 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical gap in herringbone and π-stacked crystals of [1]benzothieno[3,2-b]benzothiophene and its brominated derivative†
Vijay S.
Vyas
a,
Rico
Gutzler
a,
Jürgen
Nuss
a,
Klaus
Kern
ab and
Bettina V.
Lotsch
*acd
aMax Planck Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany. E-mail: b.lotsch@fkf.mpg.de
bInstitut de Physique de la Matière Condensée, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland
cDepartment of Chemistry, Ludwig-Maximilans-Universität München, Butenandtstr. 5-13, 81377 München, Germany
dNanosystems Initiative Munich (NIM) and Center for Nanoscience, Schellingstr. 4, 80799 München, Germany
First published on 14th May 2014
Abstract
The optical gap of the organic semiconductor [1]benzothieno[3,2-b]benzothiophene and its 2,7-dibrominated analogue is measured in solution and in the crystalline state by means of UV-vis and emission spectroscopy. Bromination leads to a change in molecular packing from herringbone to π-stacked, resulting in a marked shift in the absorption and emission spectra which is found to be in accordance with TDDFT calculations.
Organic semiconductors (OSCs) have found wide use in organic field effect transistors,1,2 organic solar cells,3,4 organic light-emitting diodes,5–7etc. The performance of these devices crucially depends on the solid state packing of the OSCs1,8,9 and continuing efforts are underway to understand the effect of molecular structure on molecular packing in (hetero)acenes.1,2 Rational molecular design allows the tuning of intermolecular interactions, leading to different crystal packing motifs, and is used to optimize optical and electronic properties in organic semiconductor devices.10 Notably, halogenation of OSCs not only offers a viable pathway to enforce stacked crystal packing in OSCs that otherwise crystallize in the herringbone arrangement, it also increases environmental stability through larger ionization potentials and reduces the HOMO/LUMO gap of the molecule.8 The effect of herringbone vs. π-stacked crystal structure is a topic of ongoing research, as the packing concomitantly affects electronic and optical properties of the crystal.1,8,10–12 The preferred assembly of halogenated OSCs in π-stacked crystals is used here to investigate the effect of molecular packing on the electronic structure in the crystalline state. The known OSC [1]benzothieno[3,2-b]benzothiophene (BTBT; 1, Fig. 1a) has been reported as a prototypical p-type organic semiconductor, as the 2,7-dialkyl substituted BTBTs show high charge carrier mobility and good air stability.13,14 However, owing to the absence of a viable direct synthetic route to functionalize BTBT,15 its functionalization is achieved during the synthesis of the BTBT core, which makes the overall synthesis tedious.16,17 Herein, we overcome this limitation and report a one-step direct bromination of BTBT to form 2,7-dibromo[1]benzothieno[3,2-b]benzothiophene (2, Fig. 1b) and show that the bromination of 1 significantly alters the packing in the crystal, resulting in a transition from a herringbone to a π-stacked structure. Optical spectra in solution and in the solid-state (UV-vis and emission) of 1 and 2 are measured to compare the optical gap of monomers and in the crystal.
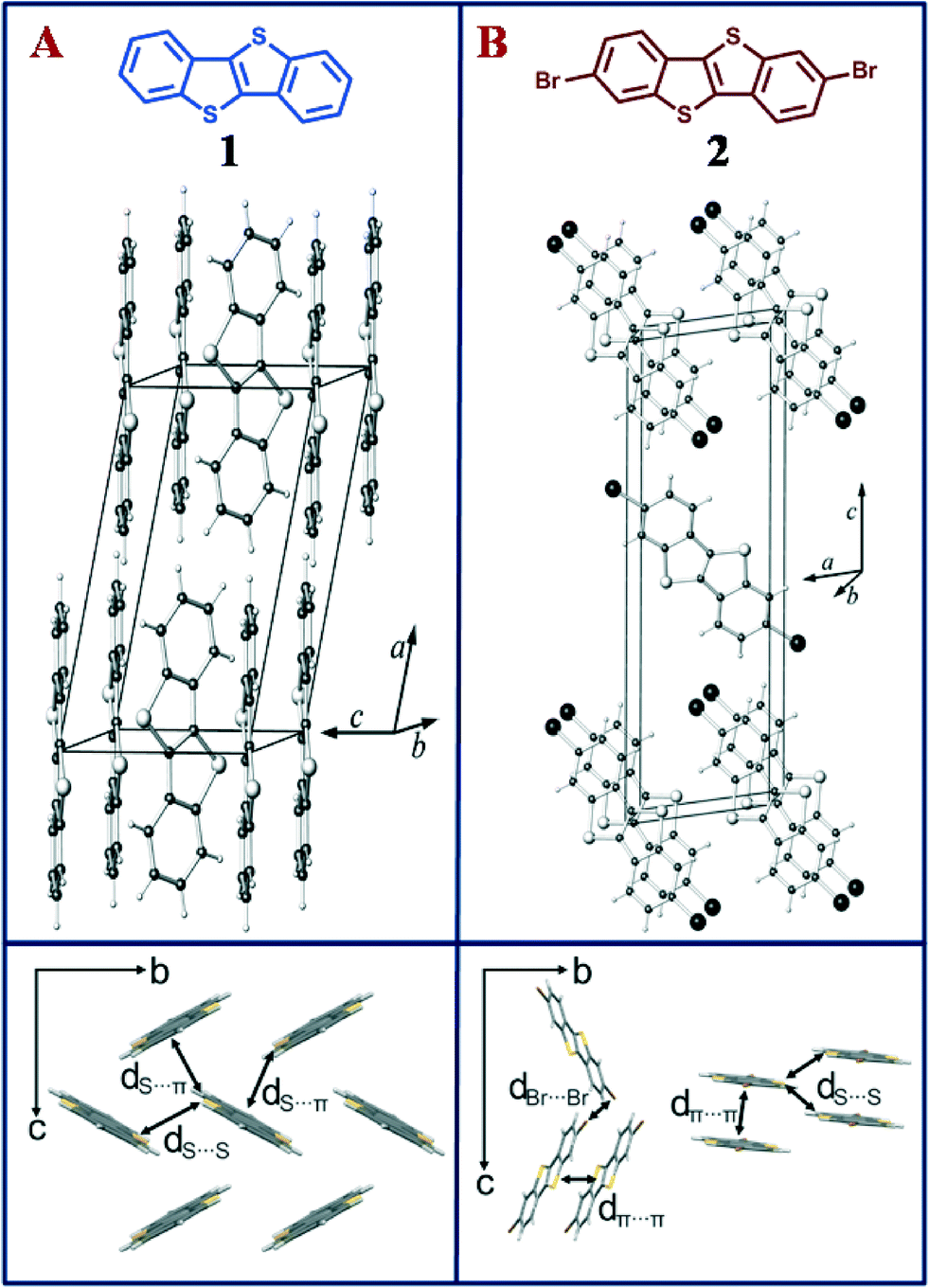 | ||
| Fig. 1 (Top) Perspective view of the crystal structure of (a) C12H8S2 (1) and (b) of C12H6S2Br2 (2), with unit cell. (Bottom) Close contacts labelled with d between adjacent molecules in the crystals (packing structure of 1 perpendicular to bc plane and packing structure of 2 perpendicular to bc plane and arbitrary direction). | ||
BTBT 1 was functionalized using bromine under ambient conditions to prepare the brominated BTBT 2 (see ESI†). In order to gain insight into the impact of halogenation on the intermolecular packing, single crystals of 2 were obtained by slow evaporation of its solution in chloroform. The resulting needle shaped crystals were studied by single-crystal X-ray diffraction. Interestingly, while single crystals of 2,7-substituted BTBT derivatives have been reported,18 we did not find any literature evidence of the crystal structure of the parent BTBT. Therefore, single crystals of 1 were obtained from a solution of dichloromethane and methanol. X-ray diffraction analyses indicate that 1 crystallizes in the well-known herringbone arrangement that many OSCs adopt (Fig. 1a). The brominated compound 2 crystallizes in a π-stacked molecular arrangement with an interplanar distance of ~3.5 Å (Fig. 1b), as has previously been observed in similar flat OSCs.8 Herringbone and coplanar crystal packing go hand-in-hand with a changing number of close contacts between atoms of adjacent molecules (14 contacts in 1 and 10 contacts per molecule in 2), which include two S⋯S and four S⋯C(sp2) short contacts per molecule in 1, and two S⋯S and four Br⋯Br short contacts per molecule in 2 (see Fig. 1 and ESI† for full list of close contacts).
Absorption spectra of 1 and 2 in dichloromethane were recorded under conditions of identical concentration (10−5 M) and temperature (22 °C) and are compiled in Fig. 2a. 2 shows structured absorption bands that shift bathochromically (λedge = 3.64 eV in 1 to 3.58 eV in 2) due to the presence of halogens (see Table 1). This trend is in accordance with time-dependent density functional theory (TDDFT) calculations at the M06-2X/def2-TZVP level of theory (Orca 3.0 (ref. 19)), which yield an S0–S1 transition energy of 4.44 eV for 1 and 4.34 eV for 2. TDDFT with the M06-2X functional is reported to produce reasonable results for excitation energies.20 In contrast, the absorption spectra of the crystalline powder recorded in diffuse reflectance mode (dashed lines, Fig. 2b) shows a marginal red shift in 2 (Eedge = 3.23 eV) when compared with 1 (Eedge = 3.25 eV). A comparison of solution and solid-state absorption spectrum reflects the reduction of the optical gap upon crystallization for both compounds. However, in the herringbone structure of 1, the gap is reduced by 0.39 eV (from 3.64 to 3.25 eV), while in π-stacked 2, the gap is reduced by 0.35 eV (from 3.58 to 3.23 eV). This trend is opposite to what is observed in emission spectra (λexc = 300 nm) of 1 and 2 recorded under dilute conditions (10−6 M solution in dichloromethane) at room temperature (22 °C). As shown in Fig. 2b, the structured emission bands at 341 and 356 nm (λmax) in 1 are replaced by one broad spectrum of 2 at 363 nm (λmax). The emission spectra of 1 and 2 recorded in the crystalline state exhibited marked bathochromic shifts as similarly observed in the UV-vis spectra. Thus, the solid state emission maximum of 2 is observed at 407 nm, thereby exhibiting a red shift of 44 nm (0.37 eV) in comparison to the emission maximum in solution. The crystalline state emission spectrum of 1 was observed at 376 nm and hence shows a smaller red shift of only 20 nm (0.19 eV) from that in solution.
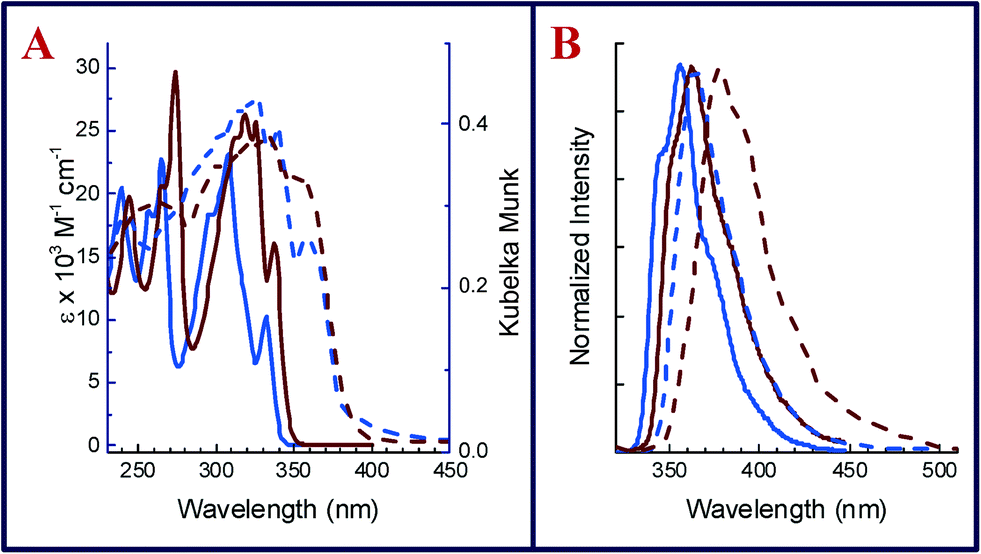 | ||
| Fig. 2 (a) Absorption spectra of 1 (blue) and 2 (brown) recorded as dichloromethane solutions (solid lines) and in the crystalline state (dashed lines). (b) Emission spectra of 1 (blue) and 2 (brown) recorded as dichloromethane solutions (solid lines) and in crystalline state (dashed lines). | ||
To validate the influence of molecular packing on excitation energies extending on the commonly discussed dimer model,11,21,22 time-dependent DFT calculations (M06-2X/def2-TZVP) were carried out on clusters of different sizes whose geometry was taken from the experimental crystal data of 1 and 2. TDDFT gives insight into relative excitation energies and transition moments of molecular clusters even though the absolute values of these quantities do not necessarily coincide with experimental values. Fig. 3 shows the oscillator strength of the lowest lying excitation energies for hexamers and the monomers 1 and 2. The cluster geometries were chosen by taking into account different close contacts in the crystal structure. For 1, the clusters were defined by the crystallographic axes c and b, respectively, both involving S⋯S and S⋯C contacts (Fig. 3b and c). In case of 2, cluster (f) involves Br⋯Br contacts between stacks (also Fig. 1. bottom right), while cluster (g) is a stack of six molecules. Similarly, cluster (h) includes short S⋯S contacts between two π-stacks.
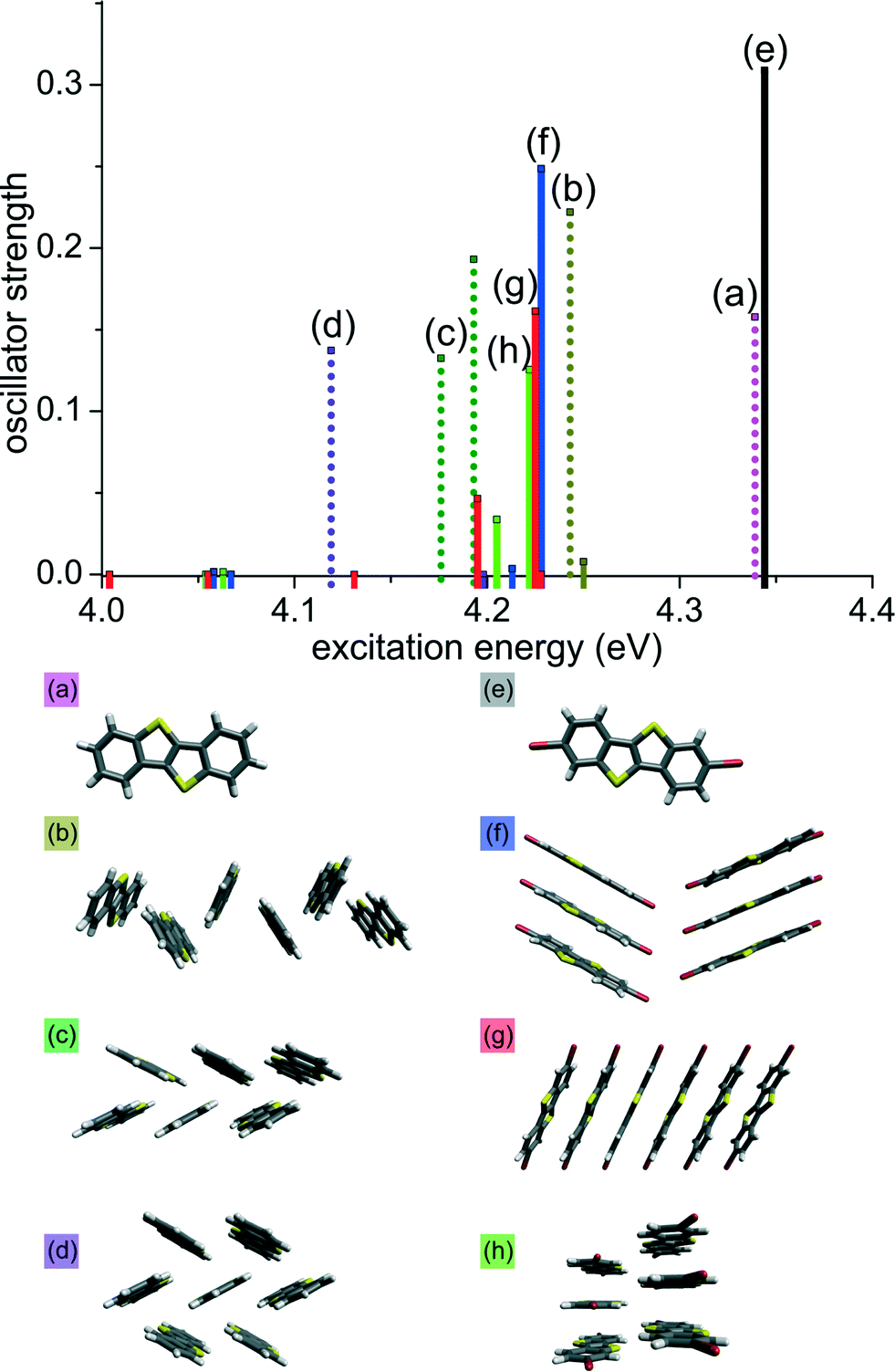 | ||
| Fig. 3 TDDFT excitation energies of different clusters of 1 (dotted bars, (a) to (d)) and 2 (solid bars, (e) to (h)). Geometric structures are taken from the experimental crystal data. | ||
As shown in Fig. 3, the stacked arrangement of 2 has the lowest excitation energy just above 4.0 eV (red solid lines; g) while its oscillator strength is negligible. For hexamers of 2, the first transition with appreciable oscillator strength is found at ~4.20 eV (f–h; blue, red and green solid lines), that corresponds to the fourth lowest transition (S0 to S4). All lower-lying transitions have small oscillator strengths due to the cancellation of transition dipole moments within the stack. On the other hand, for the typical arrangement of molecules in the herringbone structure (c; green dotted line) we observe a transition at 4.18 eV, which is lower when compared with the stacked clusters (f–h). Calculations on a heptamer cluster of 1 (violet dotted line d) results in even lower transition energy of 4.12 eV.‡ Thus, in the herringbone arrangement, the transition with largest oscillator strength is the one with lowest excitation energy due to constructive interactions of transition dipole moments. We infer that this trend extends to the crystals and hence small transition dipole moments for the lowest excitation energies in the stacked arrangement in 2 are responsible for the smaller observed shift in the solid state absorption spectra when compared to 1, in which dipole moments do not cancel each other. The calculations also explain the observance of smaller emission energy for 2, as compared to 1 in the emission spectra, asserting internal conversion (Sn to S1) in 2 before emission. The smaller emission energy of 2 can be explained by invoking Kasha's rule: internal conversion from Sn to S1 in 2 occurs before emission due to vibronic coupling.23 A full list of excitation energies and dipole moments of all clusters is given in ESI.†
Conclusions
The observance of a large bathochromic shift in the solid-state vs. solution absorption spectra of the BTBT-type organic semiconductors 1 and 2 is a crystal packing effect, in which the interaction of molecular orbitals leads to a reduction of the optical gap for large cluster sizes (cf.Fig. 3). The herringbone arrangement of 1 allows for efficient contraction of the optical gap due to constructive interactions of transition dipole moments. In the stacked crystal of 2, however, the lowest excitation energies are accompanied by vanishing transition dipole moments, which do not significantly contribute to the experimental absorption spectra. The generation of excitons thus requires less energy in the parent compound BTBT compared to its brominated counterpart. How free electron–hole pairs in herringbone vs. stacked crystals behave in transport experiments is yet another interesting topic, which shall be addressed in future experiments.Notes and references
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- M. O'Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566–584 CrossRef CAS PubMed.
- H. Ma, H. L. Yip, F. Huang and A. K. Y. Jen, Adv. Funct. Mater., 2010, 20, 1371–1388 CrossRef CAS.
- A. Y. Amin, A. Khassanov, K. Reuter, T. Meyer-Friedrichsen and M. Halik, J. Am. Chem. Soc., 2012, 134, 16548–16550 CrossRef CAS PubMed.
- M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- G. Heimel, I. Salzmann, S. Duhm and N. Koch, Chem. Mater., 2011, 23, 359–377 CrossRef CAS.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS.
- J. Cornil, D. Beljonne, J. P. Calbert and J. L. Brédas, Adv. Mater., 2001, 13, 1053–1067 CrossRef CAS.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- C. Liu, T. Minari, X. Lu, A. Kumatani, K. Takimiya and K. Tsukagoshi, Adv. Mater., 2011, 23, 523–526 CrossRef CAS PubMed.
- K. Takimiya, H. Ebata, K. Sakamoto, T. Izawa, T. Otsubo and Y. Kunugi, J. Am. Chem. Soc., 2006, 128, 12604–12605 CrossRef CAS PubMed.
- B. Košata, V. Kozmík and J. Svoboda, Collect. Czech. Chem. Commun., 2002, 67, 645–664 CrossRef.
- M.-C. Um, J. Kwak, J.-P. Hong, J. Kang, D. Y. Yoon, S. H. Lee, C. Lee and J.-I. Hong, J. Mater. Chem., 2008, 18, 4698–4703 RSC.
- C. Ruzié, J. Karpinska, A. R. Kennedy and Y. H. Geerts, J. Org. Chem., 2013, 78, 7741–7748 CrossRef PubMed.
- T. Izawa, E. Miyazaki and K. Takimiya, Adv. Mater., 2008, 20, 3388–3392 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- D. Jacquemin, E. A. Perpète, I. Ciofini, C. Adamo, R. Valero, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2071–2085 CrossRef CAS.
- J. Cornil, D. A. dos Santos, X. Crispin, R. Silbey and J. L. Brédas, J. Am. Chem. Soc., 1998, 120, 1289–1299 CrossRef CAS.
- J. Gierschner, M. Ehni, H. J. Egelhaaf, B. Milián Medina, D. Beljonne, H. Benmansour and G. C. Bazan, J. Chem. Phys., 2005, 123, 144914 CrossRef PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of 2, crystal description of 1 and 2. CCDC 975935 and 975934 for structures 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00752b |
| ‡ In the π-stacked heptamer, the lowest excitation energy is 4.00 eV with negligible oscillator strength. The first transition with appreciable oscillator strength is found at 4.20 eV, which is the same value as in the π-stacked hexamer and larger than in the herringbone heptamer (d). |
| This journal is © The Royal Society of Chemistry 2014 |