 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Soft-oxometalates beyond crystalline polyoxometalates: formation, structure and properties
Soumyajit
Roy
Eco-Friendly Applied Materials Laboratory (EFAML), Materials Science Centre, Department of Chemical Sciences, Indian Institute of Science Education and Research-Kolkata (IISER-Kolkata), Mohanpur Campus, Mohanpur-741252, India. E-mail: s.roy@iiserkol.ac.in; roy.soumyajit@googlemail.com
First published on 24th March 2014
Abstract
Polyoxometalates (POMs), as the name suggests, are single molecular charged or uncharged clusters comprising many metal centres and oxygen atoms. They are crystalline. On the other hand, recently, a class of macroionic, superstructured assemblies of POMs has been found which is reminiscent of soft matter and has been proposed to be called Softoxometalates (SOMs). This highlight gives a personal account of our work with SOMs. Starting with a brief background and history of SOMs, we explore the reasons for their formation. Thereafter, we discuss the charge regulation mechanism for the stabilization of SOMs. A few case studies for the directed formation of large surface area, mesoscopic SOMs are also discussed. Thereafter, we discuss the effects of sound and light on SOMs. This highlight finally ends with a discussion on self-assembled pattern formation with oxometalates.
 Soumyajit Roy | Soumyajit Roy is at present an Assistant Professor (2011–till date) at the Indian Institute of Science Education & Research, (IISER) Kolkata, India. Soumyajit completed his Ph.D. with a summa cum laude from University of Bielefeld, Germany with Prof. Achim Müller (2005). He later moved to the University of Utrecht's van't Hoff Laboratory for Physical & Colloid Chemistry, in the Netherlands, to work with Prof. Willem Kegel as a post-doctoral fellow (2005–2007). In 2007, he moved to BASF-ISIS, Strasbourg, France to work as a researcher. Before joining IISER-Kolkata, he was a Professor at Changshu Institute of Technology, Jiangsu, China. |
1. Introduction
The chemistry of crystalline metal oxides is a field of diverse research interests.1 In recent times, a class of metal–oxide based clusters, called polyoxometalates (POMs), has gained significant interest due to their applications in catalysis, and in materials science in general.2–8 Polyoxometalates, as the name suggests, comprise many metals, and many oxygen atoms and are usually charged and crystalline. Single molecules of POMs are usually large, of the order of 1–3 nanometers, and dissolve in polar solvents such as water and exist as discrete clusters in the solvent. Very recently, it has been discovered that such single molecules of POMs self-assemble to form large entities with soft-matter properties.9–17 Such self-assembled entities form a dispersed phase in a dispersing phase (solvent, usually water). They also scatter light and have a diffuse boundary. Hence, following de Gennes definition of soft-matter,18 these soft-state oxometalates have been proposed to be called soft-oxometalates (SOMs) (Fig. 1).19 This nomenclature facilitates the systematization and understanding of a burgeoning body of literature from the view point of soft-matter or colloids. By applying the existing knowledge of soft-matter, it would be possible to understand and predict the behaviour of SOMs. It is now perhaps apt to mention how we can understand the behaviour of SOMs that are beyond the crystalline regime of POMs. SOMs, for instance, are not point charges. Hence, the Debye–Hückel approximation does not hold for SOMs.20 On the other hand, their behaviour can be understood by the application of principles of short range repulsion and long range attraction, as proposed by Derjaguin–Landau–Verwey–Overbeek's (DLVO) theory of stabilization of colloids.21 The theory states that soft-states of matter, like colloids for instance, are stabilized by the local primary and/or the secondary minima created due to the competition of repulsive electrostatic interactions and attractive van der Waals interactions in colloids. Hence, it is reasonable to believe that SOMs should be charge stabilized dispersions of oxometalates. In addition to the DLVO theory, there is another modality of stabilization that can be envisaged with SOMs: the depletion interaction. The depletion interaction is an entropic stabilization of a colloidal system comprising a colloid–polymer mixture.22 Hence, SOMs that are comprised of polymers would be stabilized along the lines of the depletion interaction. There is another view point for understanding the existence of SOMs. We know that in soft-matter physics, colloids can be considered as soft-atoms which interact in a density dependent manner to give rise to various states in soft-matter. Along these lines, we can envisage a number density-dependent phase continuum of soft oxometalates. In this continuum, at the lower extreme lie the liquid or gas-like SOMs, where the volume fraction is ϕ < 0.2 or alike. In the higher extreme of that continuum lies the crystalline territory of oxometalates or crystalline POMs (for ϕ > 0.5 or alike). From such a perspective, it is possible to understand crystallization in POMs as a phenomenon of the assembly of single molecular colloidal oxometalate units (or soft-atoms23 of soft-matter) governed by a certain potential (like a Baxter-type potential).24 Although such a view point is convenient to understand how POMs crystallize, it is challenging to answer why the oxometalate units self-assemble to form SOMs in lower densities or volume fractions? What drives the formation of vesicle-like SOMs from symmetric single molecules of POMs? We address this question in the next section. To answer this question, we need to take into account the crystal structure of POMs. | ||
| Fig. 1 An overview of the POMs forming SOMs. Adapted with permission from ref. 20. Copyright, ACS, Washington, 2010. | ||
2. Understanding the formation of SOMs from the crystal structures of POMs
An intriguing aspect of the self-assembly of POMs into SOMs at rather low volume fractions is what drives such assembly from already symmetric POMs to higher order sheet-like structures in SOMs that fold to form giant vesicle-like structures. This is so because, although the spontaneous assembly of species like surfactants, lipids, and semiconductor nanoparticles into higher order structures is ubiquitous in nature, such spontaneous assembly is attributed to their anisotropic shapes and in the case of surfactants, to their amphiphilicity.25–28 Likewise, in the case of semiconductors like cadmium telluride nanoparticles, their truncated tetragonal shape coupled with hydrophobic and dipolar interactions lead to the formation of sheet-like nanocrystals in CdTe nanocrystals. Hence, in all the above cases, the tendency to form higher order structures can be traced to the anisotropic shapes of their constituent units and/or the directional nature of the interactions among those units. So the obvious question is: what drives the formation of higher order structures in SOMs from POMs? To answer this question, a closer look into the crystal structure of the starting POMs reveals that indeed there is an intrinsic anisotropy in the mode of packing of the clusters, like that of [Mo72Fe30] POMs29 in the crystal lattice. This anisotropy stems from the directional nature of the hydrogen bonding between Fe–O⋯H–O–Fe linkages in the crystals.29 Can such anisotropy in crystals hold the key to understanding the formation of SOMs from the corresponding [Mo72Fe30] POMs? A simulation study was performed with this end in view, where a patchy spherical model was proposed to understand the formation of SOMs.30 Since each POM unit of the SOM has 30 Fe–O sites, a model with 30 patches was proposed where each particle interacts with another particle by a single patch (Fig. 2). Each such patch size was chosen such that the transition temperature corresponds to the energy scale of the hydrogen bonding (∼5–10 kT). It was also obvious from this model that a narrow patch would induce self-assembly at a higher attraction strength, while a wide patch would lead to overlap of nearby patches, destroying the point symmetry of the POM cluster or, as in this case, that of the sphere with the patches. The patches take care of the hydrogen bonding interaction and are represented in the model as an orientation dependent interaction. The attractive interaction between the particles is represented by a square well potential in combination with the above orientation dependent interaction. Furthermore, a reduced temperature is defined as T′ = kT/ε, and the simulation is performed with 30 or 50 patchy particles in a cubic box of significantly higher length using periodic boundary conditions.30 Initially, random configurations are used at a higher reduced temperature (T′ = 0.2) and slowly the temperature is reduced using a well-defined cooling scheme. At a higher reduced temperature, a gas-like configuration of the patchy spheres is observed with no order. As the temperature is reduced to T′ = 0.112, corresponding to ε = 8.9 kT, it is observed that the patchy particles spontaneously self-assemble into a sheet-like structure for the SOM to be formed (Fig. 3). These sheet-like structures are rather stable and it is envisaged that the entire 3D crystal is slowly formed by the self-assembly of these 2D sheets in proper orientations, formed by the patchy particles or the POM units. This structure, predicted by the patchy model, is furthermore consistent with the rhombohedral crystal structure observed for [Mo72Fe30], where 2D sheets stack perpendicular to the C2 axis, as is exactly observed to be the case from the simulation studies. When subjected to suitable synthetic conditions, these sheets furthermore fold to form SOM spheres of [Mo72Fe30]. In the simulation study, more such 2D sheets were found to be formed when a repulsive screened Coulomb interaction was added between the patchy particles. The reason for such an addition was that experimentally, for the stabilization of SOM vesicles, it was said that the constituent POMs should carry some charge. Being charged, these sheets repel each other to form crystals whereas they have ample time to fold into SOM vesicles. Such folding is also energetically favoured as it reduces the number of dangling bonds along the edges. The above explanation in short explains how SOM vesicle formation can be understood by taking a closer look at the crystal structure of the starting POM and validating the formation by a patchy model, where the patches are reminiscent of directional hydrogen bonding. We now ask the question, how are these SOM vesicles stabilized? Can we understand their stabilization from simple physical principles?31 In the next section we answer this question.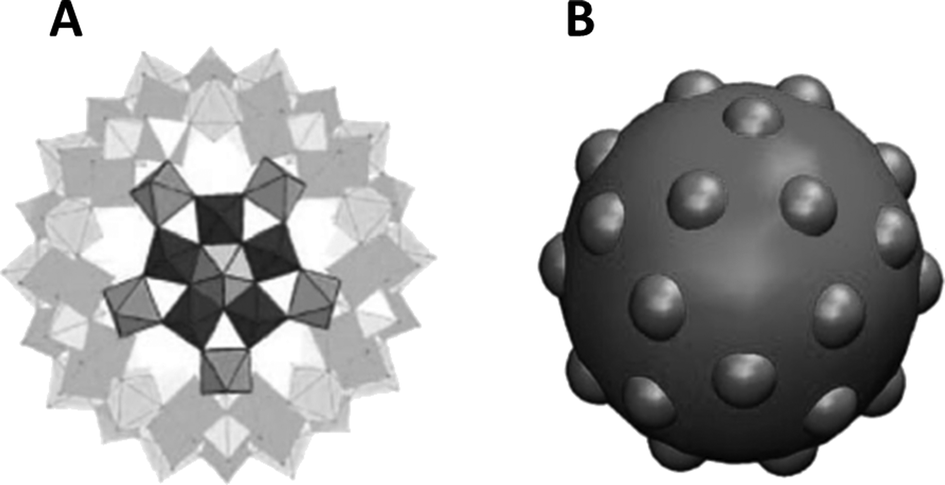 | ||
| Fig. 2 The patchy particle, as observed from the single crystal X-ray structure of [Mo72Fe30] (A). The patchy particle where icosidodecahedral patches are inserted on the sphere to emulate a [Mo72Fe30] cluster (B). Reprinted with permission from ref. 30. Copyright, 2012, AIP Publishing LLC. | ||
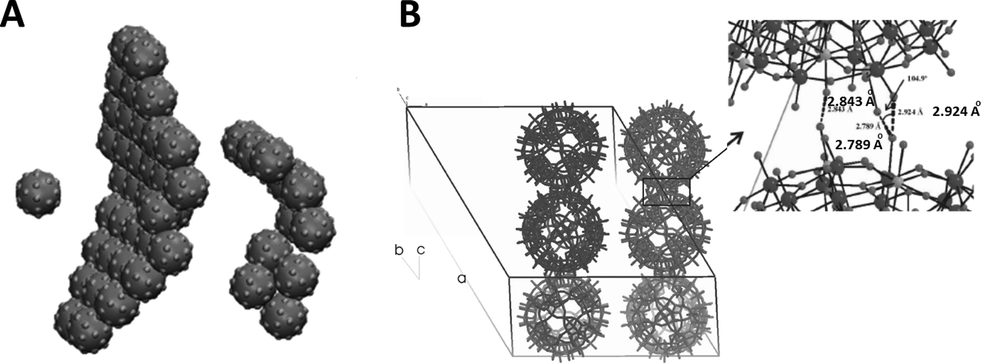 | ||
| Fig. 3 The 2D sheet observed from snapshots in the simulation (A) and that observed in the crystal formed by differential hydrogen bonding (B). The corresponding hydrogen bonding distances between the clusters, as seen from single crystal X-ray diffraction which is shown on the extreme right. | ||
3. Properties of SOMs and their stabilization mechanism
SOMs have certain interesting properties: 1. They scatter light; 2. They have a diffuse or mobile boundary; 3. They are responsive to the change in the dielectric constant of the medium. Recently, such responsiveness has been found to be linear: the radii of SOM-blackberries have been found to vary inversely with the dielectric constant of the medium in the cases of [Mo72Fe30] and [Mo132] as model systems (Fig. 4).32 This observation is explained by a simple model. This model identifies: 1. The driving force for the formation of such SOM-blackberries to be a pair-wise additive attraction between the constituent POMs in the SOMs; 2. It is also found that their equilibrium size is determined by their renormalized charge density, which in turn is controlled by counter-ion condensation. It is also possible from this model to find the interaction energy (cohesive/binding energy) that glues the POM units (each POM unit) of the SOM-blackberries together. Such energy is found to be approximately 15 kJ mol−1 (at 300 K). This cohesive/binding energy is comparable to the strength of a moderate X⋯H⋯X type hydrogen bond and is thus ‘soft’ or supramolecular in nature.33 It can also be said that these SOM-blackberries may be justified in being called ‘soft’ not only because of their mobile, diffuse boundary but because of the ‘soft’ supramolecular nature of the interaction parameter, which is comparable in magnitude to that of a moderate hydrogen bond, which in turn glues the POM units in the SOM.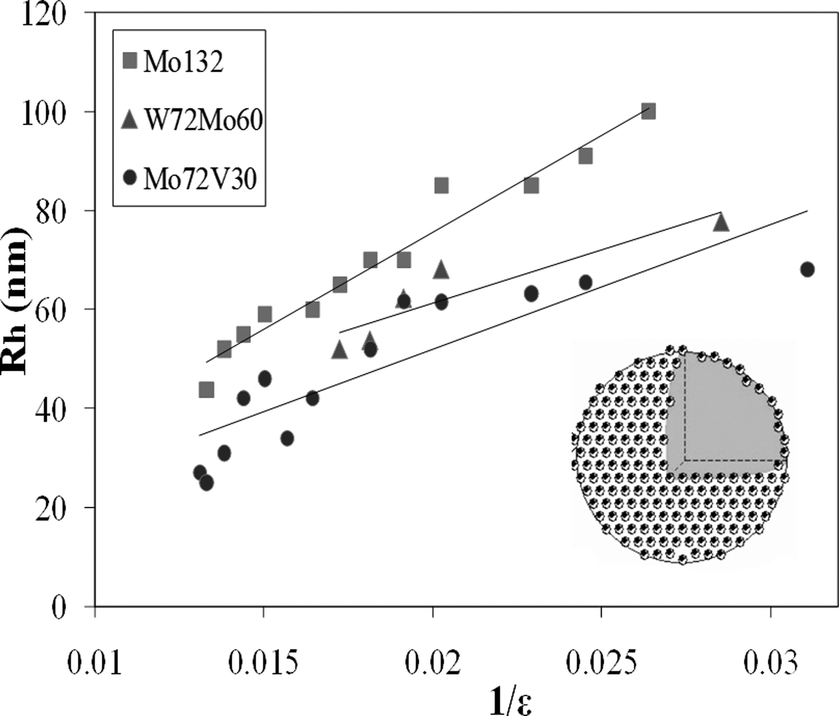 | ||
| Fig. 4 SOM-blackberry formed by three different clusters and the variation of their sizes with changing dielectric constant. Adapted with permission from ref. 20. Copyright, ACS, Washington, 2010. | ||
We now explain this charge regulation model in more detail. By assuming the free energy, G, of a SOM-shell depends on two variables that are fluctuating and dependent, viz., the aggregation number, as manifested in the radius R, and the effective charge Z, we can write,
| G/kT = 4πγr2 + 4π(2K + K′) + λZ2/[2R(1 + κR)] − ψZ | (1) |
In eqn (1), the first term with the surface tension, γ, is extensive in the aggregation number and it is expected not to show up in the equilibrium equation, when we assume that the average area occupied by a POM unit in the SOM shell does not depend on R. The second term in eqn (1) with the bending elastic modulus, K, and the Gaussian modulus, K′, is the curvature contribution from the Helfrich expansion of a spherical vesicle-like object.34 The third and fourth terms, where ψ denotes the zeta potential, regulate the effective charge of the aggregate SOM. The third term originates from the screened-Coulomb interactions on a uniformly charged sphere in the background of an electrolyte characterized by a Debye screening length of 1/κ, within the Debye–Hückel approximation, see: ref. 35. This particular term is supposed to be correct so long as we can neglect the counter ions inside the SOM-shells i.e., the case for R ≤ 1/κ. The fourth term determines the extent of the escape of ions from the narrow Gouy layer surrounding the SOM-shell. This term corresponds to a Legendre transformation from a constant charge – to a constant potential ensemble. See also: ref. 36.
By minimizing eqn (1) with Z, we obtain the renormalized charge on the SOM-shell as,
| Z = ψR(1 + κR)/λ | (2) |
Now, on substituting eqn (2) in eqn (1) and minimizing the free energy per unit area, we get the expression for R,
| R = 16πλ(2K + K′)/ψ2 | (3) |
Since, λ = e2/4πε0ε kT and putting λ = 56/ε nm, where ε is the dielectric constant of the solvent, we get Rα1/ε, which explains the experimental observation of the inverse variation of the SOM-shell radius with the dielectric constant of the solvent (Fig. 4). Now, from Eulers theorem, we obtain for SOM-shells, independent of their size, at least 12 monomers on the C5 axis of the SOM-shell are required to sit next to the predominantly present monomers on the C6 axis. This in turn implies that each SOM-shell lacks at least 12 times the cohesive bond energy, u, that monomer pairs have on the SOM-shell surface. Assuming this term to be the prime contribution, or K′ > K, we can equate the curvature energy with the cohesive energy,
| 4π(2K + K′) = −12u | (4) |
Substituting eqn (4) in eqn (3) we get,
| R = 48λu/ψ2 | (5) |
Thus u, the cohesive energy, can be obtained from the plot of R against 1/ε and the cohesive energy so obtained is around 5–7 kT for the [Mo72Fe30] and [Mo132] POMs forming the respective SOMs. This value is in close agreement with the cohesive energy obtained from the critical aggregation constant of the POMs and thereby implies the operation of a charge regulation mechanism by counter-ion condensation in the stabilization of the SOMs. This in turn implies that it is possible to control the size of the SOMs by changing the dielectric constant of the solvent. We ask in the next section: is it possible to control the overall shape and topology of the SOMs?
4. Directed formation of SOMs in dispersion: a few examples
We turn from spontaneously generated SOMs that are held by soft, supramolecular interactions, to the numerous examples where such soft, supramolecular interactions have been employed to design soft oxometalates in this section. Several design strategies have been employed. Examples include: the sol–gel method,37 surfactant encapsulation,38 Langmuir Blodgett method,39 layer-by-layer technique,40 solvent casting,41 intercalation between layered hydroxides.42 These are only a few types. We have synthesized a class of SOMs by exploiting the electrostatic interactions between suitably charged colloidal templates/layered lattices/structured surfaces and POMs. It should also be noted that a large body of literature exploring the catalytic activity of POMs has employed (similar) chemical means, as mentioned above, for designing high surface area SOMs and the methods have been known as methods for the heterogenization of POM catalysts.43 Some examples of such heterogenization can be recalled using, Rh(0), Ir(0),44 Au(0)45 clusters, silica,46 MOFs (Metal organic frameworks),47 dendrimer polyelectrolytes,48 and super-critical CO2 (ref. 49) as supports. The catalytic activity of several SOMs have also been reported.43b In fact, it is perhaps apt to say that heterogenized POMs, being dispersed oxometalates, can be considered as soft-state oxometalates. Thus, such heterogenized POMs can also be treated as SOMs. More precisely, SOMs can act as model systems to understand the phenomenon of heterogenization in catalysis involving polyoxometalates. It has been proposed that such ‘supported POMs’ with a large surface area could act as a ‘bridge’ between surface catalysts and the ‘pseudoliquid phase’ of bulk catalysts.50 Likewise, the question as to how to obtain such high-surface area POMs in a controlled way in an aqueous solution is important. In this section, we summarize the use of electrostatic and hydrogen bond interactions to form such SOMs, as has been performed in our laboratory in recent times. We have used charged colloidal entities as structure directing agents to control the overall shape of the SOMs. By varying the shape of the colloidal cast, we have been able to change the shape of the SOM. We have also shown that it is possible to use various types of POMs to make such SOMs, thereby demonstrating the applicability of the method with various POMs. Here, we summarize two such cases and three examples of SOM formation with the aid of a colloid.Controlling the size and morphology of POMs in the mesoscopic regime (in the range of 100–900 nm) remains a daunting challenge. The reasons are manifold and range from: difficulty in manoeuvring the chemistry of multiple metal centers, control of the pH to the overall control of the redox state of the complete system. Hence, in this regime, techniques that bypass the complex chemical crossroad and resort to the exploitation of the electrostatic interactions between preformed colloidal entities as templates/scaffolds for the design of mesoscopic architectures are more successful. Such a technique is evidently supramolecular (in the sense that it involves electrostatic interactions ‘beyond the chemistry of molecules’). This method also provides a platform to ‘glue’ molecules to form a mesoscopic supramolecular architecture in the SOMs. We propose to call this technique of using a colloidal template to form large surface area SOMs ‘colloidal casting’. The requirements for successful colloidal casting are as follows: (1) Complementary charge between the colloidal templates and the POM (e.g., positively charged gibbsite platelets as templates and anionic Keggins as POMs); (2) a common solvent (e.g., water). We further tested the applicability of the concept by using POMs like phosphomolybdate Keggin and [Mo72Fe30].51 The complementarity of the charge between the negatively charged [Mo72Fe30] and positively charged gibbsite platelets further prompted this choice. We now explain the charge complementarity. The pH of discrete [Mo72Fe30] clusters upon dissolving in water is around 4.5 while the isoelectric point or the point of zero charge of gibbsite is quite high, i.e., around pH 10.1. Consequently, at a pH of around 4.5, the surface of the gibbsite platelet is positively charged, (see the following equilibrium). The charged gibbsite platelet in turn acts as a platform for the attachment of anionic [Mo72Fe30]:
| Al–OH2+ + H2O ↔ Al–OH + H3O+ |
Consequently at a pH of 4.5, the complementary charges on gibbsite platelets and [Mo72Fe30] clusters act as glue to bind them together to form hexagonal platelets of [Mo72Fe30] clusters.
It is also possible to change the templates from hexagonal plates to spheres. For example, by using a spherical prefabricated cationic vesicle as a structure directing agent, it is possible to glue simple anionic oxomolybdates via electrostatic interactions and hydrogen bonds to form large SOM super-spheres.52 By using this method of colloidal casting, complexity can be deliberately induced in the resulting structure, either through the scaffold or by means of the oxometalate. There is a high degree of control in the matter of the size and morphology of the resulting SOMs, which makes this method attractive from a synthetic standpoint. For instance, it is possible to alter the SOM topology just by changing the shape of the vesicle and similar such synthetic avenues can be explored. This specific synthesis was performed by adding an appropriate amount of heptamolybdate to an already prepared DOTAP (a cationic fatty acid, 1,2-dioleol-3-trimethylammonium-propane) vesicle dispersion. There is a narrow window of heptamolybdate/DOTAP (M/D) concentration for the formation of a stable dispersion. But, beyond this window, the dispersion becomes unstable and then it is stable again. Such a phenomenon in the formation of a stable–unstable–stable dispersion was followed experimentally by electrophoretic mobility measurements, and such experiments point to the operation of a charge inversion mechanism as the M/D concentration is varied. This is explained as follows.
The positive charge on positively charged DOTAP (D) vesicles decreases as anionic molybdates (M) are added to it and finally instability is induced for certain concentration ratios of M/D (1.5 > M/D > 0.6). The dispersion becomes almost zero charge and thus becomes unstable. M has a charge of 6− while D has a charge of 1+. Thus, if all the added Ms reside at the Ds, this instability should manifest at M/D = 0.16. In practice, much higher values of M/D are required to bring about this phase instability and this indicates the presence of free Ms in the dispersion. Hence, an extra amount of M (heptamolybdate) is needed to reach the unstable regime. On further addition of M (i.e., for ratios of 10 > M/D > 3), the dispersion again undergoes charge inversion and is now negatively charge stabilized. We can deduce analytically the interface structure of an M–D SOM (molybdate–DOTAP SOM) super-sphere from this ratio. On closer inspection of the experimental results, we observe that the surface charge density of both the DOTAP vesicle and that of the M–D SOM super-sphere (for M/D ≈ 3 and higher) is the same but they have opposite signs (i.e., +5 μm cm V−1 s−1 in DOTAPs and −5 μm cm V−1 s−1 in the composite). It is known that a DOTAP molecule carries a unit positive charge, whereas heptamolybdate has a charge of −6. From the experimental results (i.e., taking charge inversion at M/D ≈ 3 and higher), it follows that in the M–D SOM super-sphere for every three DOTAP molecules, there is only one heptamolybdate. This picture matches well with the surface area of DOTAP53 and heptamolybdate.2 So, the M–D SOM super-sphere is a vesicle of DOTAP covered with a monolayer of heptamolybdate. The monolayer of heptamolybdate is positioned in such a way that every unit of the monolayer is fluxionally coordinated to three DOTAP units of the vesicle. All these SOMs are characterized by various techniques, like cryo-TEM (Transmission Electron Microscopy), TEM/EDX (TEM with Energy Dispersive X-Ray analyses), ATR-IR (Attenuated Total Reflection-Infra Red), Raman spectroscopy, static and dynamic light scattering, small angle X-ray scattering, electrophoretic mobility measurements, potentiometric titrations, etc.
Having shown that it is possible to control the topology of the SOMs in a directed manner in dispersions, we look back at spontaneously formed SOMs and ask, how does a POM interact with sound to form SOMs? We address this question in the next section.
5. Sonication and SOMs
It has been recently demonstrated that complex and large single-molecule POM clusters may even spontaneously form SOMs of colloidal size (i.e., on the order of 10–100 nm).20 Of course, complexity can lead to complexity but can simple precursors lead to complex colloidal entities? Now we address this question: can very simple sparingly soluble salts of polyoxometalates, such as the ammonium salt of a phosphododecamolybdate Keggin,54 show comparable SOM superstructure formation?100 What happens when we sonicate a dilute solution of the Keggin? It is known that dilute solutions of this Keggin salt tend to scatter light,55 and this points to the presence of objects on colloidal-length scales in the solution or more correctly in the dispersion. Recently, a phosphododecatungstate Keggin has been used in combination with AOT microemulsions and also as a template to synthesize fibrous, star-like, and other interesting architectures.56 The colloidal nature of thet Keggin is in fact not entirely unknown. Around the 1930s, complex structure formation with “phosphatide coacervates” was observed.57 Moreover, though it was known that the ammonium salt of the phosphododecamolybdate Keggin forms a colloidal dispersion in water,55 the nature of the particles of this dispersion have not been investigated until now. However, a lot of fundamental work has been done with the ammonium phosphomolybdate Keggin.58–72 Extensive investigations have also been carried out to explore the nature of POMs in solution66–72 and can be traced back to 1783 and to the efforts of Berzelius.73 However, the nature of the colloidal objects in an aqueous dispersion of the ammonium phosphomolybdate Keggin was not investigated. Hence, for us to address the question of the nature of the colloidal objects in an aqueous dispersion of the [PMo12] Keggin ([PMo12O40]3−, Keggin), we started our investigation with a very dilute sonicated dispersion of the ammonium salt of the POM. This investigation reveals that a sonicated aqueous colloidal dispersion of the [PMo12] Keggin shows spontaneous formation of small spheres of the [PMo12] Keggin and its lacunary analogues (5–50 nm radii). These nano-spheres ripen in an Ostwald ripening-like regime and finally after 2–3 days, generate stable micrometer sized “peapod”-like mesoscopic SOM-particles (Fig. 5). These peapods are structurally heterogeneous and are comprised of [P2Mo] ([P2MoO11]6−) spheres sheathed by a MoO3 nanorod. Upon acidification, the spheres leech out, leaving behind only rods of MoO3. This entire investigation was carried out using time-resolved dynamic light scattering (DLS), transmission electron microscopy (TEM), and scanning TEM (STEM) with a high-angle annular dark field detector (HAADF) for energy dispersive X-ray (TEM/EDX) elemental analyses.100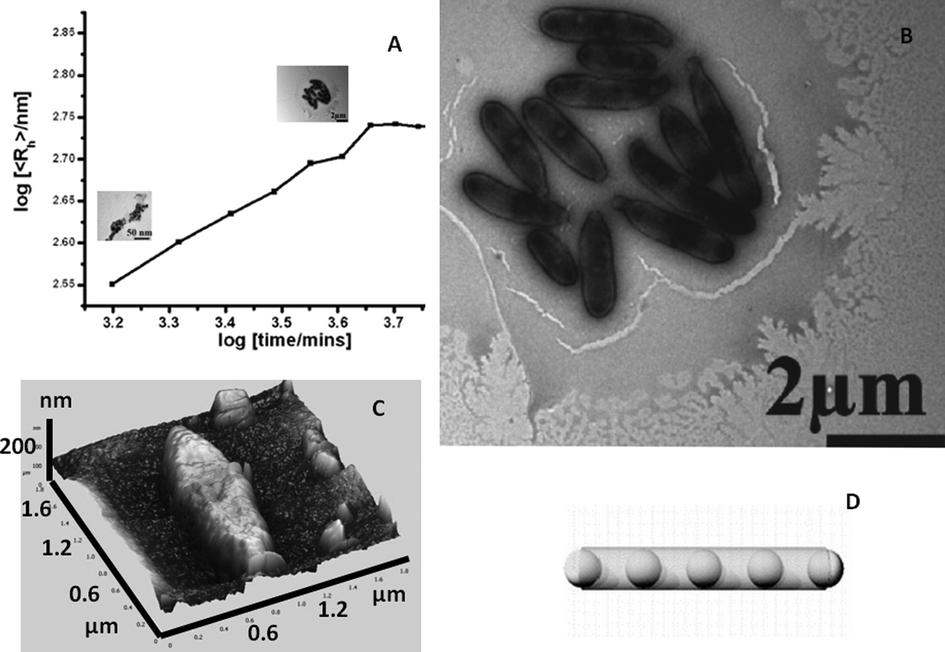 | ||
| Fig. 5 a) Ripening of the [PMo12] spheres in a sonicated dispersion into peapods with time. b) TEM image of the peapods. c) AFM image of a peapod. d) A model of a peapod. | ||
We now speculate on why peapods are formed. It is relatively easy from an energy requirement point of view to understand the formation of a cylindrical morphology rather than a sphere. This is because in the case of a cylinder, unlike a sphere, there is no requirement for the generation of 12 C5 axes and the breaking of 12 contacts therein. The next question as to why spherical [P2Mo] forms spheres and remains wrapped in a sheet of MoO3 is yet to be answered. The story is more interesting from a single molecule chemistry point of view. From such a chemical point of view, it is intriguing to see how upon sonication, the less-soluble ammonium salt of the two component [PMo12] Keggin goes back to its two starting components, a MoO3 sheet and PO43− spheres, in [P2Mo] via a series of shape transitions. Though we do not understand the exact mechanism of this shape transition, we may still allude to an architectural concept for stress analysis. It is known that any architecture or any structure breaks along the weaker lines of its construction when it is subjected to a yielding force. Similarly, we may say that the phenomenon as outlined here traces out the weaker fault lines of the [PMo12] Keggin's molecular construction. Needless to say, these fault-lines in the Keggin link the central tetrahedral phosphate with the four surrounding [Mo3] caps. More precisely, these are the μ3-Os along which the Keggin decomposes, forming macroscopic peapods. The reason for the stress along this fault-line we believe is due to the two different intrinsic curvatures preferred by two types of chemical motifs, viz., the PO4 and MoO3 caps. Moreover, the Mo–O–Mo angular strain in the starting Keggin is released as thermodynamically more stable spherical [P2Mo] species are formed within the peapods. We also believe that this strain on a molecular scale leads to the thermodynamic instability of the starting Keggin and ultimately results in the formation of a thermodynamically stable macroscopic composite, the peapods. However, for this stress to be more active, it is essential that the starting molecule is insoluble. Since the phenomenon described here leads to the formation of new shapes (on macroscopic length scales) as a result of degeneration (on a molecular scale), we propose naming this phenomenon as “degenerative morphogenesis”. In contrast to the larger clusters, the smaller and less-soluble salts of POMs, such as the ammonium salt of the phosphomolybdate Keggin discussed here, do not show spherical SOM-shell-like superstructures, instead, they show peapod-shaped SOM formation, as shown here. Now we ask: can we induce controlled motion in these peapods, whose constitution is known down to the last atom? In the next section we answer this question.
6. SOMs in motion with light
Living systems use motor proteins to actively transport ingredients over large distances.74 Clearly synthetically emulating such a process would require two steps: (1) controlled generation of mesoscopic objects starting from well-defined precursors; (2) using physical means to induce controlled motion in such mesoscopic objects. This is where SOMs, especially SOM peapods, can come into play. Being endowed with an optical axis, it can be responsive to variations in external optical fields. It is hence reasonable to envisage that a SOM-peapod with a component responsive to an external optical perturbation can be a synthetic model system, showing controlled motion comparable to biological systems. Furthermore, could we actually move the SOM in a complex pre-designed path by known amounts? To answer this question, we designed such a path using optical forces, and an optically responsive SOM-peapod was made to move along that path in our model system. The optical forces were exerted by optical tweezers. Optical tweezers can confine single mesoscopic particles and can apply controlled forces ranging from a few to several hundred pN.75 It was thus an ideal candidate to induce controlled motion in SOMs. Translation of trapped SOMs linearly by translating the optical trap is easy and can be done but translation along more complex paths, which may be required to emulate biological processes, are not simple, as shown by us. In our method, the trapped particle is moved by changing the angle of polarization of the input trapping beam (linearly polarized). This enables us to completely control the motion both in terms of stopping the particle or changing its velocity. We are also able to rotate the particles by exploiting spin–orbit interactions of light affecting the distribution of the electric field inside the sample chamber (Fig. 6).76 The enhanced spin–orbit interaction can be induced in the sample chamber using thicker cover-slips (thickness 250 μm) than the conventional ones used in optical tweezers (130–160 μm). Since peapods are asymmetric birefringent particles with a preferred optic axis, they can line up with the polarization of the trapping beam. We have also designed a rather exotic optical potential in our optical trap in order to induce controlled micro-optomechanics on individual peapod SOMs. The details on the design of optical potential are reported by us elsewhere.77 We ask, when using a light-SOM interaction, what else might be possible? SOMs have LMCT (Ligand–Metal Charge Transition) transitions. Is it possible to exploit the responsiveness of SOMs to light to self-assemble them and write patterns using light? In the next section, we address this question. | ||
| Fig. 6 Snap shots of the rotation of peapods in an optical trap. | ||
7. Self-assembly and patterning of SOMs by light
The beauty of self-assembly is often the level of complexity and high specificity that can be obtained in the final structures with a minimum dissipation of energy, thus ensuring high efficiency. Self-assembly strategies in nature have been extensively studied and applied by scientists on the meso-scale for diverse applications in nano-technology,78–82 molecular electronics,83etc. Inducing self-assembly by an external stimulus is especially interesting,84 as it allows control of the final structures by alteration of the parameters of the applied stimulus. Light, and SOMs for this reason, are an ideal choice in this matter. This is because as an external stimulus, light can be tuned. SOMs are responsive to light and thus the entire light-induced SOM assembly, if generated, can be tuned altogether. It is worth noting that light assisted self-assembly has led to novel materials85–87 for sensing,88 delivery,89 and optics90 applications. We were able to prepare SOM nanotubes with LMCT transitions that enable them to be responsive to stimulation by a laser light of 1064 nm and by exploiting this responsiveness, we were able to pattern SOMs to form higher ordered crystalline structures to be described here.Before we proceed further, it is worth reviewing the literature on reversible and irreversible self-assembly by light in a bit more detail. Our patterning of SOMs by light is an irreversible patterning induced by light. Induced self-assembly has two types: reversible self-assembly (where the assembly is lost on removal of the stimulus) and irreversible (where a permanent self-assembled structure is formed). Concentration-dependent reversible and irreversible assemblies of nanoparticles have been shown.91 Multi-scale patterning using directed fields has also been achieved recently, though continuous patterning has proven elusive.92 For instance, patterned chains and networks of gold nanocrystals have been formed. Likewise, the formation of colloidal crystals by a directed electric field using bubble-mediated nucleation93 or more recently, single crystals of glycine94 that have been grown from solution, have been achieved due to the formation of a ‘hot spot’ or high temperature region produced by a laser beam focused on the gold surface. We ask, is it possible to create similar hot spots and induce a bubble-mediated SOM assembly that would ultimately self-assemble and crystallize forming patterns at our volition?95
To do so, we synthesize SOM nanotubes with LMCT transitions tuned to the wavelength of our thermo-optic trapping laser. We focus the thermo-optic trapping laser on a dispersion of SOM nanotubes. Due to LMCT type transitions, the laser irradiation excites the dispersion and creates a bubble with SOMs (Fig. 7). Due to buoyant forces, this bubble levitates to the base of the sample chamber. Due to the difference in the surface energy between the surface and the base of the bubble, a convection current is established, which draws the SOM nanotubes from the bulk dispersion to the base of the bubble. Now we move the sample chamber by moving the microscope stage and this leads to two possibilities for the bubble, viz., the generation of a new bubble or the migration of the generated bubble with the laser. In fact the second option is energetically more favoured and consequently the bubble moves with the laser, depositing SOMs on the base of the sample chamber which later undergo nucleation to give crystals of oxometalates. Hence, by moving the sample chamber, or more precisely the microscope stage, we can write any continuous pattern we want with SOMs, which in turn nucleates forming patterns of crystalline oxometalates. In this way, we have formed patterns using: 1) soft-oxometalate nanotubes95 having comparatively high absorbance at λ = 1064 nm resulting from a Ligand Metal Charge Transfer (LMCT) type transition, and 2) paracetamol, fluorescent dyes (such as perylene, where the pattern can be illuminated under light) and carbon nanotubes (CNTs) loaded on the SOMs, where the SOM helps in inducing nucleation. We observe that continuous patterns can indeed be formed using the SOMs, at much lower powers than those typically employed in laser-induced nucleation.96–98 Patterns are also formed using organic molecules anchored on the SOMs, and we observe assisted nucleation by exploiting the excitation of the SOM core due to a LMCT-type transition when exposed to the intense trapping beam. The organic molecules are chosen keeping in mind the presence of hydrogen bonding and coordination sites. This technique is much simpler, easily controllable and fast for any optical patterning scheme and provides a facile way for forming SOM or oxometalate-based arrays for various catalytic and materials science applications. In short, starting from crystalline POMs, we can make SOMs, self-assemble them under light and write patterns of crystalline oxometalates thereof.
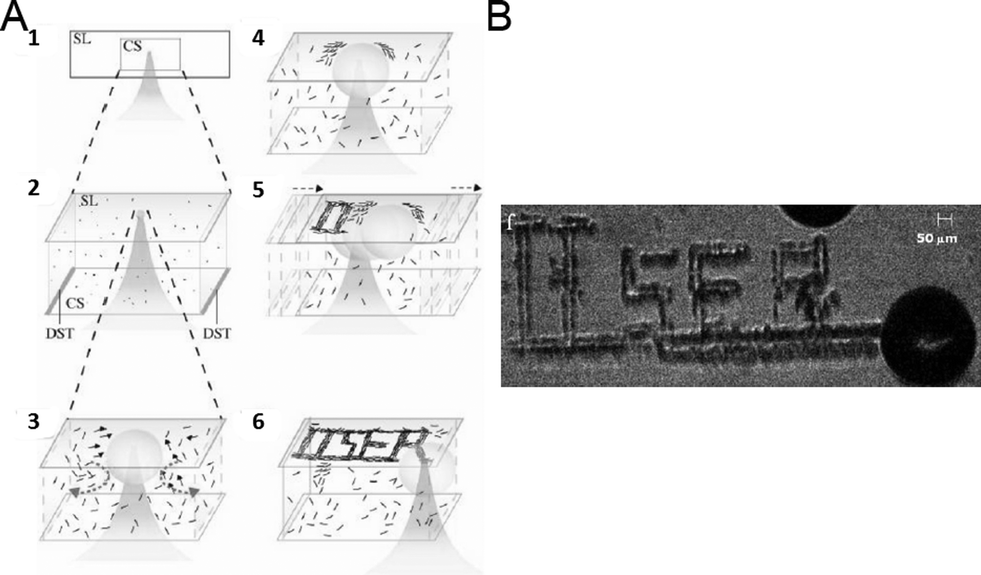 | ||
| Fig. 7 Thermo-optic trapping of SOM nanotubes: (A) focusing of the laser in the SOM dispersion to form the hot-spot is shown (1). The hot-spot leads to the formation of the bubble (2). Gibbs–Marangoni convection of the SOMs from the dispersion to the base of the bubble takes place (3). Accumulation of SOMs at the base of the bubble or on the surface of the glass slide takes place (4). Moving the laser focus moves the bubble to a new spot and the same phenomena as shown in Fig. 1–4 is repeated (5). In this way, patterns can be written. The pattern ‘IISER’ written on the glass slide is shown schematically (6). The real pattern of ‘IISER’ from the experiment is shown on the right (B). | ||
8. Conclusion
To conclude, we have shown that starting from crystalline POMs, we can transcend the crystalline boundary and enter into the territory of liquid/soft-matter by making SOMs. SOMs can be considered as units of oxometalates with a diffuse boundary constituting oxometalates in a soft/liquid state. As we increase the volume fraction/concentration/number density of SOMs, it is possible to induce a phase transition from a liquid to crystalline regime. In this way, we can envisage crystallization of POMs, which is still not well-understood, by invoking the SOM model to describe it.99 We have also shown that at a very low concentration regime, SOMs are self-assembled and their assembly can be understood from a patchy model constructed from the consideration of the crystal structure of the corresponding POMs. Self-assembly of SOMs is not confined to complex POM precursors but they can be assembled from simple POM units as well. We have shown the sonication-induced self-assembly of SOM peapods and have shown their controlled motion in an optical field. We have further shown the exploitation of SOM-light interactions in making self-assembled patterns of SOMs with light, which in turn undergo nucleation and crystallization. Starting from crystalline POMs, we have explored the world of soft-matter with SOMs and have patterned them with light in a controlled way to get back to crystalline oxometalates to conclude our journey. Needless to say, the journey with SOMs is just beginning.Acknowledgements
The author thanks Preethi Thomas and Subharanjan Biswas for their help and Prof. Tianbo Liu for kindly providing the raw materials for Fig. 1 and 4 and Dr. Ethayaraja Mani for providing the raw materials for Fig. 2. DST fast-track, BRNS-DAE grants and IISER-Kolkata are thanked for financial support. This paper is dedicated to Professor Achim Müller.Notes and references
- J. L. G. Fierro, Metal oxides: Chemistry & Applications, CRC Press, Boca Raton, 2006 Search PubMed.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 Search PubMed.
- Chem. Rev. Thematic Issue on Polyoxometalates, ed. C. L. Hill, ACS, Washington, USA, 1998, vol. 98 Search PubMed.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef.
- Inorganic Synthesis, ed. A. P. Ginsberg, John Wiley & Sons, Inc., New Jersey, USA, 1990, vol. 27, p. 71 Search PubMed.
- Comprehensive Coordination Chemistry, ed. L. Cronin, J. A. McCleverty and T. J. Meyer, Elsevier, Amsterdam, 2004 Search PubMed.
- Polyoxometalate Chemistry for Nanocomposite Design, ed. T. Yamase and M. T. Pope, Kluwer Academic Publishers, New York, 2002 Search PubMed.
- Introduction to Polyoxometalate Chemistry: From Topology via Self-Assembly to Applications, ed. M. T. Pope and A. Müller, Kluwer Academic Publishers, New York, 2001 Search PubMed.
- (a) A. Müller, S. K. Das, V. P. Fedin, E. Krickemeyer, C. Beugholt, H. Bögge, M. Schmidtmann and B. Z. Hauptfleisch, Z. Anorg. Allg. Chem., 1999, 625, 1187 CrossRef; (b) A. M. Müller and C. Serain, Acc. Chem. Res., 2000, 33, 2 CrossRef PubMed.
- A. Müller, E. Krickemeyer, H. Bögge, M. Schmidtmann and F. Peters, Angew. Chem., Int. Ed., 1998, 37, 3360 Search PubMed.
- A. Müller, S. Sarkar, S. Q. N. Shah, A. X. Trautwein and V. Schünemann, Angew. Chem., Int. Ed., 1999, 38, 3238 CrossRef.
- B. Botar, P. Kögerler and C. L. Hill, Chem. Commun., 2005, 3138 RSC.
- A. M. Todea, A. Merca, H. Bögge, J. van Slageren, M. Dressel, L. Engelhardt, M. Luban, T. Glaser, M. Henry and A. Müller, Angew. Chem., Int. Ed., 2007, 46, 6106 CrossRef CAS PubMed.
- (a) S. S. Mal and U. Kortz, Angew. Chem., Int. Ed., 2005, 44, 3777 CrossRef CAS; (b) D. Jabbour, B. Keita, L. Nadjo, U. Kortz and S. S. Mal, Electrochem. Commun., 2005, 7, 841 CrossRef CAS PubMed; (c) M. S. Alam, V. Dremov, P. Müller, A. V. Postnikov, S. S. Mal, F. Hussain and U. Kortz, Inorg. Chem., 2006, 45, 2866 CrossRef CAS PubMed.
- A. Müller, E. Beckmann, H. Bögge, M. Schmidtmann and A. Dress, Angew. Chem., Int. Ed., 2002, 41, 1162 CrossRef.
- R. C. Howell, F. G. Perez, S. Jain, W. H. Dew, A. L. Rheingold and L. C. Francesconi, Angew. Chem., 2001, 113, 4155 CrossRef.
- (a) A. Müller, E. Diemann, C. Kuhlmann, W. Eimer, C. Serain, T. Tak, A. Knöchel and P. K. Pranzas, Chem. Commun., 2001, 1928 RSC; (b) T. Liu, E. Diemann, H. Li, A. Dress and A. Müller, Nature, 2003, 426, 59 CrossRef CAS.
- P.-G. de Gennes, Soft Interfaces, Cambridge University Press, Cambridge, 1997 Search PubMed.
- S. Roy, Comments Inorg. Chem., 2011, 32, 113 CrossRef CAS.
- T. Liu, Langmuir, 2010, 26, 9202 CrossRef CAS PubMed.
- H. Lyklema, Fundamentals of Interface and Colloid Science, Elsevier Academic Press, Wageningen, 2004, vol. 1–5 Search PubMed.
- A. Vrij, Pure Appl. Chem., 1976, 48, 471 CrossRef CAS.
- W. Poon, Science, 2004, 304, 830 CrossRef CAS PubMed.
- P. Prinsen and T. Odijk, J. Chem. Phys., 2004, 121, 6525 CrossRef CAS PubMed.
- A. Patti and M. Dijkstra, Phys. Rev. Lett., 2009, 102, 128301 CrossRef.
- J. Israelachvili, Intermolecular and Surface Forces, Academic, San Diego, CA, 1992 Search PubMed.
- Z. Tang, Z. Zhang, Y. Wang, S. C. Glotzer and N. Kotov, Science, 2006, 314, 274 CrossRef CAS PubMed.
- Z. Zhang and S. C. Glotzer, Nano Lett., 2004, 4, 1407 CrossRef CAS.
- A. Müller, Angew. Chem., Int. Ed., 1999, 38, 3238 CrossRef.
- E. Mani, E. Sanz, S. Roy, M. Dijkstra, J. Groenewold and W. K. Kegel, J. Chem. Phys., 2012, 136, 144706 CrossRef PubMed.
- A. A. Verhoeff, M. L. Kistler, A. Bhatt, J. Pigga, J. Groenewold, M. Klokkenburg, S. Veen, S. Roy, T. Liu and W. K. Kegel, Phys. Rev. Lett., 2007, 99, 066104 CrossRef.
- A. Müller and S. Roy, Oxomolybdates: From Structures to Functions in a New Era of Nanochemistry, in The Chemistry of Nanomaterials: Synthesis, Properties and Applications, ed. C. N. R. Rao, A. Müller and A. K. Cheetham, Wiley-VCH, Weinheim, 2005, p. 452 Search PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- W. Helfrich, Z. Naturforsch., C: Biochem., Biophys., Biol., Virol., 1973, 28, 693 CAS.
- R. J. Hunter, Foundations of Colloid Science, Oxford University Press, New York, 2nd edn, 2001 Search PubMed.
- R. P. Feynman, R. B. Leighton and M. L. Sands, The Feynman Lectures on Physics—Commemorative Issue, Addison-Wesley, Redwood CA, 1989, vol. II Search PubMed.
- S. Polarz, B. Smarsley, C. Göltner and M. Antonietti, Adv. Mater., 2000, 12, 1503 CrossRef CAS.
- (a) D. G. Kurth, P. Lehmann, D. Volkmer, H. Cölfen, M. J. Koop, A. Müller and A. Du Chesne, Chem. – Eur. J., 2000, 6, 385 CrossRef CAS; (b) D. Volkmer, A. Du Chesne, D. G. Kurth, H. Schnablegger, P. Lehmann, M. J. Koop and A. Müller, J. Am. Chem. Soc., 2000, 122, 1995 CrossRef CAS; (c) D. G. Kurth, P. Lehmann, D. Volkmer, A. Müller and D. Schwahn, J. Chem. Soc., Dalton Trans., 2000, 3989 RSC.
- (a) M. Clemente-León, C. Mingotaud, B. Agricole, C. J. Gómez-García, E. Coronado and P. Delhaès, Angew. Chem., Int. Ed. Engl., 1997, 36, 1114 CrossRef; (b) M. Clemente-León, B. Agricole, C. Mingotaud, C. J. Gómez-García, E. Coronado and P. Delhaès, Langmuir, 1997, 13, 2340 CrossRef.
- S. W. Keller, H.-N. Kim and T. E. Mallouk, J. Am. Chem. Soc., 1994, 116, 8817 CrossRef CAS.
- I. K. Song, M. S. Kaba, G. Coulston, K. Kourtakis and M. A. Barteau, Chem. Mater., 1996, 8, 2352 CrossRef CAS.
- T. Kwon and T. J. Pinnavaia, J. Mol. Catal., 1992, 74, 23 CrossRef CAS.
- (a) Catalysts for Fine Chemicals Synthesis: Catalysis by Polyoxometalates, ed. I. V. Kozhevnikov, J. Wiley & Sons, Chichester, 2002, vol. 2 Search PubMed; (b) Journal of Molecular and Engineering Materials, Special Issue on Soft-oxometalates (SOMs): towards new oxometalate based materials, ed. S. Roy, World Scientific, Singapore, 2014, vol. 2 Search PubMed; (c) N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS; (d) R. Neumann and A. M. Khenkin, Chem. Commun., 2006, 2529 RSC.
- R. G. Finke and S. Özkar, Coord. Chem. Rev., 2004, 248, 135 CrossRef CAS PubMed.
- Y. Wang, A. Neyman, E. Arkhangelsky, V. Gitis, L. Meshi and I. A. Weinstock, J. Am. Chem. Soc., 2009, 131, 17412 CrossRef CAS PubMed.
- (a) N. M. Okun, T. M. Anderson and C. L. Hill, J. Am. Chem. Soc., 2003, 125, 3194 CrossRef CAS PubMed; (b) N. M. Okun, M. D. Ritorto, T. M. Anderson and C. L. Hill, Chem. Mater., 2004, 16, 2551 CrossRef CAS.
- (a) C.-Y. Sun, S.-X. Liu, D.-D. Liang, K.-Z. Shao, Y.-H. Ren and Z.-M. Su, J. Am. Chem. Soc., 2009, 131, 1883 CrossRef CAS PubMed; (b) A. Corma, H. García and F. X. L. Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS.
- (a) L. Plault, A. Hauseler, S. Nlate, D. Astruc, J. Ruiz, S. Gatard and R. Neumann, Angew. Chem., Int. Ed., 2004, 43, 2924 CrossRef CAS PubMed; (b) S. Nlate, D. Astruc and R. Neumann, Adv. Synth. Catal., 2004, 346, 1445 CrossRef CAS.
- G. Maayan, B. Ganchegui, W. Leitner and R. Neumann, Chem. Commun., 2006, 2230 RSC.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS.
- (a) S. Roy, M. C. D. Mourad and M. T. Rijneveld-Ockers, Langmuir, 2007, 23, 399 CrossRef CAS PubMed; (b) S. Roy, H. J. D. Meeldijk, A. V. Petukhov, M. Versluijs and F. Soulimani, Dalton Trans., 2008, 2861 RSC.
- S. Roy, L. C. A. M. Bossers, H. J. D. Meeldijk, B. W. M. Kuipers and W. K. Kegel, Langmuir, 2008, 24, 666 CrossRef CAS PubMed.
- J. Generosi, C. Castellano, D. Pozzi, A. C. Castellano, R. Felici, F. Natali and G. Fragneto, J. Appl. Phys., 2004, 96, 6839 CrossRef CAS PubMed.
- (a) J. F. Keggin, Nature, 1933, 131, 908 CrossRef CAS; (b) J. F. Keggin, Proc. R. Soc. London, Ser. A, 1934, 144, 75 CrossRef CAS.
- A. I. Vogel, Qualitative Inorganic Analysis, Longman Scientific and Technical, Essex, England, 1987 Search PubMed.
- (a) M. Li and S. Mann, Langmuir, 2000, 16, 7088 CrossRef CAS; (b) D. Rautaray, S. R. Sainkar and M. Sastry, Langmuir, 2003, 19, 10095 CrossRef CAS; (c) S. Mandal, D. Rautaray and M. Sastry, J. Mater. Chem., 2003, 13, 3002 RSC; (d) Z. Xin, J. Peng, T. Wang, B. Xue, L. Li and E. Wang, Inorg. Chem., 2006, 45, 8856 CrossRef CAS PubMed.
- H. G. B. de Jong, Colloid Science, ed. H. R. Kruyt, Elsevier, Amsterdam, 1949 Search PubMed.
- T. Ueda, T. Toya and M. Hojo, Inorg. Chim. Acta, 2004, 357, 59 CrossRef CAS.
- X. López, J. Maestre, C. Bo and J.-M. Poblet, J. Am. Chem. Soc., 2001, 123, 9571 CrossRef PubMed.
- M. Janik, B. Bardin, R. Davis and M. J. Neurock, J. Phys. Chem. B, 2006, 110, 4170 CrossRef CAS PubMed.
- C. Rocchiccioli-Deltcheff, A. Aouissi, M. Bettahar, S. Launay and M. J. Fournier, J. Catal., 1996, 164, 16 CrossRef CAS.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS PubMed.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS PubMed.
- C. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407 CrossRef CAS.
- X. López, C. Nieto-Draghi, C. Bo, J. Avalos and J.-M. Poblet, J. Phys. Chem. A, 2005, 109, 1216 CrossRef PubMed.
- A. Müller and C. Seraine, Acc. Chem. Res., 2000, 33, 2 CrossRef PubMed.
- A. Müller and S. Roy, Coord. Chem. Rev., 2001, 245, 153 CrossRef.
- M. Pope and G. Varga Jr., Inorg. Chem., 1966, 5, 1249 CrossRef CAS.
- M. Baker, P. Lyons and S. Singer, J. Am. Chem. Soc., 1955, 77, 2011 CrossRef CAS.
- I. Validzic, G. van Hooijdonk, S. Oosterhout and W. Kegel, Langmuir, 2004, 20, 3435 CrossRef CAS.
- T. Kurucsev, A. Sargeson and B. West, J. Phys. Chem., 1957, 61, 1567 CrossRef CAS.
- K. Tytko and U. Trobisch, Gmelin Handbook of Inorganic Chemistry, ed. H. Katscher, Springer-Verlag, Berlin, 1986, vol. B3a, p. 40 Search PubMed.
- J. Berzelius, Ann. Phys., 1826, 6, 1206 Search PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 2002 Search PubMed.
- T. T. Perkins, Laser Photonics Rev., 2008, 3, 203 CrossRef.
- B. Roy, N. Ghosh, S. Dutta Gupta, P. Panigrahi, S. Roy and A. Banerjee, Phys. Rev. A: At., Mol., Opt. Phys., 2013, 87, 043823 CrossRef.
- B. Roy, N. Ghosh, P. Panigrahi, A. Banerjee, A. Sahasrabudhe, B. Parasar and S. Roy, J. Mol. Eng. Mater., 2014, 2, 1440006, DOI:10.1142/S2251237314400061.
- T. Leong, Z. Gu, T. Koh and D. H. Gracias, J. Am. Chem. Soc., 2006, 128, 11336 CrossRef CAS PubMed.
- J. C. Love, A. R. Urbach, M. G. Prentiss and G. M. Whitesides, J. Am. Chem. Soc., 2003, 125, 12696 CrossRef CAS PubMed.
- Z. Gu, Y. Chen and D. H. Gracias, Langmuir, 2004, 20, 11308 CrossRef CAS PubMed.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450 CrossRef CAS PubMed.
- J. S. Lindsey, New J. Chem., 1991, 15, 153 CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278 CrossRef CAS.
- M. Fialkowski, K. J. M. Bishop, R. Klajn, S. K. Smoukov, C. J. Campbell and B. A. Grzybowski, J. Phys. Chem. B, 2006, 110, 2482 CrossRef CAS PubMed.
- A. J. Kim, P. L. Biancaniello and J. C. Crocker, Langmuir, 2006, 22, 1991 CrossRef CAS PubMed.
- U. Dassanayake, S. Fraden and A. van Blaaderen, J. Chem. Phys., 2000, 112, 3851 CrossRef CAS PubMed.
- M. Oh and C. A. Mirkin, Nature, 2005, 438, 651 CrossRef CAS PubMed.
- F. Ecole, T. P. Davisa and R. A. Evans, Polym. Chem., 2010, 1, 37 RSC.
- D. L. J. Vossen, D. Fific, J. Penninkhof, T. van Dillen, A. Polman and A. van Blaaderen, Nano Lett., 2005, 5, 1175 CrossRef CAS PubMed.
- R. Klajn, K. J. M. Bishop and P. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591 CrossRef CAS PubMed.
- S. J. Zhen, Z. Y. Zhang, N. Li, Z. D. Zhang, J. Wang, C. M. Li, L. Zhan, H. L. Zhuang and C. Z. Huang, Nanotechnology, 2013, 24, 055601 CrossRef PubMed.
- T. Uwada, S. Fuji, T. Sugiyama, A. Usman, A. Miura, H. Masuhara, K. Kanaizuka and M. Haga, ACS Appl. Mater. Interfaces, 2012, 4, 1158 CAS.
- S. Fujii, K. Kanaizuka, S. Toyabe, K. Kobayashi, E. Muneyuki and M.-A. Haga, Langmuir, 2011, 27, 8605 CrossRef CAS PubMed.
- B. Roy, M. Arya, P. Thomas, J. K. Jürgschat, K. V. Rao, A. Banerjee, C. M. Reddy and S. Roy, Langmuir, 2013, 29, 14733 CrossRef CAS.
- T. Sugiyama, K. Yuyama and H. Masuhara, Acc. Chem. Res., 2012, 45, 1946 CrossRef CAS PubMed.
- B. A. G. X. Sun and A. S. Myerson, Cryst. Growth Des., 2006, 6, 684 Search PubMed.
- J. M. J. Zaccaro, A. S. Myerson and B. A. Garetz, Cryst. Growth Des., 2001, 1, 5 CAS.
- D. Rosenbaum, P. C. Zamora and C. F. Zukoski, Phys. Rev. Lett., 1996, 76, 150 CrossRef CAS.
- S. Roy, M. Rijneveld-Ockers, J. Groenewold, B. Kuipers, H. Meeldijk and W. K. Kegel, Langmuir, 2007, 23, 5292 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |