 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemical synthesis and characterisation of two new bismuth metal organic frameworks†
L.
Tröbs
,
M.
Wilke
,
W.
Szczerba
,
U.
Reinholz
and
F.
Emmerling
*
BAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de; Fax: +49 30 8104 1139; Tel: +49 30 8104 1133
First published on 11th April 2014
Abstract
Two metal organic structures composed of the ligands benzene-1,4-dicarboxylate and pyridine-2,5-dicarboxylate and bismuth cations are presented: (H2Im)[Bi(1,4-bdc)2] (1) and [Bi(pydc)(NO3)2(H2O)2]·H2O (2) (bdc = benzenedicarboxylate, H2Im = imidazole cation, pydc = pyridinedicarboxylate). Both compounds were synthesised via grinding and the crystal structure of compound (2) was solved based on its powder diffraction pattern. Compound 1 crystallised isostructurally to the dimethyl ammonium containing compound (dma)[Bi(1,4-bdc)2]. Raman spectroscopy and extended X-ray absorption fine structure (EXAFS) measurements provided additional information about the two mechanochemically synthesised metal organic compounds.
Introduction
The synthesis and thorough characterisation of novel metal organic compounds and metal organic frameworks (MOFs) are of considerable scientific interest due to their manifold properties.1–4 Possible applications of these compounds range from gas storage and gas separation to drug-carrier, magnetism, and catalysis.4–8 Metal organic compounds consist of different tunable molecular structures and have accordingly a large variety of possible architectures and topologies. Typically, transition metals are employed as metal centres in MOFs, whereas examples including main group metals are scarce.9–11 In this context, MOFs containing bismuth as the metal centre are currently investigated in greater detail.12 Connected via organic ligands, bismuth cations offer a wide range of structural diversity. Bi3+ cations are known for their stereoactive lone pair leading to interesting and versatile coordination geometries.13 Moreover, bismuth shows low toxicity even though its position in the periodic table suggests contrary findings. The pharmaceutical value of bismuth and its complexes are well documented, e.g. bismuth subsalicylate complexes have been used successfully for the treatment of gastritis and similar stomach diseases.14,15Dicarboxylate ligands are widely used to link metal centres in metal organic frameworks. In combination with Bi3+ cations as inorganic metal centres, the structural diversity is even higher. Recently, a number of metal organic compounds including bismuth centres and dicarboxylate ligands have been reported.12,16–21 Small changes in the synthesis of these compounds can lead to different crystalline structures. Thirumurugan et al. reported four different MOF structures including Bi3+ cations and benzene-1,4-dicarboxylate synthesised by changing the metal precursor or the reducing agent.12 Recently, Wibowo et al. presented solvothermal and hydrothermal syntheses of three different bismuth–pyridine-2,5-dicarboxylate structures by varying the applied base from potassium hydroxide to sodium hydroxide.16 These syntheses require a certain amount of solvents and reaction times up to several days. Here, mechanochemical syntheses are an efficient and fast alternative.22–27 Mechanochemistry prevents the use of large amounts of solvents and proceeds at ambient temperatures. Typically, pure products are obtained in high yields within minutes.
In the present work, two metal organic compounds containing Bi3+ cations as metal centres and benzene-1,4-dicarboxylate ((H2Im)[Bi(1,4-bdc)2] (1)) or pyridine-2,5-dicarboxylate ([Bi(pydc)(NO3)2(H2O)2]·H2O (2)) as ligands are presented. Both compounds were synthesised mechanochemically. Compound (1) is the solvent free analogue to the dma containing MOF reported by Thirumurugan et al. The crystal structure of (2) was solved and refined from powder diffraction data. The characterisation of the crystal structures was carried out by Raman spectroscopy and EXAFS measurements.
Results and discussion
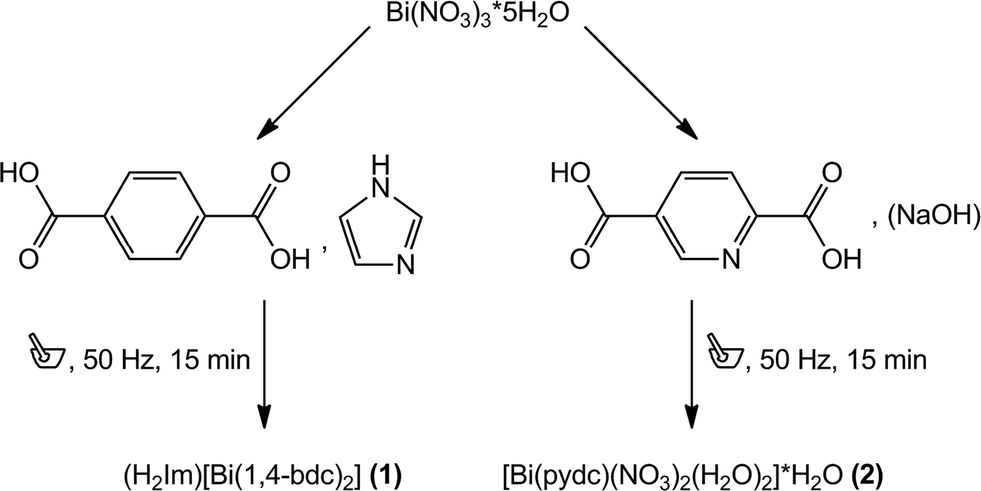
Two metal carboxylates including Bi3+ cations and benzene-1,4-dicarboxylate ((H2Im)[Bi(1,4-bdc)2] (1)) or pyridine-2,5-dicarboxylate ([Bi(pydc)(NO3)2(H2O)2]·H2O (2)) have been synthesised mechanochemically. For both syntheses, bismuth(III) nitrate pentahydrate was used as the precursor. Compound (1) was synthesised by grinding bismuth(III) nitrate pentahydrate together with the ligand benzene-1,4-dicarboxylic acid and imidazole as the proton acceptor in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 (Scheme 1). Under mechanochemical conditions, bismuth nitrate decomposes and HNO3 evaporates from the reaction mixture after opening the grinding jar. The completeness of the reaction was confirmed by powder X-ray diffraction measurements (Fig. 1, top). A comparison with database entries revealed a good agreement of the XRD pattern of (1) with the diffraction pattern of (dma)[Bi(1,4-bdc)2] (dma = dimethylammonium) (Fig. 1, top).12 It is reasonable that the protonated imidazole cations replace the dma cations in the structure obtained under mechanochemical conditions leading to an isotypic structure.
:2:4 (Scheme 1). Under mechanochemical conditions, bismuth nitrate decomposes and HNO3 evaporates from the reaction mixture after opening the grinding jar. The completeness of the reaction was confirmed by powder X-ray diffraction measurements (Fig. 1, top). A comparison with database entries revealed a good agreement of the XRD pattern of (1) with the diffraction pattern of (dma)[Bi(1,4-bdc)2] (dma = dimethylammonium) (Fig. 1, top).12 It is reasonable that the protonated imidazole cations replace the dma cations in the structure obtained under mechanochemical conditions leading to an isotypic structure.
 | ||
| Scheme 1 Reaction scheme of the syntheses of (H2Im)[Bi(1,4-bdc)2] (1) (left) and [Bi(pydc)(NO3)2(H2O)2]·H2O (2) (right). | ||
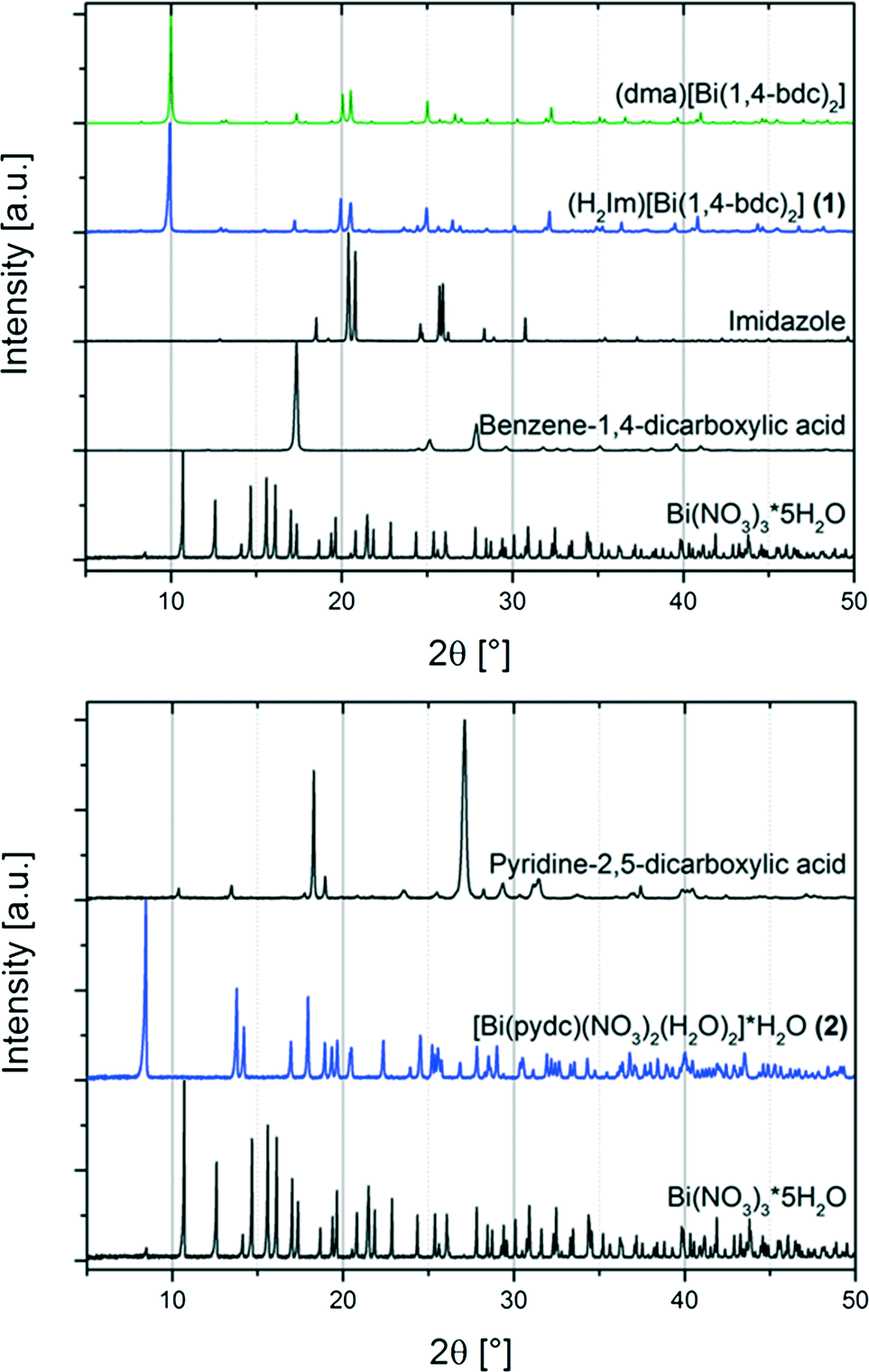 | ||
| Fig. 1 Top: powder XRD patterns of the metal organic compound (H2Im)[Bi(1,4-bdc)2] (1) (blue) and starting materials (black) bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O), benzene-1,4-dicarboxylic acid, and imidazole; and the powder XRD pattern for comparison of (dma)[Bi(1,4-bdc)2] (green) simulated based on its crystal structure (CCDC 776715, CSD-XAFBOX).12 Bottom: powder XRD patterns of the synthesised metal organic structure [Bi(pydc)(NO3)2(H2O)2]·H2O (2) (blue) and starting materials (black) bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O) and pyridine-2,5-dicarboxylic acid. | ||
For the synthesis of compound (2) bismuth(III) nitrate pentahydrate, pyridine-2,5-dicarboxylic acid and a small amount of sodium hydroxide solution (200 μL, 1 M) were ground together for 15 minutes. Sodium hydroxide acts as the reducing agent during the reaction.
The comparison of the powder diffraction patterns of bismuth(III) nitrate pentahydrate, pyridine-2,5-dicarboxylic acid and the product (2) indicates the completeness of the reaction (see Fig. 1, bottom). The diffraction pattern of (2) could not be assigned to any database entry. The structure was solved from the powder diffraction pattern leading to the first description of compound [Bi(pydc)(NO3)2(H2O)2]·H2O.
Structure of (H2Im)[Bi(1,4-bdc)2] (1)
The mechanochemically synthesised three-dimensional metal organic framework (H2Im)[Bi(1,4-bdc)2] (1) crystallises in the rhombohedral space group R-3c. The crystal data of this structure are summarised in Table 1. The asymmetric unit cell consists of two different Bi3+ cations, one benzene-1,4-dicarboxylate anion, and one imidazole cation. The two Bi3+ cations Bi1 and Bi2 are present in the ratio of 1(Bi1) to 2(Bi2). Both are holodirected with a stereochemically inactive lone pair. Bi1 is twelve-coordinated and Bi2 is nine-coordinated by six different benzene-1,4-dicarboxylate molecules. The connection between the benzene-1,4-dicarboxylate molecules and the bismuth polyhedra results in a three-dimensional framework (Fig. 2).1,3,12| Structure parameter | (H2Im)[Bi(1,4-bdc)2] (1) | [Bi(pydc)(NO3)2(H2O)2]·H2O (2) |
|---|---|---|
| Empirical formula | C19H13N2O8Bi | C7H9N3O13Bi |
| Formula weight (g mol−1) | 606.30 | 552.14 |
| Crystal system | Rhombohedral | Triclinic |
| Space group | R-3c | P1 |
| a (Å) | 17.6932(7) | 5.10416(9) |
| b (Å) | 17.6932(7) | 6.8558(3) |
| c (Å) | 29.7350(9) | 10.8069(2) |
| α (°) | 90 | 104.328(1) |
| β (°) | 90 | 90.119(1) |
| γ (°) | 120 | 104.302(1) |
| V (Å3) | 8061.42(68) | 354.23(1) |
| Z | 18 | 1 |
| D calc (g cm−3) | 2.163 | 2.557 |
| R wp | 10.4 | |
| R Bragg | 5.156 | |
| GOF | 1.64 |
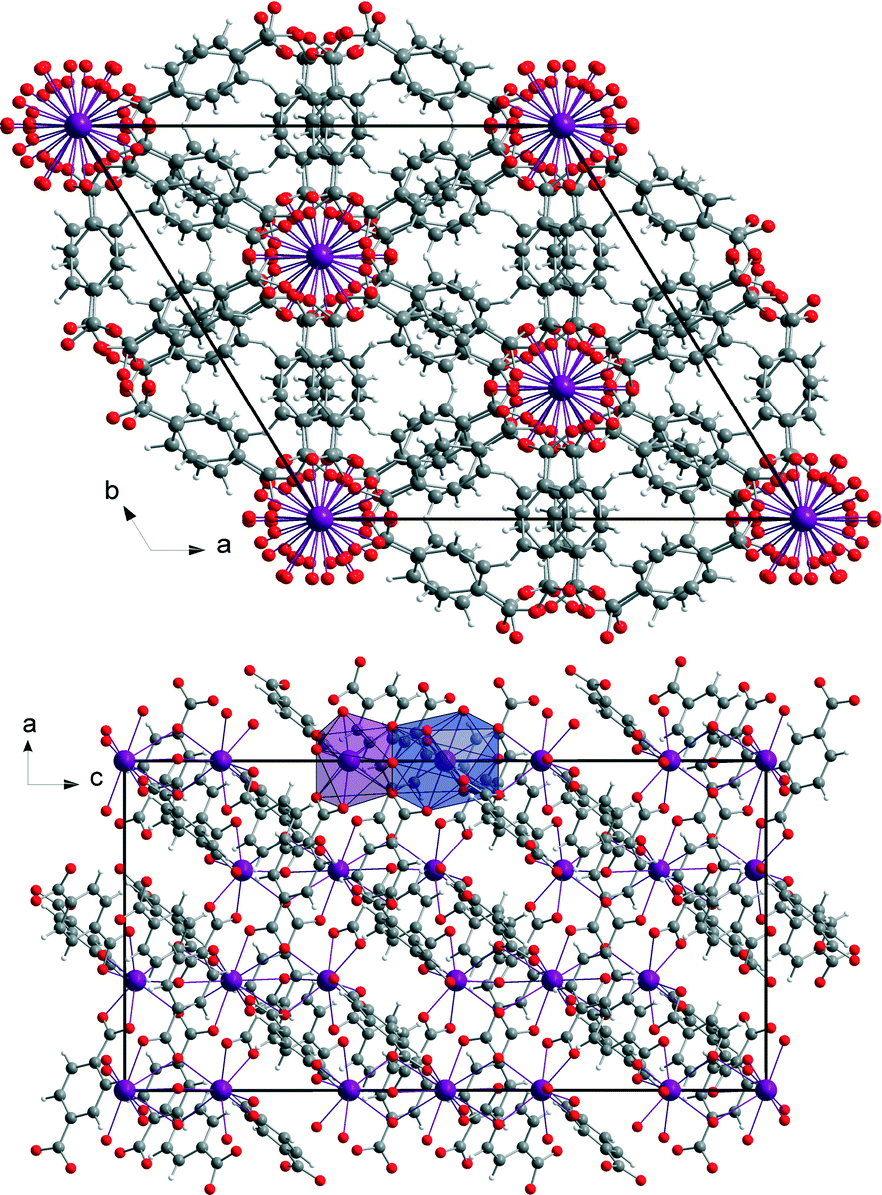 | ||
| Fig. 2 View of the three-dimensional structure (H2Im)[Bi(1,4-bdc)2] (1) seen along the c-axis (top) and the b-axis (bottom). The coordination polyhedra of both Bi3+ cations are given in blue (Bi1, twelve-coordinated) and violet (Bi2, nine-coordinated) in the view of the a–c plane (bottom). | ||
EXAFS measurements at the Bi L3-edge were performed for compound (1). These measurements provide information about the coordination environment of the Bi3+ cations. EXAFS data of structure (1) shown in real space are given in Fig. 3, left and the corresponding fit parameters are summarized in Table 2. The fitted comparison data were calculated based on the crystal structure. For the evaluation, single-scattering paths of the first coordination shell of the Bi centres were used. A good agreement of the experimental data with the fit results could be proven. The root mean square error (RMSE) of the atom distances of structure (H2Im)[Bi(1,4-bdc)2] (1) is 0.145 Å. This proves a good agreement of the theoretical atom distances with the experimentally obtained distances. Thus, the crystal structure and especially the coordination spheres of the two Bi3+ cations can be proven by EXAFS measurements.
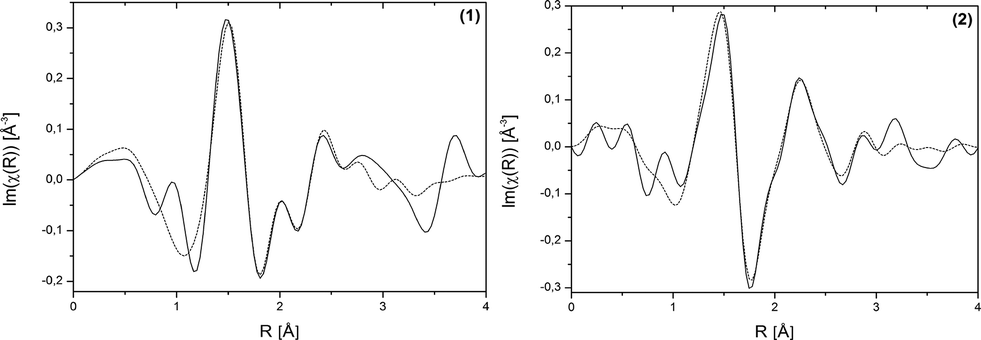 | ||
| Fig. 3 Bi L3-edge EXAFS data shown in real space of (H2Im)[Bi(1,4-bdc)2] (1) and [Bi(pydc)(NO3)2(H2O)2]·H2O (2). Experimental data – solid line, fit results – dotted line. | ||
| Sample | Scattering path | R model [Å] | R fit [Å] | R diff 2 [Å2] |
|---|---|---|---|---|
| (1) | Bi1–O1 | 2.49 | 2.35 | 0.0169 |
| Bi1–O2 | 3.04 | 3.10 | 0.0036 | |
| Bi1–C1 | 3.13 | 3.37 | 0.0576 | |
| Bi2–O3 | 2.31 | 2.21 | 0.0081 | |
| Bi2–O4 | 2.61 | 2.54 | 0.0036 | |
| Bi2–O2 | 2.67 | 2.75 | 0.0049 | |
| Bi2–C8 | 2.81 | 3.04 | 0.0529 | |
| (2) | Bi1–O6 | 2.34 | 2.26 | 0.0064 |
| Bi1–O10 | 2.53 | 2.26 | 0.0729 | |
| Bi1–N6 | 2.53 | 2.13 | 0.1681 | |
| Bi1–O13 | 2.57 | 2.36 | 0.0441 | |
| Bi1–O5 | 2.59 | 2.49 | 0.0100 | |
| Bi1–O2 | 2.65 | 2.57 | 0.0064 | |
| Bi1–O2 | 2.71 | 2.87 | 0.0256 | |
| Bi1–O11 | 2.79 | 3.10 | 0.0961 | |
| Bi1–O8 | 2.80 | 2.77 | 0.0009 | |
| Bi1–N2 | 2.96 | 3.02 | 0.0036 | |
| Bi1–O1 | 3.11 | 3.32 | 0.0441 | |
| Bi1–N1 | 3.17 | 3.49 | 0.0961 | |
| Bi1–N1 | 3.20 | 3.63 | 0.1849 | |
| Bi1–C3 | 3.24 | 3.17 | 0.0064 | |
| Bi1–C5 | 3.34 | 3.65 | 0.0961 | |
| Bi1–C1 | 3.47 | 3.83 | 0.1296 |
Structure of [Bi(pydc)(NO3)2(H2O)2]·H2O (2)
The crystal structure of [Bi(pydc)(NO3)2(H2O)2]·H2O (2) was calculated based on its powder diffraction data. Fig. 4 shows the result of the Rietveld refinement illustrating the good agreement of the simulated powder pattern with the measured one. The refinement converged at Rwp = 10.4%. The crystal data are given in Table 1. [Bi(pydc)(NO3)2(H2O)2]·H2O (2) crystallises in the triclinic space group P1, Z = 1, with unit cell parameters of a = 5.10416(9) Å, b = 6.8558(3) Å, c = 10.8069(2) Å, α = 104.328(1)°, β = 90.119(1)°, γ = 104.302(1)°, and V = 354.23(1) Å3. The asymmetric unit cell contains one Bi3+ cation, one pyridine-2,5-dicarboxylate anion, two nitrate anions and three water molecules. The Bi3+ cation is nine-coordinated and holodirected with a stereochemically inactive lone pair of electrons. The Bi3+ cation coordinates bidentate to the nitrogen atom and one carboxylate oxygen atom of the pyridine-2,5-dicarboxylate molecule. The coordination environment is completed by two water molecules, two nitrate units coordinating bidentate, and a third nitrate anion coordinating monodentate. In addition, one uncoordinated water molecule is near the second carboxylate unit of pyridine-2,5-dicarboxylate. The Bi–N bond length is 2.533 Å and the Bi–O bond lengths are in the range of 2.337–2.798 Å. The monodentate coordinated nitrate anion connects this complex to the adjacent Bi3+ cations. Consequently, a one-dimensional chain of nine-coordinated Bi3+ cations is formed (Fig. 5). The chains are arranged parallel to each other running along the a-axis.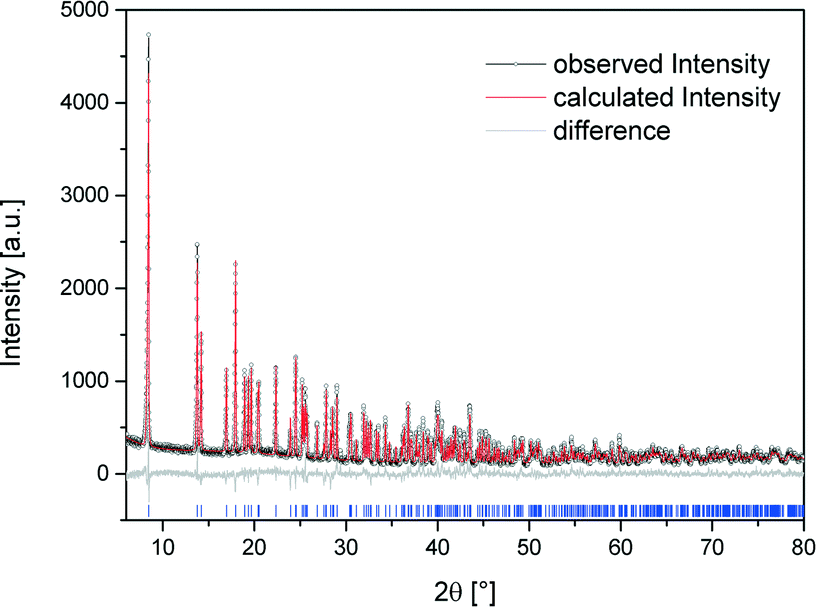 | ||
| Fig. 4 Scattered X-ray intensity of structure (2) under ambient conditions as a function of diffraction angle 2θ. The observed pattern (circles), the best Rietveld fit profile (red line), the reflection positions (blue tick marks), and the difference curve (grey line) between observed and calculated profiles are shown. The wavelength is λ = 1.54056 Å (Cu-Kα1). The R-values are Rp = 8.0%, Rwp = 10.4%; for Rp and Rwp, refer to the Rietveld criteria of fit for profile, weighted profile, and structure factor, as defined by Langford and Louer.28 | ||
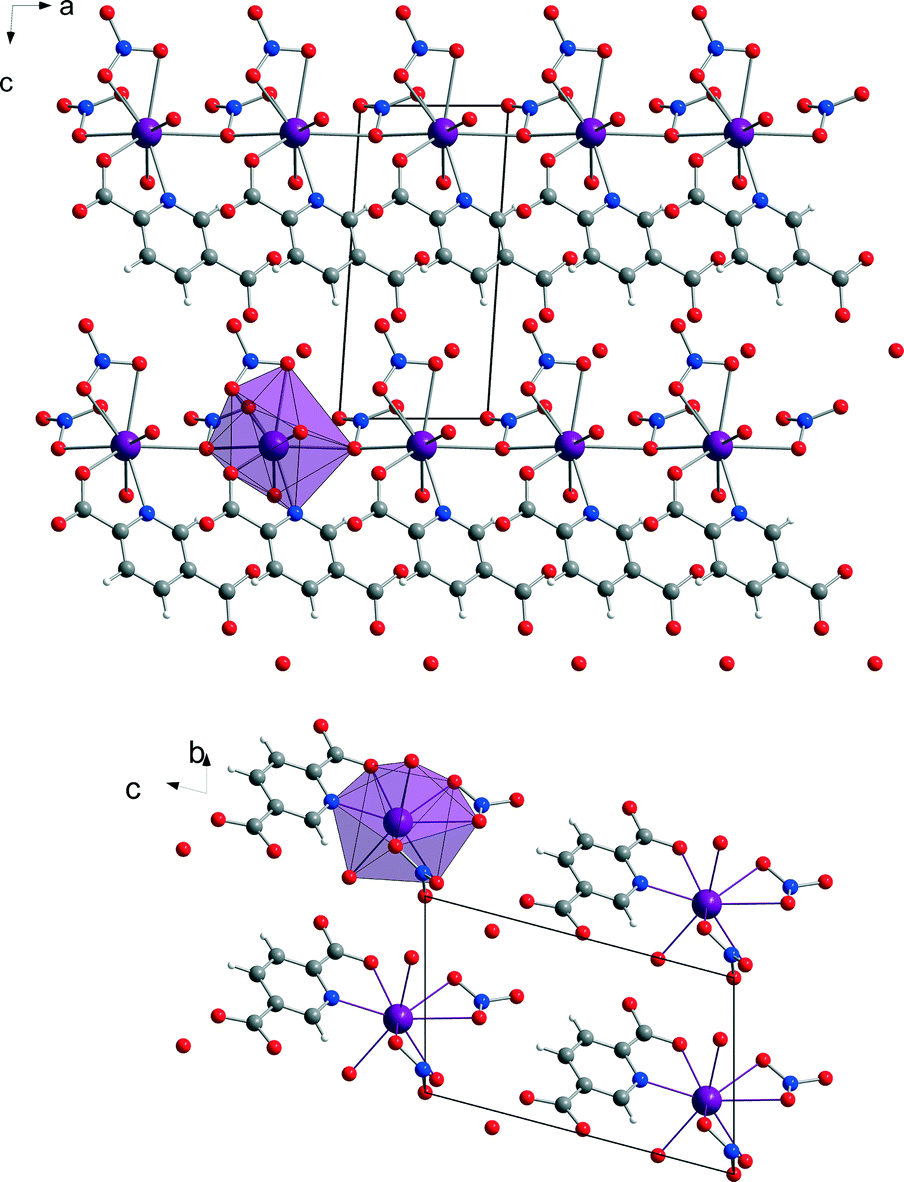 | ||
| Fig. 5 View of the chain structure [Bi(pydc)(NO3)2(H2O)2]·H2O (2) along the b-axis (top). Representation of the packing of the molecules in a projection along the a-axis (bottom). For each projection, the polyhedron around one Bi3+ cation is shown in violet. The hydrogen atoms of the water molecules are omitted for clarity. | ||
The coordination sphere around the Bi3+ cation and thereby the crystal structure of compound (2) could be supported by EXAFS measurements conducted at the Bi L3-edge. EXAFS data shown in real space and the fit parameters are given in Fig. 4, right and Table 2. The fitted comparison data, based on the crystal structure from the powder diffraction pattern, showed a good agreement with the experimental data. The first scattering path was used for evaluation. The RMSE of the atom distances of structure [Bi(pydc)(NO3)2(H2O)2]·H2O (2) is 0.249 Å. This proves a good agreement of the calculated atom distances with the experimental distances.
Like other bismuth carboxylates described in the literature, compounds (1) and (2) show the ability of bismuth to adopt a high coordination environment; typical coordination numbers are eight or nine.29–32 The Bi–O distances in these structures range between 2.21 and 3.13 Å. The Raman spectra of both structures (H2Im)[Bi(1,4-bdc)2] (1) and [Bi(pydc)(NO3)2(H2O)2]·H2O (2) as well as of the starting materials are shown in Fig. 6. Compounds (1) and (2) show characteristic Raman bands and comparison to the Raman spectra of the respective starting materials bismuth(III) nitrate pentahydrate, pyridine-2,5-dicarboxylic acid, benzene-1,4-dicarboxylic acid and imidazole proves the completeness of the reaction. Both structures (1) and (2) show a few similar Raman bands to the ligands benzene-1,4-dicarboxylic acid and pyridine-2,5-dicarboxylic acid but they are clearly shifted in the products due to deprotonation and new coordination spheres of the ligands in the final products. For the synthesis of compound (1) the protonation of the added imidazole molecule can be verified based on the Raman spectra. Two intensive bands at 1220 cm−1 and 1445 cm−1 characterize the spectrum of the imidazolium ion (H2Im).33 These bands are clearly detectable in the Raman spectrum of compound (1).
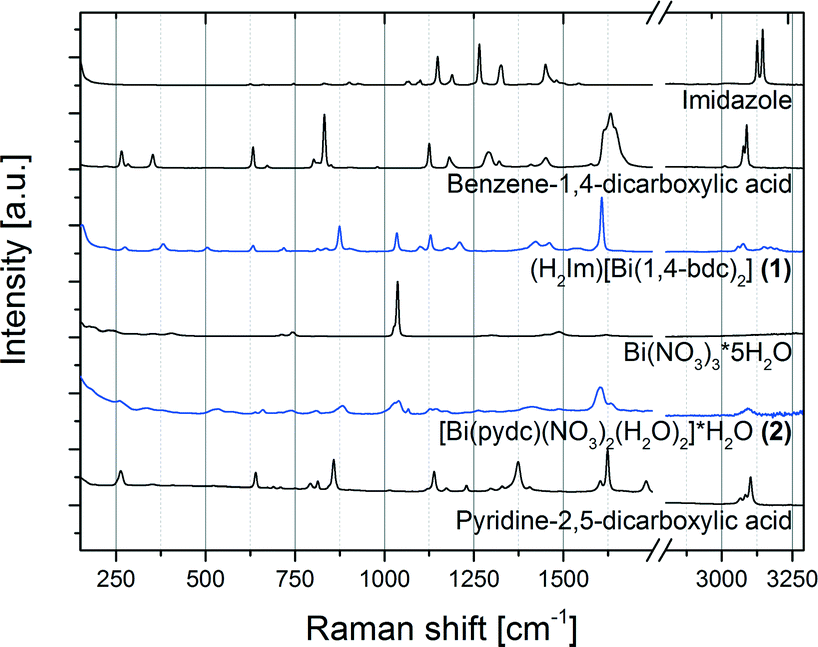 | ||
| Fig. 6 Raman spectra of the products (H2Im)[Bi(1,4-bdc)2] (1) and [Bi(pydc)(NO3)2(H2O)2]·H2O (2) (blue) in comparison with the Raman spectra of the starting materials pyridine-2,5-dicarboxylic acid, bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O), benzene-1,4-dicarboxylic acid and imidazole (black). | ||
Experimental
Materials
Bismuth(III) nitrate pentahydrate Bi(NO3)3·5H2O (98%, Alfa Aesar, Karlsruhe, Germany), benzene-1,4-dicarboxylic acid (terephthalic acid) C6H4(COOH)2 (98+%, Alfa Aesar, Karlsruhe, Germany), imidazole C3H4N2 (99%, Alfa Aesar, Karlsruhe, Germany) and pyridine-2,5-dicarboxylic acid C5NH3(COOH)2 (for synthesis, Merck, Hohenbrunn, Germany) were used without further purification. A 1 M sodium hydroxide solution was prepared using solid sodium hydroxide (for analysis, Merck, Darmstadt, Germany) and MilliQ water (18.2 MΩ, ultrapure water system seralpur Pro 90 CN, Seral, Ransbach-Baumbach, Germany).Sample preparation
Methods
Conclusions
Two metal organic compounds containing bismuth and benzene-1,4-dicarboxylate or pyridine-2,5-dicarboxylate as ligand were synthesised mechanochemically. The crystal structure of compound (1) was identified based on its powder diffraction data. The crystal structure of the bismuth and pyridine-2,5-dicarboxylate containing compound (2) could be calculated from its powder diffraction pattern. The mechanochemical synthesis pathway revealed a fast, efficient, and solvent free access to both compounds. For both structures, Raman spectra and EXAFS measurements revealed a good agreement of the calculated crystalline structures and the bismuth coordination spheres.Notes and references
- S. Bauer and N. Stock, Chem. Unserer Zeit, 2008, 42, 12–19 CrossRef CAS.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS PubMed.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- L. F. Ma, Y. Y. Wang, L. Y. Wang, D. H. Lu, S. R. Batten and J. G. Wang, Cryst. Growth Des., 2009, 9, 2036–2038 CAS.
- B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- C. Janiak, Dalton Trans., 2003, 2781–2804 RSC.
- G. Scholz, F. Emmerling, M. Dreger and E. Kemnitz, Z. Anorg. Allg. Chem., 2013, 639, 689–693 CrossRef CAS.
- S. R. Miller, E. Alvarez, L. Fradcourt, T. Devic, S. Wuttke, P. S. Wheatley, N. Steunou, C. Bonhomme, C. Gervais, D. Laurencin, R. E. Morris, A. Vimont, M. Daturi, P. Horcajada and C. Serre, Chem. Commun., 2013, 49, 7773–7775 RSC.
- R. Babarao and J. W. Jiang, Langmuir, 2008, 24, 6270–6278 CrossRef CAS PubMed.
- A. Thirumurugan and A. K. Cheetham, Eur. J. Inorg. Chem., 2010, 3823–3828 CrossRef CAS.
- W. H. Bi, N. Leblanc, N. Mercier, P. Auban-Senzier and C. Pasquier, Chem. Mater., 2009, 21, 4099–4101 CrossRef CAS.
- V. Andre, A. Hardeman, I. Halasz, R. S. Stein, G. J. Jackson, D. G. Reid, M. J. Duer, C. Curfs, M. T. Duarte and T. Friscic, Angew. Chem., Int. Ed., 2011, 50, 7858–7861 CrossRef CAS PubMed.
- P. C. Andrews, G. B. Deacon, C. M. Forsyth, P. C. Junk, I. Kumar and M. Maguire, Angew. Chem., Int. Ed., 2006, 45, 5638–5642 CrossRef CAS PubMed.
- A. C. Wibowo, M. D. Smith and H. C. zur Loye, Cryst. Growth Des., 2011, 11, 4449–4457 CAS.
- A. C. Wibowo, M. D. Smith, J. Yeon, P. S. Halasyamani and H. C. zur Loye, J. Solid State Chem., 2012, 195, 94–100 CrossRef CAS PubMed.
- A. Thirumurugan, W. Li and A. K. Cheetham, Dalton Trans., 2012, 41, 4126–4134 RSC.
- S. Busch, I. Stein and U. Ruschewitz, Z. Anorg. Allg. Chem., 2012, 638, 2098–2101 CrossRef CAS.
- M. Feyand, M. Koppen, G. Friedrichs and N. Stock, Chem. – Eur. J., 2013, 19, 12537–12546 CrossRef CAS PubMed.
- S. R. Sushrutha and S. Natarajan, Cryst. Growth Des., 2013, 13, 1743–1751 CAS.
- M. Klimakow, P. Klobes, A. F. Thunemann, K. Rademann and F. Emmerling, Chem. Mater., 2010, 22, 5216–5221 CrossRef CAS.
- M. Klimakow, P. Klobes, K. Rademann and F. Emmerling, Microporous Mesoporous Mater., 2012, 154, 113–118 CrossRef CAS PubMed.
- A. Delori, T. Friscic and W. Jones, CrystEngComm, 2012, 14, 2350–2362 RSC.
- T. Friscic, I. Halasz, V. Strukil, M. Eckert-Maksic and R. E. Dinnebier, Croat. Chem. Acta, 2012, 85, 367–378 CrossRef CAS.
- S. Heiden, L. Tröbs, K. J. Wenzel and F. Emmerling, CrystEngComm, 2012, 14, 5128–5129 RSC.
- V. Strukil, L. Fabian, D. G. Reid, M. J. Duer, G. J. Jackson, M. Eckert-Maksic and T. Friscic, Chem. Commun., 2010, 46, 9191–9193 RSC.
- J. I. Langford and D. Louer, Rep. Prog. Phys., 1996, 59, 131–234 CrossRef CAS.
- P. C. Andrews, G. B. Deacon, W. R. Jackson, M. Maguire, N. M. Scott, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 2002, 4634–4638 RSC.
- P. C. Andrews, G. B. Deacon, P. C. Junk, I. Kumar and M. Silberstein, Dalton Trans., 2006, 4852–4858 RSC.
- T. Hatanpaa, M. Vehkamaki, M. Ritala and M. Leskela, Dalton Trans., 2010, 39, 3219–3226 RSC.
- P. C. Andrews, V. L. Blair, R. L. Ferrero, P. C. Junk and I. Kumar, Chem. Commun., 2013, 49, 2870–2872 RSC.
- L. M. Markham, L. C. Mayne, B. S. Hudson and M. Z. Zgierski, J. Phys. Chem., 1993, 97, 10319–10325 CrossRef.
- T. V. 2.0, Bruker AXS, Karlsruhe (Germany), 2000 Search PubMed.
- J. Laugier and B. Bochu, Chekcell, 2001 Search PubMed.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- H. Riesemeier, K. Ecker, W. Gorner, B. R. Muller, M. Radtke and M. Krumrey, X-Ray Spectrom., 2005, 34, 160–163 CrossRef CAS.
- W. Gorner, M. P. Hentschel, B. R. Muller, H. Riesemeier, M. Krumrey, G. Ulm, W. Diete, U. Klein and R. Frahm, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467, 703–706 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503–5513 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC-978120 contains the supplementary crystallographic data for [Bi(pydc)(NO3)2(H2O)2]·H2O (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce42633e |
| This journal is © The Royal Society of Chemistry 2014 |