
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-state photoreactivity of 9-substituted acridizinium bromide salts†
Samuel A.
Stratford
a,
Mihails
Arhangelskis
a,
Dejan-Krešimir
Bučar
*ab and
William
Jones
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: wj10@cam.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: d.bucar@ucl.ac.uk
First published on 15th October 2014
Abstract
A series of 9-X-acridizinium bromide salts (where X = F, Cl, Br, I, CH3, C(CH3)3) was synthesised, and their solid-state [4 + 4] photoreactivity was investigated using crystallographic and spectroscopic methods, while their thermal stability was studied using thermal gravimetry and differential-scanning calorimetry. The study aimed to identify how the nature and size of the substituent on the acridizinium cation affected the regioselectivity of the [4 + 4] cycloaddition reactions, the reaction yields and relative reaction rates. Computational methods were utilised to rationalize the unexpectedly high regioselectivity of the solid-state photodimerisation of the 9-Cl-acridizinium bromide salt.
Introduction
Acridizinium cations are highly conjugated, rigid anthracene-like molecules (Scheme 1a), which upon exposure to UV irradiation undergo a reversible [4 + 4] cycloaddition reaction, both in solution and the solid state.1–3 Acridizinium salts have been shown to be useful as molecular switches,4 fluorescent probes5,6 and sensors.7,8 Furthermore, acridizinium-based dendrimers were revealed as promising materials in the context of optical data-storage devices (e.g. Blu-ray Disc technology).9 | ||
| Scheme 1 Molecular structures of: a) an acridizinium cation, b) the studied 9-substituted acridizinium cations and c) possible regioisomers of the acridizinium photodimer. The nomenclature of the regioisomers was adapted from Ihmels et al.1,2 for clarity and comparison purposes. | ||
While acridizinium derivatives have been reported to photodimerise in solution with low regioselectivity, it has been demonstrated that the regioselectivity of the solution-based reaction can be improved through appropriate derivatisation of the acridizinium cation. A study by Ihmels et al. showed that UV-irradiation of 9-Cl-, 9-Br- and 9-OCH3-substituted acridizinium cations gives rise to four regioisomers, namely syn-hh, anti-hh, syn-ht, anti-ht (where ht = “head-to-tail” and hh = “head-to-head”, Scheme 1c), while the 9-NH2 derivative yields only two.10 The solid-state [4 + 4] photodimerisation, on the other hand, exhibits further improved regioselectivity.2
Initial reports of photoreactivity studies of polycrystalline acridizinium salts suggested that only the anti-ht photodimers form.11 Later studies, however, showed that polycrystalline acridizinium salts yield mixtures of all four possible regioisomers.3,12 Single crystals of the corresponding salts, on the other hand, yield exclusively the anti-ht photodimer,12,13 thus indicating that the photodimerisation is affected by the orientation of the molecules within the single crystal (i.e. the reaction is topochemical). The fact that a high degree of regioselectivity can only be achieved within single crystals (possibly because of the high surface area associated with polycrystalline materials and the inability to control the surface) presents a limitation in terms of functionality. Specifically, the growth of single crystals may not be straightforward and they are also susceptible to mechanical stress and, thus, may be difficult to be processed and incorporated into functional devices.
We assessed in a previous study how the nature of the counter ion in the acridizinium salt affects the reaction yields and the regioselectivity of the solid-state [4 + 4] photodimerisation.13,14 It was shown that larger anions (e.g. picrate) and those with a tetrahedral geometry (e.g. perchlorate, hexafluorophosphate, tetrafluoroborate) distort the packing of the acridizinium ions to affect the photoreactivity in two ways: they either lower the reaction rates (e.g. perchlorate, tetrafluoroborate), or they render the solid photostable (e.g. hexafluorophosphate). Spherical anions (e.g. bromide and iodide) were found to improve reaction kinetics. The effect of the counterion was also shown to be different in the case of the 9-methylacridizinium cation, as compared to the 9-bromoacridizinium cation.14 While the perchlorate and the bromide salts reacted very quickly, the iodide salt reacted very slowly and exhibited a long induction period. In a related study, Ihmels et al. also showed that 9-bromoacridizinium bromide resulted in exclusively the anti-ht isomer.1 This contrasted with the regioselectivity of 8-bromoacridizinium bromide, a solid that was reported to yield all four regioisomers upon UV-irradiation in the solid state.2
The work reported here focuses on how the substituents on the acridizinium framework influence the solid-state photoreactivity of the salts. With this in mind, we have synthesised a series of 9-X-acridizinium bromide salts (9-X) (where X = F, Cl, Br, I, CH3, C(CH3)3; Scheme 1b) in order to study how the nature and the size of the substituent controls reaction rates and regioselectivity. We chose to study 9-substituted acridizinium salts with bromide as the counterion, as it has been previously shown that related acridizinium bromide salts yield photodimers in a regioselective fashion and in high yields.1
Results and discussion
1. Structural analyses of the 9-substituted acridizinium salts
The diffraction data for compounds 9-X were collected at room temperature (i.e. 298 K) in order to study their structural features at conditions under which the photoreactivity studies were conducted. Room temperature diffraction experiments were considered to be necessary, as the photoreactivity is temperature dependent (low temperatures are reported to induce shifts of acridizinium cations in the crystal lattice, or even phase transitions).2Single crystal and powder X-ray diffraction studies showed that the structures of all studied single crystals are identical to those of the corresponding bulk materials. All the 9-X-acridizinium bromides were found to crystallise as hydrates. The acridizinium cations in the majority of the studied salts exhibit a preference to arrange in an anti-ht fashion. This is attributed to the repulsion forces occurring between the quaternary nitrogen atoms, as well as to the donor–acceptor π⋯π interactions between the electron rich phenyl moiety and the pyridinium moiety. Based on the alignment of the acridizinium cations in their crystal lattices and assuming topochemical control, it was anticipated that the majority of solids would yield only anti-ht photodimers upon UV-irradiation. The cations in 9-F, 9-Cl and 9-C(CH3)3, however, display different packing modes and were thus expected to yield other photodimers. Specifically, the 9-Cl cations are arranged in an anti-ht and syn-ht fashion, while the 9-F cations stack in both an anti-ht and anti-hh manner (as a consequence of molecular disorder). The photoreactions of 9-F and 9-Cl were, therefore, expected to be less regioselective than those of the other salts. 9-C(CH3)3, on the other hand, displays stacks wherein the cations are exclusively aligned in a syn-ht fashion for the formation of only one photodimer.
Our studies have also revealed the unexpected formation of 9-I triiodide. It should be noted that this compound is not a major constituent of the bulk material, but rather appears as an impurity in the bulk of 9-I bromide. It is presumed that the triiodide counter anion was formed at some point during the synthesis of 9-I. The crystal structure of the triiodide stands out because it does not include a water molecule, thus, suggesting that the triiodide might exhibit greater thermal stability than other acridizinium salts described herein. The structural features of all solids are described below in succession.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.50), thus resulting in an anti-ht and anti-hh arrangement of the acridizinium cation pairs (Fig. 1a). The reaction centres in the acridizinium pair are separated by 3.99 Å. The pairs are connected by four-component centrosymmetric bromide-water hydrate assemblies via C–H⋯Br and C–H⋯O interactions, and form a one-dimensional array (highlighted in Fig. 1b). The four-component bromide-water assembly is sustained by O–H⋯Br interactions (highlighted in Fig. 1b). The one-dimensional molecular array is extended into a planar two-dimensional structure via C–H⋯F forces. The two-dimensional sheets are stacked (Fig. 1b) and held together by Br⋯π forces.
:0.50), thus resulting in an anti-ht and anti-hh arrangement of the acridizinium cation pairs (Fig. 1a). The reaction centres in the acridizinium pair are separated by 3.99 Å. The pairs are connected by four-component centrosymmetric bromide-water hydrate assemblies via C–H⋯Br and C–H⋯O interactions, and form a one-dimensional array (highlighted in Fig. 1b). The four-component bromide-water assembly is sustained by O–H⋯Br interactions (highlighted in Fig. 1b). The one-dimensional molecular array is extended into a planar two-dimensional structure via C–H⋯F forces. The two-dimensional sheets are stacked (Fig. 1b) and held together by Br⋯π forces.
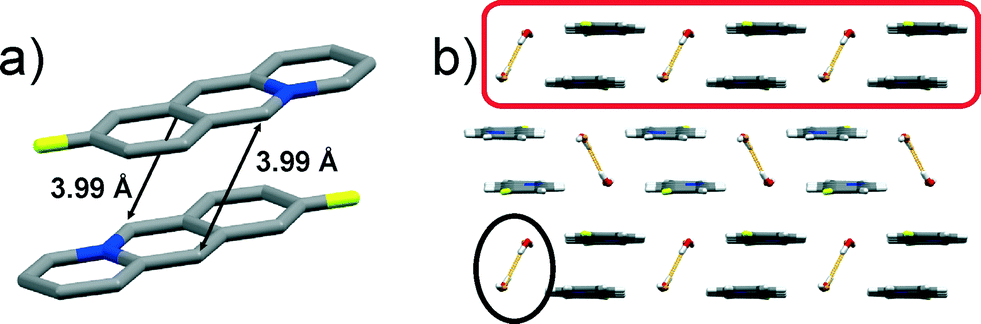 | ||
| Fig. 1 X-ray crystal structure of: a) two stacked 9-F cations in an anti-ht arrangement (hydrogen atoms are omitted for clarity, only one occupancy is shown), and b) crystal packing of 9-F viewed along a*. | ||
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit consists of two 9-chloroacridizinium cations, two bromide counter anions and two molecules of water. The cations stack infinitely in an offset manner leading to anti-ht and syn-ht packing arrangements. The reaction centres of anti-ht arranged cations in the stack are separated by 3.80 Å, while the reaction centres of the syn-ht arranged cations are separated by 3.63 and 3.83 Å (Fig. 2a). While the reaction centres in the anti-ht and syn-ht acridizinium pairs display similar separation distances, their alignment differs noticeably. More specifically, the anti-ht acridizinium cations align in a parallel offset fashion, while the acridizinium cations of the syn-ht pairs display an angular offset (Fig. 2b). The acridizinium stacks are separated by four-component centrosymmetric bromide-water assemblies via C–H⋯Br and C–H⋯O interactions. The bromide-water assemblies are held together by O–H⋯Br hydrogen bonds. The crystal lattice of 9-Cl (Fig. 2c) is sustained by C–H⋯Br interactions.
space group. The asymmetric unit consists of two 9-chloroacridizinium cations, two bromide counter anions and two molecules of water. The cations stack infinitely in an offset manner leading to anti-ht and syn-ht packing arrangements. The reaction centres of anti-ht arranged cations in the stack are separated by 3.80 Å, while the reaction centres of the syn-ht arranged cations are separated by 3.63 and 3.83 Å (Fig. 2a). While the reaction centres in the anti-ht and syn-ht acridizinium pairs display similar separation distances, their alignment differs noticeably. More specifically, the anti-ht acridizinium cations align in a parallel offset fashion, while the acridizinium cations of the syn-ht pairs display an angular offset (Fig. 2b). The acridizinium stacks are separated by four-component centrosymmetric bromide-water assemblies via C–H⋯Br and C–H⋯O interactions. The bromide-water assemblies are held together by O–H⋯Br hydrogen bonds. The crystal lattice of 9-Cl (Fig. 2c) is sustained by C–H⋯Br interactions.
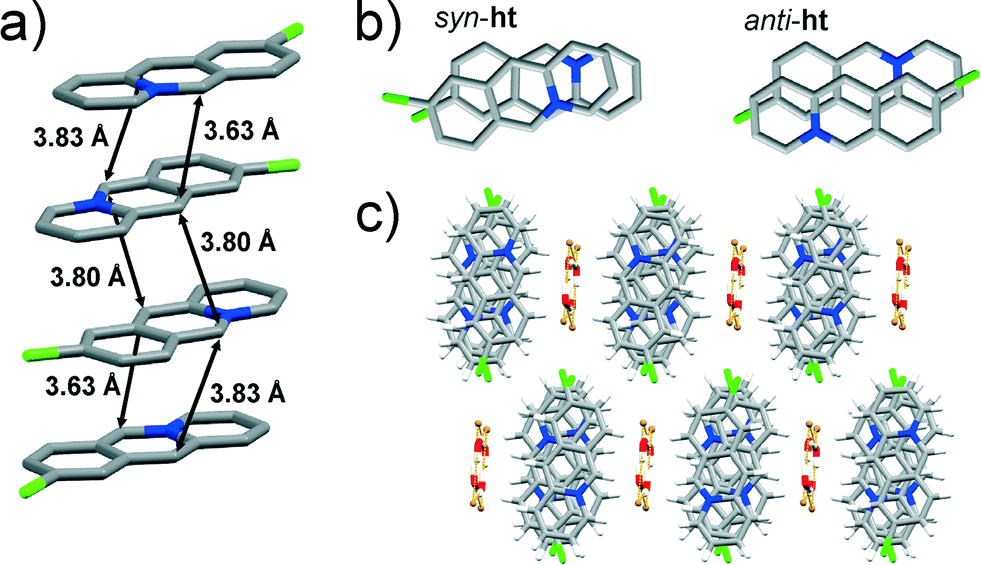 | ||
| Fig. 2 X-ray crystal structure of: a) an infinite stack of 9-Cl cations (hydrogen atoms are omitted for clarity), b) parallel and angular offsets in pairs of syn-ht and anti-ht pairs of acridizinium cations, and c) crystal packing of 9-Cl viewed along the crystallographic plane (−110). | ||
The diffraction data suggested that the studied 9-Cl single crystal was partially dehydrated (more specifically, one of the two water molecules in the asymmetric unit exhibited a partial occupancy of ~50%). Considering the low dehydration temperature of the solid (see Table 2), the fact that the diffraction data was collected at room temperature, and that all other studied acridizinium salts crystallise as monohydrates, it is likely that 9-Cl in fact crystallises as a (thermally unstable) monohydrate.
Another polymorph of 9-Cl was earlier identified by Ihmels et al. (CCDC reference code: CIPQUN, Form I).1 Unlike the 9-Cl polymorph described herein (Form II), Form I features 9-chloroacridizinium cations being arranged exclusively in an anti-ht fashion, similarly to the 9-bromoacridizinium cations in the structure of 9-Br (described below).
space group with one 9-bromoacridizinium cation, one bromide anion and one water molecule in the asymmetric unit. The acridizinium cations pack pair-wise in an anti-ht manner forming infinite stacks. The reaction centres in the cation pair are separated by 3.78 Å, while the reaction centres of cations belonging to neighbouring pairs are separated by 3.95 Å (Fig. 3a).
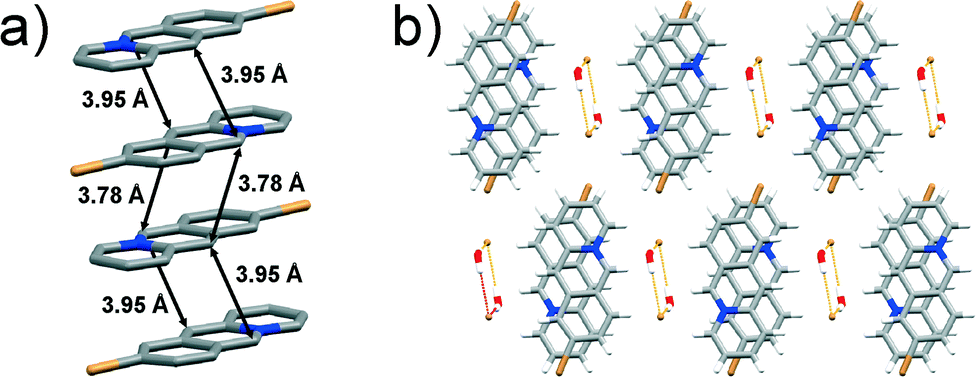 | ||
| Fig. 3 X-ray crystal structure of: a) an infinite stack of 9-Br cations (hydrogen atoms are omitted for clarity), and b) crystal packing of 9-Br viewed along the crystallographic axis a. | ||
The stacks of 9-bromoacridizinium cations are separated by bromide-water hydrate assemblies (as seen in the previously described structures), and held together by C–H⋯Br and C–H⋯O interactions to form a two-dimensional molecular array. These molecular arrays further stack and are held together by van der Waals forces (Fig. 3b). The herein reported room-temperature crystal structure of 9-Br is identical to one determined at low temperature by Ihmels et al. (CCDC reference code: CIPQOH).1
Colourless single crystals corresponding to the bulk material were identified as 9-iodoacridizinium-bromide monohydrate (9-I-Br). The compound crystallises in the monoclinic P21/c space group with one 9-iodoacridizinium cation, one bromide anion and one water molecule in the asymmetric unit. The structure features pairs of acridizinium cations that stack infinitely in an anti-ht manner. The reaction centres of the cations within the pairs are separated by 3.81 Å, while the reaction centres of cations belonging to neighbouring acridizinium pairs are separated by 4.04 Å (Fig. 4a). Neighbouring stacks are, as in the other described structures, separated by hydrogen-bonded bromide-water hydrate assemblies. The crystal structure of 9-I-Br is sustained by C–H⋯O, C–H⋯Br and I⋯O and interactions (Fig. 4b).
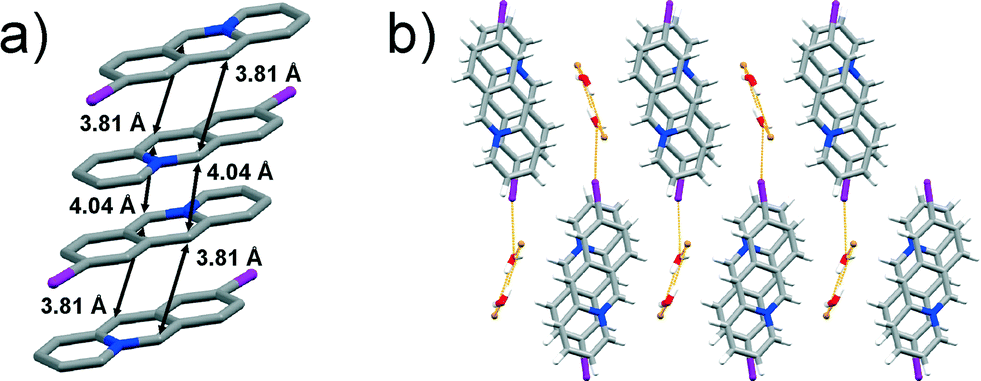 | ||
| Fig. 4 X-ray crystal structure of: a) an infinite stack of 9-I cations (hydrogen atoms are omitted for clarity), and b) crystal packing of 9-I-Br viewed along the crystallographic axis a. | ||
The brown single-crystalline blocks were identified as an anhydrous form of 9-iodoacridizinium triiodide, 9-I-I3. The solid crystallises in the triclinic space group P, with one 9-iodoacridizinium cation and one triiodide anion in the asymmetric unit. The cations stack pair-wise in an anti-ht fashion. The cation pairs are isolated by triiodide anions. The reaction centres within each cation pair are separated by 3.79 Å (Fig. 5a). The crystal structure of 9-I-I3 is held together by C–H⋯I, I⋯π and I⋯I interactions (Fig. 5b).
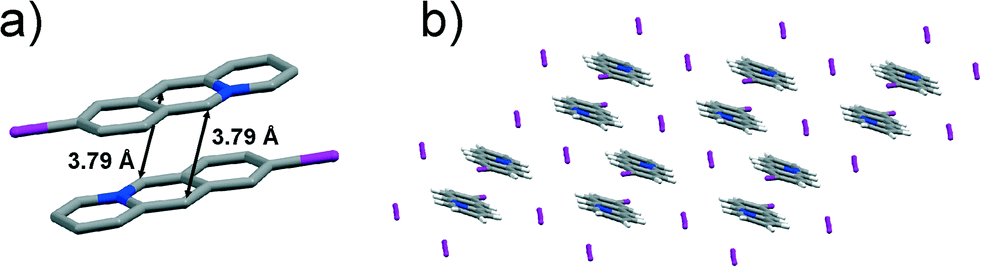 | ||
| Fig. 5 X-ray crystal structure of: a) an infinite stack of 9-I cations (hydrogen atoms are omitted for clarity), and b) crystal packing of 9-I-I3 viewed along the crystallographic plane (−101). | ||
space group with one 9-methylacridizinium cation, one bromide anion and one water molecule in the asymmetric unit. The cations pack pair-wise in an anti-ht manner forming infinite slanted stacks. The reaction centres of the cations within the pairs are separated by 3.69 Å, whereas the reaction centres of cations belonging to the neighbouring cation pairs are separated by 4.53 Å (Fig. 6a), i.e. a distance that is believed not to support a [4 + 4] photodimerisation in the solid state.15 The stacks of 9-CH3 cations are segregated by bromide-water hydrate assemblies, and held together by C–H⋯O and C–H⋯Br interactions (Fig. 6b) to form a two-dimensional molecular array. The molecular arrays stack and are held together by van der Waals forces, as seen in 9-Cl (Fig. 3b).
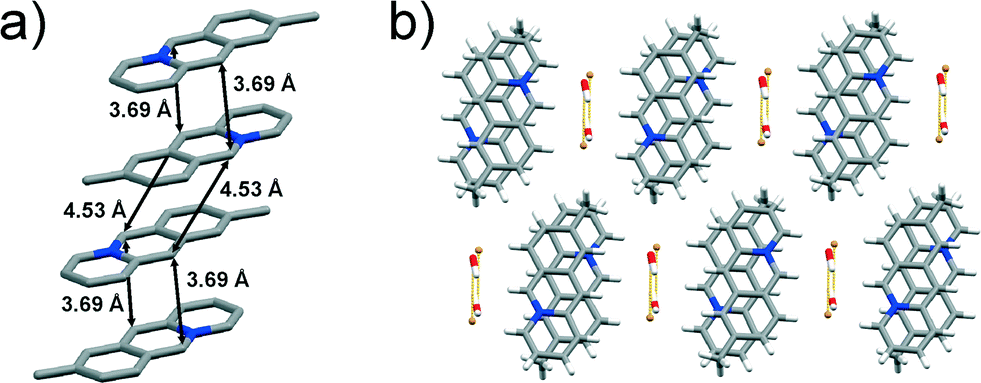 | ||
| Fig. 6 X-ray crystal structure of: a) an infinite stack of 9-CH3 cations (hydrogen atoms are omitted for clarity), and b) crystal packing of 9-CH3 viewed along the crystallographic axis a. | ||
The 9-C(CH3)3 monomer crystallises in the orthorhombic Pca21 space group with one acridizinium cation, one bromide anion and one water molecule in the asymmetric unit. The slightly twisted acridizinium cations are stacked in a syn-ht fashion (Fig. 7a) whereby neighbouring cations exhibit an angular offset (Fig. 7b). The reaction centres of the cations are separated by 3.42 and 3.95 Å and are thus conforming to the topochemical postulates. The stacks are separated by infinite zigzag-shaped bromide-water chains, which are held together by O–H⋯Br interactions (Fig. 7b). The chains interact with the acridizinium cations via C–H⋯Br and C–H⋯O interactions.
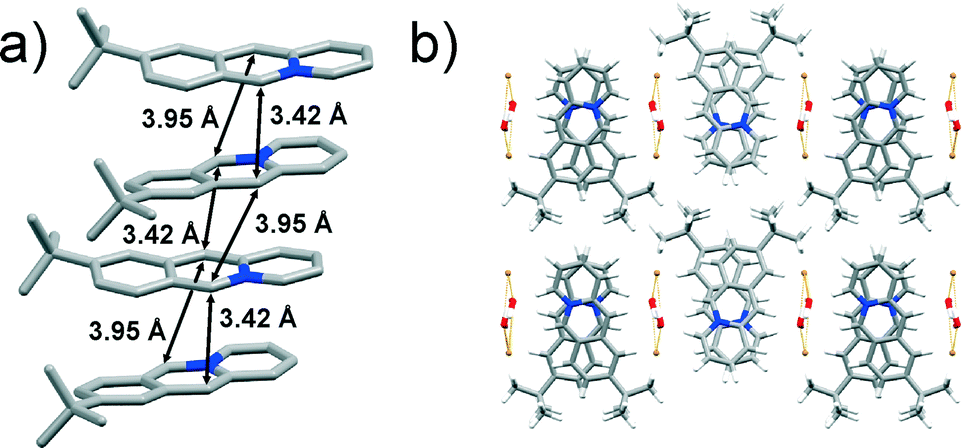 | ||
| Fig. 7 X-ray crystal structure of: a) an infinite stack of 9-C(CH3)3 cations (hydrogen atoms are omitted for clarity), and b) crystal packing of 9-C(CH3)3 viewed along the crystallographic axis c. | ||
2. Photoreactivity studies – yields and regioselectivity
Powder samples of salts 9-Cl, 9-Br and 9-CH3 formed anti-ht photodimers in almost quantitative yields after 5 hours of irradiation (9-Br: 95.7%, 9-Cl: 95.4%, 9-CH3: 97.4%; the overall yields of photoproducts are >99% in all three solids). Solids 9-F, 9-I-Br and 9-C(CH3)3 photoreacted to a lesser extent. In particular, UV irradiation of 9-I-Br for 24 hours yielded the anti-ht photodimer in only 8.6% yield, despite the fact that the alignment of the monomers conforms to Schmidt's topochemical postulate.15 The photoreaction of 9-I-Br also yielded other regioisomers as by-products with a 4.4% yield. Salt 9-C(CH3)3 generated a mixture of different regioisomers in 25.9% yield (including the anti-ht dimer). The 9-F salt was UV-irradiated for 24 hours to undergo the formation of a mixture of regioisomers in 58.4% yield (see ESI†).The high regioselectivity of the photodimerisation of 9-Br and 9-CH3 was not surprising considering that their respective cations are exclusively aligned in an anti-ht fashion (Fig. 3a and 6a). The high regioselectivity of the 9-Cl (Form II) photodimerisation (>95%), on the other hand, is unexpected bearing in mind that the cations in this structure are aligned in both an anti-ht and syn-ht manner, and that the separation distances of the reactive centres in the anti-ht and syn-ht cation pairs in Form II both conform to Schmidt's topochemical postulate. It is, therefore, reasonable to expect that the photodimerisation reaction of Form II would be regioselective than that of 9-Br and 9-CH3. The high regioselectivity of the Form II photoreaction suggested that Form II potentially transformed into its initially reported polymorph Form I (which is expected to yield exclusively the anti-ht isomer1) either before or while the solid was UV irradiated. This proposition was supported by the finding that an unreacted portion of the initially prepared Form II batch transformed into Form I after being in storage for more than six months (see ESI,† Fig. S21). Attempts to confirm whether the initially obtained Form II actually yields one regioisomer in >95% yield, or whether the solid undergoes a phase transition prior to or during the UV-irradiation, have initially failed, as Form II could not be reproduced. The inability to prepare a crystal form that has previously been synthesised (or to prepare elusive crystal forms16,17) is a frustrating issue that solid-state scientists occasionally face.18 Although the crystallization conditions for the consistent production of a ‘disappearing polymorph’ can be determined as a result of systematic and copious crystallization studies,19 our efforts to obtain the initially observed polymorph Form II through recrystallization were unsuccessful after numerous attempts. The initially observed polymorph Form II was finally obtained from a freshly synthesised batch of the 9-chloroacridizinium cation. We believe that the formation of Form II eventually succeeded through heteronuclear seeding20,21 by an unidentified material in the reaction mixture. After Form II was reproduced and structurally characterized, the solid was immediately subjected to UV irradiation for five hours to produce the anti-ht regioisomer in 96% yield. Ex situ analysis of a Form II bulk after only 30 min of UV irradiation confirmed that the majority of the unreacted bulk in the solid corresponds to Form II, which suggests that the Form II might not convert into Form I while being exposed to UV irradiation (see ESI,† Fig. S21).
Preliminary computational studies (see ESI†) point to the fact that the high regioselectivity of the photodimerisation reaction might be attributed to a more efficient molecular orbital overlap between the cations in the anti-ht pair, as compared to the cations in the syn-ht pair. More specifically, DFT calculations showed that the electron distribution in the LUMO of the anti-ht cation pair is localised in the area between two cations where carbon–carbon single bonds are expected to form upon UV irradiation. The LUMO of the syn-ht pair, on the other hand, does not feature any electron distribution that could correspond to the formation of the carbon–carbon single bonds, thus indicating that the [4 + 4] photodimerisation is not likely to occur between the cations of the syn-ht pair (Fig. 8).
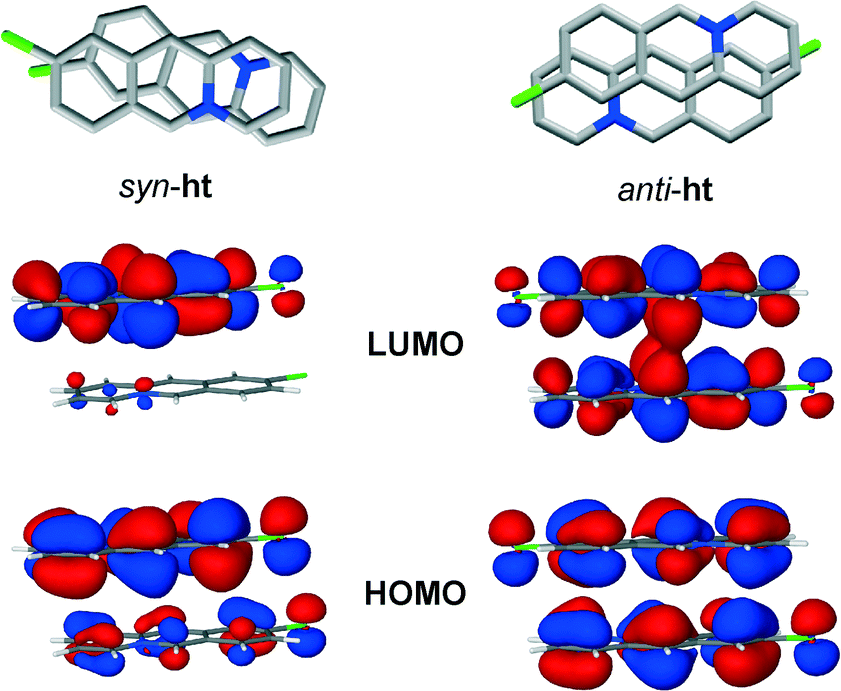 | ||
| Fig. 8 HOMO and LUMO orbitals of the syn-ht (left) and anti-ht (right) pairs of 9-chloroacridizinium cations in 9-Cl. | ||
The results of the photoreactivity studies are summarised in Table 1. The NMR spectra of the acridizinium-based solids are shown in the ESI† document. The assignment of the 1H NMR signals to the dominant regioisomers of the photodimers is detailed in the ESI.†
| Salt | d(C⋯C)/Å | Arrangements | Overall yield (irradiation time) | Main isomer (yield) |
|---|---|---|---|---|
| 9-F | 3.99 | anti-hh, anti-ht | 58.4% (24 h) | anti-ht (18.2%) |
| 9-Cl | 3.63, 3.83 | anti-ht, syn-ht | >99.0% (5 h) | anti-ht (96.2%) |
| 9-Br | 3.78, 3.95 | anti-ht | >99.0% (5 h) | anti-ht (95.7%) |
| 9-I-Br | 3.81, 4.04 | anti-ht | 13.0% (24 h) | anti-ht (8.6%) |
| 9-CH3 | 3.69, 4.53 | anti-ht | >99.0% (5 h) | anti-ht (97.4%) |
| 9-C(CH3)3 | 3.42, 3.95 | syn-ht | 25.9% (5 h) | syn-ht (17.0%) |
3. Photoreactivity studies – reaction rates
Photoreactivity studies were also carried out using a UV source (366 nm) of significantly lower intensity (namely 10 W) to study the relative rates of the [4 + 4] photodimerisation reaction involving 9-Cl, 9-Br and 9-CH3. Salts 9-F, 9-I-Br and 9-C(CH3)3 were not considered in this part of the study owing to the low yields and the low regioselectivity of their photodimerisation reaction. The relative conversion rates were obtained by measuring the integrals of 1H NMR signals corresponding to monomers and dimers of the studied acridizinium cations (see ESI†).Irradiation of the salts was again shown to result in the formation of the anti-ht regioisomer. Fig. 9 shows that salt 9-Cl undergoes photodimerisation with the fastest reaction rate in nearly quantitative yields (i.e. 93%) after low-intensity UV irradiation for 60 days. The reaction of salts 9-Br proceeded somewhat slower in the same timeframe to react up to 87% yield, whereas 9-CH3 reacted at the lowest rate up to yield 67%. The relative conversion rates for solids are shown in Fig. 9. The low-intensity UV-irradiation of the three studied solids surprisingly revealed that the regioselectivity of the dimerisation had diminished. A two-fold increase in the formation of the syn-ht 9-Cl and 9-Br photodimers was observed, while a four-fold increase of the syn-ht photodimer formation was observed in the case of 9-CH3. The increased formation of the syn-ht photodimer during the low-intensity UV-irradiation studies might be attributed to the low thermal stability of the solids, which were found to dehydrate at relatively low temperatures (i.e. 40–98 °C, Table 2). The low thermal stability of the salts is thought to lead to structural rearrangement of the acridizinium cations in their respective crystal lattices, which feasibly enables the increased formation of the syn-ht isomers. In addition, long UV-irradiation times might lead to partial melting of the solids, which facilitates the alignment of the acridizinium cations favouring the formation of other regioisomers.
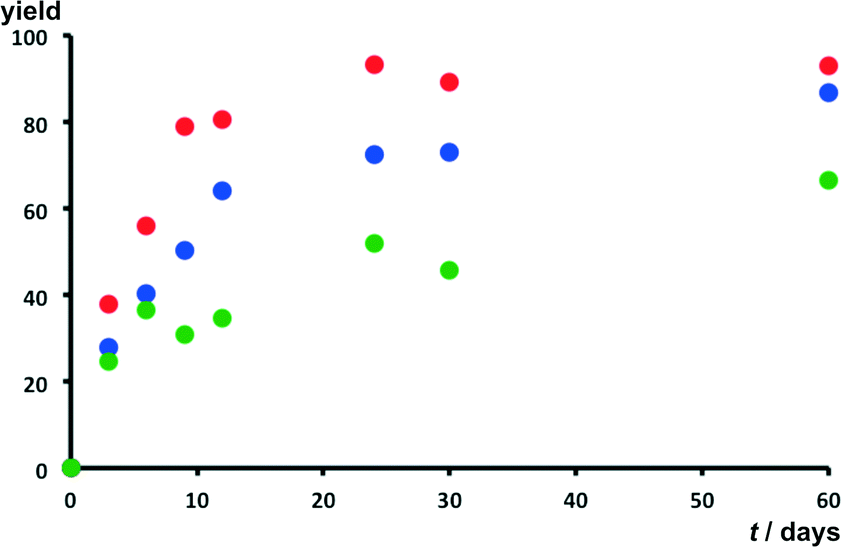 | ||
| Fig. 9 Relative rates of conversions for salts 9-Cl (red), 9-Br (blue), and 9-CH3 (green) using a low intensity single wavelength UV lamp. | ||
| Salt | t d/°C | Determined mass loss/% | Calculated mass loss/% | t m/°C |
|---|---|---|---|---|
| 9-F | 45 | 4.20 | 6.00 | 230 |
| 9-Cl | 40 | 4.33 | 5.76 | 249 |
| 9-Br | 98 | 5.17 | 5.04 | 270 |
| 9-I-Br | 67 | 3.70 | 4.46 | 257 |
| 9-CH3 | 55 | 6.15 | 6.16 | 179 |
| 9-C(CH3)3 | 45 | 4.65 | 5.38 | 145 |
In addition to the observed differences in reaction yields, reaction rates and thermal behavior, we note that the acridizinium salts exhibit distinct hydration behavior, as well as various degrees of crystallinity upon exposure to UV irradiation. Studies are underway to explain the complex relationship between the solid-state reactivity (i.e. reaction yields and rates), the reaction conditions (e.g. humidity) and the molecular dynamics of the acridizinium cations within the solid during UV irradiation.
Conclusions
A series of 9-X-acridizinium bromide salts (where X = F, Cl, Br, I, CH3, C(CH3)3) (9-X) was synthesised and characterised using crystallographic, thermal, spectroscopic and computational methods. The structural studies revealed that the salts mainly pack in an anti-ht fashion in their crystal structures. Exceptions to such packing behaviour are 9-F, 9-Cl and 9-C(CH3)3, wherein the cations pack in either an anti-ht and anti-hh fashion (9-F), a syn-ht and anti-ht manner (9-Cl), or a syn-ht fashion (9-C(CH3)3). All studied bromide salts were shown to crystallise as hydrates.All structurally characterised acridizinium salts were found to conform to Schmidt's topochemical postulate. Photoreactivity studies showed that the anti-ht photodimer was formed in 97% yield for 9-CH3 and 96% yield for 9-Cl and 9-Br. The overall photodimerisation yields (i.e. the extent of the formation of all possible regioisomers) for these salts was >99%. The photodimerisation reactions of salts 9-I-Br, 9-F and 9-C(CH3)3 were less regioselective and also yielded less photoproducts. Only one crystal structure of a photodimer, namely that of the anti-ht 9-C(CH3)3 photodimer, was determined.
Thermal investigations showed that all studied acridizinium salts exhibit similar thermal stabilities with respect to dehydration. Salt 9-Cl was found to have the lowest stability towards dehydration, while the 9-Br salt exhibited the highest stability.
Acknowledgements
DKB acknowledges the Royal Society for a Newton International Fellowship and University College London for a UCL Excellence Fellowship. DKB and WJ thank the Isaac Newton Trust (Trinity College, University of Cambridge) for funding. EPSRC is thanked for a studentship to MA. A part of this work was performed using the Darwin Supercomputer of the University of Cambridge High Performance Computing Service (http://www.hpc.cam.ac.uk/), provided by Dell Inc. using Strategic Research Infrastructure Funding from the Higher Education Funding Council for England and funding from the Science and Technology Facilities Council.Notes and references
- H. Ihmels, D. Leusser, M. Pfeiffer and D. Stalke, J. Org. Chem., 1999, 64, 5715–5718 CrossRef CAS.
- H. Ihmels and J. Luo, J. Photochem. Photobiol., A, 2008, 3–9 CrossRef CAS PubMed.
- T. Wolff, C. Lehnberger and D. Scheller, Heterocycles, 1997, 45, 2033–2039 CrossRef.
- D. Kuckling, I. G. Ivanova, H.-J. P. Adler and T. Wolff, Polymer, 2002, 43, 1813–1820 CrossRef CAS.
- A. Granzhan and H. Ihmels, Org. Lett., 2005, 7, 5119–5122 CrossRef CAS PubMed.
- A. Bergen, A. Granzhan and H. Ihmels, Photochem. Photobiol. Sci., 2008, 7, 405–407 CAS.
- R. Jagt, M. Kheibari and M. Nitz, Dyes Pigm., 2009, 81, 161–165 CrossRef CAS PubMed.
- M. Tian and H. Ihmels, Chem. Commun., 2009, 3175–3177 RSC.
- B. Lohse, R. Vestberg, M. T. Ivanov, S. Hvilsted, R. H. Berg, C. J. Hawker and P. S. Ramanujam, Chem. Mater., 2008, 20, 6715–6720 CrossRef CAS.
- H. Ihmels, B. Engels, K. Faulhaber and C. Lennartz, Chem. – Eur. J., 2000, 6, 2854–2864 CrossRef CAS.
- C. Bradsher, L. Beavers and J. Jones, J. Org. Chem., 1957, 22, 1740–1741 CrossRef CAS.
- H. Ihmels, D. Leusser, M. Pfeiffer and D. Stalke, Mol. Cryst. Liq. Cryst., 2001, 356, 433–441 CrossRef CAS.
- W.-N. Wang and W. Jones, Tetrahedron, 1987, 43, 1273–1279 CrossRef CAS.
- W.-N. Wang and W. Jones, Mol. Cryst. Liq. Cryst., 1994, 242, 227–240 CrossRef.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- P. Naumov, N. Yasuda, W. M. Rabeh and J. Bernstein, Chem. Commun., 2013, 49, 1948–1950 RSC.
- D.-K. Bučar, G. M. Day, I. Halasz, G. G. Z. Zhang, J. R. G. Sander, D. G. Reid, L. R. MacGillivray, M. J. Duer and W. Jones, Chem. Sci., 2013, 4, 4417–4425 RSC.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- J.-O. Henck, J. Bernstein, A. Ellern and R. Boese, J. Am. Chem. Soc., 2001, 123, 1834–1841 CrossRef CAS PubMed.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS.
- A. D. Bond, K. A. Solanko, S. Parsons, S. Redder and R. Boese, CrystEngComm, 2011, 13, 399–401 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details relating to crystallographic, spectroscopic and thermal studies. CCDC 975287–975293 and 992871. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce01622j |
| This journal is © The Royal Society of Chemistry 2014 |