
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe phosphaethynolate anion reacts with unsaturated bonds: DFT investigations into [2+2], [3+2] and [4+2] cycloadditions†
Liu Leo
Liu
ac,
Jun
Zhu
*b and
Yufen
Zhao
*ad
aDepartment of Chemistry, College of Chemistry and Chemical Engineering, Key Laboratory for Chemical Biology of Fujian Province, Xiamen University, Xiamen 361005, Fujian, China. E-mail: yfzhao@xmu.edu.cn; Web: http://chem.xmu.edu.cn/group/yfzhao/zhao-home.html
bState Key Laboratory of Physical Chemistry of Solid Surfaces and Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, Fujian, China. E-mail: jun.zhu@xmu.edu.cn; Web: http://junzhu.chem8.org
cDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093-0343, USA
dKey Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China
First published on 1st August 2014
Abstract
Density functional theory (DFT) calculations were carried out to investigate the [2+2], [3+2] and [4+2] cycloadditions of the phosphaethynolate anion (PCO−). The results reveal that the electronic properties of different unsaturated compounds play a crucial role in reactivity and regioselectivity.
Since the discovery of H–C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P in 1961,1 the number of derivatives of phosphaalkynes (R–CP) has increased.2 Among them, the simplest isolated phosphaethynolate anion (PCO−), the phosphorus-containing analogue of cyanate (NCO−), was first reported by Becker and co-workers in 1992.3 Based on natural resonance theory (NRT) calculations, this anion is mainly described as a hybrid between the phosphaethynolate (51.7%) and phosphaketenide (40.2%) resonance structures (Fig. 1),4 and was also found to be an ambident nucleophile depending on reaction conditions.5
P in 1961,1 the number of derivatives of phosphaalkynes (R–CP) has increased.2 Among them, the simplest isolated phosphaethynolate anion (PCO−), the phosphorus-containing analogue of cyanate (NCO−), was first reported by Becker and co-workers in 1992.3 Based on natural resonance theory (NRT) calculations, this anion is mainly described as a hybrid between the phosphaethynolate (51.7%) and phosphaketenide (40.2%) resonance structures (Fig. 1),4 and was also found to be an ambident nucleophile depending on reaction conditions.5
 | ||
| Fig. 1 The resonance structures of PCO−. | ||
However, the chemistry of phosphaethynolate has been quite limited owing to the technical difficulties associated with its synthesis. Recently, Grützmacher and co-workers found that PCO− reacted with an imidazolium salt as a P− transfer reagent with concomitant loss of CO.6 Furthermore, Goicoechea and Jupp obtained the first phosphorus-containing analogue of urea by treatment of PCO− with NH4+.7 More interestingly, several phosphorus-containing heterocycles were generated by [2+2], [3+2] and [4+2] cycloadditions of PCO− with different unsaturated bonds (Scheme 1).8 Thus, one can realize that PCO− is indeed a valuable building block with great potential for the construction of structurally sophisticated organophosphorus compounds. Very recently, Grützmacher and co-workers reported a simple one-pot procedure to prepare larger quantities of pure Na(PCO)(dioxane)2.5,9 which could readily combine with CO2 followed by dimerization. This was shown to serve as a rare example of a dianionic transfer species (P2C2O2)2−. The mechanistic study indicated that the CO2-triggered dimerization does not proceed in a concerted fashion, but is stepwise and initiated by a nucleophilic attack of the phosphorus centre of PCO−. This poses an interesting question as to whether the reported reactions between PCO− and unsaturated bonds proceed in a formal stepwise fashion or by concerted cycloaddition. Moreover, with unsymmetrical alkynes or different types of double bonds, the reaction proceeds with high regioselectivity and sometimes removal of CO occurs (Scheme 1). However, the mechanisms of these reactions are not well understood.
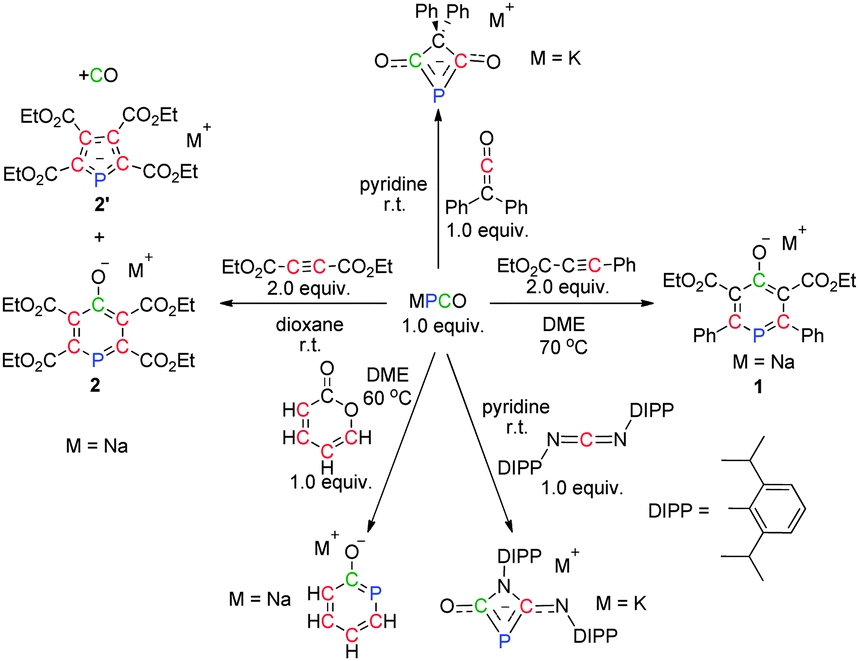 | ||
| Scheme 1 Cycloaddition chemistry of PCO−. | ||
Inspired by the intriguing results,8 we performed theoretical calculations to investigate the detailed reaction mechanisms of the cycloaddition chemistry of PCO− (Scheme 1), including the reactivity and selectivity of substrates with different unsaturated bonds. Our findings may open a new avenue for further development of PCO− chemistry.
On the basis of the experimental results, the cycloadditions of PCO− with different unsaturated compounds proceed with high regioselectivity. For example, Na(PCO) was treated with 2.0 equiv. of an asymmetrical alkyne (EtO2CCCPh) to form 1 as the only product (Scheme 1, right).8b More importantly, the more electron-deficient alkyne (EtO2CCCCO2Et) did not generate only 2, but also 2′ with the loss of CO (Scheme 1, left).8b Significant uncertainties exist concerning the observed chemical selectivities, in particular on the high regioselectivity and the process of removal of CO from the formed heterocycle. Thus, we first investigated the cycloadditions of PCO− and alkynes through DFT calculations at the M06-2X/6-311++G(2d,p)//B3LYP-D/6-31+G(d) level (see the ESI† for details).
The proposed reaction pathway for the first [2+2] cycloaddition of EtO2CCCPh and PCO−, including the computed free energies is shown in Fig. 2. All efforts made towards locating a transition state for a concerted [2+2] cycloaddition reaction failed. Instead, a nucleophilic attack of the phosphorus atom in PCO− on C1 and C2 of the alkyne could be identified. Interestingly, the nucleophilic attack on C1 (TS1A′, 27.1 kcal mol−1) is significantly higher than that on C2 (TS1A, 19.4 kcal mol−1), which could be mainly attributed to the electronic effects (Fig. 3). For instance, natural population analysis (NPA) indicates that the charges of C1 and C2 in EtO2CCCPh are −0.10 and 0.07e, respectively, whereas that of P in PCO− is −0.44e, indicating that PCO− is more favoured to attack C2. This is in line with the experimental observations that only 1 was formed (Scheme 1).8b It is important to note that the charge of oxygen in PCO− is −0.70e, indicating that the oxygen might attack C2. However, no stable intermediate of C2–O bond formation could be identified.10 Indeed, the HOMO of PCO− is mainly localized at the π orbital of the P centre. The absolute maximum of the coefficient at P (0.51) is much larger than that at O (0.30). Therefore, attacking alkynes via the P centre could lead to favorable orbital overlap between the PCO− π orbital and the alkyne π* orbitals (LUMOs).
 | ||
| Fig. 2 Free energy profile for the first [2+2] cycloaddition of EtO2CCCPh and PCO−. The values are given in kcal mol−1. | ||
 | ||
| Fig. 3 HOMO of PCO− and LUMOs of unsaturated compounds (isovalue = 0.05). NPA partial charges are given in e. Absolute maxima of the coefficients at the reacting termini are given in parentheses. | ||
Subsequently, IN1A is formed with the CO2Et group on the same side of PCO, which is sterically unfavourable for the next C–C bond formation. Thus, an isomerization step was located with an activation energy of 6.6 kcal mol−1. From IN2A (15.7 kcal mol−1), a four-membered transition state TS3A (18.4 kcal mol−1) occurred, leading to the first [2+2] cycloaddition product IN3A (−13.6 kcal mol−1).
Fig. 4 depicts the second [2+2] cycloaddition of EtO2CCCPh and PCO−. In a similar way, very stable 1 (−88.9 kcal mol−1) is generated via nucleophilic attack of the P atom, isomerization and C–C bond formation. It is important to note that the final process is highly exergonic (TS6A → 1). According to the computed NICS(1)zz11 value of 1 (−13.4 ppm), the significant stability is mainly attributed to the release of ring strain and the gain of aromaticity in 1.
 | ||
| Fig. 4 Free energy profile for the second [2+2] cycloaddition of EtO2CCCPh and PCO−. The values are given in kcal mol−1. | ||
We next turned our attention to the loss of CO, which is observed by using the more electron-deficient alkyne (EtO2CCCCO2Et) as the substrate (Scheme 1, left). Similar to 1, a six-membered aromatic product 2 is formed (−100.5 kcal mol−1) (see the ESI† for details). Direct removal of CO from 2 is found to be too energy demanding to take place (Fig. 5, right). Alternatively, from IN5B, a formal [3+2] cycloaddition with the loss of CO could be located via two steps (Fig. 5, left). The activation barriers for the transformation are only 2.2 and 0.3 kcal mol−1. The free energies of TS5B and TS7B are −34.0 and −35.2 kcal mol−1, which are 3.4 and 2.2 kcal mol−1 higher than that of IN5B, respectively, indicating that both processes could readily occur, in agreement with the experimental observations that 2 and 2′ were generated. In addition, when EtO2CCCPh was used as the substrate (Fig. 4), the activation energy of C3 attacking C5 is 6.0 kcal mol−1 (TS6A′), which is approximately 2 times higher than that of C3 attacking C4 (TS6A, 2.2 kcal mol−1), explaining why 1 was formed exclusively during the reaction.
 | ||
| Fig. 5 Free energy profile for the CO removal process using EtO2CCCCO2Et and PCO− as substrates. The values are given in kcal mol−1. | ||
To gain more insight into the cycloaddition chemistry of PCO−, the regioselectivities8a of its outcome with Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO and DIPPNCNDIPP were also studied (see the ESI† for details). The results showed that these two transformations do not proceed in a concerted fashion, but are stepwise and initiated by a nucleophilic attack of the phosphorus centre of PCO−, which is similar to what was discussed above. The regioselectivities were determined by the nature of electronic properties of substrates as illustrated in Fig. 3. For example, it is much easier for PCO− to attack C7 (0.77e) over C6 (−0.37e). The activation energy is 4.6 kcal mol−1, which is significantly lower than that of attacking C6 (37.0 kcal mol−1).
CO and DIPPNCNDIPP were also studied (see the ESI† for details). The results showed that these two transformations do not proceed in a concerted fashion, but are stepwise and initiated by a nucleophilic attack of the phosphorus centre of PCO−, which is similar to what was discussed above. The regioselectivities were determined by the nature of electronic properties of substrates as illustrated in Fig. 3. For example, it is much easier for PCO− to attack C7 (0.77e) over C6 (−0.37e). The activation energy is 4.6 kcal mol−1, which is significantly lower than that of attacking C6 (37.0 kcal mol−1).
Finally, we examined the [4+2] cycloaddition between 2H-pyran-2-one and PCO− (Fig. 6).8b According to the experimental study, a distinct gas evolution (CO2) is observed. Very interestingly, the transformation involves two concerted steps, including a Diels–Alder-type cycloaddition and a rearrangement involving the removal of CO2. The electronic effects also play a key role in this reaction. The favourable reactive sites lead to the lower barrier process (TS1E, 20.6 kcal mol−1). Our calculations showed that the NICS(1)zz value of IN2E is −15.2 ppm and the entropy change of the rearrangement step is 43.3 cal mol−1 K−1 (IN1E → IN2E), indicating that the resulting aromaticity and entropy increase are the driving force for the transformation.
 | ||
| Fig. 6 Free energy profile for the [4+2] cycloaddition using 2H-pyran-2-one and PCO− as substrates. The values are given in kcal mol−1. | ||
We have computationally characterized the mechanisms for the [2+2], [3+2] and [4+2] cycloaddition chemistry of PCO− with different unsaturated compounds, including alkynes, ketenes, carbodiimides and 2H-pyran-2-one. The results showed that the [2+2] and [3+2] cycloaddition of PCO− favoured stepwise processes, whereas [4+2] cycloaddition is a concerted process. More importantly, electronic effects play a key role in the regioselectivities of cycloadditions. Our findings can serve as a clue for further development of PCO− chemistry.
This work was supported by the National Basic Research Program of China (2012CB821600, 2013CB910700 and 2011CB808504), the Chinese National Natural Science Foundation (21232005, 21375113, and 21103142), the Program for New Century Excellent Talents in University (NCET-13-0511), and the Program for Changjiang Scholars and Innovative Research Team in University and the Fundamental Research Funds for the Central Universities (2012121021). Thanks are also given to the China Scholarship Council for a Graduate Fellowship (L.L.). L. L. thanks the D. A. Ruiz from UCSD for valuable discussion.
Notes and references
- T. E. Gier, J. Am. Chem. Soc., 1961, 83, 1769 CrossRef CAS.
- F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578 CrossRef CAS PubMed.
- G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72 CrossRef CAS PubMed.
- S. Alidori, D. Heift, G. Santiso-Quinones, Z. Benkő, H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. – Eur. J., 2012, 18, 14805 CrossRef CAS PubMed.
- D. Heift, Z. Benko and H. Grützmacher, Dalton Trans., 2014, 43, 5920 RSC.
- A. M. Tondreau, Z. Benko, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545 RSC.
- A. R. Jupp and J. M. Goicoechea, J. Am. Chem. Soc., 2013, 135, 19131 CrossRef CAS PubMed.
- (a) A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064 CrossRef CAS PubMed; (b) X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkő, Z. Li, G. Becker, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641 CrossRef CAS PubMed; (c) D. Heift, Z. Benkő and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 6757 CrossRef CAS PubMed.
- D. Heift, Z. Benko and H. Grützmacher, Dalton Trans., 2014, 43, 831 RSC.
- All efforts failed to locate an intermediate of C2–O bond formation. The bond breaks up into EtO2CCCPh and PCO− immediately during the optimization process.
- (a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS; (b) H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Org. Lett., 2006, 8, 863 CrossRef CAS PubMed; (c) J. Zhu, K. An and P. v. R. Schleyer, Org. Lett., 2013, 15, 2442 CrossRef CAS PubMed; (d) K. An and J. Zhu, Eur. J. Org. Chem., 2014, 2764 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details. See DOI: 10.1039/c4cc04610b |
| This journal is © The Royal Society of Chemistry 2014 |