 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn activatable, polarity dependent, dual-luminescent imaging agent with a long luminescence lifetime†
Marcus T. M.
Rood
a,
Maria
Oikonomou
b,
Tessa
Buckle
a,
Marcel
Raspe
c,
Yasuteru
Urano
d,
Kees
Jalink
c,
Aldrik H.
Velders
ab and
Fijs W. B.
van Leeuwen
*ab
aInterventional Molecular Imaging Laboratory, Department of Radiology, Leiden University Medical Center, Leiden, The Netherlands. E-mail: f.w.b.van_leeuwen@lumc.nl
bLaboratory of BioNanoTechnology, Wageningen University, Wageningen, The Netherlands
cDivision of Cell Biology I, Netherlands Cancer Institute, Amsterdam, The Netherlands
dLaboratory of Chemical Biology & Molecular Imaging, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
First published on 4th July 2014
Abstract
In this proof-of-concept study, a new activatable imaging agent based on two luminophores and two different quenching mechanisms is reported. Both partial and total activation of the luminescence signal can be achieved, either in solution or in vitro. Bond cleavage makes the compound suitable for luminescence lifetime imaging.
Luminescence imaging is widely used in molecular cell biology and the technology is more and more explored in the clinical setting e.g. for surgical guidance.1 While the emission of luminophores can be used directly, the (photo)physical interactions between different luminescent compounds also have value in diagnostic applications.2 Uniquely, disturbance of the interaction between a luminophore and a quencher, or chemical modification of a luminophore, can generate disease-specific signals.3 Generally such an activatable approach provides a measure of enzymatic activity.4
Organic dyes, which are most commonly explored as activatable imaging agents, are prone to interferences from autofluorescence. This disturbance can be minimized by tailoring the wavelengths towards the far-red and near-infrared window, where autofluorescence is minimal.5 Alternatively, luminophores may be designed to have a large Stokes shift (>100 nm) to obtain a peak intensity that lies beyond the spectral range where autofluorescence generally occurs.1 The luminescence lifetime may also help to separate exogenous from endogenous signals.6
We reasoned that the specificity of an activatable imaging agent can be improved by exploiting luminescence lifetimes that exceed those of endogenous molecules (0.1–7 ns).7 Added advantages of phosphorescent transition complexes are their high photostability, large Stokes shift, and inability to undergo self-quenching.8 Previously some efforts have been made to use ruthenium or iridium complexes for imaging,5,9 and a few ruthenium complexes have been investigated with a change in the luminescence lifetime (τ > 10 ns).10 Iridium complexes allow two types of quenching: first, Förster Resonance Energy Transfer (FRET) or triplet–triplet energy transfer from an iridium complex to an organic moiety, thereby quenching the iridium-based phosphorescence.11,12 Second, iridium atoms can induce quenching via spin–orbit coupling on other luminophores.12 In contrast to effective distances in FRET (up to 10 nm),13 distances in spin–orbit coupling effects are confined to 1 nm.14 Although spin–orbit coupling is considered a drawback in the efficiency of LEDs containing transition metals,15 we aim to exploit this effect, in combination with FRET, as a basis to generate a new class of activatable imaging agents.
The combination of luminescence signal activation and luminescence lifetime imaging was examined using an Ir(ppy)3 complex and a Cy5 dye; we combined both imaging strategies in a dual-luminescent imaging agent.
After synthesis of the Ir(ppy)3-complex (1), a suitable linker was attached (2–3). Excess linker was used to minimize dimer formation (Scheme 1, see ESI† for the detailed Experimental section). This resulted in yields of 42% (2) and 56% (3). Cy5 was chosen as a FRET acceptor due to its spectral overlap with Ir(ppy)3 (Fig. 1A) and high extinction coefficient (ε = 2.5 × 105). Compounds 5 and 6 were made to provide both a stable and an activatable derivative of the conjugate. Conjugation with Cy5 (4) was achieved using standard peptide coupling chemistry. After purification, in both cases a blue/green solid was obtained in yields of 64% (5) and 33% (6).
 | ||
| Scheme 1 Synthesis of Ir(ppy)3-Cy5 compounds using different linkers. | ||
 | ||
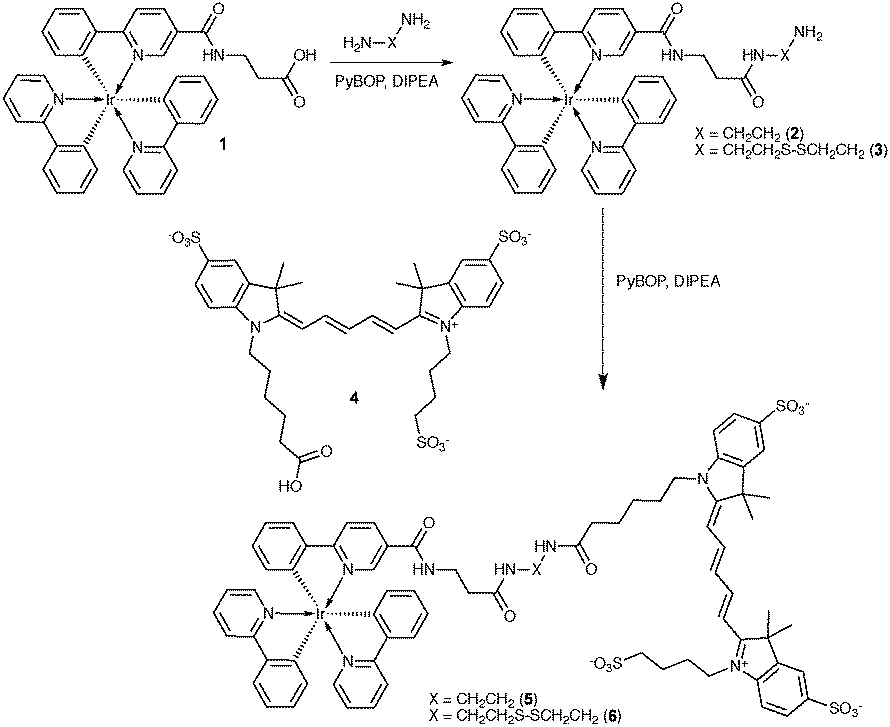
| Fig. 1 (A) Normalized absorption and emission spectra of Ir(ppy)3 and Cy5 in PBS showing the spectral overlap. (B) Emission of equimolar solutions of 1, 5 and 6 with 457 nm excitation and (C) emission of equimolar solutions of 4, 5 and 6 with 627 nm excitation in PBS. | ||
When Ir(ppy)3 (1) is excited, it undergoes rapid inter-system crossing to a triplet excited state and from there emits phosphorescent light (ε = 9 × 103; Φ = 0.12 (DMSO)).16 FRET from Ir(ppy)3 to Cy5 prevented Ir(ppy)3 emission in 5 and 6 in phosphate buffered saline (PBS) (Fig. 1B). Independent of the solvent, the Ir(ppy)3 emission remained fully quenched indicating that energy transfer occurs from Ir(ppy)3 to the Cy5 singlet excited state by FRET; we calculated the Förster distance between these luminophores to be 4.8 nm (see ESI,† p. 5).17 The donor–acceptor distances in our compounds fall well within this distance range, allowing for efficient quenching.
The spin–orbit coupling induced by the iridium atom was used to quench the emission of Cy5 efficiently (Fig. 1C) by allowing energy transfer from the singlet excited state of Cy5 to a non-emissive triplet excited state of Cy5 (Fig. S1, ESI†). Regardless of the excitation wavelength (405 or 633 nm), at 77 K the emission spectra of 5 showed two peaks at 760 and 840 nm (Fig. S3, ESI†). These peaks correspond to the previously reported triplet state emission of Cy5.18 In 5 and 6 the emission of both luminophores is substantially quenched (>99.7%), hence we state that in PBS the luminescence is in the off-state when the luminophores are conjugated.
The difference in distance dependence between the two quenching mechanisms was used to largely mitigate spin–orbit coupling, while leaving FRET intact. Using MeOH as co-solvent increased the solvation of 5 and 6 and this resulted in Cy5 singlet emission at 670 nm upon excitation of Ir(ppy)3 (Fig. S2, ESI†). In the absorption spectra of Cy5, changing to a more apolar solvent led to a decrease in the peak (610 nm) that indicates stacking interactions (Fig. S4B, ESI†). This conformational change resulted in a 30-fold increase of Cy5 fluorescence intensity (Fig. S4C, ESI†), while the Ir(ppy)3 emission remained quenched. The rotational freedom of the molecules, however, seems to prevent complete signal restoration. The triplet emission caused by spin–orbit coupling in 5 (observed at 77 K in H2O) disappeared upon reduction in polarity, also indicating a change in the interaction between Cy5 and the Ir atom (Fig. S3A, ESI†). Lastly ROESY spectra in CD3OD did not reveal close distance correlations between Cy5 and Ir(ppy)3 (ESI,† Appendix).
Similar to the use of MeOH, micelles of SDS were able to increase the Cy5 luminescence intensity, providing a model system for interactions with the cell membrane. Only above the critical micelle concentration of 1 mM (ref. 19) an increase of Cy5 fluorescence intensity was observed (Fig. S5, ESI†). In line with these findings, interaction of 5 (the uncleavable derivative; Fig. 2) or 6 (Fig. S6, ESI†) with cell membranes provided detectable Cy5 emission; Ir(ppy)3 emissions remained quenched.
 | ||
| Fig. 2 (A) Schematic representation of partial activation by the conformational change of the probe. (B–D) Confocal microscope image of 4T1 cells after 1 h incubation with 5 at 4 °C. (B) Differential interference contrast, (C) Cy5, (D) overlay. | ||
In 6, the quenching of both Ir(ppy)3 and Cy5 can be fully undone by cleavage of the connective bond between the two dyes (Fig. 3A). To study the full activation, the disulfide bond of 6 (a model system for cleavage) was initially cleaved using cysteamine (Scheme S2, ESI†). In MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PBS 4:1, after 1 h at RT, the cleavage reaction induced by an excess of cysteamine yielded an intensity increase of both Ir(ppy)3 phosphorescence (100-fold increase, Fig. 3C) and Cy5 fluorescence (a further 3-fold increase on the 30-fold increase caused by the solvent, making a total 90-fold, Fig. 3D). In PBS a 200-fold increase of Cy5 fluorescence intensity was observed (Fig. S7, ESI†). After cleavage the end products were analyzed using HPLC and mass spectrometry (Fig. S8 and S9, ESI†).
:PBS 4:1, after 1 h at RT, the cleavage reaction induced by an excess of cysteamine yielded an intensity increase of both Ir(ppy)3 phosphorescence (100-fold increase, Fig. 3C) and Cy5 fluorescence (a further 3-fold increase on the 30-fold increase caused by the solvent, making a total 90-fold, Fig. 3D). In PBS a 200-fold increase of Cy5 fluorescence intensity was observed (Fig. S7, ESI†). After cleavage the end products were analyzed using HPLC and mass spectrometry (Fig. S8 and S9, ESI†).
 | ||
| Fig. 3 (A) Schematic representation of disulfide cleavage, (B) difference in luminescence decay before and after cleavage in solution, (C) increase of luminescence of Ir(ppy)3 and (D) Cy5 upon cleavage. Insets in (C) and (D) show the change in peak height over time. | ||
In vitro evaluation of the disulfide bond cleavage was performed in a 4T1 murine breast tumor cell line. After passive cellular internalization at 37 °C, Ir–S–S–Cy5 (6) was confined in the lysosomes of the cell. Here the disulfide bond was reduced by a redox enzyme or an intracellular thiol.20 We observed activation of both Ir(ppy)3 and Cy5 in the lysosomes (Fig. 4 and Fig. S10, ESI†) and even when a high concentration (5 μM) of 6 surrounded the cells, only the cleaved components were visible (Fig. S11, ESI†). No Ir(ppy)3 signal activation was observed when cells were incubated at 37 °C with 5 (Fig. S12, ESI†).
 | ||
| Fig. 4 Confocal microscope images of 4T1 cells after 24 h incubation with 6 at 37 °C. (A) Cy5 in red, (B) Ir(ppy)3 in green, (C) differential interference contrast, (D) overlay of all channels. Yellow indicates overlay of red and green. | ||
Luminescence lifetime measurements showed minor differences between the iridium complexes 1, 2, and 3 (Table 1). Conjugation with Cy5 (4) drastically shortened the lifetime. A short-lived species (τ < 1 ns), representative of residual Cy5 emission, accounted for a large part of the total emission at 600 nm; 89% in 5 and 95% in 6 (Fig. S13, ESI†). After cleavage, there is a relatively large increase of long-lifetime emission (Fig. 3B), indicating re-activation of Ir(ppy)3 phosphorescence. Activation effects observed in solution were confirmed in vitro by fluorescence lifetime imaging microscopy (FLIM) using two different cell lines, 4T1 and U2OS. Results were independent of the cell type (Fig. 5 and Fig. S14, ESI†). With 5 (uncleavable), the lifetime was short, while after activation of 6 (cleavable) the lifetime increased to 90 ns, similar to control experiments with the same Ir(ppy)3 complex without Cy5 (Fig. S14A, ESI†). The change in the lifetime seen with FLIM provides a clear measure of the in vitro activation. Unfortunately, the suboptimal filter cube in FLIM also allowed for some background emission, giving an average of the lifetime signal. A time-gated approach, which was not possible in our set-up, might prevent this.
:PBS 4:1
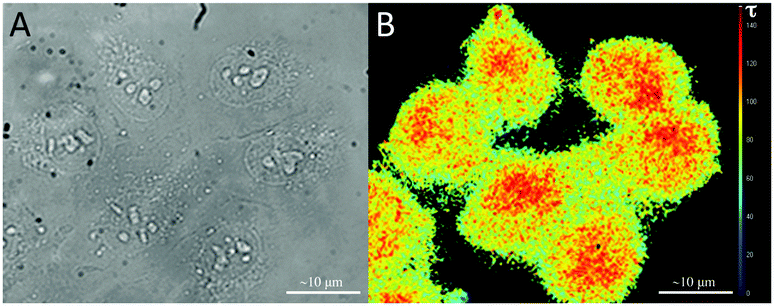 | ||
| Fig. 5 Fluorescence lifetime microscopy images of U2OS cells after 24 h incubation with 6 at 37 °C using a CFP/YFP filter cube (excitation 436/12 and 500/20, dichroic 445 and 515, emission 467/37 and 545/45). (A) Transmission, (B) lifetime, the scale of the lifetimes is depicted on the right side (0–150 ns). | ||
The lifetime technology proves to be a promising tool to analyze variations in cellular function related to disease progression.7,21 The here-described disulfide cleavage procedure is not disease specific, every cell is able to cleave disulfide bonds,20 but it can act as a model system. In the future, activatable lifetime imaging agents derived from this system can in theory be used to detect expression levels of disease-related enzymes. When more disease-specific probes for FLIM are available, the scope of lifetime imaging may be expanded from in vitro to in vivo applications and maybe even to applications in image guided surgery.22
To conclude, two different quenching mechanisms were used to generate a dual-luminescent activatable (long) lifetime imaging agent based on Cy5 and Ir(ppy)3 (6). In our view activatable long lifetime imaging agents provide a promising tool for future molecular imaging applications related to disease progression, which can be complementary to intensity-based fluorescence detection.
This research was supported by an NWOnano-Grant (STW 11435; F.v.L.). The authors would like to thank A. Bunschoten, D. van Willigen and P. Steunenberg for kindly supplying compounds. We also thank P. Navarro, P. Chin, S. van der Wal and H. Tanke for fruitful discussion.
Notes and references
- P. T. K. Chin, M. M. Welling, S. C. J. Meskers, R. A. V. Olmos, H. Tanke and F. W. B. van Leeuwen, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, 1283 CrossRef CAS PubMed.
- R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123 CrossRef CAS PubMed.
- Y. Urano, Curr. Opin. Chem. Biol., 2012, 16, 602 CrossRef CAS PubMed; J. F. Lovell and G. Zheng, J. Innovative Opt. Health Sci., 2008, 1, 45 CrossRef.
- C. W. Huang, Z. B. Li and P. S. Conti, Bioconjugate Chem., 2012, 23, 2159 CrossRef CAS PubMed; C. H. Tung, U. Mahmood, S. Bredow and R. Weissleder, Cancer Res., 2000, 60, 4953 Search PubMed; Y. Urano, M. Sakabe, N. Kosaka, M. Ogawa, M. Mitsunaga, D. Asanuma, M. Kamiya, M. R. Young, T. Nagano, P. L. Choyke and H. Kobayashi, Sci. Transl. Med., 2011, 3, 110ra119 Search PubMed.
- G. Zhang, H. Zhang, Y. Gao, R. Tao, L. Xin, J. Yi, F. Li, W. Liu and J. Qiao, Organometallics, 2014, 33, 61 CrossRef CAS.
- R. Alford, M. Ogawa, M. Hassan, A. H. Gandjbakhche, P. L. Choyke and H. Kobayashi, Contrast Media Mol. Imaging, 2010, 5, 1 CAS.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641 CrossRef CAS PubMed.
- A. Ruggi, C. Beekman, D. Wasserberg, V. Subramaniam, D. N. Reinhoudt, F. W. B. van Leeuwen and A. H. Velders, Chem. – Eur. J., 2011, 17, 464 CrossRef CAS PubMed.
- P. Steunenberg, A. Ruggi, N. S. van den Berg, T. Buckle, J. Kuil, F. W. B. van Leeuwen and A. H. Velders, Inorg. Chem., 2012, 51, 2105 CrossRef CAS PubMed; L. Murphy, A. Congreve, L.-O. Palsson and J. A. G. Williams, Chem. Commun., 2010, 46, 8743 RSC; L. Xiong, Q. Zhao, H. Chen, Y. Wu, Z. Dong, Z. Zhou and F. Li, Inorg. Chem., 2010, 49, 6402 CrossRef PubMed; G. Li, Y. Chen, J. Wu, L. Ji and H. Chao, Chem. Commun., 2013, 49, 2040 RSC; A. Ruggi, F. W. B. van Leeuwen and A. H. Velders, Coord. Chem. Rev., 2011, 255, 2542 CrossRef PubMed.
- W. Zhong, P. Urayama and M. A. Mycek, J. Phys. D: Appl. Phys., 2003, 36, 1689 CrossRef CAS; E. Baggaley, M. R. Gill, N. H. Green, D. Turton, I. V. Sazanovich, S. W. Botchway, C. Smythe, J. W. Haycock, J. A. Weinstein and J. A. Thomas, Angew. Chem., Int. Ed., 2014, 53, 3367 CrossRef PubMed.
- H. Y. Shiu, M. K. Wong and C. M. Che, Chem. Commun., 2011, 47, 4367 RSC.
- R. D. Costa, F. J. Cespedes-Guirao, H. J. Bolink, F. Fernandez-Lazaro, A. Sastre-Santos, E. Orti and J. Gierschner, J. Phys. Chem. C, 2009, 113, 19292 Search PubMed; A. A. Rachford, R. Ziessel, T. Bura, P. Retailleau and F. N. Castellano, Inorg. Chem., 2010, 49, 3730 CrossRef CAS PubMed.
- E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387 CrossRef CAS PubMed.
- M. Rae, A. Fedorov and M. N. Berberan-Santos, J. Chem. Phys., 2003, 119, 2223 CrossRef CAS PubMed.
- C. Rothe, S. King and A. Monkman, Nat. Mater., 2006, 5, 463 CrossRef CAS PubMed.
- J. Kuil, P. Steunenberg, P. T. K. Chin, J. Oldenburg, K. Jalink, A. H. Velders and F. W. B. van Leeuwen, ChemBioChem, 2011, 12, 1896 Search PubMed.
- G. Horvath, M. Petras, G. Szentesi, A. Fabian, J. W. Park, G. Vereb and J. Szollosi, Cytometry, Part A, 2005, 65A, 148 CrossRef CAS PubMed.
- Z. X. Huang, D. M. Ji, A. D. Xia, F. Koberling, M. Patting and R. Erdmann, J. Am. Chem. Soc., 2005, 127, 8064 CrossRef CAS PubMed.
- R. J. Williams, J. N. Phillips and K. J. Mysels, Trans. Faraday Soc., 1955, 51, 728 RSC.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199 CrossRef CAS.
- W. Becker, J. Microsc., 2012, 247, 119 CrossRef CAS PubMed.
- Y. Sun, J. E. Phipps, J. Meier, N. Hatami, B. Poirier, D. S. Elson, D. G. Farwell and L. Marcu, Microsc. Microanal., 2013, 19, 791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, in vitro studies, Förster distance calculation, and supplementary images. See DOI: 10.1039/c4cc04015e |
| This journal is © The Royal Society of Chemistry 2014 |