH4)2, LiZn2(BH4)5, NaZn(BH4)3, NaZn2(BH4)5, LiSc(BH4)4, NaSc(BH4)4, Li4Al3(BH4)13, and so on.12,14,15 It was found that the decomposition temperature of metal borohydrides can be substantially lowered by combining a less electronegative metal with a more electronegative metal.12,14–16 From a practical point of view, the dual- or multiple-cation derivatives of LiBH4, Mg(BH4)2 and Ca(BH4)2, are even more attractive due to their high hydrogen content and potentially favorable thermodynamic properties. However, attempts on the synthesis of the single-phase (Li,Mg), (Li,Ca) and (Mg,Ca) dual-cation borohydrides are still unsuccessful, possibly due to their strong tendency to decompose to the corresponding monometallic borohydrides.10,17Recent research revealed that NH3 can easily adduct to metal borohydrides to form ammoniates with extensive N–H⋯H–B dihydrogen bonds existing in the lattice.18–22 A range of borohydride ammoniates relevant to hydrogen storage applications have been synthesized and characterized, including LiBH4·NH3, Mg(BH4)2·2NH3, Ca(BH4)2·2NH3, Al(BH4)2·6NH3, Zn(BH4)2·2NH3 and Y(BH4)3·4NH3, etc.19,21–24 Moreover, the ammonia-containing derivatives of the (Li,Al) and (Na,Zn) dual-cation borohydrides, Li2Al(BH4)5·6NH3 and NaZn(BH4)3·2NH3, were also disclosed recently.25 These observations stimulate us to synthesize the novel mixed-cation borohydrides by using NH3 as an “assistant” to stabilize the coexistence of different metal cations. Excitingly, the successful observation of LiMg(BH4)3·2NH3 provides a strong evidence of the feasibility of this notion.26 Unfortunately, the resultant product in that work is a mixture of LiMg(BH4)3·2NH3 and LiBH4 instead of the pure single-phase LiMg(BH4)3·2NH3.26 Moreover, the formation mechanism of the compounds of this type and, especially, the role played by NH3 are still unclear. In addition, the preparation of a dual-cation borohydride ammoniate of (Li,Ca) failed by reacting Ca(BH4)2·NH3 and LiBH4, the underlying reaction of which has not been studied and elucidated yet.27
Herein, we report the synthesis, crystal structure and thermal decomposition behavior of a novel ammonia-containing mixed-cation borohydride, Li2Mg(BH4)4·6NH3, by employing Mg(BH4)2·6NH3 as the NH3 supplier to understand the formation mechanism of the multi-cation borohydride ammoniates. It is found that the NH3 groups play a critical role in stabilizing the coexistence of Li and Mg cations in borohydrides. Very short dihydrogen bonds were identified in this novel compound and the structural dependence of the decomposition behavior was elucidated. The new insight into the formation mechanism of the mixed-cation borohydride ammoniates is very useful as a guideline for the design and synthesis of novel B–N-based materials with high hydrogen content.
Experimental
All handling of the samples was conducted in either a Schlenk apparatus or a glovebox (MBRAUN 200B) equipped with a circulation purifier which can keep the concentration of O2 and H2O bellow 0.1 ppm during operation.Materials and sample preparation
The commercially available chemicals, sodium borohydride (NaBH4, Alfa Aesar, 98%),28 anhydrous magnesium chloride (MgCl2, Alfa Aesar, 99%), lithium borohydride (LiBH4, Sigma Aldrich, 95%), potassium borohydride (KBH4, Alfa Aesar, 98%) and anhydrous calcium chloride (CaCl2, Sinopharm Chemical, 96%) were purchased and used as received. Anhydrous diethyl ether and tetrahydrofuran were delivered from Sinopharm Chemical and further dried with calcium hydride (CaH2). Anhydrous ammonia gas (NH3, 99%) was also used as received. Magnesium borohydride (Mg(BH4)2, 96%) was synthesized via the metathesis reaction between sodium borohydride and magnesium chloride in diethyl ether, as described in our previous report.29 Calcium borohydride (Ca(BH4)2, 95%) was prepared through the metathesis reaction between sodium borohydride and calcium chloride in tetrahydrofuran (THF). Magnesium borohydride hexaammoniate (Mg(BH4)2·6NH3) was obtained by ball milling Mg(BH4)2 under 6 bar of ammonia atmosphere, and Mg(BH4)2·3NH3, Mg(BH4)2·2NH3 and Mg(BH4)2·NH3 were prepared by ball milling the mixtures of Mg(BH4)2·6NH3 and Mg(BH4)2 with corresponding molar ratios in Ar atmosphere as reported previously.20 The samples of Mg(BH4)2·6NH3–xLiBH4, Mg(BH4)2·3NH3–xLiBH4, Mg(BH4)2·2NH3–xLiBH4, Mg(BH4)2·NH3–xLiBH4, Mg(BH4)2·6NH3–2NaBH4, Mg(BH4)2·6NH3–2KBH4, and Mg(BH4)2·6NH3–2CaBH4 with different molar ratios were prepared through mechanochemical reactions of the corresponding chemicals on a planetary ball mill (QM-3SP4) rotating at 500 rpm for 24 h. A gas valve that can be connected to a pressure gauge for measuring the inside pressure was mounted on the cover of the milling jar. To ensure even mixing and milling and to limit the temperature rise during milling, the mill was set to rotate for 0.2 h in one direction, followed by a pause of 0.1 h and subsequent rotation in the reverse direction. No obvious temperature increase was observed owing to the ventilation device inside the compartment of the mill.Structural characterization
Phase identification was conducted using powder X-ray diffraction (XRD) on a Phillips X′ Pert Pro X-ray diffractometer with Cu Kα radiation at 40 kV and 40 mA. Data were collected from 3 to 90° (2θ) with step increments of 0.02° at ambient temperature. A specially designed sample container was adopted to protect samples from contamination from the oxygen and moisture. The structure of the as-synthesized compound was solved by direct-space methods using simulate annealing. First-principles molecular dynamics simulated annealing were performed to confirm the BH4 and NH3 configuration with the lowest energy. Rietveld structural refinement on the optimal structural candidate was performed using the GSAS package30 on the XRD data.Vibrational characteristics of the B–H and N–H bonds were determined by a JDbin-yvon LabRam HRUV Raman spectroscopy and a Bruker Tensor 27 Fourier transform infrared spectroscopy. The samples (∼20 mg) were transferred into a homemade optical cell which was then sealed with vacuum grease in the glovebox. Afterwards, the cell was transferred to the apparatus for Raman spectroscopic analysis. The measurement was carried out using the green line of an argon ion laser (λ = 514.5 nm) with a resolution of 1 cm−1. The FTIR spectra of all the samples (as KBr pellet with a KBr-to-sample weight ratio of about 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were acquired in the range of 4000–400 cm−1, and the transmission mode was adopted with a resolution of 4 cm−1.
:1) were acquired in the range of 4000–400 cm−1, and the transmission mode was adopted with a resolution of 4 cm−1.
Solid-state 11B MAS NMR experiments were performed at room temperature on a Bruker Avance II 300 MHz spectrometer at frequencies of 96.216 MHz. The samples were packed into 7 mm ZrO2 rotors and sealed with tightly fitting Kel-F caps inside an Ar-filled glovebox. All spectra were obtained after 1024 scans with an acquisition time of 38 ms per scan and a repetition delay time of 1 s. Chemical shifts (δ) were reported in parts per million (ppm) externally referenced to solid NaBH4 at −41 ppm for 11B nuclei.
Property evaluation
Temperature-programmed decomposition (TPD) measurements were performed on a home-made apparatus attached with an online mass spectrometer (MS, Hiden QIC-20). About 40 mg of sample was loaded into a specially designed tube reactor which allows the purge gas (pure argon) to get through upon heating. The temperature was gradually elevated from room temperature to 600 °C at a ramping rate of 2 °C min−1.Simultaneously, differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) examinations were conducted to determine the heat flow and weight loss of Li2Mg(BH4)4·6NH3 under a gas flow of 50 ml Ar min−1 using a Netzsch STA 449 F3 thermal analyser. About 2 mg of sample was loaded into an Al crucible covered by a lid. The crucible was then put onto the equipment and heated from room temperature to 600 °C at 2 °C min−1.
The hydrogen storage properties of Li2Mg(BH4)4·6NH3 were quantitatively evaluated with a home-made Sieverts-type apparatus. Typically, about 80 mg of the sample was loaded into a stainless steel reactor in the glove box and then connected to the Sieverts-type apparatus. After evacuating the system to 10−3 bar, the sample was heated to a desired temperature at 2 °C min−1 (initially in vacuum) for dehydrogenation and 1 °C min−1 (initially in 100 bar H2) for hydrogenation. The pressure and temperature data were recorded automatically. The amounts of desorbed–absorbed hydrogen were calculated according to the pressure change in the system using the equation of state.
Results and discussion
Structural characteristics of as-synthesized Li2Mg(BH4)4·6NH3
The ammoniates of Mg(BH4)2 (Mg(BH4)2·xNH3, x = 1, 2, 3, and 6) were utilized as NH3 sources to control the precious content of NH3 introduced into the (Li,Mg) dual-cation borohydride systems. Firstly, mixtures of magnesium borohydride hexaammoniate (Mg(BH4)2·6NH3) and lithium borohydride (LiBH4) with different molar ratios (1:0.5−1:6) were mechanochemically reacted and the products were characterized using XRD (Fig. S1, ESI†). A new series of diffraction peaks emerges in the XRD profile of the Mg(BH4)2·6NH3−2LiBH4 sample after ball milling, implying the formation of a novel compound, i.e., Li2Mg(BH4)4·6NH3, by the following reaction: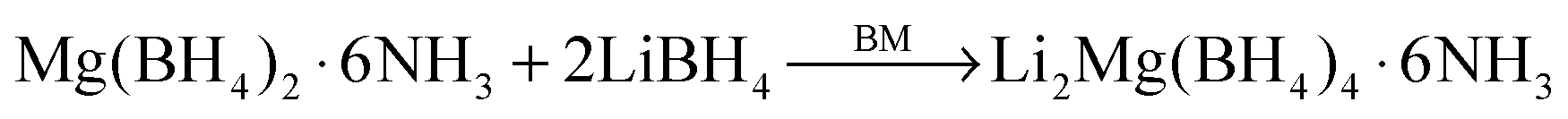 | (1) |
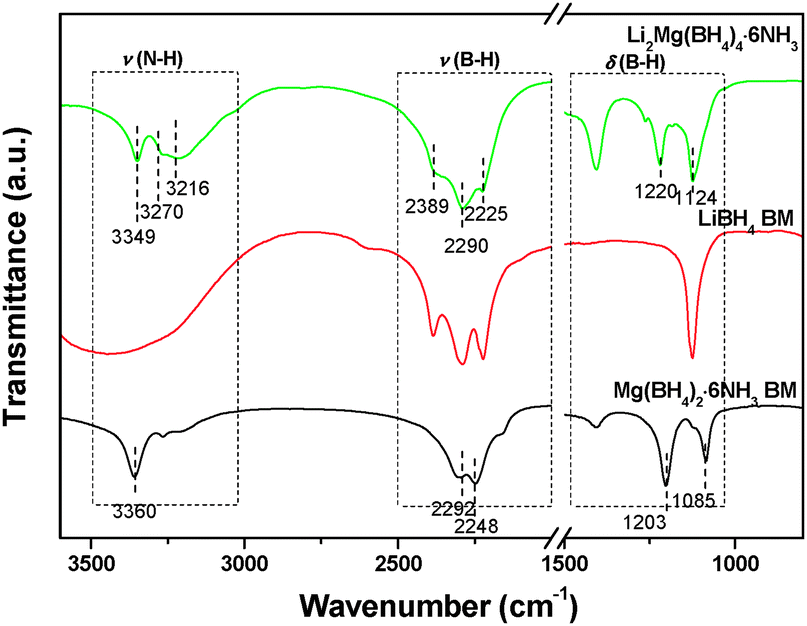 | ||
| Fig. 1 FTIR spectra of Mg(BH4)2·6NH3, LiBH4 and Li2Mg(BH4)4·6NH3. | ||
NMR measurements provide further evidence of the different chemical environment of [BH4]− in Li2Mg(BH4)4·6NH3 as shown in Fig. 2. The 11B NMR spectrum exhibits only one resonance at −36.94 ppm for Li2Mg(BH4)4·6NH3, which is in the region of tetracoordinated boron nuclei, further confirming the persistence of the tetrahedral [BH4]− anions. Moreover, there is a downfield shift for the chemical shift of 11B in Li2Mg(BH4)4·6NH3 in comparison with Mg(BH4)2·6NH3, Mg(BH4)2 and LiBH4, suggesting the more deshielded 11B nuclei in Li2Mg(BH4)4·6NH3.
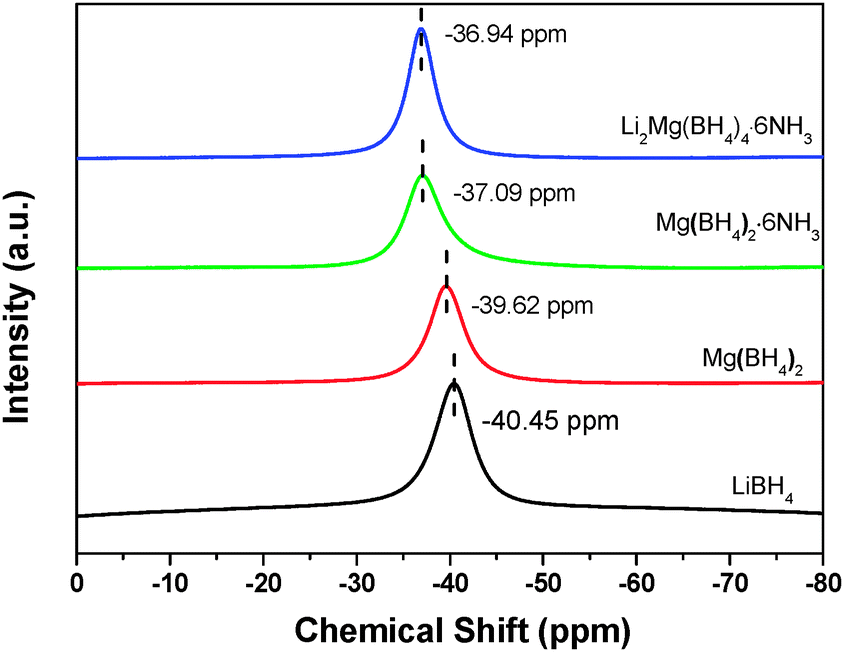 | ||
| Fig. 2 11B NMR spectra of Li2Mg(BH4)4·6NH3, Mg(BH4)2·6NH3, Mg(BH4)2 and LiBH4. | ||
The structure of Li2Mg(BH4)4·6NH3 was solved and refined from the XRD data (Fig. 3). Tables S2 and S3 (ESI†) list the details of the structure refinement and the atomic positions from refinement, respectively. The majority of XRD reflections in the pattern of Li2Mg(BH4)4·6NH3 were indexed to a tetragonal structure with lattice parameters of approximately a = b = 10.7771 Å, c = 13.8620 Å, and V = 1610 Å3. Assessment of the extinction symbol associated with the space group of the new phase indicated P43212 as the most probable. A probable composition of the new phase was surmised from a comparison of the lattice volumes of the precursors, LiBH4 and Mg(BH4)2·6NH3, which have volumes per formula unit of 54 and 316 Å3, respectively. The new phase has a volume per formula unit of 402.5 Å3 (assuming four formula units per unit cell, which is consistent with the extinction symbol P43212), and is approximately the sum of the values for two LiBH4 and one Mg(BH4)2·6NH3 (424 Å3), suggesting that the new phase may possess a Li2Mg(BH4)4·6NH3 stoichiometry. The [BH4]− and NH3 groups were kept as rigid bodies with common refined bond lengths and bond angles constrained as reasonable values due to the inadequate number of observations. B–H distances were defined as 1.23 Å. The N–H distances were constrained as 1.03 Å. The refinement based on laboratory X-ray data cannot provide highly accurate atomic coordinates, especially for H, hence only positions and not orientations of [BH4]− and NH3 units were refined. Isotropic displacement parameters were constrained for the same type of atoms or rigid bodies. The cell parameters for the P43212 structure are: a = b = 10.7656(8) Å, c = 13.843(1) Å and V = 1604.4(3) Å3. The agreement factors are: Rwp = 0.0545, Rp = 0.0409 and GOF = 2.316.
 | ||
| Fig. 3 Experimental (circles), fitted (line), and difference (line below observed and calculated patterns) XRD profiles for Li2Mg(BH4)4·6NH3 at 25 °C. Vertical bars indicate the calculated positions of Bragg peaks. | ||
Fig. 4 illustrates the crystal structure of Li2Mg(BH4)4·6NH3. Each Mg2+ ion in Li2Mg(BH4)4·6NH3 is surrounded by six NH3 groups with Mg–N distances of 2.128–2.298 Å, forming a hexaaminemagnesium(II) (Mg[NH3]62+) complex cation. The Mg–N distances in Li2Mg(BH4)4·6NH3 are quite similar to those in Mg(NH3)6Cl2,19 in which the Mg2+ ions have the same coordination environment, but differ from those in Mg(BH4)2·2NH3, Mg(NH3)2Cl2, Mg(NH3)2Br2 and Mg(NH3)2I2, in which Mg2+ ions are coordinated by both NH3 groups and the corresponding anions.19,31 Li+ ions in this compound are coordinated by four [BH4]− units with Li–B distances of 2.455–2.587 Å, which are close to those in LiBH4 (2.374–2.567 Å)32 and Li(NH3)BH4 (2.52–2.57 Å).21 The Li[BH4]4 tetrahedra are edge-shared with each other in a zigzag fashion, forming a Li2[BH4]42− anionic complex along [001] direction. Each Li2[BH4]42− anionic complex chain is surrounded by adjacent Mg[NH3]62+ cations in [100] and [010] directions, and vice versa, as shown in Fig. 4a and b. Therefore, Li2Mg(BH4)4·6NH3 is comprised of the ordered arrangement of Li2[BH4]42− anionic complex chains and Mg[NH3]62+ cationic complexes in an alternating manner along [110] direction. As shown in Fig. 4c and Table S4 (ESI†), the N–H⋯H–B distances in Li2Mg(BH4)4·6NH3 were determined to be in the range of 1.868–2.406 Å, most of which are more than twice shorter than the van der Waals radius of the H atoms (1.2 Å),33 evidencing the formation of dihydrogen bonding in the structure. Moreover, the closest N–H⋯H–B intermolecular distance is only 1.868 Å, exhibiting the shortest H⋯H distance in all known metal borohydride ammoniates.19–22,26 The short H⋯H distances manifest a strong N–H⋯H–B interaction which links the adjacent Mg[NH3]62+ cations and Li2[BH4]42− anionic chains, and are thus primarily responsible for the stability of the crystal structure of Li2Mg(BH4)4·6NH3. Furthermore, such short N–H⋯H–B dihydrogen bonds are expected to have a significant impact on the dehydrogenation properties of Li2Mg(BH4)4·6NH3 since the driving force of dehydrogenation mostly originates from the combination of Hδ+ in NH3 groups and Hδ− in [BH4]− groups.
 | ||
| Fig. 4 Crystal structure of Li2Mg(BH4)4·6NH3. The Li[BH4]43− tetrahedra are highlighted in yellow and Mg[NH3]62+ hexagons in blue. Li, Mg, B, N and H atoms are donated by yellow, pink, green, blue and white spheres, respectively. (a) (001) view. (b) Off-(100)-view. (c) B–H⋯H–N dihydrogen bonds. The B–H⋯H–N distances were measured in angstroms. | ||
Formation mechanisms of Li2Mg(BH4)4·6NH3 and relevant compounds
For elucidating the role played by NH3 in the formation of Li2Mg(BH4)4·6NH3, an intermediate was collected during the mechanochemical preparation process of Li2Mg(BH4)4·6NH3 for XRD examination. The result is shown in Fig. 5. LiBH4·NH3 is unambiguously identified in the intermediate collected after ball milling for 6 h at 100 rpm in addition to the starting chemical of LiBH4, the nominal Mg(BH4)2·4NH3 and the resultant product of Li2Mg(BH4)4·6NH3. This observation implies that in the mechanochemical synthesis process, a part of NH3 in Mg(BH4)2·6NH3 was first transferred to LiBH4 to form LiBH4·NH3 and a lower-coordinated ammoniate of Mg(BH4)2. Then, they further reacted with each other to produce Li2Mg(BH4)4·6NH3. Thus, the NH3 group plays a critical role in the formation of Li2Mg(BH4)4·6NH3 with the combination of LiBH4 and Mg(BH4)2.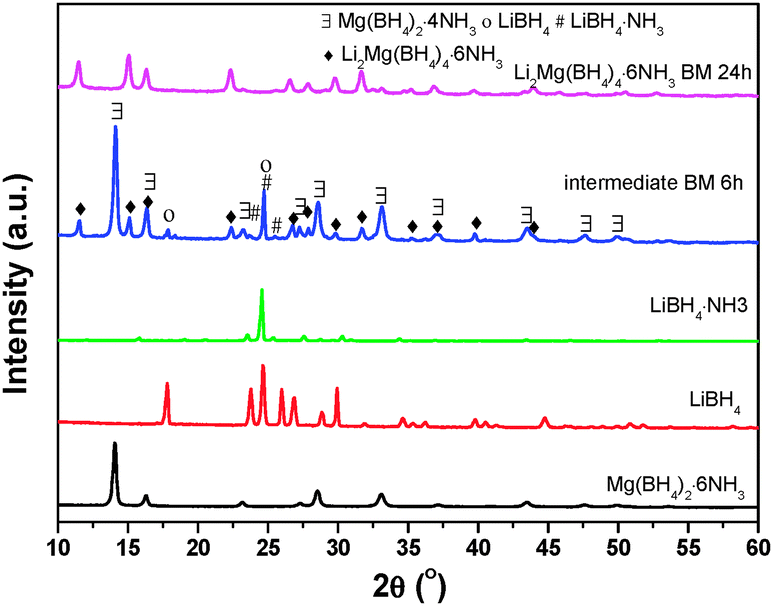 | ||
| Fig. 5 XRD patterns of the as-prepared Li2Mg(BH4)4·6NH3 and the corresponding intermediate collected in the synthesis process. The patterns of Mg(BH4)2·6NH3, LiBH4 and LiBH4·NH3 are added as reference compounds. | ||
Further understanding of the role played by NH3 in compounds of this type was enhanced by synthesizing a series of new ammonia-stabilized (Li,Mg) mixed-cation borohydrides with a single-phase structure, including Li2Mg(BH4)4·3NH3 and LiMg(BH4)3·2NH3 with different magnesium borohydride ammoniates as NH3 sources (Fig. S1–S3, ESI†). It is noticed that more LiBH4 could be incorporated with the Mg(BH4)2 with increasing the NH3 amount. However, an attempt to synthesize the monoammoniate of (Li,Mg) mixed-cation borohydride failed. It is attributed to the insufficient amount of NH3 as stabilizing groups (Fig. S4, ESI†). A similar phenomenon was also observed in the Ca(BH4)2·NH3–LiBH4 composite.27 It was reported that no (Li,Ca) mixed-cation borohydride ammoniate was formed during the mechanochemical process, but a detailed reason was not provided. According to the findings in the present study, we believe that this phenomenon is also closely related to the insufficient amount of NH3 groups in Ca(BH4)2·NH3.
In addition, three designed mixtures with compositions Mg(BH4)2·6NH3–2NaBH4, Mg(BH4)2·6NH3–2KBH4 and Mg(BH4)2·6NH3–2Ca(BH4)2 were subjected to mechanochemical processing to examine the combination possibility of Mg with other light metals such as Na, K and Ca in the presence of NH3.
As shown in Fig. S5 (ESI†), structural investigations revealed that an ammonia-stabilized (Mg,Ca) dual-cation borohydride MgCa2(BH4)6·6NH3 was also obtained. However, no new compound was found while Mg(BH4)2·6NH3 and NaBH4 or KBH4 were mechanochemically reacting, which is possibly due to NaBH4 and KBH4 not being ammoniate-forming species. More interestingly, directly ammoniating K2Mg(BH4)4 and K3Mg(BH4)5 produced Mg(BH4)2·6NH3 and KBH4 as the resultant products instead of the K–Mg mixed-cation ammoniates (Fig. S6, ESI†). These facts suggest that NH3 functionalizes as a “stabilizing group” to assist the formation of mixed-cation borohydride ammoniates. This finding is very useful for guiding the design and synthesis of novel ammonia-stabilized mixed-cation borohydrides.
Thermal decompositions of Li2Mg(BH4)4·6NH3
The thermal decomposition behavior of Li2Mg(BH4)4·6NH3 was determined by TPD-MS and DSC as shown in Fig. 6. TPD-MS results show that the decomposition of Li2Mg(BH4)4·6NH3 under a dynamic flow mode is a three-step process including one deammoniation step and two dehydrogenation steps. Hydrogen dominates the gaseous products although the NH3 signal is also discernable. Moreover, no B2H6 byproduct was detected in the testing temperature range (Fig. S7, ESI†). Observations on the MS signals and the DSC heat flow revealed that deammoniation and the second step of dehydrogenation peaked at 398 °C exhibit an endothermic nature while the first step of dehydrogenation is exothermic. This fact implies that hydrogen desorption from Li2Mg(BH4)4·6NH3 should be partially thermodynamically reversible.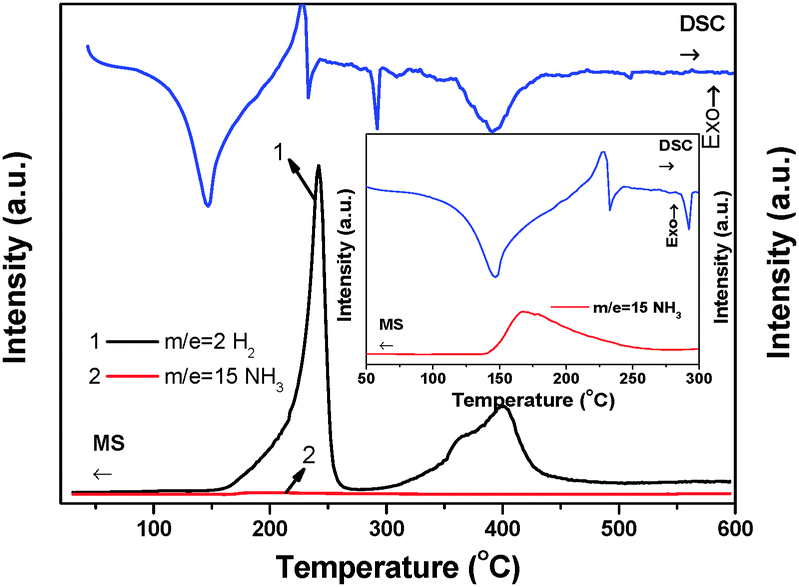 | ||
| Fig. 6 MS and DSC curves of Li2Mg(BH4)4·6NH3. The inset is an enlarged view of the DSC and MS (NH3) signals in the temperature range of 50–300 °C. | ||
Fig. 7 demonstrates the volumetric release and TGA curves of Li2Mg(BH4)4·6NH3 and Mg(BH4)2·6NH3 with temperature. It is clear that the on-set desorption temperature is about 80 °C for Li2Mg(BH4)4·6NH3, which is distinctly lower than that of Mg(BH4)2·xNH3 (x = 1, 2, 3 and 6), Mg(BH4)2 and LiBH4 as reported previously.6,20,34 TGA results show that the weight loss of Li2Mg(BH4)4·6NH3 is 37.30 wt% while heated from room temperature to 600 °C, while it is 51.99 wt% for Mg(BH4)2·6NH3 under the same conditions. Further volumetric measurements reveal that the quantities of gas desorption are 14.09 mol and 10.91 mol per mol of Li2Mg(BH4)4·6NH3 and Mg(BH4)2·6NH3, respectively. Since only H2 and NH3 are released from Li2Mg(BH4)4·6NH3 and Mg(BH4)2·6NH3, the decreased weight loss and the increased volumetric release mean improved purity of hydrogen desorbed from Li2Mg(BH4)4·6NH3. On the basis of TGA and volumetric release results, it is determined that in total 11.02 moles of H2 (equivalent to 11.1 wt%) and 3.07 moles of NH3 are evolved from one mole of Li2Mg(BH4)4·6NH3, exhibiting significantly improved dehydrogenation properties.
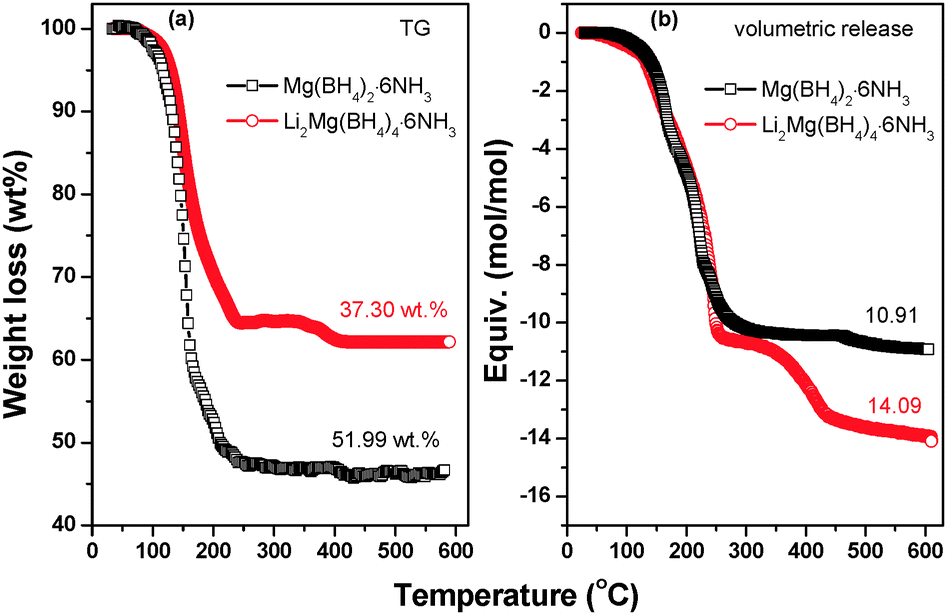 | ||
| Fig. 7 (a) TGA and (b) volumetric release results of Li2Mg(BH4)2·6NH3 and Mg(BH4)2·6NH3. | ||
Thermal decomposition mechanisms of Li2Mg(BH4)4·6NH3
Fig. 8a illustrates XRD patterns of the decomposed Li2Mg(BH4)4·6NH3 samples at different stages. The results indicate that after decomposition at 195 °C, only the characteristic reflections of Li2Mg(BH4)4·3NH3 were observed with the disappearance of Li2Mg(BH4)4·6NH3. With the decomposition temperature elevating to 270 °C, the newly formed Li2Mg(BH4)4·3NH3 is invisible, and MgB2N3 and LiBH4 were identified. For the sample decomposing at 310 °C, only LiBH4 was detected with distinctly decreased diffraction intensities. With further increasing the decomposition temperature to 450 °C, the metallic Mg dominates the XRD profile, and meanwhile BN and LiH were also found although their peak intensities were rather weak. Combining the TPD-MS and DSC results with structural analyses, it can be concluded that upon heating, Li2Mg(BH4)4·6NH3 first decomposed to evolve NH3 and form another new (Li,Mg) ammonia-stabilized mixed-cation borohydride Li2Mg(BH4)4·3NH3. And then, the pure hydrogen was released from the newly developed Li2Mg(BH4)4·3NH3 to produce MgB2N3 and LiBH4. Finally, the chemical reaction between LiBH4 and MgB2N3 proceeds to release hydrogen and generate the resultant products of LiH, Mg, and BN. Since there are 4 mol B atoms and 3 mol N atoms in one Li2Mg(BH4)4·3NH3 unit, the elemental B should exist in the full decomposition sample from a compositional-equilibrium point of view. It is well known that in general, the crystallinity of B obtained by thermal decomposition of metal borohydrides is rather poor,7 which is responsible for its non-detection by XRD. Thus, the chemical events occurring during thermal decomposition of Li2Mg(BH4)4·6NH3 can be described with the following reaction: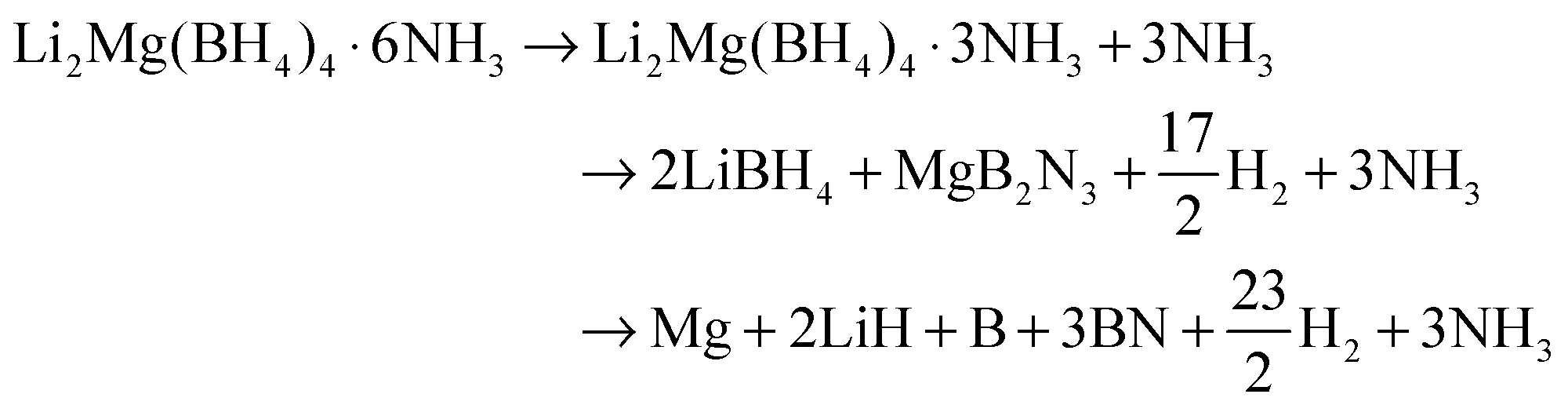 | (2) |
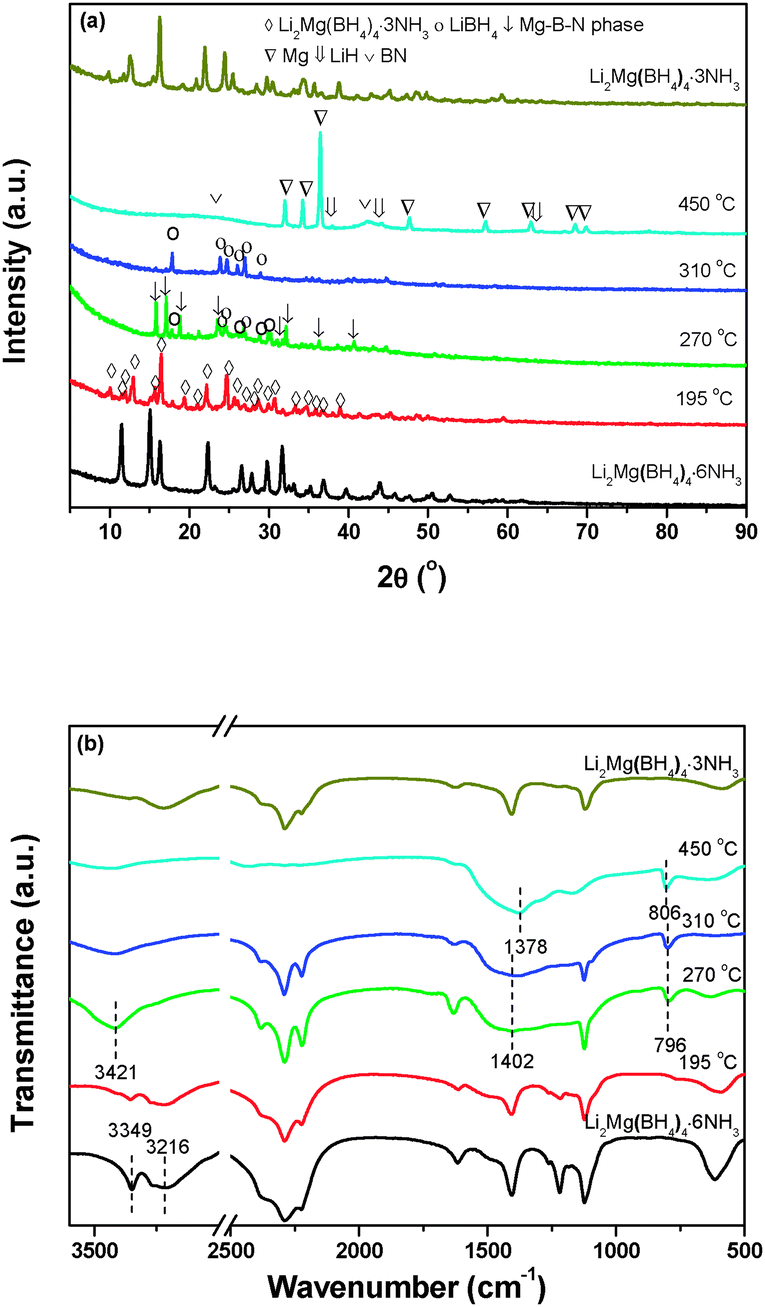 | ||
| Fig. 8 (a) XRD patterns and (b) FTIR spectra of Li2Mg(BH4)4·6NH3 heated to different temperatures. | ||
Relationship between thermal decomposition behavior and structural characteristics of Li2Mg(BH4)4·6NH3
It is noteworthy that ammonia release was depressed significantly for Li2Mg(BH4)4·6NH3 with respect to Mg(BH4)2·6NH3. As reported previously, the ammonia release from borohydride ammoniates correlates closely to the molar ratio of NH3/[BH4]− and the strength of M–NH3 bond (M: metal).20 Raman spectra were compared to elucidate the reason for depressed ammonia release from Li2Mg(BH4)4·6NH3. Fig. 9 shows the Raman spectra of Mg(BH4)2·6NH3 and Li2Mg(BH4)4·6NH3. The identical features of the N–H vibration were observed for Mg(BH4)2·6NH3 and Li2Mg(BH4)4·6NH3 as both of them exhibit three N–H vibrations at around 3178, 3270 and 3346 cm−1, suggesting an identical local environment for Mg atoms in these two compounds, which are hexacoordinated by six NH3 units.19 However, there is an obvious difference in the B–H vibrations at 2000–2500 cm−1. Two Raman peaks at 2365 and 2266 cm−1 were detected for Li2Mg(BH4)4·6NH3 while only one Raman peak at 2279 cm−1 was observed for Mg(BH4)2·6NH3, which is possibly attributed to the fact that [BH4]− units in Mg(BH4)2·6NH3 coordinate to Mg cations but those in Li2Mg(BH4)4·6NH3 coordinate to Li cations. Interestingly, as shown in the inset in Fig. 9, a blue shift for the Mg–N bond was found from 321 cm−1 in Mg(BH4)2·6NH3 to 326 cm−1 in Li2Mg(BH4)4·6NH3, implying a strengthened Mg–N bonding in the dual-cation Li2Mg(BH4)4·6NH3 with respect to the monometallic Mg(BH4)2·6NH3. Such bonding characteristics provides an additional restriction for the NH3 groups and consequently facilitates the local combination of Hδ− in the [BH4]− groups and Hδ+ in the NH3 groups, as reported in our previous work.20 On the other hand, as mentioned above, LiBH4 was separated from the (Li,Mg)-containing compound after the first step of dehydrogenation, which means that the [BH4]− groups in LiBH4 did not contribute to the Hδ+–Hδ− combination reaction. Therefore, the strengthened Mg–N bond instead of the increased molar ratio of NH3/[BH4]− in Li2Mg(BH4)4·6NH3 is reasonably responsible for decreased ammonia release.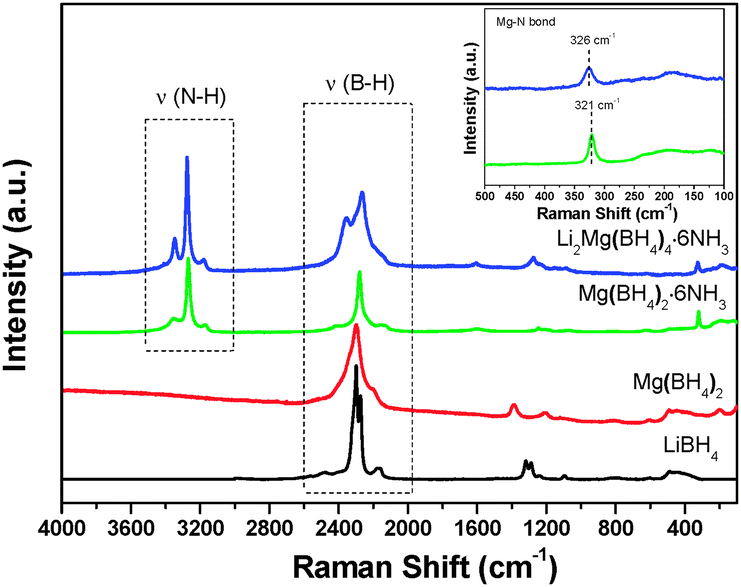 | ||
| Fig. 9 Raman spectra of Li2Mg(BH4)4·6NH3, Mg(BH4)2·6NH3 and the pristine Mg(BH4)2 and LiBH4. The inset is an enlarged view of the Raman spectra at 100–500 cm−1. | ||
A close examination on the decomposition behavior of Li2Mg(BH4)4·6NH3 under dynamic heating conditions reveals a minor hydrogen release before initiation of ammonia release (Fig. S9, ESI†). This phenomenon, which should correlate closely with the unique structural characteristics of Li2Mg(BH4)4·6NH3, has not been discovered in the decomposition of other metal borohydride ammoniates.19–22,25,26 Further isothermal MS measurements at 120 °C indicated that there is an induction period of 36 min for the initial decomposition reaction of Li2Mg(BH4)4·6NH3 as shown in Fig. 10. Interestingly, minor hydrogen release was also observed in this induction period and at even lower temperatures. As mentioned above, three dihydrogen bonds of H4⋯H11 (1.8680 Å), H8⋯H10 (1.8360 Å) and H8⋯H14 (1.8781 Å) are much shorter than other N–H⋯H–B distances in Li2Mg(BH4)4·6NH3. Such short Hδ+ and Hδ− distances favor the local combination of the corresponding Hδ+ and Hδ− upon heating, consequently inducing hydrogen release at lower temperatures. Moreover, this process consumes selectively three NH3 groups of Li2Mg(BH4)4·6NH3 and produces Li2Mg(BH4)4·3NH3. The newly developed Li2Mg(BH4)4·3NH3 probably plays a critical role as the nucleation centers in speeding up the first-step decomposition of Li2Mg(BH4)4·6NH3. This notion was supported further by the fact that directly adding a small amount of Li2Mg(BH4)4·3NH3 can remarkably shorten the induction period of the initial deammoniation of Li2Mg(BH4)4·6NH3 to 13 min (Fig. 10). It is therefore believed that the structural characteristics of Li2Mg(BH4)4·6NH3 are responsible for its low decomposition temperature.
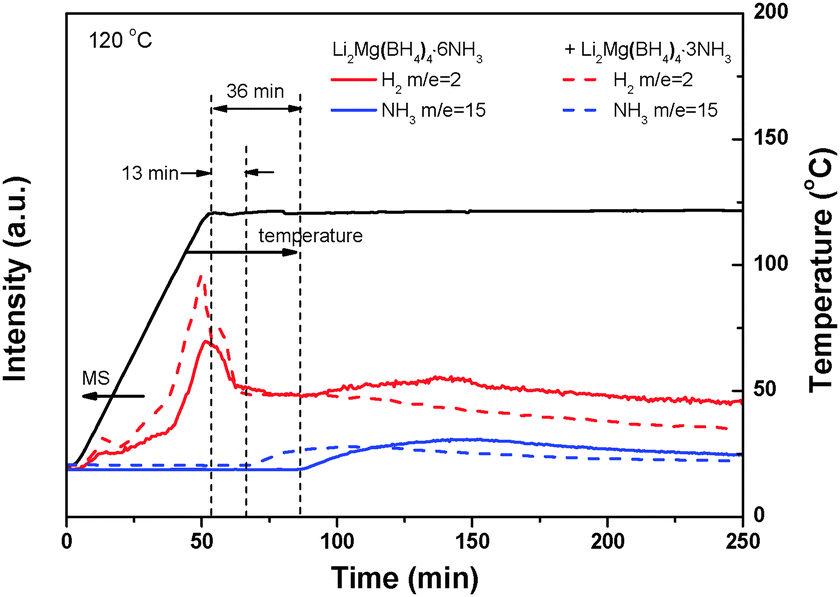 | ||
| Fig. 10 Isothermal MS spectra of Li2Mg(BH4)2·6NH3 with and without extra addition of Li2Mg(BH4)4·3NH3. | ||
Reversibility for hydrogen storage in Li2Mg(BH4)4·6NH3
Hydrogen storage reversibility was further evaluated by hydrogenating the fully dehydrogenated Li2Mg(BH4)4·6NH3. Fig. 11 shows the temperature dependence of hydrogenation–re-dehydrogenation behavior of Li2Mg(BH4)4·6NH3. It can be seen that under 100 bar of hydrogen pressure, the fully decomposed product starts to absorb hydrogen at about 70 °C, and hydrogen uptake amounts to about 4 wt% as it is heated to 450 °C, exhibiting partial hydrogen storage reversibility. The follow-up re-dehydrogenation experiment confirms a 4 wt% reversible hydrogen capacity. Such phenomenon has not been reported in other known metal borohydride ammoniates.19–22,26 | ||
| Fig. 11 (a) Hydrogenation and (b) re-dehydrogenation curves of Li2Mg(BH4)4·6NH3 with temperature. | ||
To understand the reversible hydrogen storage process, the rehydrogenated samples at different stages were collected for XRD and FTIR analyses. Fig. S10 (ESI†) presents the XRD patterns and FTIR spectra of the re-hydrogenated Li2Mg(BH4)4·6NH3 samples at different stages. The results indicate that the sample hydrogenated at 230 °C consists mainly of MgH2, LiH and BN. After hydrogenation at 450 °C, LiBH4 can be unambiguously identified in the XRD and FTIR profiles, and a new set of diffraction peaks was also observed in the XRD pattern along with absence of MgH2. Further FTIR examinations show a new B–H absorbance at 2484 cm−1, which can be assigned to the [B12H12]− anion as reported previously.35 As a result, it is believed that a new Mg–B–H compound containing the [B12H12]− anion may also be formed in the hydrogenation product.
Conclusions
In this paper, ammonia was found to play a “stabilizer” role in forming the ammonia-stabilized mixed-cation borohydride for the first time, and a series of novel ammonia-stabilized mixed-cation borohydrides were synthesized including Li2Mg(BH4)4·6NH3, Li2Mg(BH4)4·3NH3, LiMg(BH4)3·2NH3, and MgCa2(BH4)6·6NH3. Of them, Li2Mg(BH4)4·6NH3 crystallizes in a tetragonal P43212 (No. 96) structure with an ordered arrangement of Mg[NH3]62+ ammine complexes and Li2[BH4]42− complex anions. The extremely short dihydrogen bonds (1.868–1.9818 Å) were found in the Li2Mg(BH4)4·6NH3, which facilitates the local combination of Hδ+ in the NH3 groups and Hδ− in the BH4 groups, consequently increasing hydrogen release and decreasing ammonia evolution. While heating Li2Mg(BH4)4·6NH3 from room temperature to 600 °C, in total 11.02 moles of H2 (equivalent to 11.1 wt%) and 3.07 moles of NH3 were released via a three-step reaction. Mechanistic investigations revealed that upon heating, Li2Mg(BH4)4·6NH3 first decomposed to evolve NH3 and form Li2Mg(BH4)4·3NH3. And then, the newly developed Li2Mg(BH4)4·3NH3 decomposed to produce MgB2N3 and LiBH4. Finally, the chemical reaction between LiBH4 and MgB2N3 proceeds to release hydrogen and generate the resultant products of LiH, Mg and BN. The formation of Li2Mg(BH4)4·3NH3 in the first step of decomposition plays a critical role as the nucleation centre in speeding up the first-step decomposition of Li2Mg(BH4)4·6NH3. The findings in this work provide insights into the formation mechanism of dual-cation borohydride ammoniates, which will guide the design and synthesis of the novel B–N-based materials with high hydrogen content.Acknowledgements
We gratefully acknowledge financial support received from the Ministry of Science and Technology of China (2010CB631304), the National Natural Science Foundation of China (51222101, 51025102, 51171170), the Fundamental Research Funds for the Central Universities (2013QNA4009), and the Science and Technology Department of Zhejiang Province (2011R10017 and 2010R50013).References
- J. A. Ritter, A. D. Ebner, J. Wang and R. Zidan, Mater. Today, 2003, 6, 18 CrossRef CAS; I. Jain, Int. J. Hydrogen Energy, 2009, 34, 7368 CrossRef PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef CAS PubMed.
- W. Li, C. Li, H. Ma and J. Chen, J. Am. Chem. Soc., 2007, 129, 6710 CrossRef CAS PubMed; B. Bogdanović and M. Schwickardi, J. Alloys Compd., 1997, 253, 1 CrossRef; P. Chen, Z. T. Xiong, J. Z. Luo, J. Y. Lin and K. L. Tan, Nature, 2002, 420, 302 CrossRef PubMed; Y. F. Liu, K. Zhong, K. Luo, M. X. Gao, H. G. Pan and Q. D. Wang, J. Am. Chem. Soc., 2009, 131, 1862 CrossRef PubMed; A. Züttel, S. Rentsch, P. Fischer, P. Wenger, P. Sudan, P. Mauron and C. Emmenegger, J. Alloys Compd., 2003, 356, 515 CrossRef.
- H.-W. Li, Y. G. Yan, S.-i. Orimo, A. Züttel and C. M. Jensen, Energies, 2011, 4, 185 CrossRef CAS.
- K. Chłopek, C. Frommen, A. Léon, O. Zabara and M. Fichtner, J. Mater. Chem., 2007, 17, 3496 RSC; J. H. Kim, S. A. Jin, J. H. Shim and Y. W. Cho, J. Alloys Compd., 2008, 461, L20 CrossRef CAS PubMed; G. Severa, E. Rönnebro and C. M. Jensen, Chem. Commun., 2010, 46, 421 RSC.
- A. Züttel, P. Wenger, S. Rentsch, P. Sudan, P. Mauron and C. Emmenegger, J. Power Sources, 2003, 118, 1 CrossRef.
- S.-i. Orimo, Y. Nakamori, G. Kitahara, K. Miwa, N. Ohba, S.-i. Towata and A. Züttel, J. Alloys Compd., 2005, 404, 427 CrossRef PubMed.
- J. E. Chen, Y. Zhang, Z. T. Xiong, G. T. Wu, H. L. Chu, T. He and P. Chen, Int. J. Hydrogen Energy, 2012, 37, 12425 CrossRef CAS PubMed; A. Al-Kukhun, H. T. Hwang and A. Varma, Int. J. Hydrogen Energy, 2012, 37, 17671 CrossRef PubMed.
- J. J. Vajo, S. L. Skeith and F. Mertens, J. Phys. Chem. B, 2005, 109, 3719 CrossRef CAS PubMed.
- E. G. Bardají, Z. Zhao-Karger, N. Boucharat, A. Nale, M. J. van Setten, W. Lohstroh, E. Röhm, M. Catti and M. Fichtner, J. Phys. Chem. C, 2011, 115, 6095 Search PubMed.
- M. Fichtner, Z. Zhao-Karger, J. Hu, A. Roth and P. Weidler, Nanotechnology, 2009, 20, 204029 CrossRef PubMed.
- E. A. Nickels, M. O. Jones, W. I. F. David, S. R. Johnson, R. L. Lowton, M. Sommariva and P. P. Edwards, Angew. Chem., Int. Ed., 2008, 47, 2817 CrossRef CAS PubMed.
- L. H. Rude, E. Groppo, L. M. Arnbjerg, D. B. Ravnsbaek, R. A. Malmkjaer, Y. Filinchuk, M. Baricco, F. Besenbacher and T. R. Jensen, J. Alloys Compd., 2011, 509, 8299 CrossRef CAS PubMed.
- D. Ravnsbaek, Y. Filinchuk, Y. Cerenius, H. J. Jakobsen, F. Besenbacher, J. Skibsted and T. R. Jensen, Angew. Chem., Int. Ed., 2009, 48, 6659 CrossRef CAS PubMed.
- H. Hagemann, M. Longhini, J. W. Kaminski, T. A. Wesolowski, R. černý, N. Penin, M. H. Sørby, B. C. Hauback, G. Severa and C. M. Jensen, J. Phys. Chem. A, 2008, 112, 7551 CrossRef CAS PubMed; R. Cerny, G. Severa, D. B. Ravnsbaek, Y. Filinchuk, V. D'Anna, H. Hagemann, D. r. Haase, C. M. Jensen and T. R. Jensen, J. Phys. Chem. C, 2009, 114, 1357 Search PubMed; D. B. Ravnsbaek, L. H. Sørensen, Y. Filinchuk, F. Besenbacher and T. R. Jensen, Angew. Chem., Int. Ed., 2012, 51, 3582 CrossRef PubMed.
- Y. Nakamori, K. Miwa, A. Ninomiya, H. Li, N. Ohba, S.-i. Towata, A. Züttel and S.-i. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045126 CrossRef.
- S. H. Lee, V. R. Manga and Z. K. Liu, Int. J. Hydrogen Energy, 2010, 35, 6812 CrossRef CAS PubMed; J. Y. Lee, D. Ravnsbaek, Y.-S. Lee, Y. Kim, Y. Cerenius, J.-H. Shim, T. R. Jensen, N. H. Hur and Y. W. Cho, J. Phys. Chem. C, 2009, 113, 15080 Search PubMed; A. Nale, M. Catti, E. G. Bardají and M. Fichtner, Int. J. Hydrogen Energy, 2011, 36, 13676 CrossRef PubMed; T. Durojaiye, A. A. Ibikunle and A. J. Goudy, Int. J. Hydrogen Energy, 2010, 35, 10329 CrossRef PubMed; A. A. Ibikunle and A. J. Goudy, Int. J. Hydrogen Energy, 2012, 37, 12420 CrossRef PubMed; Y. G. Yan, A. Remhof, P. Mauron, D. Rentsch, Z. Łodziana, Y.-S. Lee, H.-S. Lee, Y. W. Cho and A. Züttel, J. Phys. Chem. C, 2013, 117, 8878 Search PubMed.
- E. A. Sullivan and S. Johnson, J. Phys. Chem., 1959, 63, 233 CrossRef CAS.
- G. Soloveichik, J. H. Her, P. W. Stephens, Y. Gao, J. Rijssenbeek, M. Andrus and J. C. Zhao, Inorg. Chem., 2008, 47, 4290 CrossRef CAS PubMed.
- Y. J. Yang, Y. F. Liu, Y. Li, M. X. Gao and H. G. Pan, Chem.–Asian J., 2013, 8, 476 CrossRef CAS PubMed.
- S. R. Johnson, W. I. F. David, D. M. Royse, M. Sommariva, C. Y. Tang, F. Fabbiani, M. O. Jones and P. P. Edwards, Chem.–Asian J., 2009, 4, 849 CrossRef CAS PubMed.
- H. L. Chu, G. T. Wu, Z. T. Xiong, J. Guo, T. He and P. Chen, Chem. Mater., 2010, 22, 6021 CrossRef CAS; Y. H. Guo, X. B. Yu, W. W. Sun, D. L. Sun and W. N. Yang, Angew. Chem., Int. Ed., 2011, 50, 1087 CrossRef PubMed; Y. H. Guo, Y. X. Jiang, G. L. Xia and X. B. Yu, Chem. Commun., 2012, 48, 4408 RSC.
- Q. F. Gu, L. Gao, Y. H. Guo, Y. B. Tan, Z. W. Tang, K. S. Wallwork, F. W. Zhang and X. B. Yu, Energy Environ. Sci., 2012, 5, 7590 CAS.
- F. Yuan, Q. F. Gu, Y. H. Guo, W. W. Sun, X. W. Chen and X. B. Yu, J. Mater. Chem., 2012, 22, 1061 RSC.
- Y. H. Guo, H. Wu, W. Zhou and X. B. Yu, J. Am. Chem. Soc., 2011, 133, 4690 CrossRef CAS PubMed; G. L. Xia, Q. F. Gu, Y. H. Guo and X. B. Yu, J. Mater. Chem., 2012, 22, 7300 RSC.
- W. W. Sun, X. W. Chen, Q. F. Gu, K. S. Wallwork, Y. B. Tan, Z. W. Tang and X. B. Yu, Chem.–Eur. J., 2012, 18, 6825 CrossRef CAS PubMed.
- Z. W. Tang, Y. B. Tan, Q. F. Gu and X. B. Yu, J. Mater. Chem., 2012, 22, 5312 RSC.
- Certain commercial suppliers are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the NIST, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
- Y. F. Liu, Y. J. Yang, Y. F. Zhou, Y. Zhang, M. X. Gao and H. G. Pan, Int. J. Hydrogen Energy, 2012, 37, 17137 CrossRef CAS PubMed.
- A. C. Larson, R. B. Von Dreele, in General Structure Analysis System; Report LaUR 86-784; Los Alamos National Laboratory: NM, 1994.
- A. Leineweber, M. W. Friedriszik and H. Jacobs, J. Solid State Chem., 1999, 147, 229 CrossRef CAS.
- Y. Filinchuk, D. Chernyshov and R. Cerny, J. Phys. Chem. C, 2008, 112, 10579 CAS.
- F. H. Stephens, V. Pons and R. Tom Baker, Dalton Trans., 2007, 2613 RSC.
- Y. J. Yang, M. X. Gao, Y. F. Liu, J. H. Wang, J. Gu, H. G. Pan and Z. X. Guo, Int. J. Hydrogen Energy, 2012, 37, 10733 CrossRef CAS PubMed.
- E. Muetterties, R. Merrifield, H. Miller, W. Knoth and J. Downing, J. Am. Chem. Soc., 1962, 84, 2506 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data in CIF; crystallographic details, XRD, MS, FTIR results, including Fig. S1–S10 and Tables S1–S4. CCDC 964912. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cp54099e |
| This journal is © the Owner Societies 2014 |