 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guest control of structure in porous organic cages†
Marc A.
Little
,
Samantha Y.
Chong
,
Marc
Schmidtmann
,
Tom
Hasell
and
Andrew I.
Cooper
*
Department of Chemistry and Centre for Materials Discovery, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: aicooper@liv.ac.uk; Web: http://www.liv.ac.uk/cooper-group/
First published on 10th July 2014
Abstract
Two porous organic cages with different thermodynamic polymorphs were induced by co-solvents to interchange their crystal packing modes, thus achieving guest-mediated control over solid-state porosity. In situ crystallography allows the effect of the co-solvent guests on these structural interconversions to be understood.
Porous molecular solids1 comprise non-covalent intermolecular interactions that are weaker and often less directional than the coordination or covalent bonds that define zeolites,2 metal–organic frameworks (MOFs),3 and covalent organic frameworks (COFs).4 Structural polymorphism is therefore common, and this raises challenges for the targeted design of solid-state function.
We have developed porous organic cage molecules via [4+6] cycloimination reactions of 1,3,5-triformylbenzene (TFB) with vicinal aliphatic diamines such as (1R,2R)-diaminocyclohexane (CC3-R) and (1R,2R)-diaminocyclopentane (CC4-R) (Fig. 1a).5 The molecular solubility of organic cages6 allows crystallisation to be decoupled from synthesis, unlike MOFs and COFs where these two processes constitute the same step.
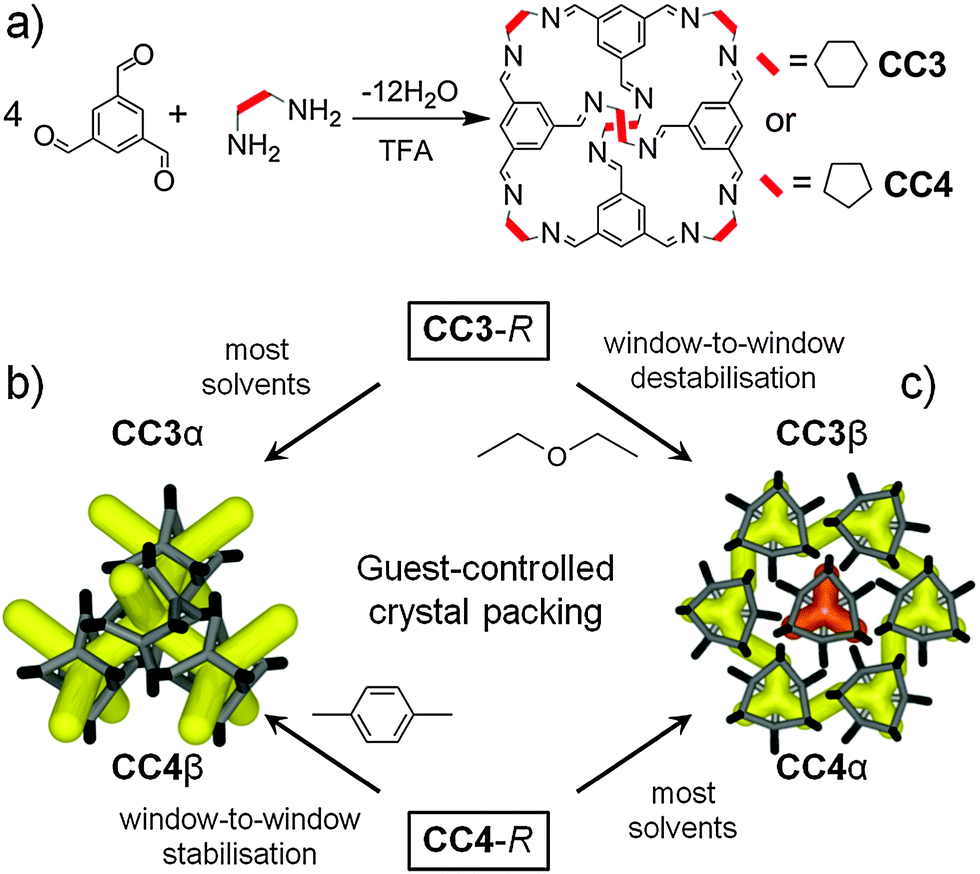 | ||
| Fig. 1 (a) Synthesis of organic cage molecules CC3 and CC4. Scheme showing interchangeable crystal packings for cages CC3 and CC4, (b) 3-D diamondoid pore channels, and (c) 2-D layered pore structure with formally disconnected voids. Orange = disconnected voids; yellow = interconnected pores. | ||
CC3-R prefers to pack in a window-to-window arrangement,5,7 which generates an interconnected diamondoid pore network (Fig. 1b). This thermodynamic polymorph, CC3α, has an apparent BET surface area (SABET) of ∼410 m2 g−1 in the crystalline state.7 Both crystal structure predictions (CSP)8 and DFT ‘cage dimer’ calculations7 rationalise this crystal packing. By contrast, CC4-R packs via a window-to-arene interaction, despite its close structural similarity with CC3-R, and this results in a 2-D pore structure in its thermodynamic polymorph, CC4α, rather than a 3-D diamondoid pore structure (Fig. 1c).
In this study, we show that these two thermodynamic ‘alpha’ crystal packing modes can be interchanged by using specific co-solvents to direct the crystal packing, such that CC3-R packs like CC4-R, and vice versa (Fig. 1b and c). We showed recently that 1,4-dioxane can direct three different tetrahedral cage molecules (CC1, CC2 and CC13)9 to crystallise isostructurally with CC3α, providing that the cages comprise a racemic mixture.
Here we expand this concept to the homochiral cage molecules CC3-R and CC4-R. We direct these two cages into isostructural packings, and hence achieve control over pore topology, pore volume, and surface area. Computational CSP can map the energy landscape for solvated crystal structures11 or for solvent-free organic cage crystals,8,10 but this is not yet computationally affordable for cage solvates. Our strategy, therefore, was to combine intuitive concepts of shape-direction,9 solvent screening, and CSP calculations for solvent-free crystals.10
CC3-R is readily soluble in CH2Cl2 but it is insoluble in Et2O. However, the addition of excess of an antisolvent, Et2O, to solutions of CC3-R in CH2Cl2 did not result in direct precipitation of the cage, even when a twentyfold volumetric excess of Et2O was added. Instead, slow evaporation of the resultant homogenous solution affords hexagonal, needle-shaped single crystals of (CC3-R)·(Et2O)3·CH2Cl2 (Fig. S1, ESI†), which crystallise in the trigonal space group R3. The asymmetric unit comprises one third of a CC3-R molecule positioned on a threefold rotation axis, plus one CH2Cl2 molecule in the intrinsic cage cavity and one well-ordered Et2O solvent molecule in the window site (Fig. 2). Overall, Et2O occupies three of the four cage window sites, with one hydrophobic methyl terminus directed toward the hydrophobic cage cavity, sharing the cage void with CH2Cl2.
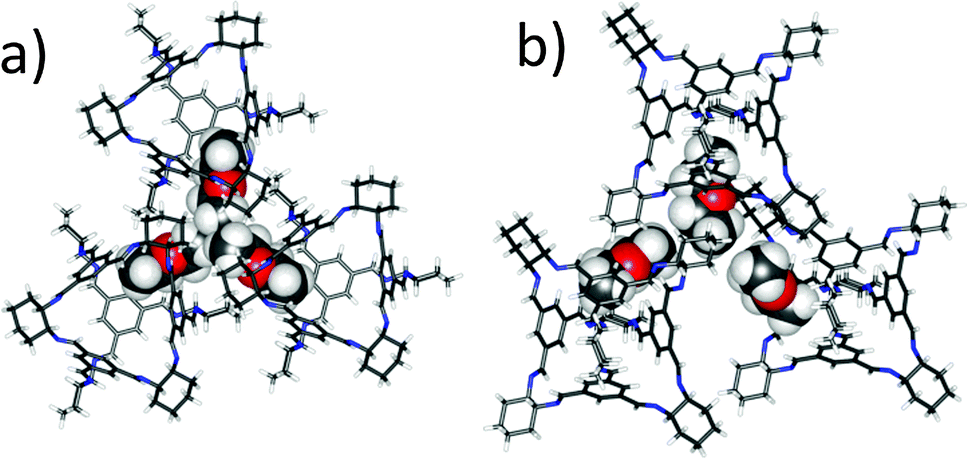 | ||
| Fig. 2 Single crystal structure for CC3-R·(Et2O)3·CH2Cl2, viewed parallel to [001] axis (a) and off axis (b). Et2O solvent molecules are shown in space filling representation, with the CH2Cl2 molecules in the cage cavities omitted for clarity. | ||
The inclusion of Et2O during crystallisation disrupts the otherwise favourable window-to-window CC3-R cage pairing, as found in CC3α, which has a cage–cage intermolecular interaction energy calculated to be around 150 kJ mol−1 (DFT-D3).7 Hence, the three cage windows pack in a relatively inefficient manner, forming three extrinsic cavities into which the methyl termini of the Et2O solvent molecules are directed (Fig. 2). The fourth cage window is occupied by the aromatic face of a neighbouring cage: this window-to-face interaction has a binding energy that was calculated to be 55 kJ mol−1 less favourable than the window-to-window interaction,7 but this is evidently compensated by favourable cage–solvent interactions.
Stacks of CC3-R molecules are formed as a result of this window-to-face packing (Fig. 3). This packing is very similar to that reported previously for crystalline CC4-R.12 For CC4-R, we observed a de-symmetrisation upon desolvation (R3 to P3) as a result of the CC4-R molecules undergoing a screw-type rotation to afford what is referred to hereafter as CC4α (Fig. 1c). Similarly, desolvation of (CC3-R)·(Et2O)3·CH2Cl2 affords a new solvent-free, metastable polymorph of CC3-R, CC3β. This is also accompanied by screw-type rotation of the cage molecules in the crystal lattice (Fig. 3; Section 1.3, Fig. S2–S11, ESI†). Single-crystal-to-single-crystal transformations have been reported for discrete host molecules such as metallocycles,13 and also for extended frameworks,14 but preservation of single crystallinity is not typically observed when there is a substantial structural rearrangement. For CC3, loss of the guest solvent causes the cages to pack in a more frustrated arrangement in CC3β, causing a significant contraction of the unit cell volume (5%).
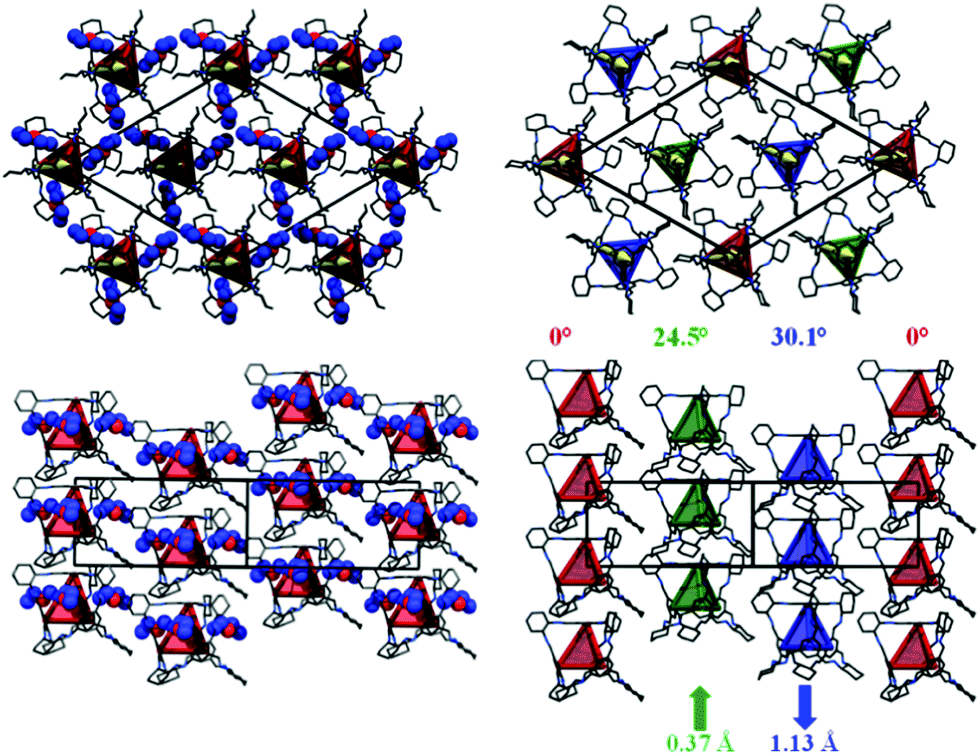 | ||
| Fig. 3 Single crystal structure for CC3-R·(Et2O)3·CH2Cl2 collected at 100 K (left), viewed along [001] (above), along the direction of the cage stacks, and [110] (below), perpendicular to the cage stacks. Et2O solvent molecules shown as space filling representation, carbon atoms in blue. Intra-cavity CH2Cl2 solvent molecules and hydrogen atoms are omitted for clarity. Red tetrahedra represent the connectivity between four aromatic faces of the crystallographically equivalent CC3-R molecules. Single crystal structure for desolvated CC3β collected at 300 K (right). Red, green and blue tetrahedra represent the connectivity between the aromatic faces of the three crystallographically distinct CC3-R molecules. Magnitude of rotation and translation of the cage molecules upon desolvation are indicated. | ||
For CC3β, the angle of cage rotation upon desolvation was defined as the angle rotated relative to the red cage (Fig. 3), which remains static and which, for convenience, has been located on the cell origin. For CC3, these rotation angles were determined to be 24.5° and 30.1°, respectively, for the green and blue cages shown in Fig. 3. There is also a 0.37 Å and 1.13 Å shift, respectively, along the z-direction for the green and blue cages in a convergent manner. Unlike the thermodynamic desolvated polymorph CC3α, CC3β does not display any window-to-window packing of the cage molecules. CC3α is predicted to be the lowest energy solvent-free form of CC3-R,8 and it is the most prevalent experimentally, being obtained from most solvents tested. CC3α also exhibits the most efficient packing between cages and has the highest density. We suggest, therefore, that CC3β is a kinetically trapped polymorph, as supported by the occasional observation of the β-form when CC3-R is rapidly precipitated from solvent by rotary evaporation.
Gas sorption analysis shows that CC3β is porous to nitrogen, with an apparent SABET of 555 m2 g−1, and a microporous type I adsorption isotherm (Fig. 4). The nitrogen uptake and porosity for CC3β is higher than for CC3α, which can be rationalised by its lower density (0.922 g cm−3 for CC3β vs. 0.973 g cm−3 for CC3α). Hence, CC3β has extrinsic lattice sites that can accommodate additional N2 molecules. The CC3β polymorph is also porous to hydrogen and carbon dioxide, in both cases with a higher gas uptake than was found for the original CC3α polymorph (Fig. S12 and S13, ESI†). Even though the crystal packings for CC4α and CC3β are essentially isostructural, the gas sorption isotherms are strikingly different. Gas-induced transformations are known for organic solids.15 For CC4α, there are low-pressure adsorption steps12 that are not observed for CC3β. Also, CC4α also adsorbs significantly more N2 than CC3β at higher relative pressures (Fig. 4). In situ powder XRD data indicates a sharp, low-pressure structural transition (Section 1.5, Fig. S14–S17, ESI†). Analogous in situ powder XRD data for CC3β did not show any low-pressure transition (Fig. S18, ESI†). The observation of a low-pressure structural transition for CC4α, but not for CC3β, might be due to the smaller cycloalkane vertex functionality in CC4. The more compact cyclopentyl vertices in CC4-R might allow small rotational rearrangements of the cages at low N2 pressures, enabling the guest to gain more extensive access to the pore network at higher N2 pressures. By contrast, we speculate that the analogous rearrangements are hindered in CC3β by the bulkier cyclohexyl substituents, and a hence a classic type I isotherm is observed with no structural transition steps (Fig. 4).
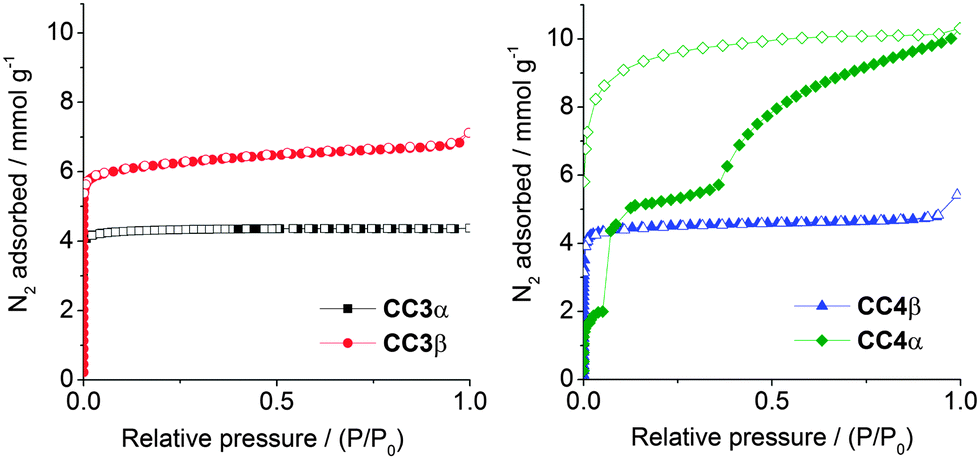 | ||
| Fig. 4 Nitrogen sorption isotherms, at 77 K, for CC3α and -β (left), and CC4α and -β (right). Adsorption isotherm branches are shown as solid symbols, and desorption branches as open symbols. | ||
Having directed CC3-R to pack in the typical CC4-R fashion (Fig. 1c), the reverse challenge was to direct CC4-R to pack isostructurally with CC3α (Fig. 1b). A recent computational study predicted a polymorph, CC4β, as the global lattice energy minimum for the homochiral CC4-R.10CC4β is isostructural with CC3α, suggesting initially that the same window-to-window cage pairing is also thermodynamically preferred for CC4-R. However, for CC4-R, the rigid-molecule constraint used in the CSP limits the reliability of the structure searches. Specifically, the experimental CC4α structure involves close intermolecular contacts that distort the molecular geometry, and hence geometry-constrained CSP calculations lead to artificially high lattice energies for CC4α. This was resolved by periodic DFT-D calculations, which showed the known CC4α polymorph to have a formation energy that is 8.19 kJ mol−1 more stable than CC4β when molecular flexibility is taken into account. In the context of lattice energies, 8.19 kJ mol−1 is a relatively small difference, suggesting that CC4-R might be directed to pack as CC4β by inclusion of an appropriate directing solvent.
A screen of 30 different crystallisation co-solvents was used in an attempt to access the CC4β polymorph that was suggested by CSP (Section 1.6, Table S4, ESI†). PXRD data showed that only one of the 30 co-solvents tested, para-xylene, produced a crystalline form that was distinct from CC4α (Fig. S19, ESI†), again supporting the conclusion that CC4α is the thermodynamically most stable polymorph, as predicted computationally using DFT-D.10 We showed recently that para-xylene can occupy the interstitial site between two cage windows in the crystal lattice of CC3-R,16 and also that it can bridge adjacent windows in 1-D cage catenane chains.17 Here, para-xylene ‘pegs’ adjacent CC4-R cage windows together in a similar fashion. Single crystals of this new CC4-R phase were grown from a layered CH2Cl2–para-xylene solution (see Section 1.7, ESI†). Single crystal X-ray diffraction revealed a new phase, CC4-R·(C8H10)3·(H2O)2 had crystallised in the chiral orthorhombic space group P212121, where each of the four cage windows are penetrated by the methyl terminus of a para-xylene solvent molecule (Fig. 5a). The linear shape of the para-xylene molecule would therefore seem to be important, as observed for 1,4-dioxane in our previous study.9 This is supported by the fact that the structural isomers of this solvent, meta- and ortho-xylene (Table S4, ESI†), did not produce the CC4β polymorph (cf., 1,3-dioxane in our previous work9).
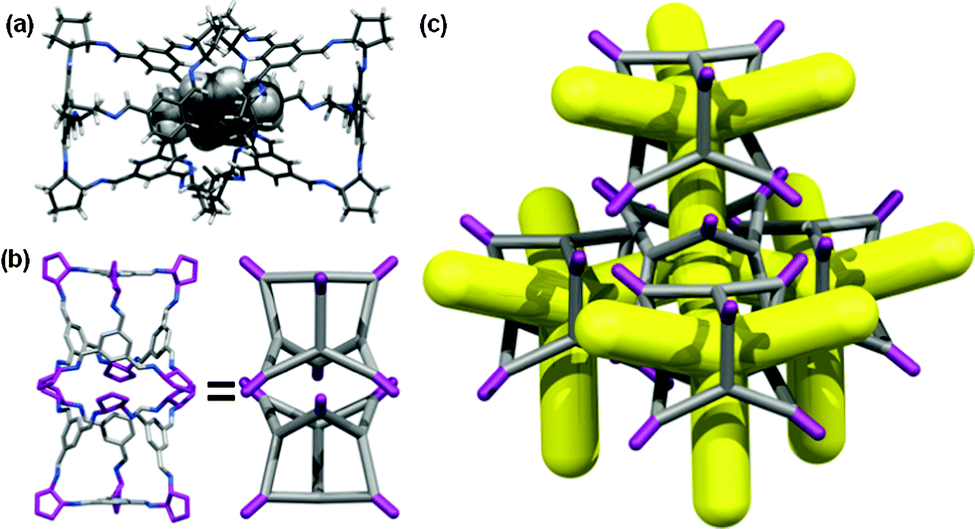 | ||
| Fig. 5 (a) para-Xylene ‘pegged’ CC4-R molecules from the single crystal structure CC4-R·(C8H10)3·(H2O)2; (b) direct window-to-window pairing arrangement displayed in CC4β, generating (c) an interconnected diamondoid pore network upon desolvation of the xylene solvate. | ||
In this structure, four para-xylene molecules are shared equally between two adjacent CC4-R molecules. Extending this window pairing arrangement in three dimensions generates a diamondoid network filled with para-xylene solvent molecules (Fig. S20 and S21, ESI†), reminiscent of the known CC3-R para-xylene solvate.16 However, unlike the CC3-R solvate, one additional para-xylene solvent molecule per cage unit is located in extrinsic 1-D channels in the CC4-R solvate (Fig. S21, ESI†). As a result of this additional solvent molecule, desolvation to a symmetrical diamondoid pore network requires significant anisotropic contraction of the unit cell parameters, potentially resulting in a transformation to an alternate pore topology or loss of crystallinity. Remarkably, gradual heating of a solvate crystal of CC4-R·(C8H10)3·(H2O)2 (Section 1.7 & Fig. S22, ESI†) resulted in preservation of single crystallinity and isolation of CC4β (Fig. 5b and c), which was formed after a transformation of the crystal symmetry to the chiral cubic space group, F4132. This is structure is isostructural both with CC3α and with the computationally predicted structure obtained for CC4 in CSP studies, as reported previously.10 This single-crystal-to-single-crystal transformation was accompanied by a large contraction of the cell volume per CC4-R molecule of around 13%. The bulk CC4β material also remains crystalline after desolvation under heating and vacuum, and the resultant gas sorption properties very similar to those of CC3α, as expected from the isostructural packing. Indeed, the nitrogen sorption isotherms for CC3α and CC4β are almost identical (SABET for CC3α = 409 m2 g−1; SABET for CC4β = 387 m2 g−1; Fig. 4). The preservation of single crystallinity upon desolvation is remarkable, given the large anisotropic contraction in the unit cell. The ‘pegged’ orientation of the CC4-R molecules in the para-xylene solvate probably facilitates the preservation of this window-to-window packing after desolvation and prevents the cages from rearranging to form CC4α. Transformation to CC4α would involve substantial and presumably high-energy reorganisation from the para-xylene solvate in order to generate the window-to-arene packing mode found in the thermodynamic polymorph.
Two organic cages, CC3-R and CC4-R, were induced to interchange their low-energy packing modes by using directing solvents. This shows that polymorphism in porous molecular solids can to some extent be controlled by combining CSP calculations, intuitive design, and high-throughput crystallisation screens. We can identify specific co-solvents that either reinforce (para-xylene in CC4β) or disrupt (Et2O in CC3β) the solid-state window-to-window packing arrangement for these porous cages. Without the use of these directing co-solvents, the lowest energy α-polymorph for CC3-R is isostructural with the higher energy β-polymorph of CC4-R, and vice versa (Fig. 1b).
Isostructural crystal packings, however, can lead to quite different gas sorption properties: for example, CC4α has marked steps in its isotherm, whereas CC3β does not. By contrast, the sorption isotherms for CC3α and CC4β are almost identical.
This represents a further step toward controlling the functional properties of porous molecular crystals by design. In the future, the a priori prediction of these solvent effects might also be possible, although at present this is prohibited by the computational expense of the relevant CSP methods.
We thank Diamond Light Source for access to beamline I11 (EE7040) that contributed to the results presented here and Prof. C. Tang, Dr P. Adamson, and Dr S. Thompson for their assistance during the experiment. We thank the EPSRC (EP/H000925/1) and the ERC under FP7 (321156) for funding.
Notes and references
- J. Tian, P. K. Thallapally and B. P. McGrail, CrystEngComm, 2012, 14, 1909–1919 RSC; M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042–5053 CrossRef CAS PubMed; L. J. Barbour, Chem. Commun., 2006, 1163–1168 RSC; J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000–1002 CrossRef PubMed; G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–702 CrossRef CAS PubMed.
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS; A. K. Cheetham, G. Férey and T. Loiseau, Angew. Chem., Int. Ed., 1999, 38, 3268–3292 CrossRef; S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef PubMed; D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273–282 CrossRef PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- M. Mastalerz, M. W. Schneider, I. M. Oppel and O. Presly, Angew. Chem., Int. Ed., 2011, 50, 1046–1051 CrossRef CAS PubMed; M. W. Schneider, I. M. Oppel, A. Griffin and M. Mastalerz, Angew. Chem., Int. Ed., 2013, 52, 3611–3615 CrossRef PubMed; A. Avellaneda, P. Valente, A. Burgun, J. D. Evans, A. W. Markwell-Heys, D. Rankine, D. J. Nielsen, M. R. Hill, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2013, 52, 3746–3749 CrossRef PubMed.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS PubMed.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- T. Hasell, J. L. Culshaw, S. Y. Chong, M. Schmidtmann, M. A. Little, K. E. Jelfs, E. O. Pyzer-Knapp, H. Shepherd, D. J. Adams, G. M. Day and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 1438–1448 CrossRef CAS PubMed.
- E. O. Pyzer-Knapp, H. P. G. Thompson, F. Schiffmann, K. E. Jelfs, S. Y. Chong, M. A. Little, A. I. Cooper and G. M. Day, Chem. Sci., 2014, 5, 2235–2245 RSC.
- A. J. Cruz-Cabeza, S. Karki, L. Fabian, T. Friscic, G. M. Day and W. Jones, Chem. Commun., 2010, 46, 2224–2226 RSC; A. J. Cruz-Cabeza, G. M. Day and W. Jones, Phys. Chem. Chem. Phys., 2011, 13, 12808–12816 RSC.
- T. Mitra, X. Wu, R. Clowes, J. T. A. Jones, K. E. Jelfs, D. J. Adams, A. Trewin, J. Bacsa, A. Steiner and A. I. Cooper, Chem. – Eur. J., 2011, 17, 10235–10240 CrossRef CAS PubMed.
- L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2005, 128, 698–699 CrossRef PubMed; G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2005, 127, 13134–13135 CrossRef PubMed.
- E. Y. Lee and M. P. Suh, Angew. Chem., Int. Ed., 2004, 43, 2798–2801 CrossRef CAS PubMed; H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844–15851 CrossRef PubMed; S. K. Ghosh, W. Kaneko, D. Kiriya, M. Ohba and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 8843–8847 CrossRef PubMed.
- P. K. Thallapally, B. Peter McGrail, S. J. Dalgarno, H. T. Schaef, J. Tian and J. L. Atwood, Nat. Mater., 2008, 7, 146–150 CrossRef CAS PubMed.
- T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Nat. Chem., 2013, 5, 276–281 CrossRef CAS PubMed.
- T. Hasell, X. Wu, J. T. A. Jones, J. Bacsa, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2012, 2, 750–755 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic, gas sorption and crystallographic details. CCDC 991214–991219. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc04158e |
| This journal is © The Royal Society of Chemistry 2014 |