 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnoyl acyl carrier protein reductase (FabI) catalyzed asymmetric reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C double bond of α,β-unsaturated ketones: preparation of (R)-2-alkyl-cyclopentanones†
C double bond of α,β-unsaturated ketones: preparation of (R)-2-alkyl-cyclopentanones†
Ji
Liu
a,
Jinchuan
Wu
b and
Zhi
Li
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585. E-mail: chelz@nus.edu.sg; Fax: +65 67791936; Tel: +65 65168416
bInstitute of Chemical and Engineering Sciences, Agency for Sciences, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833
First published on 4th July 2014
Abstract
Enoyl-ACP reductase (FabI) was identified as a non-OYE ‘ene’-reductase for asymmetric reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of α, β-unsaturated ketones. Reduction of several 2-alkylidenecyclopentanones with A-FabI and E-FabI gave (R)-2-alkylcyclopentanones in 95–90% and 70–81% ee, respectively. The product ee was improved to 99–98% in high yield by subsequent one-pot biooxidation.
C double bond of α, β-unsaturated ketones. Reduction of several 2-alkylidenecyclopentanones with A-FabI and E-FabI gave (R)-2-alkylcyclopentanones in 95–90% and 70–81% ee, respectively. The product ee was improved to 99–98% in high yield by subsequent one-pot biooxidation.
Enzymatic asymmetric reduction of C
C double bonds is a green and useful method for the preparation of enantiopure fine chemicals, pharmaceuticals, and aroma materials.1 Thus far, old yellow enzymes (OYEs) have been used as the primary reductases for this type of reactions,1,2 with their own substrate specificity and selectivity. Recently a few non-OYE reductases have been reported, including medium chain dehydrogenases/reductases (MDRs) and enoate reductases (EnoRs).1c The MDRs prefer the substrates containing strong electron-withdrawing groups such as enals and nitroalkenes,3 and they are plant or mammalian enzymes. The EnoRs were discovered from anaerobes,4 and they are oxygen sensitive. Therefore, the discovery of non-OYE reductases with novel substrate specificity, high enantioselectivity and easy utilization has remained a challenging and very important task. Here we report the identification of enoyl acyl carrier protein reductase (FabI) as a useful non-OYE reductase with unique substrate specificity and high enantioselectivity for the asymmetric reduction of the CC double bond of α,β-unsaturated ketones.
Previously, we found Acinetobacter sp. RS1 catalyzing the asymmetric reduction of 2-alkylidenecyclopentanones 1a–b to give the corresponding (R)-2-alkylcyclopentanones 2a–b (Scheme 1), with good activity and enantioselectivity.5 The reduction of substrates 1a–c could not be achieved with OYEs, and the products were important intermediates for the preparation of chiral lactones, hydroxyl acids, and alcohols.6 To identify the unique ‘ene’-reductase from this strain, we purified the enzyme by using FPLC on anion exchange, hydrophobic interaction, and gel filtration columns. The purified enzyme displayed a specific activity of 110 U g−1 protein for the reduction of 1b, with a purification factor of 370. The ee of the produced (R)-2b was 83%, similar to the ee value obtained from the same reduction with a wild type strain.5 The purified enzyme fraction had 60% purity, which is not pure enough for the determination of the terminal amino acid sequence. Nevertheless, MALDI-TOF analysis suggested a molecular weight of 27.5 KDa for the major protein in the active enzyme fraction.
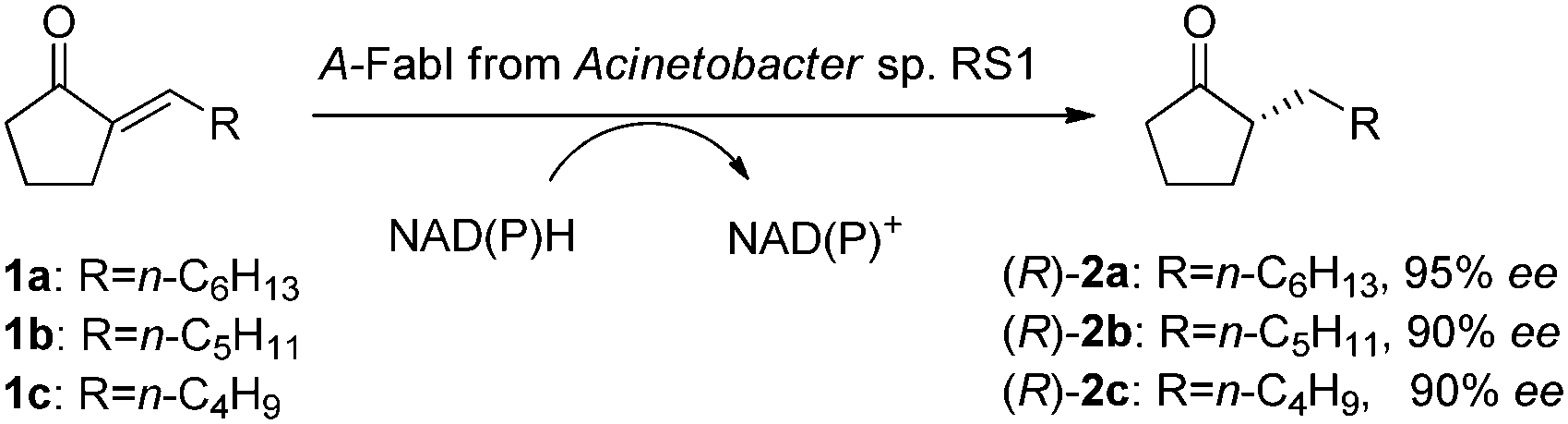 | ||
| Scheme 1 A-FabI-catalyzed asymmetric reduction of 2-alkylidenecyclopentanones 1a–c to produce (R)-2-alkyl-cyclo-pentanones 2a–c. | ||
The genome of Acinetobacter sp. RS1 was then sequenced. By using bioinformatics tools, 7 possible CC or CN double bond reductases were predicted. Out of these reductases, the enoyl-ACP reductase (A-FabI) had a molecular weight of 28.6 kDa and thus was cloned. E. coli (A-FabI) expressing his-tagged A-FabI was engineered to produce the enzyme, and the enzyme was purified by affinity chromatography with 95% purity (ESI†). The purified A-FabI catalyzed the reduction of 1b to give (R)-2b in 88% ee with a specific activity of 300 U g−1 protein. Both NADH and NADPH were found to be the necessary cofactor for the reduction. A-FabI and the partially purified ‘ene’-reductase from Acinetobacter sp. RS1 showed the same enantioselectivity, same cofactor dependence, and similar activity for the reduction, thus A-FabI was identified as the enzyme in Acinetobacter sp. RS1 responsible for the asymmetric reduction of the CC double bond of 1b.
The purified A-FabI was further examined in detail for the reduction of 2-alkylidenecyclopentanones 1a–c, in the presence of NADH or NADPH. As listed in Table 1, A-FabI gave a specific activity of 200–400 U g−1 protein and a conversion of 61–100% for the reduction of 10 mM substrates 1a–c, and it demonstrated high enantioselectivity to give (R)-2a–c in 90–95% ee. For practical applications, we are currently working on the development of immobilized A-FabI coupled with immobilized glucose dehydrogenase (GDH) for in vitro bioreduction with cofactor recycling.
| Enzymea | Sub. | Conc. (mM) | Prod. | Conv. (%) | Spec. Act.b (U g−1) | ee (%) |
|---|---|---|---|---|---|---|
a Biotransformation was conducted in 3 mL of Tris buffer (50 mM; pH = 7.5) containing 3 mg of purified FabI as well as the substrate and NADH in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 at 30 °C and 300 rpm for 1 h.
b Specific activity was determined for the first 30 min reaction and given in U g−1 protein. :1.5 at 30 °C and 300 rpm for 1 h.
b Specific activity was determined for the first 30 min reaction and given in U g−1 protein.
|
||||||
| A-FabI | 1a | 10 | (R)-2a | 63 | 300 | 92 |
| 1a | 30 | (R)-2a | 27 | 400 | 95 | |
| 1b | 10 | (R)-2b | 61 | 300 | 88 | |
| 1b | 30 | (R)-2b | 31 | 300 | 90 | |
| 1c | 10 | (R)-2c | 100 | 200 | 90 | |
| 1c | 20 | (R)-2c | 75 | 200 | 89 | |
| E-FabI | 1a | 40 | (R)-2a | 100 | 800 | 79 |
| 1a | 50 | (R)-2a | 72 | 1200 | 81 | |
| 1b | 40 | (R)-2b | 100 | 1200 | 70 | |
| 1b | 50 | (R)-2b | 81 | 1500 | 70 | |
| 1c | 15 | (R)-2c | 100 | 600 | 76 | |
| 1c | 30 | (R)-2c | 53 | 900 | 75 | |
Inspired by the discovery of A-FabI, another FabI enzyme was searched for the same type of reductions. The FabI enzyme (E-FabI) from Escherichia coli was found to show 60% sequence identity with A-FabI, and it was thus cloned, overexpressed in E. coli, and purified (ESI†). The purified E-FabI was found to catalyze the reductions of 1a–c as well (Table 1), in the presence of NADH or NADPH. The reactions were also enantioselective, giving (R)-2a–c in 70–81% ee. Although E-FabI showed lower enantioselectivity than A-FabI, it gave much higher specific activity (900–1500 U g−1 protein) and fully converted 40 mM 1a–b and 15 mM 1c to the corresponding products, respectively, within 1 h. The kinetics of E-FabI-catalyzed reduction of 1a–c was determined (ESI†). The obtained Km (31–49 μM) and kcat (1.2–2.2 s−1) were similar to those for the reduction of the natural enoyl thioester substrates (enoyl-ACP and enoyl-CoA).7 The catalytic efficiency (kcat/Km) of 2.6–6.5 × 104 s−1 M−1 suggested good application potential for E-FabI.
FabI belongs to metal-independent divergent short chain dehydrogenases/reductases. It is a key enzyme in biological type II fatty acid synthesis and has been a major target for anti-bacterial drug design.7,8 The enzyme was firstly discovered from E. coli and more than 60 X-ray structures have been determined for the FabI enzymes from 10 different sources. So far the natural substrates (enoyl-ACP and enoyl-CoA) are the only reported substrates for the enzyme.7 In contrast to the FMN-mediated trans-addition of OYEs,1c the catalysis of FabI does not require any prosthetic group. A hydride from the cofactor and a proton are directly added to the CC double bond via cis-addition.9 The natural substrates of FabI are enoyl thioesters with an ACP or a CoA moiety as the directing group in the enzyme active site. It was believed that the absence of such a directing group would eliminate the catalytic activity.7 Meanwhile, the thioester group of the substrates forms hydrogen bonds within the active site to enhance the substrate binding.7,9 However, non-natural substrates 1a–c have no such functional groups. Molecular docking of 1a to the homology structure model of A-FabI and the X-ray structure of E-FabI gave some insight into the binding of the substrates in the enzyme active site. As shown in Fig. 1, A-FabI and E-FabI have very similar binding pockets despite of only 60% sequence identity. 1a adopted a similar ‘U-shape’ conformation as that recorded for the natural C16 substrate in the crystal structure of the FabI from Mycobacterium tuberculosis.10 Due to the hydrophobic interaction, the side chain of 1a settles down in a hydrophobic pocket consisting of Leu102, Met 156, Ile203, Phe206 and Met 209 in A-FabI (Fig. 1a). The carbonyl group of 1a forms a hydrogen bond with Tyr159, which could stabilize the catalytic intermediate.9 The plane of the conjugated CO and CC double bonds of 1a is parallel to the nicotinamide ring of the cofactor NAD+/NADH. A hydride is to be transferred from the reduced cofactor to the Cβ of the CC double bond with a distance of 4.1 Å (Fig. 1a), while a proton from the solvent is to be added to the Cαvia the reported cis-addition9 to give (R)-2a. This explains the enantioselectivity outcome. The cis-addition of the enzyme is in contrast to the well-known trans-addition of OYEs.1c Similar explanations can be drawn from the active binding pose of 1a in E-FabI (Fig. 1b) and of other substrates 1b–c in A-FabI and E-FabI, respectively (ESI†). No other alternative active substrate pose was generated due to the incompatible hydride transfer distance (>5.0 Å) in all docking cases of 1a–c. Moreover, no alternative substrate pose that leads to the (S)-products was found in the docking of 1a and b in A-FabI and E-FabI as well as 1c in A-FabI. Docking of 1c in E-FabI gave a substrate pose that would lead to the formation of (S)-2c. However, the distance for hydride transfer from the cofactor to the CC double bond was 5.6 Å, which indicates a non-active pose (ESI†).
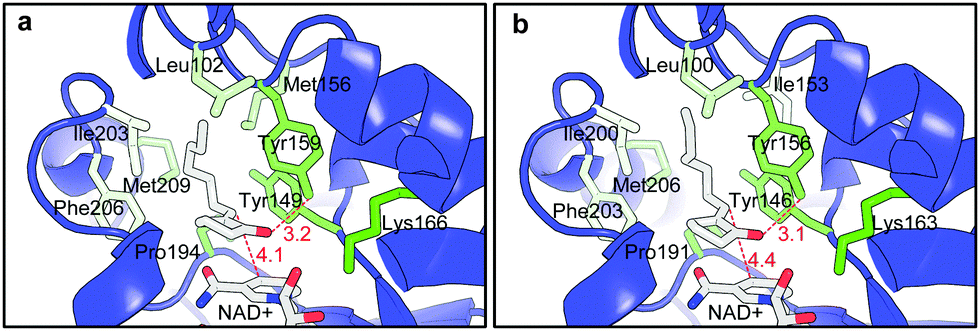 | ||
| Fig. 1 Active docking pose of 1a onto (a) the structure model of A-FabI and (b) the X-ray structure of E-FabI co-crystallized with NAD+ (1MFP in PDB database). The color of the side chains of the amino acid residues indicates their degree of hydrophobicity from green (hydrophilic) to pale green (hydrophobic). | ||
To enhance the product ee from E-FabI-catalyzed reduction, methods such as protein engineering could be applied.11 Here we developed a cascade biocatalysis to improve the product ee by introducing a subsequent S-enantioselective Baeyer–Villiger oxidation in one pot to convert the minor S-enantiomer of ketones 2a–d into the corresponding lactones 3a–d (Scheme 2). E. coli (E-FabI–GDH) co-expressing E-FabI and GDH was engineered as a whole-cell biocatalyst for the reduction of 1a–d with co-factor regeneration, and E. coli (CHMO–GDH) co-expressing cyclohexanone monooxygenase (CHMO) and GDH was constructed as the second whole-cell biocatalyst for the Baeyer–Villiger oxidation of 2a–d with co-factor regeneration as well. The cascade reduction and oxidation were conducted in a sequential manner to avoid the oxidation of 1a–d with CHMO. As shown in Table 2, cascade reduction and oxidation of 40 mM 1a–c gave (R)-2a–c in 99–98% ee and 89–72% analytical yield. In the case of 1d, the reduction with E-FabI showed low enantioselectivity to give (R)-2d in only 35% ee. Nevertheless, by introducing the subsequent Baeyer–Villiger oxidation, (R)-2d was obtained in 92% ee. For all these cascade reactions, the produced (R)-2a–d could be easily separated from the mixtures by fractional distillation due to the significant difference of the boiling points of ketones and lactones according to a previous study.6a
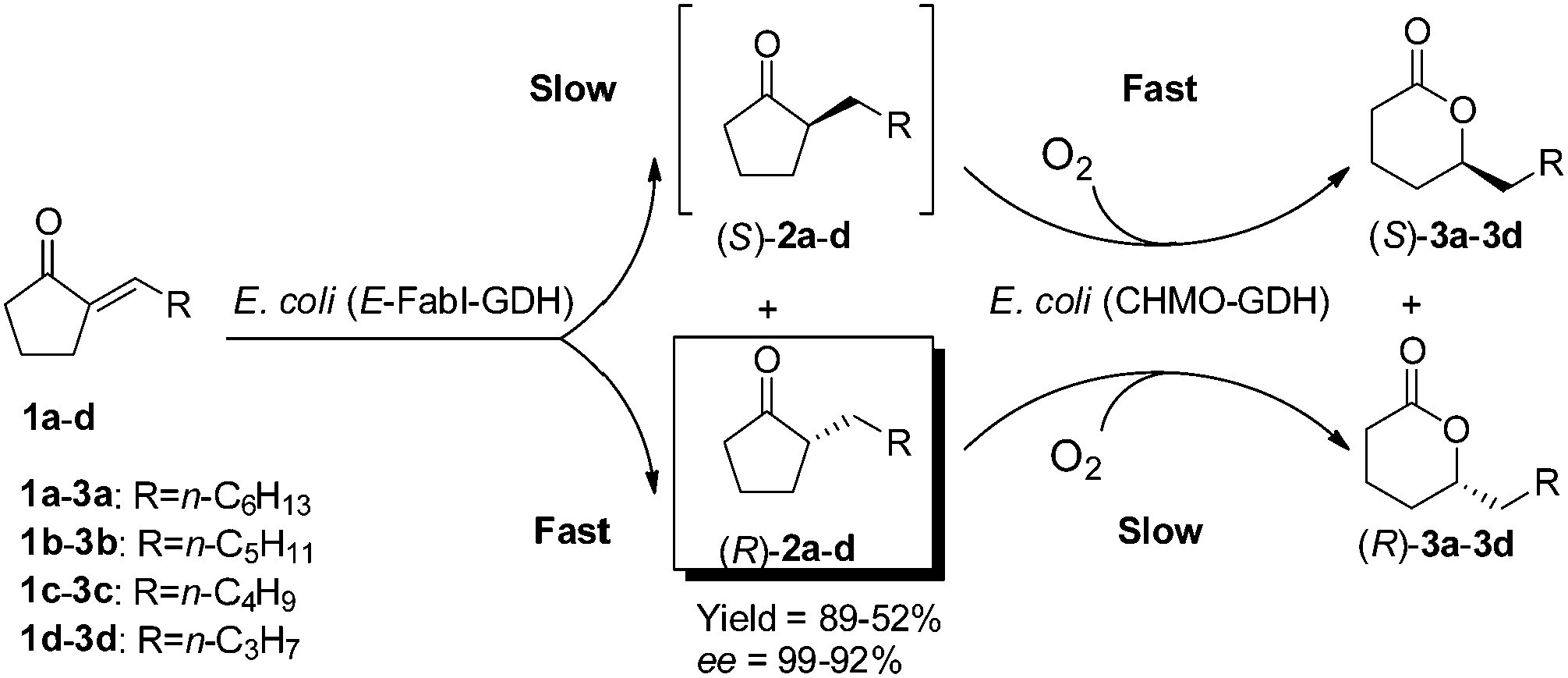 | ||
| Scheme 2 One-pot sequential cascade biotransformation of 2-alkylidenecyclopentanones 1a–dvia E-FabI-catalyzed R-enantioselective reduction and CHMO-catalyzed S-enantioselective Baeyer–Villiger oxidation to prepare (R)-2-alkyl-cyclopentanones 2a–d in high ee. | ||
| Sub. | Conc. (mM) | Catalyst Aa | Vol. (mL) | Time (min) | Catalyst Bb | Vol.c (mL) | Time (min) | Prod. | Yieldd (%) | ee (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Cell density of 20 g cdw L−1. b Catalyst B was added after the first step reduction. c The volume of the buffer containing catalyst B (8 g cdw L−1) added after the first step reduction. d Analytical yield. | ||||||||||
| 1a | 40 | E. coli (E-FabI–GDH) | 5 | 60 | E. coli (CHMO–GDH) | 1 | 60 | (R)-2a | 89 | 99 |
| 1b | 40 | E. coli (E-FabI–GDH) | 5 | 60 | E. coli (CHMO–GDH) | 1 | 60 | (R)-2b | 82 | 98 |
| 1c | 40 | E. coli (E-FabI–GDH) | 5 | 60 | E. coli (CHMO–GDH) | 5 | 60 | (R)-2c | 78 | 99 |
| 1d | 30 | E. coli (E-FabI–GDH) | 5 | 180 | E. coli (CHMO–GDH) | 5 | 60 | (R)-2d | 52 | 92 |
The time course of the one-pot sequential cascade biotransformation of 1a to produce (R)-2a in high ee is shown in Fig. 2. The product concentration in the first reduction step with the resting cells of E. coli (E-FabI–GDH) increased linearly within 50 min, and all substrate 1a was fully converted to (R)-2a in 80% ee after 50 min. In the subsequent oxidation with the resting cell of E. coli (CHMO–GDH), (S)-2a was rapidly oxidized to (S)-3a due to the S-enantioselectivity of CHMO. Consequently, the ee of (R)-2a increased with the time and reached 99% at 45 min. 89% analytical yield was achieved at 1 h. Similar reaction courses were also observed for the cascade biotransformation of 1b–d with the same catalysts (ESI†).
 | ||
| Fig. 2 Time course of the one-pot sequential cascade biotransformation of 2-heptylidenecyclopentanone 1a to produce (R)-2-heptylcyclopentanone 2a in high ee. (a) Reduction of 1a with E. coli (E-FabI–GDH). (b) Oxidation of 2a with E. coli (CHMO–GDH). | ||
In conclusion, enoyl-ACP reductase (FabI), known for its role in cellular fatty acid synthesis, was discovered for the first time for the asymmetric reduction of CC double bonds of non-natural substrates. As a non-OYE reductase A-FabI showed different substrate specificity to OYEs, reducing 2-alkylidene-cyclopentanones 1a–c to give (R)-2-alkylcyclopentanones 2a–c in 95–90% ee with high activity. E-FabI was also identified as a non-OYE reductase to catalyze the asymmetric reduction of 1a–c, giving (R)-2a–c in 81–70% ee and showing even higher activity. Although the enantioselectivity of E-FabI is not excellent for these reductions, the product ee could be simply improved to 99–98% in 89–78% yield by introducing a subsequent S-enantioselective CHMO-catalyzed oxidation in one pot. Docking of substrates 1a–c onto the structural model of A-FabI and the X-ray structure of E-FabI revealed similar binding patterns to that of natural thioester substrates in the FabI of M. tuberculosis, although 1a–c do not contain the binding structure motifs of the natural substrates. Analysis of the active docking pose gave insight into the R-enantioselectivity. The discovery of FabI reductase for asymmetric reduction of CC doubles bond of non-natural substrates could expand the toolbox for this challenging type of bioreduction in asymmetric synthesis. Currently we are developing immobilized FabI and GDH for in vitro reduction with cofactor regeneration as well as recombinant biocatalyst co-expressing A-FabI and GDH using a suitable host for whole-cell biotransformation. Further development of new FabI via protein engineering and/or enzyme evolution to achieve higher enantioselectivity and different substrate specificity is under way.
This work was supported by the Science & Engineering Research Council of A*STAR, Singapore, through a research grant (project No. 1021010026).
Notes and references
- (a) R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 CrossRef CAS PubMed; (b) C. K. Winkler, G. Tasnadi, D. Clay, M. Hall and K. Faber, J. Biotechnol., 2012, 162, 381–389 CrossRef CAS PubMed; (c) H. S. Toogood and N. S. Scrutton, Curr. Opin. Chem. Biol., 2014, 19, 107–115 CrossRef CAS PubMed.
- (a) Y. Yanto, C. K. Winkler, S. Lohr, M. Hall, K. Faber and A. S. Bommarius, Org. Lett., 2011, 13, 2540–2543 CrossRef CAS PubMed; (b) Y. A. Pompeu, B. Sullivan, A. Z. Walton and J. D. Stewart, Adv. Synth. Catal., 2012, 354, 1949–1960 CrossRef CAS; (c) E. Burda, T. Reß, T. Winkler, C. Giese, X. Kostrov, T. Huber, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2013, 52, 9323–9326 CrossRef CAS PubMed.
- (a) K. Durchschein, S. Wallner, P. Macheroux, W. Schwab, T. Winkler, W. Kreis and K. Faber, Eur. J. Org. Chem., 2012, 4963–4968 CrossRef CAS; (b) D. J. Mansell, H. S. Toogood, J. Waller, J. M. X. Hughes, C. W. Levy, J. M. Gardiner and N. S. Scrutton, ACS Catal., 2013, 3, 370–379 CrossRef CAS.
- (a) F. Rohdich, A. Wiese, R. Feicht, H. Simon and A. Bacher, J. Biol. Chem., 2001, 276, 5779–5787 CrossRef CAS PubMed; (b) M. Gall, M. Thomsen, C. Peters, I. V. Pavlidis, P. Jonczyk, P. P. Grünert, S. Beutel, T. Scheper, E. Gross, M. Backes, T. Geißler, J. P. Ley, J. Hilmer, G. Krammer, G. J. Palm, W. Hinrichs and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2014, 53, 1439–1442 CrossRef CAS PubMed.
- J. Liu and Z. Li, ACS Catal., 2013, 3, 908–911 CrossRef CAS.
- (a) G. Lardelli, V. Lamberti, W. T. Weller and A. P. de Jonge, Recl. Trav. Chim. Pays-Bas, 1967, 86, 481–503 CrossRef CAS; (b) M. A. Ogliaruso and J. F. Wolfe, ed. S. Patai and Z. Rappoport, Synthesis of lactones and lactams, Wiley, Chichester, UK, 1993, ch. 1, pp. 1–268 Search PubMed; (c) T. Yamamoto, M. Ogura, A. Amano, K. Adachi, T. Hagiwara and T. Kanisawa, Tetrahedron Lett., 2002, 43, 9081–9084 CrossRef CAS; (d) L. A. Saudan, C. M. Saudan, C. Debieus and P. Wyss, Angew. Chem., Int. Ed., 2007, 46, 7473–7476 CrossRef CAS PubMed.
- S. Rafi, P. Novichenok, S. Kolappan, X. J. Zhang, C. F. Stratton, R. Rawat, C. Kisker, C. Simmerling and P. J. Tonge, J. Biol. Chem., 2006, 281, 39285–39293 CrossRef CAS PubMed.
- S. W. White, J. Zheng, Y. Zhang and C. O. Rock, Annu. Rev. Biochem., 2005, 74, 791–831 CrossRef CAS PubMed.
- K. L. Fillgrove and V. E. Anderson, Biochemistry, 2000, 39, 7001–7011 CrossRef CAS PubMed.
- D. A. Rozwarski, C. Vilcheze, M. Sugantino, R. Bittman and J. C. Sacchettini, J. Biol. Chem., 1999, 274, 15582–15589 CrossRef CAS PubMed.
- Y. Yang, J. Liu and Z. Li, Angew. Chem., Int. Ed., 2014, 53, 3120–3124 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary Information (ESI) available: Experimental details of enzyme purification, identification, cloning and catalysis; molecular docking and HPLC chromatograms. See DOI: 10.1039/c4cc04150j |
| This journal is © The Royal Society of Chemistry 2014 |