
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced charge separation in ordered self-assemblies of perylenediimide–graphene oxide hybrid layers†
Mustafa
Supur
,
Kei
Ohkubo
and
Shunichi
Fukuzumi
*
Department of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370; Tel: +81-6-6879-7368
First published on 10th September 2014
Abstract
Remarkably fast photoinduced charge separation in well-ordered self-assemblies of perylenediimide–graphene oxide (TAIPDI–GO) hybrid layers was observed in aqueous environments. Slow charge recombination indicates an effective charge migration between the self-assembled layers of PDI–GO hybrids following the charge separation.
Graphene oxide (GO), a chemically exfoliated graphene derivative, offers new composite materials for photochemical applications.1 Effective visible light harvesting on GO sheets has been achieved by decorating with appropriate organic dye molecules via non-covalent interactions with the π-surface of GO bearing oxygen-containing functional groups. Many hybrids of GO have been formed by employing various organic dyes with hydrophilic substituents (pyrenes, perylenes, porphyrins, coronenes, and semiconducting oligomers and polymers) in aqueous environments.1 The efforts have been, so far, concentrated on obtaining only single or a few dye-functionalised GO or graphene layers and their photoinduced events.2 The studies on the self-assemblies of the well-ordered dye–GO hybrid layers are still limited3 and photoinduced charge generation in these structures is yet to be studied.
In this communication, we report the formation of ordered self-assemblies of GO layers functionalised with N,N′-di(2-(trimethylammoniumiodide) ethylene)perylenediimide (TAIPDI–GO, Fig. 1 and Fig. S1, ESI†) and their photoinduced electron-transfer processes. Perylenediimide (PDI) is an ideal building block for nanohybrids of GO because its strong absorption in the visible region and the low reduction potential make it a powerful light harvester and a good electron acceptor during a possible photoinduced electron-transfer process with the GO sheet.4 TAIPDI bears cationic groups enhancing the electrostatic interactions with the functional groups of GO, such as –COOH, and its large aromatic plane establishes strong π–π interactions with the π-surface of the GO sheet. As a result of these interactions, TAIPDI–GO hybrids are expected to form self-assembled organizations in water. Charge migration in the self-assembled layers of TAIPDI–GO following the initial electron-transfer from the GO sheet to closely interacting TAIPDI molecules is proposed upon the excitation of the TAIPDIs (Fig. 1).
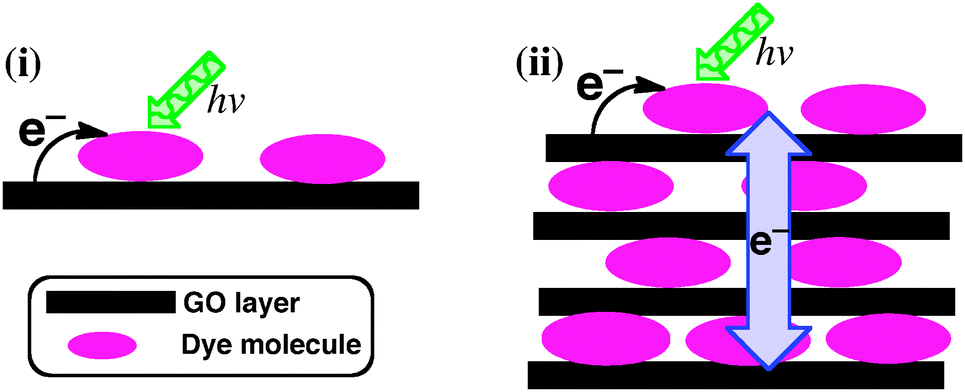 | ||
| Fig. 1 (i) Photoinduced charge separation in single or a few layers of dye–GO hybrids examined in previous studies. (ii) Proposed photoinduced charge separation and interlayer charge migration in ordered self-assemblies of TAIPDI–GO hybrid layers investigated in this communication. | ||
Self-assembly of TAIPDIs with GO layers was first detected by UV-vis spectroscopy. Absorption maxima of PDI at 501 and 537 nm were shifted to 510 and 548 nm, respectively. Hybrid formation also caused the quenching of TAIPDI fluorescence emission (Fig. 2). Significant spectral changes in the absorption of TAIPDI–GO and emission quenching indicate π–π interactions between the π-surfaces of PDI and GO, which typically favours the photoinduced electron-transfer processes due to the very close distance as observed in the non-covalently functionalized carbon materials with large π-surfaces.2g,5
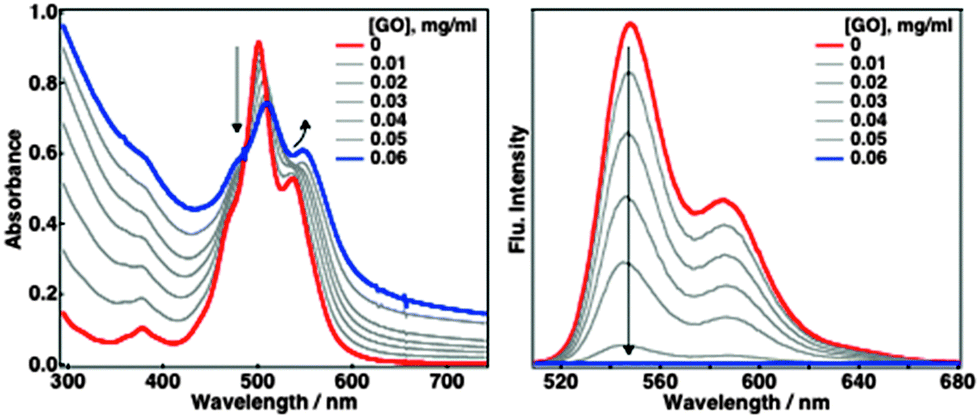 | ||
| Fig. 2 Absorption (left) and fluorescence emission (right) spectral changes during the addition of GO to the aqueous solution of 0.025 mM TAIPDI, λexc = 510 nm. | ||
Self-assembly of TAIPDI–GO layers leads to supramolecular gel formation in water under mild conditions (see Experimental section and Fig. S2–S4, ESI†). Apparently, cationic TAIPDI6 acts as a gelator, enhancing the π–π stacking. Powder X-ray diffraction (PXRD) was utilized to analyze the structural organization in the self-assemblies. GO showed a sharp peak at 2θ = 10.2° correlated with a d-spacing of 8.66 Å between the GO layers, which is very close to previously reported values.3 Incorporation of TAIPDI molecules into the GO layers increases the d-spacing to a value of 10.34 Å as the corresponding peak shifts to 2θ = 8.6° in the PXRD patterns of dried TAIPDI–GO hybrid gel (Fig. 3).
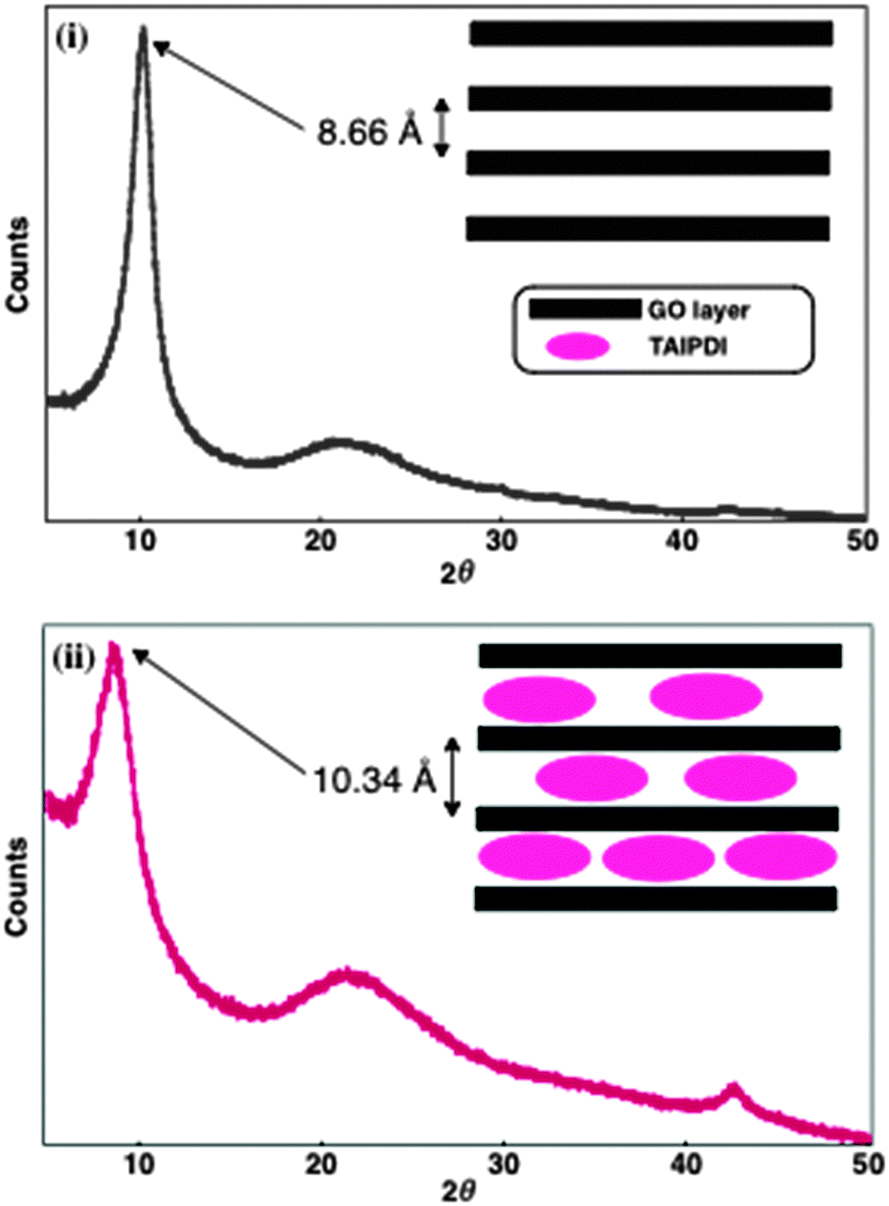 | ||
| Fig. 3 Powder X-ray diffraction patterns of dried (i) GO and (ii) TAIPDI–GO supramolecular gel. Inset: representation of GO layers with and without TAIPDIs. | ||
The free energy change of the proposed photoinduced charge separation (ΔGCS) in PDI–GO hybrid was calculated according to eqn (1),7
| ΔGCS = e(Eox − Ered) − ES | (1) |
Photoinduced electron-transfer dynamics of self-assemblies of PDI–GO hybrids were investigated by using femtosecond laser-induced transient absorption spectroscopy. The femtosecond transient spectra of aggregates of TAIPDI–GO (0.25 mM TAIPDI and 0.6 mg ml−1 GO) in water displayed a very fast formation of broad transient absorption at around 720 and 960 nm after the selective excitation of PDI at 510 nm (Fig. 4). These transient traits agree very well with those of the radical anion of PDI (PDI˙−),9,10 providing a valid proof for photoinduced electron transfer from GO to 1PDI*. By fitting the increase of the transient absorption at 720 nm to a curve of an exponential function, the rate of charge separation (kCS) was estimated to be 3.6 × 1011 s−1 (τ = 2.8 ps, Fig S8, ESI†). The fast formation rate suggests that the charge separation is very efficient, which rules out the other quenching mechanisms such as energy transfer. The decay at the same wavelength comprises two components, whose lifetime values are extracted by fitting to a sum of two decaying exponentials (Fig. 4, inset). The former component of the fitting curve gives a lifetime for the first charge recombination of 31 ps (kCR1 = 3.2 × 1010 s−1). The lifetime of the eventual charge-separated state is calculated from the latter component to be 417 ps (kCR2 = 2.4 × 109 s−1, Fig. 4, inset). Such a two-component decay was also observed in the π-stacks of PDI hosting porphyrins via π–π and electrostatic interactions in water. Slow decay accounted for the presence of the electron migration mechanism among the π-stacks of PDI.9
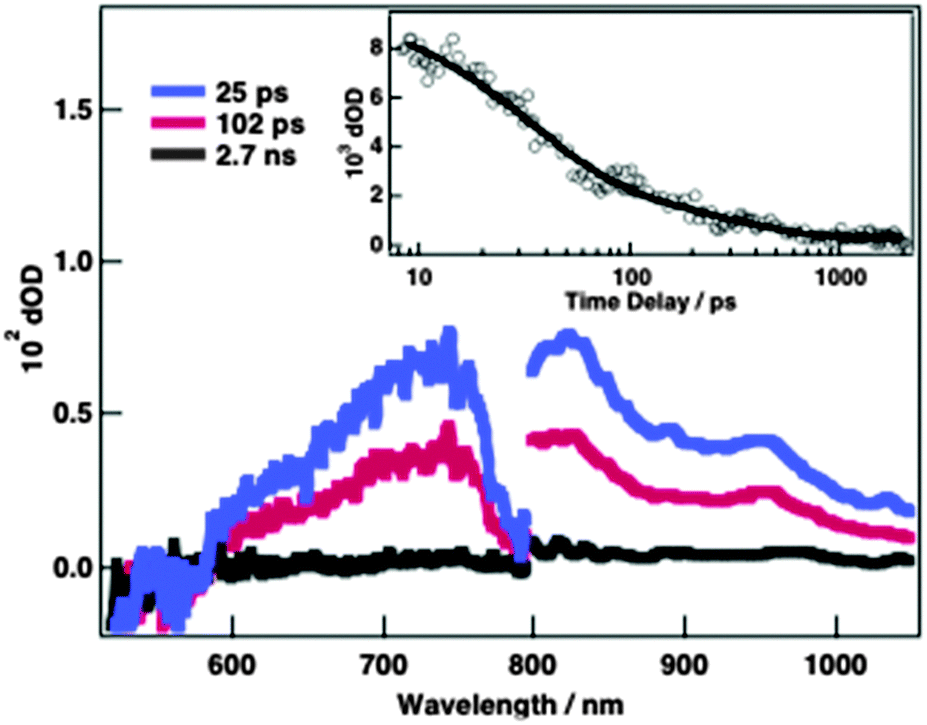 | ||
| Fig. 4 Femtosecond laser-induced transient absorption spectra of 0.25 mM TAIPDI and 0.6 mg ml−1 GO in water at indicated time delays, λexc = 510 nm. Inset: time profile at 720 nm. | ||
To view the effect of concentration of GO, it is increased to 0.9 mg ml−1 by maintaining the concentration of TAIPDI at 0.25 mM, which means a lower number of TAIPDI molecules is deposited between the GO layers. Consequently, the lifetime values of 21 and 400 ps were, respectively, obtained from the two-component decay at 720 nm (Fig. S9, ESI†). The slight decline indicates that charge migration is mainly realized through the GO sheets whereas the contribution of TAIPDI molecules is limited. In the case of low concentrations of GO (0.3 mg ml−1) with 0.15 mM TAIPDI, the first and second components of the decay at 720 nm give lifetimes for charge separation of 2.3 and 48 ps, respectively (Fig S10, ESI†). This can be explained by the lack of adequate supramolecular organization of TAIPDI and GO. The concentration change of the GO did not have a significant impact on the formation rates of charge separation.
To test the effect of self-assembly on the rates of charge separation, TAIPDI–GO was dispersed in an aqueous polyethylene glycol (PEG 200) solution, in which the gel formation was not observed under the same conditions (∼0.6 mg ml−1 GO and 0.25 mM TAIPDI). The femtosecond laser-induced transient absorption spectra of well-dispersed GO–PDI hybrids in the PEG 200 solution exhibited the excited state absorption peak at around 710 nm with bleaching at 550 nm after laser excitation at 510 nm (Fig. S11, ESI†). These transient spectra match with those of 1PDI*.9a The decay profile of absorbance at around 710 nm indicates that the lifetime of 1PDI* (τS) is remarkably reduced. There was no transient signal to be attributed to the electron-transfer products (i.e., the radical ion pair) probably because of rapid back electron transfer.10 The rate of charge separation of PDI–GO hybrids (kCS) in the PEG 200 solution was estimated to be 2.3 × 109 s−1, which is significantly slower than that of the self-assembled form of TAIPDI–GO in neat water.11 As a result, the polymer chains of PEG 200 in the solution impede the supramolecular organization of TAIPDI–GO. Although the charge separation is not efficient no other transient trait was obtained to be assigned to the energy transfer mechanism in the femtosecond transient absorption spectra.
Formation of the charge-separated states in the self-assemblies of PDI–GO hybrids in aqueous media was verified by the electron paramagnetic resonance (EPR) measurements, in which the EPR spectrum of PDI˙− displays a powder pattern without hyperfine splitting (g = 2.0041) in frozen media at 4 K after photoexcitation (Fig. S12, ESI†).
The hole (i.e. radical cation) formed after the photoinduced charge separation is expected to easily migrate on the smooth π-surface of a graphene sheet, resulting in slower charge recombination despite the fact that the electron-transfer distance between non-covalently-functionalised, electron-donating graphene and the electron-accepting dye molecule is very close due to π–π interactions.2g,12 In the case of PDI–GO dispersed in the solution of PEG 200, the fast charge recombination can be rationalized from the close distance between the electron donor and acceptor and the lack of the effective hole delocalization on the surface of the GO because of the defects on the honeycomb structure,13 resulting from the chemical oxidation.14 The presence of the functional groups on the GO sheet is practical for the non-covalent functionalisation of the GO by the dye molecules; however, they cause the interruption of the continuous conjugation of the π-surface. Despite the fact that the charge migration on the π-surface is severely inhibited by the defects it can be regenerated between the adjacent π-stacks of PDI–GO hybrid layers in the self-assemblies as indicated by the slow component of the decay time profile (Fig. 4, inset, Fig S9 and S10, ESI†). Migration through the π-stacks is likely to promote a “pull effect” for fast charge separation. Dispersion of these hybrid stacks in a polymer environment seems to block such an interlayer charge-hopping mechanism due to the disruption of the supramolecular organization, causing the fast back electron transfer from PDI˙− to oxidized GO.
In summary, non-covalent functionalisation of GO with visible light-absorbing PDI was achieved through the π–π and electrostatic interactions in water. TAIPDI–GO hybrids reveal a supramolecular self-assembly as recognized by the PXRD patterns of gel formation in an aqueous environment. Our findings have demonstrated that π-stacking is necessary for fast charge separation and slow charge recombination, which is facilitated by an interlayer charge migration mechanism within the ordered self-assemblies of the PDI–GO hybrids during the photoexcitation.
This work was supported by Grant-in-Aid for Scientific Research (No. 266201541 and 26288037), an ALCA project from JST. M.S. thanks JSPS for a postdoctoral fellowship.
Notes and references
- (a) D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027 CrossRef CAS PubMed; (b) I. V. Lightcap and P. V. Kamat, Acc. Chem. Res., 2013, 46, 2235 CrossRef CAS PubMed; (c) H. Chang and H. Wu, Energy Environ. Sci., 2013, 6, 3483 RSC; (d) Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541 RSC; (e) S. Naficy, R. Jalili, S. H. Aboutalebi, R. A. Gorkin III, K. Konstantinov, P. C. Innis, G. M. Spinks, P. Poulin and G. G. Wallace, Mater. Horiz., 2014, 1, 326 RSC.
- (a) Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191 CrossRef CAS; (b) A. Wojcik and P. V. Kamat, ACS Nano, 2010, 4, 6697 CrossRef CAS PubMed; (c) J. Geng and H.-T. Jung, J. Phys. Chem. C, 2010, 114, 8227 CrossRef CAS; (d) Z. D. Liu, H. X. Zhao and C. Z. Huang, PLoS One, 2012, 7, e50367, DOI:10.1371/journal.pone.0050367; (e) W. He, C.-E. He, R. Peng and Y. Yang, Adv. Mater. Res., 2012, 534, 118 CrossRef CAS; (f) N. Karousis, J. Ortiz, K. Ohkubo, T. Hasobe, S. Fukuzumi, Á. Sastre-Santos and N. Tagmatarchis, J. Phys. Chem. C, 2012, 116, 20564 CrossRef CAS; (g) K. Dirian, M. Á. Herranz, G. Katsukis, J. Malig, L. Rodriguez-Perez, C. Romero-Nieto, V. Strauss, N. Martín and D. M. Guldi, Chem. Sci., 2013, 4, 4335 RSC; (h) S. Roy, D. K. Maiti, S. Panigrahi, D. Basak and A. Banerjee, Phys. Chem. Chem. Phys., 2014, 16, 6041 RSC.
- S. Srinivasan, S. H. Je, S. Back, G. Barin, O. Buyukcakir, R. Guliyev, Y. Jung and A. Coskun, Adv. Mater., 2014, 26, 2725 CrossRef CAS PubMed.
- (a) F. Würthner, Chem. Commun., 2004, 1564 RSC; (b) M. R. Wasielewski, J. Org. Chem., 2006, 71, 5051 CrossRef CAS PubMed; (c) M. Supur and S. Fukuzumi, ECS J. Solid State Sci. Technol., 2013, 2, M3051 CrossRef CAS.
- (a) M. Ohtani and S. Fukuzumi, Chem. Commun., 2009, 4997 RSC; (b) Z. Zhang, H. Huang, X. Yang and L. Zang, J. Phys. Chem. Lett., 2011, 2, 2897 CrossRef CAS; (c) C. Bikram K. C., S. K. Das, K. Ohkubo, S. Fukuzumi and F. D’Souza, Chem. Commun., 2012, 48, 11859 RSC; (d) O. Ito and F. D’Souza, ECS J. Solid State Sci. Technol., 2013, 2, M3063 CrossRef CAS.
- The effect of cationic heads of PDI on binding was also tested. There was no shift in the absorption maxima of a PDI derivative with alkyl chains (N,N’-diheptadecan-9-ylperylenediimide, HHPDI) during the titration with GO (Fig. S5, ESI†) and no gel formation was observed, which declares the need of cationic substituents for effective self-assembly. Besides, fluorescence quenching of HHPDI was incomplete when titrated with the same amount of GO as in the titration of PDI with cationic heads in water (Fig. S6, ESI†).
- A. Weller, Z. Phys. Chem., 1982, 133, 93 CrossRef CAS.
- It should be noted that the oxidation potential of GO can vary with the degree of oxidation of reactant graphite during the synthesis of GO, see: K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38 CrossRef CAS.
- (a) M. Supur and S. Fukuzumi, J. Phys. Chem. C, 2012, 116, 23274 CrossRef CAS; (b) M. Supur and S. Fukuzumi, Phys. Chem. Chem. Phys., 2013, 15, 2539 RSC.
- M. Supur, Y. M. Sung, D. Kim and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 12438 CAS.
- The rate of charge separation of PDI–GO hybrids (kCS) in polymer solutions was estimated from the quenching of the singlet-excited states of PDI by using eqn:10kCS = (1/τq) − kS where τq is the lifetime of quenched 1PDI* due to photoinduced electron transfer and kS is the rate constant of 1PDI* without any quenching.
- J. Malig, N. Jux and D. M. Guldi, Acc. Chem. Res., 2013, 46, 53 CrossRef CAS PubMed.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc05694a |
| This journal is © The Royal Society of Chemistry 2014 |