 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoisomerization of the cyanoacrylic acid acceptor group – a potential problem for organic dyes in solar cells†
Burkhard
Zietz
*a,
Erik
Gabrielsson
b,
Viktor
Johansson
c,
Ahmed M.
El-Zohry
a,
Licheng
Sun
b and
Lars
Kloo
*c
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 523, SE-751 20 Uppsala, Sweden. E-mail: Burkhard.zietz@kemi.uu.se
bOrganic Chemistry, KTH Royal Institute of Technology, Teknikringen 30, SE-100 44 Stockholm, Sweden
cApplied Physical Chemistry, KTH Royal Institute of Technology, Teknikringen 30, SE-100 44 Stockholm, Sweden. E-mail: Larsa@kth.se
First published on 28th November 2013
Abstract
Organic solar cell dyes containing the most common anchoring group, cyanoacrylic acid, are shown to be photolabile and undergo photoisomerization. This may have significant consequences for dye-sensitized solar cells, as isomerisation competes with electron injection and leads to modifications of the dye and surface arrangement.
In the global ambitions to move towards a sustainable society, renewable energy systems will become essential. Considering the massive inflow of solar energy, it is more than likely that the future energy systems will be based on the conversion of solar light into either electricity or energy-rich substances using a multitude of strategies and technologies.1 In this aspect the direct conversion of light into electricity, by solar cells, will have an important role.
In 1991, O'Regan and Grätzel converted the photoelectrochemical cell from a purely academic research interest to one of the potential solar cell technologies for the future.2 The breakthrough by O'Regan and Grätzel involved the use of a nanostructured semiconductor substrate (titania), increasing the effective surface for light harvesting by nearly a factor of 1000. As a consequence, conversion efficiencies were improved from about 1% to 7–8% in one step.
The photoelectrochemical cell is also referred to as a Grätzel cell or dye-sensitized solar cell (DSC). The latter denotation emerges since the semiconductor substrate in itself only absorbs solar light at very short wavelengths, making over-all conversion efficiencies quite low. Therefore, the nanostructured semiconductor substrate is sensitized by a dye that can absorb light and inject electrons into the semiconductor substrate.
The commonly used sensitizing dyes are metal–organic or organic dye molecules, but other sensitizers such as quantum dots have also been used.3 Among the metal–organic ones, ruthenium polypyridine complexes are most frequently used. However, there are many directions of development aiming at new types of sensitizing molecules, with the most active direction towards organic dye molecules, motivated by the ambition to avoid precious and rare metals as part of the DSC. Another advantage with organic dyes is that they often show more intense absorption (higher molar extinction coefficients) than the organometallic ones. The organic dyes are generally based on three separate components: a donor, a linker and an acceptor group (the D–π–A concept) offering charge separation upon excitation with relatively long-lived excited states as a consequence, assuming that no photochemical processes are competing. Several hundreds of new sensitizing organic dyes have been reported, and they are typically classified according to the type of their donor group. The most extensively studied type of organic dyes is based on a triarylamine donor, introduced by Yanagida and co-workers4 and later significantly extended and improved by Sun and collaborators.5 By combining the acceptor group with an anchoring group for the semiconductor substrate, the stage is set for efficient injection of electrons from the excited state, mainly residing on the acceptor group, to the semiconductor conduction band. Clearly dominating among the anchoring groups is the cyanoacrylic acid (Fig. 1), combining the electron withdrawing properties of the cyano-unit with the binding motif of the carboxylic group. In a recent review on DSCs, more than half (54%) of the surveyed organic molecules contained the cyanoacrylic acid as a binding group. This single most commonly used binding group and its photochemistry is the focus of the present work.
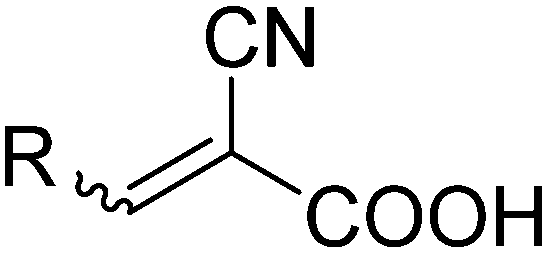 | ||
| Fig. 1 The cyanoacrylic acid acceptor/anchoring group. | ||
The concept of anchoring is based on the presumption of a self-assembled monolayer, where the anchoring group binds in an orderly fashion to the semiconductor surface and the dye molecules align up to form an adsorbed, regular monolayer. However, this presumption has recently been challenged on the basis of direct experimental investigations on the adsorbed dye layers at different coverage levels, and the mode of anchoring is not obvious.6–8
Nevertheless, the anchoring group has an essential function in the dye molecules, and is thus central to the dye function as a sensitizer in the DSC devices. We have therefore chosen the simplest triarylamine dye molecule of D–π–A type, the so-called L0 dye (Fig. 2), as a system for fundamental studies on dye adsorption, anchoring and organization in various titania model surfaces. From a spectroscopic aspect, the –CN and –COOH groups both offer potential insights into the chemistry of the dye molecule upon anchoring and function in a DSC. However, all elements in an organic molecule tend to be light, thus giving only weak contrasts in any type of X-ray-based experiment, such as diffraction or absorption spectroscopy. Therefore, we have synthesized an analogue to the L0 dye, by exchanging the cyano group for a single bromine atom, L0Br (Fig. 2). Although the L0Br molecule is likely to work less well as a sensitizer in a DSC, the Br works as a ‘spy’ atom providing clearer information from X-ray-based experiments.
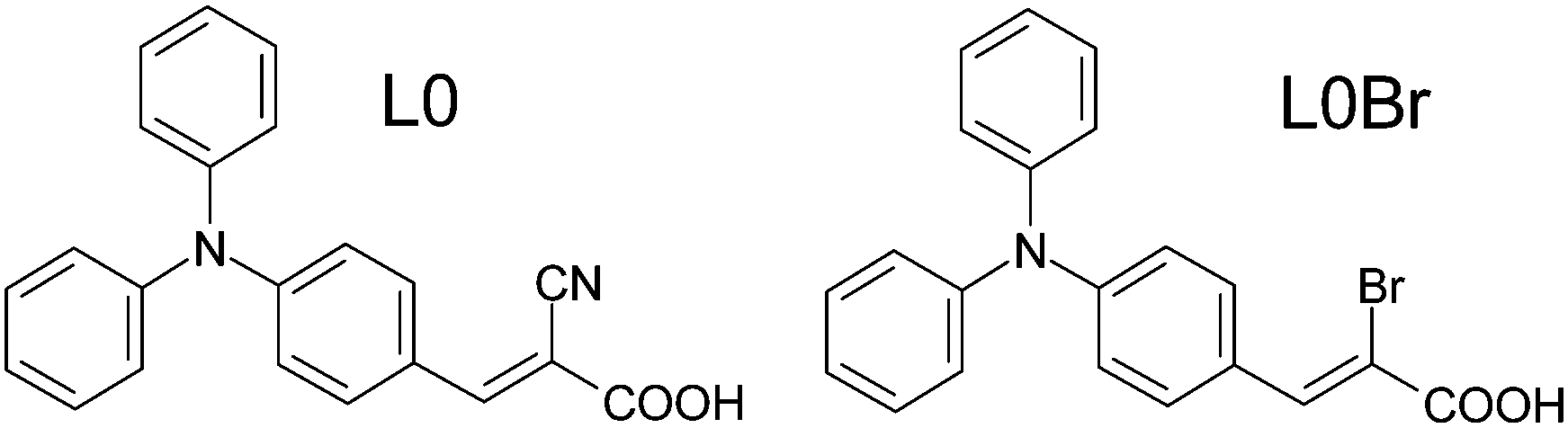 | ||
| Fig. 2 The L0 (left) and L0Br (right) dye molecules in trans configuration.9 | ||
While the double bond present in the cyanoacrylic acid group fills the important function of bridging the π-systems of the donor and anchor/acceptor, it also carries a weakness. The potential energy curves in the excited (triplet and singlet) states in e.g. ethylene lead to a twisting of the double bond and to the formation of geometrical isomers.10 Transition from the Franck–Condon-region to a 90°-distorted bond can proceed over a barrier, as in the case for the singlet excited state of stilbene and its derivatives.11,12 Alternatively, isomerization can proceed through the triplet state. Intersystem crossing is usually inefficient and sensitization instead of direct excitation is required.13 While these textbook examples point towards a possible singlet mechanism, it should be kept in mind that the excited state in the L0Br dye does not correspond to a direct π–π* excitation of the double bond, but rather a charge-transfer of the complete molecule. The mechanistic details for this case, including the singlet vs. triplet mechanism, therefore require further investigation. It has already been noted that photoisomerization is a potential complication in dye molecules used in DSCs.14,15 In the synthetic procedure used for both L0 and L0Br, the trans isomer is obtained almost exclusively (see the synthetic procedure of L0Br in the ESI†).
The first indications that photoisomerization problems may appear in dye molecules were obtained accidentally. While recording the UV/vis-spectra of solutions of L0Br, a change in the spectra over time was observed. This change could be attributed to a reversible trans–cis-photoisomerization and could also be detected later in the parent molecule L0 – it is thus a general phenomenon of D–π–A dyes containing a cyanoacrylic acid group. The reason why this has not previously been noted is that the change in absorption spectra of L0Br is significant, whereas it is much less obvious for L0. The exchange of the –CN group for –Br thus made a general but overseen phenomenon clearly visible.
Photoisomerization in the acceptor/anchoring group can have significant effects on the presumed adsorption mode, the surface dye organization and the mechanism of electron injection of a very large number of known organic dye molecules. Therefore, in this work we report on the photoisomerization of the cyanoacrylic acid group (of course, in L0Br the anchoring unit is a bromoacrylic acid, but for the sake of simplicity we will refer to the anchoring unit as the cyanoacrylic acid) in the dyes L0 and L0Br.
Because of the clear spectral changes observed for the L0Br molecule, it is natural to use a strategy of investigation starting with the effects observed for L0Br and subsequently confirm the same or similar behaviour for L0.
When irradiating a dilute solution of L0Br in a polar solvent (such as ethanol, acetonitrile or DMSO) into the main absorption band (S0 → S1 transition, ca. 360 nm), marked changes in the absorption can be observed (see Fig. 3): a decrease of the main band and an increase in absorption between ca. 300 and 340 nm with a clear isosbestic point (344 nm in ethanol). The existence of an isosbestic point confirms that a transition between only two species is taking place. Upon prolonged irradiation, the changes become smaller and a photostationary state is reached. The absorption thus consists of two contributions, from the original trans-L0Br and from the newly formed isomer cis-L0Br:
| Amix = xAtrans + yAcis, |
| Acis = (Amix − xAtrans)/y |
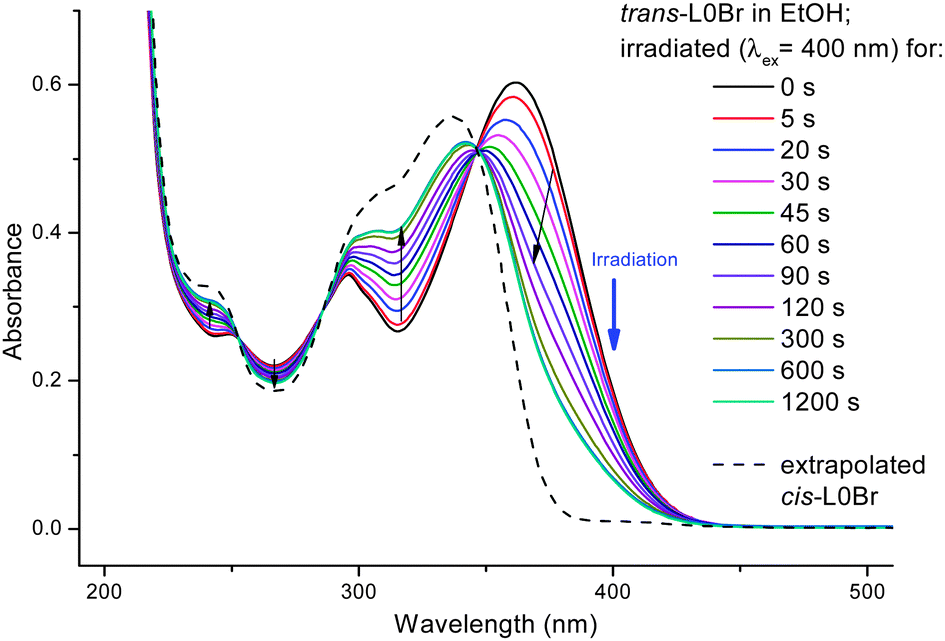 | ||
| Fig. 3 UV/vis-spectra of trans-L0Br in ethanol upon irradiation at 400 nm. For the spectrum of cis-L0Br, see text. | ||
The isosbestic point is evidence for a transformation between two species, but not necessarily for a photoisomerization (any photodegradation could lead to a similar behavior). After having reached a photostationary state, we have chosen to change the excitation wavelength to a region where an increase in absorption was observed and irradiated at 323 nm. The results (Fig. 4) show a reversal of the previously observed trend: an increase of the main band (around 360 nm) and a decrease of absorption between ca. 280 and 350 nm. Due to the reversibility we are able to attribute the spectral changes to a photoisomerization (prolonged irradiation of L0Br with shorter wavelength UV light leads to slight decomposition and loss of isosbestic points).
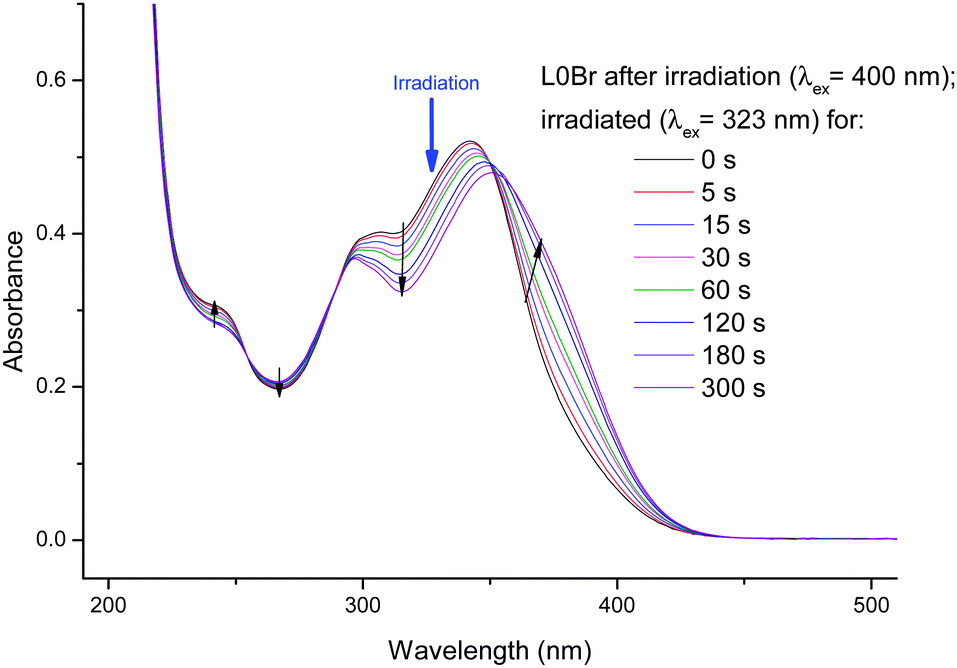 | ||
| Fig. 4 UV/vis-spectra of previously irradiated (400 nm) L0Br in ethanol upon irradiation at 323 nm. | ||
Isomerization is further evidenced by 1H-NMR spectra of the solutions, which clearly show that changes in UV/vis spectra can be correlated to NMR-peaks attributed to the trans- and cis-forms of L0Br (Fig. 5).
 | ||
| Fig. 5 1H-NMR spectra of trans-L0Br in DMSO-d6 solution (top), after irradiation at 405 nm for 5 min (middle) and extrapolated cis-L0Br (bottom). | ||
Interestingly, the trans-to-cis photoisomerization for L0Br is even observed in a solid polymethylmethacrylate (PMMA) matrix (Fig. S3, ESI†), where the dye is expected to experience considerable spatial restraints. Furthermore, we have tested the thermal stability of the product: absorption spectra of the irradiated sample recorded over the course of 12 hours at room temperature showed a change of <1% (data not shown), indicating that the cis-L0Br has a half-life of at least one month under the given conditions, allowing for considerable accumulation.
Performing the same experiment on L0 also gave systematic changes in the absorption spectra with a decrease of the main band (see Fig. S2, left, ESI†). Even in this case, a clear isosbestic point is present (383 nm). Importantly, though, no shift of the absorption maximum was observed: the band maximum remained unchanged at 405 nm (in ethanol). This is a likely reason why this phenomenon previously escaped observation. Subsequent irradiation with light at 344 nm partially reverted the changes (Fig. S2, right, ESI†), again representing a clear proof of reversible photoisomerization. In the case of L0, no decomposition is observed upon irradiation with UV light at a short-wavelength (344 nm), and the sharp isosbestic points are retained. It should be noted that the 1H-NMR spectra indicate that the photostationary state is shifted more towards the starting trans-form, due to the larger spectral overlap compared to the L0Br case (Fig. S4, ESI†). It is clear that the investigations on L0Br have helped us to detect a general phenomenon, which so far has and otherwise also very well could have continued to escape detection.
The question if the observed isomerization also takes place in surface-adsorbed dyes will be of direct relevance to DSC devices. Therefore, we have tested the photochemical behavior of L0Br adsorbed to a mesoporous ZrO2 surface. This semiconductor has a similar surface binding pattern16 but a higher band gap than TiO2,17,18 preventing electron injection from the excited dye and therefore facilitating a detailed investigation of the electronically excited state of the adsorbed dyes. The results of irradiation of L0Br on ZrO2 are shown in Fig. 6. As observed from the data of L0Br in solution and in PMMA, a systematic decrease of the main S1 band is seen, with an increase in absorption in the trough around 315 nm, and an isosbestic point at 335 nm. Consequently, the data clearly show the same trend upon irradiation as in solution and in PMMA, proving that the congestion of neighbouring molecules does not prevent photoisomerization. The photostationary state may be reached at a smaller conversion rate compared to solution, which could hint at several pools of molecules, some constrained while others are free to rotate. In the case of PMMA-embedded molecules, isomerization can take place by twisting of the small carboxylic group alone. Here, the anchor group is supposed to be locked to the semiconductor, requiring either twisting of the complete triphenylamine unit or some space-saving isomerization mechanism.
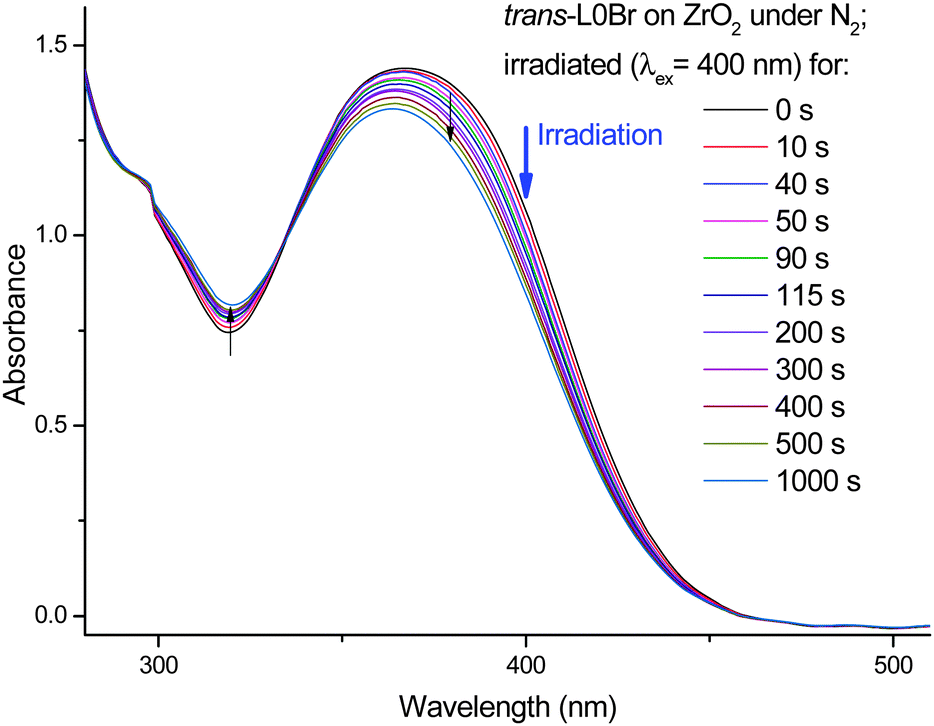 | ||
| Fig. 6 Changes in the UV/vis absorption of trans-L0Br adsorbed onto nanostructured ZrO2 in inert gas upon irradiation at 400 nm. | ||
Irradiation of the photoconverted sample using UV light (315 nm) leads to an increase of the S1 band (back-isomerization), but after a short while degradation sets in and a new species is formed (data not shown). This process, already observed in solution, is possibly enhanced by direct excitation of the semiconductor acting as a photocatalyst.18
Time-dependent fluorescence, time-correlated, single-photon counting (TCSPC) experiments of dilute solutions of L0Br and L0 are depicted in Fig. S7 (ESI†) and show relatively short lifetimes. A fit of the exponential decays gave: τ1 = 130 ps (97.7%) and τ2 = 1.85 ns (1.4%) for L0Br and τ = 750 ps (100%) for L0. The bi-exponential nature observed for L0Br can possibly be explained by a monomer–dimer-equilibrium. In any case, these short (sub-ns) lifetimes point towards a singlet mechanism, as intersystem crossing (a forbidden process) is highly unlikely to proceed at such a high rate, especially in the case of L0. Independent of the exact mechanism (which we are investigating in detail at the moment), photoisomerization has been proven to occur under a range of different conditions from the S1 state for both L0Br and L0.
The rotational barrier of the cyanoacrylic group was investigated at the hybrid density-functional level including dispersion interactions.19 The geometry was optimized in the trans configuration, representing the global minimum for both L0 and L0Br. The dihedral angle over the cyanoacrylic group is very close to planar, denoted as 0° in the following. These structures were subsequently used to construct a potential-energy map (PEM) of the rotation around the cyanoacrylic double bond in the singlet ground state (S0), the first excited singlet state (S1) and the first excited triplet state (T1). The details of the calculation strategy and the PEMs (Fig. S1, ESI†) are given in the ESI.† The main results show that the S0 and S1 states of both molecules are essentially degenerate at 90° and 270° rotation around the double bond (avoided crossing because of symmetry), clearly highlighting a spin-conserved (singlet), low-barrier mechanism for system cross-over from the excited S1 state to the S0 ground state in excellent agreement with the experimental results in terms of photoisomerization and sub-ns excited state lifetimes. The PEMs also show a S0–T1-intersection, although the necessary spin-flip is expected to prevent a cross-over, especially considering the steep curvature of the S0 state resulting in a strong potential directing the molecules away from the crossing. This is in line with the experimental finding showing only short-lived species.
The barrier of trans-to-cis rotation is estimated to be 47 kJ mol−1 for L0 and 22 kJ mol−1 for L0Br. These values are expected to be overestimated and would be reduced in a relaxed potential energy scan, but they at least give an indication that the barriers of rotation are low and that they are lower for L0Br than for L0.
Conclusions
In conclusion, we here report on the photoisomerization in solution, in a plastic matrix and on a semiconductor surface of a class of widely used organic dyes involving the cyanoacrylic acid group as the acceptor/anchoring component. Photoisomerization of the dye molecules may seriously affect the performance of the sensitizer in a DSC device, including the lifetime of the excited state, electron injection rate, rate of regeneration, as well as rate of dye-mediated recombination losses to the electrolyte components. Furthermore, photoisomerization may represent a competing dye relaxation; a concern that may gain some support from recent results on the dye D149, where electron injection was shown to be as slow as 30 ps.20 It should also be noted that the photoisomerization phenomenon, now when it has become known to the present authors, has been observed for more complex and more commonly used organic dyes of the same type as L0, such as D35.21The main conclusion from the results of this work is the necessity to avoid (exocyclic) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds as the D–π–A linker in future organic dye structures. Although many fundamental consequences remain to be investigated, such as the exact mechanism of photoisomerization, the current results stress the importance of DSC preparation procedures in terms of how the dye molecules, the dye baths and dyed photoelectrodes are handled.
C double bonds as the D–π–A linker in future organic dye structures. Although many fundamental consequences remain to be investigated, such as the exact mechanism of photoisomerization, the current results stress the importance of DSC preparation procedures in terms of how the dye molecules, the dye baths and dyed photoelectrodes are handled.
Acknowledgements
This work has been supported by the Swedish Research Council, the Swedish Energy Agency and the Knut & Alice Wallenberg Foundation.Notes and references
- International Energy Agency, Solar Energy Perspectives, OECD/IEA, Paris, 2011 Search PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- P. V. Kamat, K. Tvrdy, D. R. Baker and J. G. Radich, Chem. Rev., 2010, 110, 6664–6688 CrossRef CAS PubMed.
- T. Kitamura, M. Ikeda, K. Shigaki, T. Inoue, N. A. Anderson, X. Ai, T. Lian and S. Yanagida, Chem. Mater., 2004, 16, 1806–1812 CrossRef CAS.
- D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. Sun, Chem. Commun., 2006, 2245–2247 RSC.
- H. Pettersson, K. Nonomura, L. Kloo and A. Hagfeldt, Energy Environ. Sci., 2012, 5, 7376–7380 CAS.
- L. Ellis-Gibbings, V. Johansson, R. B. Walsh, L. Kloo, J. S. Quinton and G. G. Andersson, Langmuir, 2012, 28, 9431–9439 CrossRef CAS PubMed.
- J.-S. Ni, C.-Y. Hung, K.-Y. Liu, Y.-H. Chang, K.-C. Ho and K.-F. Lin, J. Colloid Interface Sci., 2012, 386, 359–365 CrossRef CAS PubMed.
- Here, trans is used although it formally does not apply to the L0Br dye with its heavier substituent Br.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Kluwer Academic/Plenum Publishers, New York, 2000 Search PubMed.
- H. Meier, Angew. Chem., Int. Ed. Engl., 1992, 31, 1399–1420 CrossRef.
- L. Sun and H. Corner, J. Phys. Chem., 1993, 97, 11186–11193 CrossRef CAS.
- B. Wardle, Principles and Applications of Photochemistry, Wiley, 2009 Search PubMed.
- A. El-Zohry, A. Orthaber and B. Zietz, J. Phys. Chem. C, 2012, 116, 26144–26153 CAS.
- A. M. El-Zohry and B. Zietz, J. Phys. Chem. C, 2013, 117, 6544–6553 CAS.
- S. Thyagarajan, E. Galoppini, P. Persson, J. M. Giaimuccio and G. J. Meyer, Langmuir, 2009, 25, 9216–9226 CrossRef PubMed.
- K. Kalyanasundaram and M. Grätzel, Coord. Chem. Rev., 1998, 177, 347–414 CrossRef CAS.
- Nanostructured and Photoelectrochemical Systems for Solar Photon Conversion, ed. M. D. Archer, Imperial College Press, London, 2008 Search PubMed.
- M. J. Frisch, and et al., Gaussian 09, Rev. A02, Gaussian Inc., 2009 Search PubMed.
- M. Juozapavicius, M. Kaucikas, J. J. van Thor and B. C. O'Regan, J. Phys. Chem. C, 2013, 117, 116–123 CAS.
- E. Gabrielsson, personal communication.
Footnote |
| † Electronic supplementary information (ESI) available: Information and results obtained from synthesis, spectroscopy and calculations including additional figures. See DOI: 10.1039/c3cp54048k |
| This journal is © the Owner Societies 2014 |