Probing the stability of neutral and anionic transition-metal-doped golden cage nanoclusters: M@Au16 (M = Sc, Ti, V)
Hui-Fang
Li
and
Huai-Qian
Wang
*
College of Engineering, Huaqiao University, Quanzhou, 362021, China. E-mail: hqwang@hqu.edu.cn
First published on 17th October 2013
Abstract
The golden Au16q (q = 0, −1) cage is doped systematically with an external atom of different valence electrons: Sc, Ti, and V. The structural, electronic, and magnetic properties of the doped clusters, M@Au16q (M = Sc, Ti and V; q = 0, −1) are investigated using the Saunders “Kick” (SK) global search technique combined with density-functional theory (DFT) calculations (SK-DFT). It is found that the closeness of the calculated vertical/adiabatic detachment energy for Ti-doped and V-doped (3.09/3.16 eV for Ti-doped, and 3.31/3.38 eV for V-doped) is consistent with the negligible geometry change between the anionic and neutral ground state structures. The characteristics of the Sc@Au16− cluster includes its remarkably high average binding energy and doping energy, which reflects its high stability. The different spectral features between doped M@Au16− and pure Au16− clusters indicate endohedral structures with larger distortion from the parent Au16− cage for the doped clusters. The s electrons of the Au16 cage are observed to transfer to Sc, Ti and V atom for doped M@Au16q clusters by natural population analysis (NPA). The magnetic moment of the impurity Sc/Ti/V atom is somewhat quenched. Furthermore, the electron localization function analysis does not reveal strong interactions. The current work shows that the electronic properties of the golden cage can be systematically tuned through doping.
Introduction
Nanoclusters exhibit a rich array of new properties, which are unlike their bulk and can change suddenly even doped with an additional atom. This is especially in the case of gold clusters: while macroscopic bulk gold is known as the most chemically inert metal in the periodic table, nanometre-sized gold particles are exceptionally active as catalysts in a wide range of chemical transformations.1,2 The influence of transition metal impurities on the electric transport in gold nanocontacts is an example,3 among many others, of interesting items in the study of gold clusters doped with transition-metal atoms. Recent experimental and theoretical work has demonstrated that the introduction of a transition metal dopant atom in the gold cluster can change its structure and electromagnetic properties significantly.4–12 On the one hand, the stable endohedral structures of doped gold cage could be used as building blocks for assemblies forming new nanomaterials. On the other hand, the atomic clusters provide a unique medium for exploring local magnetism as the cluster size and the local structures can be easily controlled and changed after by transition metal doping.13 During a recent photoelectron spectroscopy (PES) and density functional theory (DFT) study of the structures of Aun− clusters,14 Au16− was found to be a highly stable hollow cage cluster with a large internal volume analogous to the fullerenes.15 The diameter of the Au16− cage (the golden buckyball) was shown to be ∼5.5 Å,14 suggesting possible endohedral doping to form a new class of golden cages with tailored properties similar to the endohedral fullerenes.16 This study spurred immediate theoretical interests, suggesting possible endohedral doping by a single atom M (M = Si, Al, Fe, Co, Ni, Ag, Zn, In and Mg).7,13,17,18 Since the structure of a cluster is intimately linked to its properties, an understanding of the cluster geometry is of fundamental interest. Unfortunately, there are no experimental techniques that can currently determine the structure of a cluster directly and unambiguously. While this can be achieved using theoretical methods, the limitations in current theoretical approach do not make it easy to predict the ground state geometry with absolute certainty not only because there is no exact theory that can treat large systems, but because clusters can have numerous low lying isomers often protected by energy barriers. As cluster size increases, the numerous structural isomers on the potential energy surface increases exponentially and search for the ground state geometry becomes a very difficult task.There have been many efforts to deal with problems of geometry optimization for cluster using automated procedures. For example, R. Car and M. Parrinello's well-known “dynamic simulated annealing” combines molecular dynamics (MD) and DFT.19 Searches employing “simulated annealing”, “Monte Carlo metropolis search”, and various “genetic algorithm techniques” are also well-documented.19–22 However, most of the structures obtained this way could be anticipated easily, since only the relative positions of the atoms change. Babadova-Parvanova et al. employed the density functional tight binding level to search for low-energy isomers using the “single parent genetic algorithm” (DFTB/SPGA) method.23 R. O. Jones has employed density functional methods for many years to study the structure and bonding of elemental as well as mixed element clusters.24–26 Starting geometries were generated by either MD simulated annealing or density functional tight binding or selected from DFT-based computations or experimental results. Choosing starting geometries becomes more and more difficult when the number of atoms increases. Even though their energies were lowest or competitive, hollow cage isomers of gold clusters were not located in a recent genetic algorithm search.27 Alexandrova et al. proposed genetic algorithm-based techniques on randomly generated structures to search for global minima.28 D. J. Wales and J. P. K. Doye in 1997 proposed a global optimization technique using “basin-hopping” in which the potential energy surface is transformed into a collection of interpenetrating staircases.29 A major problem which baffles any search method is that the potential energy surface at lower theoretical levels, employed initially for practical considerations, may differ from the potential energy surface at higher, more accurate levels. We have encountered cases where structures, important on higher level potential energy surfaces, do not exist as minima at lower levels! Fortunately, Saunders's fast, comprehensive, and automated “kick” method produces isomers and conformers simply and effectively with little thought or effort.30 This stochastic method generates structures randomly and facilitates the thorough exploration of unknown isomers much more easily than manual methods. In this work, we report a number of anionic and neutral doped golden cage isomers using Saunders “Kick” (SK) global search technique combined with DFT calculation (SK-DFT). Very recently, we have successfully employed the SK-DFT method for global minimum searches of relatively small clusters.31–35
Any isomer lying within a very small energy range of the lowest energy structure is considered a potential candidate for the ground state geometry. The correct ground state structure is then identified by finding out which of the isomers in this narrow energy range accurately predicts properties in agreement with experiments. Currently there are three experimental techniques, namely, ion mobility,36,37 PES,38–40 and trapped ion electron diffraction (TIED),41,42 which are used to shed light on the geometries of size-selected charged clusters. While ion mobility and TIED experiments can be carried out on both anionic and cationic clusters, PES experiments are confined only to anions due to low photon energies of the lasers used. In the PES experiment one measures the adiabatic detachment energy (ADE) (which provides the energy difference between the anion and neutral ground states), vertical detachment energy (VDE) (which provides the energy difference between the anion ground state and the neutral state at the anion geometry) as well as the photoelectron spectra. In the TIED approach, on the other hand, one compares the simulated diffraction pattern of theoretically generated candidate geometries with experiment. The advantage of the later approach is that one compares the entire simulated and experimental diffraction pattern. Note that in the PES experiment, it is difficult to calculate, within DFT, the entire photoelectron spectra and hence one is limited to comparing a few selected data such as ADE, VDE, and the first several peaks in the PES.43 In addition, experimental determination of the ADE becomes difficult if the geometries of the ground states of the anion and neutral clusters are very different and are protected by significant energy barrier. Nevertheless, the above experimental techniques complement each other and the theoretical structure that explains all available experiments is considered to be the preferred structure of the charged cluster. A fourth experimental technique that provides information on the structure of neutral clusters is the infrared absorption spectroscopy44 that measures the vibrational spectra of clusters and directly reflects the symmetry of the particle. However, all the above experiments rely on theoretical calculations of geometries of possible low lying isomers. Recently, we have used small vanadium monoxide/dioxide cluster as a test case to provide a comprehensive analysis of the ability of current methods to determine the geometry of the ground state of clusters.31,33
Despite the progress achieved, there are still many open questions for 3d transition-metal-doped gold cluster: M@Au16q (M = Sc, Ti and V; q = 0, −1). For example, no direct experimental information is available on the structures of these systems. The parent Au16q (q = 0, −1) clusters are empty cages, and many different types of atoms could be used as dopants to form new endohedral gold clusters. It would be particularly interesting to dope transition-metal atoms inside these gold cages to create magnetic gold clusters as these may exhibit new, physical, chemical, and catalytic properties that are distinct from the pure gold clusters. To the best of our knowledge, systematic investigation of the neutral and anionic golden cage Au16 clusters doped by 3d transition-metal (Sc, Ti and V) atoms, M@Au16q have not been reported. Thus, an accurate first principles calculation is important for understanding the structural and electronic properties of those clusters. Most previous ab initio calculations on doped gold clusters were limited by the presumed symmetric constraints. An unconstrained global search on the cluster potential energy surface is needed. In this work, we report a number of anionic and neutral doped golden cage isomers from SK global search technique.30 These metastable isomers are further evaluated at the PBEPBE/Au/SDD+2f/M/6-311+G*//PBEPBE/LANL2DZ level to determine the lowest-energy structures. Here, we study the evolution of the binding energy, doping energy, highest occupied and lowest unoccupied molecular orbit (HOMO–LUMO) gap as well as the first ADE/VDE were calculated and compared with experimental results. This work should be interesting for future theoretical and experimental chemists, especially those designing new materials. The rest of this article is organized as follows: section “Computational methods” briefs involved computational details and theoretical backgrounds, section “Results and discussions” introduces and compares various geometric structure, simulated PES, magnetism and electron localization function (ELF), the conclusions are given in the last section.
Computational methods
The calculations in this work began with unbiased searches for the low-lying structures of M@Au16q (M = Sc, Ti and V; q = 0, −1) using the Saunders “Kick” (SK) global search technique30 combined with DFT geometric optimization (SK-DFT) within the generalized gradient approximation in the Perdew–Burke–Ernzerhof (PBE)45 functional form using the GAUSSIAN09 program.46 This SK stochastic method generates structures randomly and facilitates the thorough exploration of unknown isomers much more easily than manual methods. Specifically, all the atoms are placed at the same point initially and then are “kicked” randomly within a sphere of radius R (R is the maximum kick distance), e.g., a hollow sphere of inside radius 10 Å. The method developed further in this work generates up to 500 unbiased starting geometries and then submits them automatically for optimization with GAUSSIAN09. We recently have successfully employed the SK-DFT method for global minimum searches on a series of bimetallic cluster NbnAl−,0, Cr/Mn@Au16−,0 and vanadium oxides.31–35 The scheme of this research consists of the following four steps. Firstly, the initial SK global minimum search was performed using the DFT method known in the literature as “PBEPBE” functional45 with a scalar relativistic effective core potential and LANL2DZ basis set implemented in the GAUSSIAN09 package. Many structural isomers were generated for each cluster. The kick method runs at the PBEPBE/LANL2DZ level some 500 times until no new minima appeared. If this procedure is repeated enough times, eventually all isomeric structures for the cluster will be found. Secondly, the distinct isomers were ranked according to their relative energies at the PBEPBE/LANL2DZ level. Meanwhile, the effect of spin multiplicity is also taken into account in the present study. Thirdly, among the top-several isomers, those with their energy value within 0.3 eV from the lowest-energy isomer were all regarded as potential candidate lowest-energy structures to be further evaluated at higher level. Relative energies of these candidate isomers with respect to the lowest-energy isomer were further evaluated by using the PBEPBE exchange–correlation functional with the large basis set Au/SDD+2f/M/6-311+G* at the geometries optimized at PBEPBE/LANL2DZ (PBEPBE/Au/SDD+2f/M/6-311+G*//PBEPBE/LANL2DZ). Here “SDD+2f” denotes the Stuttgart/Dresden ECP valence basis47,48 augmented by two sets of f polarization functions (exponents = 1.425, 0.468) for Au and 6-311+G*49 for transition-metal atom M (M = Sc, Ti and V). Finally, vibrational frequency calculations were performed at the same level of theory to verify the nature of the stationary points. Although other energetically more favorable structures could not be ruled out strictly, we believe that the lowest-energy structures of M@Au16q (Sc, Ti and V; q = 0, −1) found here are at least powerful candidates for their ground states, which are hoped to be verified in the future experiments and calculations at more accurate levels of theory.The accuracy of LANL2DZ basis set for transition-metal atoms doped inside gold cage clusters was validated by many recently published papers13,17,50–52 using the same exchange–correlation functional (PBEPBE). Compared the calculated vertical detachment energies (PBEPBE/LANL2DZ) with measured results by PES experiment for the M@Au16− (M = Fe, Co, Ni and Zn),13,17 PBEPBE/LANL2DZ level of theory gives very good agreement with the experimental observations, and therefore the same level has been selected as the method of choice for M@Au16q (Sc, Ti and V; q = 0, −1) species as well. Here, the geometries are regarded as optimized when the maximum force, the root-mean-square (RMS) force, the maximum displacement of atoms, and the RMS displacement of atoms have magnitudes less than 0.00045, 0.0003, 0.0018, and 0.0012 a.u., respectively. Furthermore, because the computed zero-point energy (ZPE) corrections of the isomers of a specific cluster size are small and almost the same, they are not expected to affect the relative energy ordering. Hence, the relative energies of isomers are obtained from the total electronic energies (excluding ZPE). Next, we calculated the first VDE (which is defined as the energy difference between the neutral clusters at optimized anion geometry clusters and optimized anion clusters) and ADE (which is the energy difference between the optimized anion geometry and the optimized neutral geometry). The binding energies of deeper orbitals were added to the first VDE to give the VDEs of the excited states. This procedure is along the lines of the so-called “generalized Koopman's theorem” (GKT).53 Each VDE was fitted with a full width at half-maximum (FWHM) of 0.09 eV to yield the simulated PES spectra.
Results and discussions
Three important types of structures of neutral and anionic Au16 clusters are shown in Fig. 1. The formation of lowest-energy structure of pure Au16 cluster is given in Fig. 2. The optimized ground-state geometries and selected low-lying isomers of M@Au16q (M = Sc, Ti and V; q = 0, −1) species at PBEPBE/LANL2DZ level are presented in Fig. 3. To determine the appropriate order of the isomers we performed single-point energy calculations at a higher level of theory (PBEPBE/Au/SDD+2f/M/6-311+G*//PBEPBE/LANL2DZ) for all isomers lying within 0.3 eV of the global minimum at the PBEPBE/LANL2DZ level of theory. Table 1 gives various structural and energetic characteristics of doped M@Au16q and parent gold clusters. The calculated first ADE and VDE of the top-3 lowest-energy isomers shown in Fig. 3 for the three M@Au16− clusters are compared with calculated values of the host cluster Au16− anion, which are given in Table 2. Magnetic moment (μB) of 3d, 4s, and 4p states for guest M atom, natural charge (e), total magnetic moment (μB) of the M atom, and total magnetic moment of the lowest-energy M@Au16q clusters are listed in Table 3. The simulated PES for the three low-lying isomers of M@Au16− as well as the experimental PES14 for host gold cluster Au16− are shown in Fig. 4–6. Fig. 7 depicts their electron structure characteristic by electron localization function (ELF) at PBEPBE/Au/SDD+2f/M/6-311+G*//PBEPBE/LANL2DZ level of theory.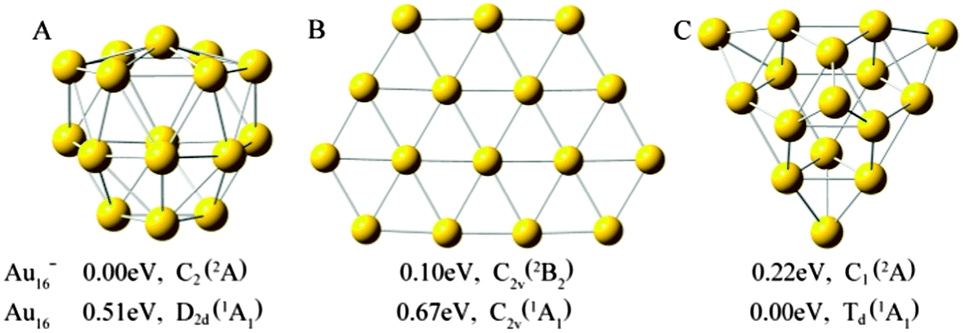 | ||
| Fig. 1 Optimized structures for Au16−,0 using density functional theory at the PBEPBE/LANL2DZ level. | ||
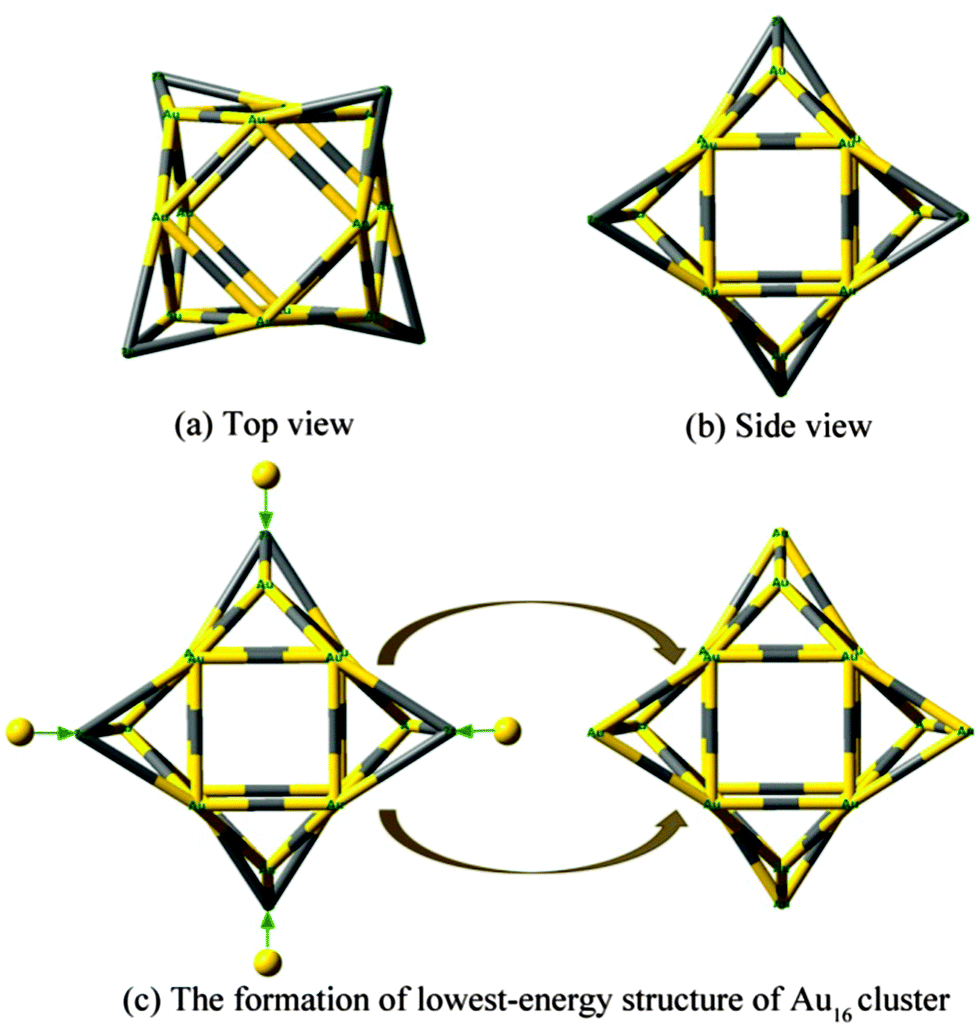 | ||
| Fig. 2 Structure of pure Au12 cluster: (a) top view and (b) side view. (c) The formation of lowest-energy structure of pure Au16 cluster. The additional gold atoms are represented by yellow spheres. Au12 consists of four tetragonal pyramid-shaped, while for Au16, four additional Au atoms are capping on four triangular faces of Au12, respectively. | ||
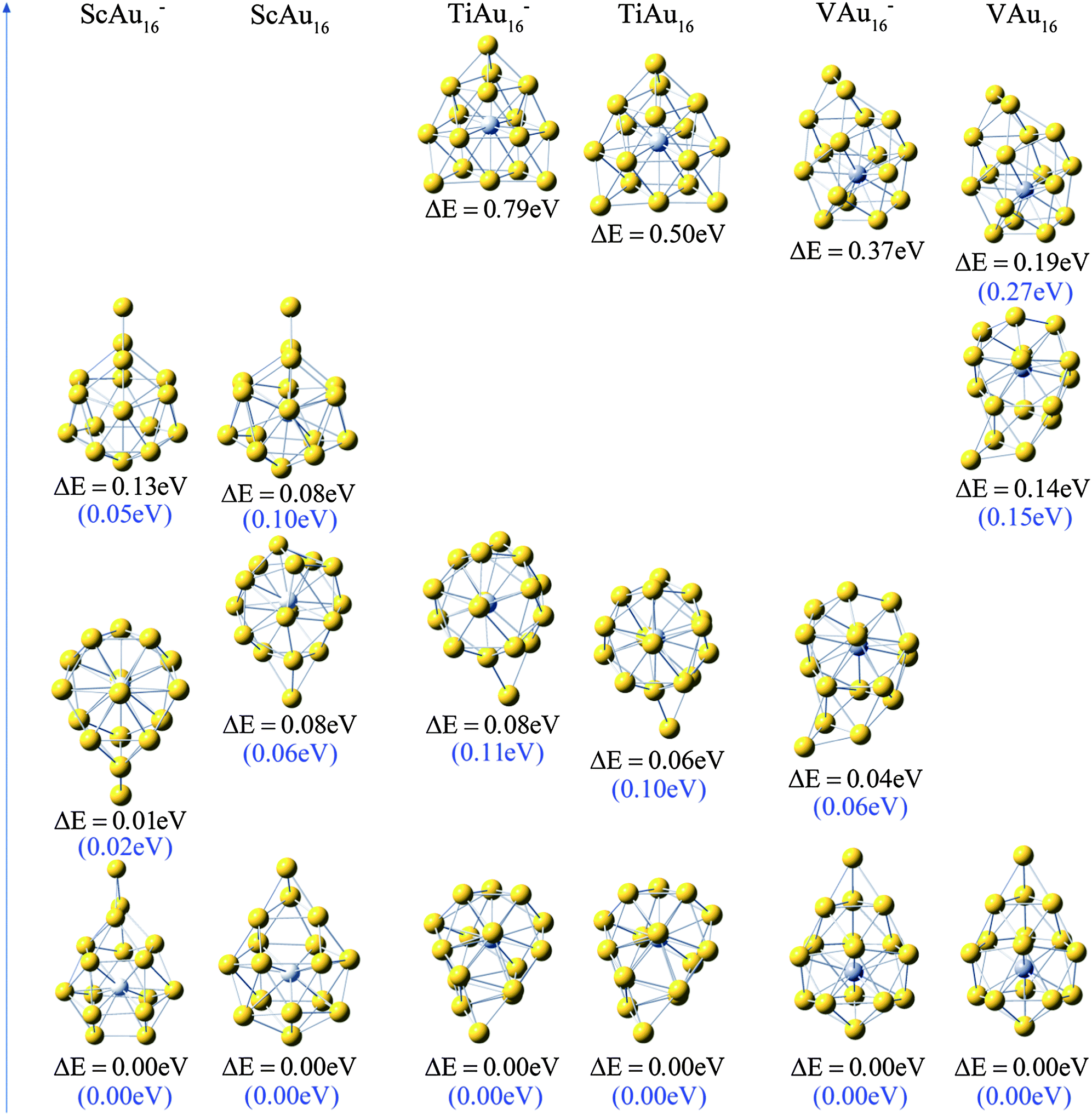 | ||
| Fig. 3 The global minimum and two low-lying isomers of M@Au16q (M = Sc, Ti and V; q = 0, −1). The dopant atoms are shown in color (Sc in light-gray, Ti in mid-gray, and V in dark-gray). The relative energies ΔE for all isomers are at the PBEPBE/LANL2DZ level of theory, and the values in the parentheses denotes the relative energies (RE) at PBEPBE/Au/SDD+2f/M/6-311+G*//PBEPBE/LANL2DZ level. | ||
| Cluster | The lowest-energy isomer | Low lying isomer | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Structure | SM | Sym | BE (eV) | DE (eV) | E gap (eV) | Structure | ΔEa,b (eV) | Structure | ΔEa,b (eV) | |
| a At PBEPBE/LANL2DZ level. b The parentheses at PBEPBE/SDD+2f//PBEPBE/LANL2DZ level. c Ref. 58. d Ref. 14. | ||||||||||
| Au16 | C | 1 | T d | 2.15 | 1.48 | A | 0.51 | B | 0.67b | |
| Au16− | A | 2 | C 2 | 2.22 | 0.14 | B | 0.10 | C | 0.22b | |
| Au17c | a | 2 | C 2v | 2.22 | b | 0.15 | c | 0.09 | ||
| Au17−d | a | 1 | C 2v | 2.30 | b | 0.09 | c | 0.07 | ||
| Sc@Au16 | I | 2 | C 1 | 2.49 | 0.46 | 0.16 | II | 0.08 (0.06) | III | 0.08 (0.10) |
| Sc@Au16− | I | 1 | C 1 | 2.68 | 0.47 | 1.29 | II | 0.01 (0.02) | III | 0.13 (0.05) |
| Ti@Au16 | I | 1 | C 1 | 2.46 | 0.43 | 0.72 | II | 0.06 (0.10) | III | 0.50 |
| Ti@Au16− | I | 2 | C 1 | 2.60 | 0.41 | 0.36 | II | 0.08 (0.11) | III | 0.79 |
| V@Au16 | I | 2 | C 1 | 2.39 | 0.37 | 0.24 | II | 0.14 (0.15) | III | 0.19 (0.27) |
| V@Au16− | I | 3 | C 1 | 2.51 | 0.36 | 0.42 | II | 0.04 (0.06) | III | 0.37 |
| Anion | REb | ADEc | VDEc |
|---|---|---|---|
| a Experimental ADE and VDE energies for the host cluster anion Au16− are 3.99 ± 0.03 and 4.03 ± 0.03, respectively. b The values in the parentheses denotes the relative energies (RE) at PBEPBE/Au/SDD+2f/M/6-311+G*//PBEPBE/LANL2DZ level. c ADE, VDE at PBEPBE/LANL2DZ level. | |||
| Au16−(Td)a | 0.00 | 3.89 | 4.18 |
| ScAu16−-I (1) (C1) | 0.00 (0.00) | 3.58 | 3.94 |
| ScAu16−-II (1) (C1) | 0.01 (0.02) | 3.67 | 3.83 |
| ScAu16−-III (1) (Cs) | 0.13 (0.05) | 3.46 | 3.51 |
| TiAu16−-I (2) (C1) | 0.00 (0.00) | 3.09 | 3.16 |
| TiAu16−-II (2) (C1) | 0.08 (0.11) | 3.08 | 3.15 |
| TiAu16−-III (4) (C1) | 0.79 (0.82) | 2.80 | 2.84 |
| VAu16−-I (3) (C1) | 0.00 (0.00) | 3.31 | 3.38 |
| VAu16−-II (3) (C1) | 0.04 (0.06) | 3.42 | 3.50 |
| VAu16−-III (5) (C1) | 0.37 (0.44) | 3.13 | 3.19 |
| Cluster | Charge (e) | M moment (μB) | Molecule (μB) | |||
|---|---|---|---|---|---|---|
| Total | M-(3d) | M-(4s) | M-(4p) | |||
| Sc@Au16 | −4.50 | 0.21 | 0.20 | 0.00 | 0.01 | 1 |
| Sc@Au16− | −4.41 | 0.00 | 0.00 | 0.00 | 0.00 | 0 |
| Ti@Au16 | −4.11 | 0.00 | 0.00 | 0.00 | 0.00 | 0 |
| Ti@Au16− | −3.94 | 0.77 | 0.75 | 0.00 | 0.02 | 1 |
| V@Au16 | −3.70 | 1.79 | 1.77 | 0.00 | 0.02 | 1 |
| V@Au16− | −3.48 | 2.15 | 2.14 | −0.01 | 0.02 | 2 |
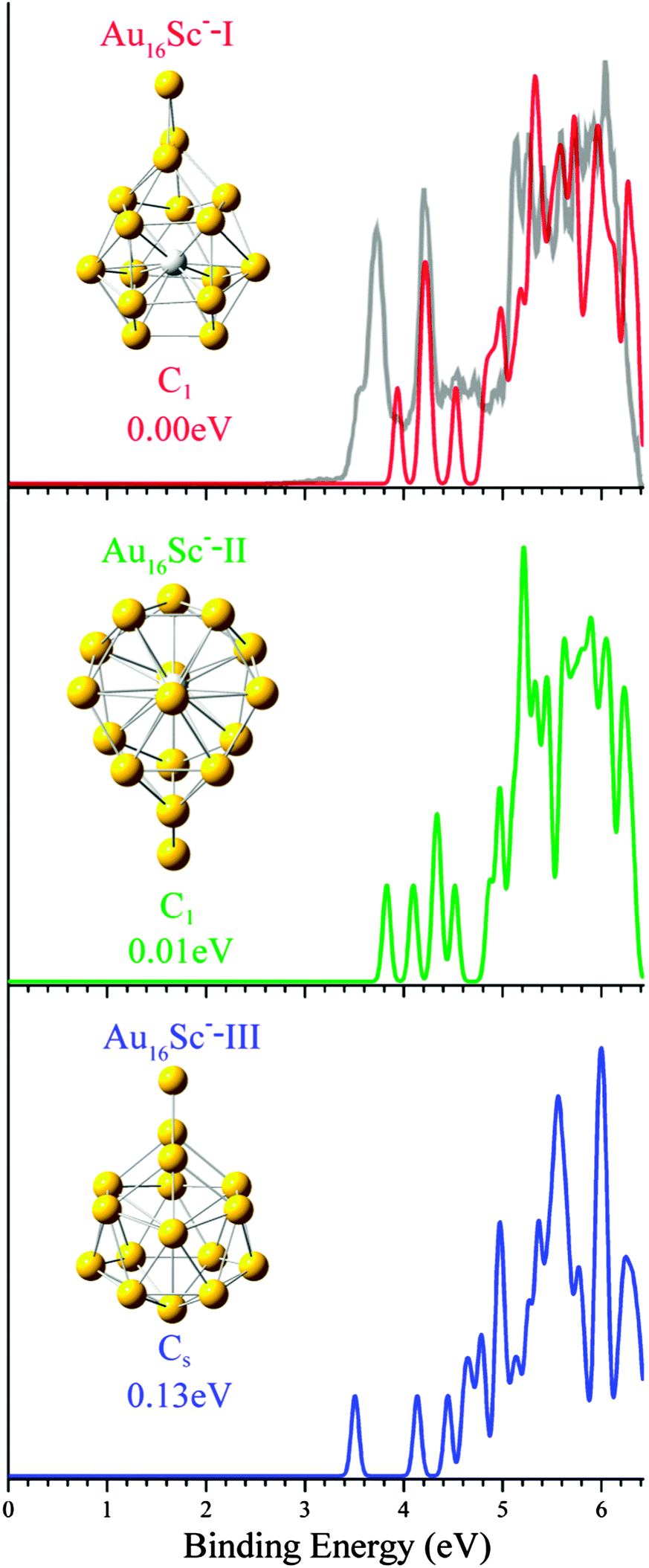 | ||
| Fig. 4 Structures, relative energies, and simulated photoelectron spectra for the three low-lying isomers of Au16Sc−. The gray curve is for the experimental PES for host gold cluster Au16−. The experimental PES spectra are cited from ref. 14. | ||
 | ||
| Fig. 5 Structures, relative energies, and simulated photoelectron spectra for the three low-lying isomers of Au16Ti−. The gray curve is for the experimental PES for host gold cluster Au16−. The experimental PES spectra are cited from ref. 14. | ||
 | ||
| Fig. 6 Structures, relative energies, and simulated photoelectron spectra for the three low-lying isomers of Au16V−. The gray curve is for the experimental PES for host gold cluster Au16−. The experimental PES spectra are cited from ref. 14. | ||
 | ||
| Fig. 7 (a)–(f) The global minimum structures of M@Au16q (M = Sc, Ti and V; q = 0, −1) on the left, electron localization function (ELF) on the right-hand side. The values of ELF are in the range of (0, 1) with 1 (red) meaning complete localization and 0.5 (green) corresponds to a delocalized (metallic) electron gas (for more details see ref. 60). | ||
Geometric structure and stability
We have obtained many low-lying isomers and determined the lowest-energy structures for M@Au16q (M = Sc, Ti and V; q = 0, −1) using the computation scheme SK-DFT described in section 2. Here, in order to discuss the effects of doped impurity atom on pure gold clusters, we carried out an additionally SK-DFT calculation on pure Au16−,0 clusters. Many isomers were found and three structures, representing the three structural types, are given in Fig. 1. As for pure neutral and anionic Au16−,0 clusters, three important types of neutral structures are similar to the anionic structures though the order of isomers is reversed in this case. For the pure Au16− cluster, a low-symmetric cage with C2 symmetry structure was obtained for Au16−, which can be derived from the previously discovered Td Au20− pyramid39 by removing its four apex atoms and allowing subsequent pop-up of the four face-center atoms. It should be point out that our global minimum structure of Au16− cluster (Fig. 1A) with C2 symmetry is about 0.48 eV more stable than the highly symmetric cage with tetrahedral (Td) symmetry structure obtained by Bulusu et al.14 It is worth to note that the second lowest-energy planar isomer B of Au16− found by Jena's group54 is less stable than the A cage structure by 0.1 eV. The third low-lying isomer C is less stable 0.22 eV than the isomer A. For the neutral Au16 cluster, a new Td compact structure C is the lowest-energy configuration. The most stable isomer of Au120/− is a planar structure which is validated by Häkkinen55 and Fernández.56 The fourth low-lying isomer of Au12 reported by Häkkinen consists of four tetragonal pyramids, and the lowest-energy configuration of Au16 can be viewed as capping four gold atoms to on four triangular faces of Au12, respectively. The formation of the new Td structure C is presented in Fig. 2. Our calculated structure on neutral Au16 is in line with the results of Jena et al.54 The second metastable isomer A is less stable 0.51 eV than the ground state isomer C. The planar isomer B exhibits C2v symmetry and is 0.67 eV higher in energy than the Td-symmetry compact structure C.The ground-state structures and some low-lying metastable isomers of M@Au16q (M = Sc, Ti and V; q = 0, −1) species are shown in Fig. 3, where a gray sphere refers to the impurity atom M. Remarkably, our calculations show that the three candidate lowest-energy isomers for M@Au16q are “endohedral cages” with a dopant atom trapping inside the cage and the point group symmetry is C1. The endohedral structures with relatively high symmetry (C3v) for M@Au16q (M = Sc, Ti and V) are also found in the present work but at high energy relative to the lowest-energy structures. These unstable structures were less stable than the global minimum structure by about 0.46–1.08 eV.
The structures with exohedral doping are all higher-lying isomers (>3 eV), this case is in line with the results of other transition-metal atoms (e.g. Fe, Co, Ni, Zn)13,17 doping in golden cage cluster Au16−. Relative to Au16−,0, due to the M–Au local interactions, the lowest-energy endohedral structures of M@Au16q have a significant distortion to the parent Au16−,0 cage. It is also completely different from the doping of a group IV atom (Si, Ge and Sn) into the Au16− cage,52 forming the exohedral gold cluster M@Au16−. The exohedral structure also occurred for K- and Ag-doped clusters, which was given previously by Fa and Dong.57 As can be seen from Fig. 3, three low-lying isomers of M@Au16− (M = Ti and V) resemble closely the shape of neutral M@Au16 clusters, even the order are completely similar to each other. For Sc@Au16−,0, we present three isomers in the energy range about 0.10 eV, the other two low-lying structures are 0.06 and 0.10 eV above the lowest-energy structure for neutral cluster and 0.02 and 0.05 eV for anionic ones, respectively. Similarly, the low-lying metastable isomers of Ti@Au16−,0 lies higher in energy than the global minimum structure by 0.11 and 0.82 eV for anionic Ti@Au16− and 0.10 and 0.53 eV for neutral Ti@Au16, respectively. As for V@Au16−,0, the low-lying isomers with low or without symmetry were found to be less stable than lowest-energy structures by 0.27–0.44 eV.
It is interesting to know if and how the structure and stability of the clusters will be changed when an early 3d transition-metal atom is doped into Au16 cluster compared with the case of by doping late 3d transition-metal atoms. Here, the main features of M@Au16− are present for all of the 3d transition metals as follows: (1) transition-metal-encapsulated structures are formed for all of the 3d transition metals, the additional transition-metal atom is located considerable from the center of the parent cages for the ground-state of 3d (from Sc to Ni)-doped clusters, while the late 3d (Cu and Zn) atom is located in the center with little distortion from the parent Au16− cage for the doped clusters; (2) depending on the metal, the ground states of M@Au16− possess low symmetry (C1) for the early transition metals (Sc to Mn), whereas with the exception of Ni@Au16−, the relatively high symmetry is formed for the late transition metals (C2 point group symmetry for Fe@Au16− and Co@Au16−, Cs for Cu@Au16− and Td for Zn@Au16−). It seems that the more the d-electron becomes, the higher the point group symmetry is. This may be attributed to Jahn–Teller distortion. Based on above discussion, we can conclude that a partially filled d shell of the transition-metal atoms plays a crucial role in determining the structures and properties of M@Au160,− clusters.
Table 1 gives various structural and energetic characteristics of M@Au16q clusters. It is well-known that the binding energy (BE) of a given cluster is a measure of its thermodynamic stability, which is defined as the difference between the energy sum of all the free atoms constituting the cluster and the total energy of the cluster. It can be seen from Table 1 that the binding energies per atom for the ground state of M@Au16q (M = Sc, Ti and V; q = 0, −1) clusters are higher than those of pure Au16−,0 and Au17−,0 clusters, indicating that all doped transition-metal atoms with open d shells can stabilize the pure hollow-cage structures. To further study the stabilities of M@Au16q clusters, we will discuss the doping energy (DE) of the ground-state structure, which is defined as the energy sum of the Au16−,0 and the M atom minus the total energy of the M@Au16q. From Table 1 we can see that the Sc@Au16− cluster has the highest DE of 0.47 eV among the clusters considered in this study. So, Sc@Au16− should be the most stable one among these clusters. The highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO–LUMO) energy gap is another useful quantity for examining the kinetic stability of clusters. A large energy gap corresponds to a high strength required to perturb the electronic structure. Comparing the difference between energy gap of pure and doped clusters in Table 1 one can see that, for M = Sc, it decreases (increases) for the neutral (anionic) cluster. Analogous behavior is shown by the M = V and Ti clusters. Specifically, the energy gaps of the clusters for Sc@Au16−, Ti@Au16 and V@Au16− with even-electron are larger than those of other clusters with odd-electron species. That systematic behavior indicates typical electronic odd-even effects. We compare the Egap's of all M@Au16−,0 and also find the Egap of Sc@Au16− with 1.29 eV is the largest one, larger than the Egap of the pure Au16− cluster, and than that of other clusters. This suggests that Sc@Au16− is relatively more chemically stable than the neighboring clusters.
Comparison of the PES spectrum of M@Au16− and Au16−
To facilitate the future comparison with further experiments, we have drawn the simulated PES of the three lowest-lying isomers of M@Au16− (M = Sc, Ti and V) in Fig. 4–6. Furthermore, the experimental PES14 of host gold cluster anion Au16− is also presented for comparison. The PES is simulated by adding the occupied orbital energies relative to the HOMO (SOMO for opened-shell systems) to the VDE and fitting them with a FWHM of 0.09 eV. The comparison of calculated first ADE and VDE energies between the doped cluster anions M@Au16− (M = Sc, Ti and V) and the host cluster Au16− is presented in Table 2. The VDEs of each cluster anion corresponds to the first peak maximum of each spectrum in Fig. 4–6. As shown in Table 2, the adiabatic and vertical detachment energies (ADE/VDE) are 3.58/3.94, 3.09/3.16, and 3.31/3.38 eV, respectively, for M = Sc, Ti, and V, which are smaller than those of Au16− cage (theoretical: 3.89/4.18 eV, experimental: 3.99 ± 0.03/4.03 ± 0.03 eV, see Table 1). The calculated ADE and VDE for Ti-doped or V-doped (3.09 and 3.16 eV for Ti-doped, 3.31 and 3.38 eV for V-doped) is very close to each other. This is due to the negligible geometry changes between the anionic and neutral ground state structures. It should be mentioned that Bulusu et al.14 predicted that there may exist a minor change between the structures of neutral Au16 and anionic Au16− by observing the PES of Au16−. Recently, Jena et al. used DFT calculations found that the structure of the neutral Au16 is obviously different from that of anionic Au16−.54 Indeed, our results support this view. However, the calculated ground state structures of Au16−,0 are difficult to get a reasonable explanation from the experimental PES.As discussed previously,14,51 the Au16− spectrum is unique: it has a very high electron binding energy and does not exhibit a large energy gap like other even-sized gold clusters,39 which is because the tetrahedral Au16 cage is open shell with two unpaired electrons and two extra electrons are needed to make a closed-shell 18-electron Au162− cage. Similarly, our theoretical calculations confirmed the spectrum of the ground-state M@Au16− (M = Sc and V) (isomer-I) is similar to that of the Au16− cage with a small energy gap. The neutral Ti@Au16 cluster possesses 20 valence electrons and is closed-shell, as evident from the PES spectra of Ti@Au16−-I which exhibit a sizable HOMO–LUMO gap of ∼0.6 eV, i.e., ∼0.4 eV larger compared to the simulated PES of M@Au16−-I (M = Sc and V) (Fig. 4–6). For the top-three lowest-lying Sc@Au16− isomers, the shape of the simulated PES spectra are different from each other, reflecting their geometric structure with a large change. Each spectrum reveals a fairly small X–A energy gap (between the first and second VDEs). The spectrum of lowest-energy structure Sc@Au16−-I (see Fig. 3) presents three well-resolved peaks in the binding energy range between 3.8 and 4.7 eV followed by the more congested spectral features beyond 5.0 eV. The first peak displays parallel move 0.2 eV toward right due to the effect of the impurity Sc atom. The second lowest-lying isomer-II is only about 0.01 eV above the global minimum structure. Considering the uncertainty of the DFT energies for such systems, we could not conclude which isomer structure should be assigned as the global minimum for Sc@Au16−. The PES of the doped Sc@Au16−-II isomer with the inside Sc atom present different features with that of the parent hollow Au16− cage, suggesting that its geometric and electronic structures alter remarkably due to the addition of Sc atom. Our optimization results also support this point of view, indeed, there are significant differences in their geometric structure compared with the pure truncated tetrahedron cage. The simulated PES spectrum of isomer III of Sc@Au16− seems to be closest to the experimental spectrum of the pure Au16− cage, and it is likely to be the concomitant anion in the experimental Au16− distribution. For the ground-state Ti@Au16− (isomer-I), four apparent peaks appear nearby each of the first two major peaks of the parent Au16− cage. However, the simulated PES for the three low-lying isomers of Ti@Au16− show slightly larger X–A energy gaps than the bare hollow Au16− cage, suggesting the fact that the neutral cluster Ti@Au16 is more stable than the parent Au16. The lowest-lying isomers II and III of Ti@Au16− have very different configurations, their PES spectra have some resemblances in the energies below 4.2 eV, possessing a wider gap between the X and A peaks (about 0.6 eV) and similar calculated first ADE/VDE of about 3.1 eV just like the cases of Zn@Au16−.13 The second lowest-lying isomer-II is about 0.08 eV above the global minimum structure. So we could conclude that the isomer-I should be assigned as the global minimum for Ti@Au16−. Furthermore, the simulated PES of V@Au16− cluster show smaller X–A energy gap and more congested spectral features beyond 4.4 eV compared with that of the parent hollow Au16− cage, suggesting that its geometric and electronic structures alter remarkably due to the addition of V atom. The small difference between ADE and VDE (3.31/3.38, 3.42/3.50, 3.13/3.19 eV) suggested that there is very little geometrical change between the low lying isomers of V@Au16− and V@Au16. It will be very interesting if the most proper structure could be detected by the future PES experiment for validating the existence of the Sc-, Ti- and V-doped Au16 clusters.
At this stage, although other energetically more favorable structures could not be ruled out strictly, we believe that the lowest-energy structures of M@Au16q found here are at least powerful candidates for their ground states, which are hoped to be verified in the future experiments and calculations at more accurate levels of theory.
Magnetic moment
Unfortunately, there are no Stern–Gerlach experiment for present species until now, however, the reliability of the present DFT (PEBPEB) was validated by performing calculations on the first VDEs and ADEs on pure Au16− and many other analogous doped M@Au16− (M = Fe, Co, Ni and Zn)13,17 clusters. Previous studies show that the dopant–Au interactions are crucial in determining the structures of the doped golden cages. Cu and Zn, which have closed 3d shells, primarily donate their 4s electrons to the cage, forming charge-transfer complexes with very little distortions to the cage.13,51 However, for the open 3d shell transition metal dopant, e.g. Cr, Mn, Fe, Co, and Ni, they have more significant interactions with Au and they distort the golden cages and form other new kinds of structures.17,35 The 3d orbitals obtain electrons from the gold cage and the magnetic moment of the impurity Mn/Cr atom is slightly quenched. In what follows, we will discuss the magnetic moments of open 3d transition-metal-doped gold clusters M@Au16q (M = Sc, Ti and V; q = 0, −1). Magnetic moment (μB) of 3d, 4s, and 4p states for guest M atom, natural charge (e), total magnetic moment (μB) of the M atom, and total magnetic moment of the lowest-energy M@Au16q clusters are listed in Table 3. It is interesting to describe certain trends of the magnetic moments of M@Au16q in this work as follows. (1) Table 3 shows that Sc@Au16, Ti@Au16− and V@Au160,− are magnetic clusters, and the magnetic moments of Sc@Au16, Ti@Au16− and V@Au160,− clusters are 1, 1, and 1, 2 μB, respectively. Sc@Au16− and Ti@Au16 species present a non-magnetic ground state. (2) For Sc@Au16 cluster, Au16 carries most of the magnetic moments. With the increasing of d-electron, for Ti@Au16− and V@Au160,−, the magnetism of the clusters considered in this study is mainly located on the transition-metal atoms, only very small magnetic moments are found on Au16. This is quite consistent with the result of Fe and Co doped gold cluster anions. For Sc–Au and Ti–Au clusters, Au16 contribute positive magnetic moment to the total moment, while for V–Au cluster, they carry a small negative magnetic moment. (3) The total magnetic moments of M@Au16− increase gradually along with d-electron increasing from Sc- to V-doped. Although there is considerable interaction between the transition-metal atoms and the host gold cage, the 3d states of the dopant atoms remain largely localized and the atomic like magnetism is maintained in the doped clusters, e.g. the anionic M@Au16− (M = Cr, Mn, Fe, Co) clusters preferred higher spin multiplicities (SM) (SM = 6 for M = Cr, SM = 7 for M = Mn, SM = 6 for M = Fe and SM = 5 for M = Co) and carried large magnetic moments (5 μB for Cr@Au16−, 6 μB for Mn@Au16−, 5 μB for Fe@Au16− and 4 μB for Co@Au16−).17,35 Unlike Fe and Co-doped gold cage systems,17 where the 4s electrons of metals transfer to the Au16 cage, the s electrons of the Au16 cage transferred to Sc, Ti and V atom for M@Au16q (M = Sc, Ti and V; q = 0, −1) clusters. The bonding in the doped cluster anions can be viewed as an Au163+ interacting with a Sc4−/Ti4− core or Au162+ interacting with a V3− core. The neutral M@Au16 clusters can be described as an Au165+ interacting with a Sc5− core or Au164+ interacting with a Ti4−/V4− core, where 3d orbitals obtain electrons from the gold cage and the dopant possesses d0.2, d0 and d1.8 valence configurations, respectively, which means the magnetic moment of the impurity transition-metal (Sc, Ti and V) atom is quenched.Electron localization function (ELF)
In order to obtain deep insight into reliable electronic structure information, the electron localization function (ELF) was investigated using the Multiwfn program59 and is sketched in Fig. 7. The global minimum structures of M@Au16q (M = Sc, Ti and V; q = 0, −1) clusters are “endohedral cages” with a dopant atom trapping inside the cage, so we plotted the ELF diagrams with 200, 200 grids in two dimensions (2D) defined by three atoms. The ELF introduced by Becke and co-workers describes how much the Pauli repulsion is efficient at a point in the real molecular space.60 ELF provides a rigorous basis for the analysis of interaction. The local maxima of ELF define localization domains and they correspond to chemically interesting regions. The values of ELF are in the range of (0, 1) with 1 (red) meaning complete localization and 0.5 (green) corresponds to a delocalized (metallic) electron gas.60 It can be seen that very low value occurs between each boundary gold and M (Sc, Ti and V) atom as well as in some regions outside of the central atoms. This observation suggests that the bonding between gold and dopant atoms is weak. However, ELF in some areas is a bit high, for example, around the center of transition-metal atoms; on the whole, these bonding can be expected to be very weak. Note that the order of ELF is Sc-doped > Ti-doped > V-doped clusters, this trend is consistent with the doping energy per atom. The ELF analysis of M@Au16q (M = Sc, Ti and V; q = 0, −1) clusters show that strong interactions are not revealed in this study.Conclusions
In summary, we have investigated the structural, electronic, and magnetic properties of the doped clusters M@Au16q (M = Sc, Ti and V; q = 0, −1), with the aid of the density functional theory (DFT). Stochastic search procedure by Saunders “Kick” method combined with extensive DFT calculations are used for global minimum searches (SK-DFT). Optimized geometries of the ground-state and low-lying energy states of both the anion and neutral species are reported using SK-DFT and the results are compared with experimental literature PES data. The global minimum search revealed that the endohedral cages with no symmetry represent the global minimum structure for the doped gold clusters M@Au16q. The Sc@Au16− cluster has the largest binding energy and doping energy, indicating it to be the most stable among M@Au16q species. The doped endohedral structures exhibit lager distortion from the parent Au16− cage for the doped clusters. The study of magnetic moment indicates that the magnetic moment of the impurity transition-metal (Sc, Ti and V) atom is quenched. The order of ELF is Sc-doped > Ti-doped > V-doped clusters, this trend is consistent with the doping energy per atom. The ELF analysis of M@Au16q (M = Sc, Ti and V; q = 0, −1) clusters show that strong interactions are not revealed in this study. It will be very interesting if the most proper structure could be detected by the future PES experiment for validating the existence of the Sc-, Ti- and V-doped Au16 clusters.Acknowledgements
This work was supported by the National Natural science Foundation of China (No. 11247257), by the Natural Science Foundation of Fujian Province of China (No. 2012J05005), and by the Fundamental Research Funds for the Central Universities (JB-ZR1201).Notes and references
- G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Imperial College Press, London, 2006 Search PubMed.
- Gold: Progress in Chemistry, Biochemistry and Technology, ed. H. Schmidbaur, Wiley, Chichester, 1999 Search PubMed.
- K. Palotás, B. Lazarovits, L. Szunyogh and P. Weinberger, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 134421 CrossRef.
- H. Q. Wang, X. Y. Kuang and H. F. Li, Phys. Chem. Chem. Phys., 2010, 12, 5156 RSC.
- H. Q. Wang, X. Y. Kuang and H. F. Li, J. Phys. Chem. A, 2009, 113, 14022 CrossRef CAS PubMed.
- J. Autschbach, B. A. Hess, M. P. Johansson, J. Neugebauer, M. Patzschke, P. Pyykkö, M. Reiher and D. Sundholm, Phys. Chem. Chem. Phys., 2004, 6, 11 RSC.
- M. Walter and H. Häkkinen, Phys. Chem. Chem. Phys., 2006, 8, 5407 RSC.
- Y. Zhao, Z. Li and J. Yang, Phys. Chem. Chem. Phys., 2009, 11, 2329 RSC.
- H. Tanaka, S. Neukermans, E. Janssens, R. E. Silverans and P. Lievens, J. Chem. Phys., 2003, 119, 7115 CrossRef CAS.
- H. Tanaka, S. Neukermans, E. Janssens, R. E. Silverans and P. Lievens, J. Am. Chem. Soc., 2003, 125, 2862–2863 CrossRef CAS PubMed.
- M. Zhang, L. M. He, L. X. Zhao, X. J. Feng and Y. H. Luo, J. Phys. Chem. C, 2009, 113, 6491 CAS.
- X. Li, B. Kiran, L. F. Cui and L. S. Wang, Phys. Rev. Lett., 2005, 95, 253401 CrossRef.
- L. M. Wang, R. Pal, W. Huang, X. C. Zeng and L. S. Wang, J. Chem. Phys., 2009, 130, 051101 CrossRef PubMed.
- S. Bulusu, X. Li, L. S. Wang and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8326 CrossRef CAS PubMed.
- H. W. Kroto, J. R. Heath, S. C. O'Brian, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162 CrossRef CAS.
- Y. Chai, T. Guo, C. Jin, R. E. Haufler, L. P. F. Chibante, J. Fure, L. Wang, J. M. Alford and R. E. Smalley, J. Phys. Chem., 1991, 95, 7564 CrossRef CAS.
- L. M. Wang, J. Bai, A. Lechtken, W. Huang, D. Schooss, M. M. Kappes, X. C. Zeng and L. S. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 033413 CrossRef.
- Y. Gao, S. Bulusu and X. C. Zeng, ChemPhysChem, 2006, 7, 2275 CrossRef CAS PubMed.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS.
- T. Back, Evolutionary Algorithm in Theory and Practice: Evolutionary Programming Genetic Algorithms, Oxford University Press, Oxford, 1990 Search PubMed.
- P. Chaudhuri and S. P. Bhattacharyya, Chem. Phys., 1999, 241, 313 CrossRef.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087 CrossRef CAS.
- P. Babadova-Parvanova, K. A. Jackson, S. Srinivas, M. Horoi, C. Kohler and G. Seifert, J. Chem. Phys., 2002, 116, 3576 CrossRef.
- R. O. Jones, G. Gantefor, S. Hunsicker and P. Pieperhoff, J. Chem. Phys., 1995, 103, 9549 CrossRef CAS.
- R. O. Jones, A. I. Lichtenstein and J. Hutler, J. Chem. Phys., 1997, 106, 4556 Search PubMed.
- R. O. Jones, J. Chem. Phys., 1999, 110, 5189 CrossRef CAS.
- J. Wang, J. Jellinek, J. Zhao, Z. Chen, R. B. King and P. v. R. Schleyer, J. Phys. Chem. A, 2005, 109, 9265 CrossRef CAS PubMed.
- A. Alexandrova, A. Boldyrev, Y. J. Fu, X. Yang, X. B. Wang and L. S. Wang, J. Chem. Phys., 2004, 121, 5709 CrossRef CAS PubMed.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111 CrossRef CAS.
- M. Saunders, J. Comput. Chem., 2004, 25, 621 CrossRef CAS PubMed.
- H. Q. Wang and H. F. Li, J. Chem. Phys., 2012, 137, 164304 CrossRef PubMed.
- H. Q. Wang and H. F. Li, Chem. Phys. Lett., 2012, 554, 231 CrossRef CAS PubMed.
- H. Q. Wang, H. F. Li and X. Y. Kuang, Phys. Chem. Chem. Phys., 2012, 14, 5272 RSC.
- H. Q. Wang, H. F. Li, J. X. Wang and X. Y. Kuang, J. Mol. Model., 2012, 18, 2993 CrossRef CAS PubMed.
- H. Q. Wang, H. F. Li and L. X. Zheng, J. Magn. Magn. Mater., 2013, 344, 79 CrossRef PubMed.
- F. Furche, R. Ahlrichs, P. Weis, C. Jacob, S. Gilb, T. Bierweiler and M. M. Kappes, J. Chem. Phys., 2002, 117, 6982 CrossRef CAS.
- S. Gilb, P. Weis, F. Furche, R. Ahlrichs and M. M. Kappes, J. Chem. Phys., 2002, 116, 4094 CrossRef CAS.
- M. Ji, X. Gu, X. Li, X. G. Gong, J. Li and L. S. Wang, Angew. Chem., Int. Ed., 2005, 44, 7119 CrossRef CAS PubMed.
- J. Li, X. Li, H. J. Zhai and L. S. Wang, Science, 2003, 299, 864 CrossRef CAS PubMed.
- W. Huang and L. S. Wang, Phys. Rev. Lett., 2009, 102, 153401 CrossRef.
- X. Xing, B. Yoon, U. Landman and J. H. Parks, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 165423 CrossRef.
- M. P. Johansson, A. Lechtken, D. Schooss, M. M. Kappes and F. Furche, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 77, 053202 CrossRef.
- M. Walter and H. Häkkinen, New J. Phys., 2008, 10, 043018 CrossRef.
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- M. J. Frisch, et al., GAUSSIAN09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS.
- P. Schwerdtfeger, M. Dolg, W. H. E. Schwarz, G. A. Bowmaker and P. D. W. Boyd, J. Chem. Phys., 1989, 91, 1762 CrossRef CAS.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS.
- S. Zorriasatein, K. Joshi and D. G. Kanhere, J. Chem. Phys., 2008, 128, 184314 CrossRef PubMed.
- L. M. Wang, S. Bulusu, H. J. Zhai, X. C. Zeng and L. S. Wang, Angew. Chem., Int. Ed., 2007, 46, 2915 CrossRef CAS PubMed.
- L. M. Wang, S. Bulusu, W. Huang, R. Pal, L. S. Wang and X. C. Zeng, J. Am. Chem. Soc., 2007, 129, 15136 CrossRef CAS PubMed.
- D. J. Tozer and N. C. Handy, J. Chem. Phys., 1998, 109, 10180 CrossRef CAS.
- G. Chen, Q. Wang, Q. Sun, Y. Kawazoe and P. Jena, J. Chem. Phys., 2010, 132, 194306 CrossRef PubMed.
- H. Häkkinen, B. Yoon, U. Landman, X. Li, H. J. Zhai and L. S. Wang, J. Phys. Chem. A, 2003, 107, 6168 CrossRef.
- E. M. Fernández and L. C. Balbás, Phys. Chem. Chem. Phys., 2011, 13, 20863 RSC.
- W. Fa and J. Dong, J. Chem. Phys., 2008, 128, 144307 CrossRef PubMed.
- S. Bulusu and X. C. Zeng, J. Chem. Phys., 2006, 125, 154303 CrossRef PubMed.
- T. Lu, Multiwfn, version 3.0. http://multiwfn.codeplex.com/.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS.
| This journal is © the Owner Societies 2014 |