Electrofluorescence switching from a multilayer thin film by spin-assisted layer-by-layer assembly of an anionic fluorescent conjugated polyelectrolyte with poly(diallyldimethylammonium chloride)†
Cheng-Po
Kuo
a and
Man-kit
Leung
*ab
aDepartment of Chemistry, National Taiwan University, 1, Roosevelt Road Section 4, Taipei, Taiwan 106, Republic of China. E-mail: mkleung@ntu.edu.tw
bInstitute of Polymer Science and Engineering, National Taiwan University, 1, Roosevelt Road Section 4, Taipei, Taiwan 106, Republic of China
First published on 25th October 2013
Abstract
An electrofluorochromic conjugated polyelectrolyte, PFTSO3Na as a polyanion, was synthesized and used in the fabrication of layer-by-layer (LbL) assembled multilayer film with poly(diallyldimethylammonium chloride) (PDDA) as a polycation. The electrofluorescent device (EFD) fabricated from the multilayer thin film of PFTSO3Na/PDDA emits blue light under UV excitation. The fluorescence intensity can be switched off upon electrochemical oxidation. The electrofluorescent behavior of the EFD is reversible and can be switched between the non-fluorescent (oxidized) state and the fluorescent (neutral) state with a maximum contrast ratio (If/If0) of 5.20.
Introduction
Reversible changes in the optical properties by electrochemical or photochemical conversion of UV-vis or photoluminescence spectra have been an attractive field of investigation due to their potential applications in smart windows, displays, bioanalysis, signaling recognition events, and optical memories.1–6 Electrofluorescence (EF) that refers to the electrically driven reversible optical changes in the fluorescence (“electrofluorochromism”)7–19 is a promising method to achieve these research demands. A pioneering example of an electrofluorescent device (EFD) that switches the fluorescence on and off by changing the redox states of tetrazine molecules, between their neutral state and an anion-radical state, was reported by Kim et al.12 In particular, fluorescent conjugated polymers are promising candidates for application in EFDs due to their intrinsic optoelectronic properties. Compared with inorganic or molecular organic materials, the advantage of organic π-conjugated polymers is their ability to amplify the sensitivity to the fluorescence responses that has been attributed to the rapid transport of excitons and efficient electronic delocalization along the π-conjugated backbone.20–23 Hence polymeric EFDs have also been reported; in these devices, the fluorescence emission can be switched on and off through redox reactions of the fluorescent conjugated polymers. For example, n-type doping poly(oxadiazole) (POD) enables easy fabrication of the EFD.18 The POD shows a reversible change in the fluorescence properties by repetitive transformation between its reduced state (non-fluorescent) and the neutral state (fluorescent) with a maximum on/off ratio of 2.5.Thin films of polymeric materials are commonly fabricated via spin-coating, solution-casting, electropolymerization, or surface polymerization by chemical means. An alternative, straightforward approach for fabrication of nanostructured thin films is the layer-by-layer (LbL) assembly technique, which is based on the sequential adsorption of polycations and polyanions (or hydrogen-bond donors and acceptors) from dilute aqueous solution onto a solid substrate.24–37 The thickness of the thin film is controlled by varying the number of bilayers. In addition, control of the layer thickness, composition, and topography can be achieved through adjustment of the assembly parameters such as ionic strength, deposition temperature, concentration, and the pH value of the dipping solutions. The conventional dip-assisted LbL assembly is based on a repeated solution-dipping process. However, it is a time-consuming process in which deposition of a polymer layer contains numerous steps, including adsorption, diffusion, and rearrangement. To accelerate the LbL assembly process, multilayer films are manufactured by the spin-assisted LbL assembly through intermolecular interactions between two materials as well as by centrifugal force or air shear force. Moreover, the spin-assisted LbL assembly has drawn much attention because of its own virtues: significantly decreasing LbL assembly time, minimizing solvent usage, and achieving an ordered internal structure due to limited interpenetration between polymer layers.
In our previous work, we have reported the development of the novel p-type doping triphenylamine-based polymer of EFD, in which the fluorescent behavior can be reversibly switched between a non-fluorescent (oxidized) state and a strong fluorescence (neutral) state.38 Furthermore, we have demonstrated a white-light emitting EFD from electrochemically convertible yellow and blue fluorescent conjugated polymers.39 Since the fluorescent conjugated polymers have their emission simultaneously quenched under the low working potential, the EFD could show white fluorescence state and dark state inter-conversion. The promising results obtained encouraged us to further develop PFTSO3Na that can be used as a polyanion for the EFD by spin-assisted LbL assembly with poly(diallyldimethylammonium chloride) (PDDA) as the counter polycation (Scheme 1). The significant advantage of utilizing the LbL assembly technique for the EFD is that hypothetically charged species might be incorporated into the multilayer electrolytic thin film more efficiently with relatively low activation barrier, which may be a concept to improve switching time.
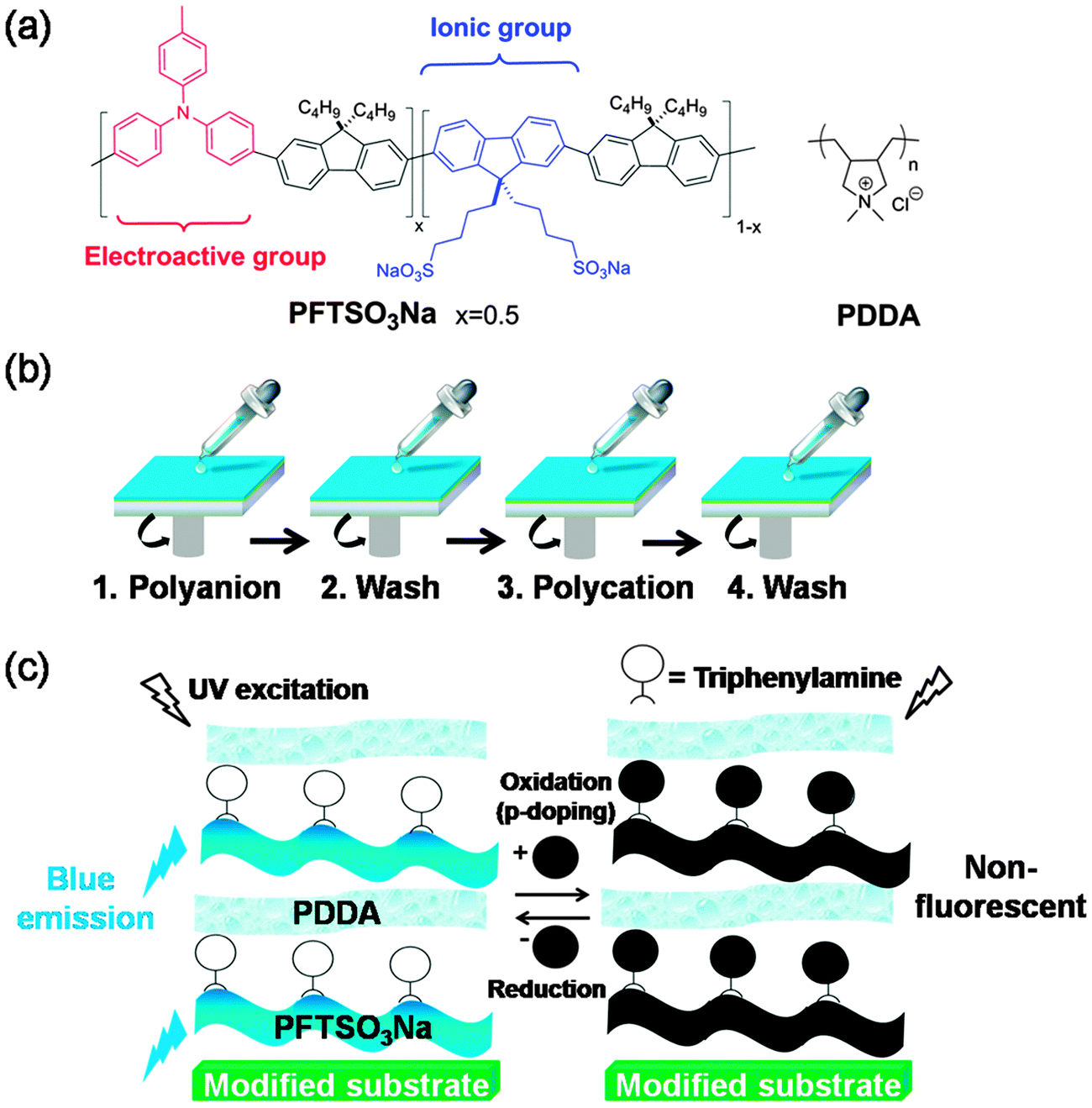 | ||
| Scheme 1 (a) Chemical structures of PFTSO3Na and PDDA. (b) A schematic representation of the process used to fabricate multilayer films by spin-assisted LbL assembly. Multilayer films are formed by repeating steps 1 to 4 in a cyclic fashion. (c) A schematic illustration of the creation of a blue-light EF switching. | ||
Fluorene-based conjugated polymers (PFs) are of great interest as light-emitting materials due to their high photoluminescence (PL) efficiency and excellent thermal stability in the solid state.40–51 Moreover, it would be an appropriate choice to adopt triarylamine (TAA) as a switching unit to facilitate the fluorescence on/off through a redox mechanism. Two basic properties of the TAA units are beneficial for the switching functions: (1) TAA can be easily oxidized to form stable radical cations; (2) the radical cations have good ability for the positive-charge transport through hopping mechanisms.52–64 Under the influence of an electric field, positive charges are transported with high drift-mobility up to 10−3 cm2 (V−1 s−1).65
In the present article, we report the synthesis of the polyanion PFTSO3Na (Scheme 2) and fabrication of an electrochemically-convertible blue light emitting multilayer thin-film EFD by LbL assembly of PFTSO3Na with PDDA. Their multilayer buildup, optical, electrochemical performance, and electrofluorescent characteristics were investigated by UV/vis spectroscopy, photoluminescence (PL) spectroscopy, and cyclic voltammetry (CV).
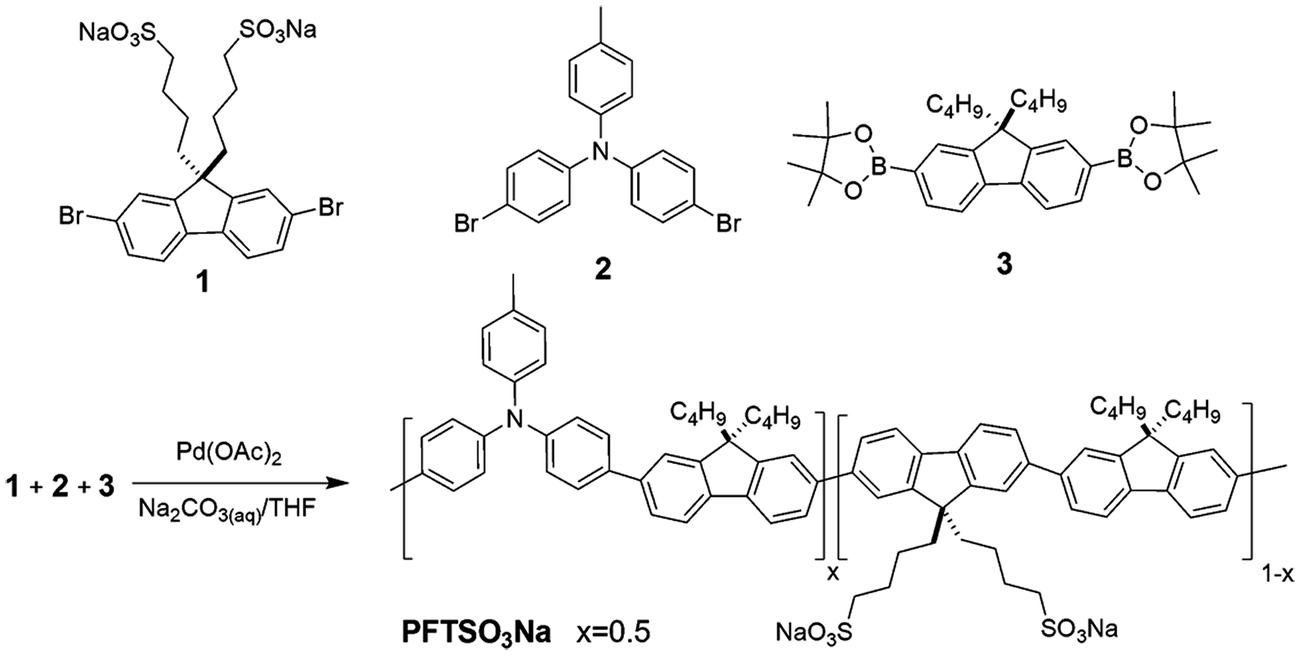 | ||
| Scheme 2 Synthetic route of PFTSO3Na. | ||
Experimental section
Materials
All reagents, including PDDA (low molecular weight, 20% weight in water, Mw ∼ 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000, available from Aldrich Chemical Co.), poly(propylene carbonate) (PPC) (average Mn ∼ 50000 by GPC, available from Aldrich Chemical Co.), LiClO4, propylene carbonate (PC), and Na2CO3 were commercially available.
000–200000, available from Aldrich Chemical Co.), poly(propylene carbonate) (PPC) (average Mn ∼ 50000 by GPC, available from Aldrich Chemical Co.), LiClO4, propylene carbonate (PC), and Na2CO3 were commercially available.
Monomer synthesis
The 2,7-dibromo-9,9-bis(4′-sulfonatobutyl)fluorene sodium salt (1),66 4-bromo-N-(4-bromophenyl)-N-p-tolylaniline (2),38 and 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dibutylfluorene (3)67 were synthesized according to literature procedures. All of the monomers were carefully purified prior to use in the polymerization reaction.Synthesis of copolymer PFTSO3Na
2,7-Dibromo-9,9-bis(4′-sulfonatobutyl)-fluorene sodium salt (1) (0.60 g, 0.94 mmol), 4-bromo-N-(4-bromophenyl)-N-p-tolylaniline (2) (0.39 g, 0.94 mmol), and 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dibutylfluorene (3) (1.00 g, 1.89 mmol), Na2CO3 (4.00 g, 37.8 mmol) and Pd(OAc)2 (0.10 g, 1 mol%) were added into a flask under nitrogen, then oxygen-free distilled water (20 mL) and THF (30 mL) were injected into the mixture. After stirring at 80 °C for 72 h, the mixture was poured into acetone (1 L). The precipitate was collected and re-dissolved in distilled water for dialysis for three days, using a membrane to cut off the oligomers with molecular weight less than 6000–8000. The obtained high-molecular-weight polymer in water was concentrated under vacuum. The polymer was first rinsed in a Soxhlet apparatus with hexane and CH2Cl2 for removal of any organic soluble impurities, followed by extraction with methanol. The polymer extracts in methanol were concentrated under reduced pressure to afford PFTSO3Na (0.55 g, 45%). 1H NMR (400 MHz, CD3OD, δ, ppm): 7.88–7.60 (m, 18H), 7.51–7.33 (br, 2H), 2.60–2.58 (m, 8H), 2.22–2.10 (m, 14H), 1.63 (br, 9H), 1.28–1.15 (m, 8H), 0.85–0.70 (m, 24H). 13C NMR (100 MHz, CD3OD, δ, ppm): 154.18, 153.08, 152.54, 151.88, 142.89, 142.34, 142.01, 141.77, 141.66, 140.76, 131.55, 127.60, 122.48, 122.28, 121.52, 121.32, 56.72, 56.55, 56.45, 52.66, 52.58, 41.44, 41.13, 27.53, 26.52, 26.40, 24.66, 24.51, 24.35, 24.16, 14.45, 14.39.Layer-by-layer self-assembly of electrofluorescent films
The electrofluorescent polymer films were formed by the spin-coating method onto an ITO glass slide. PFTSO3Na was dissolved in MeOH at a concentration of 2 mg mL−1. An aqueous solution of PDDA was treated at a concentration of 0.1 g mL−1. First, the ITO glass was treated with N-(3-triethoxysilylpropyl)ethylenediamine to make the substrate surface hydrophilic. Subsequently, the substrate was spun in a PFTSO3Na solution at a speed of 2000 rpm for 20 s, followed by two washing steps in MeOH and Milli-Q water (deionized) at the same spinning speed, respectively. The substrate was then spun in a PDDA solution at a speed of 2000 rpm for 20 s, followed by a washing step in Milli-Q water at the same spinning speed. These processes were repeated to obtain multilayer films. The number of the repeating cycles n was denoted as the subscript in the expression of (PFTSO3Na/PDDA)n.Fabrication of an electrofluorescent switching device
The electrofluorescent polymer film was formed by the LbL assembly method, as described above, onto a piece of ITO-glass. In our study, this layer is responsible for the EFD functions. Another layer of a highly transparent and conductive gel electrolyte based on poly(propylene carbonate) (PPC) and LiClO4, plasticized with propylene carbonate (PC), was spread on top. The gel electrolyte was prepared from PPC (1.0 g), LiClO4 (0.15 g) and PC (3.0 g), and heated at 60 °C to obtain a homogeneous and transparent gel. The device was then sandwiched with another piece of ITO electrode and sealed with epoxy resin.Measurements
1H and 13C NMR spectra were recorded on a Varian Unity Plus 400 MHz spectrometer at room temperature. Thermogravimetric analysis (TGA) was determined under nitrogen by measuring weight loss while heating at a rate of 10 °C min−1. Photoluminescence spectra were obtained on a Hitachi F-4500 luminescence spectrometer. The thickness of the thin film was measured using an alpha step (TENCOR INSTRUMENTS, Alpha-step 200). PL quantum yields were estimated using the Hitachi F-7000 fluorescence spectrophotometer with an integrating sphere. The atomic force microscopy (AFM) image was taken on a precursor modified ITO glass using a Digital Instrument (NS3a controller with D3100).Results and discussion
Synthesis and characterization of PFTSO3Na
The polyelectrolyte PFTSO3Na was synthesized by Pd(OAc)2-catalyzed Suzuki coupling reaction (Scheme 2).68 The polymerization was carried out in a mixture of THF (60%), aqueous sodium carbonate solution (2 M) (40%), and Pd(OAc)2 (1 mol%) under stirring conditions at 80 °C for 3 days. In the synthesis of PFTSO3Na, a feeding ratio of 0.5:0.5:1 for 1, 2, and 3 was respectively adopted. The obtained PFTSO3Na is soluble in methanol, DMF, and DMSO, but insoluble in non-polar organic solvents such as chloroform, toluene, and dichloromethane.
Thermal property of the PFTSO3Na was evaluated by thermogravimetric analysis (TGA). The TGA analysis of PFTSO3Na shows onset decomposition temperature at around 300 °C and with 5% weight-loss at 402 °C, indicating that the polyelectrolyte is thermally stable (Fig. 1).
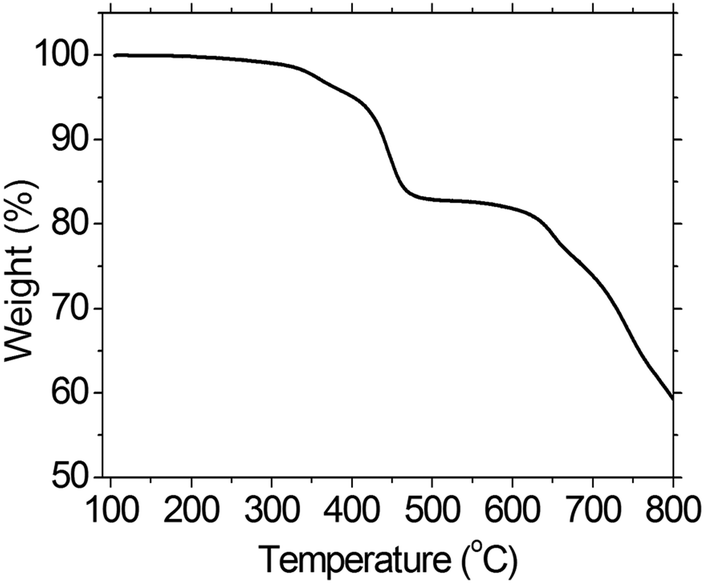 | ||
| Fig. 1 TGA of PFTSO3Na. | ||
Optical properties of PFTSO3Na
The UV-vis and PL spectra of PFTSO3Na in a MeOH solution and in a thin-film on a quartz plate are shown in Fig. 2. The UV-vis spectrum of PFTSO3Na in a MeOH solution exhibits an absorption band peaked at 366 nm, which is corresponding to the π–π* electronic transitions of conjugated chromophores (Fig. 2). Moreover, PFTSO3Na displays a shoulder at about 310 nm, which is attributed to the absorption of the triphenylamine moieties. Compared with the maximum absorption band in solution, the absorption maximum is slightly red-shifted by 8 nm in the solid film state.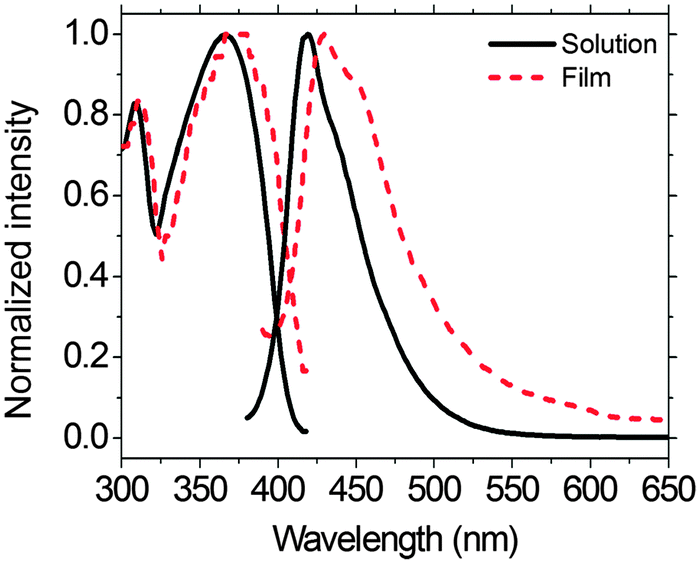 | ||
| Fig. 2 UV-vis absorption and PL spectra of PFTSO3Na in MeOH solution and the solid-film state. | ||
Under UV irradiation, PFTSO3Na shows strong PL in MeOH solution. The PL spectrum of PFTSO3Na in MeOH solution displays an emission maximum at 419 nm, which is originated from fluorene segments. On the other hand, the emission maximum occurs at 430 nm in the solid-film state, which is red-shifted by 11 nm. This may be due to the intermolecular aromatic π-stacking interactions in the solid state that favor to have the poly(fluorene) aligned in a coplanar fashion. This would enhance the intramolecular π-conjugation, leading to the red-shift phenomenon in the UV-Vis absorption as well as in the fluorescence spectra. By comparing against Coumarin 1, a solution fluorescence standard, in MeOH (ΦPL = 0.38),69 and poly(9,9-dioctylfluorene) thin-film standard (ΦPL = 0.55),70 the PL quantum yields (ΦPL) of 0.65 and 0.53 for PFTSO3Na in solution and film state were respectively estimated.
Characterization of multilayer ultrathin LbL film
As shown in Fig. 3, a linear correlation was found between the layer thickness and the LbL growth cycles of PFTSO3Na/PDDA films. The average thickness of 1.87 nm for each bilayer was estimated. The LbL multilayer film buildup was also evidenced by UV/Vis absorption spectroscopy (Fig. 4). The PFTSO3Na/PDDA film showed an increase in the absorbance peaked at 383 nm (Fig. 4a), which is attributed to the π–π* electronic transitions of the conjugated chromophore in the PFTSO3− skeleton. Fig. 4b is the correlation plot that demonstrates the linear relationship between the number of bilayers in the film and absorbance intensity. The absorption intensity of the spectra linearly increased as the number of bilayers increased, indicating that the layer by layer formation of the film was efficient. The explanation for the slightly bathochromic shift of the absorption maximum of the LbL film of PFTSO3Na/PDDA in comparison to that of the single film of PFTSO3Na at 374 nm is not immediately obvious. We tentatively suggest that the effective conjugation length of PFTSO3Na may be elongated by the electrostatic interactions with the counter polyelectrolyte of PDDA.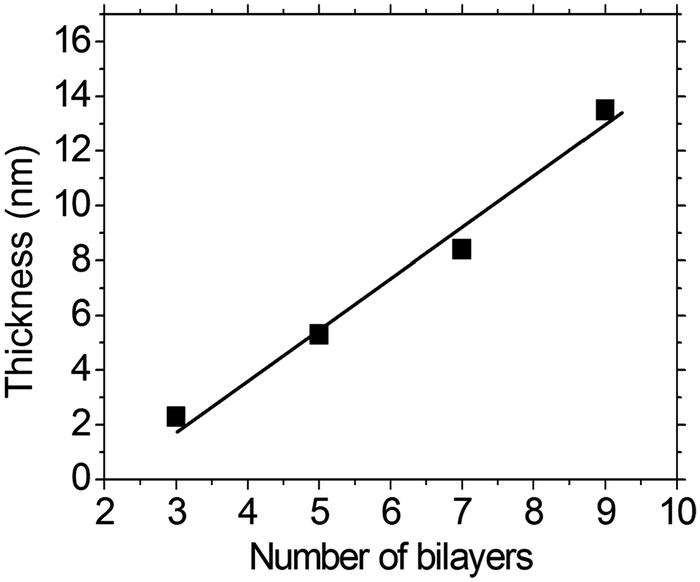 | ||
| Fig. 3 Correlation of film thickness to layer pair number for the PFTSO3Na/PDDA series. | ||
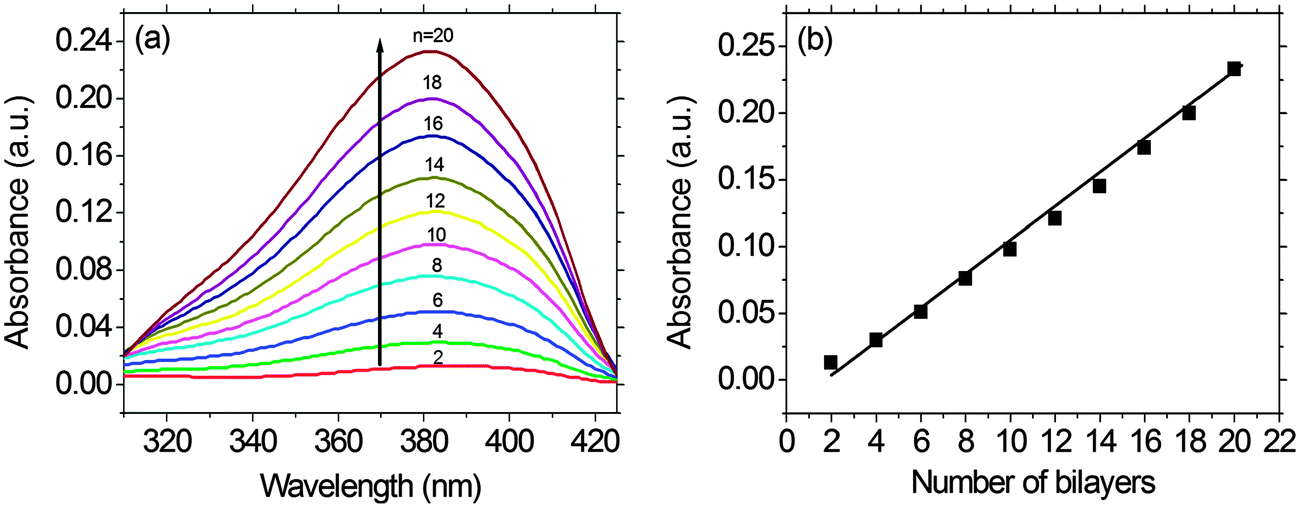 | ||
| Fig. 4 (a) UV-vis absorbance of multilayer assembly from PFTSO3Na/PDDA LbL films (n = 2–20) on a precursor modified substrate. (b) The absorption intensity changes of multilayer assembly from PFTSO3Na/PDDA LbL films (n = 2–20) at 383 nm versus the number of bilayers. | ||
The surface morphology of the multilayer thin film of PFTSO3Na/PDDA was investigated by AFM as shown in Fig. 5. The AFM image displays the surface topography of 5.0 × 5.0 μm2 dimensions of (PFTSO3Na/PDDA)10 film on a precursor modified substrate. The surface was smooth and RMS (root mean square) roughness was calculated to be about 1.46 nm.
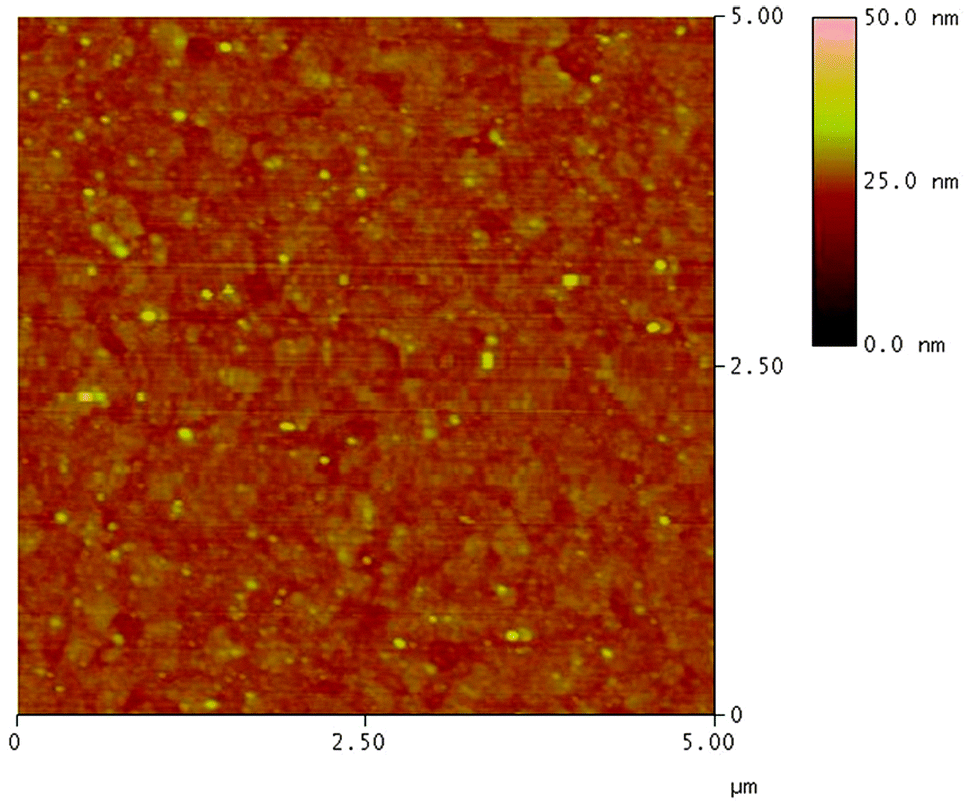 | ||
| Fig. 5 AFM image of (PFTSO3Na/PDDA)10 film deposited on a precursor modified substrate of 5.0 μm × 5.0 μm in size. | ||
The multilayer buildup on an ITO electrode was also performed by cyclic voltammetry (CV) in an electrolyte of LiClO4/MeCN (0.1 M), using Pt wire as the counter electrode, and an Ag/AgCl couple as the reference electrode (Fig. 6). The scan rate being adopted was 100 mV s−1. Monitoring of the electrochemical data of the PFTSO3Na/PDDA film as a function of film thickness for up to 9 bilayers revealed that the electroactivity of PFTSO3Na/PDDA increased with the number of bilayers. The films exhibited a redox response despite the insulating nature of the interstitial PDDA layer. This result suggests the presence of an interpenetrating polymer network as opposed to discrete bilayers within the film. As shown in Fig. 6, the films of PFTSO3Na/PDDA show two anodic waves: one starts at around 0.96 V with a sharp oxidative peaked at about 1.18 V, which is characteristic for the oxidation of the triphenylamine units to form the corresponding cation-radicals (Scheme 3). Another one arising at 1.30 V corresponds to the fluorene oxidation in the polymer main chain.
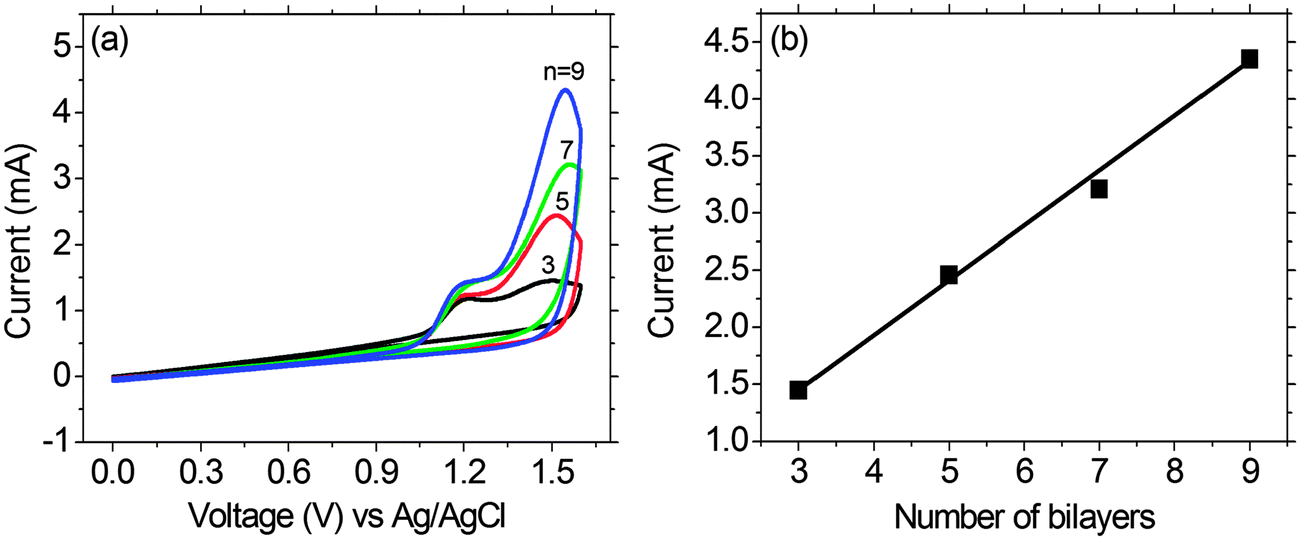 | ||
| Fig. 6 (a) Cyclic voltammograms of multilayer assembly from PFTSO3Na/PDDA LbL films (n = 3–9) on a precursor modified substrate, at a scan rate of 100 mV s−1, between 0.0 V and 1.6 V. (b) The peak current vs. the number of bilayers on a precursor modified substrate. | ||
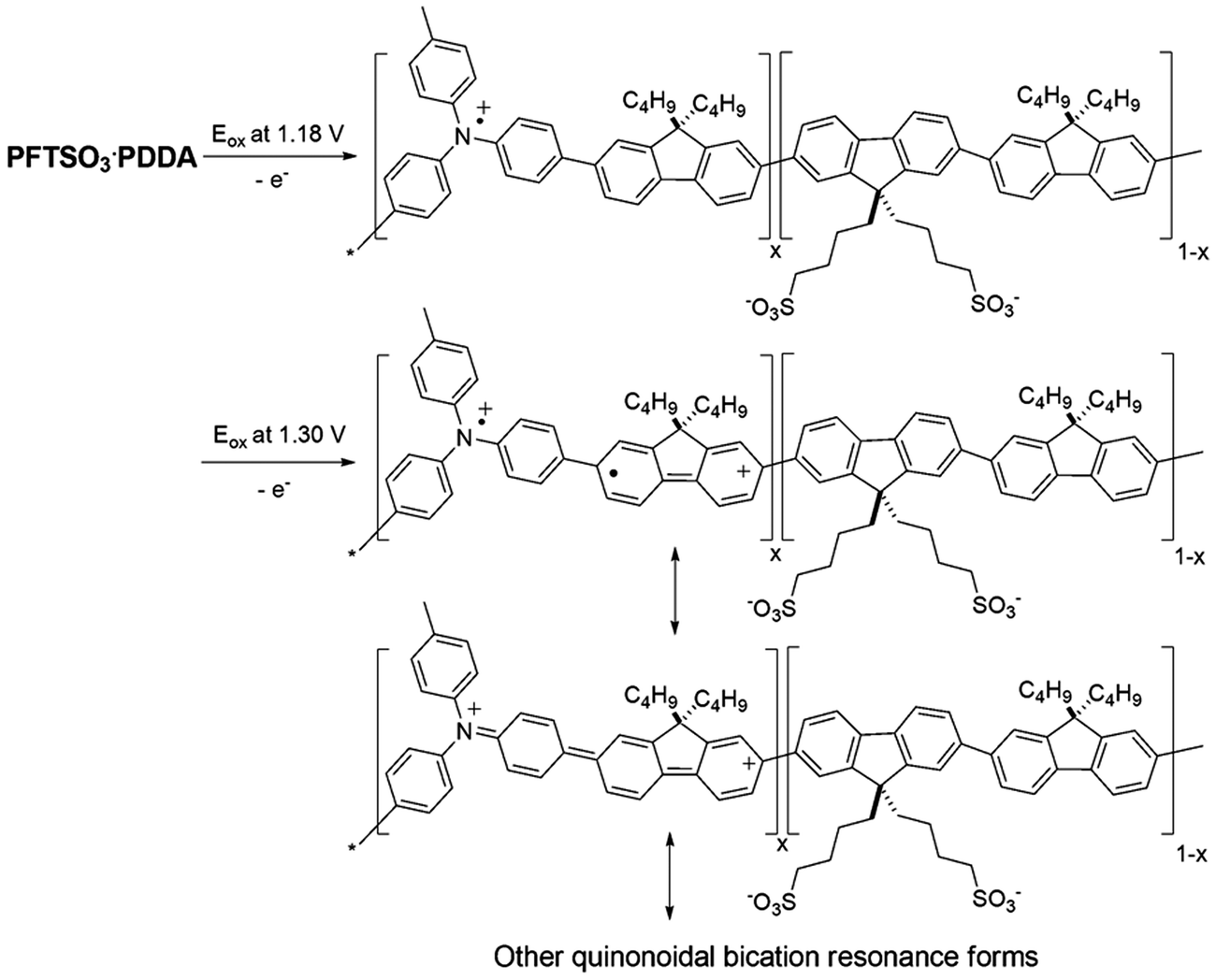 | ||
| Scheme 3 Proposed mechanisms for the electrochemical oxidation of PFTSO3Na. | ||
It is noteworthy to point out that the electroactivity of the first wave is less responsive to the layer number. This is probably due to the slow diffusion rate of the triphenylaminyl cation-radicals that confines the electroactivity at the interfacial layer. On the other hand, the second wave shows a linear increase in the layer number, implying that the migration rate of the bications should be much higher than the radical cations. We tentatively attribute this to the formation of quinonoidal bications under the high doped conditions that enhances the conductivity of the polymer matrix.
Electrochemical fluorescence switching in an electrofluorescent device (EFD)
As shown in Fig. 7, we fabricated a prototype of a two-layered EFD. An electrofluorescent film is fabricated using the LbL method, which involves the spin-coating of a modified ITO glass substrate in dilute solutions of PFTSO3Na (polyanion) and PDDA (polycation) in an alternating manner. Herein the gel electrolyte based on poly(propylene carbonate) (PPC) and LiClO4 was plasticized with propylene carbonate (PC) to form a highly transparent and conductive gel. PPC is of particular interest in lithium-conducting polymeric electrolytes; the repeating unit of PCC, depicted as –(CH2–CH2–CH2–O–CO–O)–, has a similar chemical structure to the low-molecular-weight carbonates such as propylene carbonate, ethylene carbonate and dimethyl carbonate that are dipolar aprotic solvents with high dielectric constant. The EFD of PFTSO3Na/PDDA emits blue light under UV excitation. In our preliminary study, the EFD was used in the experiments of fluorescence intensity changes with the applied electrical positive potentials (Fig. 8).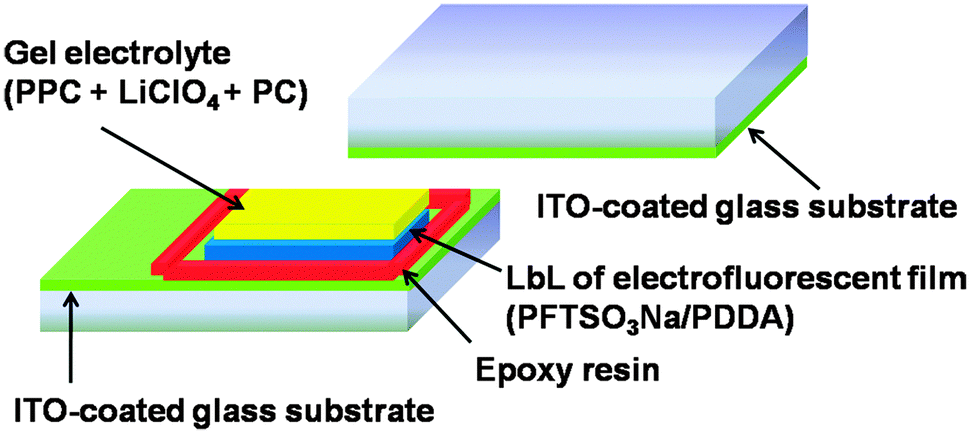 | ||
| Fig. 7 Schematic diagram of the EFD sandwich cell. | ||
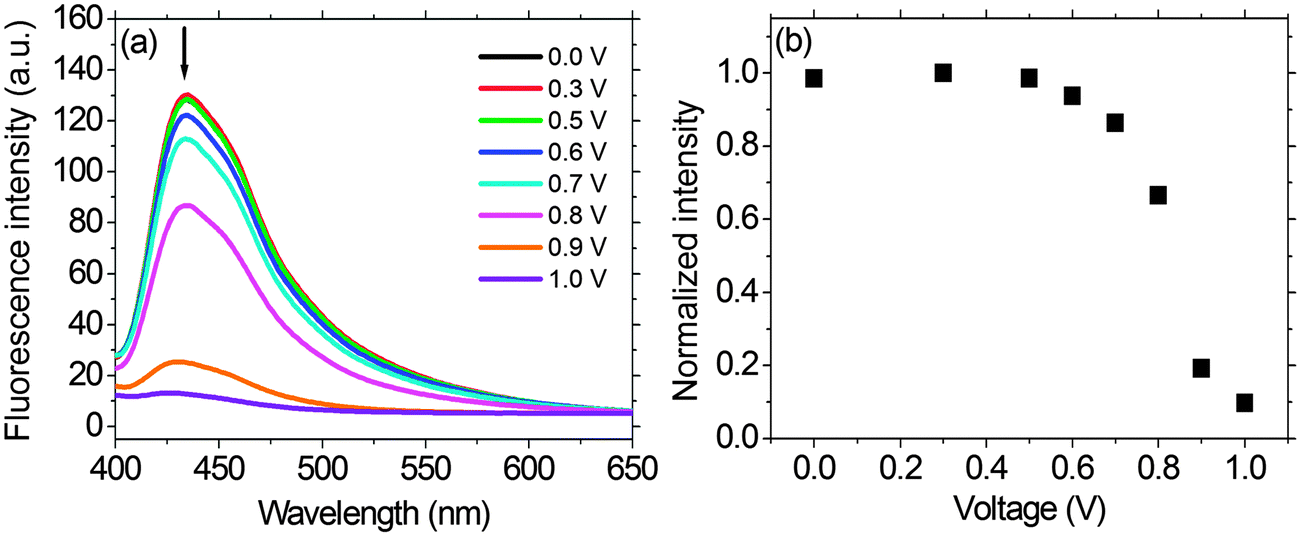 | ||
| Fig. 8 (a) Fluorescence intensity changes (λexc = 380 nm) of the EFD of (PFTSO3Na/PDDA)20 at different applied potentials. Each spectrum was obtained after applying the target potential for 30 s. (b) Fluorescence intensity changes of the EFD of (PFTSO3Na/PDDA)20 at 435 nm versus the applied electrical potential (V). | ||
Upon applying positive electrical potentials beyond 0.9 V, the EFD of (PFTSO3Na/PDDA)20 was significantly extinguished and became almost completely quenched after 1.0 V (Fig. 8). The fluorescence intensity changes occurred without a shift of the emission band along with the potential changes, indicating that the fluorescence quenching originated from the electrochemical oxidation of the triphenylamine unit to its cation-radical form, without production of any other emissive side products.
Proposed mechanisms for the oxidative fluorescence quenching process of the EFD of (PFTSO3Na/PDDA)20 are displayed in Scheme 4. Our previous study revealed that when a positive electrical potential of 1.0 V was applied, oxidation selectively occurred on the triphenylamine moieties and gave rise to the corresponding TAA radical cations. Since the TAA radical cation is known to have an absorption band at about 500 nm,38,39 it acts as an effective fluorescence quencher so that the fluorescence of PFTSO3Na is efficiently quenched. When a reverse sweep was applied, the triphenylamine radical cations in PFTSO3Na were annihilated and the fluorescence at a neutral state of PFTSO3Na was recovered.
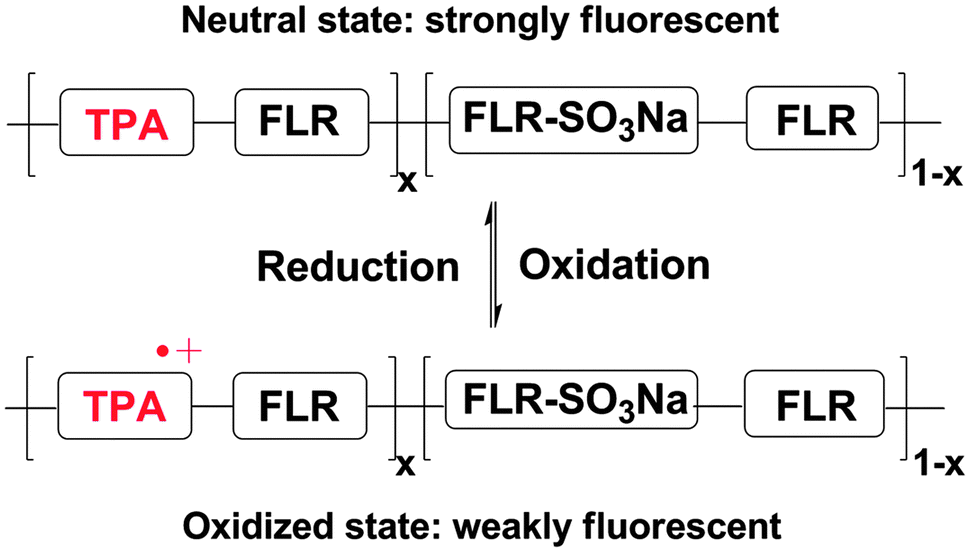 | ||
| Scheme 4 Illustration of the EFD based on the polyelectrolyte PFTSO3Na. | ||
To elucidate if a similar EF switching mechanism does operate in the EFD of (PFTSO3Na/PDDA)20, spectroelectrochemistry was employed to monitor the redox intermediates that were generated in the EFD. Fig. 9 shows the UV-Vis spectra of the EFD of (PFTSO3Na/PDDA)20 being collected under different applied electrical potentials for 150 s. The absorbance enhancement at about 425–500 nm is less obvious below 1.0 V and gradually increased when the applied electrical voltage was set to be higher than 1.5 V. All these support our assumption of the TAA radical cations and quinonoidal bications formation in the polymer matrix.
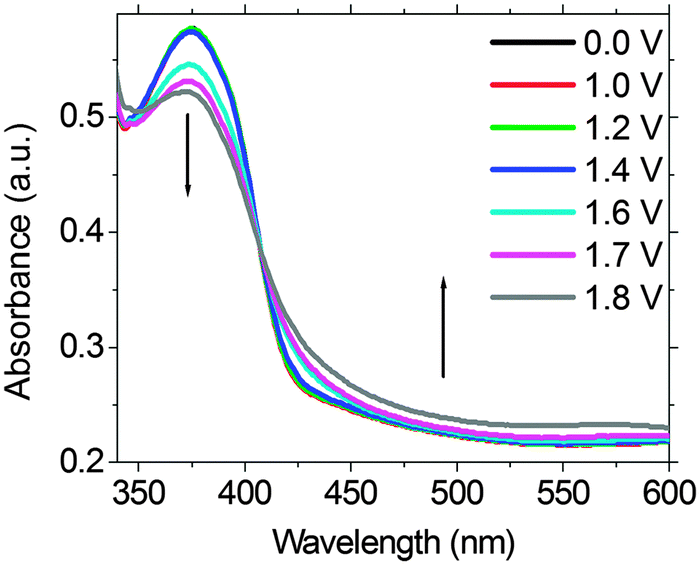 | ||
| Fig. 9 Spectroelectrochemistry of the EFD of (PFTSO3Na/PDDA)20. | ||
Fig. 10a demonstrates the fluorescence switching responses of the solid-state EFD of (PFTSO3Na/PDDA)20. Redox pulse-cycles were carried out for the EFD of (PFTSO3Na/PDDA)20. Fixed potentials at 1.0 V (for oxidation) and −1.0 V (for reduction) were used with step duration times of 10 and 5 seconds at each potential. The EFD of (PFTSO3Na/PDDA)20 shows the reversible EF switching by repetitive transformation between its oxidized state (non-fluorescent) at 1.0 V and the neutral state (fluorescent) at −1.0 V. The fluorescence switching responses for the EFD of (PFTSO3Na/PDDA)20 were monitored at 435 nm and exhibited switching performance of contrast ratios (If/If0) of 5.20 and 3.78 with step duration times of 10 and 5 seconds at each potential, respectively. In this experiment, the potential was quickly scanned without waiting for completion of the redox reaction of the triphenylamine units; the on/off ratio was therefore smaller than that expected from the spectral changes in Fig. 8. Moreover, the switching time was estimated at 90% of the full switch. The EFD of (PFTSO3Na/PDDA)20 showed an on-to-off switching time of 3.49 s at 1.0 V and an off-to-on switching time of 1.88 s at −1.0 V (Fig. 10b), which are faster than the switching times for our previously reported electrofluorescent polymer films39 (see ESI†).
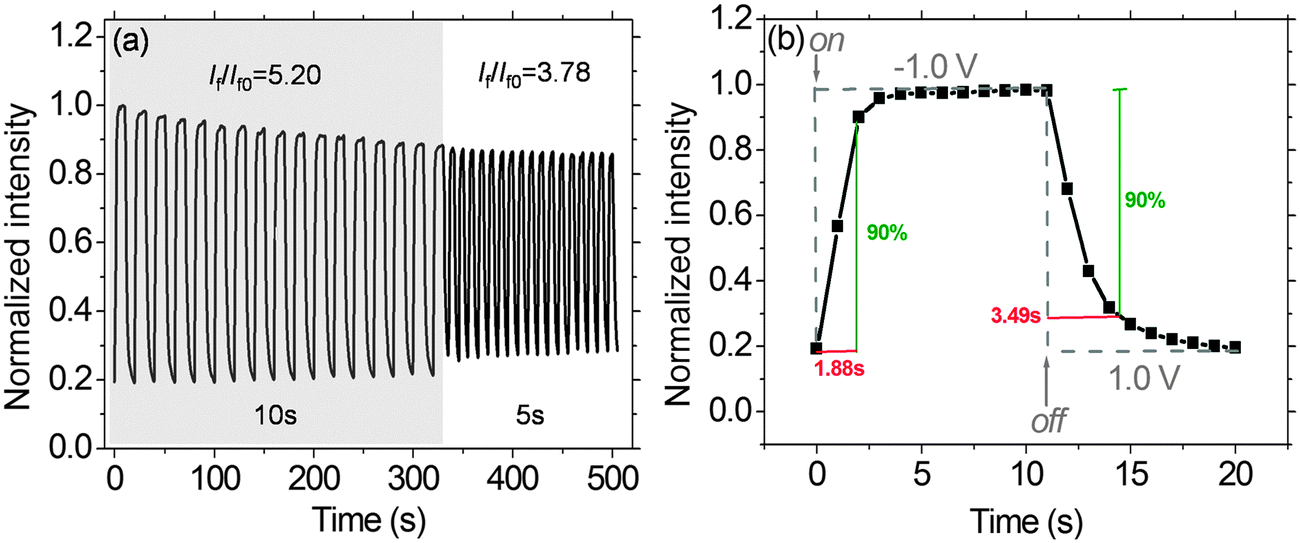 | ||
| Fig. 10 (a) Fluorescence switching responses (λexc = 360 nm) of the EFD of (PFTSO3Na/PDDA)20 monitored at 435 nm. Fixed potentials at 1.0 V (for oxidation) and −1.0 V (for reduction) were used with step duration times of 10 and 5 seconds at each potential. (b) Estimation of fluorescence switching time at 435 nm at the applied potential. | ||
As shown in Fig. 11, we had successfully fabricated a prototype of the EFD of (PFTSO3Na/PDDA)20. When a voltage of 1.0 V was applied, the fluorescence of the EFD disappeared because of electrochemical oxidation. When a reverse sweep was applied, the EFD was de-doped and turned back to the fluorescent neutral state.
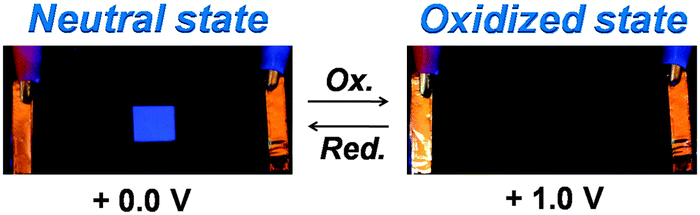 | ||
| Fig. 11 Photos of the EFD of (PFTSO3Na/PDDA)20 as the electrofluorescent film. | ||
Conclusions
We have fabricated a multilayer electrofluorescent thin film by spin-assisted LbL assembly technique using PFTSO3Na as a polyanion and PDDA as a polycation. Furthermore, we have successfully established a design of robust materials for the EFD, which can be switched on and off through the electrochemical potential applied to the fluorescent conjugated polyelectrolyte PFTSO3Na itself. In our design, we incorporated the fluorene units by taking advantage of the high fluorescence quantum yield. In addition, the electrochemically active triphenylamine-core played an important role in controlling the emissive properties of PFTSO3Na. The EFD of (PFTSO3Na/PDDA)20 from a fluorescent neutral state to a non-fluorescent cation radical state gave rise to a maximum contrast ratio (If/If0) of 5.20.Acknowledgements
The present work was supported by the Ministry of Education and National Taiwan University, National Science Council (NSC-101-2113-M-002-010-MY3, 98-2119-M-002-006-MY3 and 99-2119-M-002-007), and Ministry of Economic Affairs of Taiwan (102-EC-17-A-08-S1-183).References
- J. Kim, J. You and E. Kim, Macromolecules, 2010, 43, 2322 CrossRef CAS.
- R. A. Bissell, A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire and K. R. A. S. Sandanayake, Chem. Soc. Rev., 1992, 21, 187 RSC.
- Y. Kim, Y. Kim, S. Kim and E. Kim, ACS Nano, 2010, 4, 5277 CrossRef CAS PubMed.
- C Yun, S. Seo and E. Kim, J. Nanosci. Nanotechnol., 2010, 10, 6850 CrossRef CAS PubMed.
- C. Yun, J. You, J. Kim, J. Huh and E. Kim, J. Photochem. Photobiol., C, 2009, 10, 111 CrossRef CAS PubMed.
- X. Huang, Y. Xiao and M. Lang, Macromol. Res., 2011, 19, 113 CrossRef CAS PubMed.
- B. Wang, L.-H. Bi and L.-X. Wu, J. Mater. Chem., 2011, 21, 69 RSC.
- G. D. Santis, L. Fabbrizzi, M. Licchelli, N. Sardone and A. H. Velders, Chem.–Eur. J., 1996, 2, 1243 CrossRef.
- R. Martínez, I. Ratera, A. Tárraga, P. Molina and J. Veciana, Chem. Commun., 2006, 3809 RSC.
- R. Zhang, Y. Wu, Z. Wang, W. Xue, H. Fu and J. Yao, J. Phys. Chem. C, 2009, 113, 2594 CAS.
- M. D. Casa, L. Fabbrizzi, M. Licchelli, A. Poggi, D. Sacchi and M. Zema, J. Chem. Soc., Dalton Trans., 2001, 1671 RSC.
- Y. Kim, E. Kim, G. Clavier and P. Audebert, Chem. Commun., 2006, 3612 RSC.
- G. Zhang, D. Zhang, X. Guo and D. Zhu, Org. Lett., 2004, 6, 1209 CrossRef CAS PubMed.
- J. You, Y. Kim and E. Kim, Mol. Cryst. Liq. Cryst., 2010, 520, 128 Search PubMed.
- J. Yoo, T. Kwon, B. D. Sarwade, Y. Kim and E. Kim, Appl. Phys. Lett., 2007, 91, 241107 CrossRef.
- G. Hennrich, H. Sonnenschein and U. Resch-Genger, J. Am. Chem. Soc., 1999, 121, 5073 CrossRef CAS.
- W. R. Browne, M. M. Pollard, B. D. Lange, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 12412 CrossRef CAS PubMed.
- S. Seo, Y. Kim, J. You, B. D. Sarwade, P. P. Wadgaonkar, S. K. Menon, A. S. More and E. Kim, Macromol. Rapid Commun., 2011, 32, 637 CrossRef CAS PubMed.
- S. Seo, Y. Kim, Q. Zhou, G. Clavier, P. Audebert and E. Kim, Adv. Funct. Mater., 2012, 22, 3556 CrossRef CAS.
- T. M. Swager, Acc. Chem. Res., 1998, 31, 201 CrossRef CAS.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 5321 CrossRef CAS.
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 7017 CrossRef CAS.
- S. Wang, B. S. Gaylord and G. C. Bazan, J. Am. Chem. Soc., 2004, 126, 5446 CrossRef CAS PubMed.
- G. Decher, J. D. Hong and J. Schmitt, Thin Solid Films, 1992, 210, 831 CrossRef.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS.
- A. L. Becker, A. P. R. Johnston and F. Caruso, Small, 2010, 6, 1836 CAS.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Moehwald, Science, 1998, 282, 1111 CrossRef CAS.
- Z. Tang, Y. Wang, P. Podsidlo and N. A. Kotov, Adv. Mater., 2006, 18, 3203 CrossRef CAS.
- P. Lavalle, C. Gergely, F. J. G. Cuisinier, G. Decher, P. Schaaf, J. C. Voegel and C. Picart, Macromolecules, 2002, 35, 4458 CrossRef CAS.
- Y. Lvov, G. Decher and G. Sukhorukov, Macromolecules, 1993, 26, 5396 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS PubMed.
- J. H. Fendler, Chem. Mater., 1996, 8, 1616 CrossRef CAS.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed.
- T. Michinobu, T. Nakanishi, J. P. Hill, M. Funahashi and K. Ariga, J. Am. Chem. Soc., 2006, 128, 10384 CrossRef CAS PubMed.
- M. Ruben, J. M. Lehn and P. Muller, Chem. Soc. Rev., 2006, 35, 1056 RSC.
- C.-P. Kuo, Y.-S. Lin and M.-k. Leung, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5068 CrossRef CAS.
- C.-P. Kuo, C.-N. Chuang, C.-L. Chiou, M.-k. Leung, H.-Y. Lian and K. C.-W. Wu, J. Mater. Chem. C, 2013, 1, 2121 RSC.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS.
- M. Kreyenschmidt, G. Klaerner, T. Fuhrer, J. Ashenhurst, S. Karg, W. D. Chen, V. Y. Lee, J. C. Scott and R. D. Miller, Macromolecules, 1998, 31, 1099 CrossRef CAS.
- Q. Pei and Y. Yang, J. Am. Chem. Soc., 1996, 118, 7416 CrossRef CAS.
- C.-F. Shu, R. Dodda, F.-I. Wu, M. S. Liu and A. K.-Y. Jen, Macromolecules, 2003, 36, 6698 CrossRef CAS.
- Y. Li, J. Ding, M. Day, Y. Tao, J. Lu and M. D'iorio, Chem. Mater., 2004, 16, 2165 CrossRef CAS.
- S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Müllen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946 CrossRef CAS PubMed.
- R. Tang, Z. Tan, Y. Li and F. Xi, Chem. Mater., 2006, 18, 1053 CrossRef CAS.
- Y.-H. Kim, Q. Zhao and S.-K. Kwon, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 172 CrossRef CAS.
- Q. Zhao, Y.-H. Kim, T. T. M. Dang, D.-C. Shin, H. You and S.-K. Kwon, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 341 CrossRef CAS.
- L. Chen, P. Li, H. Tong, Z. Xie, L. Wang, X. Jing and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2854 CrossRef CAS.
- H.-M. Shih, R.-C. Wu, P.-I. Shih, C.-L. Wang and C.-S. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 696 CrossRef CAS.
- H. Tan, J. Yu, Y. Wang, J. Li, J. Cui, J. Luo, D. Shi, K. Chen, Y. Liu, K. Nie and W. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 149 CrossRef CAS.
- K. Ogino, A. Kanegae, R. Yamaguchi, H. Sato and J. Kurjata, Macromol. Rapid Commun., 1999, 20, 103 CrossRef CAS.
- W.-L. Yu, J. Pei, W. Huang and A. J. Heeger, Chem. Commun., 2000, 681 RSC.
- M.-Y. Chou, M.-k. Leung, Y. O. Su, C. L. Chiang, C.-C. Lin, J.-H. Liu, C.-K. Kuo and C.-Y. Mou, Chem. Mater., 2004, 16, 654 CrossRef CAS.
- S.-H. Cheng, S.-H. Hsiao, T.-H. Su and G.-S. Liou, Macromolecules, 2005, 38, 307 CrossRef CAS.
- G.-S. Liou, S.-H. Hsiao, N.-K. Huang and Y.-L. Yang, Macromolecules, 2006, 39, 5337 CrossRef CAS.
- G.-S. Liou, S.-H. Hsiao, W.-C. Chen and H.-J. Yen, Macromolecules, 2006, 39, 6036 CrossRef CAS.
- C.-W. Chang, G.-S. Liou and S.-H. Hsiao, J. Mater. Chem., 2007, 17, 1007 RSC.
- G.-S. Liou, S.-H. Hsiao and H.-W. Chen, J. Mater. Chem., 2006, 16, 1831 RSC.
- F. Liang, Y.-J. Pu, T. Kurata, J. Kido and H. Nishide, Polymer, 2005, 46, 3767 CrossRef CAS PubMed.
- H.-M. Wang and S.-H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 337 CrossRef CAS.
- Y. Li and T. Michinobu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2111 CrossRef CAS.
- H.-j. Yen, S.-m. Guo, J.-m. Yeh and G.-S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3637 CrossRef CAS.
- Y.-C. Kung and S.-H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3475 CrossRef CAS.
- M. Thelakkat, Macromol. Mater. Eng., 2002, 287, 442 CrossRef CAS.
- F. Huang, X. Wang, D. Wang, W. Yang and Y. Cao, Polymer, 2005, 46, 12010 CrossRef CAS PubMed.
- F. Huang, L. Hou, H. Shen, J. Jiang, F. Wang, H. Zhen and Y. Cao, J. Mater. Chem., 2005, 15, 2499 RSC.
- X. Zhu, Y. Xie, X. Li, X. Qiao, L. Wang and G. Tu, J. Mater. Chem., 2012, 22, 15490 RSC.
- M. V. Rusalov, S. I. Druzhinin and B. M. Uzhinov, J. Fluoresc., 2004, 14, 193 CrossRef CAS.
- H.-J. Su, F.-I. Wu, Y.-H. Tseng and C.-F. Shu, Adv. Funct. Mater., 2005, 15, 1209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53912a |
| This journal is © the Owner Societies 2014 |