Fabrication of graphene–carbon nanotube papers decorated with manganese oxide nanoneedles on the graphene sheets for supercapacitors†
Myeongjin
Kim
,
Yongseon
Hwang
and
Jooheon
Kim
*
School of Chemical Engineering & Materials Science, Chung-Ang University, Seoul 156-756, Korea. E-mail: jooheonkim@cau.ac.kr; Fax: +82-2-812-3495; Tel: +82-2-820-5763
First published on 20th November 2013
Abstract
Herein, 3D nanohybrid architectures consisting of MnO2 nanoneedles, carbon nanotubes (CNTs) and graphene sheets are fabricated. Nanostructured ternary hybrid papers in which MnO2 nanoneedles formed on the outermost graphene layer and CNTs intercalated between graphene layers by using the amide bonds are fabricated using the direct paper dipping method. The intercalated CNTs can separate the graphene layers and thus create the effective surface area which is associated with large electrochemically active sites as well as form the electronic conductive channel inside the nanohybrid paper. Moreover, the homogeneous dispersion of nanometer-thick MnO2 on the outermost graphene layer can maximize the surface area which can form pores for ion-buffering reservoirs to improve the diffusion rate of electrolyte ions and enable convenient participation in the pseudo-capacitive reaction. These nanostructured ternary hybrid papers exhibit enhanced specific capacitances compared with graphene-only or graphene–CNT papers. The proposed nanohybrid architectures are expected to lay the foundation for the design and fabrication of high-performance electrodes.
1. Introduction
Over the past few years, the rapid development of renewable energy (e.g., solar and wind energy) has increased the need for sustainable energy storage technologies in order to address the challenges of increasing energy demand and the cyclic nature of renewable energy sources.1–4 Due to their high power density, moderate energy density, good operational safety, and long cycling life, electrochemical double layer capacitors or supercapacitors have been extensively explored and have become a promising technique for various emerging energy applications in high-power electronic devices, electric vehicles, and hybrid electric vehicles.5–7 Due to the large surface area (∼2630 m2 g−1), excellent electronic conductivity, outstanding chemical stability, and good physical properties of graphene, many studies up to now have focused on graphene-based composites for supercapacitor applications;8–10 however, it should be noted that most of the working electrodes reported in the literature have been binder-enriched electrodes produced by conventional slurry-coating technology. In this process, the binder decreases the electrical conductivity of the electrode materials, hindering potential applications in high-performance supercapacitors.11 In order to obtain ideal electrochemical performance, it is very important to develop novel, flexible, and binder-free electrode materials for supercapacitors.12–14Typical electrode materials for EDLCs include high surface area carbon, which can store energy in a double layer formed on surfaces such as graphene, activated carbon, carbon nanotubes (CNTs), and carbon aerogels.15–17 To produce flexible and binder-free films combining graphene and CNTs, the Huang group developed graphene–CNT paper by taking advantage of the stacking properties of graphene oxide (GO) sheets.18 They reported that graphene–CNT12.5 wt% paper showed a specific capacitance of 151 F g−1 at a current density of 0.5 A g−1, which is a much higher value than that of graphene paper (118 F g−1 at the same current density). These improvements arise from the synergistic effects of the increased electronic conductivity and effective surface area associated with large electrochemical active sites due to the presence of intercalated CNTs. However, the specific capacitance is relatively low compared with carbon-based material/metal oxide material hybrid composites. Metal oxide electrode materials, which are a kind of pseudo-capacitive material, have attracted considerable interest due to their large capacitance and fast redox kinetics. Among these, manganese oxide (MnO2) is generally considered to be the most promising transition metal oxide for the next generation of supercapacitors by virtue of its high energy density, low cost, environmental friendliness, and natural abundance.19,20
To obtain a high specific capacitance, the Li group suggested graphene/birnessite-type MnO2 composite paper and reported a specific capacitance of 256 F g−1 at a current density of 500 mA g−1.11 However, in order to achieve ideal capacitive behavior, three improvements are needed with respect to selecting the crystallographic structure of MnO2 and the nano-structure design of graphene paper. They are: (i) the Chen group prepared MnO2 with different crystallographic forms (α, γ) and morphologies (needles, rods, and spindles), and investigated the electrochemical performance.21 It was reported that the needle-like sample showed a higher specific capacitance due to the high specific area of MnO2, which can form pores for ion-buffering reservoirs to improve the diffusion rate of electrolyte ions. (ii) The MnO2 particles are placed between the graphene layers in graphene/birnessite-type MnO2 paper prepared by the Li group. However, in order to maximize the surface area and pseudo-capacitive reaction of MnO2, it is beneficial that MnO2 must be formed not only between the graphene layers but also on the outermost graphene layer of graphene–CNT paper. The MnO2 which is located on the outermost graphene layer is more favorable to participate in the redox reaction than the MnO2 which isplaced between the graphene layers because it is easy for electrolyte ions to reach the MnO2 located on the outermost graphene layer. Moreover, the homogeneous dispersion of nanometer-thick MnO2 on the outermost graphene layer is also an important factor, since it is well known that the pseudo-capacitive reaction of MnO2 is a surface reaction, in that only the surface (or a very thin surface layer of the oxide) can participate in the pseudo-capacitive reaction.22 Thus, nanometer-thick MnO2 deposits deliver very high specific capacitances, ranging from 700 to 1380 F g−1.23 Therefore, the outermost graphene layer must be homogeneously decorated with MnO2 with nanometer thickness. (iii) There are no CNTs which can separate the graphene layers in graphene/birnessite-type MnO2 paper prepared by the Li group. If the CNTs don't exist in the graphene paper, the interlayer distance of graphene paper is too small for ions to access the inner region of the graphene sheets. Therefore, CNTs must be embedded between the graphene layers in order to physically separate the graphene layers and thus create a well-defined porous sandwich structure that can increase the electrolyte–electrode contact area and facilitate the transport of electrolyte ions and electrons into the inner region of the electrode.18,24
To this end, we herein develop an efficient and facile strategy to fabricate nanostructured ternary hybrid electrodes for the development of high-performance electrochemical capacitors in which MnO2 nanoneedles are formed on the outermost graphene layer and CNTs are intercalated between the graphene layers. The synthesis process of the hybrid electrode involves two steps. First, GO–CNT paper (C-GCP) is synthesized by forming amide bonds between GO and functionalized CNTs. To obtain the optimum capacitive characteristics of the weight percent of CNTs, GO–CNT paper is thermally reduced to graphene–CNT paper (C-rGCP) in order to investigate the relationship between the capacitance behavior and the CNT content. In the second stage, MnO2 nanoneedles are formed only on the outer-most GO layers in the C-GCP, which obtained optimum CNT content using various oxygen-containing functional groups on the GO sheets (C-GCP–MnO2), and then, graphene–CNT/MnO2 papers were obtained by a thermal reduction process (C-rGCP–MnO2).
2. Experimental
2.1 Preparation of graphene oxide
GO was synthesized using the modified Hummers method.25 All reagents were used without further purification. In a typical synthesis, 3 g of graphite powder was placed into a mixture containing 360 mL of concentrated sulfuric acid and 40 mL of concentrated phosphoric acid. The resulting mixture was stirred in an ice bath for 4 h. After homogeneous dispersion of the graphite powder in the solution, KMnO4 (18 g) was added slowly to the solution over 1 h in an ice bath at 0 °C. The mixture was then stirred for 12 h at 50 °C in an oil bath. After the reaction, the mixture was poured into 400 mL of DI water containing 3 mL of H2O2. The mixture was washed several times and purified with HCl and DI water. The graphite oxide was collected and then dispersed in DI water by ultrasonication to exfoliate the graphite oxide to GO, which was dried in a vacuum oven at 50 °C for 2 days. The GO obtained had particles with average lateral dimensions of several micrometers and thicknesses of 4.5 nm.2.2 Preparation of surface-treated GO and CNTs
GO with carboxylic acid groups was dispersed in SOCl2 using an ultrasonicator for 1 h at room temperature to form a suspension. Chlorinated GO was then filtered through a 0.4 μm membrane after refluxing and stirring for 12 h at 65 °C. The filtered powders were then washed with THF and dried for 12 h under vacuum at ambient temperature.The CNTs were purified and functionalized with carboxylic acid groups by heat treating the CNTs (1 g) in 800 mL of H2SO4 and HNO3 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 by volume) in an ultrasonicator bath (Bransonic, MT-1510, 42 KHz) for 8 h at room temperature to form a suspension. The suspension was then heated to 50 K and stirred for 24 h, and then filtered through a nylon membrane. The filtered cake was washed thoroughly with water, and this process was repeated several times until the filtrate was neutral. The HNO3 treatment produced carboxylic groups on the surfaces of the CNTs without greatly decreasing the length of the CNTs. This reaction also eliminated impurities that could impart unexpected thermal properties to the composite. Carboxylated CNTs were then immersed in SOCl2 using an ultrasonicator at room temperature for 1 h and stirred for 12 h at 65 °C to convert the carboxylic acid groups on the surfaces of the CNTs to acid chlorides. The suspension was vacuum-filtered through a membrane, washed with THF, and dried for 12 h under vacuum at ambient temperature. Then, acid-chloride-activated CNTs were added to ethylenediamine in DMF and refluxed at 120 °C for 24 h with stirring. Finally, the reaction mixture was filtered through a nylon membrane and washed with DI water, and the remaining solvent was dried in a vacuum oven at 100 °C for 5 h.
:2 by volume) in an ultrasonicator bath (Bransonic, MT-1510, 42 KHz) for 8 h at room temperature to form a suspension. The suspension was then heated to 50 K and stirred for 24 h, and then filtered through a nylon membrane. The filtered cake was washed thoroughly with water, and this process was repeated several times until the filtrate was neutral. The HNO3 treatment produced carboxylic groups on the surfaces of the CNTs without greatly decreasing the length of the CNTs. This reaction also eliminated impurities that could impart unexpected thermal properties to the composite. Carboxylated CNTs were then immersed in SOCl2 using an ultrasonicator at room temperature for 1 h and stirred for 12 h at 65 °C to convert the carboxylic acid groups on the surfaces of the CNTs to acid chlorides. The suspension was vacuum-filtered through a membrane, washed with THF, and dried for 12 h under vacuum at ambient temperature. Then, acid-chloride-activated CNTs were added to ethylenediamine in DMF and refluxed at 120 °C for 24 h with stirring. Finally, the reaction mixture was filtered through a nylon membrane and washed with DI water, and the remaining solvent was dried in a vacuum oven at 100 °C for 5 h.
2.3 Preparation of C-GCP and C-rGCP
The composite was fabricated by solvent mixing and hot-pressing to obtain sheet-type hybrid papers. In the first step, 0.05 g of surface-treated GO was suspended in 10 mL of THF with various amounts of surface-treated CNTs (5, 10, 15, 20, and 30 wt%) under ultrasonication for 1 h, and stirred for 3 h at 65 °C. The suspension was then filtered through a 0.4 μm membrane and hot-pressed at 100 psi and 80 °C for 2 h to obtain well-aligned C-GCP. Then, thermal reduction was carried out to obtain C-rGCP by annealing at 300 °C for 30 min in a N2 gas atmosphere. The products were denoted as C-GCP0, C-GCP5, C-GCP10, C-GCP15, C-GCP20 and C-GCP30 for the weight percent of CNTs of 0, 5, 10, 15, 20 and 30 wt%, respectively.2.4 Preparation of C-GCP10–MnO2 and C-rGCP10–MnO2
The amount of CNT with GO was fixed at 10 wt%, and MnO2 was then added at different feeding ratios. The proposed formation mechanism for MnO2 nanoneedle structures can be illustrated by the following reaction:26| 2KMnO4 + 3MnCl2 + 2H2O → 5MnO2 + 2KCl + 4HCl |
For example, when the feeding ratio of C-rGCF10–MnO2 was 1:2, the typical process was as follows: C-GCF10 (0.06 g) was immersed in isopropyl alcohol (100 mL) with MnCl2·4H2O (0.327 g). Next, the reactor was heated to approximately 85 °C in a water-cooled condenser with mild stirring (150 rpm). After which KMnO4 (0.174 g) dissolved in 10 mL of DI water was added to the boiling solution while maintaining mild stirring at 85 °C for 30 min. The resulting C-GCF–MnO2(2) was then washed in DI water and dried in a vacuum oven at 60 °C. Finally, C-rGCF–MnO2 was obtained by annealing at 300 °C for 30 min in a N2 gas atmosphere. When the feeding ratios of MnO2–C-rGCP10 are 2/1 and 6/1, the products are denoted as C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6), respectively.
2.5 Characterization methods
Fourier transform infrared (FT-IR) spectra were obtained at a resolution of 4.0 cm−1 on an FT-IR spectrophotometer (PerkinElmer Spectrum100, USA) at 25 °C in the range of 4000–400 cm−1. The X-ray diffraction (XRD) patterns were collected (New D8-Advance/Bruker-AXS) at a scan rate of 1° s−1 with 2θ range 5°–70° and CuKα1 radiation (0.154056 nm). Field emission scanning electron microscopy (FE-SEM, SIGMA, Carl Zeiss) was used to examine the morphology of C-CGP and C-CGP–MnO2. X-ray photoelectron spectroscopy (XPS) analysis was carried out using an ESCA2000 system (VGMicrotech) with a spectrometer, an Mg Kα X-ray source (1253.6 eV), and a hemispherical analyzer. For curve fitting, the Gaussian peak widths were constant in each spectrum. Thermogravimetric analyses (TGA) were conducted under a N2 and air atmosphere using a CI Electronics microbalance (MK2-MC5). The sheet resistance was measured using a four-point probe method (ChangMin, LTD, CMT-SR2000N, Korea). The inner resistance was measured by AC impedance spectroscopy (IM-6ex, Zahner) between 0.1 kHz and 1 MHz. The samples were sandwiched rapidly between two Pt electrodes.2.6 Preparation and characterization of the supercapacitor
A three-electrode cell was employed for electrochemical tests, where an Ag/AgCl (KCl-saturated) electrode and platinum foil were used as the reference and counter electrodes, respectively. The papers were pressed (4000 psi) onto nickel foam current collectors as the working electrode. The loading mass of each electrode was about 4.6–4.9 mg. The measurements were carried out in a 1 M Na2SO4 aqueous electrolyte at room temperature. Cyclic voltammetry (CV), galvanostatic charge–discharge characterization, and electrochemical impedance spectroscopy (EIS) were carried out using a CHI 660C electrochemical workstation. EIS was performed under the following conditions: an AC voltage amplitude of 5 mV, a frequency range of 105 to 0.1 Hz, and open circuit potential.3. Results and discussion
Fig. 1 illustrates the entire procedure for preparing chemically bonded GO–CNT hybrid paper (C-GCP), which consists of the following three steps: (i) surface modification of GO and CNT, (ii) chemical bonding between functionalized GO and CNT, and (iii) hot-pressing of C-GCP. This kind of paper-like carbon material is free-standing and flexible, in which amide bonding exists between chlorinated GO and amine-treated CNT.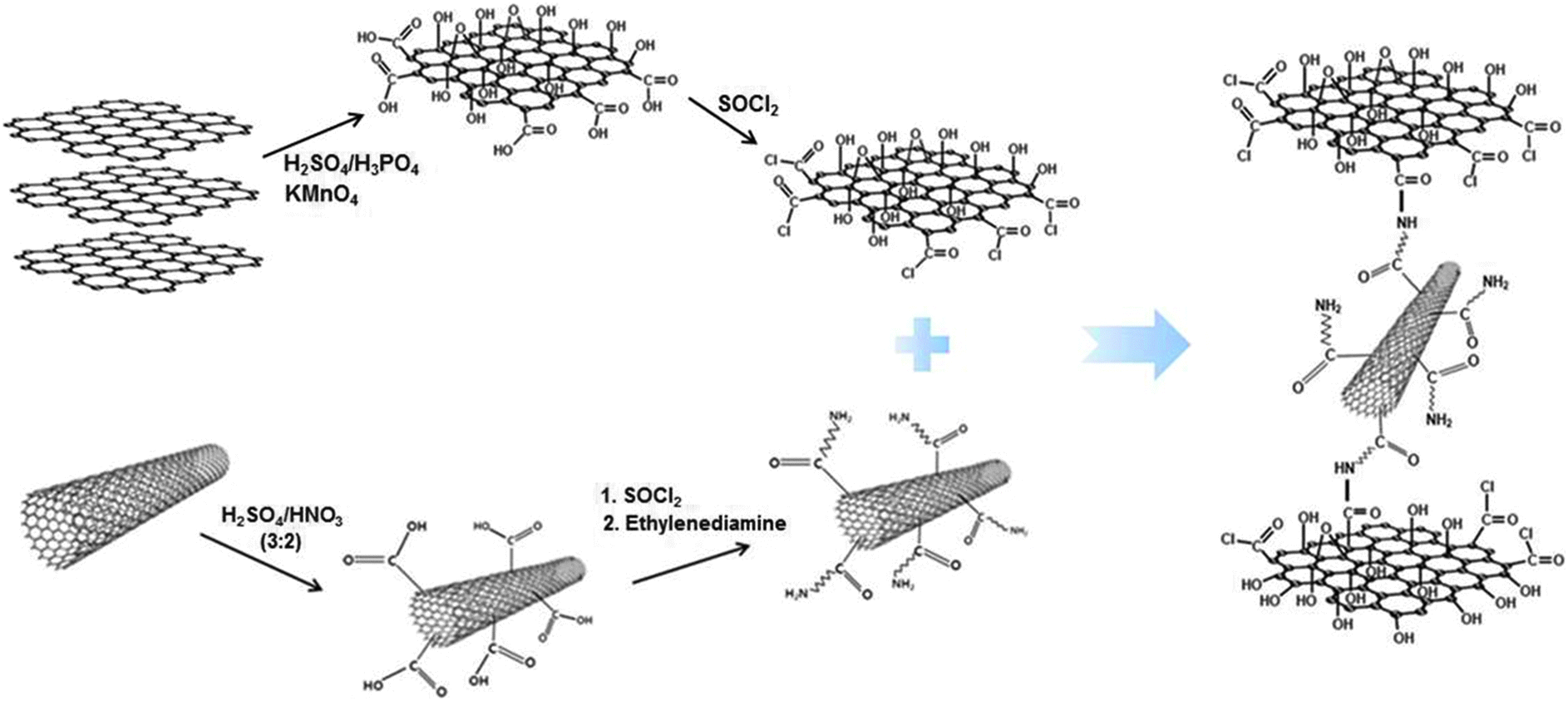 | ||
| Fig. 1 Chemical route for the fabrication of C-GCP. | ||
Fig. 2(a) and (b) shows the characterization of surface-treated GO and CNT by TGA and FT-IR. In the case of GO, a sharper mass loss near 150 °C was observed in the TGA curve (Fig. 2(a)) because of the pyrolysis of oxygen-containing functional groups from the GO surface.27 Although the TGA curve for chlorinated GO featured behavior similar to that of GO, the mass loss of chlorinated GO near 150 °C decreased by about 11 wt% because the –OH atoms in the carboxyl groups on the GO edge were converted to chlorine atoms. The raw CNTs were stable under a flowing N2 atmosphere over the entire temperature range investigated, and lost only a small percentage of mass, which was associated with absorbed humidity.28 Mass loss occurred with acid-treated CNTs up to 800 °C due to the thermal decomposition of COOH and the release of CO2. The thermal loss of ethylenediamine covalently bound to the CNTs was observed to occur from 250 °C, with a mass loss of about 10 wt% compared with raw CNTs, which was in agreement with the results of other studies.29 The FT-IR results provided clear evidence of surface-treated GO and CNTs (Fig. 2(b)). The spectrum of carboxylated CNT –COOH and GO is presented for comparison with the aminated CNT–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)NH(CH2)2NH2 and chlorinated GO. The GO surface was grafted with hydroxyl and carbonyl groups, which were formed during the oxidation process in the chemical exfoliation. In addition, carboxyl groups were attached to the CNT in the carboxylated CNT. Thus, typical O–H stretching vibrations (3420 cm−1) and CO stretching vibrations (1720 cm−1) were detected in the spectrum of GO and carboxylated CNT.30 After surface modification of GO and the carboxylated CNTs, other peaks were observed in the spectrum. Chlorinated GO was observed in the peak at 1230 cm−1, which was related to the formation of COCl groups.31 For the aminated CNTs, the band at 2911 cm−1 represents stretching of the methylene group –CH2– from the ethylenediamine molecules.32 In addition, the peaks at 1258 cm−1, corresponding to C–N stretching, suggest the presence of amide functional groups –(CO)NH– on the CNTs, thereby demonstrating that NH2 functional groups were covalently attached to the carboxylated CNTs.33,34 The band at 1080 cm−1 represents the C–N stretching vibrations of the amine molecules –CH2NH2.35 XPS analyses were conducted to investigate C-GCP10. Fig. 2(c) shows de-convoluted C 1s peaks, at 284.4 (C–C/CC), 286.4 (C–O/C–O–C), 288.3 (CO), 289 (OC–OH) and 287.7 eV (N–CO), which were attributable to the formation of amide bonds between the chlorinated GO and the primary amine of aminated CNTs. To provide clearer evidence of amide bonding in the C-GCP10, N1s peaks were fitted to two component peaks, as shown in Fig. 2(d). The lower binding energy peak at 399.9 eV was assigned to the amine nitrogen (CH2–NH2) bond associated with un-reacted amine groups on the CNT surfaces. The higher binding energy peak at 401.5 eV was assigned to the amide nitrogen (N–CO), and represented the bond formation between chlorinated carbon in the GO and free amines, making the amide nitrogen electropositive, resulting in higher binding energy.36 Moreover, the FE-SEM images of C-GCP0, C-GCP5, C-GCP10, C-GCP15, C-GCP20 and C-GCP30 are represented in Fig. S1 (ESI†).
O)NH(CH2)2NH2 and chlorinated GO. The GO surface was grafted with hydroxyl and carbonyl groups, which were formed during the oxidation process in the chemical exfoliation. In addition, carboxyl groups were attached to the CNT in the carboxylated CNT. Thus, typical O–H stretching vibrations (3420 cm−1) and CO stretching vibrations (1720 cm−1) were detected in the spectrum of GO and carboxylated CNT.30 After surface modification of GO and the carboxylated CNTs, other peaks were observed in the spectrum. Chlorinated GO was observed in the peak at 1230 cm−1, which was related to the formation of COCl groups.31 For the aminated CNTs, the band at 2911 cm−1 represents stretching of the methylene group –CH2– from the ethylenediamine molecules.32 In addition, the peaks at 1258 cm−1, corresponding to C–N stretching, suggest the presence of amide functional groups –(CO)NH– on the CNTs, thereby demonstrating that NH2 functional groups were covalently attached to the carboxylated CNTs.33,34 The band at 1080 cm−1 represents the C–N stretching vibrations of the amine molecules –CH2NH2.35 XPS analyses were conducted to investigate C-GCP10. Fig. 2(c) shows de-convoluted C 1s peaks, at 284.4 (C–C/CC), 286.4 (C–O/C–O–C), 288.3 (CO), 289 (OC–OH) and 287.7 eV (N–CO), which were attributable to the formation of amide bonds between the chlorinated GO and the primary amine of aminated CNTs. To provide clearer evidence of amide bonding in the C-GCP10, N1s peaks were fitted to two component peaks, as shown in Fig. 2(d). The lower binding energy peak at 399.9 eV was assigned to the amine nitrogen (CH2–NH2) bond associated with un-reacted amine groups on the CNT surfaces. The higher binding energy peak at 401.5 eV was assigned to the amide nitrogen (N–CO), and represented the bond formation between chlorinated carbon in the GO and free amines, making the amide nitrogen electropositive, resulting in higher binding energy.36 Moreover, the FE-SEM images of C-GCP0, C-GCP5, C-GCP10, C-GCP15, C-GCP20 and C-GCP30 are represented in Fig. S1 (ESI†).
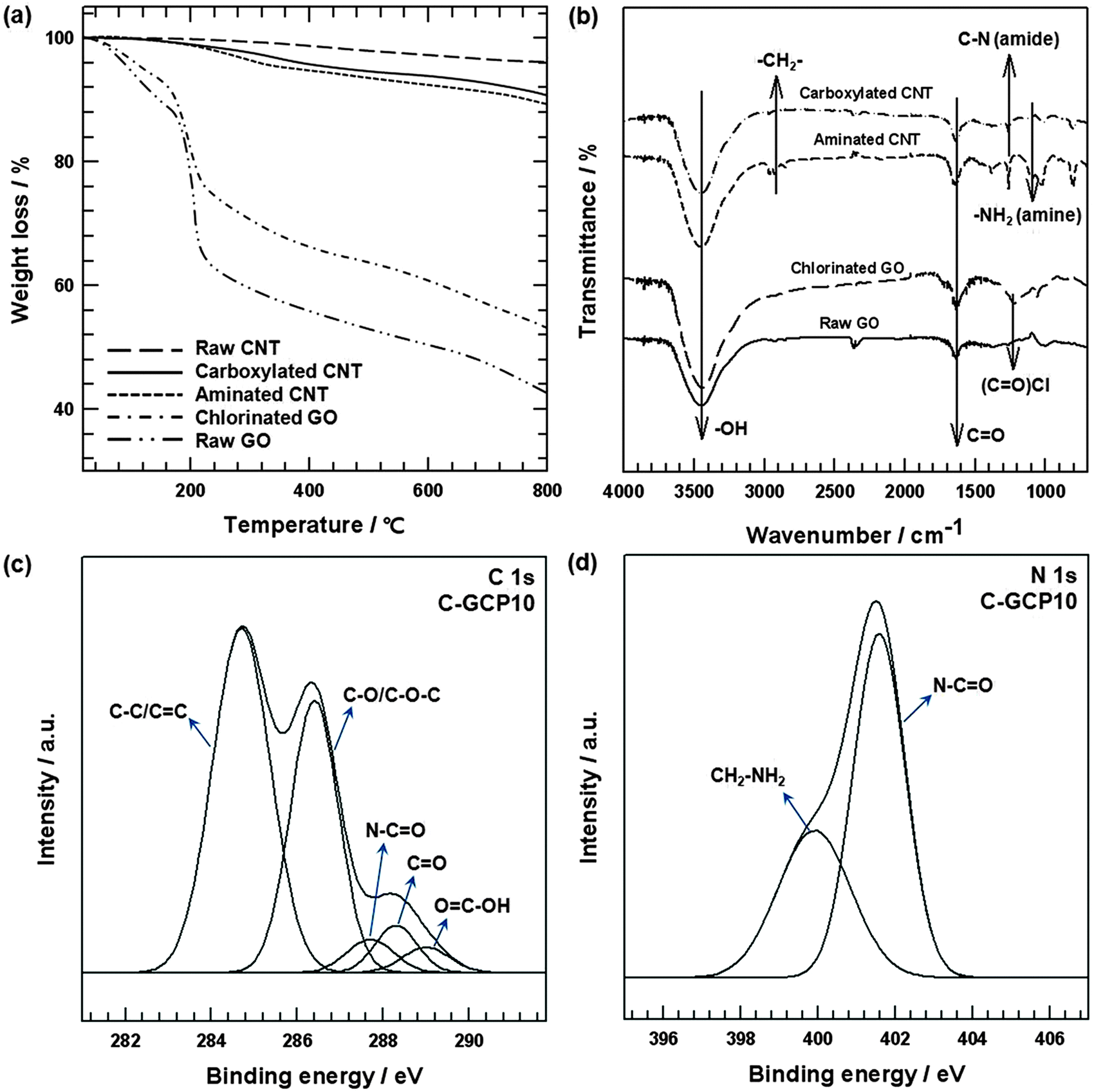 | ||
| Fig. 2 (a) TGA of raw GO, raw CNTs, chlorinated GO, carboxylated CNTs and aminated CNTs. (b) FT-IR spectra of raw GO, chlorinated GO, carboxylated CNTs and aminated CNTs. (c) XPS C 1s spectrum of C-GCP10. (d) XPS N 1s spectrum of C-GCP10. | ||
In order to obtain the optimum capacitive characteristics of C-rGCP, the relationship between the specific capacitance (Cs) from the CV curve and the weight percent of CNT can be studied in Fig. 3(a). The specific capacitances (Cs) were calculated from the CV curves according to the following equation:
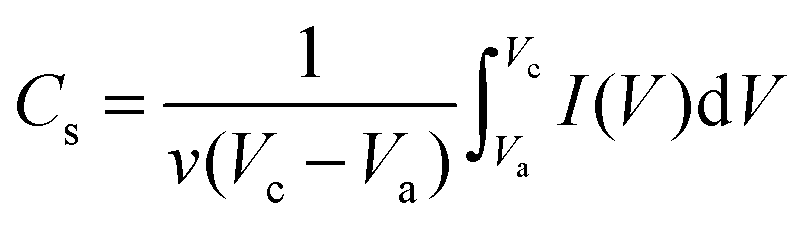
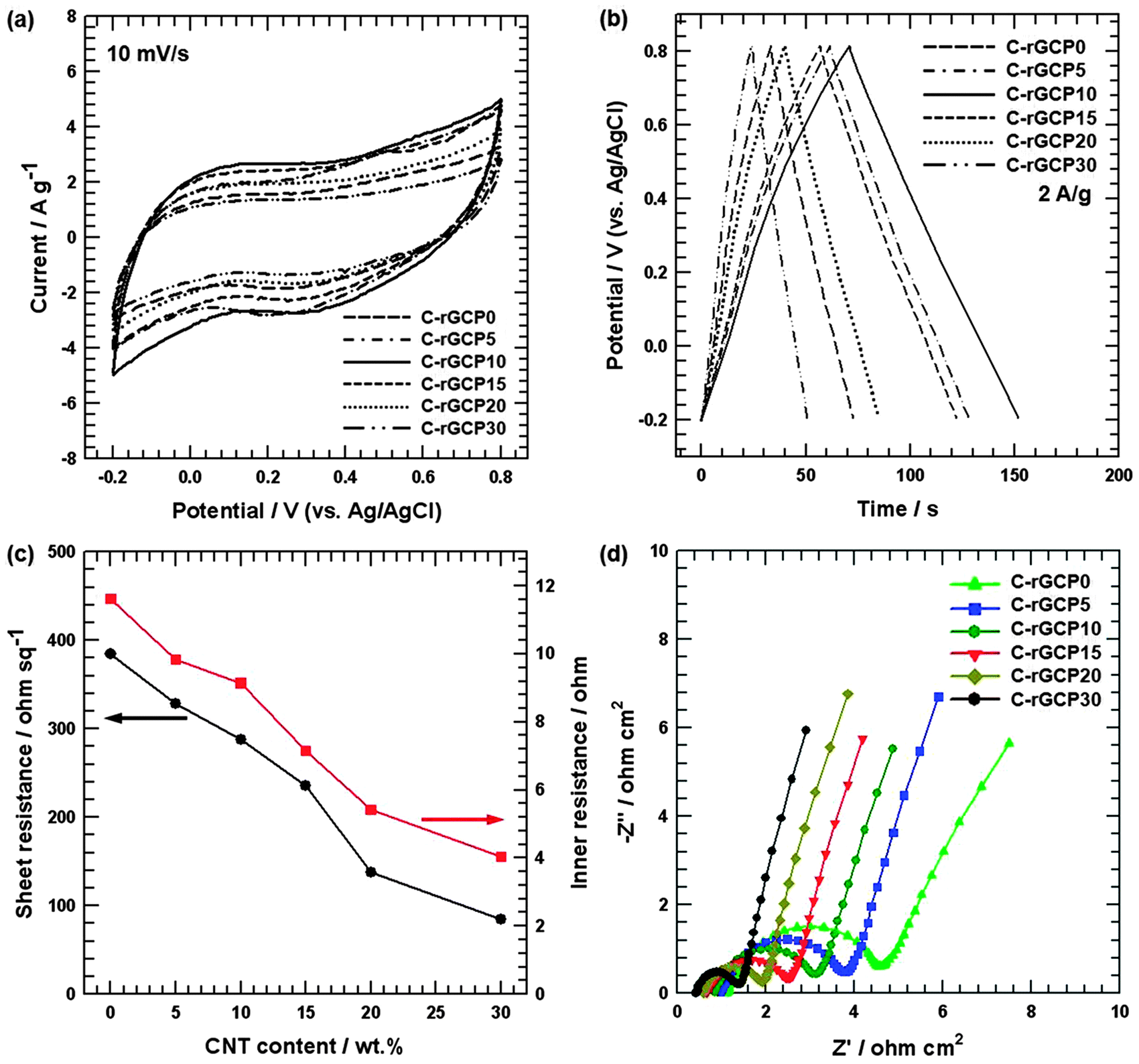 | ||
| Fig. 3 (a) CV curves of C-rGCP electrodes with different CNT contents at a scan rate of 10 mV s−1. (b) Galvanostatic charge–discharge curves of C-rGCP electrodes with different CNT contents at a current density of 2 A g−1. (c) Sheet resistance and inner resistance of C-rGCP as a function of CNT content. (d) Nyquist plots of C-rGCP electrodes with different CNT contents. | ||
As shown in Fig. 4, the formation procedure of chemically bonded graphene–CNT hybrid paper decorated with MnO2 nanoneedles (C-rGCP–α-MnO2) consisted of the following three steps: (i) synthesis of C-GCP, (ii) formation of homogeneously dispersed MnO2 nanoneedle structures using oxygen-containing functional groups present on the outermost GO layer surface of C-GCP using the direct paper dipping method in an isopropyl alcohol–water solution system, and (iii) thermal reduction of C-GCP–MnO2 to obtain C-rGCP–MnO2.
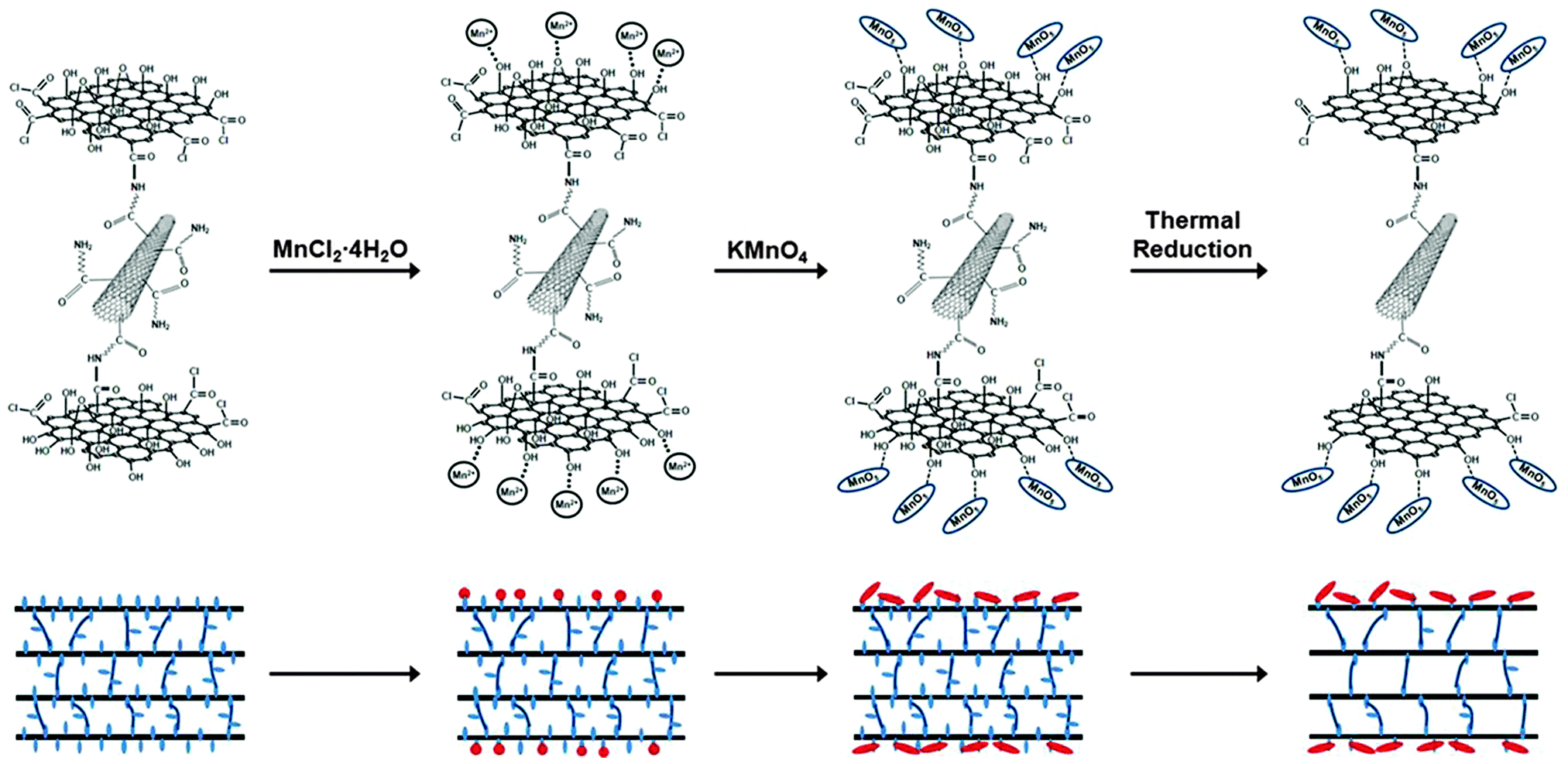 | ||
| Fig. 4 Chemical route for the fabrication of C-rGCP–MnO2. | ||
The formation of nanoneedle structure of C-rGCP10–MnO2 was confirmed by XRD analysis (Fig. 5(a)). The XRD patterns of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) are shown in Fig. 5(a). The diffraction peaks of as-synthesized C-rGCP10–MnO2 were similar to those of nanotetragonal-phase α-MnO2 (JCPDS 44-0141, a = 9.7845 Å, c = 2.8630 Å), where the (0 0 2) and (1 0 0) reflection peaks of C-rGCP10 have almost disappeared.21,26,41 Furthermore, the diffraction peaks show a gradual increase in intensity with an increase in MnO2 content in the composites. In addition, detailed surface information on C-rGCP10–MnO2 was collected by XPS in order to confirm the formation of C-rGCP10–MnO2, and the corresponding wide scan and de-convoluted Mn 2p peaks of C-rGCP10–MnO2 are presented in Fig. 5(b) and (c). Fig. 5(b) shows the XPS survey spectrum of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6). The survey spectrum of C-rGCP10 consisted of only three elements, namely, C, N, and O. However, an Mn signal (2p3/2, 2p1/2) emerged in the spectrum of C-rGCP10–MnO2, implying that MnO2 nanoneedle structures were successfully formed on the outermost graphene layer surface in C-rGCP10. Moreover, the Mn 2p3/2 and Mn 2p1/2 peaks show a gradual increase in intensity with the increase in MnO2 content in the composites, which is in accordance with XRD analysis. As shown in Fig. 5(c), the Mn 2p3/2 peak was centered at 642.6 eV and the Mn 2p1/2 peak was centered at 654.2 eV. Although the observed peak positions were slightly different, a consistent spin-energy separation of 11.6 eV was observed between the Mn 2p3/2 and Mn 2p1/2 peaks. These results were also in accordance with previously reported data for Mn 2p3/2 and Mn 2p1/2 in MnO2 nanoneedles.41 Moreover, to investigate the degree of reduction, the composition of heterocarbon was analyzed based on the deconvoluted C 1s core-level spectra of C-GCP10–MnO2(6) and C-rGCP10–MnO2(6). In comparison with the heterocarbon component, C-rGCP10–MnO2(6) (24.2%) was markedly decreased compared with that of C-GCP10–MnO2(6) (49.9%), indicating that thermal reduction effectively removed oxygen functional groups (Fig. S2, ESI†). The compositions of as-prepared C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) composites were further investigated by TGA (shown in Fig. 5(d)). Experiments were performed up to a temperature of 800 °C under air flow at a heating rate of 10 °C min−1. Under these conditions, C-rGCP10 burned up while MnO2 was transformed into Mn2O3.42Fig. 5(d) shows representative TGA curves of MnO2 nanoneedle structures of (Mn), C-rGCP10–MnO2(2), C-rGCP10–MnO2(6), and C-rGCP10. The weight losses of Mn nanoneedles, C-rGCP10–MnO2(2), C-rGCP10–MnO2(6), and C-rGCP10, were found to be 12.5%, 30.3%, 21.8%, and 93.2%, respectively. Accordingly, the mass ratios of MnO2/(C-rGCP10) for C-rGCP10/MnO2(2) and C-rGCP10/MnO2(6) were 3.53/1 and 7.67/1, respectively.
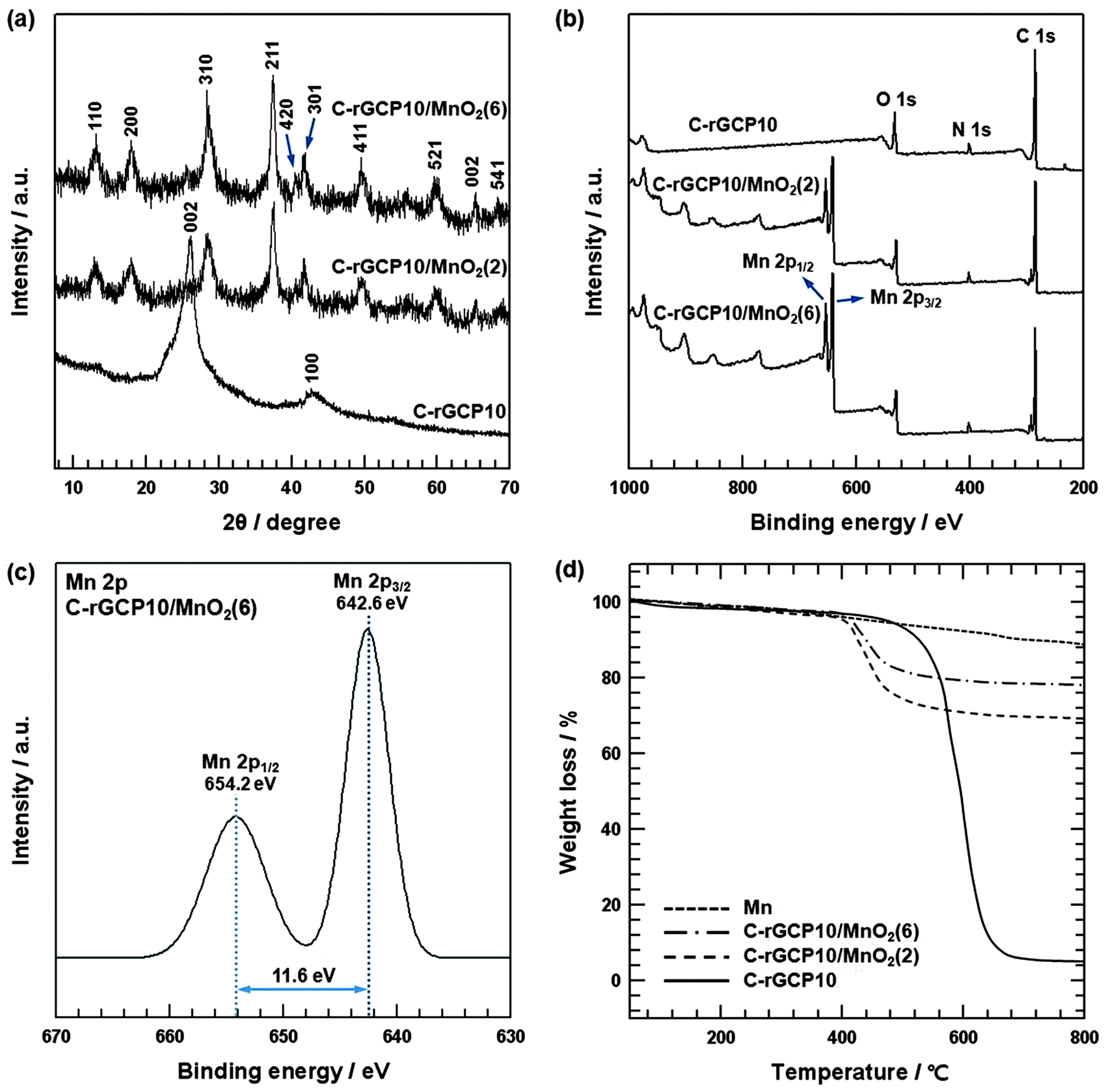 | ||
| Fig. 5 (a) XRD patterns of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6). (b) XPS wide scan survey spectra of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6). (c) XPS Mn 2p spectrum of C-rGCP10–MnO2(6). (d) TGA curves of Mn, C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6). | ||
The morphology and structure of as-prepared C-rGCP10–MnO2(6) were further studied by SEM (Fig. 6). As shown in Fig. 6(a) and (b), MnO2 nanoneedles were uniformly decorated on the outermost layers (top and bottom) of C-rGCP10. The oxygen-containing functional groups (epoxy and hydroxyl groups on the basal planes, and carbonyl and carboxyl groups at the edges), acting as anchor sites, allowed the homogeneous formation of nanostructures attached to the outermost graphene layer surface of C-rGCP10. Furthermore, the morphology of these nanostructures was needle-like, with approximately 50 nm and lengths of 300 nm, which corresponded with other studies of needle-type MnO2.26 Recently, it has been suggested that metal oxide particles are fully distributed on the outermost graphene layers of graphene paper with surfaces on the order of hundreds of micrometers and millimeters, which is attributable to the larger specific surface area accessible to the electrolyte ions and the shorter transport–diffusion path lengths for both ions and electrons. Moreover, cross-sectional SEM images under different magnifications (Fig. 6(c) and (d)) demonstrated that the outermost graphene layer surfaces of C-rGCP10 were successfully decorated with MnO2 nanoneedles. The CNTs were randomly embedded between the graphene layers, which produced superior ion transport paths into the inner region of the electrode. The MnO2 nanoneedle crystals on the outermost graphene layer are beneficial to increase the diffusion rate of electrolyte ions and electrons, as well as pore reservoirs. Therefore, ternary hybrid electrodes consisting of graphene, CNT, and MnO2 were fabricated by direct dipping of C-GCP in isopropyl alcohol with added KMnO4. The reason for the as-prepared papers without exfoliation in the solvents can be explained by the small electrical dipole moment of isopropyl alcohol (1.58 D) compared with other solvents exhibiting large electrical dipole moments (e.g., N,N-dimethlyformamide (3.24 D)), which afford more stable conditions during the formation of MnO2 nanoneedles on the outermost graphene surfaces of C-GCP43–45 (Fig. S3, ESI†).
 | ||
| Fig. 6 FE-SEM images of C-rGCP10–MnO2(6). (a) Surface image of C-rGCP10–MnO2(6). (b) Highly magnified surface image of C-rGCP10–MnO2(6). (c) Cross-sectional image of C-rGCP10–MnO2(6). (d) Highly magnified cross-sectional image of C-rGCP10–MnO2(6). | ||
To explore potential applications of supercapacitors, samples were fabricated into supercapacitor electrodes and characterized using cyclic voltammetry and galvanostatic charge–discharge measurements. Fig. 7(a) shows the CV curves of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes at a scan rate of 10 mV s−1 in 1M Na2SO4 electrolyte. All of the C-rGCP10–MnO2 electrodes exhibited a much larger integrated area than that of the C-rGCP10 electrode, which indicated excellent electrochemical performance. This observation is attributable to the nanoneedle structure of MnO2. As illustrated in Fig. 6, the formation of MnO2 nanoneedles on the outermost graphene layers can help to increase the specific area, which can form pores for ion-buffering reservoirs to improve the diffusion rate of Na+ ions and generate well-aligned nanoneedles that, in turn, act as well-ordered tunnels for the insertion/extraction of Na+ cations both into and out of MnO2. Moreover, nanoscale MnO2 particles can greatly reduce the diffusion length over which Na+ must transfer during the charge–discharge process, thereby improving the electrochemical utilization of MnO2.26 Furthermore, the C-rGCP10–MnO2(6) electrode showed a larger integrated area than that of the C-rGCP10–MnO2(2) electrode. Therefore, it is reasonable to conclude that the capacitance behavior of the C-rGCP10–MnO2(6) electrode is much better than that of the C-rGCP10–MnO2(2) electrode due to the fact that higher MnO2 loading results in a larger pseudo-capacitive contribution. Fig. 7(b) shows the relationship between calculated specific capacitance (Cs) and the scan rate of the C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes. The calculated Cs values of the C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes at 10 mV s−1 are 228.92, 302.04 and 367.23 F g−1, respectively. The C-rGCP10–MnO2(6) electrode showed the highest specific capacitance value over the entire range of scan rates. Moreover, the Cs values of all the electrodes decreased steadily with an increase in scan rate due to the reduced access of ions to the active surface, especially with relatively slow faradic reactions.46 Galvanostatic charge–discharge measurements of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes were carried out in 1 M Na2SO4 between −0.2 and 0.8 V at a current density of 2 A g−1. As illustrated in Fig. 7(c), during the charging and discharging steps, the charge curves of these electrodes were nearly symmetric with their corresponding discharge counterparts, with slight curvature, indicating pseudocapacitive properties and double layer contributions.26 However, in comparison with the discharge curves of C-rGCP10–MnO2 composite electrodes, the C-rGCP10–MnO2(6) electrode exhibited a much longer discharge time, which was consistent with specific capacitance behavior, since discharging time is directly proportional to the specific capacitance of electrodes. Impedance measurements of the C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes were performed using EIS, as shown in Fig. 7(d). The EIS data were analyzed using Nyquist plots. The electrodes containing the MnO2 component presented a slight inclination at the initial stage of the linear part, which was attributed to the presence of pseudo-capacitance. Comparing the Nyquist plots of these electrodes, it is apparent that the values of Rct gradually increase with increasing MnO2 loading, which was confirmed by TGA analysis, as shown in Fig. 5(d). This result was partially caused by the low electrical conductivity of C-rGCP10–MnO2 materials after the formation of MnO2.47
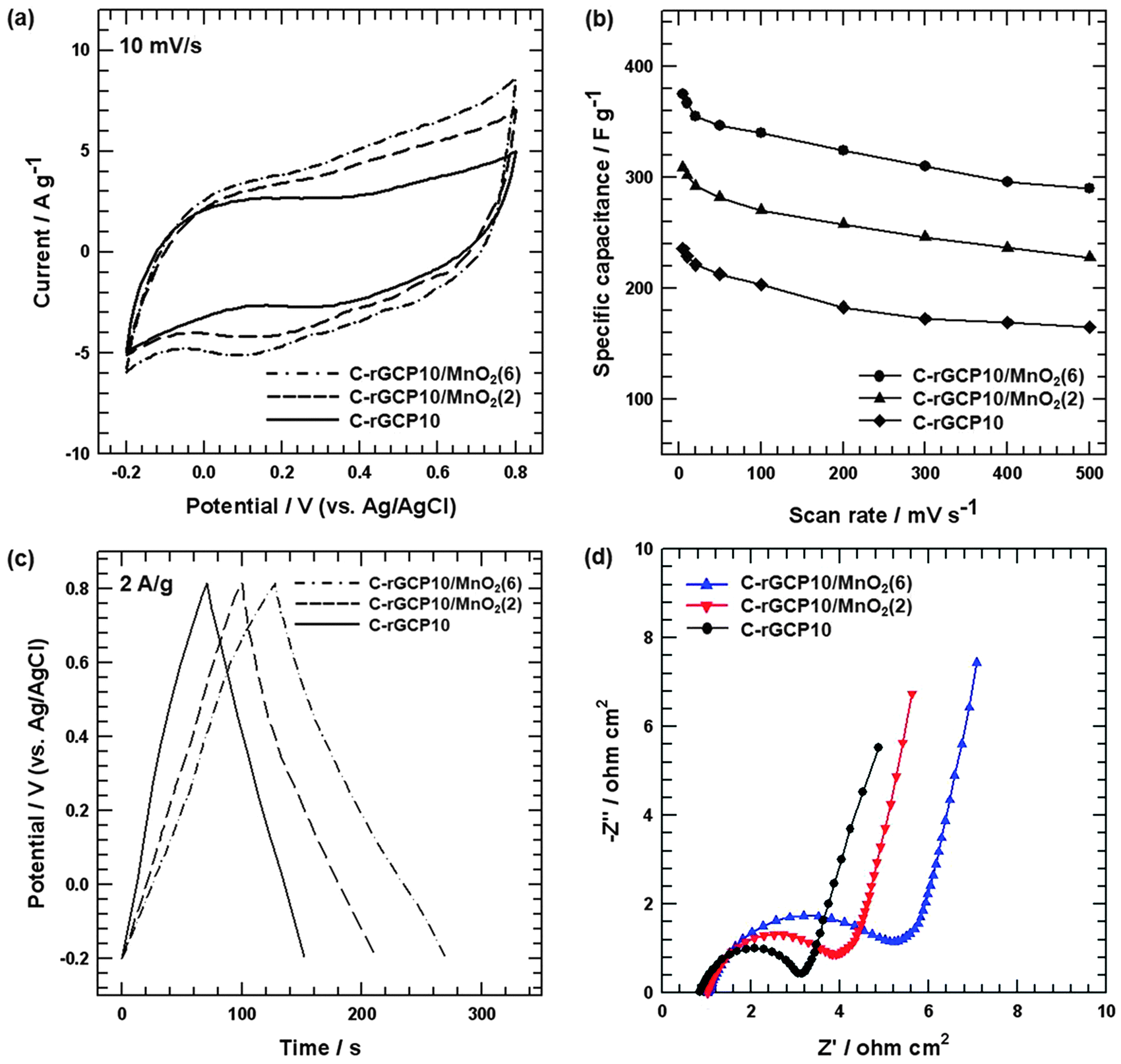 | ||
| Fig. 7 (a) CV curves of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes at a scan rate of 10 mV s−1. (b) Specific capacitance of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes at different scan rates. (c) Galvanostatic charge–discharge curves of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes at a current density of 2 A g−1. (d) Nyquist plots of C-rGCP10, C-rGCP10–MnO2(2) and C-rGCP10–MnO2(6) electrodes. | ||
Cycle lifetime is one of the most critical factors in supercapacitor applications. Typical issues associated with MnO2-based electrodes in aqueous electrolytes include mechanical expansion of MnO2 during the ion insertion–desertion process, MnO2 detachment from the electrode surfaces, and dissolution of Mn into the electrolyte.48 A cyclic stability test of over 1000 cycles of the C-rGCP10–MnO2(6) electrode at a scan rate of 10 mV s−1 was performed over a potential window ranging from −0.2 to 0.8 V. Fig. 8 shows the specific capacitance retention as a function of cycle number. The C-rGCP10–MnO2(6) electrode exhibited a loss in specific capacitance of less than 15.4% after 1000 charge–discharge cycles, indicating excellent capacity retention.
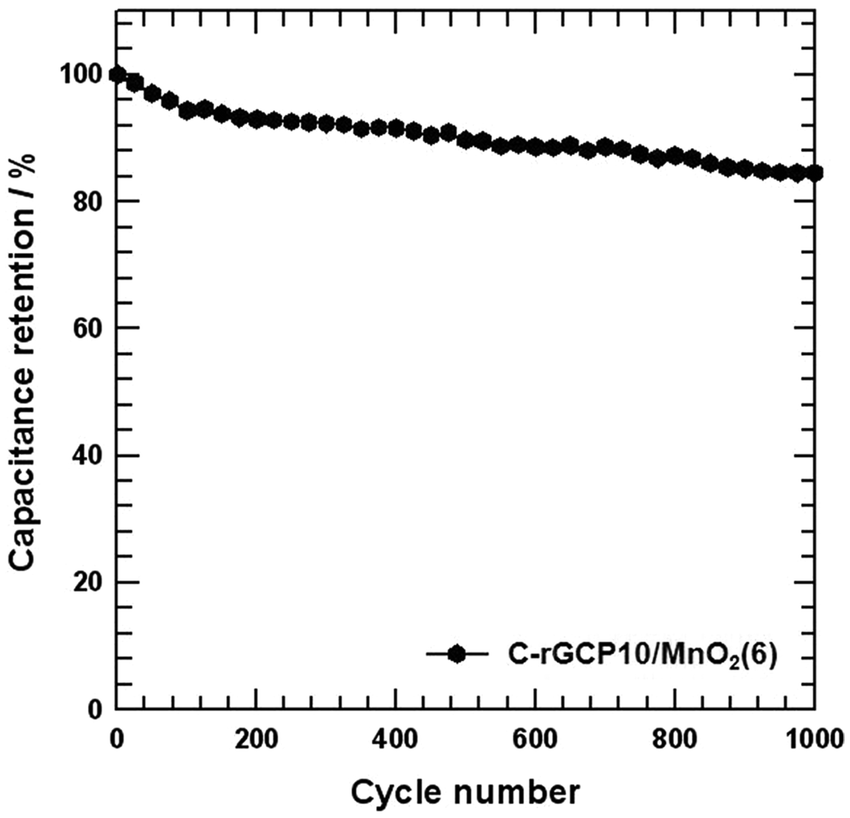 | ||
| Fig. 8 Cycling stability of the C-rGCP10–MnO2(6) electrode with different MnO2 contents measured at a scan rate of 10 mV s−1. | ||
4. Conclusions
In summary, we developed 3D nanostructured ternary hybrid papers using a direct paper dipping method, in which MnO2 nanoneedles formed on the outermost graphene layer and CNTs were intercalated by using the amide bonds between graphene layers. This graphene paper exhibited a unique structure, which was confirmed by FT-IR, XRD, XPS, TGA, and FE-SEM characterization. When used as electrodes for C-rGCP10–MnO2 and supercapacitors, the C-rGCP10–MnO2(6) fabricated in this work showed a much higher performance compared with graphene paper because the MnO2 nanoneedles formed on the outermost graphene layers increase the specific area, which can lead to the formation of pores for ion-buffering reservoirs to improve the diffusion rate of Na+ ions and generate well-aligned nanoneedles that act as well-ordered tunnels for the insertion/extraction of Na+ cations both into and out of MnO2. Moreover, chemically bonded CNTs produce the synergistic effect of increased electronic conductivity and effective surface area, which are associated with large electrochemically active sites. This unique graphene paper is promising for applications in new kinds of flexible electrodes for wearable or rolling-up devices.Acknowledgements
This work was supported by the Technological Innovation R&D Program (S2085171) funded by the Small and Medium Business Administration (SMBA, Korea).References
- V. Subramanian, H. Zhu, R. Vajtai, P. M. Ajayan and B. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef CAS PubMed.
- A. L. Mohana, R. F. Estaline, A. Imran and J. S. Ramaprabhu, Nanoscale Res. Lett., 2008, 3, 145–151 CrossRef.
- A. C.-G. Karina, L.-C. Monica, C.-P. Nieves and G.-R. Pedro, Adv. Funct. Mater., 2005, 15, 1125–1133 CrossRef.
- J. Chow, R. J. Kopp and P. R. Portney, Science, 2003, 302, 1528–1531 CrossRef PubMed.
- Z. Wen, X. Wang, S. Mao, Z. Bo, H. Kim, S. Cui, G. Lu, X. Feng and J. Chen, Adv. Mater., 2012, 24, 5610–5616 CrossRef CAS PubMed.
- R. Kotz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2496 CrossRef CAS.
- Z. Chen, Y. Qin, D. Weng, Q. Xiao, Y. Peng, X. Wang, H. Li, F. Wei and Y. Lu, Adv. Funct. Mater., 2009, 19, 3420–3426 CrossRef CAS.
- S. Y. Yang, K. H. Chang, H. W. Tien, Y. F. Lee, S. M. Li, Y. S. Wang, J. Y. Wang, C. C. M. Ma and C. C. Hu, J. Mater. Chem., 2011, 21, 2374–2380 RSC.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS PubMed.
- Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- Z. Li, Y. Mi, X. Liu, S. Liu, S. Yang and J. Wang, J. Mater. Chem., 2011, 21, 14706–14711 RSC.
- Z. S. Wu, W. Ren, D. W. Wang, F. Li, B. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS PubMed.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- Z. S. Wu, G. Zhou, L. C. Yin, W. Ren, F. Li and H. M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS PubMed.
- E. Raymundo-Pinero, V. Khomenko, E. Frackowiak and F. Beguin, J. Electrochem. Soc., 2005, 152, A229–A235 CrossRef CAS PubMed.
- K. R. Prasad and N. Miura, J. Power Sources, 2004, 135, 354–360 CrossRef CAS PubMed.
- X. Jin, W. Zhou, S. Zhang and G. Z. Chen, Small, 2007, 3, 1513–1517 CrossRef CAS PubMed.
- Z. D. Huang, B. Zhang, R. Liang, Q. B. Zheng, S. W. Oh, X. Y. Lin, N. Yousefi and J. K. Kim, Carbon, 2012, 50, 4239–4251 CrossRef CAS PubMed.
- S. B. Ma, K. W. Nam, W. S. Yoon, X. Q. Yang, K. Y. Ahn, K. H. Oh and K. B. Kim, J. Power Sources, 2008, 178, 483–489 CrossRef CAS PubMed.
- S. Komaba, A. Ogata and T. Tsuchikawa, Electrochem. Commun., 2008, 10, 1435–1437 CrossRef CAS PubMed.
- S. Chen, J. Zhu, Q. Han, Z. Zheng, Y. Yang and X. Wang, Cryst. Growth Des., 2009, 9, 4356–4361 CAS.
- S. B. Ma, K. Y. Ahn, E. S. Lee, K. H. Oh and K. B. Kim, Carbon, 2007, 45, 375–382 CrossRef CAS PubMed.
- N. Nagarajan, M. Cheong and I. Zhitomirsky, Mater. Chem. Phys., 2007, 103, 47–53 CrossRef CAS PubMed.
- X. Lu, H. Dou, B. Gao, C. Yuan, S. Yang, L. Hao, L. Shen and X. Zhang, Electrochim. Acta, 2011, 56, 5115–5121 CrossRef CAS PubMed.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1985, 80, 1339 CrossRef.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS PubMed.
- K. Kinoshita, Carbon Materials. Carbon–Electrochemical and Physicochemical Properties, Wiley, New York, 1988 Search PubMed.
- P. A. Jelliss, S. S. Graham, A. Josipovic, S. Boyko, S. D. Minteer and V. Svoboda, Polyhedron, 2013, 50, 36–44 CrossRef CAS PubMed.
- E. Castillejos, B. Bachiller-Baeza, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Carbon, 2012, 50, 2731–2740 CrossRef CAS PubMed.
- J. Wu, S. Bai, X. Shen and L. Jiang, Appl. Surf. Sci., 2010, 257, 747–751 CrossRef CAS PubMed.
- R. R. V. A. Rios, D. E. Alves, I. Dalmazio, S. Fernando, V. Bento, C. L. Donnici and R. M. Lago, Mater. Res., 2003, 6, 129–135 CrossRef CAS PubMed.
- H. Hui, B. Zhao, M. A. Hamon, K. Kamaras, M. E. Itkis and R. C. Haddeon, J. Am. Chem. Soc., 2003, 125, 14893–14900 CrossRef PubMed.
- T. Ramanathan, F. T. Fisher, R. S. Ruoff and L. C. Brinson, Chem. Mater., 2005, 17, 1290–1295 CrossRef CAS.
- J. Zhu, H. Peng, F. Rodriguez-Macias, J. L. Margrave, V. N. Khabashesku and A. M. Imam, Adv. Funct. Mater., 2004, 14, 643–648 CrossRef CAS.
- S. E. Baker, W. Cai, T. L. Lasseter, K. P. Weidkamp and R. J. Hamers, Nano Lett., 2002, 2, 1413–1417 CrossRef CAS.
- B. X. Yang, J. H. Shi, K. P. Pramoda and S. H. Goh, Compos. Sci. Technol., 2008, 68, 2490–2497 CrossRef CAS PubMed.
- M. J. Kim, Y. S. Hwang and J. H. Kim, J. Power Sources, 2013, 239, 225–233 CrossRef CAS PubMed.
- H. R. Byon, S. W. Lee, S. Chen, P. T. Hammond and Y. S. Horn, Carbon, 2011, 49, 457–467 CrossRef CAS PubMed.
- O. S. Kwon, T. J. Kim, J. S. Lee, S. J. Park, H. W. Park, M. J. Kang, J. E. Lee, J. S. Jang and H. S. Yoon, Small, 2013, 9, 248–254 CrossRef CAS PubMed.
- T. Bordjiba and D. Belanger, J. Electrochem. Soc., 2009, 156, A378–A384 CrossRef CAS PubMed.
- L. Mao, K. Zhang, H. Chan and J. Wu, J. Mater. Chem., 2012, 22, 1845–1851 RSC.
- M. N. Patel, X. Wang, B. Wilson, D. A. Ferrer, S. Dai, K. J. Stevenson and K. P. Johnston, J. Mater. Chem., 2010, 20, 390–398 RSC.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- C. A. Furtado, U. J. Kim, H. R. Gutierrez, L. Pan, E. C. Dickey and P. C. Eklund, J. Am. Chem. Soc., 2004, 126, 6095–6105 CrossRef CAS PubMed.
- J. A. Riddick, W. B. Bunger and T. K. Sakano, Organic solvents, John Wiley & Sons, New York, 4th edn, 1986 Search PubMed.
- X. Zhang, X. Sun, H. Zhang, D. Zhang and Y. Ma, Mater. Chem. Phys., 2012, 137, 290–296 CrossRef CAS PubMed.
- Z. Li, J. Wang, S. Liu, X. Liu and S. Yang, J. Power Sources, 2011, 196, 8160–8165 CrossRef CAS PubMed.
- G. Xiong, K. P. S. S. Hembram, R. G. Reifenberger and T. S. Fisher, J. Power Sources, 2013, 227, 254–259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53488j |
| This journal is © the Owner Societies 2014 |