Is π halogen bonding or lone pair⋯π interaction formed between borazine and some halogenated compounds?†
Received
22nd September 2013
, Accepted 24th October 2013
First published on 28th October 2013
Abstract
Borazine, “inorganic benzene”, exhibits some different properties from benzene although both of them are isostructural and isoelectronic. It was known that benzene is favorable to form halogen bonds with halogenated molecules. However, borazine more easily forms lone pair–π interactions with halogenated molecules, but for stronger halogen donors it can also form halogen bonds. The halogen bonds formed by borazine are stronger than the corresponding lone pair–π interactions. It was found that the pair–π interactions can be changed into halogen bonds with the increase of interaction strength. The dispersion energy plays a main role in stabilizing the weakly bonded complexes, while the electrostatic energy is dominant in the strongly bonded complexes. This is different from the nature of the respective benzene complexes.
1. Introduction
Halogen bonding, a highly directional noncovalent interaction between a covalently-bonded halogen atom and a negative site,1 has been widely investigated, due to its potential applications in molecular recognition,2 crystal engineering,3 and biological systems.4 Dihalogen molecules can form π halogen bonds with some usual π systems, such as benzene, thiophene, furan, and pyridine, and the structures and properties of these complexes have been extensively studied using experimental and theoretical methods.5–7 However, C–X⋯π halogen bonds have more important practical value in crystal engineering,8 drug design,9 and protein–ligand complexation,10 although only sporadic studies have been performed for them.11 The induction interaction plays a dominant role in stabilizing benzene–X2 (X = Cl, Br, I) complexes,12 while the dispersion interaction is primarily responsible for the stability of benzene–halocarbon complexes.13
Lone pair–π interaction between a neutral electron-rich molecule and an electron-poor π ring is another important noncovalent interaction. It has been found in a number of biological systems14–16 and supramolecular systems.17,18 A detailed analysis of the Cambridge Structure Database has shown that such an interaction is not unusual in organic compounds,19 but it has been rather unexplored,20 due to the general belief that it is mostly repulsive.21 Another possible reason is its weak interaction strength with interaction energy smaller than 3 kcal mol−1 in most cases.22 This weak strength leads to the fact that it is seldom present alone in complicated systems and it often coexists with other types of interactions.23–25 In the benzene–dimethylether complex, where only lone pair–π interaction exists, a substantial part of the stabilization stems from dispersion energy.26
Borazine (B3N3H6), isolated in 1926 by Stock and Pohland,27 exhibits some similarity with benzene in its physical properties, because it is isostructural and isoelectronic with benzene, thus it is called “inorganic benzene”.28 People have shown great interest in borazine and its derivatives due to their potentials as building blocks in materials' science. They can act as precursors for nonoxidic ceramics containing B–N bonds and boron nitride nanotubes or other nanostructures.29 They also have the potential to act as chemical H2 storage materials.30–32 Very recently, Kervyn and co-workers33 reported the first bottom-up preparation of borazine-based supermolecular architectures on metal surfaces, which are rationalized as a delicate interplay of short-range van der Waals attractions between neighboring molecules and long-range Coulomb repulsions between deprotonated charged molecules. It has been demonstrated that borazine can form π hydrogen-bonded complexes with the first-row hydrides (BH3, CH4, NH3, H2O, and HF) and electrostatic and dispersion energies play an important role in the formation of these complexes.34 Interestingly, borazine can participate not only in anion–π interactions with anions (F−, Cl−, Br−, NO3−, and BF4−)35 but also in cation–π interactions with cations (Li+, Na+, and K+).36
In the present paper, we designed some complexes of borazine and halogenated compounds. Selected halogenated compounds include XF, XCN, XCCH, and XCF3 (X = F, Cl, Br, and I), which usually act as halogen donors in halogen bonds.37–39 Bonding energy and geometrical parameters have been investigated for these complexes. Our interest is in determining the interaction type between borazine and the halogenated compounds, that is, is the main force between them π halogen bonding or lone-pair–π interaction? To answer this question, we performed natural bond orbital (NBO) and symmetry-adapted perturbation theory (SAPT) calculations. In addition, we also considered the substitution effect on the interaction type.
2. Calculation methods
The structures of complexes and monomers were determined using the MP2 method with the aug-cc-pVDZ basis set for all atoms except the I atom. For iodine, pseudopotentials with the aug-cc-pVDZ-PP basis set were used to account for relativistic effects. Symmetry constrain has been imposed for the C3,v geometries. Frequency calculations were performed at the same level for the obtained structures to confirm the minima on their potential surfaces. The interaction energy is the difference between the energy of the complex and the sum of the isolated monomers in their minima configuration. The basis set superposition error (BSSE) was obtained using the full counterpoise method proposed by Boys and Bernardi40 to correct the interaction energy. All calculations were carried out using the Gaussian09 package.41
Molecular electrostatic potentials (MEPs) on the 0.001 electrons per bohr3 contour of the electronic density have been calculated with the Wave Function Analysis-Surface Analysis Suite (WFA-SAS) program42 at the MP2/aug-cc-pVDZ level. Natural bond orbital (NBO) analyses were carried out using NBO 3.1 version43 implemented in Gaussian 09 to provide an insight into the bonding nature of these complexes. The symmetry adapted perturbation theory (SAPT) method using the SAPT2008 program44 was used to decompose the interaction energy at the MP2/aug-cc-pVDZ level.
3. Results and discussion
3.1. MEPs of borazine
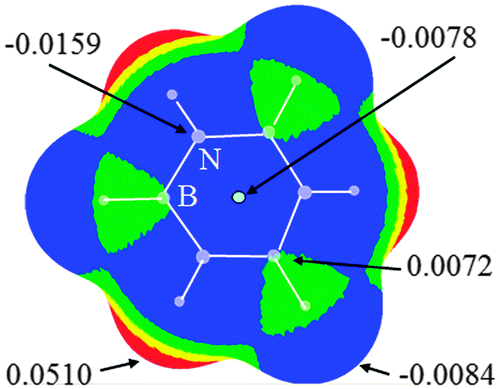
It was known that MEPs are a very effective method for predicting the possibility of intermolecular interactions.45 Thus we plotted the MEPs map of borazine in Fig. 1. One can conclude that the hydrogen atoms connected with the boron atoms are basic (blue and negative MEPs), while those connected with the nitrogen atoms are acidic (red and positive MEPs). Thus both types of hydrogen atoms in borazine can be respectively used as the proton donors and acceptors in hydrogen bonds.34 With respect to the hydrogen atom, the nitrogen atom shows greater electronegativity, while the boron atom exhibits smaller electronegativity. Thus the MEPs on the nitrogen atoms are negative (blue MEPs) and those on the boron atoms are positive (green MEPs). Additionally, the central site of the borazine ring has negative MEPs, although they are smaller than those on the N atoms. Thus both the nitrogen atoms and the ring center in borazine can be taken as basic centers in intermolecular interactions. It is also observed that the region of negative MEPs on the N atom in borazine is discrete, in agreement with the conclusion that borazine is nonaromatic.46 Comparing the MEPs of borazine and benzene (Fig. S1, ESI†), one can see that the negative MEPs at the ring center and carbon atoms in benzene are more negative than those found in borazine. This further shows that borazine is an electron-deficient π system with respect to benzene.22
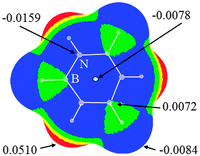 |
| | Fig. 1 Molecular electrostatic potentials of borazine. Color ranges, in eV, are: red, greater than 0.02; yellow, between 0.02 and 0.01; green, between 0.01 and 0; blue, less than 0. | |
3.2. Interactions between borazine and halogenated compounds
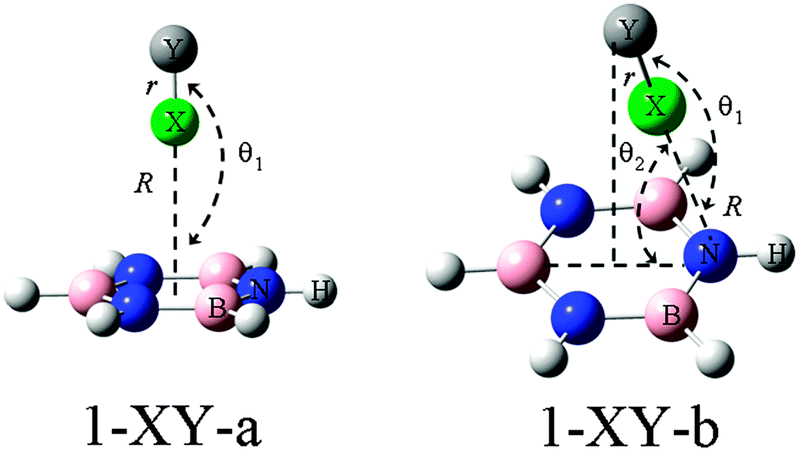
On the basis of the MEPs of borazine, more than one site of Lewis bases and acids are observed. Thus the interaction modes between borazine and halogenated compounds would be diverse. Here we are concerned only with the interactions between the ring center or N of borazine (1) and the halogen atom, although other types of complexes are also possible, which are shown in Fig. S2 in ESI.† The sketched structures of 1-XY (Y = F, CN, CCH, and CF3) are shown in Fig. 2 and their optimized structures are shown in Fig. S3 (ESI†). There are two structures for each halogen atom: 1-XY-a and 1-XY-b. The X atom in structure 1-XY-a points to the ring center of borazine with C3,v symmetry. Three F atoms are located over the three B atoms of borazine in the 1-XCF3-a complex. In structure 1-XY-b the halogen atom combines with the N atom of borazine, not being perpendicular to the ring plane. For the halogen-bonded complexes of benzene–X2, X = halogen atom, there are three types of π halogen bonds:47 the halogen atom points to the center of the benzene ring, the middle of the C–C bond, and the C atom, respectively.
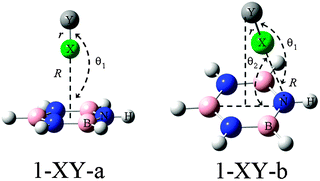 |
| | Fig. 2 Structures of complexes of borazine (1) with XY (X = F, Cl, Br, and I; Y = F, CN, CCH, and CF3). | |
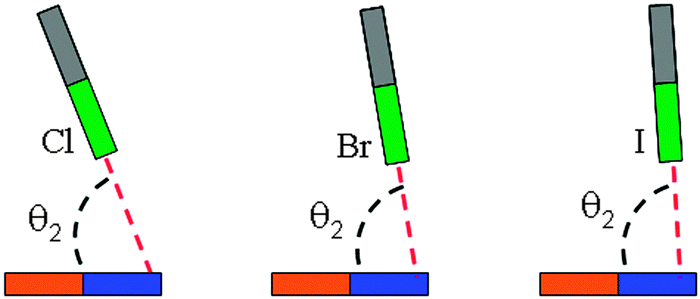
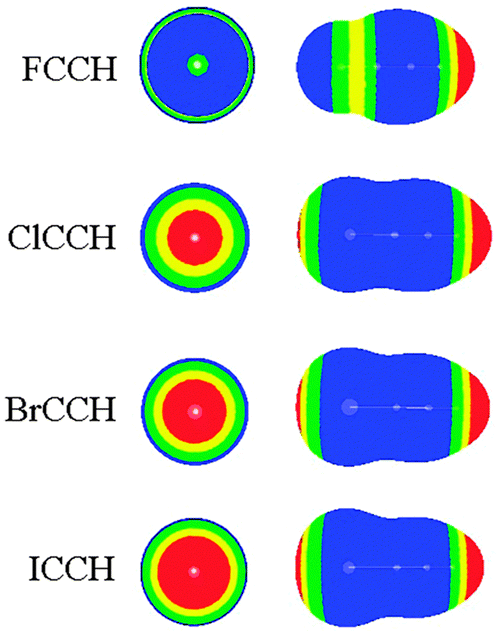
In Table 1, we summarized the structural parameters in all complexes. Here we pointed out that the planar structure of borazine has small deformation for the 1-XY-b complex. The Y-X⋯N angle (θ1) almost amounts to 180° in the complexes except for 1-ClCF3-b. For the 1-XY-b complex, the angle θ2 becomes larger with the increase of the halogen atomic number (Fig. 3). The geometrical difference in structure b can be explained with the size of the σ-hole on the interacting halogen atom. MEPs maps of XCCH (X = F, Cl, Br, and I) are shown in Fig. 4. When X varies from F to I, the region of the positive MEPs (σ-hole) on the halogen atomic surface becomes bigger, while the area of the negative MEPs on the halogen atomic surface becomes smaller. Other halogenated compounds show a similar change in the size of the σ-hole with the increase of the halogen atomic number. The negative area (lone pair electrons) on the halogen atom could have an attractive interaction with the electron-deficient ring center of borazine and this interaction becomes weaker for the heavier halogen. Simultaneously, the bigger positive region results in a stronger halogen bond between the N and X atoms. The change in the strength for both interactions is jointly responsible for the larger θ2 in the heavier halogen complex. This angle is bigger than 90° in 1-XY-b (XY = Cl2, Br2, I2) complexes but is smaller than 90° in other b complexes. For 1-XY-b (XY = Cl2, Br2, I2) complexes, the dihalogen molecules are almost perpendicular to the plane of 1. The distance between the F atom and the ring center of borazine is about 3.0 Å in the 1-FY-a (Y = F, CN, CCH, and CF3) complexes with small interaction energy (Table 2). The weak interaction in the 1-FY complex has a negligible effect on the change in the F–Y bond length and the frequency shift of the respective bond stretch vibration. The X⋯N distance shows an irregular change for the different series of 1-XY-b (X = Cl, Br, and I) complexes. With the increase of the X atomic radius, it becomes shorter in the 1-XCN-b complex and larger in the 1-XCF3-b complex, however, in the 1-XCCH-b complex, it grows in the order of Br < I < Cl and in the 1-XF-b complex in the order of Br < Cl < I. Even so, the X⋯N distances in all b complexes are smaller than the sum of van der Walls radii of the respective atoms (3.3 Å for Cl and N atoms, 3.4 Å for Br and N atoms, 3.6 Å for I and N atoms). However, the interaction energy shows a consistent change in the 1-XY-b (X = Cl, Br, and I) complex. Namely, the interaction energy becomes more negative with the increase of the halogen atomic number. This is in agreement with the most positive MEP on the halogen atomic surface.48 The interaction energy is also related to the Y substitutent and becomes more negative in the order of Y = CF3 < CCH < CN < F. It is also found that the b type complexes are more stable than the corresponding a type complexes. Both types of complexes are less stable than halogen-bonded complexes of benzene–X2.47 This further shows that borazine is a weaker Lewis base than benzene.
Table 1 Binding distance (R, Å), change in the X–Y bond length (Δr, Å), and bond angles (θ, degree) in the complexes at the MP2/aug-cc-pVDZ level
|
|
R
|
Δr |
θ
1
|
|
R
|
Δr |
θ
1
|
θ
2
|
|
1-FF-a |
2.963 |
0.002 |
180.0 |
1-FF-b |
2.672 |
0.013 |
178.6 |
84.4 |
|
1-ClF-a |
3.176 |
0.004 |
180.0 |
1-ClF-b |
2.576 |
0.030 |
179.4 |
93.4 |
|
1-BrF-a |
3.200 |
0.005 |
180.0 |
1-BrF-b |
2.548 |
0.038 |
179.2 |
95.0 |
|
1-IF-a |
3.261 |
0.007 |
180.0 |
1-IF-b |
2.644 |
0.036 |
179.2 |
96.4 |
|
1-FCN-a |
2.980 |
−0.001 |
180.0 |
1-FCN-b |
— |
— |
— |
— |
|
1-ClCN-a |
3.235 |
— |
180.0 |
1-ClCN-b |
3.250 |
0.001 |
180.0 |
76.5 |
|
1-BrCN-a |
3.252 |
— |
180.0 |
1-BrCN-b |
3.139 |
0.004 |
180.0 |
82.3 |
|
1-ICN-a |
3.330 |
0.002 |
180.0 |
1-ICN-b |
3.126 |
0.012 |
180.0 |
85.8 |
|
1-FCCH |
2.976 |
−0.002 |
180.0 |
1-FCCH-b |
— |
— |
— |
— |
|
1-ClCCH-a |
3.273 |
— |
180.0 |
1-ClCCH-b |
3.283 |
0.000 |
176.4 |
75.5 |
|
1-BrCCH-a |
3.289 |
— |
180.0 |
1-BrCCH-b |
3.189 |
0.002 |
179.6 |
79.5 |
|
1-ICCH-a |
3.376 |
0.002 |
180.0 |
1-ICCH-b |
3.197 |
0.008 |
179.4 |
82.3 |
|
1-FCF3-a |
2.976 |
−0.002 |
180.0 |
1-FCF3-b |
— |
— |
— |
— |
|
1-ClCF3-a |
3.283 |
−0.002 |
180.0 |
1-ClCF3-b |
3.312 |
−0.002 |
173.7 |
75.7 |
|
1-BrCF3-a |
3.308 |
−0.003 |
180.0 |
1-BrCF3-b |
3.230 |
−0.002 |
178.8 |
80.5 |
|
1-ICF3-a |
3.383 |
−0.003 |
180.0 |
1-ICF3-b |
3.237 |
0.000 |
179.5 |
83.8 |
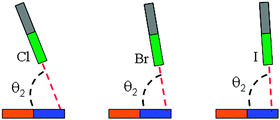 |
| | Fig. 3 Dependence of the angle θ2 on the nature of the halogen atom in the 1-XY-b complex. | |
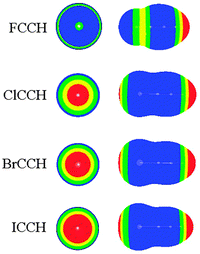 |
| | Fig. 4 Molecular electrostatic potentials of XCCH (X = F, Cl, Br, and I). Color ranges, in eV, are: red, greater than 0.02; yellow, between 0.02 and 0.01; green, between 0.01 and 0; blue, less than 0. | |
Table 2 Interaction energy corrected for BSSE (ΔE, kJ mol−1), frequency shift of X–Y stretch vibration (Δν, cm−1), and number of imaginary frequencies (N) in the complexes at the MP2/aug-cc-pVDZ level
|
|
ΔE |
Δν |
N
|
|
ΔE |
Δν |
N
|
|
1-FF-a |
−3.70 |
−10 |
2 |
1-FF-b |
−5.10 |
−63 |
0 |
|
1-ClF-a |
−8.69 |
−10 |
2 |
1-ClF-b |
−18.81 |
−92 |
0 |
|
1-BrF-a |
−10.69 |
−11 |
2 |
1-BrF-b |
−27.00 |
−76 |
0 |
|
1-IF-a |
−12.62 |
−12 |
2 |
1-IF-b |
−32.37 |
−55 |
0 |
|
1-FCN-a |
−3.91 |
0 |
0 |
1-FCN-b |
— |
— |
— |
|
1-ClCN-a |
−9.01 |
−2 |
2 |
1-ClCN-b |
−9.37 |
−4 |
0 |
|
1-BrCN-a |
−10.44 |
−1 |
2 |
1-BrCN-b |
−11.66 |
−8 |
0 |
|
1-ICN-a |
−12.23 |
−2 |
2 |
1-ICN-b |
−15.02 |
−14 |
0 |
|
1-FCCH |
−2.45 |
3 |
0 |
1-FCCH-b |
— |
— |
— |
|
1-ClCCH-a |
−6.98 |
−1 |
2 |
1-ClCCH-b |
−7.30 |
−3 |
0 |
|
1-BrCCH-a |
−8.11 |
−1 |
2 |
1-BrCCH-b |
−9.06 |
−6 |
0 |
|
1-ICCH-a |
−9.53 |
−1 |
2 |
1-ICCH-b |
−11.60 |
−11 |
0 |
|
1-FCF3-a |
−2.11 |
0 |
3 |
1-FCF3-b |
— |
— |
— |
|
1-ClCF3-a |
−6.77 |
−1 |
3 |
1-ClCF3-b |
−6.94 |
−1 |
0 |
|
1-BrCF3-a |
−7.90 |
−1 |
3 |
1-BrCF3-b |
−8.53 |
−2 |
0 |
|
1-ICF3-a |
−9.29 |
−1 |
3 |
1-ICF3-b |
−10.77 |
−3 |
0 |
The interaction strength is weak in most complexes except some XF complexes. Thus we plan to enhance it by the substitution effect. Specifically, the H atom of B–H or N–H bond in 1-BrCF3 complex is replaced with CN, Li, and CH3, respectively. The obtained complexes are represented as B-R-1-BrCF3 and N-R-1-BrCF3, where the bold atom indicates the substitution site and R represents substituents. The corresponding results are given in Table 3. The electron-withdrawing group CN leads to a weakening of the interaction strength with less negative interaction energy, while the electron-donating group Li results in an enhancement of the interaction strength with more negative interaction energy. The enhancing effect of Li is more prominent than the weakening effect of CN. The N substitution effect is more prominent whether for the Li enhancing effect or for the CN weakening effect, due to its direct participation in the interaction. The interaction energy in the N-Li-1-BrCF3 complex is about 2.5 times as much as that in the 1-BrCF3 complex. The prominent effect of Li stubstitutent has been reported for the halogen-bonded complex of HCCBr–NCH.49 A similar enhancing effect is also found for the CH3 substituent, although its effect is smaller than that of Li substitution. This indicates that the methyl group plays an electron-donating role in this interaction, which is similar to that in hydrogen bonds.50 Considering the possible Br⋯Li interaction in the N-Li-1-BrCF3 complex, the θ1 angle decreases by about 12° and the θ2 angle increases by about 40°. Both angles change a little in other substituted complexes. In general, the enhancing interaction shortens the binding distance, while the weakening interaction enlarges it. The binding distance is an exception in the N-CN–1–BrCF3 complex.
Table 3 Interaction energy corrected for BSSE (ΔE, kJ mol−1), binding distance (R, Å), change in the X–Y bond length (Δr, Å), bond angles (θ, degree), and frequency shift of the X–Y stretch vibration (Δν, cm−1) in the complexes at the MP2/aug-cc-pVDZ level
|
|
ΔE |
R
|
Δr |
θ
1
|
θ
2
|
Δν |
|
B-CN-1-BrCF3-b |
−7.01 |
3.240 |
0.001 |
173.4 |
79.8 |
−1 |
|
N-CN-1-BrCF3-b |
−6.60 |
3.213 |
0.001 |
171.7 |
82.7 |
0 |
|
B-Li-1-BrCF3-b |
−14.42 |
3.178 |
−0.005 |
174.0 |
81.5 |
−4 |
|
N-Li-1-BrCF3-b |
−21.66 |
2.888 |
0.018 |
166.6 |
120.2 |
−9 |
|
B-CH3-1-BrCF3-b |
−10.13 |
3.178 |
−0.001 |
175.5 |
81.6 |
−2 |
|
N-CH3-1-BrCF3-b |
−11.10 |
3.089 |
−0.001 |
176.0 |
86.3 |
−3 |
3.3. NBO and SAPT analyses
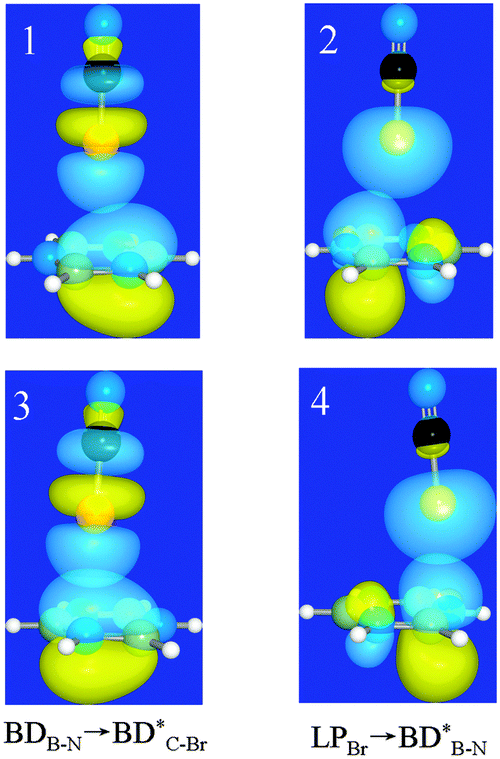
To unveil the interaction nature in the above types of complexes, we fall back on the NBO and SAPT analyses. For each type of complex, two orbital interactions are analyzed, and their strengths are estimated with second-order perturbation energy (E) given in Table 4. The two orbital interactions are BDB–N → BD*X–Y and LPX → BD*B–N, as shown in Fig. 5, and the perturbation energies are represented as E1 and E2, respectively. In the former the π bonding orbital in borazine acts as the donor orbital (Lewis bases), while in the latter the lone pair bonding orbital on the halogen atom acts as the donor orbital. The former orbital interaction corresponds to halogen bonding, while the latter orbital interaction suggests lone pair–π interactions. One can see that for the 1-XY-a complex the E2 value is larger than the E1 one, indicating that the interaction in the 1-XY-a complex should belong to lone pair–π interactions. This also goes for most 1-XY-b complexes except the 1-XF-b (X = Cl, Br, and I) complex, in which the E1 value is larger than the E2 one and the ratio of E1/E2 increases for the heavier halogen atom. This means that halogen bonding is present in the 1-XF-b (X = Cl, Br, and I) complex.
Table 4 Second-order perturbation energies due to the BDB–N → BD*X–Y (E1, kJ mol−1) and LPX → BD*B–N (E2, kJ mol−1) orbital interactions and charge transfer (CT, me) in the complexes
|
|
E
1
|
E
2
|
CT |
|
E
1
|
E
2
|
CT |
|
1-FF-a |
0.75 |
9.20 |
−4.8 |
1-FF-b |
8.94 |
22.49 |
−0.4 |
|
1-ClF-a |
2.88 |
17.18 |
−9.7 |
1-ClF-b |
70.31 |
54.00 |
33.5 |
|
1-BrF-a |
5.52 |
21.69 |
−9.8 |
1-BrF-b |
122.01 |
73.69 |
53.3 |
|
1-IF-a |
9.91 |
27.88 |
−9.4 |
1-IF-b |
138.90 |
81.13 |
48.2 |
|
1-FCN-a |
0.00 |
7.61 |
−2.4 |
1-FCN-b |
— |
— |
— |
|
1-ClCN-a |
1.13 |
12.08 |
−4.3 |
1-ClCN-b |
2.84 |
16.30 |
−4.0 |
|
1-BrCN-a |
2.63 |
15.92 |
−1.0 |
1-BrCN-b |
9.20 |
24.24 |
0.9 |
|
1-ICN-a |
5.39 |
19.86 |
−2.3 |
1-ICN-b |
21.57 |
33.36 |
6.0 |
|
1-FCCH |
0.00 |
7.82 |
−3.5 |
1-FCCH-b |
— |
— |
— |
|
1-ClCCH-a |
0.88 |
11.33 |
−6.2 |
1-ClCCH-b |
2.26 |
16.47 |
−6.4 |
|
1-BrCCH-a |
2.01 |
15.26 |
−6.6 |
1-BrCCH-b |
6.81 |
23.58 |
−5.4 |
|
1-ICCH-a |
4.01 |
18.89 |
−5.6 |
1-ICCH-b |
14.21 |
31.10 |
−1.3 |
|
1-FCF3-a |
0.00 |
9.74 |
−2.8 |
1-FCF3-b |
— |
— |
— |
|
1-ClCF3-a |
0.75 |
10.83 |
−4.4 |
1-ClCF3-b |
2.13 |
16.43 |
−4.7 |
|
1-BrCF3-a |
2.01 |
14.04 |
−4.4 |
1-BrCF3-b |
6.69 |
21.57 |
−3.2 |
|
1-ICF3-a |
4.64 |
17.35 |
−3.3 |
1-ICF3-b |
15.55 |
28.38 |
1.2 |
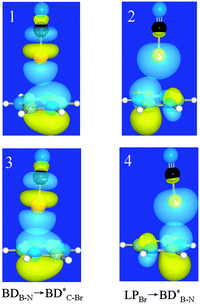 |
| | Fig. 5 Schematic diagrams of two orbital interactions in 1-BrCN-a (1 and 2) and 1-BrCN-b (3 and 4) complexes. | |
To further provide evidence for the above results, charge transfer from borazine to the halogenated molecule was also calculated. The charge transfer is negative in the 1-XY-a complex, showing that borazine gains electrons in the 1-XY-a complex. This is consistent with the electron-withdrawing role of borazine in the lone pair–π interaction. Likely, the charge transfer is also negative in most 1-XY-b complexes except 1-XF-b (X = Cl, Br, and I), 1-BrCN-b, and 1-IY-b (Y = CN, and CCH, CF3) complexes. This means that for most 1-XY-b complexes borazine also gains electrons. However, this negative charge transfer is changed to positive in the 1-XF-b (X = Cl, Br, and I), 1-BrCN-b, and 1-IY-b (Y = CN, CCH, and CF3) complexes. The positive charge transfer in the 1-BrCN-b and 1-IY-b (Y = CN, CCH, and CF3) complexes is inconsistent with the magnitude of the respective orbital interactions. We think that other interactions such as polarization interaction, besides the orbital interactions, are also responsible for the charge transfer in these complexes. The positive charge transfer in the 1-XF-b (X = Cl, Br, and I) complex supports the electron-donating role of borazine in the formation of the halogen bond. With the increase of the halogen atomic mass, the magnitude of negative charge transfer in the 1-XY-b complex becomes smaller and is even changed to positive one in some complexes.
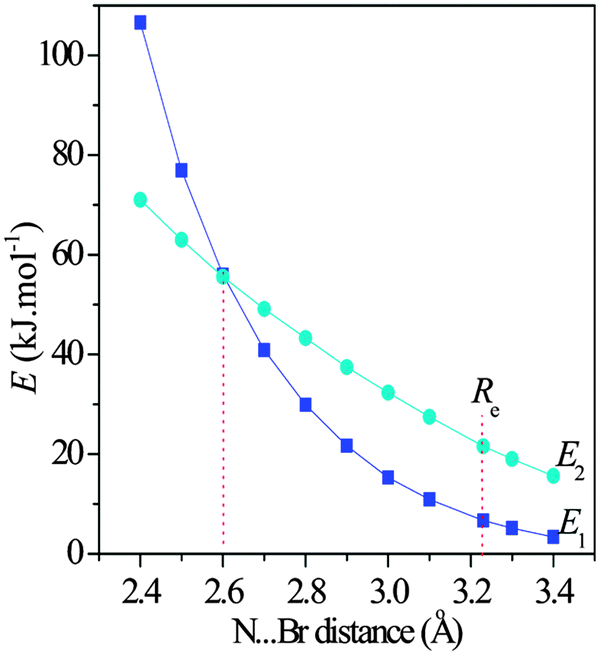
One finds the stronger halogen bonding interaction in the 1-XF-b (X = Cl, Br, and I) complex but the weaker lone pair–π interaction in other complexes. Thus we studied the dependence of the interaction type on the binding distance. One can see from Fig. 6 that with the shortening of the Br⋯N distance in the 1-BrCF3-b complex the values of E1 and E2 are both increased. The E2 value almost increases linearly, while the E1 one displays an exponential increase. However, the slope of their change is different. Interestingly, the E1 value is smaller than the E2 one with the Br⋯N distance larger than 2.6 Å, both values are equal at 2.6 Å, and the E1 value is greater than the E2 one with the Br⋯N distance shorter than 2.6 Å. Thus the lone pair–π interaction is changed into the halogen bond with the shortening of the binding distance (stronger interaction). This result indicates that the interaction strength can change the interaction type.
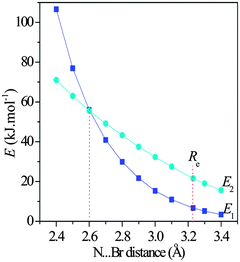 |
| | Fig. 6 Dependence of E1 and E2 on the N⋯Br distance in the 1-BrCF3-b complex. | |
The interaction energies in both types of complexes are decomposed into five terms: electrostatic energy (Eelst), exchanged energy (Eexch), induction energy (Eind), dispersion energy (Edisp), and δEint,rHF, and the related results are given in Table 5, while the detailed components are presented in Tables S1 and S2 in ESI.† For the 1-FY-a (Y = CN, CCH, and CF3) complex, the Edisp term has a greater contribution than the Eelst one and the Eind contribution is very small. A similar result is also found in the 1-ClY-a (Y = CN, CCH, and CF3) complex. For the 1-BrY-a (Y = CN, CCH, and CF3) complex, the Eelst term is close to the Edisp term and is even larger in some cases, and the Eind contribution is still smallest. However, the Eelst contribution is negative for the 1-FF-a complex. For the other dihalogen complexes, the Eelst contribution is dominant although the other two attractive terms are also important. For the 1-XY-b (X = Cl and Br; Y = CN, CCH, and CF3) complex, the Eelst contribution is still dominant, while the Eind contribution is small. Thus the physical origin of the interaction between borazine and halogenated molecule depends on the interaction type and the nature of the halogen atom. In summary, the dispersion energy is the main force responsible for the weaker interactions, while the electrostatic energy has a main contribution to the stronger interactions.
Table 5 Electrostatic energy (Eelst), exchange energy (Eexch), induction energy (Eind), dispersion energy (Edisp), and δEint,rHF. All are in kJ mol−1
|
|
E
elst
|
E
exch
|
E
ind
|
E
disp
|
δE
int,r
HF
|
| Note: Data in parentheses are for the b complex. δEint,rHF collects all third- and higher-order induction and exchange-induction terms. |
|
1-FF-a |
5.01(−24.62) |
10.85(46.69) |
−5.31(−4.26) |
−3.09(−8.23) |
−0.35(−0.98) |
|
1-ClF-a |
−12.00(−61.90) |
16.84(101.15) |
−2.68(−16.72) |
−8.94(−20.86) |
−1.92(−24.48) |
|
1-BrF-a |
−19.48(−105.04) |
26.12(154.93) |
−6.48(−45.77) |
−12.91(−30.05) |
−3.93(−34.37) |
|
1-FCN-a |
−7.90 |
27.78 |
−0.75 |
−13.21 |
−1.59 |
|
1-ClCN-a |
−9.27(−12.00) |
15.62(20.13) |
−1.30(−1.63) |
−9.86(−10.83) |
−0.96(−1.52) |
|
1-BrCN-a |
−15.76(−24.83) |
24.11(36.35) |
−3.59(−5.89) |
−13.96(−16.05) |
−2.19(−4.39) |
|
1-FCCH-a |
−1.55 |
8.80 |
−0.21 |
−5.14 |
−0.38 |
|
1-ClCCH-a |
−7.06(−9.70) |
14.56(19.24) |
−0.54(−0.84) |
−9.66(−10.70) |
−0.74(−1.21) |
|
1-BrCCH-a |
−13.00(−20.77) |
22.70(33.28) |
−2.05(−3.72) |
−13.63(−15.59) |
−1.65(−3.23) |
|
1-FCF3-a |
−2.42 |
8.78 |
0.04 |
−4.60 |
−0.36 |
|
1-ClCF3-a |
−7.77(−10.12) |
13.59(18.00) |
−0.92(−1.17) |
−8.99(−10.03) |
−0.81(−1.31) |
|
1-BrCF3-a |
−13.38(−19.90) |
20.73(30.04) |
−2.80(−4.39) |
−12.75(−14.55) |
−1.88(−3.71) |
4. Conclusions
The complexes of borazine and four halogenated molecules XF, XCN, XCCH, and XCF3 (X = F, Cl, Br, and I) have been investigated. Two structures (a and b) are studied: the halogen atom in structure a points to the ring center of borazine and combines with the N atom of borazine in structure b. The structure a is stabilized by a lone pair–π interaction although it is not located at the minima on the potential surface in most complexes. The structure b is also combined with a lone pair–π interaction in most complexes except the dihalogen systems, where a halogen bond with large interaction energy is present. NBO analysis indicates that with the increase of interaction strength between the two molecules the lone pair–π interaction may change to the halogen bonding interaction. SAPT results show that the dispersion energy plays a main role in stabilizing the weakly bonded complexes, while the electrostatic energy is dominant in the strongly bonded complexes.
Acknowledgements
This work was supported by the Outstanding Youth Natural Science Foundation of Shandong Province (JQ201006), the Program for New Century Excellent Talents in University, the National Natural Science Foundation of China (51278443), and Open Project of State Key Laboratory of Supramolecular Structure and Materials (sklssm201321).
References
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- A. Caballero, N. G. White and P. D. Beer, Angew. Chem., Int. Ed., 2011, 50, 1845–1848 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278–294 CrossRef CAS PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- S. A. Cooke, C. M. Evans, J. H. Holloway and A. C. Legon, J. Chem. Soc., Faraday Trans., 1998, 94, 2295–2302 RSC.
- Z. X. Wang, B. S. Zheng, X. Y. Yu, X. F. Li and P. G. Yi, J. Chem. Phys., 2010, 132, 164104 CrossRef PubMed.
- Y. L. Zeng, X. Y. Zhang, X. Y. Li, L. P. Meng and S. J. Zheng, ChemPhysChem, 2011, 12, 1080–1087 CrossRef CAS PubMed.
- D. S. Reddy, D. C. Craig and G. R. Desiraju, J. Am. Chem. Soc., 1996, 118, 4090–4093 CrossRef CAS.
- Y. X. Lu, Y. Wang and W. L. Zhu, Phys. Chem. Chem. Phys., 2010, 12, 4543–4551 RSC.
- K. S. Yeung, Drug Discovery Today, 2011, 15, 158 CrossRef PubMed.
- X. Liang, T. Sun and Y. B. Wang, Chem. J. Chin. Univ., 2012, 33, 541–547 CAS.
- Y. X. Lu, J. W. Zou, Y. H. Wang and Q. S. Yu, J. Chem. Phys. A, 2007, 334, 10781–10788 CrossRef PubMed.
- N. Nagels, D. Hauchecorne and W. A. Herrebout, Molecules, 2013, 18, 6829–6851 CrossRef CAS PubMed.
- M. Egli and R. V. Gessner, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 180–184 CrossRef CAS.
- S. Sarkhel, A. Rich and M. Egli, J. Am. Chem. Soc., 2003, 125, 8998–8999 CrossRef CAS PubMed.
- A. Jain, C. S. Purohit, S. Verma and R. Sankararamakrishnan, J. Phys. Chem. B, 2007, 111, 8680–8683 CrossRef CAS PubMed.
- J. Zukerman-Schpector, A. Otero-de-la-Roza, V. Luaña and E. R. T. Tiekink, Chem. Commun., 2011, 47, 7608–7610 RSC.
- A. Das, S. R. Choudhury, C. Estarellas, B. Dey, A. Frontera, J. Hemming, M. Helliwell, P. Gamez and S. Mukhopadhyay, CrystEngComm, 2011, 13, 4519–4527 RSC.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- G. A. DiLabio and E. R. Johnson, J. Am. Chem. Soc., 2007, 129, 6199–6203 CrossRef CAS PubMed.
- W. Gung, B. Y. Zou, Z. Xu, J. C. Amicangelo, S. Ma and H. C. Zhou, J. Org. Chem., 2008, 73, 689–693 CrossRef PubMed.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- G. A. DiLabio and E. R. Johnson, J. Am. Chem. Soc., 2007, 129, 6199–6203 CrossRef CAS PubMed.
- A. Nijamudheen, D. Jose, A. Shine and A. Datta, J. Phys. Chem. Lett., 2012, 3, 1493–1496 CrossRef CAS.
- C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, Cent. Eur. J.
Chem., 2011, 9, 25–34 CAS.
- J. Ran and P. Hobza, J. Chem. Theory Comput., 2009, 5, 1180–1185 CrossRef CAS.
- A. Stock and E. Pohland, Ber. Dtsch. Chem. Ges., 1926, 59, 2210–2215 CrossRef.
- E. Wiberg and A. Bolz, Ber. Dtsch. Chem. Ges., 1940, 73, 209–232 CrossRef.
- H. F. Bettinger, T. Kar and E. Sanchez-Garcia, J. Phys. Chem. A, 2009, 113, 3353–3359 CrossRef CAS PubMed , and references cited here.
- T. J. Carter, J. W. Kampf and N. K. Szymczak, Angew. Chem., Int. Ed., 2012, 51, 13168–13172 CrossRef CAS PubMed.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 509–521 CrossRef CAS.
- S. Kervyn, N. Kalashnyk, M. Riello, B. Moreton, J. Tasseroul, J. Wouters, T. S. Jones, A. D. Vita, G. Costantini and D. Bonifazi, Angew. Chem., Int. Ed., 2013, 52, 7410–7414 CrossRef CAS PubMed.
- J. Y. Wu, H. Yan, H. Chen, G. L. Dai and A. G. Zhong, Comput. Theor. Chem., 2012, 984, 51–56 CrossRef CAS PubMed.
- A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, Chem. Phys. Lett., 2012, 530, 145–150 CrossRef PubMed.
- R. Miao, G. S. Yang, C. M. Zhao, J. Hong and L. G. Zhu, THEOCHEM, 2005, 715, 91–100 CrossRef CAS PubMed.
- R. Y. Li, Z. R. Li, D. Wu, Y. Li, W. Chen and C. C. Sun, J. Phys. Chem. A, 2005, 109, 2608–2613 CrossRef CAS PubMed.
- D. J. R. Duarte, M. M. de las Vallejos and N. M. Peruchena, J. Mol. Model., 2010, 16, 737–748 CrossRef CAS PubMed.
- G. Valerio, G. Raos, S. V. Meille, P. Metrangolo and G. Resnati, J. Phys. Chem. A, 2000, 104, 1617–1620 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. A. Yazyev, J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. A. Dapprich, D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
-
R. Bukowski, W. Cencek, P. Jankowski, B. Jeziorski, M. Jeziorska, S. A. Kucharski, V. F. Lotrich, A. J. Misquitta, R. Moszynski, K. Patkowski, R. Podeszwa, S. Rybak, K. Szalewicz, H. L. Williams, R. J. Wheatley, P. E. S. Wormer and P. S. Zuchowski, SAPT2008: An Ab initio Program for Many-Body Symmetry-Adapted Perturbation Theory Calculations of Intermolecular Interaction Energies, Version 2008.2.
- J. M. Campanario, E. Bronchalo and M. A. Hidalgo, J. Chem. Edu., 1994, 71, 761–766 CrossRef CAS.
- J. J. Engelberts, R. W. A. Havenith, J. H. Lenthe, L. W. Jenneskens and P. W. Fowler, Inorg. Chem., 2005, 44, 5266–5272 CrossRef CAS PubMed.
- E. Munusamy, R. Sedlak and P. Hobza, ChemPhysChem, 2011, 12, 3253–3261 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and M. C. Concha, J. Mol. Model., 2007, 13, 643–650 CrossRef CAS PubMed.
- J. B. Cheng, R. Li, Q. Z. Li, B. Jing, Z. B. Liu, W. Z. Li, B. A. Gong and J. Z. Sun, J. Phys. Chem. A, 2010, 114, 10320–10325 CrossRef CAS PubMed.
- Q. Z. Li, G. S. Wu and Z. W. Yu, J. Am. Chem. Soc., 2006, 128, 1438–1439 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp54006e |
|
| This journal is © the Owner Societies 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.