 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A C-shaped p-sulfonatocalix[4]arene-based supermolecule exhibiting mutual-inclusion and bilayer insertion of dipicolinate†
Ahmad
Husain
and
Clive L.
Oliver
*
Centre for Supramolecular Chemistry Research, Department of Chemistry, University of Cape Town, Rondebosch 7701, South Africa. E-mail: clive.oliver@uct.ac.za; Fax: +27 21 6505195; Tel: +27 21 6503830
First published on 19th December 2013
Abstract
A p-sulfonatocalix[4]arene-based supermolecule, [Ce3(PDA)4(C4AS)2·13H2O](NO3)·25H2O 1 was prepared by slow evaporation of a solution of Ce(NO3)3·6H2O, 2,6-pyridinedicarboxylic acid (PDA) and pentasodium p-sulfonatocalix[4]arene salt. The molecular and crystal structure of 1 was determined by single crystal X-ray structural analysis and characterized by thermal methods (TG, DSC and hot-stage microscopy) and Hirshfeld analysis. The supermolecule is C-shaped with alternating calixarene and PDA ligands linked to three Ce(III) cations. The supermolecules exhibit mutual-inclusion via its PDA ligands with the remaining PDA ligands inserted into the calixarene bilayer. The bilayer consists of an “up–down” arrangement of calixarenes, with the inserted PDA ligands effecting a zig-zag propagation of the calixarenes within the bilayer.
Introduction
Due to their high degree of structural flexibility, calix[n]arenes have been extensively explored in host–guest supramolecular chemistry involving various kinds of interactions such as hydrogen bonding, metal coordination, van der Waals, π⋯π and cation⋯π interactions.1,6 Water-soluble p-sulfonatocalix[4]arene (C4AS) in its usual cone shape, and usually employed as a sodium salt, has shown remarkable inclusion properties. The C4AS anions usually assemble into bilayers as a result of an “up–down” arrangement of the calixarene anions or sometimes into more complicated supramolecular architectures afforded by an “up–up” arrangement. These structures often include guest molecules of various sizes and shapes without or with metal coordination.Raston and co-workers have produced molecular capsules which include various guest molecules such as crown ethers, aza-macrocycles, lanthanide–imidazolium functionalised carboxylate and polyphenylphosphonium ions.2 Atwood et al. reported more complicated supramolecular assemblies including cuboctahedral and icosahedral spheroidal arrays made up of twelve calixarenes.1 Dalgarno et al. produced and reviewed a number of examples based on p-sulfonatocalix[n]arenes (where n = 4–8) assembled with metal ions (amongst other components) to facilitate the assembly of large metallosupramolecular capsules or capsule-like architectures.3 Recently Crowley and co-workers presented a high-resolution crystal structure of a typical supramolecular protein–p-sulfonatocalix[4]arene complex, displaying the interaction of C4AS with the outer surface of the cytochrome c-protein.4
Usually planar aromatic guests can interact with the hydrophobic cone-like cavity of the C4AS through π⋯π and “non-classical” H-bonding interactions. Therefore, they may influence the special arrangements of the resulting complexes. We noted that the 12-calixarene assembly produced by Atwood et al. involved two p-sulfonatocalix[4]arenes and two pyridine N-oxide ligands linked via a lanthanide metal to form a “hinged” supermolecule. Keeping this in mind we have chosen 2,6-pyridinedicarboxylic acid (PDA), which may further metallate using its carboxylate functionality and may thus influence the packing of the calixarene anions. Herein we report the preparation and structure of a C-shaped supermolecule by the reaction of pentasodium p-sulfonatocalix[4]arene salt, 2,6-pyridinedicarboxylic acid and Ce(NO3)3·6H2O in aqueous media which exhibits mutual-inclusion and bilayer insertion of PDA ligands.
Experimental section
Materials and physical measurements
All chemicals were of reagent grade, purchased from commercial sources, and used without further purification. Pentasodium p-sulfonatocalix[4]arene salt was prepared according to literature methods.6Hot-stage microscopy was performed on a Nikon SMZ-10 stereoscopic microscope fitted with a Linkam THMS600 hot stage and a Linkam TP92 control unit. Samples were placed under silicone oil on a cover slip and heated at 10 °C min−1. Thermal events were monitored using a Sony Digital Hyper HAD colour video camera and captured using the Soft Imaging System program analysis.
TG/DTG measurements were performed at a heating rate of 10 °C min−1 in the temperature range 25–600 °C under a dry nitrogen flow of 60 mL min−1 on a TGA Q500 instrument. Approximately 2–5 mg of sample was placed in an open aluminum crucible.
DSC measurements were performed at a heating rate of 10 °C min−1 in the temperature range 25–400 °C under a dry nitrogen flow of 50 mL min−1 on a DSC Q200 instrument. Approximately 1–2 mg of sample was placed in an aluminum pan with a lid. A sealed and empty pan was used as a reference.
Preparation of [Ce3(PDA)4(C4AS)2·13H2O](NO3)·25H2O
2,6-Pyridinedicarboxylic acid (34 mg, 0.2 mmol) was dissolved in water–methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2 mL) by heating. An aqueous solution (2 mL) of Ce(NO3)3·6H2O (43 mg, 0.1 mmol) was then added, producing a yellow solution. HNO3 (1 mL, 1 M) was added to acidify the solution, which turns colourless. 120 mg of the pentasodium p-sulfonatocalix[4]arene salt (in 2 mL H2O) was added and the solution was then left open in air for crystallization to occur. Colourless block-like crystals formed within 2 days.
:1, 2 mL) by heating. An aqueous solution (2 mL) of Ce(NO3)3·6H2O (43 mg, 0.1 mmol) was then added, producing a yellow solution. HNO3 (1 mL, 1 M) was added to acidify the solution, which turns colourless. 120 mg of the pentasodium p-sulfonatocalix[4]arene salt (in 2 mL H2O) was added and the solution was then left open in air for crystallization to occur. Colourless block-like crystals formed within 2 days.
Single crystal X-ray diffraction analysis and structure determination
A suitable single crystal of 1 was selected and mounted on a cryoloop in oil. Data collection was carried out on a Bruker DUO APEX II CCD diffractometer using graphite monochromated Mo Kα (λ = 0.71073 Å) radiation with the crystal cooled to 173(2) K using an Oxford Cryostream-700. Data reduction and cell refinement were performed using SAINT-Plus,7 and the space group was determined from systematic absences by XPREP8 and confirmed using the program layer.9The X-ray diffraction data were corrected for Lorentz-polarization factor and scaled for absorption effects by multi-scan using SADABS.10 The structure was solved by direct methods and implemented in SHELXS-97.11 A refinement procedure by full-matrix least-squares methods based on F2 values against all reflections was performed by SHELXL-97, including anisotropic displacement parameters for all non-H atoms.
Calculations concerning the molecular geometry, the affirmation of the chosen space group and analysis of hydrogen bonds were performed using PLATON.12 Molecular graphics were produced with X-Seed13 and MERCURY.14 Crystal parameters, data collection, and refinement results are summarized in Table S1.†
CCDC 927681 contains the supplementary crystallographic data for this paper.
Results and discussion
The supermolecule crystallized from an aqueous solution of pentasodium p-sulfonatocalix[4]arene (C4AS) salt, 2,6-pyridinedicarboxylic acid (PDA) and cerium nitrate hexahydrate with the pH adjusted to 2 using 1 M HNO3 (Scheme 1).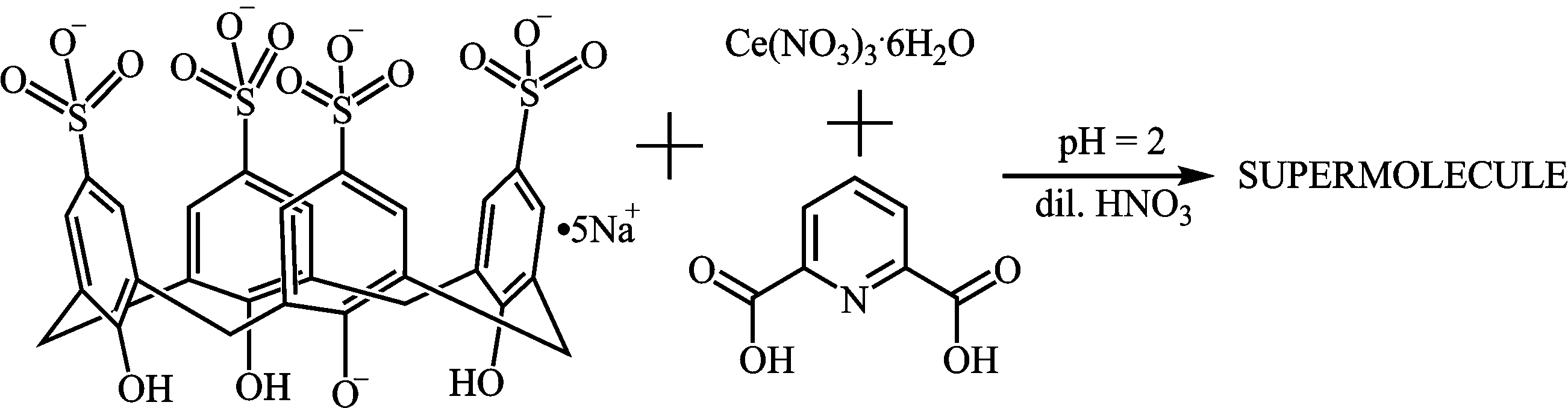 | ||
| Scheme 1 Preparation of the supermolecule. | ||
Single crystal X-ray diffraction analysis reveals that the asymmetric unit of 1 is a supermolecule (Fig. 1) which comprises three Ce(III) cations, two p-sulfonatocalix[4]arene anions (C4AS-A and C4AS-B), four PDA ligands, one nitrate anion, thirteen coordinated water molecules and twenty five uncoordinated lattice water molecules.
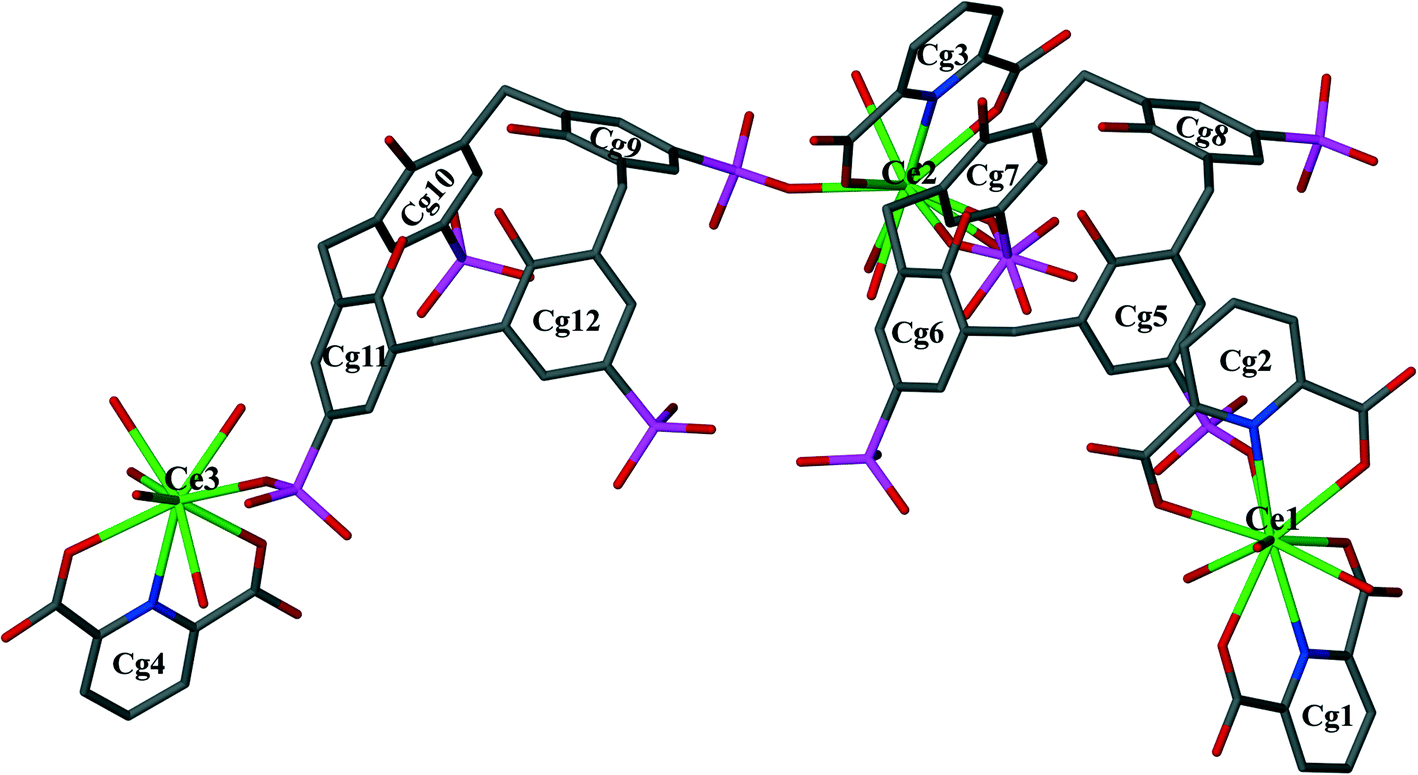 | ||
| Fig. 1 The asymmetric unit displaying C-shaped supermolecule 1. Water molecules, nitrate anion and H-atoms have been omitted for clarity. Aromatic rings have been numbered to show centre of gravity. Disorder of the sulfonate group on Cg7 is shown. | ||
The C-shaped supermolecule consists of three Ce(III) cations, two calixarenes and four tridentate PDA ligands. Ce(1) exhibits a ten-coordinate environment completed by three water molecules, four O and two N atoms of two PDA (Cg1 and Cg2) ligands and one sulfonate O atom of C4AS-A. Ce(2) is coordinated to one N and two O atoms of one PDA (Cg3) ligand, one O atom from a sulfonate group on C4AS-A and C4AS-B and five water molecules to complete a ten-coordination environment. The skeletal structure around Ce(1) and Ce(2) forms a slightly distorted bicapped square antiprism (Fig. 2). For Ce(1), the N1 and N2 atoms, which are at relatively longer distances than the O atoms from Ce(1) cation, are on the bicapped positions with the bond angle N(2)–Ce(1)–N(1) being 172.48(14)° while in the case of Ce(2), the N3 and O86 atoms are on the bicapped positions with the bond angle N(3)–Ce(2)–O(86) being 174.5(4)°. Ce(3) is coordinated to one N and two O atoms from one PDA (Cg4) ligand, one O atom of sulfonate from C4AS-B and five water molecules to form its monocapped square antiprismatic nine-coordination environment having N4 at the capped position. The Ce–O and Ce–N bond distances are in the 2.454(4)–2.736(13) Å and 2.674(5)–2.731(5) Å ranges, respectively, which compare well with literature values.15 Selected bond lengths and angles are given in Table S2.†
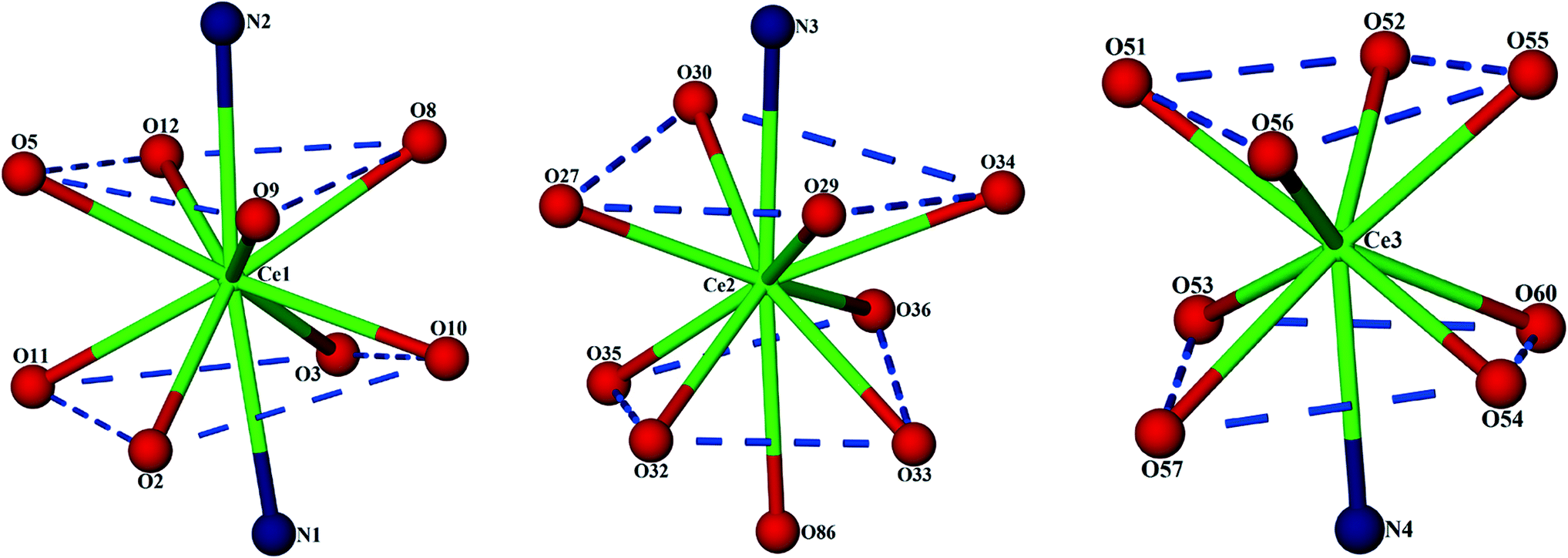 | ||
| Fig. 2 Display of distorted capped square antiprisms of all the three Ce(III) coordination environments. | ||
The p-sulfonatocalix[4]arene anion is present in its usual flattened cone conformation with C2v symmetry (cone angles between the plane of opposite aromatic rings are 30.42°, 89.26° and 42.49°, 86.63° for C4AS-A and C4AS-B, respectively). The p-sulfonatocalix[4]arenes take on an overall charge of 4− each (due to the pH of the crystallization medium being 2), with two of the opposing sulfonic groups of each C4AS bonded to a Ce(III) cation. All the carboxylic acid groups of the PDA ligands remain protonated except for O6 (Cg2) which results in a total negative charge of 9− to balance with the three Ce(III) cations to yield a supermolecule. We do note that the nitrate anion does not have a counterion in the modelled structure which is probably due to the lack of ability to model a hydronium ion from the X-ray data. Each C4AS includes a pyridyl ring from neighbouring supermolecules involving weak and slanted π⋯π interactions to the phenyl rings of the host C4AS. For C4AS-A these interactions are described by Cg4⋯Cg5 = 4.620(3) Å and Cg4⋯Cg7 = 4.789(3) Å with planar angles of 12.8(3)° and 17.6(3)°, respectively, whilst for C4AS-B these interactions are Cg1⋯Cg10 = 4.642(4) Å and Cg1⋯Cg12 = 4.402(4) Å with planar angles of 37.9(3)° and 31.2(3)°, respectively. The inclusion of the pyridyl ring in C4AS-B is slightly tilted and not orthogonal to any of the host aromatic rings, while the inclusion of a pyridyl ring in C4AS-A is nearly orthogonal to aromatic rings Cg6 and Cg8 (Fig. 3).
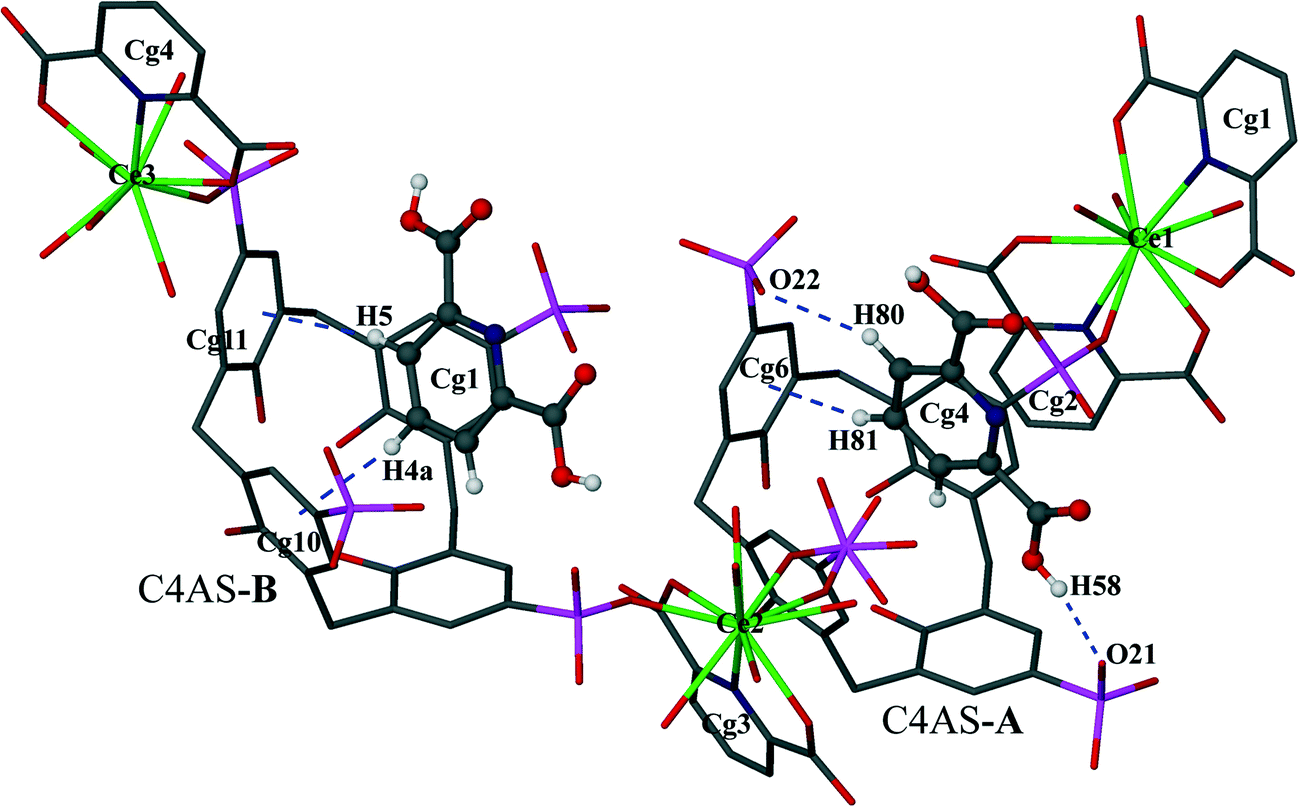 | ||
| Fig. 3 Display of inclusion of pyridyl rings of the PDA ligands by C–H⋯π, C–H⋯O and O–H⋯O interactions. Atoms of pyridyl rings inserted into the cavity are shown as spheres of arbitrary radii. Blue broken lines depict CH⋯π, CH⋯O(S) and OH⋯O(S) interactions. | ||
In the supermolecule, the inclusion of one of the pyridyl rings (Cg1) in the cavity of C4AS-B is further supported by C–H⋯π interactions involving adjacent meta and para-hydrogens (H4a, H5) to a pair of adjacent aromatic rings (Cg10 and Cg11), with C–H⋯centroid distances of 2.95 Å and 2.76 Å, respectively and C–H⋯centroid angles of 123° and 153°, respectively (Fig. 3), while the inclusion of a pyridyl ring (Cg4) in the cavity of C4AS-A is supported by one C–H⋯π interaction involving a meta hydrogen (H81) to an aromatic ring (Cg6), with a C–H⋯centroid distance of 2.59 Å and a C–H⋯centroid angle of 160° (Fig. 3). One of the meta hydrogen atoms of PDA (Cg4) also interacts through a C–H⋯O hydrogen bond to the neighbouring sulfonic group (Fig. 3) of C4AS-A, with a H(80)⋯O(24) distance of 2.53 Å (Table S3†). A strong hydrogen bond (H58⋯O21 = 1.82 Å) from a carboxylic proton to one of the sulfonic groups of C4AS-A further stabilizes the inclusion of the PDA ring (Cg4). This may be responsible for the tilting of the PDA ring (Cg4) towards the Cg6 ring (Fig. 3). The two bowl shaped cavities of C4ASs are nearly parallel but offset relative to each other, presumably to accommodate the PDA rings and for geometrical complementarity. Each unit cell contains four supermolecules (two interdigitated pairs) arranged in a centrosymmetric fashion which can be considered as a translationally repeating motif (Fig. 4a). In each interdigitated pair a pyridyl group (Cg1) from each supermolecule resides in a cavity of the other, whilst the remaining cavities of a particular pair are filled by a pyridyl group (Cg4) from neighbouring pairs to result in a mutual-inclusion packing arrangement. The remaining two pyridyl rings Cg2 and Cg3 intercalate between the C4ASs aryl rings which involve π⋯π interactions.
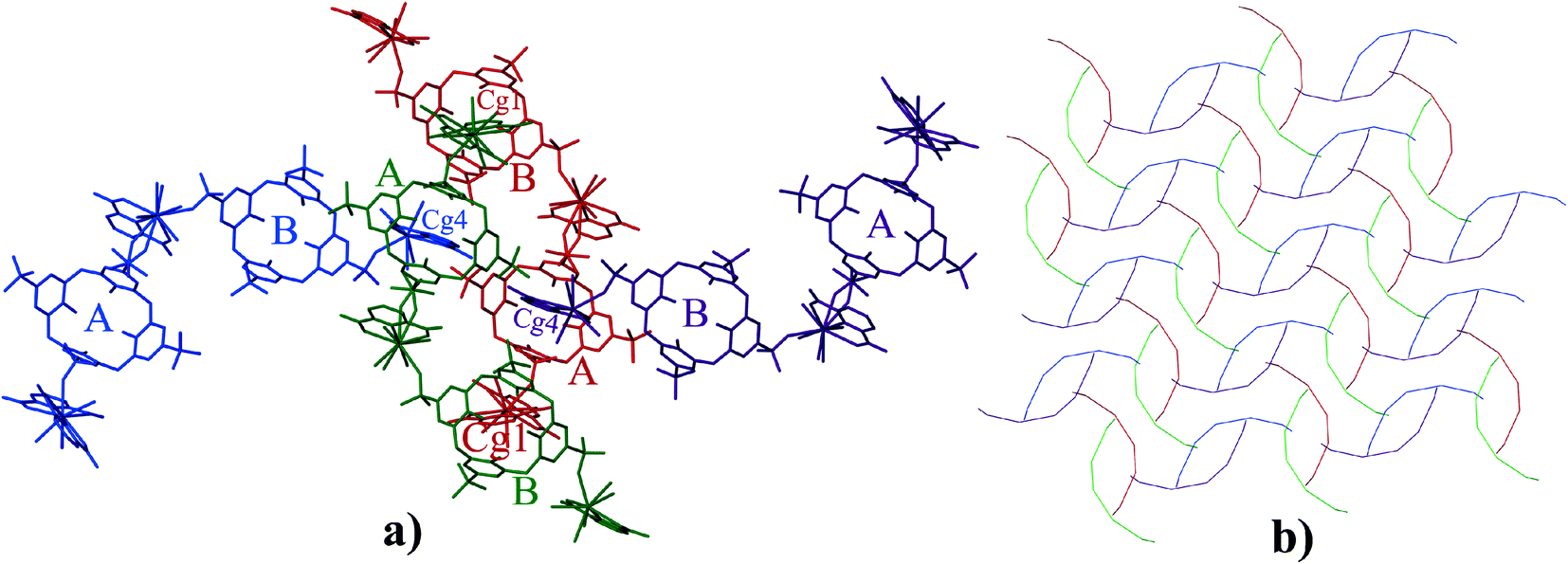 | ||
| Fig. 4 a) Projection of four supermolecules of 1 in the unit cell. The four symmetry-related molecules are represented in different colours and hydrogen atoms have been removed for clarity. b) Schematic representation of repeating pattern of the four supermolecules. | ||
The average deviation of each hydroxyl O atom (O15–O18) from the O4 mean plane in C4AS-A is 0.1679 Å while the hydroxyl O atoms (O39–O42) of C4AS-B are deviated by 0.1998 Å on average from this plane (Fig. 5). The dihedral angles between the aromatic rings and the mean plane of the four phenolic oxygens which evaluate the relative inclinations of each aryl unit relative to the O4 mean plane are 74.34(14)°, 45.66(16)°, 75.26(16)° and 43.77(14)° for Cg5, Cg6, Cg7 and Cg8, respectively, for C4AS-A whilst for C4AS-B they are 56.76(14)°, 66.81(17)°, 29.91(20)° and 70.30(13)° for Cg9, Cg10, Cg11 and Cg12, respectively. This is consistent with the aromatic rings of the C4ASs being splayed to accommodate the PDA ligands (Fig. 5). It can be clearly seen that the hydrophobic cavity in C4AS-B is more available for deeper penetration than that of the C4AS-A, which demonstrates the conformational flexibility of p-sulfonatocalix[4]arene to act as a host molecule.
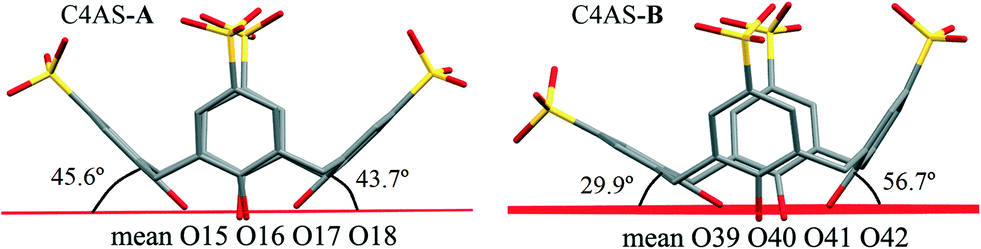 | ||
| Fig. 5 Display of two splayed p-sulfonatocalix[4]arenes in supermolecule 1. | ||
The calixarenes are assembled into bilayers with two of the unique PDA moieties intercalated into the bilayer (Fig. 6). It has been shown that in the cases of certain C4AS transition metal complexes, and recently, lanthanide complexes, an aromatic organic substrate can be intercalated into the bilayer.5,16 The C4ASs comprising the bilayer are not aligned and form a zigzag arrangement, most probably as the result of the intercalated PDA ligands. The calixarenes propagate via π-stacking (Fig. 7a and Table S4†) and OH⋯OS interactions from one calixarene to another within the bilayer in an up–down fashion along the b-axis. These chains are further extended along the c-axis with intercalating PDA ligands, also in a zig-zag arrangement through π-stacking (Fig. 6 and 7b, Table S4†) forming 2D sheets.
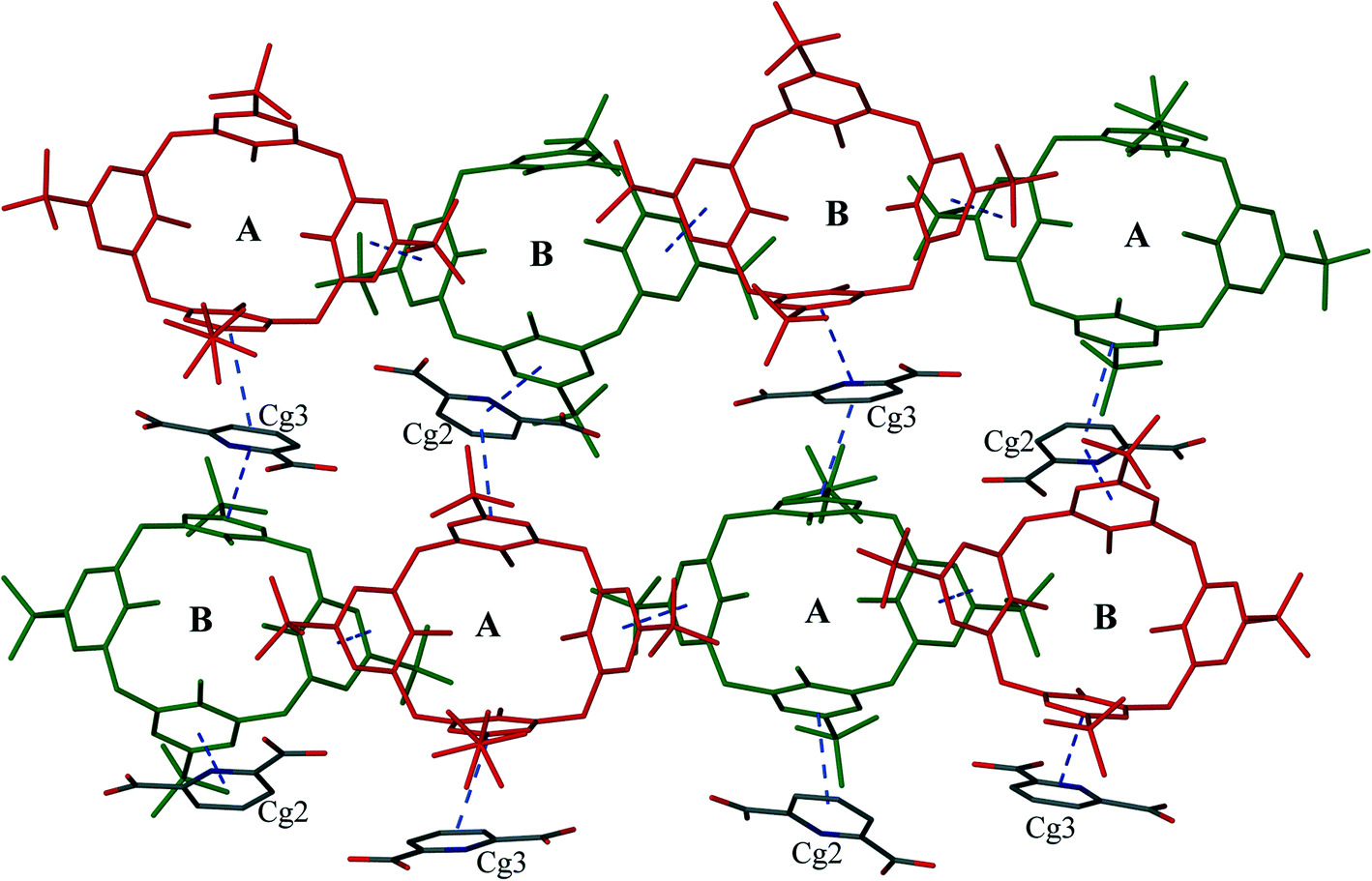 | ||
| Fig. 6 Alternately upwards and downwards arrangement of C4ASs displaying sandwiched pyridyl rings between the aryl rings of C4ASs. Broken lines depict π⋯π interactions. Upward-pointing molecules (red) and the downward-pointing molecules (green). | ||
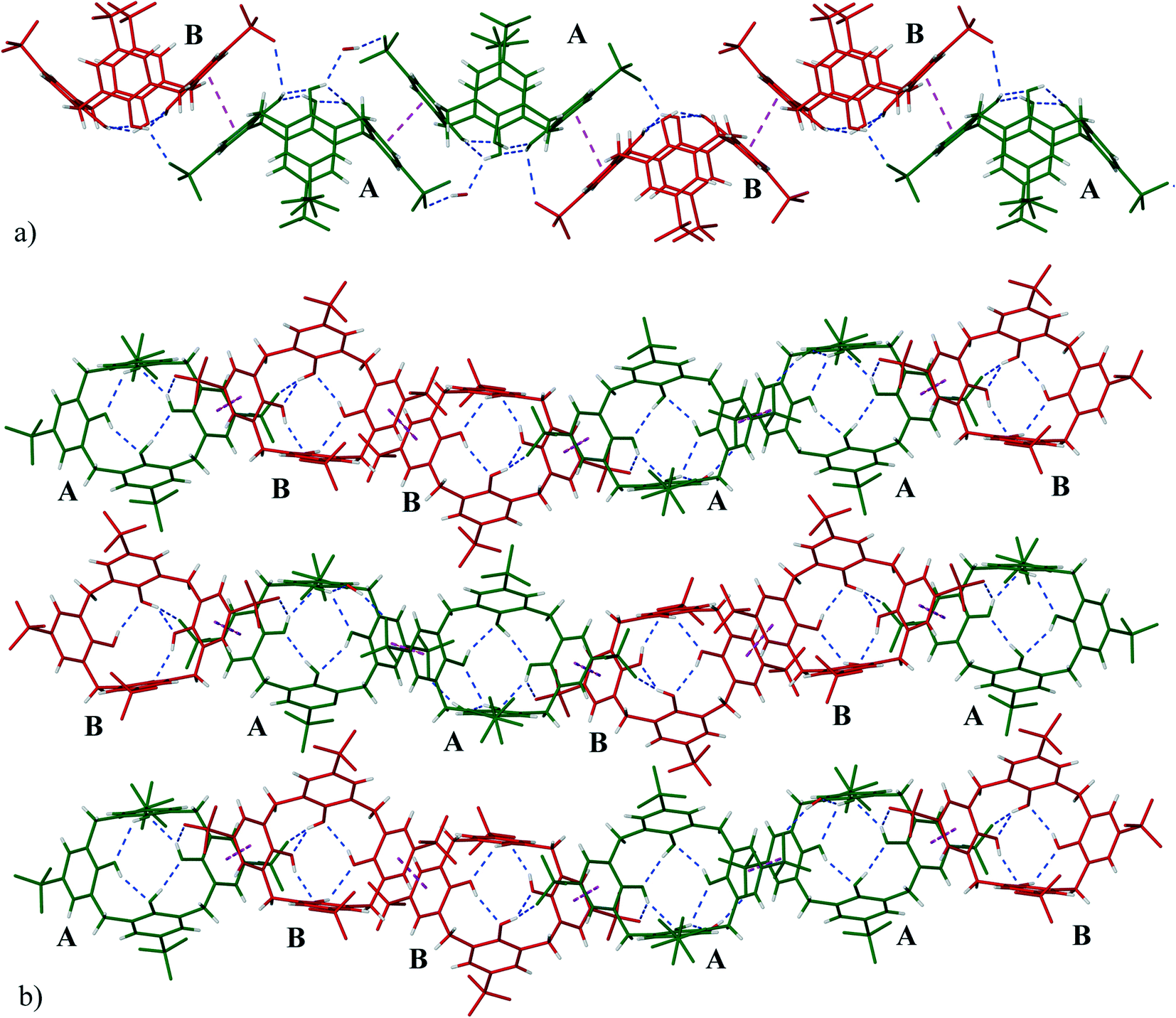 | ||
| Fig. 7 a) A view of the zig-zag chains running parallel to the b-axis (C4ASs-A in green and C4ASs-B in red). b) The extended (along c-axis) structure of 1 showing ‘up–down’ antiparallel bilayer arrangements (AABBAABB…). The hydrogen bonding between hydroxyl groups and sulfonate groups are shown as dashed blue lines while π⋯π interactions are shown as dashed pink lines. | ||
The sulfonate group of C4AS-A is hydrogen bonded to the hydroxyl group of C4AS-B (O42⋯O20, separation of 2.699 Å, Table S3†) complemented by a slightly displaced π-stacking of phenyl groups (centroid Cg8⋯Cg11 separation of 3.597(4) Å, a torsion angle of 5.1(3)° between aromatic faces). All four phenolic hydroxyl groups hydrogen bond to one another at the lower rim (Table S3†). One of the phenolic oxygen atoms (O17) of C4AS-A forms a bifurcated hydrogen bond with the neighbouring phenolic oxygen and lattice water molecule (O69) which in turn hydrogen bonds to O22 of the sulfonate of the same ring (Cg6) of the adjacent downward C4AS-A. In a similar manner O40 of C4AS-B forms a bifurcated hydrogen bond with the neighbouring phenolic oxygen and coordinated water (O34) molecule.
The water molecules are located around the sulfonate groups of the C4ASs as well as clustered near the [Ce(PDA)n·mH2O] moieties (Fig. 8). The hydrophilic and hydrophobic layers are 9.6 and 5.4 Å thick, respectively. Numerous water molecules are present in the crystal lattice; however, the X-ray data quality was not sufficient to allow all hydrogen atoms on these molecules to be located and the hydrogen bonding network is implied through numerous close O⋯O contacts ranging from 2.49 to 3.0 Å.
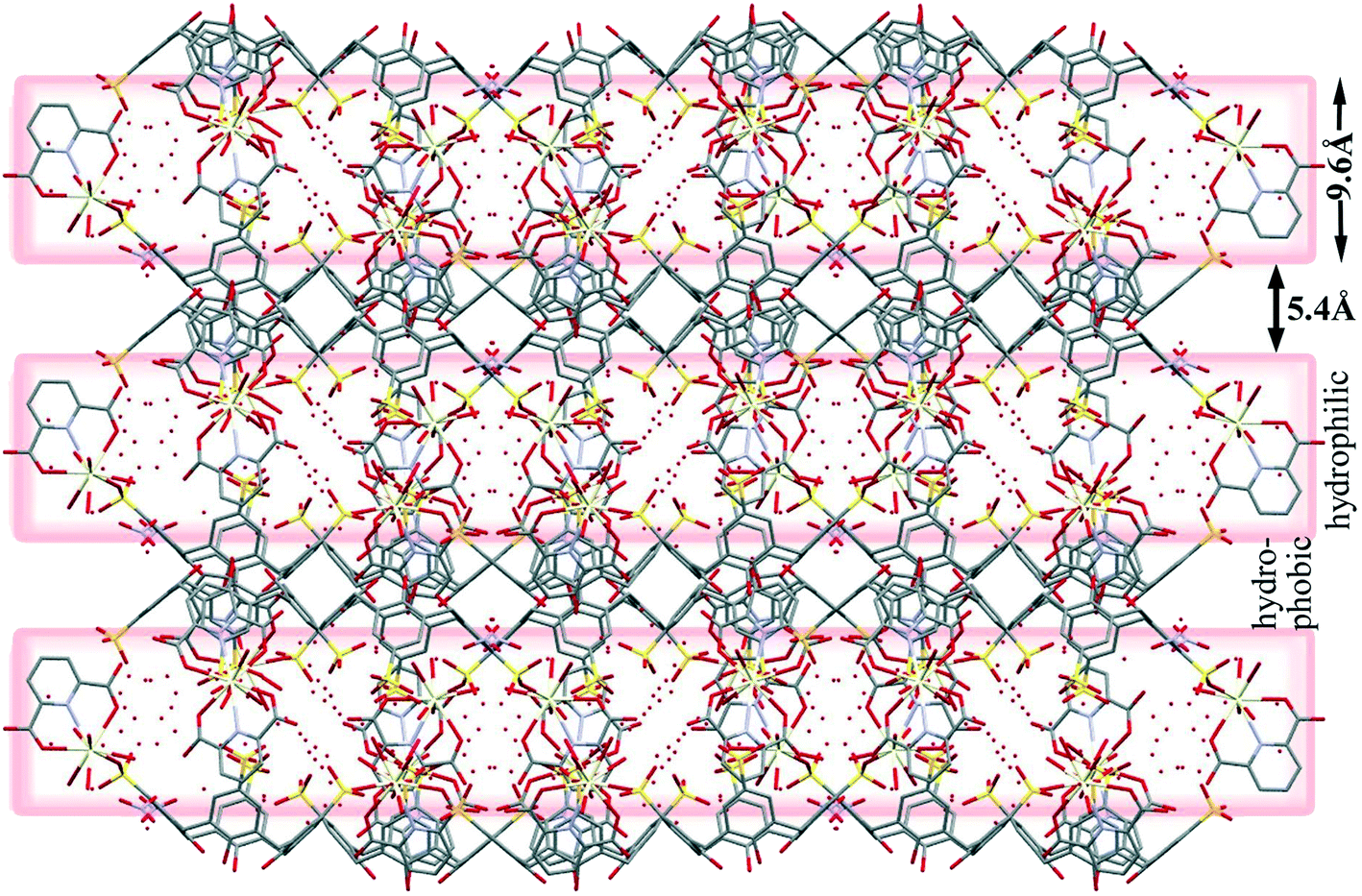 | ||
| Fig. 8 Display of hydrophilic and hydrophobic layers. | ||
Thermal analysis
The TGA data of complex 1 indicate two mass losses (Fig. S1†). The first mass loss 45–135 °C (13.54%) corresponds to the loss of all the lattice water molecules (calculated: 13.7%). A long range mass loss in the range 140–600 °C (40%) corresponds to the loss of all the coordinated water molecules along with the nitrate ion subsequently followed by structural decomposition. DSC shows only one broad endotherm peak which should be due to the loss of solvent molecules (Fig. S1†). Hot stage microscopy indicates that visual loss of water occurs for 1 at the temperature 110 °C (Fig. S2†).Hirshfeld surface analysis
Hirshfeld surface analysis is a unique17 and novel method to visualize the intermolecular interactions of molecular crystals by colour-coding short or long contacts, the colour intensity indicating the relative strength of the interactions. The Hirshfeld surfaces of the title complex are illustrated in Fig. 9, showing surfaces that have been mapped over dnorm (−1.281 to 1.417Å), while 2D fingerprint plots (plot of diversus de) which identify the occurrence of different kinds of intermolecular interactions are shown in Fig. 10.18 The function dnorm is a ratio encompassing the distances di and de. de is the distance from the point to the nearest nucleus external to the surface while di is the distance to the nearest nucleus internal to the surface. dnorm is negative or positive when the intermolecular contacts are shorter or longer than the sum of the relevant van der Waals radii, respectively. The dnorm Hirshfeld surfaces are displayed using a red–white–blue colour scheme, where red highlights are used for shorter contacts, white is used for contacts around the van der Waals radii separation and blue is for longer contacts.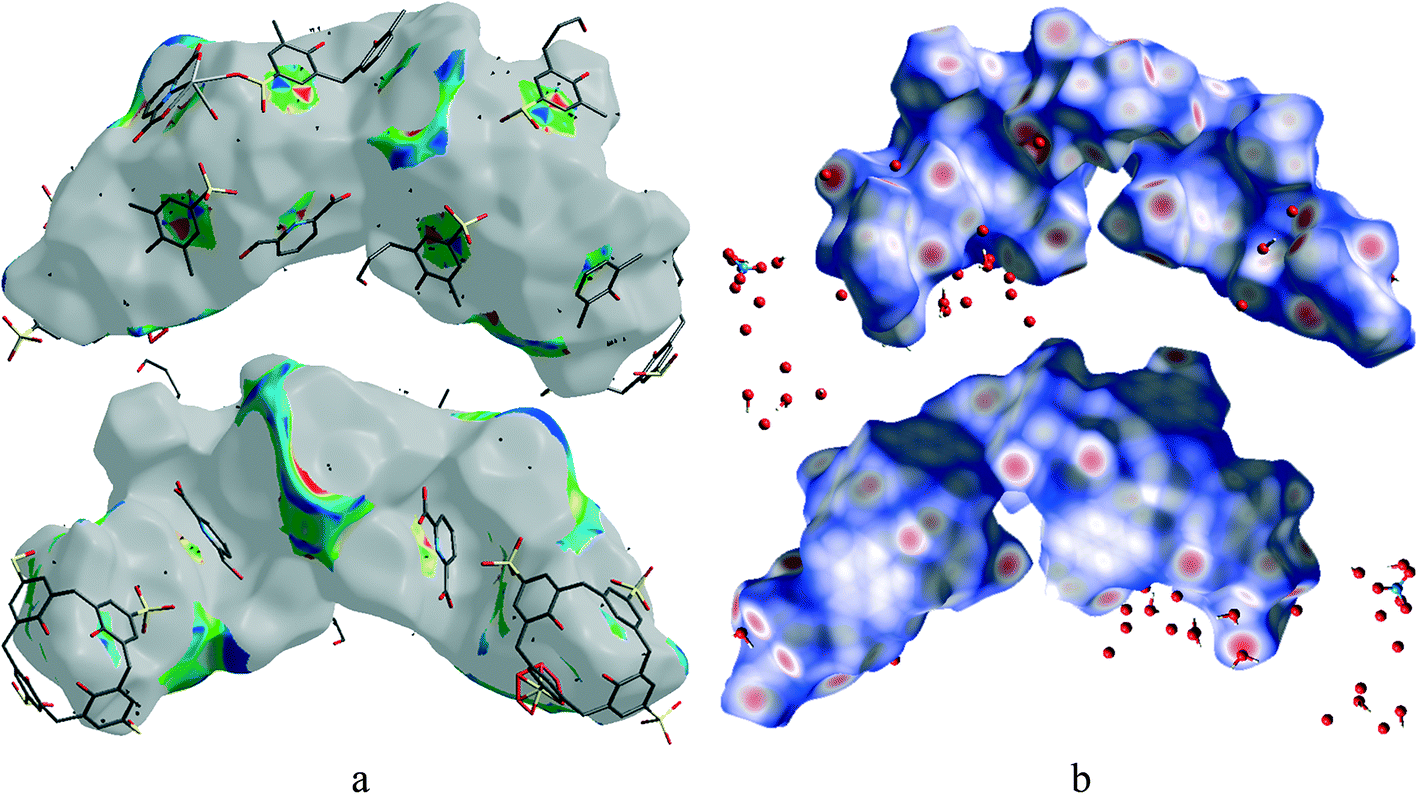 | ||
| Fig. 9 Front and back views of the Hirshfeld surface representation for 1 mapped with (a) shape-index and (b) dnorm with all types of intermolecular interactions considered. | ||
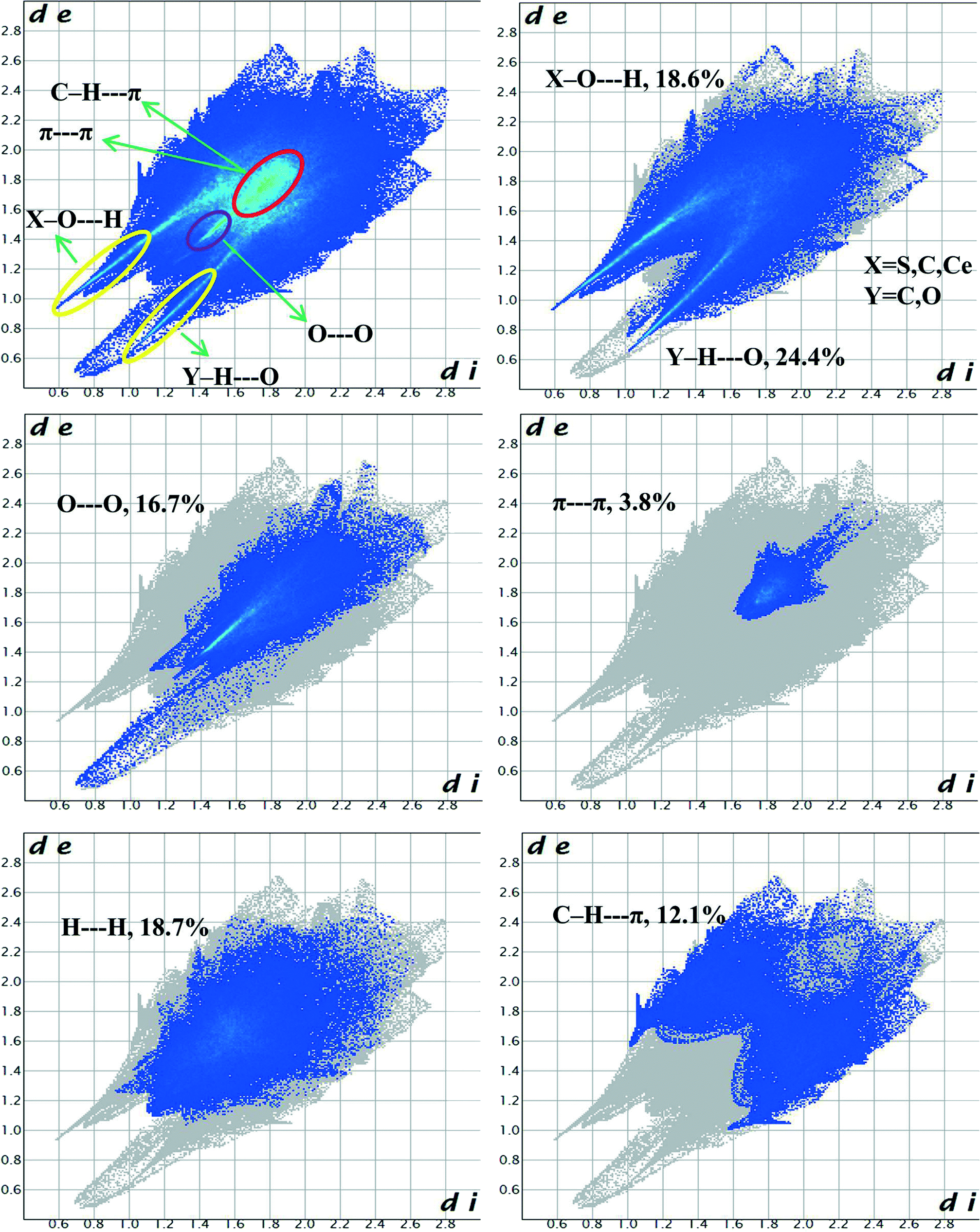 | ||
| Fig. 10 Fingerprint plots of 1: all contacts accounted for (upper left) and resolved into different intermolecular interactions showing the percentages of contacts contributing to the total Hirshfeld surface area of molecules. | ||
Fig. 9 shows that O–H⋯C interactions in 1 are dominant in the sulfonate and Ce–O regions. The proportion of O⋯H/H⋯O interactions cover 43% of the total Hirshfeld surface with two distinct spikes in the 2D fingerprint plots (Fig. 10) indicating a hydrogen bonding interaction. The O⋯H interactions are represented by a sharp small spike (de + di ~ 1.72 Å), while the H⋯O interactions are represented by a comparatively large spike (de + di ~ 1.51 Å). This is one of the closest contacts in the structures and can be viewed as numerous dark red spots on the dnorm surface (Fig. 9).
The H⋯H interactions also have a significant contribution of 18.7% of the total Hirshfeld surfaces and are reflected in the distribution of scattered points in the 2D fingerprint plot. Besides these dark spots the presence of faint red spots on the Hirshfeld surface can be assigned to O⋯O interactions with a sharp spike having (de + di) ~ 2.8 Å which comprises 16.7% of the entire Hirshfeld surfaces. Significant C–H⋯π interactions are also observed comprising 12.1% of the total Hirshfeld surfaces indicated by the wings having (de + di) ~ 3.2 Å in the 2D fingerprint plot. The specific C⋯O/O⋯C interactions also contribute significantly, 5.1% of the total Hirshfeld surfaces showing the characteristic wings with (de + di) ~ 3.1 Å corresponding to lone-pair (oxygen atoms)⋯π interactions. The shape index has been plotted on Hirshfeld surfaces with close C⋯C interactions (these generally fall in the region 3.3–3.9 Å)19 to indicate π⋯π interactions. We found that π⋯π interactions have a relatively less contribution to the entire Hirshfeld surfaces comprising 3.8% in the hydrophobic region around the aryl rings of C4ASs.
Conclusion
A unique supermolecule using p-sulfonatocalix[4]arene, Ce(III) and 2,6-pyridinecarboxylic acid was prepared and characterized. The supermolecules are C-shaped and form interdigitated pairs with each other exhibiting mutual-inclusion, whilst their PDA ligands are inserted into calixarene cavities of neighbouring supermolecules as well as into the calixarene bilayer. The bilayer consists of a zig-zag arrangement of calixarenes, resulting from the PDA ligand insertion. Despite the unique shape of the supermolecule, the usual bilayer arrangement persists which once again illustrates that this arrangement is highly favoured amongst these types of structures. Hirshfeld surface analyses indicate that the O⋯H/H⋯O interactions cover the largest proportion of the total Hirshfeld surface and that O–H⋯C interactions dominate in the sulfonate and Ce–O regions. Surprisingly, π⋯π interactions contribute relatively less to the total interactions indicating that the packing arrangement is perhaps influenced to a greater extent by O–H⋯O and other hydrogen bond interactions.Acknowledgements
A.H. and C.L.O. thank the South African National Research Foundation and the University of Cape Town for financial support.References
- (a) G. L. Zheng, Y. Y. Li, R. P. Deng, S. Y. Song and H. J. Zhang, CrystEngComm, 2008, 10, 658–660 RSC; (b) C. B. Smith, L. J. Barbour, M. Makha, C. L. Raston and A. N. Sobolev, Chem. Commun., 2006, 950–952 RSC; (c) K. Fucke, K. M. Anderson, M. H. Filby, M. Henry, J. Wright, S. A. Mason, M. J. Gutmann, L. J. Barbour, C. Oliver, A. W. Coleman, J. L. Atwood, J. A. K. Howard and J. W. Steed, Chem.–Eur. J., 2011, 17, 10259–10271 CrossRef CAS PubMed.
- (a) I. Ling, Y. Alias, B. W. Skelton and C. L. Raston, Dalton Trans., 2012, 41, 4884–4889 RSC; (b) I. Ling, Y. Alias, M. S. A. Rahim, B. W. Skelton, L. T. Byrne and C. L. Raston, Aust. J. Chem., 2012, 65, 755–762 CrossRef CAS; (c) P. J. Nichols, M. Makha and C. L. Raston, Cryst. Growth Des., 2006, 6, 1161–1167 CrossRef CAS; (d) M. J. Hardie and C. L. Raston, J. Chem. Soc., Dalton Trans., 2000, 2483–2492 RSC.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS PubMed.
- R. E. McGovern, H. Fernandes, A. R. Khan, N. P. Power and P. B. Crowley, Nat. Chem., 2012, 4, 527–533 CrossRef CAS PubMed.
- (a) J. L. Atwood, G. W. Orr, F. Hamada, R. L. Vincent, S. G. Bott and K. D. Robinson, J. Am. Chem. Soc., 1991, 113, 2760–2761 CrossRef CAS; (b) J. L. Atwood, G. W. Orr, F. Hamada, S. G. Bott and K. D. Robinson, Supramol. Chem., 1992, 1, 15–17 CrossRef CAS; (c) J. L. Atwood, G. W. Orr, K. D. Robinson and F. Hamada, Supramol. Chem., 1993, 2, 309–317 CrossRef CAS.
- G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049–1052 CrossRef CAS.
- SAINT-Plus, Version 7.12; Bruker AXS Inc.: Madison, WI, USA, 2004 Search PubMed.
- XPREP2, Version 6.14; Bruker AXS Inc.: Madison, WI, USA, 2003 Search PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 1999, 32, 351–352 CrossRef CAS.
- G. M. Sheldrick, SADABS; University of Gottingen: Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- L. J. Barbour, X-Seed - A Software Tool for Supramolecular Crystallography, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- Y. Oh, J. Y. Kim, H. J. Kim, T. Lee and S. K. Kang, Bull. Korean Chem. Soc., 2010, 31, 1058–1060 CrossRef CAS.
- (a) I. Ling, Y. Alias, A. N. Sobolev and C. L. Raston, New J. Chem., 2010, 34, 414–419 RSC; (b) S. J. Dalgarno and C. L. Raston, Chem. Commun., 2002, 2216–2217 RSC; (c) Y. Liu, D. S. Guo, H. Y. Zhang, S. Kang and H. B. Song, Cryst. Growth Des., 2006, 6, 1399–1406 CrossRef CAS.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, Crystal Explorer (Version 3.0), University of Western Australia, 2012 Search PubMed.
- A. Karmakar, A. E. Platero-Pratsb and L. Öhrström, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 251–254 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal and geometrical data, TG-DTG and DSC curves for 1. CCDC 927681 for 1. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce42358a |
| This journal is © The Royal Society of Chemistry 2014 |